
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Epibrassinolide Induces Apoptosis and Inhibits the Migration of Gastric Cancer AGS Cells by Regulating Reactive Oxygen Species-Mediated Signaling Pathways
1 Department of Food Science and Engineering, College of Food Science, Heilongjiang Bayi Agricultural University, Daqing, 163319, China
2 Agricultural Products and Processed Products Supervision and Testing Center, Ministry of Agriculture, Daqing, 163319, China
3 Department of Biochemistry and Molecular Biology, College of Life Science and Technology, Heilongjiang Bayi Agricultural University, Daqing, 163319, China
4 National Coarse Cereals Engineering Research Center, Daqing, 163319, China
* Corresponding Author: Chenghao Jin. Email:
# These authors contributed equally to this work
BIOCELL 2025, 49(3), 465-482. https://doi.org/10.32604/biocell.2025.062155
Received 11 December 2024; Accepted 05 March 2025; Issue published 31 March 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Objectives: Epibrassinolide (EBR) is a steroid hormone with anti-tumor properties. Nevertheless, its potential to inhibit gastric cancer (GC) cells remains unknown. The aim of this research was to examine the effects of EBR on GC cells and to investigate the specific mechanism of EBR. Methods: A cell counting kit-8 (CCK-8) assay was utilized to determine cell survival rates. The investigation of apoptosis, cell cycle progression, and reactive oxygen species (ROS) levels was performed using flow cytometry. To detect cell migration, a wound-healing assay was performed on AGS cells. Furthermore, western blotting assay was utilized to determine protein expression levels. Results: The CCK-8 assay demonstrated that EBR reduced the survival rates of AGS, KATO-3, and MKN-45 cells, while causing only minor toxicity to normal cells. The apoptosis assay indicated that EBR induced AGS cell apoptosis through a mitochondria-mediated pathway. Western blotting results demonstrated that EBR induced AGS cell apoptosis via mitogen-activated protein kinase (MAPK)/signal transducer and activator of transcription 3 (STAT3)/nuclear factor kappa B (NF-κB) signaling pathway. Further, after treating AGS cells with EBR, the accumulation of intracellular ROS markedly increased. EBR also induced G2/M phase cell cycle arrest in AGS cells by downregulating phospho-protein kinase B (p-AKT), cyclin-dependent kinase 1/2 (CDK1/2), and cyclin B1 expression levels, while simultaneously upregulating p21 and p27 expression levels. EBR inhibited AGS cell migration by downregulating p-AKT, phosphorylated-glycogen synthase kinase 3β (p-GSK-3β), and β-catenin expression levels and upregulating E-cadherin expression levels. However, these effects were reversed by pretreatment with N-acetylcysteine (NAC). Conclusion: EBR regulates AGS cells by inducing apoptosis and G2/M phase arrest, while also inhibiting cell migration, all of which are mediated through ROS-mediated signaling pathways. Ultimately, these effects suggest a significant role for EBR in regulating cellular processes within AGS cells.Keywords
Globally, gastric cancer (GC) holds the third position in causing cancer-related deaths, giving rise to millions of deaths yearly [1]. Currently, the primary treatments for GC include surgery, radiotherapy, and chemotherapy. Among them, chemotherapy is the most widely used treatment, but the high toxicity of current chemotherapy medications damages the human body [2]. Therefore, it is urgent to develop a drug that is less toxic, has fewer side effects and is more effective. The chemotherapy drug cisplatin is extensively used in GC. Due to its excellent therapeutic effect, it was selected as the positive control treatment in this study.
Phytoactive ingredients have been widely studied for some time by scholars for their low toxicity and low side effects. Epibrassinolide (EBR) is a highly active steroid hormone widely present throughout the plant kingdom [3]. It increases plant stress resistance and promotes plant growth and development [4]. EBR exhibits antioxidant properties and has been demonstrated to shield neuronal PC12 cells from the harmful effects of 1-methyl-4-phenylpyridinium toxicity, an effect attributed to its steroid B ring and lateral chain structure [5]. EBR inhibited the neutrophil migration of Tg(mpx:GFP)i114 larvae to injured tissue and inhibited the action of a proinflammatory chemical agent [6]. In addition, one study showed that EBR induced the expression of IGFBP-1 in liver cancer cells, thereby inducing cell apoptosis [7]. However, the pharmacological effect and specific mechanism in GC are still unclear.
Currently, induction of cell apoptosis is one of the means by which different drugs exert an anticancer effect [8]. Apoptosis promotes the regular growth of organisms by eliminating harmful cells, and it is essential for maintaining tissue homeostasis [9]. When mitochondrial membrane potential (MMP) decreases, cytochrome c (Cyto C) is released, which activates caspase-3, leading to apoptosis [10].
Reactive oxygen species (ROS), a highly active product of aerobic metabolism, are crucial to the development of cancer [11]. ROS are involved in numerous cellular processes, and their levels are tightly regulated under normal physiological conditions. However, when this delicate balance is disrupted, it can have profound implications on cellular homeostasis. The imbalance of intracellular ROS levels can cause intracellular oxidative stress, resulting in cell damage and death [12]. When the generation of ROS outpaces the cell’s antioxidant defenses, oxidative stress occurs. This imbalance ultimately compromises the integrity and function of cells. An excessive increase in intracellular ROS levels can activate apoptosis-related signaling pathways, including mitogen-activated protein kinase (MAPK), signal transducer and activator of transcription 3 (STAT3), and nuclear factor kappa B (NF-κB) pathways, thereby inducing apoptosis [13–15]. In addition, one study showed that brassinosteroids enhanced ROS accumulation in lung cancer A549 cells with an anticancer effect [16]. The accumulation of intracellular ROS was associated with mitochondrial dysfunction, which was characterized by the decline of MMP [17]. Therefore, it was an interesting question whether EBR could exert anti-GC effects through the mitochondrial pathway.
The aim of this research was to assess the inhibitory effects of EBR on GC and its specific mechanism.
EBR (purity ≥ 97.0%; Herbpurify, B-128. Chengdu, China), and cisplatin (purity ≥ 99.0%, Solarbio, D-8810. Beijing, China) were purchased for the study. AGS, KATO-3, and MKN-45 cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA). THLE-2 cell lines were obtained from Otwo Biotech (Shenzhen, China). GES-1 and 293T cell lines were obtained from Sage Biotech (Shanghai, China).
AGS, KATO-3, and MKN-45 human GC cells, as well as normal cells GES-1 (normal human gastric mucous cells), 293T (normal human renal epithelial cells), and THLE-2 (normal human hepatocyte cells), were cultured in Dulbecco’s modified Eagle medium (DMEM, Gibco, C11995500BT. Waltham, MA, USA). The culture medium formulation included the addition of 10% fetal bovine serum (FBS, Gibco, 11011-8611) and 1% penicillin/streptomycin (PS, Solarbio, P1400). Cultures were maintained in a sterile incubator with 5% CO2 at 37°C. All of the cell cultures remained free of mycoplasma contamination throughout the study period.
A cell counting kit-8 (CCK-8, Solarbio, CA1210) was used to detect cell survival rates in GC cells and normal cells after EBR treatment. The experiment commenced with seeding cells at a cell concentration of 1 × 104 cells per well in 96-well plates followed by culturing for 24 h. Following this, EBR and cisplatin were applied to the cultures at concentrations of 20, 40, 60, 80, and 100 μM for 24 h. After that, each well received 12 μL of CCK-8 solution. The OD of the sample was determined at 450 nm with a multifunctional microplate reader (Tecan, Tecan M200 PRO. Mannedorf, Switzerland). The half-maximal inhibitory concentration (IC50) was calculated by employing the GraphPad Prism (GraphPad Software, GraphPad Prism 10.0. San Diego, CA, USA). Subsequently, cells were pretreated with 62.45 µM EBR or cisplatin for 3, 6, 12, 24, and 36 h. The experimental concentrations for this part of the study were based on the previously determined IC50 value.
The experiment commenced with seeding AGS cells at a cell concentration of 1 × 105 cells per well in six-well plates followed by culturing for 24 h. Then, the cells underwent pretreatment with 62.45 µM EBR for 3, 6, 12, and 24 h. An annexin V-FITC/PI apoptosis kit (Solarbio, CA1020) was utilized to stain the cells. To evaluate EBR-induced apoptosis in AGS cells, flow cytometry (Sysmex, CyFlow Cube8. Kobe, Japan) and fluorescence microscopy (Mingmei Optoelectronics, MF52-N. Guangzhou, China) were employed.
The experiment commenced with seeding AGS cells at a cell concentration of 1 × 105 cells per well in 3.5-cm plates followed by culturing for 24 h. Then, the cells underwent pretreatment with 62.45 µM EBR for 3, 6, 12, and 24 h. The MMP was evaluated using an MMP assay kit from Solarbio (M8650) and flow cytometry to gain insights into the cellular changes.
The experiment commenced with seeding AGS cells at a cell concentration of 1 × 105 cells per well in 3.5-cm plates followed by culturing for 24 h. Then, the cells underwent pretreatment with 62.45 µM EBR for 3, 6, 12, and 24 h. The distribution of cells across the cell cycle was assessed employing a cell cycle assay kit (Solarbio, CA1510).
A western blotting assay was used to determine the protein expression levels in AGS cells after treatment with EBR or ERK inhibitor (FR180204), JNK inhibitor (SP600125), and p38 inhibitor (SB203580) (MedChemExpress, Shanghai, China), or N-Acetylcysteine (NAC, Beyotime Institute Biotechnology, S0077. Shanghai, China). AGS cells were lysed to extract proteins using a lysis buffer comprising 1 M HEPES, pH 7.3 (C0215); 5 M NaCl (ST347); 0.5% Triton X-100 (ST795); 10% glycerol (ST1348) (Beyotime Institute Biotechnology); 20 mg/mL AEBSF (HY-12821); 2 mg/mL aprotinin (HY-P0017); 0.5 mg/mL pepstatin (HY-P0018); and 0.5 mg/mL leupeptin (HY-18234A) (MedChemExpress). Coomassie brilliant blue (Beyotime Institute Biotechnology, P0017F) was added to the protein sample, and the OD at 595 nm was determined with the use of a spectrophotometer (Aoyi Instruments Co., UV-1800PC. Shanghai, China). Finally, a protein concentration of 1.5 μg/μL was obtained. The proteins were separated and electrotransferred onto nitrocellulose membranes (Pall Corporation, 66485. Port Washington, NY, USA) using 8%–12% SDS-polyacrylamide gel electrophoresis. Following a 2-h 5% skim milk blocking step, membranes were treated with primary antibodies, including protein kinase B (AKT) (Cat#sc-8312, 1:1000), p-AKT (Cat#sc-7985-R, 1:1000), Bcl-2 associated death promoter (Bad) (Cat# sc-8044, 1:1500), B-cell lymphoma-extra-large (Bcl-xl) (Cat#sc-8392, 1:1500), caspase-3 (Cat#sc-373730, 1:1500), cyclin B1 (Cat#sc-245, 1:1500), poly ADP-ribose polymerase (PARP) (Cat#sc-8007, 1:1500), Cyto C (Cat#sc-13156, 1:2000), cyclin-dependent kinase 1/2(CDK1/2) (Cat#sc-53219, 1:1500), extracellular signal-regulated kinase (ERK) (Cat#sc-154, 1:1000), p-ERK (Cat#sc-7383, 1:1000), glycogen synthase kinase 3β (GSK-3β) (Cat#sc-377213, 1:1500), p-GSK-3β (Cat#sc-373800, 1:1000), STAT3 (Cat#sc-8019, 1:1500), p-STAT3 (Cat#sc-8059, 1:1500), c-Jun N-terminal kinase (JNK) (Cat#sc-7345, 1:1500), p-JNK (Cat#sc-6254, 1:1500), NF-κB (Cat#sc-8008, 1:1500), I-κBα (Cat#sc-1643, 1:1500), p21 (Cat#sc-397, 1:1500), p27 (Cat#sc-528, 1:1500), p38 (Cat#sc-7149, 1:1500), p-p38 (Cat#sc-7973, 1:1500), β-catenin (Cat#sc-7963, 1:1000), N-cadherin (Cat#sc-59987, 1:1000), E-cadherin (Cat#sc-8426,1:1000), vimentin (Cat# sc-6260, 1:1000), and α-tubulin (Cat#sc-47778, 1:2500) (Santa Cruz Biotechnology, Dallas, TX, USA) for 10 h at 4°C. Then, the appropriate secondary antibody, goat anti-mouse IgG (ZB-2305, 1:50,000) or goat anti-rabbit IgG (ZB-2306, 1:50,000) (ZSGB-BIO, Beijing, China) was applied to the membrane for 2 h at 25°C. Using a UVP ChemStudio 515 (Analytik Jena AG, UVP Chemstudio touch. Jena, Germany), the membranes were examined after treatment with an enhanced chemiluminescence reagent (Tanon, 180-501. Shanghai, China). To analyze the bands, ImageJ software (National Institutes of Health, lmageJ 1.46r. Bethesda, MD, USA) was used.
The experiment commenced with seeding AGS cells at a cell concentration of 1 × 105 cells per well in 3.5-cm plates followed by culturing for 24 h. Then, the cells underwent pretreatment with 62.45 µM EBR for 3, 6, 12, and 24 h. The ROS levels were quantified utilizing a ROS assay kit (Beyotime Institute Biotechnology, S0033S).
The experiment commenced with seeding AGS cells at a cell concentration of 1 × 105 cells per well in six-well plates followed by culturing for 24 h. Then, a 10 µL pipette tip was employed to generate a scratch in the confluent cell layer. The cells underwent pretreatment with 62.45 µM EBR for 3, 6, 12, and 24 h, and the migration of cells was observed using a microscope (Mshot, MF52-N. Guangzhou, China).
The results of three separate experiments are presented as mean values accompanied by their standard deviations. To assess variations among groups, a one-way ANOVA was used. Tukey’s post hoc test was conducted using SPSS (IBM, SPSS 21.0. Tulsa, OK, USA). Statistical significance was defined as *p < 0.05, **p < 0.01, and ***p < 0.001, representing statistically significant variations.
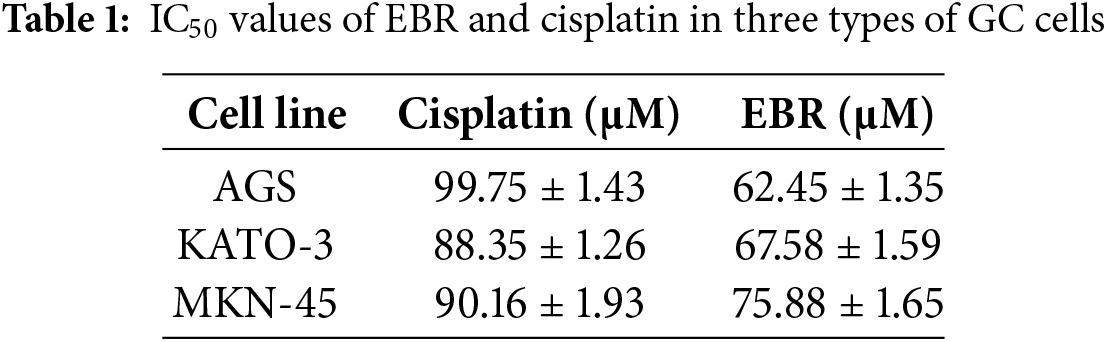
The study employed a CCK-8 assay to assess the cytotoxic effects of EBR on three GC cell types and three normal cell types. After treatment with EBR or cisplatin, three types of GC cells underwent marked cell death, and EBR displayed a significantly stronger cytotoxic effect compared to cisplatin in each of the three GC cell types (Fig. 1A and B). The above results clearly showed the different cytotoxicity of the two treatments in three types of GC cells, highlighting the relatively strong cytotoxicity of EBR. Meanwhile, EBR showed a distinct advantage in terms of toxicity in normal cells. EBR demonstrated less toxicity in three types of normal human cells, and EBR displayed a significantly lower cytotoxic effect compared to cisplatin in each of the three normal cell types (Fig. 1C and D). According to the experimental data, when the EBR concentration reached its highest level, the survival rate of normal cells remained near 90%. We believed that the IC50 values of normal cells could not be accurately determined because EBR did not show quantifiable inhibition on normal cells even at high concentrations. This phenomenon strongly supported the view in our study that EBR is relatively selective to GC cells. Table 1 provides an overview of the IC50 values of EBR and cisplatin in three types of GC cells treated. Among them, the lowest IC50 value was 62.45 μM in AGS cells, and therefore AGS cells were chosen for follow-up studies.
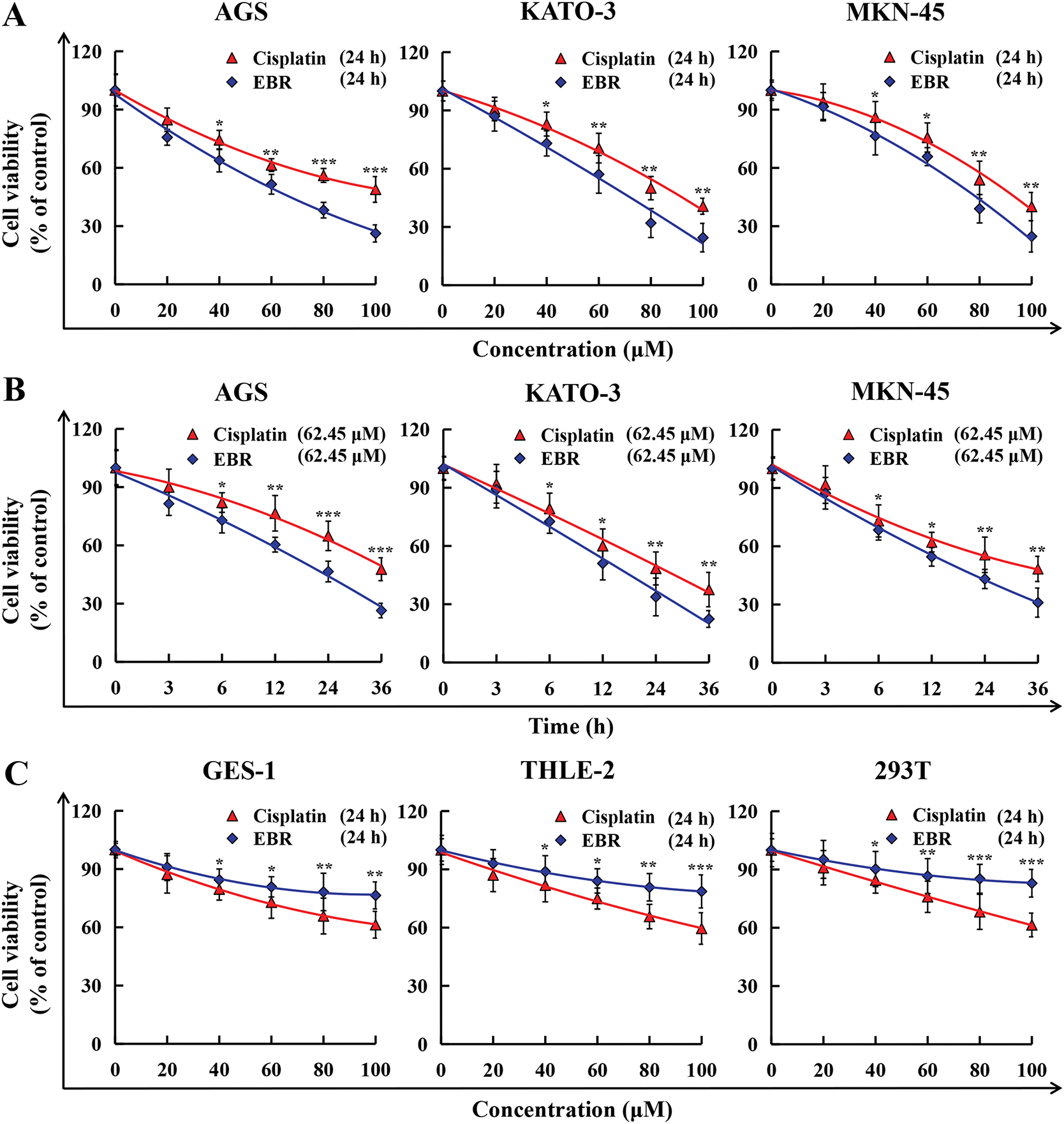
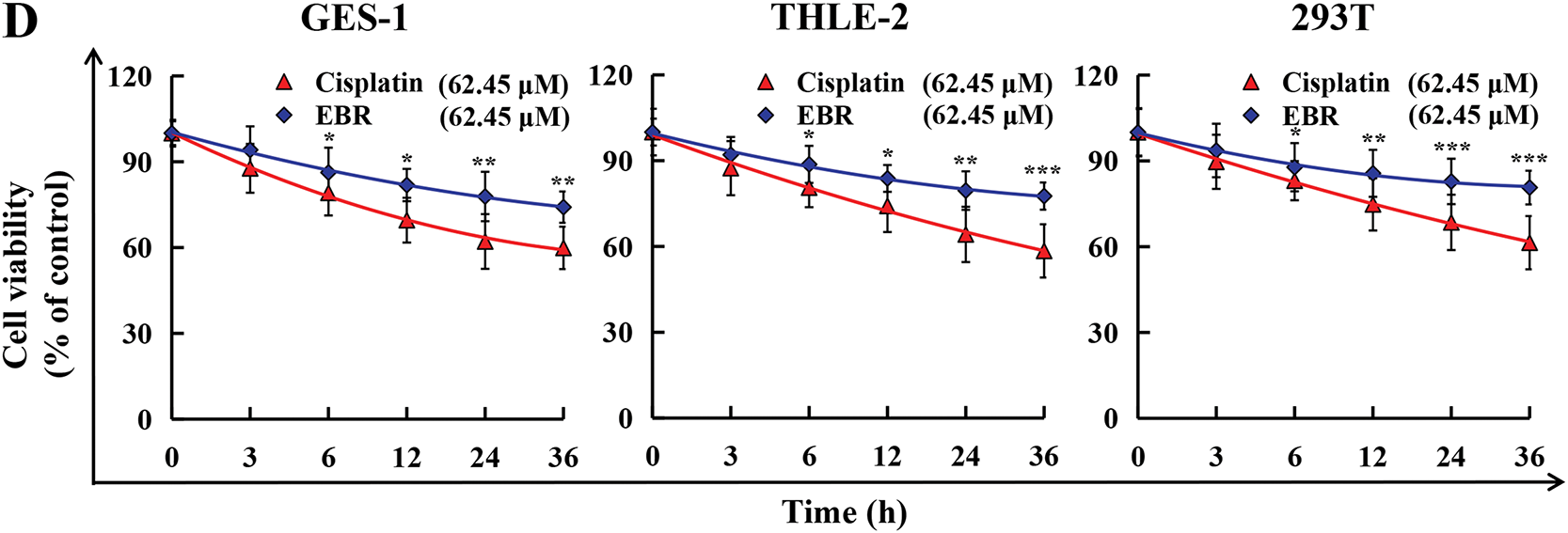
Figure 1: The cytotoxicity of EBR and cisplatin was assessed in three GC and normal cell lines. (A) GC cells were exposed to 20, 40, 60, 80, and 100 μM EBR or cisplatin for 24 h, following which survival rates were evaluated. (B) At 3, 6, 12, 24, and 36 h, the survival rates of these three GC cells were evaluated after treatment with 62.45 μM (IC50 value of AGS cells) EBR or cisplatin. (C) Normal cells were exposed to 20, 40, 60, 80, and 100 μM EBR or cisplatin for 24 h, following which survival rates were evaluated. (D) At 3, 6, 12, 24, and 36 h, the survival rates of these three normal cells were evaluated after treatment with 62.45 μM (IC50 value of AGS cells) EBR or cisplatin (*p < 0.05, **p < 0.01, ***p < 0.001 vs. cisplatin group. Data are reported as means and SD)
3.2 EBR-Induced AGS Cell Apoptosis
The study employed an annexin V-FITC/PI apoptosis detection assay to assess the relationship between survival rate and apoptosis of AGS cells affected by EBR. After treatment with EBR, AGS cells exhibited a time-dependent increase in fluorescence intensity that was higher than that of cisplatin at the same time point (Fig. 2A). The time-dependent nature of this response further underscores the dynamic impact of EBR on cellular viability and apoptosis induction. Flow cytometry results further indicated that EBR treatment for 24 h led to alterations in apoptosis rate and MMP level in AGS cells. The apoptosis rate of AGS cells increased by 40.66%, which was ascribed to the activation of the intrinsic apoptotic pathway (Fig. 2B). This activation was confirmed by a 36.8% decrease in the MMP level (Fig. 2C). The decrease in the MMP level is a hallmark of mitochondrial outer membrane permeabilization, a critical step in the intrinsic apoptotic cascade. These changes in the apoptosis rate and MMP level provided evidence of EBR-induced apoptosis in AGS cells. The western blotting results indicated a decrease in Bcl-xl protein expression, and simultaneously an increase in Bad, Cyto C, cleaved caspase-3 (cle-caspase-3), and cle-PARP expression (Fig. 2D). The changes in these protein levels were consistent with the activation of the intrinsic apoptotic pathway.
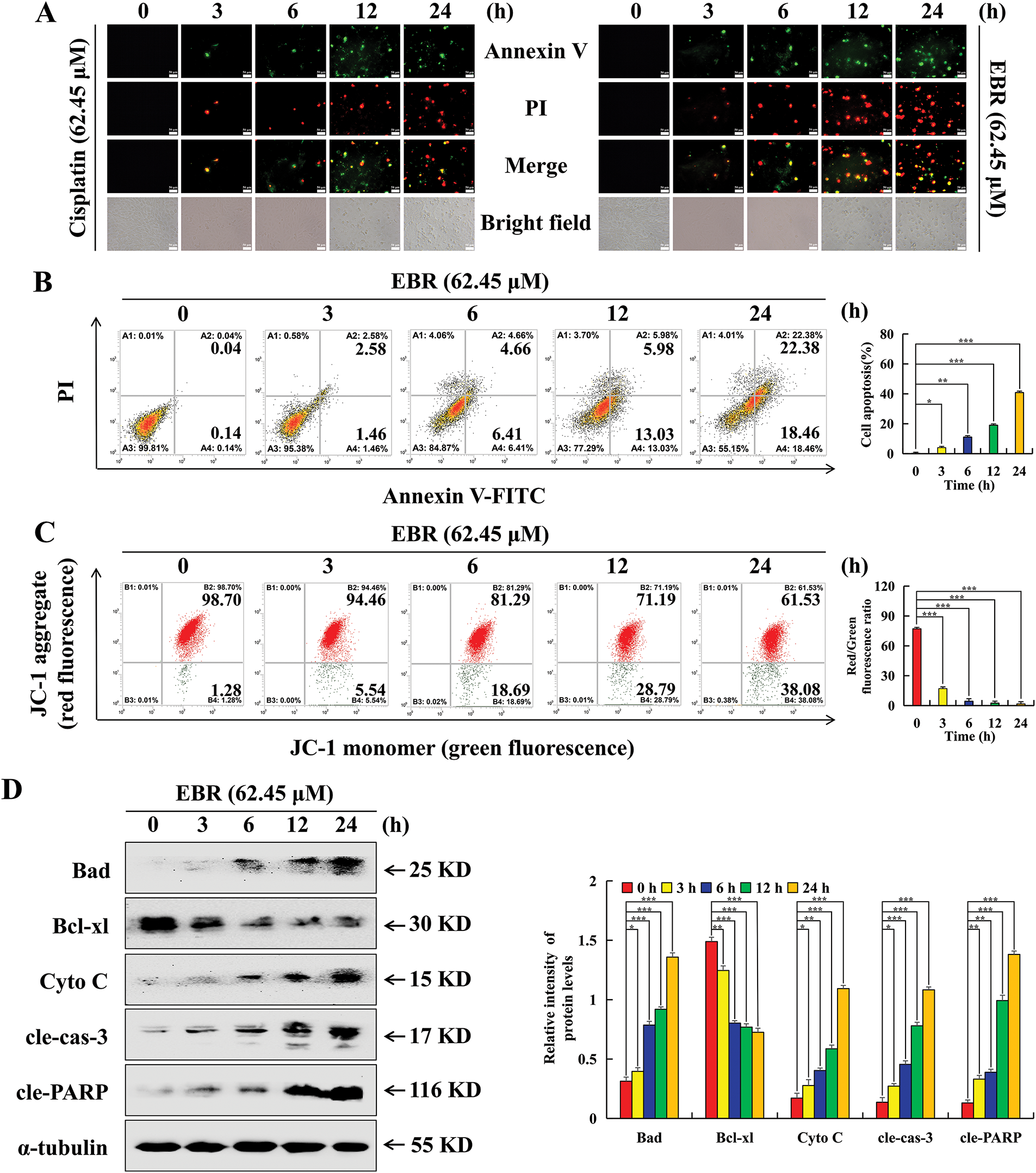
Figure 2: The apoptotic consequences that EBR exerts on AGS cells were further investigated. At 3, 6, 12, and 24 h, the AGS cells were subjected to a treatment of 62.45 μM (IC50 value of AGS cells) EBR or cisplatin. (A) The assessment of fluorescence intensity and cellular morphology changes after annexin V-FITC/PI staining was conducted, and the cells were observed at a 200× original magnification. (B) The AGS cell apoptosis rate was quantified. (C) The MMP level of AGS cells was quantified. (D) To analyze protein expression changes in AGS cells subjected to EBR treatment, western blotting was performed. For the purpose of ensuring accurate quantification in western blotting experiments, α-tubulin was selected to function as a loading control (* p < 0.05, ** p < 0.01, *** p < 0.001 vs. cisplatin group. Data are reported as means and SD)
3.3 EBR-Induced AGS Cell Apoptosis via MAPK/STAT3/NF-κB Signaling Pathway
The study employed the western blotting assay to explore the mechanisms underlying the EBR-induced AGS cell apoptosis by examining changes in proteins linked to the MAPK/STAT3/NF-κB signaling pathway. After EBR treatment, the p-ERK, p-STAT3, and NF-κB expression levels were downregulated, and the p-JNK, p-p38, and IκBα expression levels were upregulated (Fig. 3A). To validate an interaction between the MAPK and STAT3 pathways, specific inhibitors were applied to AGS cells and protein levels were measured using western blotting assay. In contrast to the EBR treatment group, the EBR + ERK inhibitor (FR180204) group inhibited the increase of cle-caspase-3 expression and reduced Bcl-xl, p-ERK, and p-STAT3 expression. (Fig. 3B). The EBR + p38 inhibitor (SB203580) group and EBR + JNK inhibitor (SP600125) group inhibited p-JNK and p-p38 expression, respectively, and inhibited the reduction of p-STAT3 expression compared to the EBR treatment group (Fig. 3C and D).
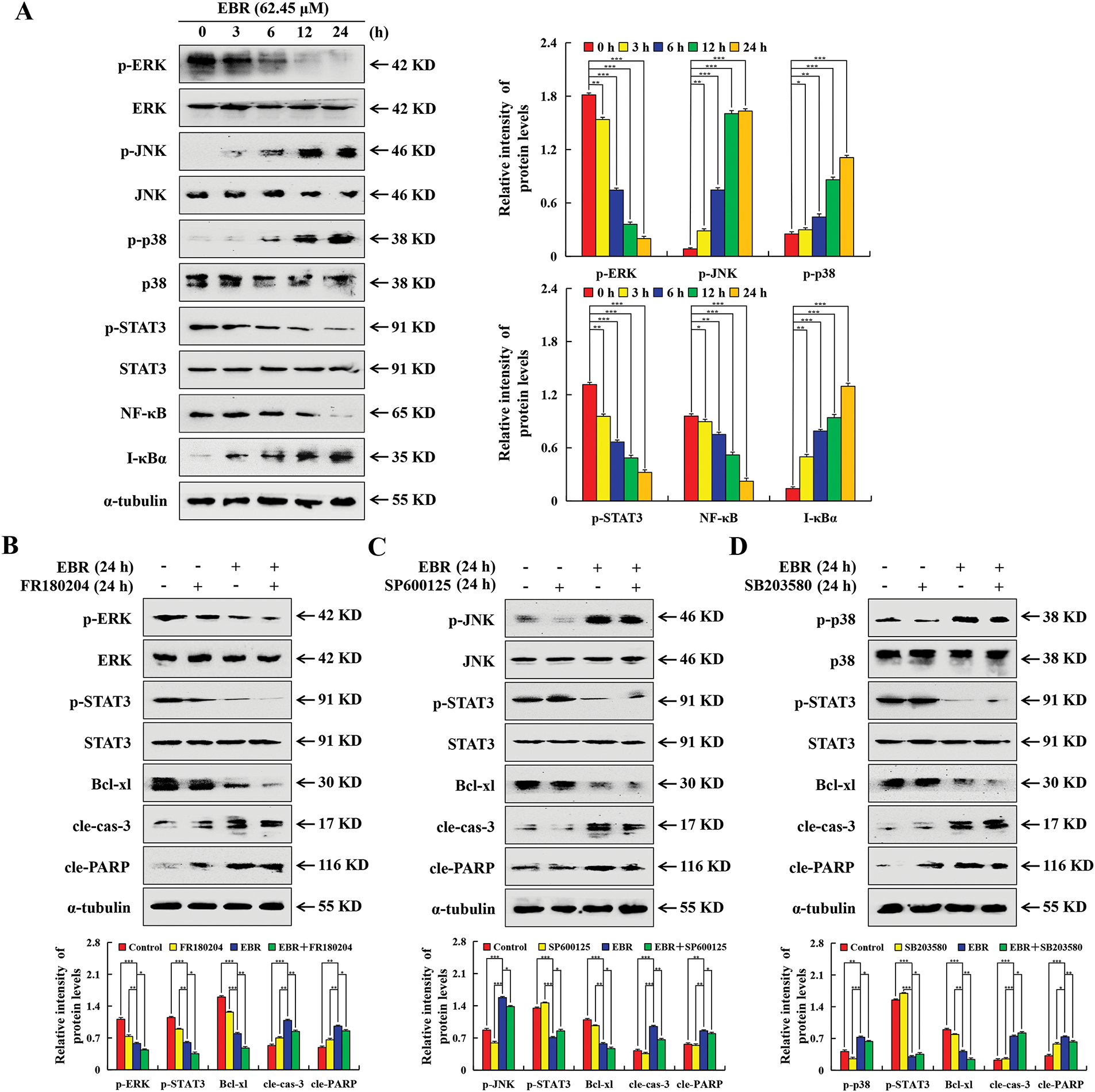
Figure 3: EBR-mediated modulation of MAPK/STAT3/NF-κB pathway in AGS cells. (A) At 3, 6, 12, and 24 h, the AGS cells were subjected to a treatment of 62.45 μM (IC50 value of AGS cells) EBR. The pathway-related proteins expression in AGS cells was determined. (B) The protein expression in AGS cells under treatment with EBR (62.45 µM) with or without an ERK inhibitor (FR180204; 10 µM) for 24 h varied significantly. (C) The protein expression in AGS cells under treatment with EBR (62.45 µM) with or without a JNK inhibitor (SP600125; 10 µM) for 24 h varied significantly. (D) The protein expression in AGS cells under treatment with EBR (62.45 µM) with or without a p38 inhibitor (SB203580; 10 µM) for 24 h varied significantly. For the purpose of ensuring accurate quantification in western blotting experiments, α-tubulin was selected to function as a loading control (*p < 0.05, **p < 0.01, ***p < 0.001 vs. 0 h or EBR + MAPK inhibitor. Data are reported as means and SD)
3.4 EBR Regulated MAPK/STAT3/NF-κB Signaling Pathway by Increasing ROS Levels
The study employed flow cytometry to explore the correlation between ROS and EBR-induced apoptosis. The levels of ROS in AGS cells were significantly increased after EBR treatment (Fig. 4A). NAC, a potent antioxidant agent, was utilized for the pretreatment of AGS cells. In comparison with the EBR treatment group, the apoptosis rate in the EBR + NAC group decreased by 27.95%, which indicated a significant inhibitory effect (Fig. 4B). In contrast to the EBR treatment group, protein expression changes of the EBR + NAC group were inhibited, which indicated that the related signaling pathways were inhibited (Fig. 4C). The findings suggested that ROS had a crucial role in EBR-induced apoptosis.
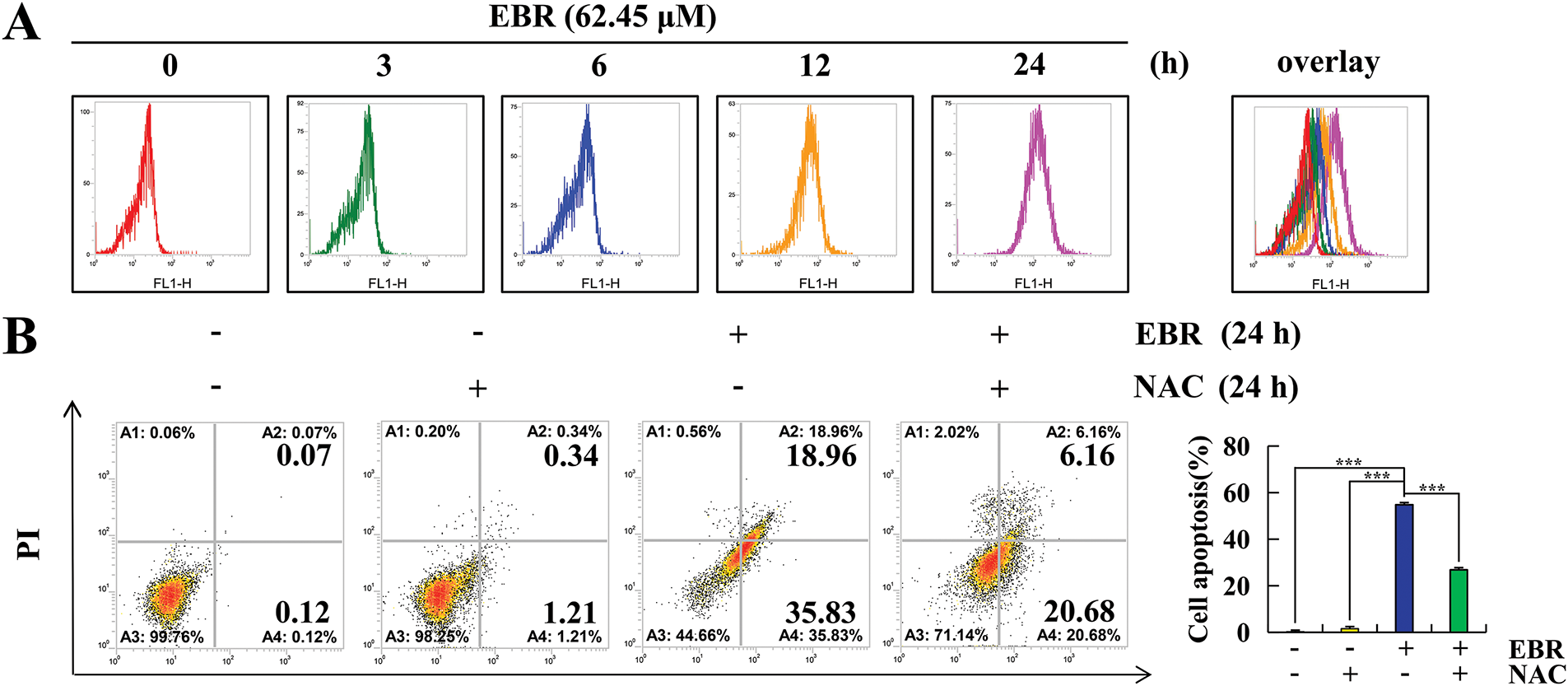
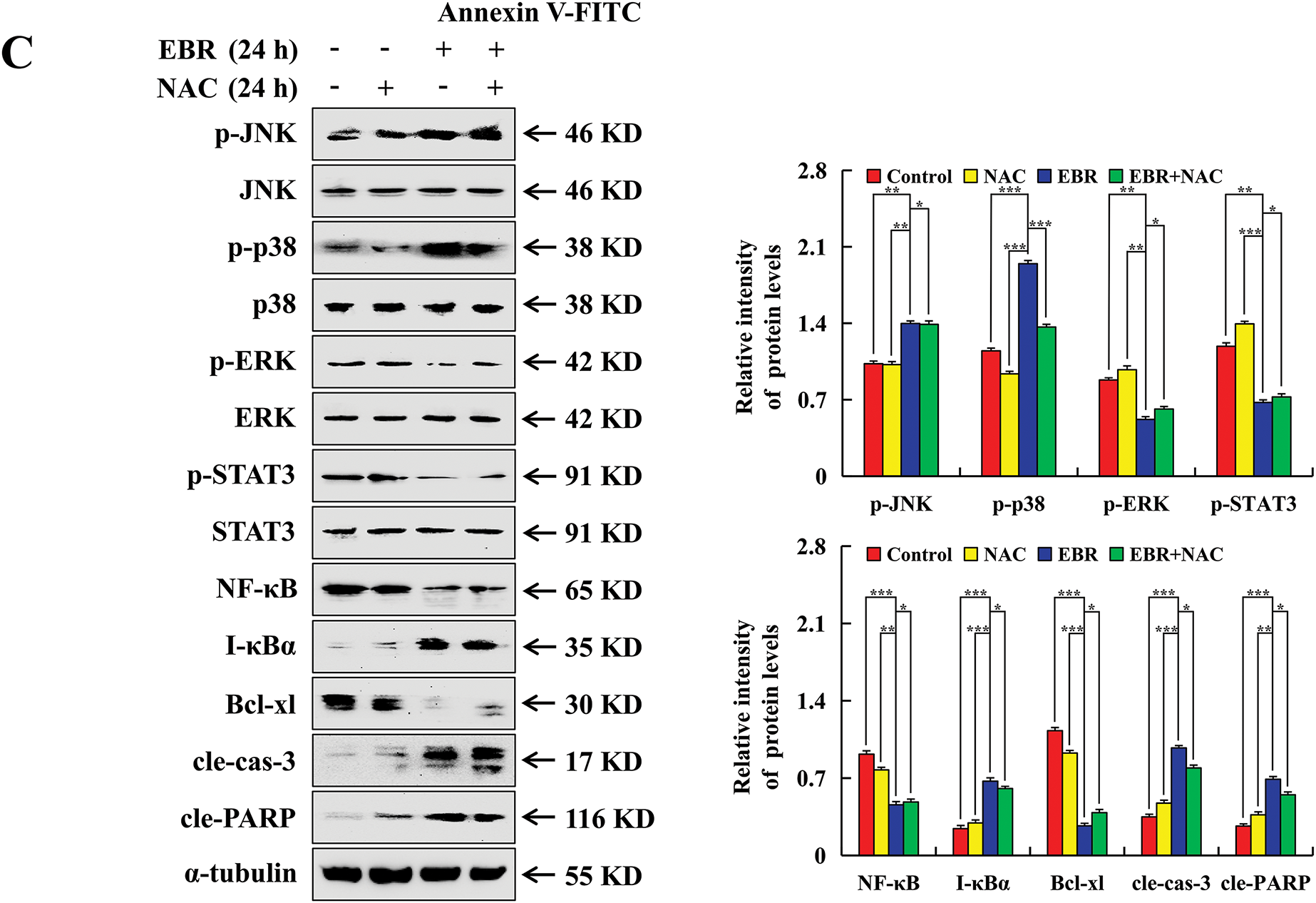
Figure 4: AGS cells were analyzed to determine the effects of EBR on ROS accumulation. (A) At 3, 6, 12, and 24 h, the AGS cells were subjected to a treatment of 62.45 μM (IC50 value of AGS cells) EBR and the ROS levels were evaluated. (B) At 24 h, the apoptosis rate was quantified under treatment with EBR (62.45 µM) with or without NAC (10 mM). (C) At 24 h, the expression of proteins was assessed under treatment with EBR (62.45 µM) with or without NAC (10 mM). For the purpose of ensuring accurate quantification in western blotting experiments, α-tubulin was selected to function as a loading control (*p < 0.05, **p < 0.01, ***p < 0.001 vs. 0 h or the NAC + EBR group. Data are reported as means and SD)
3.5 EBR Arrested AGS Cells in the G2/M Phase
The study employed flow cytometry to explore changes in the AGS cell cycle after EBR treatment by measuring the distribution of cells across different cell cycle phases. After treatment with EBR for 24 h, a reduction was observed in the G0/G1 phase cell population, decreasing from 68.24% to 59.4%, while the G2/M phase cell population increased from 30.84% to 39.37% (Fig. 5A). After treatment with EBR, p21 and p27 expression levels were upregulated. These proteins block the activity of cyclin-dependent kinases, thereby preventing cell cycle progression. In contrast, p-AKT, CDK1/2, and cyclin B1 expression was downregulated (Fig. 5B). The upregulation of p21 and p27, which are well-known CDK inhibitors, supports the notion that EBR disrupts cell cycle progression by inhibiting CDK activity. AKT signaling pathway is vital for cell survival, growth, and metabolism, and its phosphorylation status affects cell cycle progression. CDK1/2 and cyclin B1 are vital regulators of the G2-to-M phase transition. To further elucidate the relationship between ROS and its underlying mechanism, NAC was used to pretreat AGS cells. The G2/M phase cell population was significantly lower in the EBR + NAC group than in the EBR group and the related protein expression was reversed (Fig. 5C and D). This suggested that NAC effectively interfered with the EBR-mediated AGS cell cycle regulatory cascade.
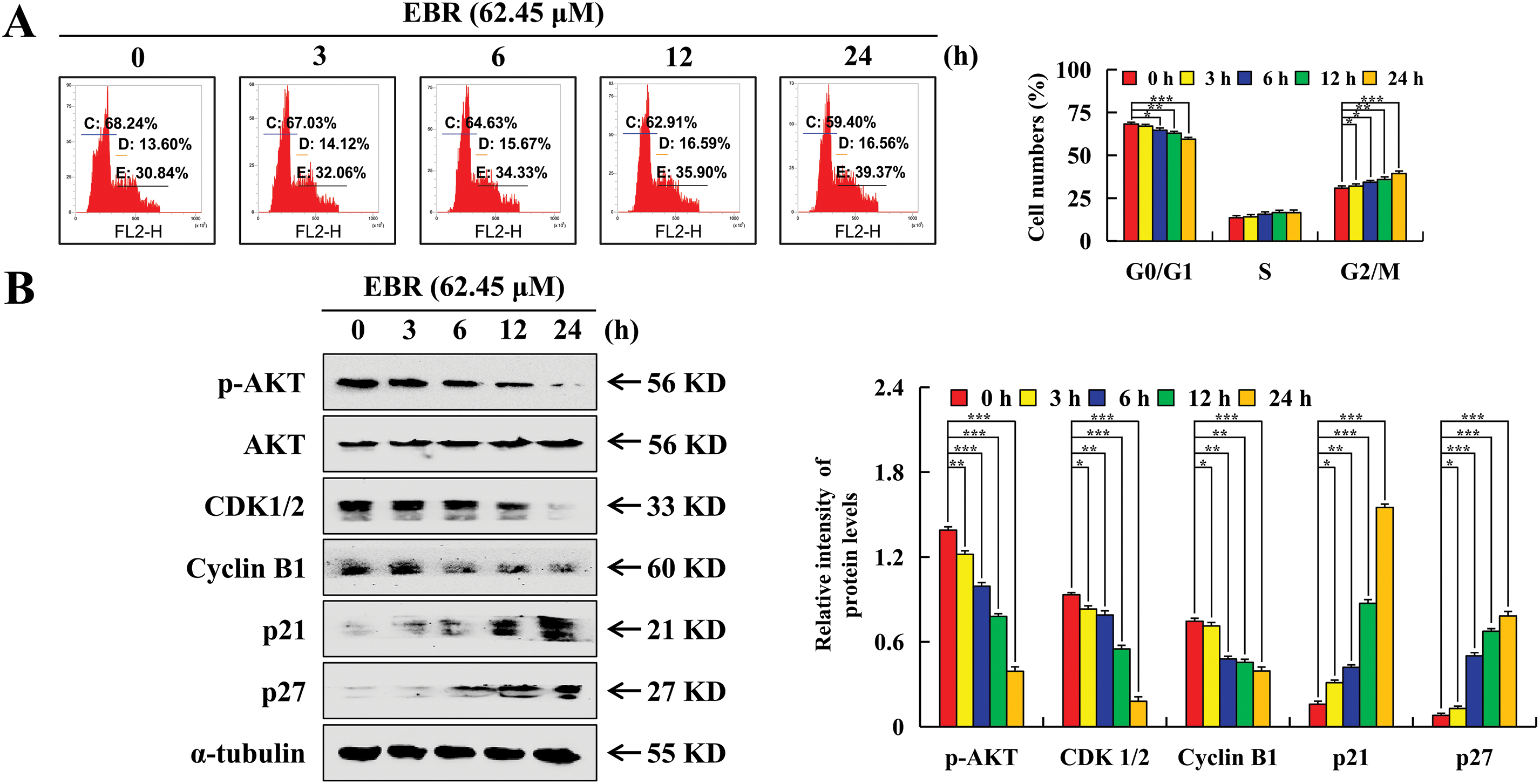
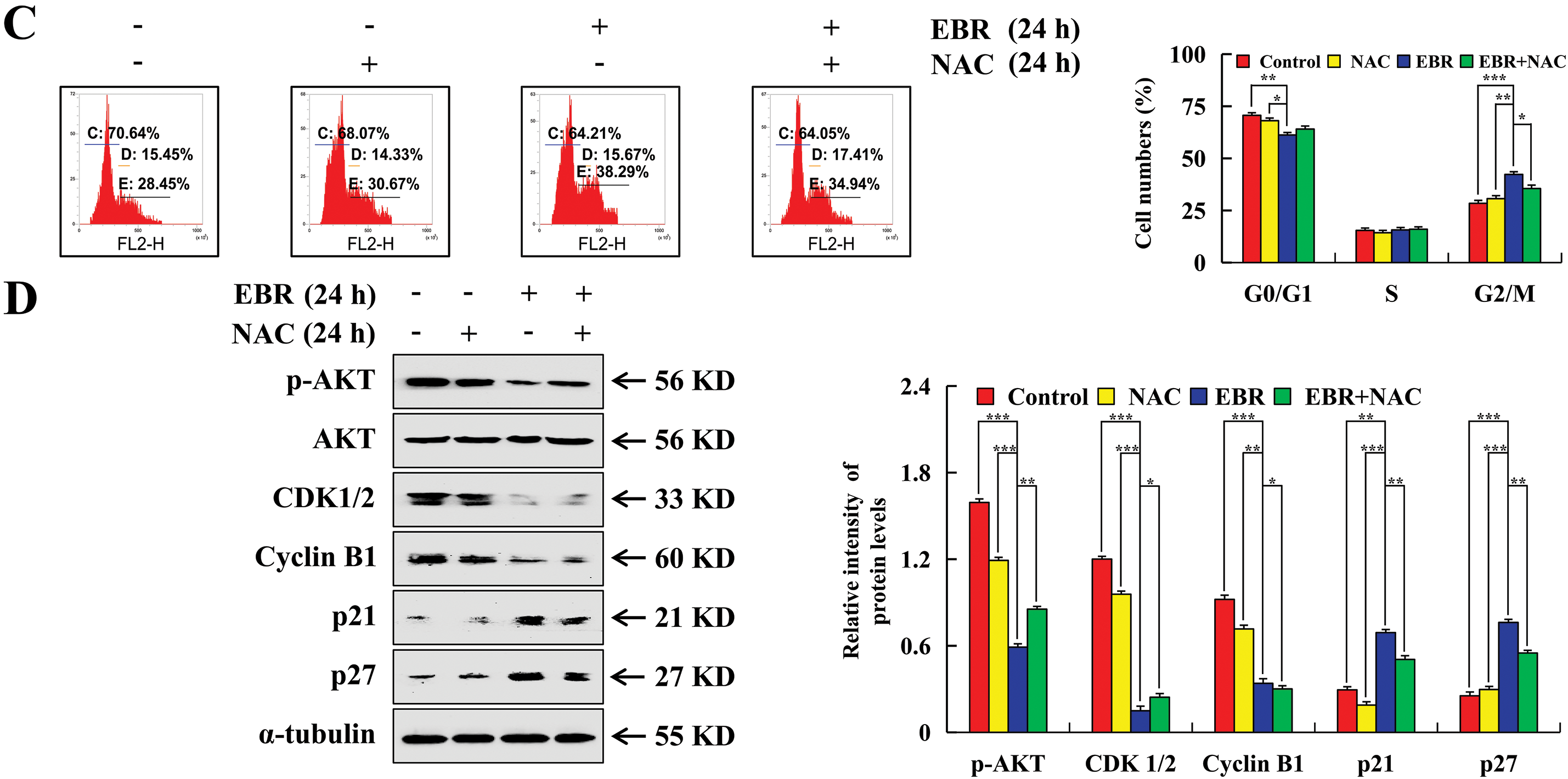
Figure 5: Cell cycle arrest effects of EBR on AGS cells. At 3, 6, 12, and 24 h, the AGS cells were subjected to a treatment of 62.45 μM (IC50 value of AGS cells) EBR. (A) The cell population at various cell cycle stages was detected. (B) G2/M phase protein expression was assessed. (C) At 24 h, the cells in various cell cycle stages were quantified under treatment with EBR (62.45 µM) with or without NAC (10 mM). (D) At 24 h, the expression of proteins was assessed under treatment with EBR (62.45 µM) with or without NAC (10 mM). For the purpose of ensuring accurate quantification in western blotting experiments, α-tubulin was selected to function as a loading control (*p < 0.05, **p < 0.01, ***p < 0.001 vs. 0 h or the NAC + EBR group. Data are reported as means and SD)
3.6 EBR Inhibited AGS Cell Migration
The study employed a wound-healing assay to explore whether EBR affected the AGS cell migration by measuring the wound closure rate. After treatment with EBR, the AGS cell migration rate decreased (Fig. 6A). The migration-related protein expression levels, including those of p-AKT, p-GSK-3β, β-catenin, N-cadherin, and vimentin, were found to be downregulated, while an increase was observed in E-cadherin expression (Fig. 6B). To further elucidate the relationship between ROS and EBR inhibition of AGS cell migration, the cells were pretreated with NAC. In contrast to the EBR group, in the EBR + NAC group, the inhibitory effect of cell migration by EBR decreased, and the migration-related protein expression levels were reversed (Fig. 6C and D).
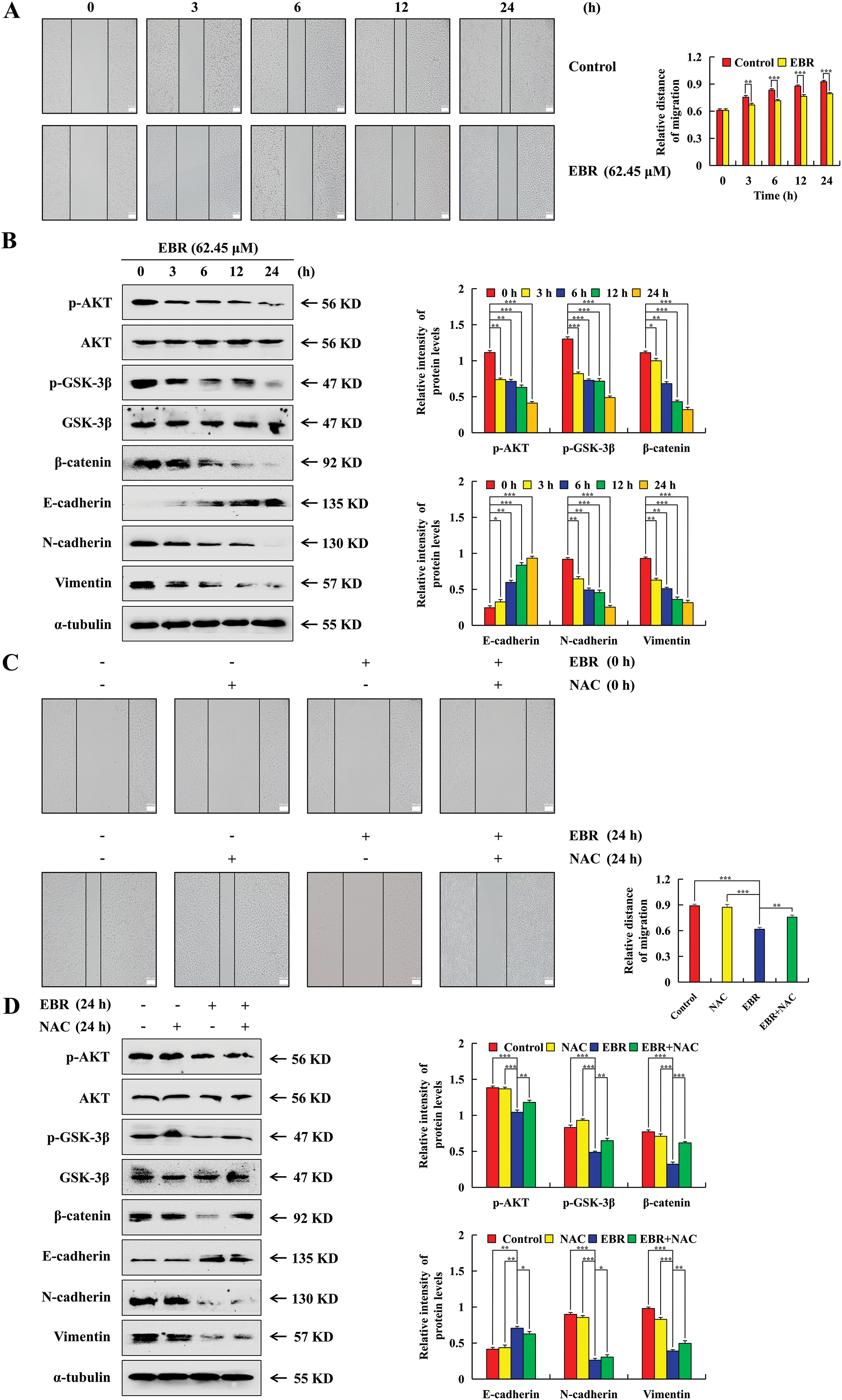
Figure 6: The migration of AGS cells was significantly inhibited by EBR treatment. At 3, 6, 12, and 24 h, the AGS cells were subjected to a treatment of 62.45 μM (IC50 value of AGS cells) EBR. (A) The assessment of cell migration after generating a linear scratch was conducted, and the results were observed at a 100× original magnification. (B) Migration-related protein expression was assessed. (C) At 0 and 24 h, the cell migration was observed under treatment with EBR (62.45 µM) with or without NAC (10 mM). (D) At 24 h, the expression of proteins was assessed under treatment with EBR (62.45 µM) with or without NAC (10 mM). For the purpose of ensuring accurate quantification in western blotting experiments, α-tubulin was selected to function as a loading control (*p < 0.05, **p < 0.01, ***p < 0.001 vs. 0 h or the NAC + EBR group. Data are reported as means and SD)
Because traditional chemotherapy drugs tend to induce strong resistance in GC treatment, a prolonged therapeutic effect is often not satisfactory [18]. This limits the long-term efficacy of treatment and increases the risk of disease recurrence and metastasis. In contrast, plant hormones are expected to be a promising alternative, with reported characteristics of low toxicity and low drug resistance [19,20]. Their unique molecular structures and mechanisms of action may interact with cancer cells in a distinct manner. Cisplatin is a chemotherapy drug that is extensively used in GC. Due to its excellent therapeutic effect, it was selected as the positive control treatment in this study. By comparing the effects of EBR and cisplatin, the potential of EBR as an anti-GC drug was rigorously evaluated. The research demonstrated that EBR treatment significantly reduced the survival rate of hepatocarcinoma cells, in which the IC50 values of EBR in HCCLM3 and HepG2 cells were 68.7 and 75.8 μM, respectively, after being treated for 24 h [7]. Here, we investigated the cytotoxic effect of EBR on GC cells. These results indicated that the IC50 values of EBR in AGS, MKN-45, and KATO-3 cells were 62.45, 75.88, and 67.58 μM. This demonstrated that EBR also exerts a cytotoxic effect on GC cells. The sensitivity of the three types of GC cell lines to EBR differed, which may be caused by the molecular differences among the three types of GC cell lines [21]. The results revealed that, compared to cisplatin, EBR exhibited a more significant cytotoxic impact on each of the three GC cells. We speculated that cisplatin exerts its anticancer effect by disrupting the cell membrane structure, whereas EBR, as a steroid hormone, is capable of directly penetrating the membranes of cancer cells. Chronopharmacology has revealed that the same dose of a drug exhibits varying effects depending on the time of administration, and time constitutes one of the crucial variables in biomedical research. Therefore, in our study, we proceeded to investigate the toxic effect of EBR on GC cells by using time as a variable. When AGS cells were treated with EBR for over 24 h, the cell apoptosis rate increased significantly, and a large number of AGS cells were observed to shed during the culture process. At this point, the evaluation ceased to be scientifically meaningful, and thus 24 h was selected as the termination time for cell experiments in this study. This time point allowed us to effectively evaluate the early-stage to mid-stage effects of EBR on GC cells without the confounding factors associated with excessive cell damage at later time points.
Apoptosis is a mechanism by which cells maintain environmental stability [22], and mitochondria, as the power source of cells, have an essential role in cell apoptosis. When mitochondrial membrane permeability is altered, the release of intracellular apoptosis-inducing factors can induce mitochondria-dependent apoptosis [23,24]. A previous study demonstrated that EBR induced apoptosis of pancreatic and colon cancer cells through endoplasmic reticulum stress pathways [25,26]. Our investigation delved into the apoptosis pathways activated in AGS cells. A decrease in the MMP, as was observed after EBR treatment, is a significant indicator of mitochondrial dysfunction and apoptosis [27]. This reduction in the MMP can be attributed to the disrupted mitochondrial membrane integrity [28]. The Bcl-2 family is essential for regulating mitochondrial-mediated apoptosis. With decreased Bcl-xl and increased Bad, the balance is tipped towards apoptosis and inducing the release of apoptotic factors [29,30]. Cle-caspase-3 is a key enzyme in the execution stage of apoptosis, and following EBR treatment, our findings indicated a marked upregulation of cle-caspase-3 expression in AGS cells. Its activation leads to the cleavage of cell substrates, including PARP, which is a signature event of apoptosis [31]. The findings demonstrated that EBR induced AGS cell apoptosis via the mitochondrial pathway. In addition, exploring potential combination therapies that leverage EBR’s apoptotic effects on AGS cells through the mitochondrial pathway could open new avenues for more effective GC treatment.
The phytoestrogen calycosin and EBR share structural similarities with vertebrate steroid hormones. Studies have indicated that GC cell apoptosis was induced by calycosin via the MAPK/STAT3/NF-κB pathway [32]. The MAPK signaling pathway was highly correlated with apoptosis in previous studies [33–35]. The interaction of STAT3 and NF-κB have been observed in numerous cancers from diverse origins [36]. We speculated whether EBR induced GC cell apoptosis via a similar signaling pathway. According to our research, western blotting results indicated that EBR triggered AGS cell apoptosis by the MAPK/STAT3/NF-κB pathway, which confirmed our conjecture. We speculated that vertebrate steroid hormones may be used to treat cancer. However, we currently lack sufficient data on their potential side effects and long-term efficacy. In addition to a thorough understanding of how these hormones affect normal physiological processes in cancer patients, long-term clinical studies are still needed to assess their therapeutic risks.
The accumulation of intracellular ROS threatens human health to a great extent and has a significant function in cell apoptosis [37,38]. EBR enhances antioxidant enzyme activity in plants [39]. However, due to large differences between cells, compounds can exhibit different antioxidant or pro-oxidative effects in different types of cells [40]. For example, other studies reported that intracellular ROS levels in pancreatic cancer and hepatocarcinoma cells increased after EBR treatment [7,25]. Similarly, we observed that ROS accumulation in AGS cells was positively correlated with EBR treatment time in this study. We speculated that the reason for this difference may be that the expression levels of antioxidant enzymes and other components of the antioxidant defense system vary among cell types. In AGS cells, EBR may inhibit the expression or activity of antioxidant enzymes. For example, EBR may interfere with transcription factors that regulate the expression of superoxide dismutase, catalase, or glutathione peroxidase genes, reducing the cell’s ability to scavenge ROS [41]. This inference needs to be further tested.
Studies have shown that many cancers result from changes in genomic structure induced by damage to DNA, and cells are capable of activating cell cycle arrest when they detect DNA damage [42,43]. Studies have shown that the G0/G1 phase cells in breast cancer increased after EBR treatment [44]. Contrary to the above findings, our study found that EBR increased the proportion of G2/M phase AGS cells. We speculated that this phenomenon may be caused by differences in signaling pathways. The downstream signaling cascade activated by DNA damage sensors in AGS cells may be different than that in breast cancer cells. This may lead to different patterns of cell cycle regulation in response to EBR [45]. Furthermore, our research revealed a strong correlation between the EBR-induced arrest of the AGS cell cycle and the accumulation of intracellular ROS. We speculated that ROS accumulation in cells can oxidize DNA bases, leading to mispairing during replication and subsequent mutations, and that this DNA damage causes cell cycle arrest [46,47]. However, this speculation requires validation in combination with molecular biology techniques in future studies.
Cell migration is one of the key elements that renders cancer treatment difficult [48]. Studies have shown that isoorientin inhibited migration of GC cells via downregulating p-AKT, p-GSK-3β, and β-catenin expression [49]. We speculated whether EBR inhibited AGS cell migration via a similar signaling pathway, and western blotting results confirmed our conjecture. Our study also showed that the inhibitory effect was regulated by ROS. Specifically, we observed that EBR-induced ROS generation led to the suppression of key migratory proteins, suggesting a redox-dependent mechanism. However, the study has several limitations. It is unclear whether EBR directly targets kinases involved in this pathway or acts indirectly through other signaling cascades. For instance, EBR might modulate the activity of PI3K, a key upstream activator of AKT, or interact with redox-sensitive proteins that indirectly regulate the pathway. Additionally, this experiment mainly used AGS cells for in vitro experiments. While in vitro studies provide valuable insights into the basic mechanisms, they may not fully recapitulate the complex in vivo environment. In future studies, we will work to address these limitations. In addition, our future goal is to investigate the potential synergistic effects of EBR with existing therapeutic agents to explore its utility in combination treatment strategies. These efforts will provide us with a more comprehensive understanding of EBR’s potential as a therapeutic agent for GC.
In summary, EBR exerted its anticancer effects in the GC AGS cell lines through ROS-mediated signaling pathways by inducing mitochondria-dependent apoptosis via the MAPK/STAT3/NF-κB pathway, triggering G2/M phase arrest via the AKT/CDK1/2/cyclin B1 pathway, and inhibiting migration via the AKT/GSK-3β/β-catenin pathway (Fig. 7). Consequently, we believe that EBR has excellent potential as a chemotherapy drug for GC treatment.
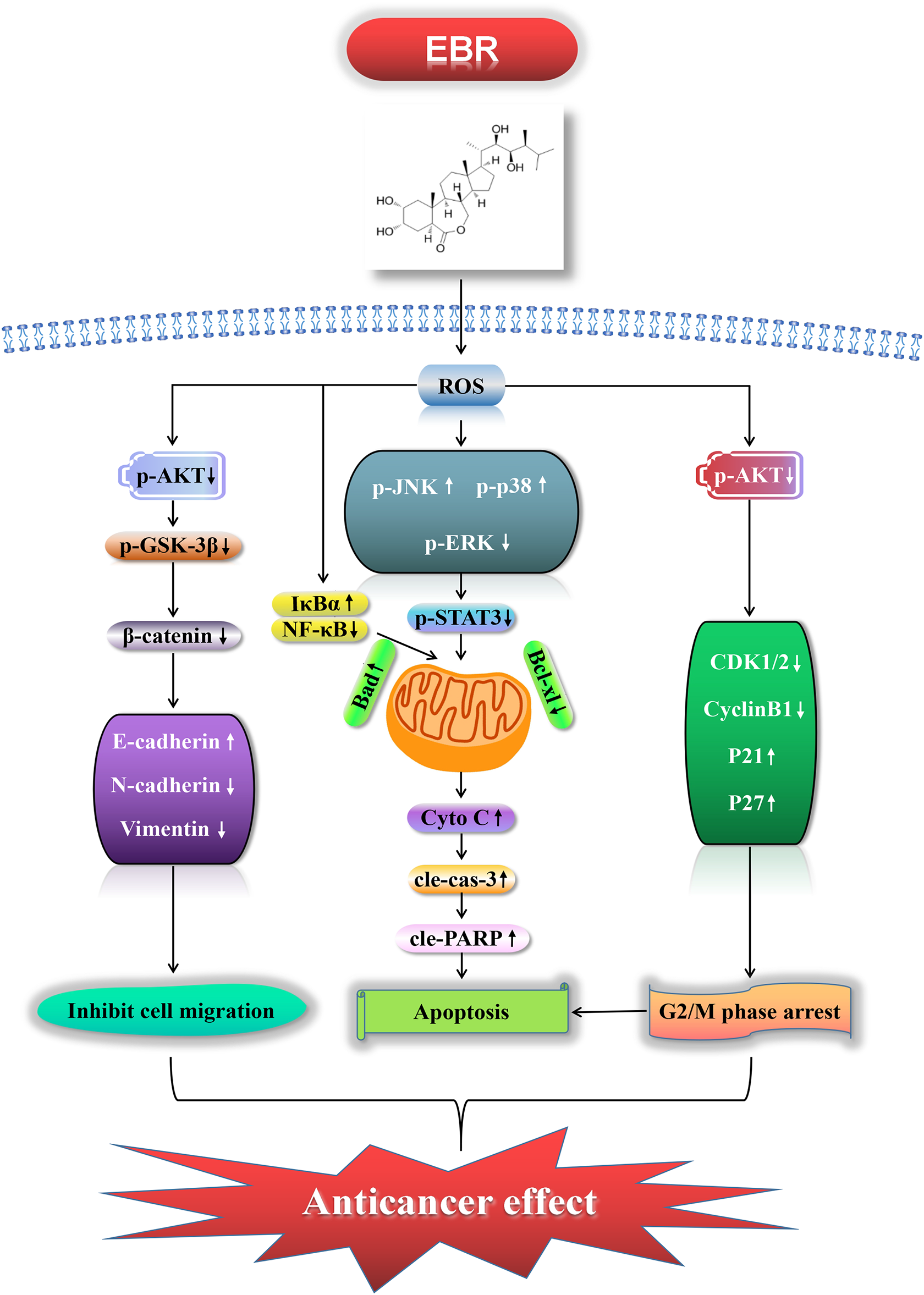
Figure 7: EBR exerted its anticancer effects in the GC AGS cell lines through ROS-mediated signaling pathways: it induced apoptosis via MAPK/STAT3/NF-κB, triggered G2/M phase arrest via AKT/CDK1/2/cyclin B1, and inhibited migration via AKT/GSK-3β/β-catenin
Acknowledgement: The authors are thankful for the financial support of the Heilongjiang Province Key Research and Development Plan Guidance Project and the Central Government Supports Local College Reform and Development Fund Talent Training Project.
Funding Statement: This work was supported by the Heilongjiang Province Key Research and Development Plan Guidance Project [Grant No. GZ20220039], and the Central Government Supports the Local College Reform and Development Fund Talent Training Project [Grant No. 2020GSP16].
Author Contributions: The authors confirm contributions to the study as follows: study conception and design: Chang Wang; data collection: Wei Sun; analysis and interpretation of results: Quan Quan and Wenshuang Hou; draft manuscript preparation: Zhi Zhang; and final draft revision: Chenghao Jin. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The datasets generated during the analyzed study are available from the corresponding author upon reasonable request.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Xu X, Chen J, Li W, Feng C, Liu Q, Gao W, et al. Immunology and immunotherapy in gastric cancer. Clin Exp Med. 2023;23(7):3189–204. doi:10.1007/s10238-023-01104-2. [Google Scholar] [PubMed] [CrossRef]
2. He Y, Zheng J, Ye B, Dai Y, Nie K. Chemotherapy-induced gastrointestinal toxicity: pathogenesis and current management. Biochem Pharmacol. 2023;216(1):115787. doi:10.1016/j.bcp.2023.115787. [Google Scholar] [PubMed] [CrossRef]
3. Avalbaev A, Yuldashev R, Fedorova K, Petrova N, Fedina E, Gilmanova R, et al. 24-epibrassinolide-induced growth promotion of wheat seedlings is associated with changes in the proteome and tyrosine phosphoproteome. Plant Biol. 2021;23(3):456–63. doi:10.1111/plb.13233. [Google Scholar] [PubMed] [CrossRef]
4. Zheng H, Ma J, Huang W, Di H, Xia X, Ma W, et al. Physiological and comparative transcriptome analysis reveals the mechanism by which exogenous 24-epibrassinolide application enhances drought resistance in potatoes (Solanum tuberosum L.). Antioxidants. 2022;11(9):1701. doi:10.3390/antiox11091701. [Google Scholar] [PubMed] [CrossRef]
5. Ismaili J, Boisvert M, Longpré F, Carange J, Le Gall C, Martinoli MG, et al. Brassinosteroids and analogs as neuroprotectors: synthesis and structure-activity relationships. Steroids. 2012;77(1–2):91–9. doi:10.1016/j.steroids.2011.10.009. [Google Scholar] [PubMed] [CrossRef]
6. Lanzarin GAB, Félix LM, Monteiro SM, Ferreira JM, Oliveira PA, Venâncio C. Anti-inflammatory, anti-oxidative and anti-apoptotic effects of thymol and 24-epibrassinolide in zebrafish larvae. Antioxidants. 2023;12(6):1297. doi:10.3390/antiox12061297. [Google Scholar] [PubMed] [CrossRef]
7. Zhou H, Zhuang W, Huang H, Ma N, Lei J, Jin G, et al. Effects of natural 24-epibrassinolide on inducing apoptosis and restricting metabolism in hepatocarcinoma cells. Phytomedicine. 2022;107:154428. doi:10.1016/j.phymed.2022.154428. [Google Scholar] [PubMed] [CrossRef]
8. Morana O, Wood W, Gregory CD. The apoptosis paradox in cancer. Int J Mol Sci. 2022;23(3):1328. doi:10.3390/ijms23031328. [Google Scholar] [PubMed] [CrossRef]
9. Obeng E. Apoptosis (programmed cell death) and its signals—a review. Braz J Biol. 2021;81(4):1133–43. doi:10.1590/1519-6984.228437. [Google Scholar] [PubMed] [CrossRef]
10. Alhoshani A, Alatawi FO, Al-Anazi FE, Attafi IM, Zeidan A, Agouni A, et al. BCL-2 inhibitor venetoclax induces autophagy-associated cell death, cell cycle arrest, and apoptosis in human breast cancer cells. Onco Targets Ther. 2020;13:13357–70. doi:10.2147/OTT.S281519. [Google Scholar] [PubMed] [CrossRef]
11. Wang Y, Qi H, Liu Y, Duan C, Liu X, Xia T, et al. The double-edged roles of ROS in cancer prevention and therapy. Theranostics. 2021;11(10):4839–57. doi:10.7150/thno.56747. [Google Scholar] [PubMed] [CrossRef]
12. Raza MH, Siraj S, Arshad A, Waheed U, Aldakheel F, Alduraywish S, et al. ROS-modulated therapeutic approaches in cancer treatment. J Cancer Res Clin Oncol. 2017;143(9):1789–809. doi:10.1007/s00432-017-2464-9. [Google Scholar] [PubMed] [CrossRef]
13. Lennicke C, Cochemé HM. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol Cell. 2021;81(18):3691–707. doi:10.1016/j.molcel.2021.08.018. [Google Scholar] [PubMed] [CrossRef]
14. Xiao L, Zhong M, Huang Y, Zhu J, Tang W, Li D, et al. Puerarin alleviates osteoporosis in the ovariectomy-induced mice by suppressing osteoclastogenesis via inhibition of TRAF6/ROS-dependent MAPK/NF-κB signaling pathways. Aging. 2020;12(21):21706–29. doi:10.18632/aging.103976. [Google Scholar] [PubMed] [CrossRef]
15. Chen W, Li P, Liu Y, Yang Y, Ye X, Zhang F, et al. Isoalantolactone induces apoptosis through ROS-mediated ER stress and inhibition of STAT3 in prostate cancer cells. J Exp Clin Cancer Res. 2018;37(1):309. doi:10.1186/s13046-018-0987-9. [Google Scholar] [PubMed] [CrossRef]
16. Kisselev PA, Panibrat OV, Sysa AR, Anisovich MV, Zhabinskii VN, Khripach VA. Flow-cytometric analysis of reactive oxygen species in cancer cells under treatment with brassinosteroids. Steroids. 2017;117(8):11–5. doi:10.1016/j.steroids.2016.06.010. [Google Scholar] [PubMed] [CrossRef]
17. Rizwan H, Pal S, Sabnam S, Pal A. High glucose augments ROS generation regulates mitochondrial dysfunction and apoptosis via stress signalling cascades in keratinocytes. Life Sci. 2020;241:117148. doi:10.1016/j.lfs.2019.117148. [Google Scholar] [PubMed] [CrossRef]
18. Song M, Cui M, Liu K. Therapeutic strategies to overcome cisplatin resistance in ovarian cancer. Eur J Med Chem. 2022;232:114205. doi:10.1016/j.ejmech.2022.114205. [Google Scholar] [PubMed] [CrossRef]
19. Chen L, Hou J, Hu XL, Zhang JZ, Wang HD. Environmental behaviors of plant growth regulators in soil: a review. Huan Jing Ke Xue. 2022;43(1):11–25. [Google Scholar] [PubMed]
20. Sadava D, Chen S. Castasterone, a plant steroid hormone, affects human small-cell lung cancer cells and reverses multi-drug resistance. Pharmaceuticals. 2023;16(2):170. doi:10.3390/ph16020170. [Google Scholar] [PubMed] [CrossRef]
21. Goyal Y, Busch GT, Pillai M, Li J, Boe RH, Grody EI, et al. Diverse clonal fates emerge upon drug treatment of homogeneous cancer cells. Nature. 2023;620(7974):651–9. doi:10.1038/s41586-023-06342-8. [Google Scholar] [PubMed] [CrossRef]
22. Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17(7):395–417. doi:10.1038/s41571-020-0341-y. [Google Scholar] [PubMed] [CrossRef]
23. Wang H, Zhang C, Li M, Liu C, Wang J, Ou X, et al. Antimicrobial peptides mediate apoptosis by changing mitochondrial membrane permeability. Int J Mol Sci. 2022;23(21):12732. doi:10.3390/ijms232112732. [Google Scholar] [PubMed] [CrossRef]
24. Morris JL, Gillet G, Prudent J, Popgeorgiev N. Bcl-2 family of proteins in the control of mitochondrial calcium signalling: an old chap with new poles. Int J Mol Sci. 2021;22(7):3730. doi:10.3390/ijms22073730. [Google Scholar] [PubMed] [CrossRef]
25. Obakan-Yerlikaya P, Mehdizadehtapeh L, Rencüzoğullari Ö, Kuryayeva F, Çevikli SS, Özağar Ş, et al. Gemcitabine in combination with epibrassinolide enhanced the apoptotic response in an ER stress-dependent manner and reduced the epithelial-mesenchymal transition in pancreatic cancer cells. Turk J Biol. 2022;46(6):439–57. doi:10.55730/1300-0152.2630. [Google Scholar] [PubMed] [CrossRef]
26. Obakan-Yerlikaya P, Arisan ED, Coker-Gurkan A, Adacan K, Ozbey U, Somuncu B, et al. Calreticulin is a fine tuning molecule in epibrassinolide-induced apoptosis through activating endoplasmic reticulum stress in colon cancer cells. Mol Carcinog. 2017;56(6):1603–19. doi:10.1002/mc.22616. [Google Scholar] [PubMed] [CrossRef]
27. Feng J, Ma Y, Chen Z, Hu J, Yang Q, Ding G. Mitochondrial pyruvate carrier 2 mediates mitochondrial dysfunction and apoptosis in high glucose-treated podocytes. Life Sci. 2019;237:116941. doi:10.1016/j.lfs.2019.116941. [Google Scholar] [PubMed] [CrossRef]
28. Sakamuru S, Zhao J, Attene-Ramos MS, Xia M. Mitochondrial membrane potential assay. Methods Mol Biol. 2022;2474:11–9. doi:10.1007/978-1-0716-2213-1. [Google Scholar] [CrossRef]
29. Czabotar PE, Garcia-Saez AJ. Mechanisms of BCL-2 family proteins in mitochondrial apoptosis. Nat Rev Mol Cell Biol. 2023;24(10):732–48. doi:10.1038/s41580-023-00629-4. [Google Scholar] [PubMed] [CrossRef]
30. Shi X, Wang P, Zhu Y, Li L, Yang T, Sun J, et al. Regulation of survivin and caspase/Bcl-2/Cyto-C signaling by TDB-6 induces apoptosis of colorectal carcinoma LoVo cells. J Gastrointest Oncol. 2022;13(5):2322–32. doi:10.21037/jgo-22-780. [Google Scholar] [PubMed] [CrossRef]
31. Zhou L, Wang S, Cao L, Ren X, Li Y, Shao J, et al. Lead acetate induces apoptosis in Leydig cells by activating PPARγ/caspase-3/PARP pathway. Int J Environ Health Res. 2021;31(1):34–44. doi:10.1080/09603123.2019.1625034. [Google Scholar] [PubMed] [CrossRef]
32. Zhang Y, Zhang JQ, Zhang T, Xue H, Zuo WB, Li YN, et al. Calycosin induces gastric cancer cell apoptosis via the ROS-mediated MAPK/STAT3/NF-κB pathway. Onco Targets Ther. 2021;14:2505–17. doi:10.2147/OTT.S292388. [Google Scholar] [PubMed] [CrossRef]
33. Pua LJW, Mai CW, Chung FF, Khoo AS, Leong CO, Lim WM, et al. Functional roles of JNK and p38 MAPK signaling in nasopharyngeal carcinoma. Int J Mol Sci. 2022;23(3):1108. doi:10.3390/ijms23031108. [Google Scholar] [PubMed] [CrossRef]
34. Guo Y, Lin D, Zhang M, Zhang X, Li Y, Yang R, et al. CLDN6-induced apoptosis via regulating ASK1-p38/JNK signaling in breast cancer MCF-7 cells. Int J Oncol. 2016;48(6):2435–44. doi:10.3892/ijo.2016.3469. [Google Scholar] [PubMed] [CrossRef]
35. Cook SJ, Stuart K, Gilley R, Sale MJ. Control of cell death and mitochondrial fission by ERK1/2 MAP kinase signalling. FEBS J. 2017;284(24):4177–95. doi:10.1111/febs.14122. [Google Scholar] [PubMed] [CrossRef]
36. Degoricija M, Situm M, Korać J, Miljković A, Matić K, Paradžik M, et al. High NF-κB and STAT3 activity in human urothelial carcinoma: a pilot study. World J Urol. 2014;32(6):1469–75. doi:10.1007/s00345-014-1237-1. [Google Scholar] [PubMed] [CrossRef]
37. Li Y, Xiu Y, Tang Y, Cao J, Hou W, Wang A et al. Sciadopitysin exerts anticancer effects on hepg2 hepatocellular carcinoma cells by regulating reactive oxygen species-mediated signaling pathways. BIOCELL. 2024;48(7):1055–69. doi:10.32604/biocell.2024.050515. [Google Scholar] [CrossRef]
38. Wang B, Wang Y, Zhang J, Hu C, Jiang J, Li Y, et al. ROS-induced lipid peroxidation modulates cell death outcome: mechanisms behind apoptosis, autophagy, and ferroptosis. Arch Toxicol. 2023;97(6):1439–51. doi:10.1007/s00204-023-03476-6. [Google Scholar] [PubMed] [CrossRef]
39. Wang X, Li H, Zhang S, Gao F, Sun X, Ren X. Interactive effect of 24-epibrassinolide and silicon on the alleviation of cadmium toxicity in rice (Oryza sativa L.) plants. Environ Technol. 2024;45(23):4725–36. doi:10.1080/09593330.2023.2283073. [Google Scholar] [PubMed] [CrossRef]
40. Wang Y, Qian J, Cao J, Wang D, Liu C, Yang R, et al. Antioxidant capacity, anticancer ability and flavonoids composition of 35 citrus (Citrus reticulata Blanco) varieties. Molecules. 2017;22(7):1114. doi:10.3390/molecules22071114. [Google Scholar] [PubMed] [CrossRef]
41. Ferreira de Oliveira JM, Costa M, Pedrosa T, Pinto P, Remédios C, Oliveira H, et al. Sulforaphane induces oxidative stress and death by p53-independent mechanism: implication of impaired glutathione recyclin. PLoS One. 2014;9:e92980. [Google Scholar] [PubMed]
42. Wu S, Bafna V, Chang HY, Mischel PS. Extrachromosomal DNA: an emerging hallmark in human cancer. Annu Rev Pathol. 2022;17(1):367–86. doi:10.1146/annurev-pathmechdis-051821-114223. [Google Scholar] [PubMed] [CrossRef]
43. Huang R, Zhou PK. DNA damage repair: historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct Target Ther. 2021;6(1):254. doi:10.1038/s41392-021-00648-7. [Google Scholar] [PubMed] [CrossRef]
44. Steigerová J, Oklešťková J, Levková M, Rárová L, Kolář Z, Strnad M. Brassinosteroids cause cell cycle arrest and apoptosis of human breast cancer cells. Chem Biol Interact. 2010;188(3):487–96. doi:10.1016/j.cbi.2010.09.006. [Google Scholar] [PubMed] [CrossRef]
45. Wang Z. Regulation of cell cycle progression by growth factor-induced cell signaling. Cells. 2021;10(12):3327. doi:10.3390/cells10123327. [Google Scholar] [PubMed] [CrossRef]
46. Srinivas US, Tan BWQ, Vellayappan BA, Jeyasekharan AD. ROS and the DNA damage response in cancer. Redox Biol. 2019;25(12):101084. doi:10.1016/j.redox.2018.101084. [Google Scholar] [PubMed] [CrossRef]
47. Clay DE, Fox DT. DNA damage responses during the cell cycle: insights from model organisms and beyond. Genes. 2011;12(12):1882. doi:10.3390/genes12121882. [Google Scholar] [PubMed] [CrossRef]
48. Yoon YJ, Han YM, Choi J, Lee YJ, Yun J, Lee SK, et al. Benproperine, an ARPC2 inhibitor, suppresses cancer cell migration and tumor metastasis. Biochem Pharmacol. 2019;163:46–59. doi:10.1016/j.bcp.2019.01.017. [Google Scholar] [PubMed] [CrossRef]
49. Zhang T, Xiu YH, Xue H, Li YN, Cao JL, Hou WS, et al. A mechanism of isoorientin-induced apoptosis and migration inhibition in gastric cancer AGS cells. Pharmaceuticals. 2022;15(12):1541. doi:10.3390/ph15121541. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools