
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

The Pathophysiologic Role of Oxidative Stress in Mitotic Cell Division
1 Department of Human Anatomy, Faculty of Basic Medical Sciences, University of Maiduguri, Maiduguri, 600233, Nigeria
2 Faculty of Medicine, Cyprus International University, Nicosia, 1010, Cyprus
* Corresponding Author: Nathan Isaac Dibal. Email:
(This article belongs to the Special Issue: Mitochondrial Dynamics and Oxidative Stress in Disease: Cellular Mechanisms and Therapeutic Targets)
BIOCELL 2025, 49(3), 419-435. https://doi.org/10.32604/biocell.2025.060565
Received 04 November 2024; Accepted 21 January 2025; Issue published 31 March 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Oxidative stress is characterized by elevated intracellular reactive oxygen species (ROS) levels. At physiological levels, ROS work as signaling molecules, helping cells go through the cell cycle normally and keeping their balance. They also balance several physiological processes. However, a shift in the delicate balance between antioxidants and ROS results in aberrant cell death and deleterious effects. Elevated ROS is implicated in many diseases and disorders like diabetes, autoimmune diseases, infertility, and cardiovascular disorders. The imbalance disrupts normal cellular functions, including cell division. ROS are important regulators of the cell cycle, exerting both favorable and harmful effects depending on their levels in the system, time of action, and cellular context. The present review article highlights the role of ROS as a predisposing factor of cell cycle arrest and its effect on various stages of the cell cycle. It also considers the role ROS plays in disorders that are caused by oxidative stress, presents the interplay between ROS and cell division and explores the therapeutic intervention beneficial in managing these disorders.Keywords
Abbreviations
| 1O2 | Singlet molecular oxygen |
| ACE | Angiotensin-converting enzyme |
| ATP | Adenosine triphosphate |
| cAMP | Cyclic adenosine monophosphate |
| CDK | Cyclin-dependent kinase |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| CVD | Cardiovascular disorders |
| DNA | Deoxyribonucleic acid |
| eNOS | Endothelial nitric oxide synthase |
| G0 | Quiescence |
| G1 | Gap 1 |
| G2 | Gap 2 |
| H2O2 | Hydrogen peroxide |
| HIT-T15 | Clonal beta-cell line |
| JNK | c-Jun-NH (2)-terminal kinase |
| LDL | Low-Density Lipoprotein |
| M | Mitosis |
| MAPK | Mitogen-activated protein kinase |
| NAC | N-acetyl-L-cysteine |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NOS | Nitric oxide synthase |
| OH | Hydroxyl radical |
| O2- | Superoxide anion radical |
| O3 | Ozone |
| RO | Akoxyl radical |
| R-O=O | Electronically excited carbonyl |
| ROO. | Peroxyl radical |
| ROOH | Hydroperoxide |
| ROS | Reactive oxygen species |
| SAC | Spindle assembly checkpoint |
| T2D | Type 2 diabetes |
Oxidative stress is a condition where an increase in reactive oxygen species (ROS) levels results in the damage of the cell membrane and organelles as well as some proteins, lipids, and nucleic acid. It is manifested when ROS production exceeds the antioxidant defense mechanisms [1]. Previous reports have shown that oxidative stress is associated with many human and animal diseases, including cancer, nephritis, diabetes, and neurological disorders. Failure of the antioxidant defense system that usually conditions all cell types to prevent oxidative stress enhances disease progression and complications [2–4]. Despite the discovery of many antioxidants in plants that play an essential role in the repair mechanisms aimed at preventing oxidative stress in biological systems, oxidative damage still causes tissue injury and cellular damage, leading to low quality of life. It is also associated with degenerative disorders [5].
ROS are radicals or molecules with unpaired electrons in their orbits, making them unstable [6]. ROS can be generated from external sources such as radiation, environmental pollutants, ultraviolet (UV) rays, alcohol, tobacco, and industrial smoke (exogenous ROS) or may be generated within the body cells (endogenous ROS) as byproducts of metabolism. The endogenous sources of ROS include mitochondria, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, xanthine oxidases, peroxisomes, endoplasmic reticulum, and endothelial nitric oxide synthase (Fig. 1) [7]. This is an indication that ROS could be generated by different tissues and organs of the body. The skeletal muscles, heart, and lungs were also reported to produce ROS, especially during exercise [8–10]. Previous studies reported that some phagocytes (macrophages and neutrophils) have enzymes responsible for the production of ROS in terms of tissue injury, and the ROS produced plays important roles in the replacement of worn-out tissues [11–13]. ROS can also be divided into non-radical and free-radical species. The non-radical species include hydrogen peroxide (H2O2), organic hydroperoxide (ROOH), singlet molecular oxygen (1O2), ozone (O3), electronically excited carbonyl (R-O=O), hypochlorous acid (HOCl), and hypobromous acid (HOBr), while the free radical ROS consists of hydroxyl radical (.OH), peroxyl radical (ROO.), superoxide anion radical (O2-), and alkoxyl radical (RO.) [14].
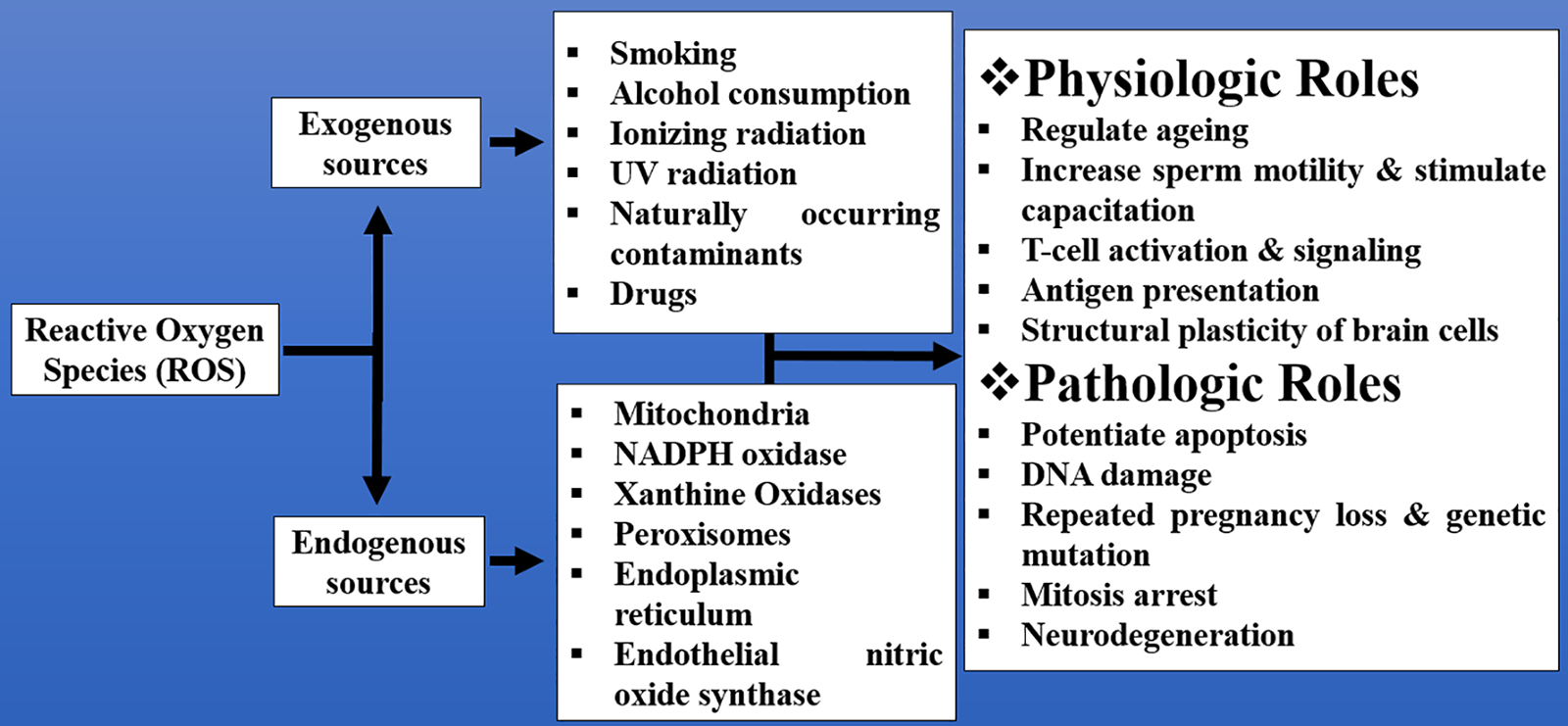
Figure 1: Examples of some exogenous and endogenous sources of ROS and their physiologic and pathologic roles that range from regulation of aging to neurodegeneration
ROS are important in the regulation of cell division; previous studies suggest that low concentrations of ROS stimulate cell proliferation while higher levels may lead to cell cycle arrest and apoptosis [15,16]. The cell cycle is a highly controlled process of cell growth, differentiation, and division that is influenced by several factors within or outside the cell [17]. ROS from different sources play a critical role in cell cycle regulation and cell division; the intracellular sources include cytochrome P450, mitochondria, p53, and peroxisomes, while the external sources include radiation and chemicals [15]. A balance between ROS and antioxidants is important in maintaining homeostasis. However, too much ROS or high antioxidants may alter the cell cycle through deoxyribonucleic acid (DNA) damage, apoptosis, and/or mitotic arrest [16]. The present review highlights the role of ROS as a predisposing factor of cell cycle arrest and its effect on various stages of the cell cycle. It also considers the role ROS plays in other disorders that are caused by oxidative stress. The review introduces the interplay between ROS and cell division and also explores the therapeutic intervention beneficial in the management of these disorders.
Mitosis is a type of cell division where eukaryotic cells give rise to two daughter cells that are genetically identical to the parent cell [18]. It is a cell cycle phase that involves equal segregation of genetic material and appropriate distribution of the cytoplasm [19]. The process is driven by microtubules, which are intracellular cytoskeletons, and cytokinesis, which brings about cell division. High levels of ROS in the body may increase chromosomal instability, leading to mitosis arrest [20,21]. Before mitosis, the cells undergo DNA and cellular component replication to produce what will be needed by the two daughter cells so that after cytokinesis, each cell can grow and divide [22]. The period is known as interphase and consists of the G1 (gap 1) phase, which is the period before DNA synthesis; the S phase, where DNA synthesis takes place; and the G2 (gap 2) phase, which is the period between DNA synthesis and mitosis. Mitosis is divided into five phases as follows: prophase, prometaphase, metaphase, anaphase, and telophase (Fig. 2). Prophase begins with chromosomal condensation into visible units, followed by spindle formation, and two pairs of centrioles move to opposite poles [23]. At prometaphase, kinetochores are formed and chromosomes are attached to the spindles at opposite poles by the microtubules. Nuclear membrane breaks. At metaphase, the centromere of chromosomes lines up at the spindle equator to produce a metaphase plate. Mitosis can be arrested at this phase with colchicine, and only cells with correctly assembled spindles move to anaphase. Hence, mitosis can be delayed at metaphase if chromosomes fail to move to the spindle equator. However, the spindle often corrects misalignment of chromosomes [24]. At anaphase, sister chromatids separate due to the breakdown of cohesion by a protease called separase. The sister chromatids that separate become chromosomes. Kinetochore microtubules shorten, and chromosomes move towards the spindle poles [25]. Mitotic division ends at telophase, where the chromosome arms contract, the nuclear membrane is formed, and the cytoplasm divides, resulting in the formation of two daughter cells [26]. The two most important functions of mitosis include serving as an asexual mode of reproduction in single-celled organisms and the replacement of worn-out/damaged tissues in multicellular organisms.
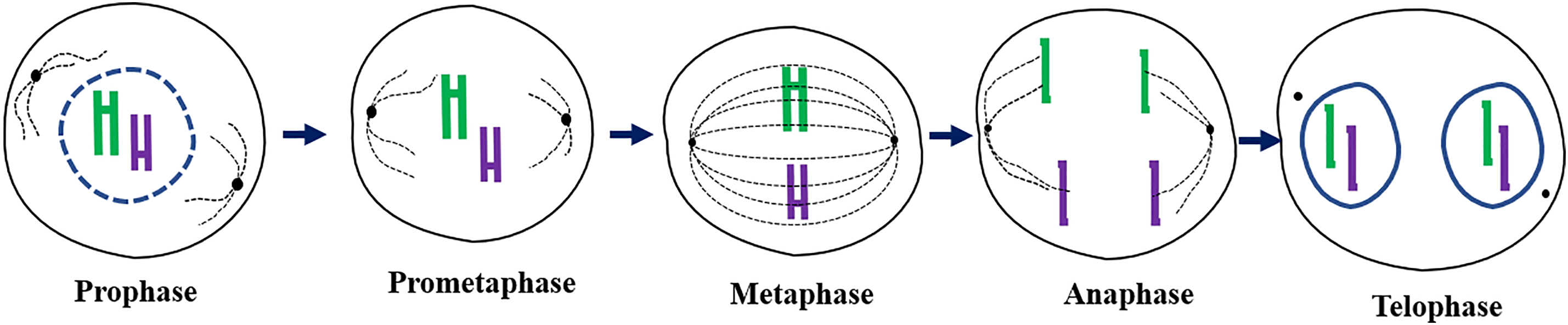
Figure 2: A schematic diagram highlighting the different phases of mitosis: prophase showing the disappearing nuclear membrane, prometaphase demonstrating the formation of kinetochores, metaphase showing the alignment of chromosomes along the equator and attachment to spindle fibers, anaphase demonstrating the separation of chromatids to opposite poles of the cell, and telophase showing the reappearance of the nuclear membrane and cytokinesis
3 Physiologic and Pathologic Role of ROS in Cell Division
ROS plays an important role in many physiologic and pathologic processes that promote growth and development, regulate homeostasis, and/or affect many biological functions such as aging, cognition, immune response, infertility, diabetes, cancer, cardiovascular disease, and neurodegeneration [7]. The exogenous and endogenous ROS as well as their physiologic and pathological roles are summarized in Fig. 1.
Aging is a biological process associated with a gradual decrease in the physiologic activities of an organism [27]. It can be influenced by environmental factors, diet, genetics, as well as increased ROS production [28]. ROS are produced as a result of endogenous and exogenous processes, with their adverse effects checkmated by antioxidant defenses. An imbalance between ROS and antioxidant defense leads to oxidative stress. The theory that oxidative stress accelerates aging is based on the hypothesis that age-related damages are attributed to ROS-induced damages [29]. These damages are caused by the oxidative damage to the intracellular macromolecules caused by ROS. In addition, ROS is implicated in many conditions that accelerate aging, including cardiovascular disorders (CVD), autoimmune disorders, neurodegenerative diseases, and cancer, to mention a few. Although the exact mechanism of the age-related effect of ROS is unclear, several studies reveal that ROS levels are high in the mitochondria of older animals [27]. Several other studies report that high ROS levels lead to cellular senescence. Senescent cells are cells that exist in a stable cell cycle arrest (remain arrested in the G1/G2 phase), become resistant to growth-promoting stimuli, do not die, but continue producing chemicals that trigger inflammation [30]. DNA lesions and cellular senescence are common in older mammals, suggesting that they are associated with the aging process [31,32].
The immune system protects plants and animals from micro-organisms and toxins by distinguishing host cells from foreign cells. To perform this task, they have to rely on both the innate and adaptive immune systems [33]. The two work together to eliminate pathogens and produce memory against future invaders/infections. ROS were reported to perform the role of second messenger for T-cell receptor activation and signaling as well as in chemotaxis and antigen presentation [34]. ROS initiates an inflammatory response by regulating cytokine production during infection. They can also trigger the defense system through autophagy, extracellular trap formation, and lymphocyte response [35]. Despite the numerous benefits of ROS in promoting immune response, they can cause pathologic effects on the immune system, leading to severe outcomes. Hydrogen peroxide, an exogenous ROS, was reported to reduce the capability of T-cells by decreasing the mitochondrial transmembrane potential followed by the release of cytochrome C [35]. ROS can also potentiate apoptosis by amplifying the release of apoptogenic factors from the mitochondria and increasing oligonucleosomal DNA fragmentation [36].
3.3 ROS and Autoimmune Diseases
ROS are formed in cells and tissues during oxidative phosphorylation in the respiratory system and/or by NADPH oxidases (NOX). NOX2 functions in a cell by mediating the oxidative burst in phagocytes [37]. On one hand, the ROS generated by NOX2 are very effective in physiological reactions; to break down ingested pathogens, ROS could accumulate within the cells. Accumulated ROS is implicated in cellular biomacromolecule damage and degenerative changes in tissues and influences various inflammatory responses if a suitable detoxification system does not balance their production. ROS also acts as a mediator between adaptive and innate immunity, influencing the antigen presentation and activating T-cells. They act as integral signaling molecules that are important in regulating autoimmune diseases and immune response [38]. Evidence from human and animal models of autoimmune disorders supports the role of ROS in preventing irregular autoimmune responses by supporting the preservation of macrophage efferocytosis and the different types of T helper cell balance. The failure of many anti-oxidation treatments indicates that ROS cannot be considered entirely harmful [38].
It is essential to point out that a precise redox balance is essential in sustaining an immune state that prevents the progress of overt autoimmunity and minimizes collateral tissue damage [37]. Studies have revealed that the absence or decrease in the level of ROS can lead to the development of immune-mediated inflammatory diseases as seen in animal models of autoimmune disorders (lupus, arthritis, and psoriasis). Results revealed ROS deficiency as a potent driver of pathogenesis [39]. On the contrary, in chronic stages, oxidative stress can still contribute to the progression of inflammation.
3.4 ROS in Fertility and Infertility
ROS is reported to increase motility, stimulate capacitation, augment sperm membrane binding to zona pellucida ZP-3 protein, and promote spermatozoa-oocyte fusion by hyperactivation through tyrosine phosphorylation [40,41]. Tyrosine phosphorylation is stimulated when ROS induces cyclic adenosine monophosphate (cAMP) in the spermatozoa to inhibit tyrosine phosphatase [42]. Ubiquinol, a reduced form of the coenzyme Q10 present in seminal fluid, is believed to decrease the level of ROS, leading to an increase in sperm count [43]. ROS is involved in physiological ovarian and uterine functions like oocyte maturation, implantation, blastocyst formation, steroidogenesis, and the maintenance of lutea in pregnancy. Furthermore, ROS modulates stromal cell physiology and ovarian germ cell integrity [44,45].
Although ROS have been implicated in the normal physiologic function of the spermatozoa, excessive production can lead to DNA damage preventing fertilization, increasing the risk of mutation in the newborns, and may lead to repeated pregnancy loss [46]. Decreased sperm motility may also occur due to lipid peroxidation in the sperm membrane decreasing the sperm flexibility and/or ROS-induced mitochondrial DNA damage that reduces adenosine triphosphate (ATP) and energy availability [47,48]. High ROS disrupts the mitochondrial membrane, causing the activation of caspase 5 and resulting in apoptosis. Cytochrome C release during apoptosis can further increase ROS levels, leading to more DNA damage and promoting apoptosis [49].
Regarding female infertility, ROS has been implicated in oocyte aging; its accumulation is reported to cause oocyte damage and result in female sub-fertility. An imbalance in ROS production and antioxidant enzymes leads to ROS accumulation resulting in reproductive disorders such as endometriosis, unexplained infertility, and polycystic ovarian syndrome [45].
3.5 ROS and Cardiovascular Disorders
CVD is described as a group of disorders that affect the blood vessels and heart. They include coronary artery disease, ischemic heart disease/angina, hypertension, stroke, myocardial infarction/heart attack, and atherosclerosis [50,51]. These diseases are caused by excessive production of ROS. CVD is one of the leading causes of death worldwide, with over 20 million deaths in 2021. More than 80% of the deaths occurred in low- and middle-income countries, where 1 in every 5 deaths is attributed to CVD [51–53].
ROS participate in cell signaling and are mediators and regulators of vascular function, acting as signaling molecules that regulate smooth muscle relaxation, contraction, and growth. They are produced by the mitochondria, NADPH, and xanthine oxidases, or during the uncoupling of nitric oxide synthase (NOS) in the adventitia, smooth muscle, and endothelial layers of vessels [51], and an imbalance brings about significant endothelial dysfunction and causes the changes in molecular pathways that promote the pathogenesis that develops into and leads to the progression of cardiovascular disorders [51]. Elevated ROS leads to decreased NO concentrations and vasoconstriction, which leads to arterial hypertension. ROS also deleteriously affects myocardial calcium handling, which leads to arrhythmia, and ROS enhances cardiac remodeling by bringing on hypertrophic signaling and apoptosis. ROS are also implicated in promoting the formation of atherosclerotic plaques [53]. The maintenance of balance between free-radical production and antioxidant defense is crucial in the prevention and treatment of CVD [51].
Type 2 diabetes (T2D) is a progressive disease characterized by sustained hyperglycemia and is frequently accompanied by hyperlipidemia [54]. In diabetic patients, chronic hyperglycemia and high levels of ROS further worsen beta-cell function and promote insulin resistance, leading to type II diabetes complications [55]. Oxidative stress is provoked, leading to the activation of the c-Jun-NH (2)-terminal kinase (JNK) pathway, which accelerates beta-cell dysfunction and subsequent insulin resistance. This occurs in a process known as glucose toxicity [56,57]. In many studies carried out, exposure of beta-cell-derived HIT-T15 (clonal beta-cell line) cells or isolated rat islet cells to oxidative stress caused a reduction in the expression of the insulin gene [58].
Other authors proposed that ROS derived from glucose metabolism acts as metabolic signaling molecules for glucose-stimulating insulin secretion in beta cells of the pancreas and that cellular accommodative response to oxidative stress challenge plays a seemingly contradictory role in pancreatic beta-cell function. Previous research suggests that antioxidant enzymes protect beta cells from oxidative injury and possible cell death. Thus, reducing oxidative stress-related impairment of insulin secretion. Another view indicates that the induction of antioxidant enzyme activation blunts glucose-triggered ROS signaling, thus resulting in reduced glucose-stimulated insulin secretion [59].
3.7 ROS and Cognitive Impairment
The hippocampus, prefrontal cortex, and cerebellum are considered the most vulnerable brain structures to oxidative stress because of their high ATP requirement, rendering them sensitive to ROS [60]. Numerous studies have reported the vulnerability of many brain regions to oxidative stress. The hippocampus was reported to undergo some biochemical changes in response to ROS and eventually lead to structural plasticity of brain cells and some level of regeneration and remodeling [61,62]. Astrocytes contribute to inflammatory responses within the central nervous system. They get inflamed in the presence of ROS, leading to astrogliosis, and the resultant effect induces neurodegeneration [63,64]. NOX2-derived ROS was reported to be the main cause of neuroinflammation in sepsis-associated encephalopathy, while NOX2 inhibition prevented glial cell activation and reduced inflammation and neurodegeneration in sepsis-associated encephalopathy [65].
ROS are formed as a by-product of normal cellular metabolism. Initial research on cancer cells showed that cancer cells had elevated levels of ROS compared with normal cells [66]. Since elevated levels of ROS cause damage to intracellular molecules: lipids, proteins, and DNA, it was believed that ROS were oncogenic, promoting genomic instability and tumorigenesis [67]. ROS, oxidative stress, and the redox state (oxidation state of an element, which determines its chemical and biological behavior, toxicity, and reactivity in the environment) may play an important role in controlling certain stages of the cell cycle [68]. Cancer is a disease where some of the body’s cells exhibit uncontrollable growth and spread to other parts of the body [69] and is characterized by uncontrolled cell proliferation. Changes in levels of ROS disrupt the redox state, leading to oxidative stress, which leads to the pathogenesis of many diseases like cancer [70].
In physiological conditions, cellular proliferation is controlled, and differentiated somatic or body cells differentiate into the organs they are fated to become. In many cases, daughter cells lose the ability to differentiate after undergoing a fixed number of cell divisions to maintain the integrity of the organism. Any changes to the normal physiological arrangement trigger programmed cell death (apoptosis) in the deviant cells [69]. Intracellular ROS levels are maintained at low levels to prevent alteration of internal homeostasis [71]. Elevated levels of ROS have been observed in cancer cells, and this has caused and accelerated rapid proliferation in cancer cells relative to normal cells [66,72,73]. The highly reactive and damaging nature of ROS in concentrations results in oxidative damage to proteins (proteotoxic stress) and nucleic acids, resulting in apoptosis and cell death [73–75]. In cancer cells, ROS concentration was found to vary during the various stages of the cell cycle. It was found to peak in mitosis, resulting in the accumulation of oxidized cysteine protein [73].
Prolonged mitotic arrest was found to increase ROS levels and further increase the amount of oxidatively damaged biomolecules in the cell, suggesting that agents that cause mitotic arrest may enhance the effect of ROS-dependent anticancer therapy [73]. ROS can also promote antitumorigenic signaling and trigger oxidative stress-induced cancer cell death [67]. Cancer cells, more than normal cells, have an altered redox intracellular environment, which makes them more sensitive to ROS levels and redox changes. Tightly regulated levels of ROS play a critical role in both normal and cancer cells as initiators and regulators of various cellular signaling pathways [73]. Cyclin-dependent kinase (CDK) inhibitors are being explored as possible remedies for cancer [76].
DNA damage and cell division were thought to be unconnected in the past because irradiation of animal cells during mitosis did not prevent the process, but the cell division continues without DNA repair [77]. However, recent studies suggest that DNA damage plays a significant role in mitosis by regulating its onset, spindle formation, correcting mismatched kinetochore microtubules, spindle checkpoint signaling, and cytokinesis completion [77]. DNA damage response may come in several ways, including DNA repair, cell cycle arrest, induction of apoptosis, and/or senescence [78]. When DNA damage occurs during mitosis as a result of oxidative stress, the cells try to repair the damaged DNA and continue with mitosis. However, if the DNA repair fails, the cell may undergo apoptosis. Apoptosis consists of a cascade of highly controlled physiological and anatomical events that involve chromosomal condensation and shrinkage of normal cells followed by membrane blebbing and disintegration of the cell into apoptotic bodies (Fig. 3). Apoptotic bodies are usually phagocytosed by macrophages. The link between ROS and mitosis could be explained by the schematic diagram in Fig. 4. A high level of ROS causes oxidative stress/damage that may lead to DNA damage; the damage will cause the cell to react in any of the following ways:
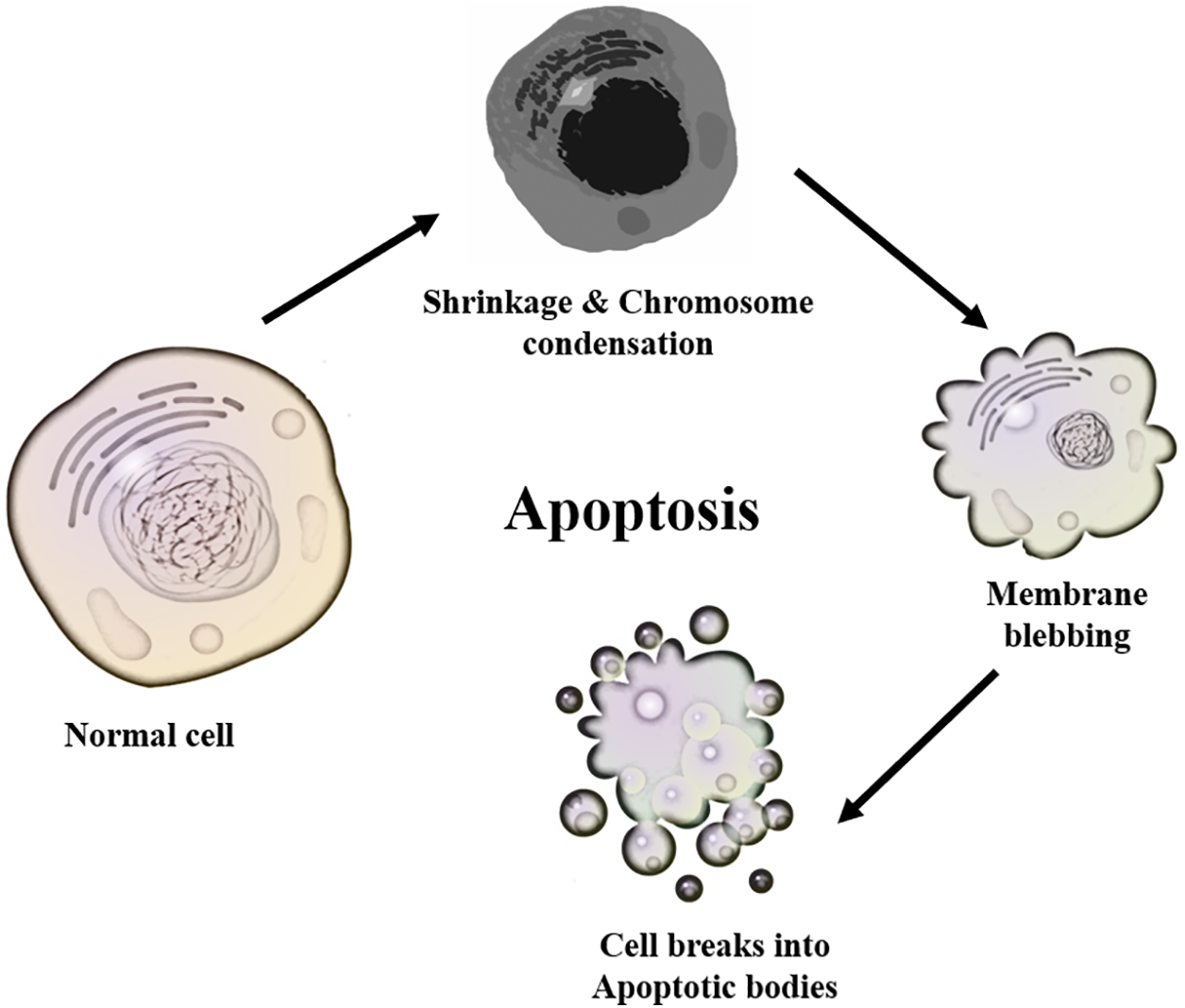
Figure 3: The apoptosis progression from the normal cell, cell shrinkage/chromosomal condensation, and membrane blebbing to the formation of apoptotic bodies
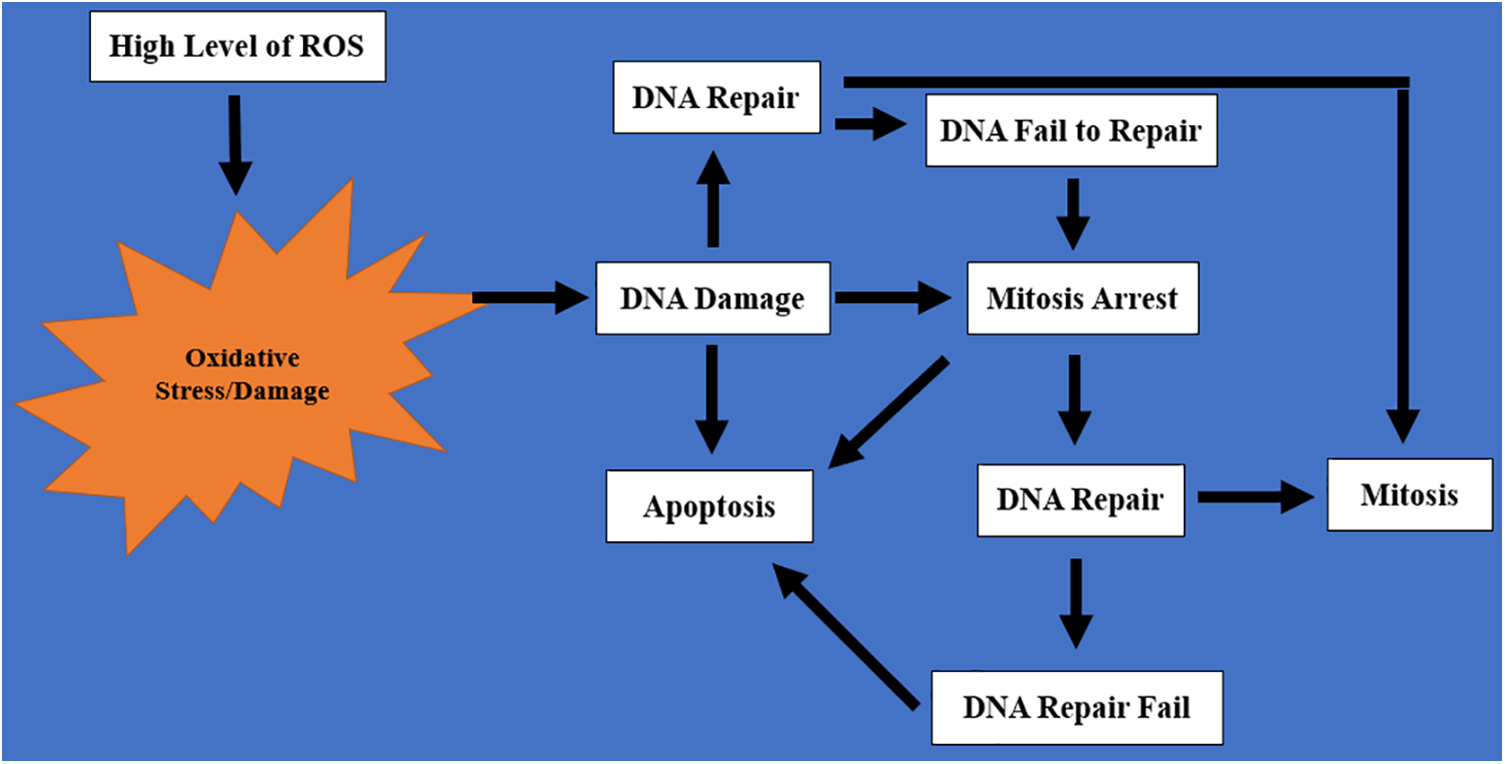
Figure 4: A schematic representation of the cascade of events that links ROS with mitosis. High levels of ROS cause oxidative stress that eventually leads to DNA damage. DNA damage will either result in DNA repair, mitosis arrest, or apoptosis. After DNA repair, mitosis resumes, but failure to repair the DNA will result in mitosis arrest and/or apoptosis
1. The cell will trigger DNA repair so that the DNA will repair itself and the cell will resume mitosis again.
2. Mitosis will be arrested, and there will be an attempt to repair the DNA. If the repair is successful, mitosis will resume. However, if the DNA repair fails after several attempts, it will result in mitosis arrest.
3. After mitosis arrest, the cell will no longer be useful in the body because of the lack of genetic material. This will result in apoptosis.
The mammalian cell cycle consists of four phases: M (mitosis), G1, S (synthesis), and G2, with a nonproliferative G0 (quiescence) phase. It also has some checkpoints as follows: G1 checkpoint between G1 and S phase, intra-S checkpoint within the S phase, and G2/M checkpoint between G2 and M phase (Fig. 5) [79]. ROS has an impact on the cell cycle at every stage, starting with the G0 phase, which is also known as the quiescence phase. Cells re-enter the cell cycle, and the process requires external signals like growth factors, hormones, and cytokines. All the external signals trigger ROS production [16]. Myc is a transcription factor encoded by the Myc proto-oncogene that controls DNA replication by stimulating quiescent cells to proliferate and activate genes responsible for cell cycle progression [80]. In the G1 phase, ROS at physiological concentrations activates pathways, assisting activation of Cyclin D1/CDKs and pRB/E2F (pRB proteins contain distinct binding sites for E2F proteins and for transcriptional repressor complexes) and eventually facilitating entry into the S phase of cell division [18]. The antioxidant N-acetyl-L-cysteine (NAC) induces G1 arrest by reducing the intracellular redox state [18]. However, in response to oxidative stress, cells arrest at the G1 restriction point or undergo apoptosis via activation of the p38 MAPK (mitogen-activated protein kinase) cascade. The p38 MAPK pathway is an important regulator of pro-inflammatory cytokine biosynthesis at the transcriptional and translational levels, which makes different components of this pathway promising targets for the treatment of autoimmune and inflammatory diseases [81]. There could also be an up-regulation of p16INK4a and the CDK inhibitors [18]. The CDKN2A gene is a tumor suppressor that encodes the CDK4/6 inhibitor p16INK4a. Loss of this tumor-suppressing gene contributes to the bypass of critical senescent signals and is related to the acceleration of malignant disease [82].
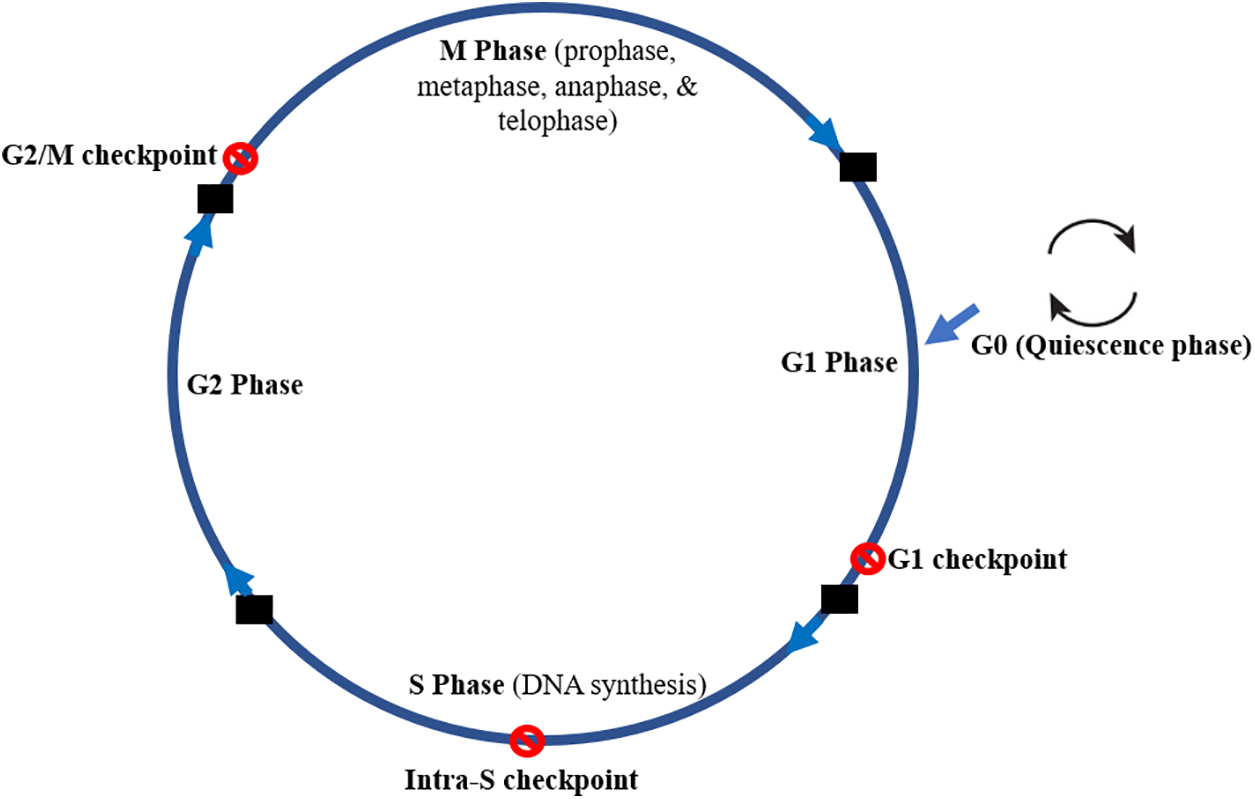
Figure 5: The mammalian cell cycle showing the different phases (M, S, G1 & G2) and the quiescence (G0) as well as the different checkpoints (G1 checkpoints, intra-S phase checkpoint, and G2/M checkpoint). The M phase is divided into prophase, metaphase, anaphase, and telophase. G0 is a phase with no proliferation while the checkpoints prevent progression to another phase when DNA is damaged
ROS levels vary following the cell cycle progression with the peak occurring at the period between S to G2/M phases of cell division [17,73]. In the S phase (synthesis phase), genetic material replication takes place, a necessary process that occurs during this phase is accurate proofreading and repairs of DNA errors to precisely duplicate DNA for transfer to daughter cells. Oxidative stress-induced DNA damage may affect the genetic material and alter the daughter cells. Several mechanisms are employed to recognize and repair the damaged DNA. CDK2 plays a vital role during the shift from the G1 to the S phase [16]. The S phase is activated by cyclin-dependent kinases (CDKs). During the G1 to S phase transition, cyclin D up-regulation displaces P16 binding to CDKs, resulting in their activation. The activated CDKs (CDK4 & CDK6) are phosphorylated; they inhibit retinoblastoma protein, causing E2F transcription factors activation that eventually initiates DNA replication [80]. p53 is a critical tumor suppressor that plays a key role in the regulating response to DNA damage. It can induce cell cycle arrest, DNA repair, and/or apoptosis when activated [82]. In oxidative stress, p53 activates genes that protect the cell from the detrimental effects of ROS. Several studies reveal that reduced p53 activity increases the accumulation of ROS [16,26].
The G2 phase is the gap between the S and M phases. It is the period where cells with replicated DNA are preparing to divide. It is very short in early embryonic and rapidly proliferating cells. The loss or downregulation of CDK activity during the G2 phase may lead to the reversal of the cell cycle [83]. The M phase is the period where the actual cell division takes place (duplicated DNA and cytoplasm divide equally to form two daughter cells). Microscopically it is identified as the period of chromosome condensation, segregation, nuclear division, and separation into two daughter cells and consisting of prophase, metaphase, anaphase, and telophase [26]. Cells that enter the M phase with damaged DNA will eventually die (mitotic catastrophe). This process is used in cancer therapy by forcing cells with damaged DNA or cells with DNA-damaging agents into the M phase [84,85]. In the G2 and M phases of the cell cycle, regulation is performed by the CDK2/cyclin A complex and also by CDKs [6,86]. ROS stimulates cell cycle progression in a context-related manner via the phosphorylation of CDKs and cell cycle regulatory molecules [6].
The G1 checkpoint prevents progression to the S phase and DNA replication when there is DNA damage. The intra-S phase checkpoint is activated to suppress CDK activity when DNA damage occurs in the S phase to limit DNA replication. The G2/M checkpoint prevents cells with DNA damage from entering mitosis [84]. However, no checkpoint is activated by DNA during mitosis in mammalian cells. The spindle assembly checkpoint (SAC) ensures appropriate chromosomal segregation. Hence, mitotic DNA damage does not usually result in cell cycle arrest unless it occurs in the telomeric or centromeric regions [79].
6 Therapeutic Strategies Involving ROS-Related Pathological Conditions
The therapeutic strategies include the usage of antioxidants, agents that increase cellular ROS levels, redox-active complexes, and regulators of redox-sensitive signaling pathways to alleviate the damage caused by ROS [16]. The therapeutic intervention involving the direct intake of exogenous antioxidants is beneficial. However, due to the fast responsiveness of oxidants, exogenous scavengers have a low performance compared to the endogenous molecules like superoxide dismutase and glutathione present in the biological system, which are more effective in removing ROS [16,87]. Fig. 6 presents the therapeutic and physiological roles of ROS and antioxidants.
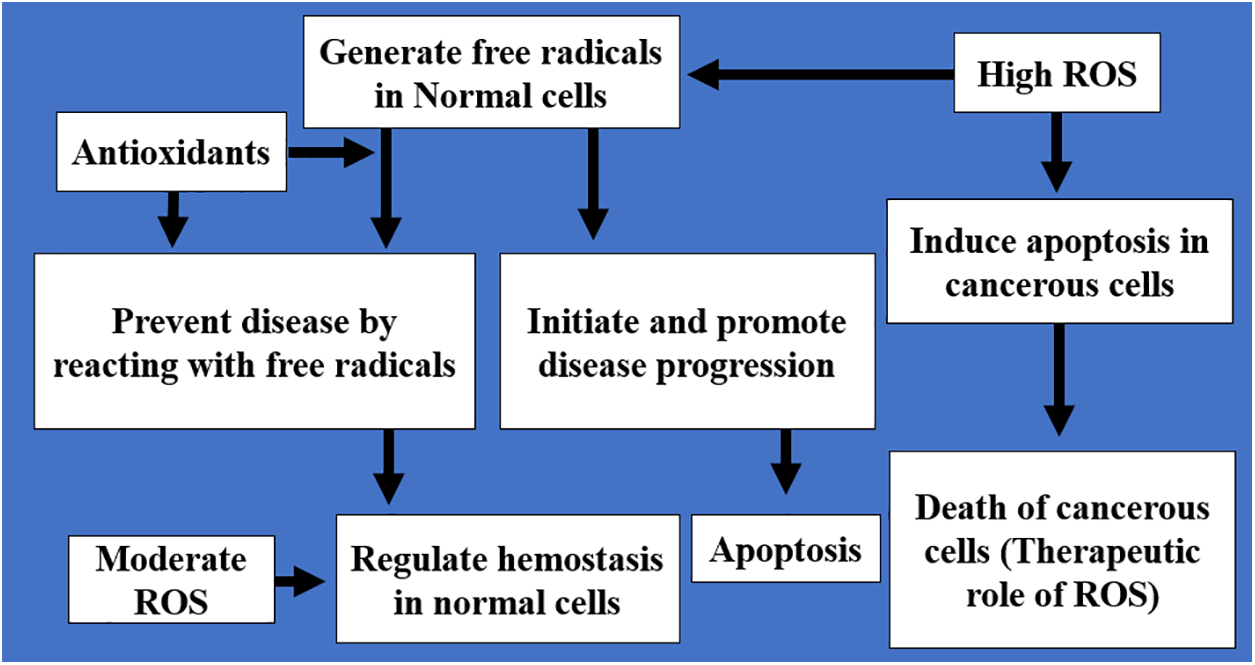
Figure 6: An illustration of the physiological and therapeutic roles of ROS and antioxidants. The therapeutic role involves either enhancing the antioxidant capacity of normal cells to reduce ROS and free radicals and prevent disease progression or promoting ROS production in cancerous cells to induce apoptosis. The physiologic role involves the regulation of hemostasis
In the management of autoimmune disorders, antioxidant therapy is helpful. However, an adjusted redox balance is essential in sustaining an immune state that prevents the progress of overt autoimmunity and attenuates chronic stages of disease [39,88]. Therapeutic strategies suggested in cases of elevated ROS in CVD are aimed at stopping the abnormal production of ROS and concomitantly improving antioxidant levels by increasing the intake of diet-rich antioxidants like vitamins C, lycopene, and quercetin, mostly found in fruits and vegetables. They have been proven to prevent the development of various types of CVD, including heart failure, ventricular remodeling, myocardial infarction, ischemia-reperfusion heart injury, and atherosclerosis [89,90]. Many natural products have been reported in several in vivo studies as rich sources of antioxidants that enhance the antioxidant activities of the cells, resulting in decreased free radical production. Hence, preventing the initiation and progression of many diseases such as diabetes, hypertension, and cardiovascular disorders at the cellular level [91–94].
Pharmacological agents like angiotensin-converting enzyme (ACE) inhibitors and statins demonstrated pleiotropic effects in reducing oxidative stress [89]. The ability of statins to ameliorate CVD is demonstrated by their ability to enhance the expression of endothelial nitric oxide synthase (eNOS). Thus, exhibiting endothelial-protecting effects mainly by reducing inflammation [53]. Factors responsible for ROS-related endothelial damage, such as oxidized low-density lipoprotein (LDL) and hypoxia, can decrease eNOS expression. However, some statins can reverse the eNOS downregulation, stressing their capacity to upgrade the vessel nitric oxide bioavailability and atherosclerotic plaque stability [95].
ACE inhibitors are drugs that are produced to treat hypertension. During arterial wall remodeling, amplified angiotensin II activity results in the thickening of the tunica media and narrowing of blood vessels, a key sign of atherosclerosis. An increase in the angiotensin II level is linked with the release of vascular superoxide anion [96]. ACE inhibitors are beneficial in treating several CVDs, including acute myocardial infarction, hypertension, heart failure, and systolic and diastolic left ventricular hypertension, and have been shown to improve the quality of life and survival rate and reduce morbidity and mortality connected to CVD [97]. Antioxidants may serve as a novel mechanism-based therapy for type II diabetes. Therapeutic intervention using antioxidants in in vivo research revealed a significant increase in beta-cell mass in diabetic rats. The intervention also suppressed beta-cell apoptosis while maintaining normal beta-cell proliferation [58]. Treatment with antioxidants also preserved insulin levels and insulin mRNA, significantly preventing insulin degranulation compared to untreated diabetic rats. The expression of the pancreatic and duodenal homeobox factor-1, which is an important transcription factor for the insulin gene, was accelerated in insulin cells after the administration of antioxidants [56–58].
A promising strategy for the elimination of cancer cells can be to target ROS levels in cancer cells [67,98]. In cancer treatment, high ROS levels have been shown to induce apoptosis in cancer cells [17]. Treatment of cancer cells with pro-oxidant agents (quercetin, elesclomol, cucurbitacin B) has been observed to selectively increase ROS levels in the cancer cells, which eventually leads to DNA damage, dysregulation of the cell cycle, mitosis arrest, apoptosis, and cell death. This creates a treatment therapy route that can be combined with other cancer therapies such as chemotherapy and radiotherapy to effectively eliminate cancerous cells in the tissues, organs, and systems [99]. Quercetin was also reported to enhance antioxidant activity and prevent oxidative stress, neurodegeneration, and steatosis [100,101].
7 Future Directions for Studying ROS
We reviewed the interplay between ROS and cell division and also explored the therapeutic intervention beneficial in the management of these disorders. There is a complex relationship between the ROS cell cycle and mitosis. Hence, there is a need to further explore the mechanism of the physiologic and pathologic changes associated with ROS as well as the role of ROS in the treatment and prevention of diseases that are linked to cell regeneration and proliferation.
The present study reviewed the physiologic and pathologic roles ROS plays and how it is associated with the regulation of mitotic division through DNA damage, repair, and remodeling, as well as its participation in several pathological conditions along with proposed therapeutic interventions. The conclusions point to the fact that the generation of moderate levels of ROS may function as signals to promote cell proliferation and survival when balanced by endogenous and exogenous antioxidants. However, a shift in the balance from redox homeostasis towards an elevated ROS level leads to disease initiation, progression state, apoptosis, and cellular death. Maintenance of redox homeostasis sustains proper cellular function and alleviates the effects of oxidative stress.
Acknowledgement: None.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Nathan Isaac Dibal and Martha Orendu Oche Attah; literature search: Nathan Isaac Dibal and Martha Orendu Oche Attah; draft manuscript preparation: Nathan Isaac Dibal; review of the manuscript for important intellectual content: Martha Orendu Oche Attah. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Ashrafizadeh M, Ahmadi Z, Samarghandian S, Mohammadinejad R, Yaribeygi H, Sathyapalan T, et al. MicroRNA-mediated regulation of Nrf2 signalling pathway: implications in disease therapy and protection against oxidative stress. Life Sci. 2020;244(4):117329. doi:10.1016/j.lfs.2020.117329. [Google Scholar] [PubMed] [CrossRef]
2. Chaudhary P, Janmeda P, Docea AO, Yeskaliyeva B, Abdull Razis AF, Modu B, et al. Oxidative stress, free radicals and antioxidants: potential crosstalk in the pathophysiology of human diseases. Front Chem. 2023;11:1158198. doi:10.3389/fchem.2023.1158198. [Google Scholar] [PubMed] [CrossRef]
3. Reddy VP. Oxidative stress in health and disease. Biomedicines. 2023;11:2925. doi:10.3390/biomedicines11112925. [Google Scholar] [PubMed] [CrossRef]
4. Stanciu GD, Bild V, Ababei DC, Rusu RN, Cobzaru A, Paduraru L, et al. Link between diabetes and alzheimer’s disease due to the shared amyloid aggregation and deposition involving both neurodegenerative changes and neurovascular damages. J Clin Med. 2020;9(6):1713. doi:10.3390/jcm9061713. [Google Scholar] [PubMed] [CrossRef]
5. dos Santos JM, Lacerda ACR, Ribeiro VGC, Figueiredo PHS, Fonseca SF, da Silva Lage VK, et al. Oxidative stress biomarkers and quality of life are contributing factors of muscle pain and lean body mass in patients with fibromyalgia. Biology. 2022;11(6):935. doi:10.3390/biology11060935. [Google Scholar] [PubMed] [CrossRef]
6. Kirova DG, Judasova K, Vorhauser J, Zerjatke T, Leung JK, Glauche I, et al. A ROS-dependent mechanism promotes CDK2 phosphorylation to drive progression through S phase. Dev Cell. 2022;57(14):1712–27. doi:10.1016/j.devcel.2022.06.008. [Google Scholar] [PubMed] [CrossRef]
7. Jomova K, Alomar SY, Alwasel SH, Nepovimova E, Kuca K, Valko M. Several lines of antioxidant defense against oxidative stress: antioxidant enzymes, nanomaterials with multiple enzyme-mimicking activities, and low-molecular-weight antioxidants. Arch Toxicol. 2024;98(5):1323–67. doi:10.1007/s00204-024-03696-4. [Google Scholar] [PubMed] [CrossRef]
8. Palma FR, Gantner BN, Sakiyama MJ, Kayzuka C, Shukla S, Lacchini R, et al. ROS production by mitochondria: function or dysfunction? Oncogene. 2024;43(5):295–303. doi:10.1038/s41388-023-02907-z. [Google Scholar] [PubMed] [CrossRef]
9. Xu H, Brown JL, Bhaskaran S, Remmen HV. Reactive oxygen species in the pathogenesis of sarcopenia. Free Radic Biol Med. 2025;227(3097):446–58. doi:10.1016/j.freeradbiomed.2024.11.046. [Google Scholar] [PubMed] [CrossRef]
10. Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev. 2008;88(4):1243–76. doi:10.1152/physrev.00031.2007. [Google Scholar] [PubMed] [CrossRef]
11. Nikolaidis MG, Jamurtas AZ. Blood as a reactive species generator and redox status regulator during exercise. Arch Biochem Biophys. 2009;490(2):77–84. doi:10.1016/j.abb.2009.08.015. [Google Scholar] [PubMed] [CrossRef]
12. Canton M, Sanchez-Rodrıguez R, Spera I, Venegas FC, Favia M, Viola A, et al. Reactive oxygen species in macrophages: sources and targets. Front Immunol. 2021;12:734229. doi:10.3389/fimmu.2021.734229. [Google Scholar] [PubMed] [CrossRef]
13. Oliveira de Almeida AJP, Lúcio de Oliveira JCP, Pontes LVP, Júnior JFS, Gonçalves TAF, Dantas SH, et al. ROS: basic concepts, sources, cellular signalling, and its implications in aging pathways. Oxid Med Cell Longev. 2022;2022:1225578. doi:10.1155/2022/1225578. [Google Scholar] [PubMed] [CrossRef]
14. Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. 2020;21(7):363–83. doi:10.1038/s41580-020-0230-3. [Google Scholar] [PubMed] [CrossRef]
15. Guo W, Xing Y, Luo X, Li F, Ren M, Liang Y. Reactive oxygen species: a crosslink between plant and human eukaryotic cell systems. Int J Mol Sci. 2023;24(17):13052. doi:10.3390/ijms241713052. [Google Scholar] [PubMed] [CrossRef]
16. Mackova V, Raudenska M, Polanska HH, Jakubek M, Masarik M. Navigating the redox landscape: reactive oxygen species in regulation of cell cycle. Redox Rep. 2024;29(1):2371173. doi:10.1080/13510002.2024.2371173. [Google Scholar] [PubMed] [CrossRef]
17. Ivanova JS, Pugovkina NA, Neganova IE, Kuzhukharova IV, Nikolsky NN, Lyublinskaya OG. Cell cycle-coupled changes in the level of reactive oxygen species support the proliferation of human pluripotent stem cells. Stem Cells. 2021;39(12):1671–87. doi:10.1002/stem.3450. [Google Scholar] [PubMed] [CrossRef]
18. Wang GF, Dong Q, Bai Y, Yuan J, Xu Q, Cao C, et al. Oxidative stress induces mitotic arrest by inhibiting Aurora A-involved mitotic spindle formation. Free Radic Biol Med. 2017;103(1):177–87. doi:10.1016/j.freeradbiomed.2016.12.031. [Google Scholar] [PubMed] [CrossRef]
19. Atalay PB, Kaluc N, Elif Ergin Cavusoglu EE. Effect of mitosis on the resistance to oxidative and osmotic stresses in yeast. SAUJS. 2020;24(1):265–71. doi:10.16984/saufenbilder.559820. [Google Scholar] [CrossRef]
20. Chen G, Li Z, Iemura K, Tanaka K. Oxidative stress induces chromosomal instability through replication stress in fibroblasts from aged mice. J Cell Sci. 2023;136(11):260688. doi:10.1242/jcs.260688. [Google Scholar] [PubMed] [CrossRef]
21. Hosea R, Hillary S, Naqvi S, Wu S, Kasim V. The two sides of chromosomal instability: drivers and brakes in cancer. Signal Transduct Target Ther. 2024;9(1):75. doi:10.1038/s41392-024-01767-7. [Google Scholar] [PubMed] [CrossRef]
22. McIntosh JR. Mitosis. Cold Spring Harb Perspect Biol. 2016;8:a023218. [Google Scholar] [PubMed]
23. Schneider MWG, Gibson BA, Otsuka S, Spicer MFD, Petrovic M, Blaukopf C, et al. A mitotic chromatin phase transition prevents perforation by microtubules. Nature. 2022;609(7925):183–90. doi:10.1038/s41586-022-05027-y. [Google Scholar] [PubMed] [CrossRef]
24. Kitamura E, Tanaka K, Komoto S, Kitamura Y, Antony C, Tanaka TU. Kinetochores generate microtubules with distal plus ends: their roles and limited lifetime in mitosis. Dev Cell. 2020;18(2):248–59. doi:10.1016/j.devcel.2009.12.018. [Google Scholar] [PubMed] [CrossRef]
25. Pati D. Role of chromosomal cohesion and separation in aneuploidy and tumorigenesis. Cell Mol Life Sci. 2024;81(1):100. doi:10.1007/s00018-024-05122-5. [Google Scholar] [PubMed] [CrossRef]
26. O’Connor C. Cell division: stages of mitosis. Nat Edu. 2008;1(1):188. [Google Scholar]
27. Maldonado E, Morales-Pison S, Urbina F, Solari A. Aging hallmarks and the role of oxidative stress. Antioxidants. 2023;12(3):651. doi:10.3390/antiox12030651. [Google Scholar] [PubMed] [CrossRef]
28. Li Y, Tian X, Luo J, Bao T, Wang S, Wu X. Molecular mechanisms of aging and anti-aging strategies. Cell Commun Signal. 2024;22(1):285. doi:10.1186/s12964-024-01663-1. [Google Scholar] [PubMed] [CrossRef]
29. Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D, et al. Oxidative stress, ageing, and diseases. Clin Interv Aging. 2018;13:757–72. doi:10.2147/CIA. [Google Scholar] [CrossRef]
30. Huang W, Hickson LJ, Eirin A, Kirkland JL, Lerman LO. Cellular senescence: the good, the bad and the unknown. Nat Rev Nephrol. 2022;18(10):611–27. doi:10.1038/s41581-022-00601-z. [Google Scholar] [PubMed] [CrossRef]
31. Lima T, Li TY, Mottis A, Auwerx J. Pleiotropic effects of mitochondria in aging. Nat Aging. 2022;2(3):199–213. doi:10.1038/s43587-022-00191-2. [Google Scholar] [PubMed] [CrossRef]
32. Niedernhofer LJ, Gurkar AU, Wang Y, Vijg J, Hoeijmakers JH, Robbins PD. Nuclear genomic instability and aging. Annu Rev Biochem. 2018;87(1):295–322. doi:10.1146/annurev-biochem-062917-012239. [Google Scholar] [PubMed] [CrossRef]
33. Bassoy EY, Walch M, Martinvalet D. Reactive oxygen species: do they play a role in adaptive immunity? Front Immunol. 2021;12:755856. doi:10.3389/fimmu.2021.755856. [Google Scholar] [PubMed] [CrossRef]
34. Tavassolifar MJ, Vodjgani M, Salehi Z, Izad M. The influence of reactive oxygen species in the immune system and pathogenesis of multiple sclerosis. Autoimmune Dis. 2020;5793817(2):1–14. doi:10.1155/2020/5793817. [Google Scholar] [PubMed] [CrossRef]
35. Martinvalet D, Walch M. Editorial: the role of reactive oxygen species in protective immunity. Front Immunol. 2022;12:832946. doi:10.3389/fimmu.2021.832946. [Google Scholar] [PubMed] [CrossRef]
36. Khan AQ, Rashid K, Al Amodi AA, Agha MV, Akhtar S, Hakeem I, et al. Reactive oxygen species (ROS) in cancer pathogenesis and therapy: an update on the role of ROS in anticancer action of benzophenanthridine alkaloids. Biomed Pharmacother. 2021;143(10152):112142. doi:10.1016/j.biopha.2021.112142. [Google Scholar] [PubMed] [CrossRef]
37. Hoffmann MH, Griffiths HR. The dual role of Reactive Oxygen Species in autoimmune and inflammatory diseases: evidence from preclinical models. Free Radic Biol Med. 2018;125:62–71. doi:10.1016/j.freeradbiomed.2018.03.016. [Google Scholar] [PubMed] [CrossRef]
38. Lin W, Shen P, Song Y, Huang Y, Tu S. Reactive oxygen species in autoimmune cells: function, differentiation, and metabolism. Front Immunol. 2021;12:1–16. doi:10.3389/fimmu.2021.635021. [Google Scholar] [PubMed] [CrossRef]
39. Kienhöfer D, Boeltz S, Hoffmann MH. Reactive oxygen homeostasis—the balance for preventing autoimmunity. Lupus. 2016;25(8):943–54. doi:10.1177/0961203316640919. [Google Scholar] [PubMed] [CrossRef]
40. Barati E, Nikzad H, Karimia M. Oxidative stress and male infertility: current knowledge of pathophysiology and role of antioxidant therapy in disease management. Cell Mol Life Sci. 2020;77(1):93–113. doi:10.1007/s00018-019-03253-8. [Google Scholar] [PubMed] [CrossRef]
41. Cito G, Becatti M, Natali A, Fucci R, Picone R, Cocci A, et al. Redox status assessment in infertile patients with non-obstructive azoospermia undergoing testicular sperm extraction: a prospective study. Andrologia. 2020;8(2):364–71. doi:10.1111/andr.12721. [Google Scholar] [PubMed] [CrossRef]
42. Miguel-Jiménez S, Pina-Beltrán B, Gimeno-Martos S, Carvajal-Serna M, Casao A, Pérez-Pe R. NADPH oxidase 5 and melatonin: involvement in ram sperm capacitation. Front Cell Dev Biol. 2021;9:655794. doi:10.3389/fcell.2021.655794. [Google Scholar] [PubMed] [CrossRef]
43. Alleva R, Scararmucci A, Mantero F, Bompadre S, Leoni L, Littarru G. The protective role of ubiquinol-10 against the formation of lipid hydroperoxides in human seminal fluid. Mol Aspects Med. 1997;18:221–8. doi:10.1016/s0098-2997(97)00040-x. [Google Scholar] [PubMed] [CrossRef]
44. Martin JH, Nixon B, Café SL, Aitken RJ, Bromfield EG, Lord T. Oxidative stress and in vitro ageing of the post-ovulatory oocyte: an update on recent advances in the field. Reprod. 2022;164:F109–24. doi:10.1530/REP-22-0206. [Google Scholar] [PubMed] [CrossRef]
45. Su Z, Ding P, Su W, Li X, Li Y, Li X, et al. Association between oxidative balance score and female infertility from the national health and nutrition examination survey 2013–2018. Front Endocrinol. 2024;15:1–14. doi:10.3389/fendo.2024.1386021. [Google Scholar] [PubMed] [CrossRef]
46. Hussain T, Kandeel M, Metwally E, Murtaza G, Kalhoro DH, Yin Y, et al. Unraveling the harmful effect of oxidative stress on male fertility: a mechanistic insight. Front Endocrinol. 2023;14:1070692. doi:10.3389/fendo.2023.1070692. [Google Scholar] [PubMed] [CrossRef]
47. Castleton PE, Deluao JC, Sharkey DJ, McPherson NO. Measuring reactive oxygen species in semen for male preconception care: a scientist perspective. Antioxidants. 2022;11:264. doi:10.3390/antiox11020264. [Google Scholar] [PubMed] [CrossRef]
48. Qamar AY, Naveed MI, Raza S, Fang X, Roy PK, Bang S, et al. Role of antioxidants in fertility preservation of sperm—a narrative review. Anim Biosci. 2023;36(3):385–403. doi:10.5713/ab.22.0325. [Google Scholar] [PubMed] [CrossRef]
49. Kowalczyk A. The role of the natural antioxidant mechanism in sperm cells. Reprod Sci. 2022;29(5):1387–94. doi:10.1007/s43032-021-00795-w. [Google Scholar] [PubMed] [CrossRef]
50. Dubois-Deruy E, Peugnet V, Turkieh A, Pinet F. Oxidative stress in cardiovascular diseases. Antioxidants. 2020;9(9):864. doi:10.3390/antiox9090864. [Google Scholar] [PubMed] [CrossRef]
51. Fei J, Demillard LJ, Ren J. Reactive oxygen species in cardiovascular diseases: an update. Explor Med. 2022;3:188–204. doi:10.37349/emed.2022.00085. [Google Scholar] [CrossRef]
52. Tsao CW, Aday AW, Almarzooq ZI, Anderson CAM, Arora P, Avery CL, et al. Heart disease and stroke statistics-2023 update: a report from the American Heart Association. Circulation. 2023;147(8):e93–621. doi:10.1161/CIR.0000000000001123. [Google Scholar] [PubMed] [CrossRef]
53. Di Cesare M, Perel P, Taylor S, Kabudula C, Bixby H, Gaziano TA, et al. The heart of the world. Glob Heart. 2024;19(1):11. doi:10.5334/gh.1288. [Google Scholar] [PubMed] [CrossRef]
54. Caturano A, D’Angelo M, Mormone A, Russo V, Mollica MP, Salvatore T, et al. Oxidative stress in type 2 diabetes: impacts from pathogenesis to lifestyle modifications. Curr Issues Mol Biol. 2023;45(8):6651–66. doi:10.3390/cimb45080420. [Google Scholar] [PubMed] [CrossRef]
55. An Y, Xu B, Wan Sr, Ma X, Long Y, Xu Y, et al. The role of oxidative stress in diabetes mellitus-induced vascular endothelial dysfunction. Cardiovasc Diabetol. 2023;22(1):237. doi:10.1186/s12933-023-01965-7. [Google Scholar] [PubMed] [CrossRef]
56. Eguchi N, Vaziri ND, Dafoe DC, Ichii H. The role of oxidative stress in pancreatic β cell dysfunction in diabetes. Int J Mol Sci. 2021;22(4):1509. doi:10.3390/ijms22041509. [Google Scholar] [PubMed] [CrossRef]
57. Bennici G, Almahasheer H, Alghrably M, Valensin D, Kola A, Kokotidou C, et al. Mitigating diabetes associated with reactive oxygen species (ROS) and protein aggregation through pharmacological interventions. RSC Adv. 2024;14(25):17448–60. doi:10.1039/D4RA02349H. [Google Scholar] [PubMed] [CrossRef]
58. Leenders F, Groen N, de Graaf N, Engelse MA, Rabelink TJ, de Koning EJP, et al. Oxidative stress leads to β-cell dysfunction through loss of β-cell identity. Front Immunol. 2021;12:690379. doi:10.3389/fimmu.2021.690379. [Google Scholar] [PubMed] [CrossRef]
59. Pi J, Zhang Q, Fu J, Woods CG, Hou Y, Corkey BE, et al. ROS signalling, oxidative stress and Nrf2 in pancreatic beta-cell function. Toxicol Appl Pharmacol. 2010;244(1):77–83. doi:10.1016/j.taap.2009.05.025. [Google Scholar] [PubMed] [CrossRef]
60. Olufunmilayo EO, Gerke-Duncan MB, Holsinger RMD. Oxidative stress and antioxidants in neurodegenerative disorders. Antioxidants. 2023;12(2):517. doi:10.3390/antiox12020517. [Google Scholar] [PubMed] [CrossRef]
61. Adusumilli VS, Walker TL, Overall RW, Klatt GM, Zeidan SA, Zocher S, et al. ROS dynamics delineate functional states of hippocampal neural stem cells and link to their activity-dependent exit from quiescence. Cell Stem Cell. 2021;28(2):300–14. doi:10.1016/j.stem.2020.10.019. [Google Scholar] [PubMed] [CrossRef]
62. McEwen BS. Understanding the potency of stressful early life experiences on brain and body function. Metabolism. 2008;57(2):11–5. doi:10.1016/j.metabol.2008.07.006. [Google Scholar] [PubMed] [CrossRef]
63. Verkhratsky A, Butt A, Li B, Zorec R, Semyanov A, Tang Y, et al. Astrocytes in human central nervous system diseases: a frontier for new therapies. Signal Transduct Target Ther. 2023;8(1):396. doi:10.1038/s41392-023-01628-9. [Google Scholar] [PubMed] [CrossRef]
64. Rizor A, Pajarillo E, Johnson J, Aschner M, Lee E. Astrocytic oxidative/nitrosative stress contributes to parkinson’s disease pathogenesis: the dual role of reactive astrocytes. Antioxidants. 2019;8(8):265. doi:10.3390/antiox8080265. [Google Scholar] [PubMed] [CrossRef]
65. Hernandes MS, D’Avila JC, Trevelin SC, Reis PA, Kinjo ER, Lopes LR, et al. The role of Nox2-derived ROS in the development of cognitive impairment after sepsis. J Neuroinflammation. 2014;11(1):36. doi:10.1186/1742-2094-11-36. [Google Scholar] [PubMed] [CrossRef]
66. Ali T, Li D, Ponnamperumage TNF, Peterson AK, Pandey J, Fatima K, et al. Generation of hydrogen peroxide in cancer cells: advancing therapeutic approaches for cancer treatment. Cancers. 2024;6(12):2171. doi:10.3390/cancers16122171. [Google Scholar] [PubMed] [CrossRef]
67. Reczek CR, Chandel NS. The two faces of reactive oxygen species in cancer. Annu Rev Cancer Biol. 2017;1(1):79–98. doi:10.1146/annurev-cancerbio-041916-065808. [Google Scholar] [CrossRef]
68. Jomova K, Raptova R, Alomar SY, Alwasel SH, Nepovimova E, Kuca K, et al. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: chronic diseases and aging. Arch Toxicol. 2023;10(10):2499–574. doi:10.1007/s00204-023-03562-9. [Google Scholar] [PubMed] [CrossRef]
69. Brown JS, Amend SR, Austin RH, Gatenby RA, Hammarlund EU, Pienta KJ. Updating the definition of cancer. Mol Cancer Res. 2023;21(11):1142–7. doi:10.1158/1541-7786.MCR-23-0411. [Google Scholar] [PubMed] [CrossRef]
70. Wu K, El Zowalaty AE, Sayin VI, Papagiannakopoulos T. The pleiotropic functions of reactive oxygen species in cancer. Nat Cancer. 2024;5(3):384–99. doi:10.1038/s43018-024-00738-9. [Google Scholar] [PubMed] [CrossRef]
71. Nakamura H, Takada K. Reactive oxygen species in cancer: current findings and future directions. Cancer Sci. 2021;112(10):3945–52. doi:10.1111/cas.15068. [Google Scholar] [PubMed] [CrossRef]
72. DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475(7354):106–9. doi:10.1038/nature10189. [Google Scholar] [PubMed] [CrossRef]
73. Patterson JC, Joughin BA, van de Kooij B, Lim DC, Lauffenburger DA, Yaffe MB. ROS and oxidative stress are elevated in mitosis during asynchronous cell cycle progression and are exacerbated by mitotic arrest. Cell Syst. 2019;8(2):163–7. doi:10.1016/j.cels.2019.01.005. [Google Scholar] [PubMed] [CrossRef]
74. Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17(10):1195–214. doi:10.1096/fj.02-0752rev. [Google Scholar] [PubMed] [CrossRef]
75. Kuczler MD, Olseen AM, Pienta KJ, Amend SR. ROS-induced cell cycle arrest as a mechanism of resistance in polyaneuploid cancer cells (PACCs). Prog Biophys Mol Biol. 2021;165(2):3–7. doi:10.1016/j.pbiomolbio.2021.05.002. [Google Scholar] [PubMed] [CrossRef]
76. Matthews HK, Bertoli C, de Bruin RAM. Cell cycle control in cancer. Nat Rev Mol Cell Biol. 2022;23(1):74–88. doi:10.1038/s41580-021-00404-3. [Google Scholar] [PubMed] [CrossRef]
77. Petsalaki E, Zachos G. DNA damage response proteins regulating mitotic cell division: double agents preserving genome stability. The FEBS J. 2020;287(9):1700–21. doi:10.1111/febs.15240. [Google Scholar] [PubMed] [CrossRef]
78. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–8. doi:10.1038/nature08467. [Google Scholar] [PubMed] [CrossRef]
79. Audrey A, de Haan L, van Vugt MATM, Rudolf de Boer H. Processing DNA lesions during mitosis to prevent genomic instability. Biochem Soc Trans. 2022;50(4):1105–18. doi:10.1042/BST20220049. [Google Scholar] [PubMed] [CrossRef]
80. Li CM, Lingeman RG, Haratipour P, Gu L, Jossart J, Perry JJP, et al. S phase. In: Bradshaw RA, Stahl PD, editors. Encyclopedia of cell biology. 2nd ed. New York: Elsevier Inc.; 2023. p. 267–84. [Google Scholar]
81. Liu X, Fan L, Lu C, Yin S, Hu H. Functional role of p53 in the regulation of chemical-induced oxidative stress. Oxid Med Cell Longev. 2020;2020(3):6039769. doi:10.1155/2020/6039769. [Google Scholar] [PubMed] [CrossRef]
82. Shi T, Dansen TB. Reactive oxygen species induced p53 activation: DNA damage, redox signalling or both? Antioxid Redox Signal. 2020;33(12):839–59. doi:10.1089/ars.2020.8074. [Google Scholar] [PubMed] [CrossRef]
83. Cornwell JA, Crncec A, Afifi MM, Tang K, Amin R, Cappell SD. Loss of CDK4/6 activity in S/G2 phase leads to cell cycle reversal. Nature. 2023;619(7969):363–70. doi:10.1038/s41586-023-06274-3. [Google Scholar] [PubMed] [CrossRef]
84. Kok YP, Guerrero Llobet S, Schoonen PM, Everts M, Bhattacharya A, Fehrmann RSN, et al. Overexpression of Cyclin E1 or Cdc25A leads to replication stress, mitotic aberrancies, and increased sensitivity to replication checkpoint inhibitors. Oncogenesis. 2020;9(10):88. doi:10.1038/s41389-020-00270-2. [Google Scholar] [PubMed] [CrossRef]
85. Sazonova EV, Petrichuk SV, Kopeina GS, Zhivotovsky B. A link between mitotic defects and mitotic catastrophe: detection and cell fate. Biol Direct. 2021;16(1):25. doi:10.1186/s13062-021-00313-7. [Google Scholar] [PubMed] [CrossRef]
86. Satyanarayana A, Kaldis P. Mammalian cell-cycle regulation: several CDKs, numerous cyclins and diverse compensatory mechanisms. Oncogene. 2009;28(33):2925–39. doi:10.1038/onc.2009.170. [Google Scholar] [PubMed] [CrossRef]
87. Lin LS, Wang JF, Song J, Liu Y, Zhu G, Dai Y, et al. Cooperation of endogenous and exogenous reactive oxygen species induced by zinc peroxide nanoparticles to enhance oxidative stress-based cancer therapy. Theranostics. 2019;9(24):7200–9. doi:10.7150/thno.39831. [Google Scholar] [PubMed] [CrossRef]
88. Ravi N, Choday S, Kumar SV, Kc A, Parisapogu A, Ojinna BT, et al. The key role of glutathione compared to curcumin in the management of systemic lupus erythematosus: a systematic review. Cureus. 2022;14(11):e31324. doi:10.7759/cureus.31324. [Google Scholar] [PubMed] [CrossRef]
89. Lu L, Ma X, Zheng J, Li L, Yang W, Kong Y, et al. Quercetin for myocardial ischemia reperfusion injury: a protocol for systematic review and meta-analysis. Medicine. 2020;99(26):e20856. doi:10.1097/MD.0000000000020856. [Google Scholar] [PubMed] [CrossRef]
90. Panth N, Paudel KR, Parajuli K. Reactive oxygen species: a key hallmark of cardiovascular disease. Adv Med. 2016;2016:9152732. doi:10.1155/2016/9152732. [Google Scholar] [PubMed] [CrossRef]
91. Mannucci C, Casciaro M, Sorbara EE, Calapai F, Di Salvo E, Pioggia G, et al. Nutraceuticals against oxidative stress in autoimmune disorders. Antioxidants. 2021;10(2):261. doi:10.3390/antiox10020261. [Google Scholar] [PubMed] [CrossRef]
92. Xie J, Dong R, Zhang T, Guo F, Li H, Chen X, et al. Natural dietary ROS scavenger-based nanomaterials for ROS-related chronic disease prevention and treatment. Chem Eng J. 2024;490:151756. doi:10.1016/j.cej.2024.151756. [Google Scholar] [CrossRef]
93. Yashin A, Yashin Y, Xia X, Nemzer B. Antioxidant activity of spices and their impact on human health: a review. Antioxidants. 2017;6(3):70. doi:10.3390/antiox6030070. [Google Scholar] [PubMed] [CrossRef]
94. Attah MOO, Jacks TW, Garba SH, Balogun SU. Leptadenia hastata Leaf Extract ameliorates oxidative stress and serum biochemical parameters in Streptozotocin-Induced diabetes in Wistar rats. J Diabetes Metab Disord. 2022;21(2):1273–381. doi:10.1007/s40200-022-01017-z. [Google Scholar] [PubMed] [CrossRef]
95. Lim SY. Role of statins in coronary artery disease. Chonnam Med J. 2013;49(1):1–6. doi:10.4068/cmj.2013.49.1.1. [Google Scholar] [PubMed] [CrossRef]
96. Laursen JB, Rajagopalan S, Galis Z, Tarpey M, Freeman BA, Harrison DG. Role of superoxide in angiotensin II-induced but not catecholamine-induced hypertension. Circulation. 1997;95(3):588–93. doi:10.1161/01.CIR.95.3.588. [Google Scholar] [PubMed] [CrossRef]
97. Nasution SA. The use of ACE inhibitor in cardiovascular disease. Acta Med Indones. 2006;38(1):60–4. [Google Scholar] [PubMed]
98. Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G, et al. ROS in cancer therapy: the bright side of the moon. Exp Mol Med. 2020;52(2):192–203. doi:10.1038/s12276-020-0384-2. [Google Scholar] [PubMed] [CrossRef]
99. Kim SJ, Kim HS, Seo YR. Understanding of ROS-inducing strategy in anticancer therapy. Oxid Med Cell Longev. 2019;2019:e5381692. doi:10.1155/2019/5381692. [Google Scholar] [PubMed] [CrossRef]
100. Dibal N, Garba S, Jacks T. Onion peel quercetin attenuates ethanol-induced liver injury in mice by preventing oxidative stress and steatosis. Biomed Res Ther. 2022;9(6):5102–12. doi:10.15419/bmrat.v9i6.745. [Google Scholar] [CrossRef]
101. Dibal NI, Chiroma SM, Attah MOO, Manye SJ. Effects of sub-acute administration of onion waste quercetin on the hippocampus of mice: a histological approach. Gazi Uni J Sci. 2024;37(2):546–54. doi:10.35378/gujs.1272285. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools