DOI: 10.32604/CHD.2021.014409
ARTICLE
Kabuki-Syndrome and Congenital Heart Disease—A Twenty-Year Institutional Experience
1Baylor College of Medicine, Houston, USA
2Section of Critical Care Medicine, Department of Pediatrics, Baylor College of Medicine, Texas Children’s Hospital, Houston, USA
3Division of Congenital Heart Surgery, Texas Children’s Hospital, Department of Surgery, Baylor College of Medicine, Houston, USA
4Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, USA
*Corresponding Author: Reghan Conrey. Email: conrey@bcm.edu
Received: 24 September 2020; Accepted: 17 November 2020
#Anders and McKenzie share senior authorship of this paper
Abstract: Background: Patients with genetic syndromes who undergo surgery to correct congenital heart defects can be at risk for increased morbidity or mortality. Surgical outcomes and postoperative courses following congenital heart surgery in patients with Kabuki-Syndrome (KS) have not been well studied. Objectives: The purpose of this study was to describe the postoperative courses and associated outcomes in the largest set of KS patients undergoing congenital heart surgery to date. Methods: Patients with a confirmed molecular diagnosis of KS and a diagnosis of a CHD admitted to Texas Children’s Hospital between January 1, 2000 and January 1, 2020 were included (n = 20). Demographics and medical histories were collected from the hospitals’ electronic health records. Results: Of 20 patients identified with KS and a CHD, 15 required surgical correction of their congenital cardiac malformation. Median age and weight at the time of surgery was 2 months and 4.1 kg, respectively. Median duration of hospital stay was 49 days for all surgeries and 151 days for the Norwood procedure. Postoperative infections and pleural effusions were detected and treated in 45.8% and 50% of patients, respectively. There was no in-hospital mortality for any surgery. Median follow up time was 5.6 years; survival at 6 years was 94%. Conclusions: Although KS patients seem to be at increased risk for a more complicated, prolonged postoperative course than that of patients without a genetic syndrome, patients with a diagnosis of a CHD and KS do not appear to be at increased risk of mortality following congenital heart surgery.
Keywords: Kabuki syndrome; genetic disorder; congenital heart disease; survival; outcome; pediatric
Congenital heart disease is often accompanied by concurring genetic syndromes that present with a constellation of extracardiac findings [1,2]. Populations with well described genetic syndromes such as Turner (TS), Noonan, DiGeorge Syndrome (DGS), and William’s syndromes have shown an increased risk for developing complicated postoperative courses after undergoing congenital heart surgery [3–5]. Less well described, Kabuki syndrome (KS) is a rare, heterogeneous, multiple congenital malformation syndrome caused by a heterozygous mutation in the KMT2D (KS-1) or KDM6A (KS-2) with variable phenotypic expression. It is characterized by mild-to-moderate intellectual delay, skeletal anomalies, and distinctive facies [6,7]. KS has been associated with an increased risk for development of feeding difficulties, autoimmune disorders, and immunodeficiency, and an increased susceptibility to respiratory infections [8–12]. Patients with KS also frequently present with a variety of skeletal and visceral anomalies including growth deficiency, cleft palate, renal and hepatic anomalies, anorectal malformations, and congenital heart defects (CHDs) [10,13–17].
Cardiac anomalies described in children with KS include left sided obstructive lesions such as aortic coarctation, bicuspid aortic valve, aortic and/or mitral stenosis, and hypoplastic left heart syndrome. These account for about 47% of CHDs in patients with KS [18–20]. Less frequently, patients are diagnosed with atrial and ventricular septal defects, patent ductus arteriosus, or patent foramen ovale [9,10,19,21]. Although these patients often require corrective surgery for their cardiac anomalies, the outcomes and postoperative course of patients with KS have not been thoroughly studied due to the rare incidence [22].
Against this background, we describe all patients with genetically proven KS and diagnosed CHDs at Texas Children’s Hospital over the course of twenty years. We describe the surgical and postoperative courses for the largest cohort to date of patients with KS undergoing congenital heart surgery.
This study was conducted as a single-center retrospective review at Texas Children’s Hospital, Houston, TX, USA. Patients were identified by an internal electronic query through the internal Heart Center database for all patients registered under a diagnosis code for KS from January 1, 2000, to January 1, 2020. Those without molecular testing to confirm the clinical suspicion of KS were excluded. As the genetic basis for KS was first identified in 2010, a number of these molecular diagnoses occurred several years after patients underwent surgery. Patients who appeared in the query but had received their cardiac surgery or postoperative care at another institution were also excluded.
We included demographic data (age, race, sex) and pre-partum and peri-partum histories. Additionally, we described information regarding the clinical presentation of non-cardiac abnormalities and the specific CHDs. For patients with CHDs, we compiled their surgical and interventional treatments and postoperative data, using preselected variables to accurately reflect the complications faced during the postoperative course. Subsequent to hospital discharge, complications other than mortality were not considered.
The Institutional Review Board at Baylor College of Medicine approved this study; informed consent was waived.
Patients were grouped by cardiac anatomic lesions and congenital cardiac surgery. Norwood and Glenn procedure subgroups included patients undergoing first and second stage palliation for Hypoplastic Left Heart Syndrome (HLHS). The “coarctation repair” group included patients undergoing coarctation repair with extended end-to-end anastomosis (n = 3), repair of aortic arch using aortic arch advancement (n = 2), and repair of residual coarctation (n = 1). Interventional surgeries that did not fall into the above categories are categorized as “other.”
For this study, pleural effusion and duration of pleural effusion are defined by moderate to severe pleural effusion reported by radiologists. Similarly, pericardial effusion was defined as moderate to severe pericardial effusion observed by cardiologists on echocardiogram. We defined glycemic homeostasis disturbance as a sudden fluctuation in blood glucose levels noted by intensivists.
Continuous values are reported as median followed by the range, minimum and maximum, in brackets. Discrete values are reported as the number of patients followed by a frequency, in parentheses. We used Kaplan-Meier estimators for survival analysis [23].
We identified 20 patients with genetically confirmed KS and concurrent CHD at our institution. The demographic distribution, clinical features, and pregnancy course of these 20 patients are described in (Tab. 1). The patient population consisted of an even distribution of males and females (11/9 = 55/45%) and most patients were white (17/20 = 85%). While receiving antenatal care, 7/20 (35%) patients were diagnosed with a cardiac malformation via ultrasound. The median birth weight was 2.93 kg [1.8–4.0 kg]. Patients demonstrated characteristic KS facies (20/20 = 100%), mild to moderate developmental delay (20/20 = 100%), immunodeficiency (15/20 = 75%), and gastrointestinal anomalies such as GERD and a persistent inability to feed (19/20 = 95%), for which 16 patients (80%) required a gastrostomy tube. Less frequently, these patients presented with renal displacement (7/20 = 35%), cleft palate (5/20 = 25%), and the hearing deficiency (9/20 = 45%).
Table 1: Demographics, clinical features, pregnancy course, and surgical interventions in all 20 patients shown to have Kabuki syndrome by genetic testing and a confirmed CHD diagnosis (n = data points recorded)


APGAR = appearance, pulse, grimace, activity, and respiration; ASD = atrial septal defect; CoA = coarctation of the aorta; CMP = cardiomyopathy; GERD = gastroesophageal reflux disease; PDA = patent ductus arteriosus; PFO = patent foramen ovale; VSD = ventricular septal defect
The most frequent CHDs identified were HLHS (5/20 = 25%), coarctation of the aorta with or without an additional defect (5/20 = 25%), and Shone’s complex (3/20 = 15%). All but five patients with CHDs underwent palliative or corrective congenital heart surgery. Patients who did not undergo surgery did not do so because surgery was not clinically indicated for their condition. These conditions, listed in (Tab. 1), included dilated cardiomyopathy, mildly dysplastic aortic and pulmonary valves, atrial septal defect, and tricuspid valve hypoplasia, as well as a combined condition consisting of a patent ductus arteriosus, patent foramen ovale, and a portosystemic shunt.
Fifteen patients with KS underwent a total of 24 congenital heart surgeries. The demographic distribution, clinical features, and pregnancy course of this subpopulation are described in (Tab. 2). Of patients who required cardiac surgery (15/20 = 75%), eight (53%) underwent only one surgery, four (27%) underwent two surgeries, and three (20%) patients underwent three surgeries. Upon surgery, 11 of the 15 patients (73%) were noted to have especially friable cardiac tissues, and seven (47%) patients were noted to have an abnormally small thymus. The individual surgeries performed are summarized in (Tab. 2).
Table 2: Demographics, clinical features, pregnancy course, and surgical interventions in the 15 patients shown to have Kabuki syndrome by genetic testing who also underwent cardiac surgery (n = data points recorded)
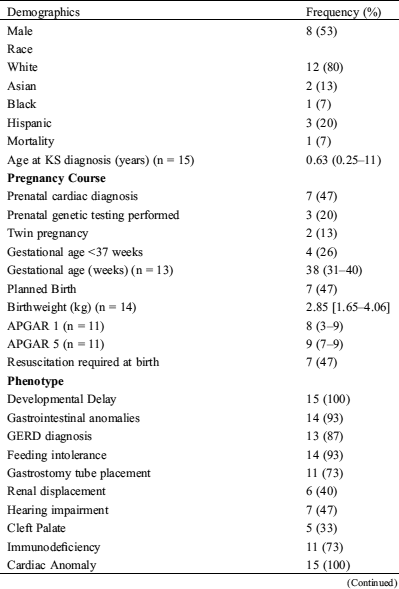

APGAR = appearance, pulse, grimace, activity, and respiration; ASD = atrial septal defect; CoA = coarctation of the aorta; GERD = gastroesophageal reflux disease; PDA = patent ductus arteriosus; PFO = patent foramen ovale; VSD = ventricular septal defect
The postoperative courses following these 24 surgeries are outlined by (Tab. 3). Median age and weight at the time of first surgery were 2 weeks and 3.1 kg, respectively. The median duration of stay in ICU for all surgeries was 39 days, and the median duration of hospital stay was 49 days. However, these values vary widely among the surgeries performed (Tab. 3). The Norwood procedure showed the longest median ICU and hospital stays, with 135 and 151 days, respectively, whereas coarctation repairs showed a median ICU stay of 8 days and a median hospital stay of 13.5 days. Either viral or bacterial infections were detected postoperatively in 45.8% of patients. The coarctation repair group had only one postoperative infection (16.7%); four of five patients who underwent the Norwood procedure developed an infection (80%), three of which were viral and one of which was bacterial. Half of the patients developed pleural effusions postoperatively that persisted for a median duration of 4.5 days, although the duration of pleural effusions following the Norwood procedure was longer at 13.5 days. One patient, previously requiring pulmonary artery banding, required temporary extracorporeal membrane oxygenation for four days due to the inability to wean off cardiopulmonary bypass following Norwood palliation. Only one patient (20%) in our single ventricle cohort underwent unplanned cardiac reintervention following Norwood palliation, requiring Blalock-Taussig shunt revision. On discharge echocardiogram following Glenn procedure, two HLHS patients showed mild tricuspid regurgitation, but no other functional abnormalities were identified. Additionally, 37.5% of patients developed a glycemic homeostasis disturbance during their time in the ICU; these patients were relatively evenly distributed among the surgical groups. Although some postoperative courses were significantly complicated, there was no in-hospital mortality up to the date of data collection, with overall survival of 94% [0.25–19 years]. Median follow up time was 5.6 years, survival at 6 years was 94%. Survival outcomes are further illustrated by (Fig. 1).
Table 3: Surgical variables and postoperative course variables for 15 KS patients undergoing 24 cardiac surgeries

*Missing values were not included in calculations
ACP = antegrade cerebral perfusion; CPB = cardiopulmonary bypass; NIV = non-invasive ventilation

Figure 1: Kaplan-Meier survival curve for 15 KS patients undergoing congenital heart surgery. Median follow up time was 5.6 years
Five of the 15 patients underwent Norwood procedures at a median age of 2 weeks and a median weight of 2.9 kg. There was no inter-stage mortality. Those patients subsequently underwent Glenn procedures at a median age of 6 months and a median weight of 5.6 kg. The median duration of stay in the ICU for patients after the Glenn procedure was 26 days and the median duration of hospital stay was 50 days (Tab. 2). All patients survived to hospital discharge; one patient died at home ten months postoperatively.
Our study is the largest to date of patients with KS undergoing congenital heart surgery. Of 20 patients with a confirmed diagnosis of KS and CHD, 15 required congenital heart surgery. Nine patients underwent only one cardiac surgery; three patients had two surgeries; three patients had 3 surgeries. There was no in-hospital mortality with long-term survival of 94%. Although we experience that these patients had longer postoperative courses than patients without a genetic condition, we did not observe an increased mortality rate.
CHDs are common findings in a variety of genetic syndromes besides KS, including Down Syndrome, TS, and DGS. These groups of patients are reported to have increased postoperative complications, including increased duration of stay, increased rate of infection, and increased duration of mechanical ventilation [4,24–26]. Only one patient had an unplanned cardiac reintervention during their postoperative course, leading us to conclude that residual cardiac lesions did not contribute significantly to their complicated postoperative courses. Patients with Down Syndrome have been found to have relatively similar rates of mortality associated with the Norwood procedure as patients without a genetic syndrome, at 14% [24]. However, patients with TS and DGS were found to have significantly increased morbidity and mortality rates when compared to a population without any chromosomal condition [4,26]. The mortality rate for patients with CHD and DGS is reported at 22.4%; mortality rates for patients with TS and generalized CHD are low, but the mortality rate for patients with TS and HLHS is reportedly as high as 75% after Glenn procedure [26]. Our population had similar rates of complications and longer postoperative courses when compared to these groups; however, the survivability of our patients is very favorable at 94% overall and 80% after undergoing the Glenn procedure.
Patients with KS and HLHS seemed to have longer postoperative courses, although not more complicated by infections or pleural effusions than patients with KS undergoing other congenital heart surgeries. The duration of ICU stay, median 135 days (range 59–221 days), for patients with KS after Norwood procedure is much longer than in infants without a genetic condition, which has been reported as median 9 days [27]. Concurrently, the total hospital length of stay for these patients, median 151 days (range 69–359 days), was significantly longer than the median duration at our institution in 2010, 21 days, for patients without a genetic condition [28]. Additionally, in an unpublished case series examining Norwood palliation in 19 HLHS patients from 2018 to 2019 without any genetic syndromes, Morales-Demori and Anders showed that both ICU (median 47 days) and hospital (median 102 days) length of stay have increased at our institution. Despite this, we observed a longer length of stay in both the ICU and the hospital length of stay in our population with KS. Although one study reports only 21.6% of patients undergoing Norwood procedures requiring intubation longer than 7 days [29], our patients had a median duration of invasive ventilation of 27 days (range 12–207 days). Additionally, 80% of our patients developed either bacterial or viral infections, requiring antibiotic treatment that lasted a median of 125 days (range 2–168 days). Despite the postoperative course being seemingly longer and more complicated than recorded in the literature for patients without a genetic condition, the outcomes for patients with KS in our cohort were excellent. Whereas in-hospital mortality rates in standard patients with HLHS who undergo the Norwood procedure have been reported between 15 and 22% [27,29,30], our patients had no in-hospital or 30-day mortality. After undergoing the Norwood procedure, an estimated 15% of infants without a genetic condition die before the Glenn procedure [31,32]. However, in our cohort, there was no inter-stage mortality. One patient died ten months after undergoing the Glenn procedure. Ultimately, our cohort of patients with KS and HLHS appears to have a lengthier and more complicated postoperative course than patients who have not been diagnosed with a genetic abnormality; nevertheless, survivability in our cohort was very favorable.
Our database review yielded limited information on a number of variables, including the cause of death for our singular patient who died after having a Glenn procedure, duration of use of inotropic agents, duration of chest tube usage, and outcomes after hospital discharge. The small number of patients within our cohort (20 overall, 15 for surgical outcomes), renders it difficult to draw statistically significant conclusions about the relationship between KS and the outcomes we observed. Regardless, we describe the largest number of patients with KS compared to the current literature, in hopes that this information combined with future research can improve the overall prognosis of these patients.
In our cohort, we found that patients with KS appear to be at an increased risk of having a prolonged postoperative course with more complications than patients without any genetic condition. We found that although the length of stay varied widely between subgroups, the rate of complications remained similar amongst patients with KS undergoing different procedures. Ultimately, although morbidities appear higher in our population, patients with a diagnosis of CHD and KS are not at increased risk for postoperative mortality.
Funding Statement: The author(s) received no specific funding for this study.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Natowicz, M., Chatten, J., Clancy, R., Conard, K., Clauser, T. et al. (1988). Genetic disorders and major extracardiac anomalies associated with the hypoplastic left heart syndrome. Pediatrics, 82(5), 698–706. [Google Scholar]
2. Alsoufi, B., McCracken, C., Oster, M., Shashidharan, S., Kanter, K. (2018). Genetic and extracardiac anomalies are associated with inferior single ventricle palliation outcomes. Annals of Thoracic Surgery, 106(4), 1204–1212. DOI 10.1016/j.athoracsur.2018.04.043. [Google Scholar] [CrossRef]
3. Michielon, G., Marino, B., Formigari, R., Gargiulo, G., Picchio, F. et al. (2006). Genetic syndromes and outcome after surgical correction of tetralogy of Fallot. Annals of Thoracic Surgery, 81(3), 968–975. DOI 10.1016/j.athoracsur.2005.09.033. [Google Scholar] [CrossRef]
4. Cramer, J. W., Bartz, P. J., Simpson, P. M., Zangwill, S. D. (2014). The spectrum of congenital heart disease and outcomes after surgical repair among children with turner syndrome: A single-center review. Pediatric Cardiology, 35(2), 253–260. DOI 10.1007/s00246-013-0766-5. [Google Scholar] [CrossRef]
5. Hemmati, P., Dearani, J. A., Daly, R. C., King, K. S., Ammash, N. M. et al. (2019). Early outcomes of cardiac surgery in patients with noonan syndrome. Seminars in Thoracic and Cardiovascular Surgery, 31(3), 507–513. DOI 10.1053/j.semtcvs.2018.12.004. [Google Scholar] [CrossRef]
6. Kuroki, Y., Suzuki, Y., Chyo, H., Hata, A., Matsui, I. (1981). A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. The Journal of Pediatrics, 99(4), 570–573. DOI 10.1016/S0022-3476(81)80256-9. [Google Scholar] [CrossRef]
7. Niikawa, N., Matsuura, N., Fukushima, Y., Ohsawa, T., Kajii, T. (1981). Kabuki make-up syndrome: A syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. Journal of Pediatrics, 99(4), 565–569. DOI 10.1016/S0022-3476(81)80255-7. [Google Scholar] [CrossRef]
8. Bögershausen, N., Wollnik, B. (2013). Unmasking Kabuki syndrome. Clinical Genetics, 83(3), 201–211. DOI 10.1111/cge.12051. [Google Scholar] [CrossRef]
9. Niikawa, N., Kuroki, Y., Kajii, T., Matsuura, N., Ishikiriyama, S. et al. (1988). Kabuki make-up (Niikawa-Kuroki) syndrome: A study of 62 patients. American Journal of Medical Genetics, 31(3), 565–589. DOI 10.1002/ajmg.1320310312. [Google Scholar] [CrossRef]
10. Kawame, H., Hannibal, M. C., Hudgins, L., Pagon, R. A. (1999). Phenotypic spectrum and management issues in Kabuki syndrome. Journal of Pediatrics, 134(4), 480–485. DOI 10.1016/S0022-3476(99)70207-6. [Google Scholar] [CrossRef]
11. Hoffman, J. D., Ciprero, K. L., Sullivan, K. E., Kaplan, P. B., McDonald-McGinn, D. M. et al. (2005). Immune abnormalities are a frequent manifestation of Kabuki syndrome. American Journal of Medical Genetics Part A, 135A(3), 278–281. DOI 10.1002/ajmg.a.30722. [Google Scholar] [CrossRef]
12. Geneviève, D., Amiel, J., Viot, G., Le Merrer, M., Sanlaville, D. et al. (2004). Atypical findings in Kabuki syndrome: Report of 8 patients in a series of 20 and review of the literature. American Journal of Medical Genetics Part A, 129A(1), 64–68. DOI 10.1002/ajmg.a.30144. [Google Scholar] [CrossRef]
13. Ewart-Toland, A., Enns, G. M., Cox, V. A., Mohan, G. C., Rosenthal, P. et al. (1998). Severe congenital anomalies requiring transplantation in children with Kabuki syndrome. American Journal of Medical Genetics, 80(4), 362–367. [Google Scholar]
14. Matsumura, M., Yamada, R., Kitani, Y., Nishi, T., Yamamoto, H. et al. (1992). Anorectal anomalies associated with Kabuki make-up syndrome. Journal of Pediatric Surgery, 27(12), 1600–1602. DOI 10.1016/0022-3468(92)90523-A. [Google Scholar] [CrossRef]
15. Kokitsu-Nakata, N. M., Vendramini, S., Guion-Almeida, M. L. (1999). Lower lip pits and anorectal anomalies in Kabuki syndrome. American Journal of Medical Genetics, 86(3), 282–284. [Google Scholar]
16. Wessels, M. W., Brooks, A. S., Hoogeboom, J., Niermeijer, M. F., Willems, P. J. (2002). Kabuki syndrome: A review study of three hundred patients. Clinical Dysmorphology, 11(2), 95–102. DOI 10.1097/00019605-200204000-00004. [Google Scholar] [CrossRef]
17. Digilio, M. C., Marino, B., Toscano, A., Giannotti, A., Dallapiccola, B. (2001). Congenital heart defects in Kabuki syndrome. American Journal of Medical Genetics, 100(4), 269–274. DOI 10.1002/ajmg.1265. [Google Scholar] [CrossRef]
18. Digilio, M. C., Baban, A., Marino, B., Dallapiccola, B. (2010). Hypoplastic left heart syndrome in patients with Kabuki syndrome. Pediatric Cardiology, 31(7), 1111–1113. DOI 10.1007/s00246-010-9773-y. [Google Scholar] [CrossRef]
19. Digilio, M. C., Gnazzo, M., Lepri, F., Dentici, M. L., Pisaneschi, E. et al. (2017). Congenital heart defects in molecularly proven Kabuki syndrome patients. American Journal of Medical Genetics Part A, 173(11), 2912–2922. DOI 10.1002/ajmg.a.38417. [Google Scholar] [CrossRef]
20. Armstrong, L., Abd El Moneim, A., Aleck, K., Aughton, D. J., Baumann, C. et al. (2005). Further delineation of Kabuki syndrome in 48 well-defined new individuals. American Journal of Medical Genetics Part A, 132A(3), 265–272. DOI 10.1002/ajmg.a.30340. [Google Scholar] [CrossRef]
21. Hughes, H. E., Davies, S. J. (1994). Coarctation of the aorta in Kabuki syndrome. Archives of Disease in Childhood, 70(6), 512–514. DOI 10.1136/adc.70.6.512. [Google Scholar] [CrossRef]
22. White, S. M., Thompson, E. M., Kidd, A., Savarirayan, R., Turner, A. et al. (2004). Growth, behavior, and clinical findings in 27 patients with Kabuki (Niikawa-Kuroki) syndrome. American Journal of Medical Genetics, 127 A(2), 118–127. DOI 10.1002/ajmg.a.20674. [Google Scholar] [CrossRef]
23. Kaplan, E. L., Meier, P. (1958). Nonparametric estimation from incomplete observations. Journal of the American Statistical Association, 53, 457–481. [Google Scholar]
24. Zakaria, D., Tang, X., Bhakta, R., ElHassan, N. O., Prodhan, P. (2018). Chromosomal abnormalities affect the surgical outcome in infants with hypoplastic left heart syndrome: A large cohort analysis. Pediatric Cardiology, 39(1), 11–18. DOI 10.1007/s00246-017-1717-3. [Google Scholar] [CrossRef]
25. Lara, D. A., Ethen, M. K., Canfield, M. A., Nembhard, W. N., Morris, S. A. (2017). A population-based analysis of mortality in patients with Turner syndrome and hypoplastic left heart syndrome using the Texas Birth Defects Registry. Congenital Heart Disease, 12(1), 105–112. DOI 10.1111/chd.12413. [Google Scholar] [CrossRef]
26. Kyburz, A., Bauersfeld, U., Schinzel, A., Riegel, M., Hug, M. et al. (2008). The fate of children with microdeletion 22q11.2 syndrome and congenital heart defect: Clinical course and cardiac outcome. Pediatric Cardiology, 29(1), 76–83. DOI 10.1007/s00246-007-9074-2. [Google Scholar] [CrossRef]
27. Stasik, C. N., Goldberg, C. S., Bove, E. L., Devaney, E. J., Ohye, R. G. (2006). Current outcomes and risk factors for the Norwood procedure. Journal of Thoracic and Cardiovascular Surgery, 131(2), 412–417. DOI 10.1016/j.jtcvs.2005.09.030. [Google Scholar] [CrossRef]
28. Shamszad, P., Gospin, T. A., Hong, B. J., McKenzie, E. D., Petit, C. J. (2014). Impact of preoperative risk factors on outcomes after Norwood palliation for hypoplastic left heart syndrome. Journal of Thoracic and Cardiovascular Surgery, 147(3), 897–901. DOI 10.1016/j.jtcvs.2013.05.012. [Google Scholar] [CrossRef]
29. Hornik, C. P., He, X., Jacobs, J. P., Li, J. S., Jaquiss, R. D. B. et al. (2011). Complications after the Norwood operation: An analysis of the Society of Thoracic Surgeons Congenital Heart Surgery Database. Annals of Thoracic Surgery, 92(5), 1734–1740. DOI 10.1016/j.athoracsur.2011.05.100. [Google Scholar] [CrossRef]
30. Tabbutt, S., Ghanayem, N., Ravishankar, C., Sleeper, L. A., Cooper, D. S. et al. (2012). Risk factors for hospital morbidity and mortality after the Norwood procedure: A report from the Pediatric Heart Network Single Ventricle Reconstruction trial. Journal of Thoracic and Cardiovascular Surgery, 144(4), 882–895. DOI 10.1016/j.jtcvs.2012.05.019. [Google Scholar] [CrossRef]
31. Hehir, D. A., Dominguez, T. E., Ballweg, J. A., Ravishankar, C., Marino, B. S. et al. (2008). Risk factors for interstage death after stage 1 reconstruction of hypoplastic left heart syndrome and variants. Journal of Thoracic and Cardiovascular Surgery, 136(1), 94–99.e3. DOI 10.1016/j.jtcvs.2007.12.012. [Google Scholar] [CrossRef]
32. Tweddell, J. S., Hoffman, G. M., Mussatto, K. A., Fedderly, R. T., Jaquiss, R. D. B. et al. (2002). Hypoplastic left heart syndrome: Lessons learned from 115 consecutive patients. Circulation, 106, 82–89. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |