
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Cell Death of Tumor Melanocytes and Treatment Options
1 Department of Biotechnology, Institute of Chemical Biology and Fundamental Medicine SB RAS, Novosibirsk, 630090, Russia
2 Department of Natural Sciences, Novosibirsk State University, Novosibirsk, 630090, Russia
3 Department of Reconstructive Surgery, The S. Fedorov Eye Microsurgery Federal State Institution, Novosibirsk, 630120, Russia
* Corresponding Author: Olga Koval. Email:
(This article belongs to the Special Issue: Cell Death and Inflammation in Signaling and Diseases)
BIOCELL 2025, 49(3), 355-379. https://doi.org/10.32604/biocell.2025.059987
Received 21 October 2024; Accepted 13 January 2025; Issue published 31 March 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Melanomas are aggressive cancers, with a high rate of metastatic disease. Cutaneous (CM) and uveal (UM) melanomas are intrinsically different diseases, and most cell death inducers effective for CM do not function for UM. This is primarily due to the fact the eye is an immunologically privileged organ, and it fails to achieve the efficacy of immune checkpoint inhibitors (ICIs) comparable to that for CM. However, approaches utilizing specific melanoma-associated antigens are being developed for metastatic forms of CM and UM. The most promising to date are gp100 and tyrosinase related protein 1 (TYRP1), primarily for the design of targeting chimeric molecules and for autologous T-/NK-cell products with a chimeric antigen receptor. The difference in the mutational profile of apoptosis-related genes in CM and UM also makes counterproductive the use of the same drugs re-activators of the intrinsic pathway of apoptosis. Therefore, the discovery of novel pathways of regulated cell death such as ferroptosis and cuproptosis may help in the development of new drugs for melanomas resistant to already available inducers of regulated cell death. Here we consistently discuss the latest advances in the therapy of melanomas, and above all-UM, which is classified as an orphan disease.Keywords
Abbreviations
| ADI | Adjuvant immunotherapy |
| APC | Antigen-presenting cells |
| BAP1 | BRCA1-associated protein-1 |
| BRAF | v-Raf murine sarcoma viral oncogene homolog B |
| CAR-T | Chimeric antigen receptor-expressing T cells |
| CM | Cutaneous melanoma |
| CSF1 | Colony-stimulating factor 1 |
| CTLA2a | Cytotoxic T lymphocyte-associated protein 2α |
| CTLA4 | Cytotoxic T-lymphocyte associated protein 4 |
| CYGB | Cytoglobin |
| CysLTR1 | Cysteinyl leukotriene receptor 1 |
| DC | Dendritic cells |
| EGFR | Epidermal growth factor receptor |
| EM | Eumelanin |
| EpCam | Epithelial cell adhesion molecule |
| FAS | Type I transmembrane protein |
| FDA | Food and Drug Administration |
| GD2 | Sialic acid-containing glycosphingolipid |
| GNA11 | Guanine nucleotide-binding protein subunit alpha-11 |
| GNAQ | Guanine nucleotide-binding protein subunit alpha |
| GPX4 | Glutathione peroxidase 4 |
| gp100 | Glycoprotein 100 |
| HER2 | Human epidermal growth factor receptor 2 |
| ICAM-1 | Intercellular Adhesion Molecule 1 |
| ICD | Immunogenic cell death |
| ICI | Immune checkpoint inhibitor |
| IFNγ | Interferon-γ |
| IL-10 | Interleukin 10 |
| IRP1 | Iron regulatory protein 1 |
| HLA | Human leukocyte antigen |
| LOX | Lipoxygenase |
| MART-1 | Melanoma antigen recognized by T cells 1 |
| MCM | Metastatic cutaneous melanoma |
| MHC | Major histocompatibility complex |
| ML-IAP | Melanoma-associated inhibitor of apoptosis |
| MUC1 | Transmembrane glycoprotein mucin 1 |
| MUM | Metastatic uveal melanoma |
| NAI | Neoadjuvant immunotherapy |
| NK | Natural killer |
| NOG | NOD/SCID IL2 receptor gamma |
| NOX | NADPH oxidase |
| OS | Overall survival |
| OXPHOS | Oxidative phosphorylation |
| PDL1 | Programmed cell death ligand 1 |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PKC | Protein kinase C |
| PLCβ4 | Phospholipase Cβ4 |
| PM | Pheomelanin |
| ROS | Reactive oxygen species |
| RSL3 | RAS-selective lethal ferroptosis activator |
| SLC3A2 | Heavy subunit 4F2hc of system Xc- |
| SLC7A11 | Cystine/glutamate antiporter solute carrier family 7 member 11 |
| STEAP3 | NADPH-dependent ferric-chelate reductase |
| TCI | T cell inhibitor molecule |
| TCR | T cell receptor |
| TCGA | The Cancer Genome Atlas |
| TIL | Tumor infiltrating lymphocyte |
| TGFβ | Transforming growth factor beta |
| TNF | Tumor necrosis factor |
| TRAIL | TNF-related apoptosis-inducing ligand |
| TYRP1 | Tyrosinase-related protein 1 |
| UM | Uveal melanoma |
| USP7 | Ubiquitin specific peptidase 7 |
| UV | Ultraviolet |
| VISTA | V-domain Ig suppressor of T cell activation |
| α-MSH | Alpha-Melanocyte stimulating hormone |
| 4-TBP | 4-Tertiary butyl phenol |
Melanomas comprise an average of 2% of all cancers [1]. This tumor type develops from pigment cells that reside in multiple regions of the body, and they are mainly located in the skin. In this regard, skin melanoma represents the major subgroup, subdivided into superficial spreading, nodular, lentigo, and acral melanomas. The second most common subtype of melanoma is eye or ocular melanoma, which develops from melanocytes of the uveal tract and makes up about 5% of all melanoma cases [2]. The epidermal melanocytes synthesize and transfer melanin to keratinocytes, while uveal melanocytes does not transfer melanin to other cells. By UM subtype, the highest incidence rates demonstrate choroidal melanomas (up to 80%), while iris and ciliary body melanoma are less common and account for up to 12 and 6 percent, respectively [3–5]. Although the diversity of melanomas is large, consideration of cutaneous and ocular melanoma separately is reasonable. First, the mutational burden of cutaneous and uveal melanomas differs significantly [6–8]. Second, because the eye is an immune privileged organ [9], the responses of cutaneous and uveal melanoma to immunologic stimuli vary. Such variation may ensure the realization of different scenarios of malignant melanocytes’ death of cutaneous and uveal melanomas. The mortality of patients with melanoma is due to metastatic disease for both cutaneous (MСM) and uveal melanoma (MUM) [10]. The highest incident rates of metastases to the lymph nodes were accounted for cutaneous melanoma and to the liver for uveal melanoma [11].
Differences in metastatic niches are because the structures of the eye lack lymphatic vessels and communication with regional lymphoid organs, therefore the spread of UM is hematogenous. In CM, both types of melanoma cells spreading are common–lymphatic and hematogenous.
Metastases in the lungs are second for both types of metastatic disease. Therefore, the main metastatic niches of UM, liver, and lungs, are relatively immunotolerant, with weak accessibility to immune cells, and metastatic uveal melanoma is an immunologically cold tumor type. This implies low T cell infiltration and resistance to ICIs [12].
Nevertheless, 2022 was a relatively “breakthrough” year for metastatic UM, as two new drugs were approved by the U.S. Food and Drug Administration (FDA) for the treatment of MUM–Tebentafusp in January 2022 and protein kinase C (PKC) inhibitor Darovasertib in May 2022 [13,14].
At the same time, all melanomas have specific features that distinguish them from other cells-an enzymatic system that ensures the formation of special organelles—melanosomes—that synthesize a specific pigment melanin [15]. Melanin acts as a protective shield against ultraviolet (UV) rays and has a reactive oxygen species (ROS)-scavenging effect. Proteins and metabolites that are involved in melanin synthesis can be specific targets for induction of tumor cell death, as we can see with the glycoprotein 100 (gp100) [16,17]. In addition, despite the differences in the mutational burden of UM and CM, tumor progression tends to enhance apoptosis resistance and may be associated with the poor responsiveness of the metastatic form of the disease to apoptosis-inducing treatments, such as chemotherapy and radiotherapy [18]. Therefore, the development of drugs that can activate regulated cell death in tumor melanocytes distinct from apoptosis, such as ferroptosis or cuproptosis, is highly desirable. In addition, there are many actual reviews on how necroptosis, pyroptosis, and autophagy-dependent cell death are involved in melanoma development, and in our work, we aimed to focus more on the role of ferroptosis and cuproptosis in melanoma treatment and progression [19–21].
Here, we will attempt to examine in detail the influence of the factors described earlier on the induction of tumor melanocyte death in CM and UM.
2 Melanomas Sensitivity to Immunotherapy and Receptor-Dependent Cell Death
Under healthy conditions, such as in the lack of carcinogens and mutagens, immunological surveillance is realized by natural killer (NK) and dendritic cells (DCs), T cells, and B-cells, which effectively eliminate tumor cells. When the tumor cell pool increases, tumor cell variants progressively lose major histocompatibility complex (MHC) class-I and -II molecules, thus relevant amounts of tumor antigens become “invisible” to the immune system [22]. This contributes to an increased pool of tumor cells that escape immunological surveillance. With aging, even in the absence of carcinogens, the probability of accumulation of cells with impaired proliferation control increases, and a parallel age-dependent attenuation of immune system function arises. For skin melanoma, the above is applicable, whereas uveal melanoma is located in the eye, an immunologically privileged organ.
What is and what properties does the immune privilege provide? For a deep understanding of the problem, you can read Streilein’s review at [9]. Briefly, the eye protects itself against sight-destroying immune responses and inflammation. This defense leaves the eye vulnerable when malignant transformation occurs. Besides the weak presentation of tumor antigens on ocular cells to eye-derived antigen-presenting cells (APCs), the elimination of lymphatic vessels in the eye forces APCs to move to the spleen through the blood vessels (Fig. 1).
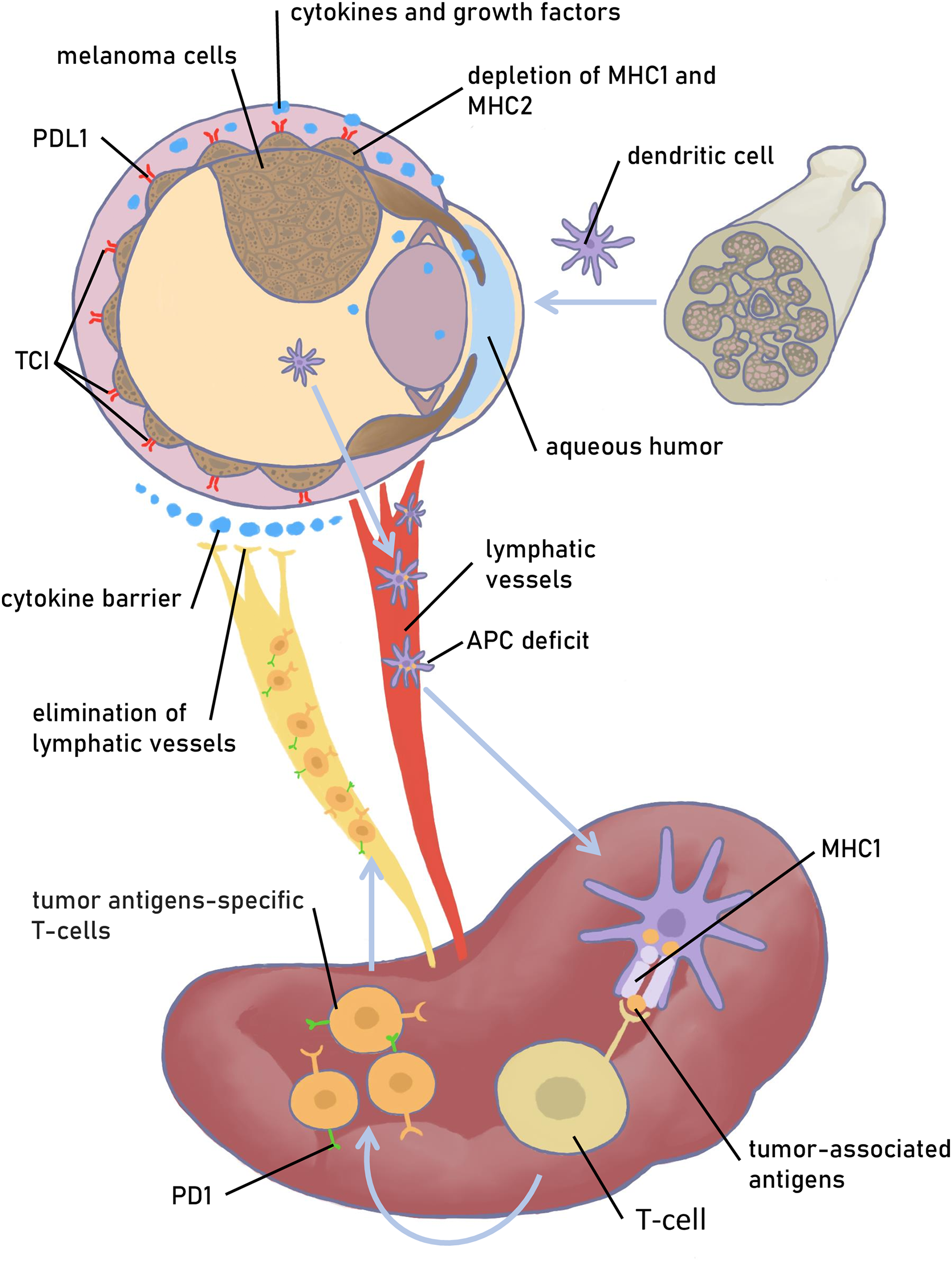
Figure 1: Physiologic and molecular mechanisms that provide immune tolerance of the eye. The melanoma is depicted as choroid melanoma. Factors that provide protection to melanoma cells from the immune system and prevent effective immunotherapy are depicted. A pathway that represses the specific T cell and B-cell immune response to tumor antigens when tumor cells are killed by cytotoxic agents is also presented. TCI—T cell inhibitor molecules, an alternative to PDL1. Figures are made in Sketchbook (2024), version 6.1.1, copyright holder-Sketchbook (https://www.sketchbook.com) (accessed on 12 January 2025)
In the spleen, naive T cells are recruited, activated, and induced to differentiate into antigen-specific Tregs, but their further activity in the eye is prevented by the local immunosuppressive microenvironment [23]. In general, the immunosuppressive and anti-inflammatory microenvironment is mediated by the secretion of immunomodulatory cytokines/small molecules and surface expression of various factors, leading to the neutralization of innate and adaptive immune effector cells. The main modulators are transforming growth factor beta (TGFβ), alpha-Melanocyte stimulating hormone (α-MSH), Interleukin 10 (IL-10), somatostatin, thrombospondin 1, apoptotic ligand FAS, tumor necrosis factor alpha-related apoptosis-inducing ligand (TRAIL) and programmed cell death 1 ligand 1 (PDL1), cytotoxic T-lymphocyte associated protein 4 (CTLA4) [24]. Other factors facilitate main modulators to protect the eye from the cytotoxic Т-cells; they are MHC class Ib, CTLA-2a, galectin-9, and V-domain Ig suppressor of T cell activation (VISTA) [25].
Immunologic approaches have been developed and used for the treatment of melanoma of the skin for quite a long time. IL-2 was approved for the treatment of cutaneous metastatic melanoma in 1998 and has remained one of the main drugs along with dacarbazine until 2011 [26]. It is an important factor for the maintenance of CD4+ regulatory T lymphocytes and responsible for the differentiation of CD4+ T cells into a variety of subsets. IL-2 can stimulate CD8+ T cell and NK cell cytotoxicity and modulate T lymphocyte lineage differentiation when interacting with antigens.
2.1 Breaking the Inhibition Signal for T Cells
For a tumor cell to be killed by T cells, at least two conditions must be met, first, the tumor cell must express a recognizable antigen, and second, it must be activated. Since tumor cells are not truly “foreign” to the body, they express special molecules or checkpoints that prevent T cell activation, such as CTLA4 and PD-L1. In solid tumors, checkpoint switching off leads to retention and amplification of tumor-infiltrating CD8+ T lymphocytes pool. Skin melanoma cells, as well as many solid tumors, are characterized by expression of CTLA4 and PD-L1 (Fig. 2). Therefore, CM is potentially suitable for therapies based on reducing the availability of CTLA4 or PD1/PD-L1 with selective inhibitors. Anti-PD1 antibodies nivolumab and pembrolizumab were approved for the treatment of metastatic melanoma [27,28].
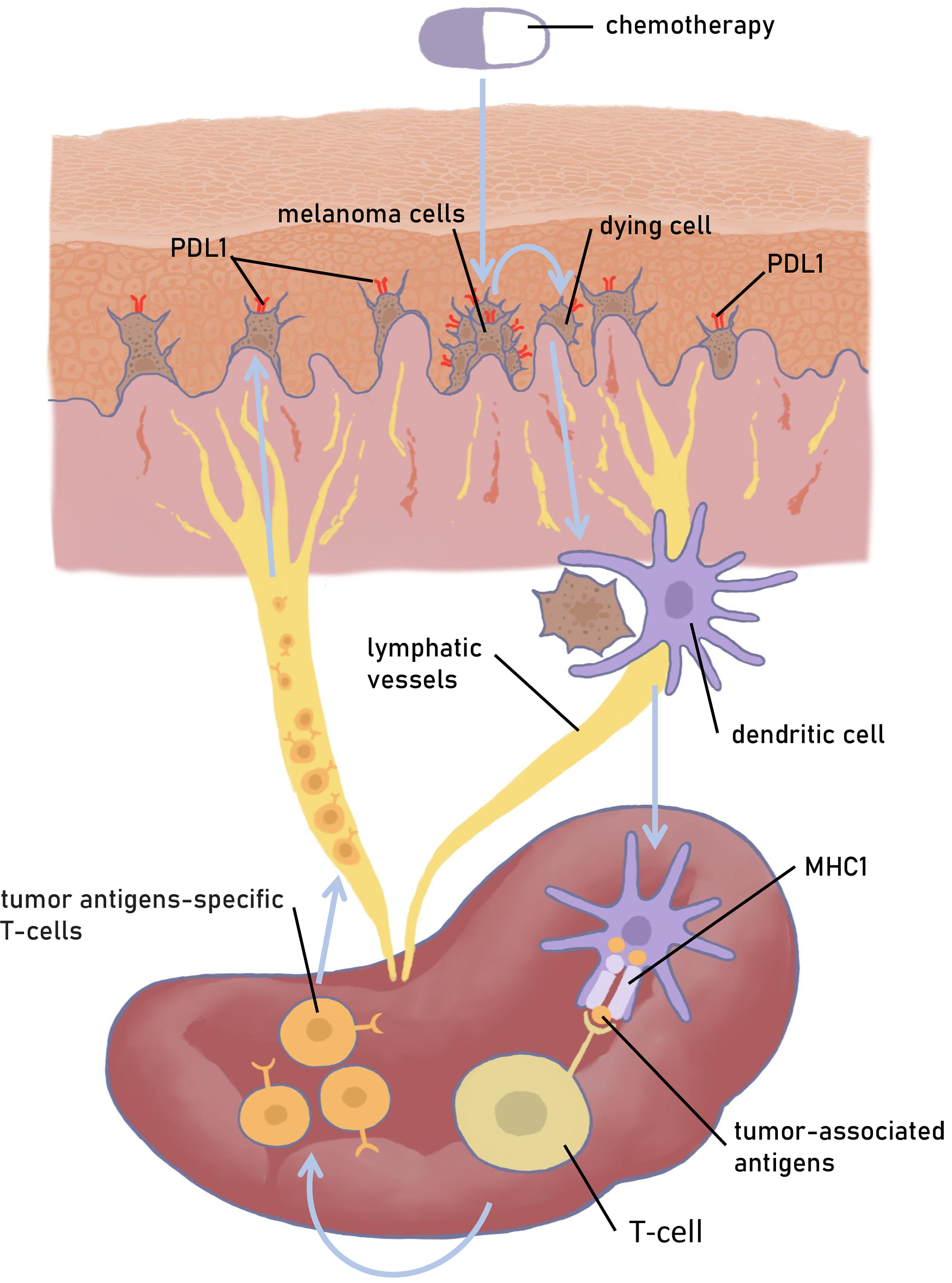
Figure 2: A model of CM interaction with the immune system. Schematically depicts the activation of the immune system under the exposure of antigens of a dying melanoma cell and the development of an antitumor immune response. Figures are made in Sketchbook (2024), version 6.1.1, copyright holder-Sketchbook (https://www.sketchbook.com) (accessed on 12 January 2025)
Anti-CTLA4 antibody ipilimumab (Yervoy) was approved in 2011 for treating advanced melanoma, especially for unresectable (Stage III) and metastatic (Stage IV) melanomas [29]. Total mRNAs analysis of melanoma patients’ biopsies with advanced-stage disease receiving nivolumab alone or in combination with ipilimumab showed that T-cell infiltration and interferon-γ (IFNγ) signaling patterns were most consistent with clinical response to therapy [30]. Thus, modern therapy with ICIs acclimates previous progress on cytokine therapy. Of greater interest are the results obtained from the comparison of tebentafusp (240 patients) vs. nivolumab plus ipilimumab (45 patients) in first-line metastatic uveal melanoma [31]. Tebentafusp provided an overall survival (OS) benefit compared with nivolumab plus ipilimumab.
Myeloid CSF1R+ cells exhibit intertumoral immune suppressive activity, inhibiting cytotoxic T cells [32]. Neubert et al. showed that Colony stimulating factor 1 (CSF1) is produced by melanoma cells, and patients with metastatic melanoma have high CSF1 in the blood [33]. They proposed the use of two antibodies simultaneously—targeting PD1 and CSF1 receptor (CSF1R). This combination of drugs caused growth inhibition of v-Raf murine sarcoma viral oncogene homolog B (BRAF)V600E driven murine melanomas. Therefore, simultaneous targeting of the above antigens may be useful in ICIs-resistant tumors. The selection of CSF1 was justified by the fact that the combination of a small CSF1R inhibitor (PLX3397) and adoptive T-cell transfer increased mouse survival compared with individual treatments in BRAFV600E-induced melanoma models [34].
Results of clinical trial of adjuvant immunotherapy (ADI) nivolumab vs. ipilimumab in resected stage III/IV melanoma showed that nivolumab ADI significantly improved recurrence-free survival and distant metastasis-free survival vs. ipilimumab [35]. Five-year rates of recurrence-free survival were 50% for nivolumab vs. 39% for ipilimumab.
The treatment regimen has been shown to have a significant impact on the outcome of therapy with the combination of ICIs. A study compared the outcome of the combination of ipilimumab plus nivolumab when four courses of ipilimumab plus nivolumab ADI therapy were given vs. two courses of neoadjuvant immunotherapy (NAI) plus two courses of ADI therapy [36]. Researchers detected a high survival rate after NAI ICIs combination: 5-year OS rates for the NAI arm was 90% vs. 70% for the ADI arm. When comparing the use of nivolumab plus ipilimumab in ADI and NAI for patients with respectable stage III melanoma in the phase 3 trial, the NAI setting has also demonstrated the best response [37]. Neoadjuvant therapy with ipilimumab plus nivolumab followed by surgery and ADI depending on response, resulted in longer event-free survival and was 83.7% compared to the “surgery followed by ADI with nivolumab” group (57.2%). It can be assumed that tumor resection is accompanied by a change in the local microenvironment, which affects the expression of immune checkpoint molecules and reduces the efficiency of ICIs therapy. Nevertheless, 10-year outcome data clearly proves the significant benefit of nivolumab plus ipilimumab combination therapy over monotherapy [38]. The median OS was 71.9 months for nivolumab plus ipilimumab, 36.9 months for nivolumab, and 19.9 months for ipilimumab.
In addition to enhancing the pool of cytotoxic T cells by ICIs, approaches to reduce the effects of other immunosuppressive mechanisms, such as the downregulation of MHC-I molecules, are being explored. Restoration of the MHC-I amount will enhance the tumor antigen presentation pathway. Tan et al. showed the impaired homeostasis of the epigenetic regulator ubiquitin-like with PHD and ring finger domains 1 (UHRF1) in cancer tissues. When phosphorylated, it is deposited in the cytosol and mediates ubiquitin-dependent proteolysis of MHC-I [39].
In a normal eye, PD-L1 is constitutively expressed in corneal epithelial cells, corneal endothelial cells, iris/ciliary body cells, retinal pigment epithelial cells, some stromal cells, and UM cells [40,41]. When the eyes are inflamed, PD-L1 is detected in excess in corneal epithelial cells, corneal endothelial cells, iris/ciliary body cells, and retinal pigment epithelial cells [41]. Such PD-L1 expression induces apoptosis of PD-1-expressing effector T cells. PD-L1 expression on human ocular cells inhibits the production of proinflammatory cytokines IFN-γ and tumor necrosis factor alpha (TNF-α) and a Th2 cytokine IL-5 by activated T cells [41,42]. The efficacy of ICIs for MUM varied greatly. The experimental group with the worst response to pembrolizumab or nivolumab was identified in the retrospective study of patients with MUM. These were patients with multiple organ metastases and elevated serum lactate dehydrogenase [43]. A meta-analysis of 41 cohorts of 1414 patients with MUM was made to analyze whether dual ICI therapy was the more plausible treatment for MUM than monotherapy. They showed the benefit of using a combination of ICIs drugs for patients with MUM [44]. Retrospective analysis of a cohort of 47 MUM patients who received two drugs—PD-1 and CTLA-4 inhibitors (nivolumab plus ipilimumab), did not show a significant increase in treatment response and progression-free survival [45]. Therefore, this combination is not recommended as a standard treatment in all MUM patients but rather as an option.
The therapeutic use of ICIs leads to adverse effects such as dry eye syndrome [25,46]. Anti-CTL4 ipilimumab has shown efficacy and safety in the treatment of MUM. However, the overall response to ICIs in uveal melanoma is low [47]. Multiple cell surface molecules, with T cell inhibitory activity in addition to PD-1, provide alternative suppression of T cells when one of them is inactivated. To find out the tumor markers that correlated with improved survival in ICIs treated UM, the analysis of 450 UM tumor samples was made using NextGen Sequencing on DNA and RNA [48]. The study included primary (10/108) or metastatic (98/108) sites of UM. It was unexpected that most tumor samples (86%, 240/279) had a PD-L1low phenotype regardless of whether specimens were from the primary tumor site or metastasis. Samples from patients who received ICIs were also largely PD-L1low (62%, 55/89).
Tebentafusp (also called IMCgp100), a bispecific T lymphocyte engager to gp100-reach melanoma cells. Tebentafusp consists of a gp100-epitope-directed T cell receptor with high affinity and a CD3-specific, humanized single-chain antibody fragment. Endo-gp100 can be presented by both MHC class I and class II, while ecto-gp100 is specifically presented by MHC class II [49]. MHC class II antigenic presentation plays a crucial role in antitumor immunity. Tebentafusp has demonstrated survival benefits for MCM and MUM in a HLA-A*02:01-positive patient [44]. Tebentafusp selectively recognizes a human leukocyte antigen (HLA)-0201-restricted gp100 peptide redirects T cells to cancer cells and activates them, resulting in tumor-cell lysis. Tebentafusp overcomes the peritumoral restriction of T cells and results in T cell trafficking in the tumor.
Despite the progress for HLA-A*02:01-positive patients, the treatment strategy for MUM in HLA-A*02:01-negative patients remains to be established. Nathan et al. have shown in clinical trials that previously untreated HLA-A*02:01-positive patients with MUM have overall survival benefit with Tebentafusp [50]. They used a cohort of 378 patients including 126 patients in the control group. OS at one year was more than 70% in the Tebentafusp group and 59% in the control group, indicating that Tebentafusp resulted in longer OS than the control. The results of the further follow-up of these patients for 3 years showed that the median OS was 21.6 months in the tebentafusp group and 16.9 months in the control group [51]. These outcomes confirmed the long-term benefit of Tebentafusp on OS among HLA-A*02:01-positive people. No treatment-related deaths occurred during the monitoring period. As mentioned above, Tebentafusp provided an OS benefit compared with nivolumab plus ipilimumab in first-line MUM [31].
Another issue is the level of gp100 representation in melanoma cells. Thus, when examining histologic specimens of skin melanoma from 230 primary melanomas and 293 MCMs, the gp100 staining was positive, however, the degree of intensity was variable [52]. A comparison of gp100 in UM and CM cell lines showed strong gp100 expression in 88% of UM cell lines and 40% of CM cell lines.
2.5 Adoptive T- and NK-Cell Therapy
Adoptive transfer of T cells modified with tumor-specific T cell receptors (TCRs) or chimeric antigen receptors (CARs) has already been successfully used against several types of solid and hematologic malignancies [53]. The success of CAR-T and CAR-NK cell therapies depends largely on the proper choice of surface proteins highly expressed in tumors but not in normal tissues. Among the most promising targets for CAR-NK cell therapy for melanoma, only gp100 and tyrosinase related protein 1 (TYRP1) can be attributed to melanoma-specific antigens [54]. Both proteins are involved in melanin biogenesis. Despite this, effective chimeric antigen receptor targeting in melanoma can utilize antigens that have already been tested and used for other cancer types: receptor tyrosine kinase c-Met, sialic acid-containing glycosphingolipid GD2, epithelial cell adhesion molecule (EpCam or CD326), epidermal growth factor receptor (EGFR), mesothelin, transmembrane glycoprotein mucin 1 (MUC1) and others [55].
Just recently the efficacy of targeting CAR to melanoma-specific TYRP1 for the treatment of cutaneous, acral, and uveal melanomas in immunocompetent mice and patient-derived models has been demonstrated [54]. TYRP1 was chosen as a target because the antitumor efficacy (disease control rate of 41%) of monoclonal antibody IMC-20D7S targeting this protein had been shown earlier in clinical trials [56]. Jilani et al. have built a drug prototype combining a clinically tested scFv (20D7S), the CD28 transmembrane domain, a immunoglobulin IgG4 hinge that gives an amplified flexibility to the CAR, and the CD28 costimulatory signal [54]. The efficiency of target cell killing by the prototype drug and IFNγ release upon co-culture with melanoma cells depends on the level of TYRP1: insufficient TYRP1 production allows tumor cells to avoid death. The greatest success in T cell therapy for MUM was demonstrated in Chandran’s et al. study [57]. A study of freshly resected liver metastases was performed to develop an adoptive therapy for UM. The study of an immunogenic subset of tumor-infiltrating lymphocytes (TIL) allowed to generation of autologous TIL cultures for intravenous infusion. Such TIL infusion supported IL-2 induced objective tumor regression in seven (35%) of twenty patients, including individuals whose disease was refractory to ICIs. It was the first report describing the ability of TIL transfer to mediate uveal melanoma regression in patients. The same strategy was used by Karlsson with colleagues for identifying and functionally validating tumor-reactive T cells in liver metastases of patients with MUM [58]. The modification of the strategy with the use of the patient-derived xenograft models allowed to make sphere cultures from these models for co-culturing with autologous or melanoma antigen recognized by T cells 1 (MART1)-specific HLA-matched allogenic TILs. TCR-seq of in vitro activated T cells allows to compare TCRs to those found in single-cell sequencing data from biopsies and answer the question which T cells in UM may be useful for cell therapy amplification. Authors identified three subtypes of tumor-reactive lymphocytes in uveal melanoma metastases.
In experiments on NOD/SCID IL2 receptor gamma (NOG) knockout mice, it has been shown that chimeric antigen receptor-expressing T cells directed against human epidermal growth factor receptor 2 (HER2) can kill uveal and cutaneous melanoma cells in vitro and in vivo [59].
Described results on the use of CAR-T/CAR-NK cells in melanomas let us look forward to benefiting patients with uveal melanoma.
The first oncolytic virus drug was approved by U.S. FDA in 2015 specifically to treat unresectable, recurrent melanoma [60]. It was talimogene laherparepvec (T-VEC; Imlygic®), derived from an attenuated herpes simplex virus 1. A number of recent studies have demonstrated the successful application of oncolytic viruses for cancers treatment [61]. Thereafter, other oncolytic viruses have been used to treat MСM. For example, a coxsackie virus A21 (V937) that infects the cells with expression of the intercellular adhesion molecule 1 (ICAM-1). Cutaneous and uveal melanomas are cancers with high ICAM-1 levels relative to normal cells. Unmodified variant coxsackie virus A21 (V937) was administrated intratumorally to 57 patients with unresectable, advanced cutaneous melanoma. Indicated treatment led to a 6-month progression-free survival rate of 39% and 12-month overall survival rate 75% [62]. Moreover, regression of melanoma was observed in non-injected lesions, which suggests that the virus reaches and lyses distant tumor sites. In almost parallel to the above study, the combined use of coxsackie virus A21 and ICI ipilimumab was evaluated in clinical trial with 50 patients [63]. Responses associated with intertumoral virus injections plus ipilimumab were robust, and the results that were obtained allowed to proceed to the use of this combination against MUM.
A phase 1b clinical trial of oncolytic coxsackievirus A21 (V937) in combination with anti-CTL4 ICI ipilimumab was done for MUM patients [64]. Eleven patients received V937 intravenous injections and intravenous ipilimumab. The best overall response was stable disease in 3 patients and progressive disease in 8 patients. Despite the manageable safety profile of the combination of V937 with ipilimumab, clinical benefit has not been observed in patients with uveal melanoma and liver metastases.
Liu et al. have constructed strain HSV-GM-CSF, oncolytic herpes simplex virus type 1 (HSV-1) encoding granulocyte-macrophage colony-stimulating factor (GM-CSF) for testing its efficacy against UM [65]. BALB/c-nude mice were implanted with MUM2B cells in the posterior chamber of the eye and HSV-GM-CSF was injected intravitreally. They have found that follow-up survivals were prolonged in the virus-treated group compared with the control group.
It can be concluded that therapy with oncolytic viruses against MUM has not been successful to date, in contrast to metastatic cutaneous melanoma. The multifactorial traumatic nature of intraocular administration of oncolytic viruses precludes any hope for the successful use of oncolytic viruses for the treatment of primary UM.
3 Mutations in Melanoma Cells Enable Escape from Apoptosis
It has been clear for a long time that high mutation burden in cutaneous melanoma is initiated by UV radiation exposure. However, for some subtypes of cutaneous melanoma, such as mucosal or acral melanoma, which is localized on the palms of the hands, soles of the feet, under the nails of the fingers or toes, both are characterized by lower mutation burdens and different genes implicated in mutagenesis [18]. CM-specific mutations are BRAF, NRAS, CDKN2A, and TP53; the combination of NRAS, BRAF, and NF1 mutations is specific for acral melanoma, and KIT mutations are detected in the samples of mucosal and acral melanomas.
3.1 p53 and Apoptosis Dysfunction in Cutaneous Melanoma
One of the most important tumor suppressor factor p53 is encoded by the gene TP53. Mutations and functional inactivation of TP53 can be observed in almost all tumors, that confirms the via p53-dependent pathway [66]. In this case, activated p53 translocates from the cytoplasm to the nucleus where it acts as a sequence-specific transcription factor. Under normal conditions, p53 is modified in complex with the E2 ubiquitin ligase MDM2. p53 realizes the transcriptional control of genes involved in the regulation of cell cycle progression and apoptosis, including p21, Bax, GADD45, Mdm2, and Cyclin G. To regulate transcription, it is necessary that p53 avoids MDM2, but also undergoes modifications, such as phosphorylation and acetylation in the N- and C-terminal domains. In the inactive state, the regulatory region of C-terminal domain interacts intramolecularly with core domain and prevents p53 interaction with DNA. Modifications of C-terminal domains change intramolecular interactions and allow the formation of p53 homotetramers, which have significantly increased affinity of binding to DNA. Modifications of the N-terminal domains lead to p53 stabilization. Thus, the activity of p53 as a specific transcription factor is regulated by its conformation [67].
Khan et al. ran a cross-database analysis of the expression levels of the p53 signaling genes in cutaneous melanoma [68]. They found that transcription factor p53 and its signaling genes (TP53, MDM2, CDKN2A, and TP53BP1) were overexpressed, while MDM4 and CDKN2B were under-expressed in cutaneous melanoma. The findings require further analysis, so as MDM2 and MDM4 are both negative regulators of p53. The inhibition of p53 by MDM2 and MDM4 is distinct, since MDM4 regulates p53 activity and E3 ubiquitin ligase MDM2 mainly regulates p53 stability [69]. MDM4 contains a p53 binding domain at the N-terminus and prevents p53 from interacting with target genes [70]. Most likely, high expression of MDM2 may promote TP53 degradation at proteasomes leading to the inhibition of TP53-dependent apoptosis. This data only partially correlates with the work, where in a study of 1015 primary and 358 metastatic melanomas was shown that metastatic disease is enriched for MDM2 and MDM4 amplifications compared to primary disease, and amplifications are associated with lower overall survival [71]. Two additional negative regulators of p53, ubiquitin specific peptidase 7 (USP7) and protein phosphatase 1 D (PPM1D), have also been altered in metastatic melanoma compared to primary disease. The authors suggest that MDM2 and MDM4, USP7 and PPM1D have the potential to be inhibited in case of drug development for the treatment of metastatic melanoma. In a recent study, Groen K. with co-workers showed that only certain p53 transcripts contribute to the pathogenesis of cutaneous melanoma [72]. They found that the C-terminally truncated p53β isoforms were the most highly expressed isoforms compared to all other p53 isoforms. Further, TAp53 isoforms, retaining both transactivation domains, were the second highest p53 isoforms expressed across all melanoma samples.
Since p53 is required for the induction of apoptosis when significant DNA damage occurs, dysregulation in the p53 signaling will be more significant for UV-related cancers such as cutaneous melanoma. It was noted that dysregulation of p53 signaling or mutated p53 are not typical of uveal melanoma [73]. Despite the fact that in in vitro experiments there are successes in the use of drugs that activate the p53 pathway in UM cells, such drugs have not yet been used in the clinic [74].
Why is uveal melanoma, unlike many other cancers, not characterized by mutations in p53? In UM, the function of wild type p53 is often inactivated by negative regulators of p53, such as MDM2 and MDM4. The p53 signaling system has been shown to be in an active state in the eye tissues during the normal embryogenesis and development [75]. In mice experiments it has been shown that during embryonic development a high level of p53 is detected in the eyeball, providing apoptosis of transformed structures. Later decrease in p53 levels coincides with the time that cells exit the cell cycle, differentiate, and become postmitotic [76]. In contrast, Mdm2 transcript levels were the highest in post-embryonal period. Thus, negative regulation of p53 both at the level of transcription and inactivation of its activity possibly protects the eye from the death of specific cells that cannot be replenished from the pool of progenitor cells with stem properties.
Heijkants et al. also studied MDM4 in UM cells as a specific target with selective inhibitor NVP-CGM097 for p53 reactivation [77,78]. CGM097 inhibits the interaction of p53 with MDM4. They showed that such MDM4 depletion inhibits survival of UM cells, which is enhanced in combination with PKCδ inhibition.
In contrast to CM, metastatic UM is characterized by major chromosomal abnormalities including the loss of chromosome 1p and chromosome 3. p73 and p63, proteins of p53 family members are located on chromosomes 1p and 3q, respectively. Both can activate p53 target genes, resulting in growth arrest, apoptosis and differentiation. However, N-terminally truncated isoforms of these proteins may act as dominant negative repressors of wild-type p53 [79]. Kilic with co-authors observed N-terminal transactivation forms of both p73 and p63 in primary and metastasis-derived UM cell lines, as well as in primary melanomas [80]. Prevalence of modified form of p73 and p63 may inhibit wild type p53 and render cells non-sensitive to apoptotic stimuli.
Molecular mechanisms of UM oncogenesis differ from those of CM [81]. The most common mutations in UM cells are mutations in GNAQ and GNA11 genes. Both genes encode subunits of the guanine nucleotide binding protein G:Gq and G11. Mutations leading to uncontrolled activation of the G-protein disrupt the control of cell proliferation. Membrane-associated G-protein activates phospholipase Cβ4 (PLCβ4), and metabolites of the enzymatic reaction of PLCβ4 with phosphatidylinositol 4,5-bisphosphate (PIP2) activate protein kinase C (PKC) that is involved in cell cycle regulation. This pathway is important for the transformation of the uveal tract’s melanocytes into melanoma cells. MUM is most often associated with mutations of BAP1, EIF1AX, GNA11, GNAQ, and SF3B1. In addition, significant chromosomal abnormalities have been shown for UM, including monosomy 3, 1p loss, 1q gain, 6q loss, 6p gain, 8p loss and 8q gain.
Сonstitutively active mutant α-subunits of the heterotrimeric G proteins Gq or G11 (Gq/11) reprograms metabolism in UM [82]. Gq/11 targeting with synthetic inhibitor FR900359 allowed to achieve attenuated glucose uptake and its methabolism in various UM cell lines resulted in led to the depression of many processes, including cell division delay. This phenomenon is important for progress in the fight against uveal melanoma since metabolic reprogramming potentially could open a new space for therapeutic discovery.
3.3 Melanoma-Associated Inhibitor of Apoptosis (ML-IAP) in CM
Progression of CM is based on the changes in the balance and functional activity of pro- and anti-apoptotic proteins, as well as compromised death receptor signaling. IAP family proteins are inhibitors of apoptosis (X-IAP, C-IAP1, and C-IAP2) and regulators of the cell cycle (survivin). Belonging to the IAP family is determined by a similar structure—the presence of tandem baculovirus IAP repeat (BIR) motifs and carboxy-terminal RING finger motif [83]. X-IAP, C-IAP1, and C-IAP2 inhibit active caspases 3 and 7 and the Apaf-1-cytochrome-c-mediated activation of caspase-9, so they are important molecules in the realization of programmed cell death. An elevated expression of melanoma-associated IAP (ML-IAP, or Livin), human IAP that contains a single BIR and RING finger motif has been reported in melanoma samples. ML-IAP is encoded by the Birc7 gene, and can inhibit both pathways of apoptosis: the extrinsic receptor-dependent and intrinsic mitochondrial [84]. In addition to melanoma, the expression of ML-IAP is typical in lymphoma cells. Treatment of melanoma cells with apoptosis inducers UVB, 4-tertiary butyl phenol (4-TBP), cisplatin, TNF, and Bax, led to cleavage of ML-IAP molecules. ML-IAP can act as a direct inhibitor of downstream effector caspases, it confers melanoma cell resistance to apoptotic stimuli and may contribute to the pathogenesis of tumor formation [85]. This statement is only partially true because the pan-caspase inhibitor zVAD-fmk blocked ML-IAP cleavage after 4-TBP treatment, but not UVB or cisplatin treatment [86]. Therefore, it is believed that proteolytic cleavage of ML-IAP in apoptotic melanoma cells is potentially mediated by a non-canonical caspase. It was shown later that ML-IAP inhibits caspase-9, and possibly possesses E3 ubiquitin ligase activity [87,88]. Knockdown of BIRC7 gene induced melanoma cell apoptosis and Livin siRNAs efficiently inhibited MCM tumor growth in vivo [89]. It has recently been shown that the BIRC7 gene is among the group of six most upregulated genes in CM cells in response to hypoxic conditions [90]. These findings suggest that Livin expression under hypoxia is directed toward avoidance of apoptosis and survival of tumor cells under oxygen-deprived conditions.
There is virtually no data on the expression and function of ML-IAP in the UM. IAP genes were expressed in the majority of primary UM and MUM and were regulated. BIRC7 (BIRC7/Livin) level was 7.0-fold (p = 0.003) higher in tumors (primary and metastatic tissue) vs. normal eye tissue, respectively [91]. Lederman et al. have analyzed the expression of IAP genes, including ML-IAP in primary uveal melanoma tumors, liver metastasis from UM, and UM cell lines [92]. They found that ML-IAP was expressed in each of the tumor samples.
4 Induction of Ferroptosis in Melanomas
Ferroptosis is a regulated cell death mode driven by iron-dependent phospholipid peroxidation (Fig. 3) [93,94]. It contributes to numerous organ injuries and degenerative pathologies. The chemical nature of ferroptosis can be defined by an imbalance between the production and neutralization of polyunsaturated phospholipid hydroperoxides (PLOOH) [95]. In the cell, the main enzymes converting lipid hydroperoxides are glutathione-dependent lipid hydroperoxidase glutathione peroxidase 4 (GPX4) and coenzyme Q (CoQ) oxidoreductase ferroptosis suppressor protein 1 (FSP1), which acts parallel to GPX4 to inhibit ferroptosis [96]. There are three major milestones that protect the cell from death by the ferroptosis pathway (Fig. 3), and they operate upstream of GPX4 [97]. The first is the activity of the cystine-glutamate antiporter system (System Xc-), which provides the cellular supply of cystine for reduced glutathione synthesis [98]. Reduced glutathione is used by GPX4 to reduce lipid hydroperoxides (LOOH) and to prevent their accumulation in the cell and ferroptosis. The next factor is the correct functioning of the system lipoxygenases (LOXs) and NADPH oxidases (NOXs) that convert lipids (PLOH) to lipid peroxides (PLOOH), and often depend on iron as a cofactor [99]. Enhanced activity of NOXs and LOXs results in aberrant accumulation of lipid peroxides and rupture of the cell membrane. Non-enzymatic lipid oxidation can also be catalyzed in iron-mediated Fenton reaction via spontaneous autoxidation in Fe2+/H2O2 systems [100]. The third milestone is the cellular availability of Fe2+ which depends on the enzymatic conversion of Fe3+ to Fe2+ in endosomes by NADPH-dependent ferric-chelate reductase (STEAP3) and the presence of iron chelators. A minor factor is the level of ROS production and iron utilization by the mitochondria and the formation of lipid peroxides in mitochondrial membranes—mtROS [101]. Lipid peroxides in mitochondria are normally neutralized by dihydroorotate dehydrogenase (DHODH) by involving CoQ10 to prevent ferroptosis. The terminal event of ferroptosis is the loss of plasma membrane integrity and rupture of the outer membrane of the mitochondria.
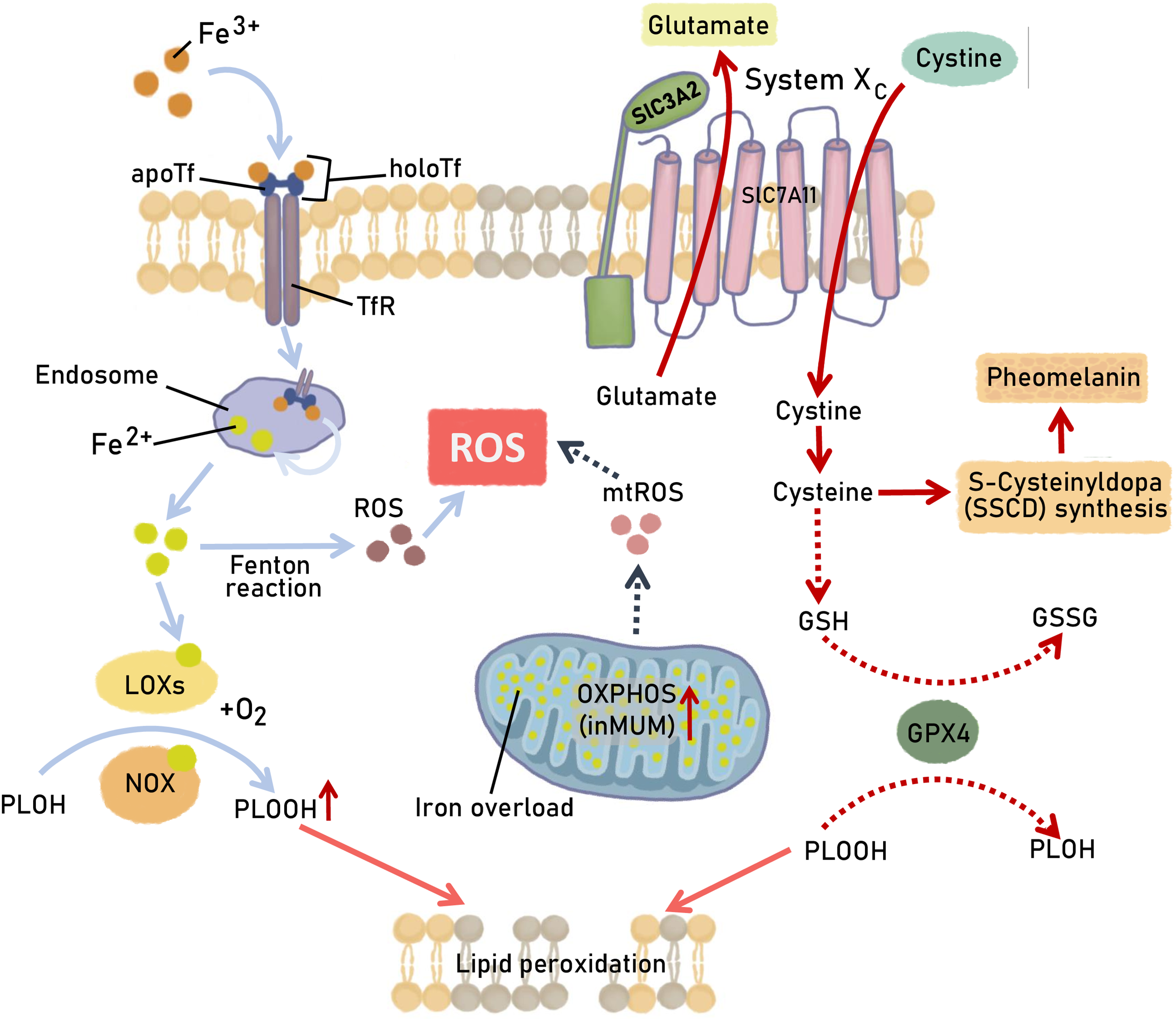
Figure 3: Ferroptosis-triggering pathways in melanoma cells. Regular mechanisms responsible for ferroptotic cell death are presented with the specific alteration in UM and CM cells and are highlighted by dashed arrows. A detailed description is provided in the text. The scheme depicts the metabolic pathways involved in glutathione peroxidase 4 (GPX4) activity, Fe3+/Fe2+ conversion, and their utilization in redox reactions producing the hydroperoxides of polyunsaturated fatty acids (PLOOH). “System Xc-” comprising of a heavy subunit 4F2hc (SLC3A2) and сystine/glutamate antiporter solute carrier family 7 member 11 (SLC7A11) −− is the transporter of cystine into the cell and glutamate out of the cell. GSH—reduced glutathione; GSSG—oxidized glutathione; LOXs—iron-dependent system lipoxygenases; NOXs—NADPH oxidase; OXPHOS—oxidative phosphorylation; TFR—Transferrin receptor; Fenton reaction: Fe2+ + H2O2 → Fe3+ + HO· + OH−. Figures are made in Sketchbook (2024), version 6.1.1, copyright holder-Sketchbook (https://www.sketchbook.com) (accessed on 12 January 2025)
It was shown that UM has one of the highest median oxidative phosphorylation (OXPHOS) levels among all solid tumors, that may shift tumor cell homeostasis toward mitochondrial ROS (mtROS) accumulation [102]. Excessive production of mtROS contributes to the accumulation of secondary oxidation products, including membrane lipids, making the cells sensitive to the induction of ferroptosis. It has been shown that among UM patients with inactivating mutations in the BRCA1-associated protein-1 (BAP1) gene, two subtypes with different levels of OXPHOS genes expression can be distinguished: OXPHOShigh BAP1 mutant UM cells utilize glycolytic and nucleotide biosynthesis pathways, whereas OXPHOSlow BAP1 mutant UM cells employ fatty acid oxidation [103]. Del Carpio et al. have found that high metabolic activity in liver-localized MUMs is associated with poor OS [104]. In all of these cases, metabolic switch can make UM cells sensitive to ferroptosis inducers. Moreover, it has been shown for CM that the increased susceptibility of cancer cells to ferroptosis occurs as a result of the dedifferentiation process of melanoma [105]. In general, dedifferentiated cancer cells become more resistant to immunotherapy and targeted therapy than before dedifferentiation [106]. Tumor melanocyte dedifferentiation can be recognized by the absence/decreased expression of the specific immunohistochemical markers and the appearance of non-melanocytic differentiation markers. Melanoma dedifferentiation downregulates eumelanin (EM) synthesis, producing a compensatory increase in pheomelanin (PM). Pheomelanin biosynthesis involves the conjugation of cysteine and dopaquinone, resulting in cysteinyldopa (SSCD). Increased incorporation of cysteine into pheomelanin in the dedifferentiated melanoma decreases cellular glutathione required for the reduction of lipid peroxidation products and modulates melanoma susceptibility to ferroptosis (Fig. 3). In the case of UM, a study of the ratio of eumelanin to pheomelanin in cultured UM cells has revealed that the EM/PM ratio (0.41) was lower than that from normal melanocytes isolated both from eyes with light-colored irises (1.31) or dark-colored irises (7.32) [107]. Thus, a significant PM content is a characteristic of UM.
Therapeutic approaches based on the induction of ferroptosis have been proposed for various types of cancers, mainly in in vitro experiments [108,109]. Ferroptosis inducers may also be useful in the treatment of melanoma, but prognostic markers are desirable for predicting sensitivity to this type of cell death. We have attempted to summarize the main hallmarks of melanoma sensitivity to ferroptosis in Table 1.

It was demonstrated that cytoglobin (CYGB), an iron hexacoordinated globin, is highly enriched in normal skin melanocytes and frequently epigenetically silenced during melanomagenesis, and CYGB silencing induced sensitivity of melanoma cells to ferroptosis [110,111]. Zou et al. produced constitutive expression or knockdown of CYGB in B16F10 mouse, G361, and A375 melanoma cell lines and tested cells’ sensitivity to ferroptosis inducers RSL3 and erastin [116]. RSL3 is a synthetic inductor of ferroptosis acting via inhibition of selenoprotein TXNRD1 in RAS-harboring cancer cells [117,118]. They confirmed the observation that melanoma cells with loss of CYGB expression were highly sensitive to ferroptosis. On the contrary, CYGB overexpression rendered melanoma much more resistant to RSL3 treatment. It can be hypothesized that CYGB expression levels can be evaluated in patients with skin melanoma to predict sensitivity to ferroptosis inducers.
In addition to CYGB, the role of iron regulatory protein 1 (IRP1) in the induction of ferroptosis was demonstrated in melanoma models. It turned out that IRP1 is required for erastin- and RSL3-induced ferroptosis [112]. IRP1 mediates iron availability in mammalian cells and controls iron homeostasis [119].
The accumulation and availability of multiple datasets on the response of melanoma patients to ICI therapy have identified 9 key immunogenic cell death (ICD) gene signatures that effectively predict ICI treatment response [120]. These signatures include both classical pathways such as apoptosis and necroptosis and rare or specific ones such as alkaliptosis, cuproptosis, entosis, lysosome-dependent cell death, oxeiptosis, and parthanatos. The authors [120] have estimated that metastatic melanoma patients with higher overlay ICD signatures are more likely to benefit from ICI therapy.
It has been shown that activated CD8+ T cells release interferon gamma (IFNγ), which downregulates the expression of components of System Xc- transporters SLC3A2 and SLC7A11 [113]. Such downregulation of SLC3A2 and SLC7A11 in tumor cells leads to death via the ferroptosis pathway [114]. Potentially, activation of such CD8+ T cells in combination with ICIs is a promising therapeutic approach.
In addition, iron nanoparticles carrying antitumor drugs can also be used for a comprehensive antitumor approach with induction of ferroptosis in CM cells, as it has been shown for hepatocellular carcinoma [121].
An attempt to identify genes associated with ferroptosis signature was made by Luo and Ma [122]. Genomes of UM patients from The Cancer Genome Atlas (TCGA) were analyzed for a total of 103 ferroptosis-related genes. A seven-gene signature was found as prognostic in UM. The expressions of ALOX12, CD44, MAP1LC3C, STEAP3, HMOX1, ITGA6, and AIFM2/FSP1 genes could accurately predict the sensitivity of UM to ferroptosis induction.
High cysteinyl leukotriene receptor 1 (CysLT1) expression correlates with poor survival of uveal melanoma patients and inhibition of CysLT1 may suppress the proliferation of UM cells [123]. Compound 1,4-dihydroxy quininib, a CysLT1 antagonist, alters UM hallmarks in vitro, ex vivo, and in vivo [124]. OMM2.5 cells were treated with 1,4-dihydroxy quininib and a proteomic analysis was made to identify differentially expressed proteins [125]. It was found that 1,4-dihydroxy quininib modulates the expression of ferroptosis markers: GPX4, biliverdin, glutamate-cysteine ligase modifier subunit (GCLM), and glutathione and lipid hydroperoxide. Public databases analysis revealed that the expression of genes inhibiting ferroptosis is significantly increased in UM patients with chromosome 3 monosomy. This data helps establish a novel ferroptosis signature correlating with UM patient survival.
Compared to ferroptosis, even less is known about cuproptosis in uveal melanoma. Cuproptosis has been discovered by Tsvetkov et al. [126]. This type of cell death is realized under excess of copper and is dependent on mitochondrial respiration. Copper directly binds to lipoylated components of the tricarboxylic acid cycle in the mitochondria and subsequent loss of Fe-S cluster protein triggers cell death [127]. Yang et al. have been trying to develop a prognostic signature of a reliable biomarker for predicting the prognosis of UM using RNA and clinical data from the TCGA database [128]. UM samples were classified into high- and low-risk groups based on cuproptosis-related prognostic signature. High expression of immune checkpoints in high-risk groups suggests significant immunotherapy potential.
6 Concluding Remarks and Potential Future Directions
Since 2010, significant progress has been made in the treatment of metastatic melanomas. However, the response rate even to such modern therapies as ICIs keeps a substantial cohort of patients without hope for a cure. For example, even for the combination of CTLA-4 and PD-1 blockade, objective response rates were 57.6% in patients with CM [129]. For uveal melanoma, the answer is even less: the overall response rate to CTLA-4 and PD-1 blockade combination therapy is 15.6% [130]. It can be concluded that a regimen of monotherapy or therapy with only one type of drug/one target does not achieve the desired efficacy. Perhaps a combination of drugs that activate different mechanisms of cell death—lymphocyte-dependent death and intrinsic pathway—will be more effective. It can be assumed that the use of IFNγ-releasing CD8+ T cells in combination with ICIs is relevant for CM but unlikely for UM, due to its low tumor infiltration by T cells. Over the past 5 years, phase phase I/II clinical trials have been completed for mono- and combination therapy for UM. Combination therapies included the ICIs nivolumab and ipilimumab (NCT02626962 [131], NCT01585194 [132]), the histone deacetylase inhibitor entinostat with pembrolizumab (NCT02697630 [133]). Monotherapy was efficient with RO7293583 bispecific molecule directed against TYRP1 and CD3 (NCT04551352 [134]), 6MHP multi-epitope vaccine, containing gp100, MelanA/MART-1, tyrosinase, MAGE-A3 and MAGE-A1,2,3,6 antigens (NCT04364230 [135]), niraparib, a PARP inhibitor for UM with mutations in the BAP1 gene (NCT03207347 [136]). In addition, radiation therapy for UM has been also tested, including brachytherapy with Ru-106 (NCT04577742 [137]) and I-125 isotopes (NCT01955941 [138]) and stereotactic radiation therapy in combination with aflibercept (NCT03712904 [139]). A phase III clinical trial of a promising approach to the therapy of UM liver metastases, liver perfusion with the chemotherapy drug melphalan, has also been completed (NCT02678572, NCT01785316 [140]).
As an opportunity for cancer therapy, iron accumulation and the implementation of ferroptosis becomes a problem in the development of cardiovascular and neurodegenerative diseases when iron accumulation is present in the heart, neurons, or glia [141]. However, given the severity and progression of MUM and MCM, one should not dismiss the possibilities based on induction of ferroptosis.
We attempted to summarize the knowledge of activated cell death pathways for different types of drugs (Fig. 4). If the tumor is not sensitive to apoptosis induction, one can try to influence it with ferroptosis or cuproptosis inducers. In this case, the mutational profile of a particular tumor should be taken into account. BAP1-deficient UMs may be less sensitive to ferroptosis than non-mutated, as BAP1 has been shown to be important for the realization of ferroptosis. BAP1 represses SLC7A11 expression with subsequent cystine uptake inhibition and ferroptotic cell death [115]. It is especially important that the potential markers of melanoma sensitivity to ferroptosis inducers have already been discovered [122].
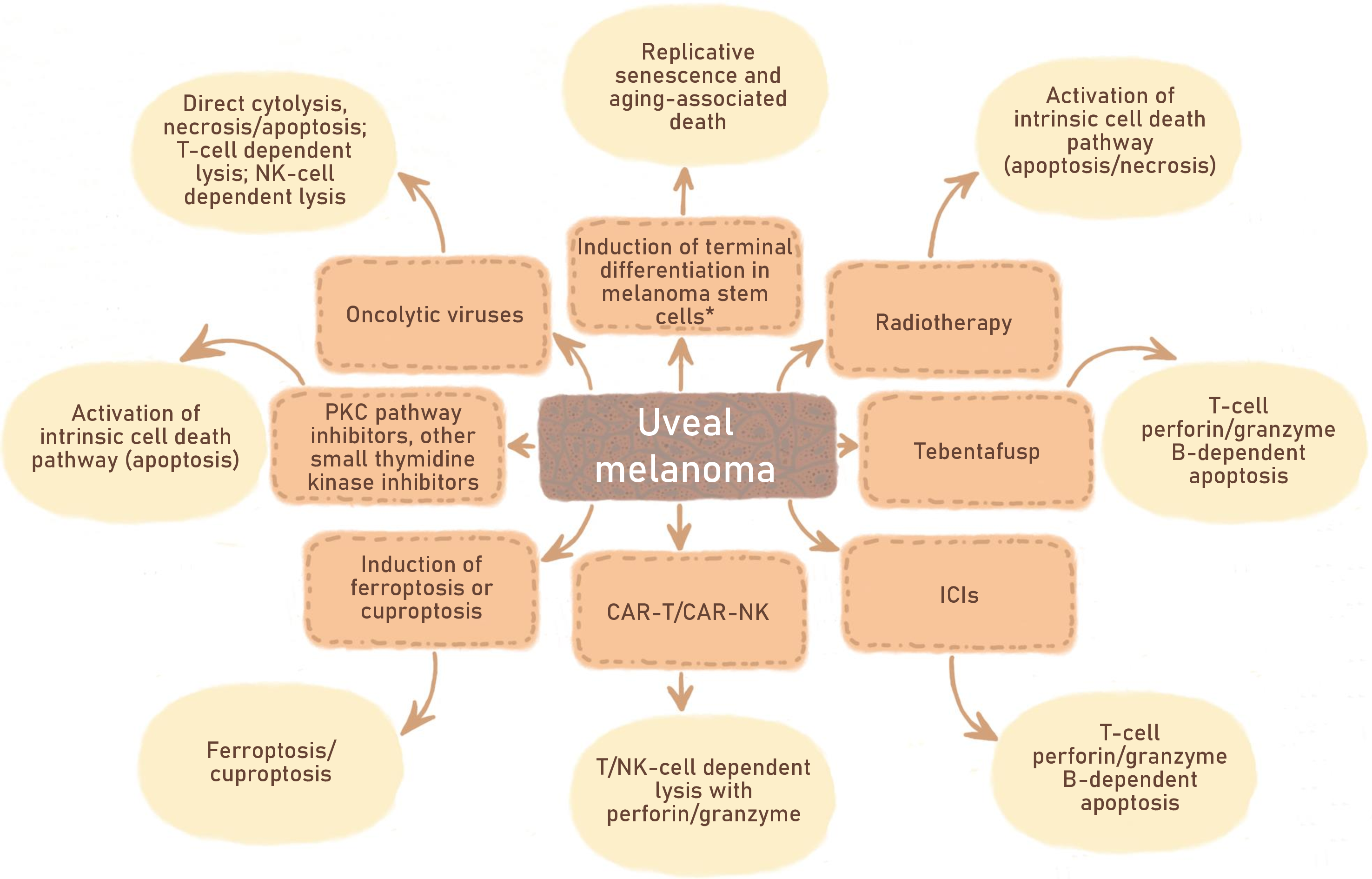
Figure 4: Antitumor drugs and therapeutic approaches being tested to combat UM. Divergences in the type of cell death activated by different inducers are indicated. *—requires additional research. Figures are made in Sketchbook (2024), version 6.1.1, copyright holder-Sketchbook (https://www.sketchbook.com) (accessed on 12 January 2025)
Moreover, in this review, we did not discuss the functionality of phagocytic cells in melanomas, but this fact should certainly be taken into account when choosing a therapy.
If we look at another extremely aggressive cancer pathology, gliomas, we can see a recently developed approach. This approach is an alternative to tumor cell killing, which is called “differentiation therapy”. The idea was that reprogramming cancer stem cells from proliferation to differentiation could correct aberrant cell division [142,143].
Since uveal melanocytes are developed from the neural crest, i.e., are of neural origin like gliomas, the question arises as to whether the said approach can be applied to deplete the stem malignant melanocyte niche in MUM. We believe that a detailed understanding of what may be considered a terminally differentiated state of malignant melanocytes is an important issue in developing such an approach. Combining this approach with the induction of replicative senescence in the remaining cells could prevent disease progression.
Acknowledgement: None.
Funding Statement: This work was supported by the Russian Science Foundation, project no. 23-14-00285 (https://www.rscf.ru) (accessed on 12 January 2025).
Author Contributions: The authors confirm their contribution to the paper as follows: review conception: Olga Koval, Maria Zhilnikova; abstract and conclusions: Olga Koval; part immunotherapy for melanomas: Mikhail Biryukov, Vasiliy Atamanov; part ferroptosis/cuproptosis and all figures: Maria Balantaeva; draft manuscript preparation: Olga Koval, Maria Zhilnikova. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Cronin KA, Scott S, Firth AU, Sung H, Henley SJ, Sherman RL, et al. Annual report to the nation on the status of cancer, part 1: national cancer statistics. Cancer. 2022;128(24):4251–84. doi:10.1002/cncr.34479. [Google Scholar] [PubMed] [CrossRef]
2. Wu M, Yavuzyiğitoğlu S, Brosens E, Ramdas WD, Kiliç E. Worldwide incidence of ocular melanoma and correlation with pigmentation-related risk factors. Invest Ophthalmol Vis Sci. 2023;64(13):45. doi:10.1167/iovs.64.13.45. [Google Scholar] [PubMed] [CrossRef]
3. Naseripoor M, Azimi F, Mirshahi R, Khakpoor G, Poorhosseingholi A, Chaibakhsh S. Global incidence and trend of uveal melanoma from 1943-2015: a meta-analysis. Asian Pac J Cancer Prev. 2022;23(5):1791–801. doi:10.31557/APJCP.2022.23.5.1791. [Google Scholar] [PubMed] [CrossRef]
4. Jamison A, Bhatti LA, Sobti MM, Chadha V, Cauchi P, Kemp EG. Uveal melanoma-associated survival in Scotland. Eye. 2019;33(11):1699–706. doi:10.1038/s41433-019-0622-9. [Google Scholar] [PubMed] [CrossRef]
5. Damato EM, Damato BE. Detection and time to treatment of uveal melanoma in the United Kingdom: an evaluation of 2384 patients. Ophthalmology. 2012;119(8):1582–9. doi:10.1016/j.ophtha.2012.01.048. [Google Scholar] [PubMed] [CrossRef]
6. Van Der Kooij MK, Speetjens FM, Van Der Burg SH, Kapiteijn E. Uveal versus cutaneous melanoma; same origin, very distinct tumor types. Cancers. 2019;11(6):845. doi:10.3390/cancers11060845. [Google Scholar] [PubMed] [CrossRef]
7. Vergara IA, Wilmott JS, Long GV, Scolyer RA. Genetic drivers of non-cutaneous melanomas: challenges and opportunities in a heterogeneous landscape. Exp Dermatol. 2022;31(1):13–30. doi:10.1111/exd.14287. [Google Scholar] [PubMed] [CrossRef]
8. Zhilnikova MV, Atamanov VV, Zvereva SP, Gayner TA, Boyarskikh UA, Stanishevskaya OM, et al. Molecular and genetic analysis of personal cultures of uveal melanoma cells. JOS. 2024;140(2S):63–71. doi:10.25276/0235-4160-2024-2s-63-71. [Google Scholar] [CrossRef]
9. Streilein JW. Ocular immune privilege: therapeutic opportunities from an experiment of nature. Nat Rev Immunol. 2003;3(11):879–89. doi:10.1038/nri1224. [Google Scholar] [PubMed] [CrossRef]
10. Carvajal RD, Schwartz GK, Tezel T, Marr B, Francis JH, Nathan PD. Metastatic disease from uveal melanoma: treatment options and future prospects. Br J Ophthalmol. 2017;101(1):38–44. doi:10.1136/bjophthalmol-2016-309034. [Google Scholar] [PubMed] [CrossRef]
11. Di Raimondo C, Lozzi F, Di Domenico PP, Campione E, Bianchi L. The diagnosis and management of cutaneous metastases from melanoma. IJMS. 2023;24(19):14535. doi:10.3390/ijms241914535. [Google Scholar] [PubMed] [CrossRef]
12. Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, et al. Cold tumors: a therapeutic challenge for immunotherapy. Front Immunol. 2019;10:168. doi:10.3389/fimmu.2019.00168. [Google Scholar] [PubMed] [CrossRef]
13. Montazeri K, Pattanayak V, Sullivan RJ. Tebentafusp in the treatment of metastatic uveal melanoma: patient selection and special considerations. Drug Des Devel Ther. 2023;17:333–9. doi:10.2147/DDDT.S368954. [Google Scholar] [PubMed] [CrossRef]
14. Cao L, Chen S, Sun R, Ashby CR, Wei L, Huang Z, et al. Darovasertib, a novel treatment for metastatic uveal melanoma. Front Pharmacol. 2023;14:1232787. doi:10.3389/fphar.2023.1232787. [Google Scholar] [PubMed] [CrossRef]
15. Cordero RJB, Casadevall A. Melanin. Curr Biol. 2020;30(4):R142–3. doi:10.1016/j.cub.2019.12.042. [Google Scholar] [PubMed] [CrossRef]
16. Buitrago E, Hardré R, Haudecoeur R, Jamet H, Belle C, Boumendjel A, et al. Are human tyrosinase and related proteins suitable targets for melanoma therapy? Curr Top Med Chem. 2016;16(27):3033–47. doi:10.2174/1568026616666160216160112. [Google Scholar] [PubMed] [CrossRef]
17. Roshardt Prieto NM, Turko P, Zellweger C, Nguyen-Kim TDL, Staeger R, Bellini E, et al. Patterns of radiological response to tebentafusp in patients with metastatic uveal melanoma. Melanoma Res. 2024;34(2):166–74. doi:10.1097/CMR.0000000000000952. [Google Scholar] [PubMed] [CrossRef]
18. Broussard L, Howland A, Ryu S, Song K, Norris D, Armstrong CA, et al. Melanoma cell death mechanisms. Chonnam Med J. 2018;54(3):135. doi:10.4068/cmj.2018.54.3.135. [Google Scholar] [PubMed] [CrossRef]
19. Wang JZ, Paulus P, Niu Y, Zhu L, Morisseau C, Rawling T, et al. The role of autophagy in human uveal melanoma and the development of potential disease biomarkers and novel therapeutic paradigms. Biomedicines. 2024;12(2):462. doi:10.3390/biomedicines12020462. [Google Scholar] [PubMed] [CrossRef]
20. Mattia G, Puglisi R, Ascione B, Malorni W, Carè A, Matarrese P. Cell death-based treatments of melanoma: conventional treatments and new therapeutic strategies. Cell Death Dis. 2018;9(2):112. doi:10.1038/s41419-017-0059-7. [Google Scholar] [PubMed] [CrossRef]
21. Zaffaroni N, Beretta GL. The Therapeutic Potential of pyroptosis in melanoma. IJMS. 2023;24(2):1285. doi:10.3390/ijms24021285. [Google Scholar] [PubMed] [CrossRef]
22. Passarelli A, Mannavola F, Stucci LS, Tucci M, Silvestris F. Immune system and melanoma biology: a balance between immunosurveillance and immune escape. Oncotarget. 2017;8(62):106132–42. doi:10.18632/oncotarget.22190. [Google Scholar] [PubMed] [CrossRef]
23. Keino H, Horie S, Sugita S. Immune privilege and eye-derived T-regulatory cells. J Immunol Res. 2018;2018(8):1–12. doi:10.1155/2018/1679197. [Google Scholar] [PubMed] [CrossRef]
24. Shechter R, London A, Schwartz M. Orchestrated leukocyte recruitment to immune-privileged sites: absolute barriers versus educational gates. Nat Rev Immunol. 2013;13(3):206–18. doi:10.1038/nri3391. [Google Scholar] [PubMed] [CrossRef]
25. Hori J, Kunishige T, Nakano Y. Immune Checkpoints contribute corneal immune privilege: implications for dry eye associated with checkpoint inhibitors. IJMS. 2020;21(11):3962. doi:10.3390/ijms21113962. [Google Scholar] [PubMed] [CrossRef]
26. Jiang T, Zhou C, Ren S. Role of IL-2 in cancer immunotherapy. OncoImmunology. 2016;5(6):e1163462. doi:10.1080/2162402X.2016.1163462. [Google Scholar] [PubMed] [CrossRef]
27. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Long-term outcomes with Nivolumab plus Ipilimumab or Nivolumab alone versus Ipilimumab in patients with advanced melanoma. J Clin Oncol. 2022;40(2):127–37. doi:10.1200/JCO.21.02229. [Google Scholar] [PubMed] [CrossRef]
28. Seth R, Agarwala SS, Messersmith H, Alluri KC, Ascierto PA, Atkins MB, et al. Systemic therapy for melanoma: ASCO guideline update. J Clin Oncol. 2023;41(30):4794–820. doi:10.1200/JCO.23.01136. [Google Scholar] [PubMed] [CrossRef]
29. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with Ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23. doi:10.1056/NEJMoa1003466. [Google Scholar] [PubMed] [CrossRef]
30. Grasso CS, Tsoi J, Onyshchenko M, Abril-Rodriguez G, Ross-Macdonald P, Wind-Rotolo M, et al. Conserved interferon-γ signaling drives clinical response to immune checkpoint blockade therapy in melanoma. Cancer Cell. 2020;38(4):500–15. doi:10.1016/j.ccell.2020.08.005. [Google Scholar] [PubMed] [CrossRef]
31. Piulats JM, Watkins C, Costa-García M, Del Carpio L, Piperno-Neumann S, Rutkowski P, et al. Overall survival from Tebentafusp versus Nivolumab plus Ipilimumab in first-line metastatic uveal melanoma: a propensity score-weighted analysis. Ann Oncology. 2024;35(3):317–26. doi:10.1016/j.annonc.2023.11.013. [Google Scholar] [PubMed] [CrossRef]
32. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–74. doi:10.1038/nri2506. [Google Scholar] [PubMed] [CrossRef]
33. Neubert NJ, Schmittnaegel M, Bordry N, Nassiri S, Wald N, Martignier C, et al. T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Sci Transl Med. 2018;10(436):eaan3311. doi:10.1126/scitranslmed.aan3311. [Google Scholar] [PubMed] [CrossRef]
34. Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, et al. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2014;74(1):153–61. doi:10.1158/0008-5472.CAN-13-1816. [Google Scholar] [PubMed] [CrossRef]
35. Larkin J, Del Vecchio M, Mandalá M, Gogas H, Arance Fernandez AM, Dalle S, et al. Adjuvant Nivolumab versus Ipilimumab in resected stage III/IV melanoma: 5-Year efficacy and biomarker results from CheckMate 238. Clin Cancer Res. 2023;29(17):3352–61. doi:10.1158/1078-0432.CCR-22-3145. [Google Scholar] [PubMed] [CrossRef]
36. Versluis JM, Menzies AM, Sikorska K, Rozeman EA, Saw RPM, Van Houdt WJ, et al. Survival update of neoadjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma in the OpACIN and OpACIN-neo trials. Ann Oncol. 2023;34(4):420–30. doi:10.1016/j.annonc.2023.01.004. [Google Scholar] [PubMed] [CrossRef]
37. Blank CU, Lucas MW, Scolyer RA, Van De Wiel BA, Menzies AM, Lopez-Yurda M, et al. Neoadjuvant Nivolumab and Ipilimumab in resectable stage III melanoma. N Engl J Med. 2024;391(18):1696–708. doi:10.1056/NEJMoa2402604. [Google Scholar] [PubMed] [CrossRef]
38. Wolchok JD, Chiarion-Sileni V, Rutkowski P, Cowey CL, Schadendorf D, Wagstaff J, et al. Final, 10-year outcomes with Nivolumab plus Ipilimumab in advanced melanoma. N Engl J Med. 2024;392(1):11–22. doi:10.1056/NEJMoa2407417. [Google Scholar] [PubMed] [CrossRef]
39. Tan L, Yin T, Xiang H, Wang L, Mudgal P, Chen J, et al. Aberrant cytoplasmic expression of UHRF1 restrains the MHC-I-mediated anti-tumor immune response. Nat Commun. 2024;15(1):8569. doi:10.1038/s41467-024-52902-5. [Google Scholar] [PubMed] [CrossRef]
40. Hori J, Wang M, Miyashita M, Tanemoto K, Takahashi H, Takemori T, et al. B7-H1-Induced apoptosis as a mechanism of immune privilege of corneal allografts. J Immunol. 2006;177(9):5928–35. doi:10.4049/jimmunol.177.9.5928. [Google Scholar] [PubMed] [CrossRef]
41. Yang W, Li H, Chen PW, Alizadeh H, He Y, Hogan RN, et al. PD-L1 Expression on human ocular cells and its possible role in regulating immune-mediated ocular inflammation. Invest Ophthalmol Vis Sci. 2009;50(1):273. doi:10.1167/iovs.08-2397. [Google Scholar] [PubMed] [CrossRef]
42. Shen L, Jin Y, Freeman GJ, Sharpe AH, Dana MR. The function of donor versus recipient programmed death-ligand 1 in corneal allograft survival. J Immunol. 2007;179(6):3672–9. doi:10.4049/jimmunol.179.6.3672. [Google Scholar] [PubMed] [CrossRef]
43. Bender C, Enk A, Gutzmer R, Hassel JC. Anti-PD-1 antibodies in metastatic uveal melanoma: a treatment option? Cancer Med. 2017;6(7):1581–6. doi:10.1002/cam4.887. [Google Scholar] [PubMed] [CrossRef]
44. Yamada K, Takeuchi M, Fukumoto T, Suzuki M, Kato A, Mizuki Y, et al. Immune checkpoint inhibitors for metastatic uveal melanoma: a meta-analysis. Sci Rep. 2024;14(1):7887. doi:10.1038/s41598-024-55675-5. [Google Scholar] [PubMed] [CrossRef]
45. Salaün H, De Koning L, Saint-Ghislain M, Servois V, Ramtohul T, Garcia A, et al. Nivolumab plus ipilimumab in metastatic uveal melanoma: a real-life, retrospective cohort of 47 patients. OncoImmunology. 2022;11(1):2116845. doi:10.1080/2162402X.2022.2116845. [Google Scholar] [PubMed] [CrossRef]
46. Ma KSK, Tsai PF, Hsieh TYJ, Chodosh J. Ocular surface complications following biological therapy for cancer. Front Toxicol. 2023;5:1137637. doi:10.3389/ftox.2023.1137637. [Google Scholar] [PubMed] [CrossRef]
47. Pham JP, On L, Ardolino L, Hurwitz J, Salaun H, Sim H-W, et al. Efficacy of immune checkpoint inhibition in metastatic uveal melanoma: a systematic review and meta-analysis. Melanoma Res. 2023;33(4):316–25. doi:10.1097/CMR.0000000000000900. [Google Scholar] [PubMed] [CrossRef]
48. Shao YF, Baca Y, Xiu J, Vanderwalde AM, In GK, Hoon DSB, et al. Immune profiling of metastatic uveal melanoma and response to immune checkpoint inhibitors. J Clin Oncol. 2022;40(16_suppl):9565–5. doi:10.1200/JCO.2022.40.16_suppl.9565. [Google Scholar] [CrossRef]
49. Lepage S, Lapointe R. Melanosomal targeting sequences from gp100 are essential for MHC Class II-restricted endogenous epitope presentation and mobilization to endosomal compartments. Cancer Res. 2006;66(4):2423–32. doi:10.1158/0008-5472.CAN-05-2516. [Google Scholar] [PubMed] [CrossRef]
50. Nathan P, Hassel JC, Rutkowski P, Baurain JF, Butler MO, Schlaak M, et al. Overall survival benefit with Tebentafusp in metastatic uveal melanoma. N Engl J Med. 2021;385(13):1196–206. doi:10.1056/NEJMoa2103485. [Google Scholar] [PubMed] [CrossRef]
51. Hassel JC, Piperno-Neumann S, Rutkowski P, Baurain JF, Schlaak M, Butler MO, et al. Three-year overall survival with Tebentafusp in metastatic uveal melanoma. N Engl J Med. 2023;389(24):2256–66. doi:10.1056/NEJMoa2304753. [Google Scholar] [PubMed] [CrossRef]
52. Mann JE, Hasson N, Su DG, Adeniran AJ, Smalley KSM, Djureinovic D, et al. GP100 expression is variable in intensity in melanoma. Cancer Immun Immunother. 2024;73(10):191. doi:10.1007/s00262-024-03776-5. [Google Scholar] [PubMed] [CrossRef]
53. Simon B, Harrer DC, Schuler-Thurner B, Schuler G, Uslu U. Arming T cells with a gp100-specific TCR and a CSPG4-specific CAR using combined DNA- and RNA-based receptor transfer. Cancers. 2019;11(5):696. doi:10.3390/cancers11050696. [Google Scholar] [PubMed] [CrossRef]
54. Jilani S, Saco JD, Mugarza E, Pujol-Morcillo A, Chokry J, Ng C, et al. CAR-T cell therapy targeting surface expression of TYRP1 to treat cutaneous and rare melanoma subtypes. Nat Commun. 2024;15(1):1244. doi:10.1038/s41467-024-45221-2. [Google Scholar] [PubMed] [CrossRef]
55. Hibler W, Merlino G, Yu Y. CAR NK cell therapy for the treatment of metastatic melanoma: potential & prospects. Cells. 2023;12(23):2750. doi:10.3390/cells12232750. [Google Scholar] [PubMed] [CrossRef]
56. Khalil DN, Postow MA, Ibrahim N, Ludwig DL, Cosaert J, Kambhampati SRP, et al. An open-label, dose-escalation phase I study of anti-TYRP1 monoclonal antibody IMC-20D7S for patients with relapsed or refractory melanoma. Clinic Cancer Res. 2016;22(21):5204–10. doi:10.1158/1078-0432.CCR-16-1241. [Google Scholar] [PubMed] [CrossRef]
57. Chandran SS, Somerville RPT, Yang JC, Sherry RM, Klebanoff CA, Goff SL, et al. Treatment of metastatic uveal melanoma with adoptive transfer of tumour-infiltrating lymphocytes: a single-centre, two-stage, single-arm, phase 2 study. Lancet Oncol. 2017;18(6):792–802. doi:10.1016/S1470-2045(17)30251-6. [Google Scholar] [PubMed] [CrossRef]
58. Karlsson JW, Sah VR, Olofsson Bagge R, Kuznetsova I, Iqba M, Alsen S, et al. Patient-derived xenografts and single-cell sequencing identifies three subtypes of tumor-reactive lymphocytes in uveal melanoma metastases. eLife. 2024;12:RP91705. doi:10.7554/eLife.91705.3. [Google Scholar] [CrossRef]
59. Forsberg EMV, Lindberg MF, Jespersen H, Alsén S, Bagge RO, Donia M, et al. HER2 CAR-T cells eradicate uveal melanoma and T cell therapy-resistant human melanoma in IL2 transgenic NOD/SCID IL2 receptor knockout mice. Cancer Res. 2019;79(5):899–904. doi:10.1158/0008-5472.CAN-18-3158. [Google Scholar] [PubMed] [CrossRef]
60. Andtbacka RHI, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene Laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–8. doi:10.1200/JCO.2014.58.3377. [Google Scholar] [PubMed] [CrossRef]
61. Kochneva G, Sivolobova G, Tkacheva A, Grazhdantseva A, Troitskaya O, Nushtaeva A, et al. Engineering of double recombinant vaccinia virus with enhanced oncolytic potential for solid tumor virotherapy. Oncotarget. 2016;7(45):74171–88. doi:10.18632/oncotarget.12367. [Google Scholar] [PubMed] [CrossRef]
62. Andtbacka RHI, Curti B, Daniels GA, Hallmeyer S, Whitman ED, Lutzky J, et al. Clinical responses of oncolytic coxsackievirus A21 (V937) in patients with unresectable melanoma. J Clin Oncol. 2021;39(34):3829–38. doi:10.1200/JCO.20.03246. [Google Scholar] [PubMed] [CrossRef]
63. Curti BD, Richards J, Hyngstrom JR, Daniels GA, Faries M, Feun L, et al. Intratumoral oncolytic virus V937 plus ipilimumab in patients with advanced melanoma: the phase 1b MITCI study. J Immunother Cancer. 2022;10(12):e005224. doi:10.1136/jitc-2022-005224. [Google Scholar] [PubMed] [CrossRef]
64. Lutzky J, Sullivan RJ, Cohen JV, Ren Y, Li A, Haq R. Phase 1b study of intravenous coxsackievirus A21 (V937) and ipilimumab for patients with metastatic uveal melanoma. J Cancer Res Clin Oncol. 2023;149(9):6059–66. doi:10.1007/s00432-022-04510-3. [Google Scholar] [PubMed] [CrossRef]
65. Liu S, Liu F, Zhao M, Zhang J. Antitumor efficacy of oncolytic herpes virus type 1 armed with GM-CSF in murine uveal melanoma xenografts. Cancer Manag Res. 2020;12:11803–12. doi:10.2147/CMAR.S274605. [Google Scholar] [PubMed] [CrossRef]
66. Zhu H, Gao H, Ji Y, Zhou Q, Du Z, Tian L, et al. Targeting p53-MDM2 interaction by small-molecule inhibitors: learning from MDM2 inhibitors in clinical trials. J Hematol Oncol. 2022;15(1):91. doi:10.1186/s13045-022-01314-3. [Google Scholar] [PubMed] [CrossRef]
67. Sullivan KD, Galbraith MD, Andrysik Z, Espinosa JM. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018;25(1):133–43. doi:10.1038/cdd.2017.174. [Google Scholar] [PubMed] [CrossRef]
68. Khan SU, Ullah Z, Shaukat H, Unab S, Jannat S, Ali W, et al. TP53 and its regulatory genes as prognosis of cutaneous melanoma. Cancer Inform. 2023;22:11769351231177267. doi:10.1177/11769351231177267. [Google Scholar] [PubMed] [CrossRef]
69. Toledo F, Wahl GM. MDM2 and MDM4: P53 regulators as targets in anticancer therapy. Int J Biochem Cell Biol. 2007;39(7–8):1476–82. doi:10.1016/j.biocel.2007.03.022. [Google Scholar] [PubMed] [CrossRef]
70. Zhang H, Hu L, Qiu W, Deng T, Zhang Y, Bergholz J, et al. MDMX exerts its oncogenic activity via suppression of retinoblastoma protein. Oncogene. 2015;34(44):5560–9. doi:10.1038/onc.2015.11. [Google Scholar] [PubMed] [CrossRef]
71. Arnoff TE, El-Deiry WS. MDM2/MDM4 amplification and CDKN2A deletion in metastatic melanoma and glioblastoma multiforme may have implications for targeted therapeutics and immunotherapy. Am J Cancer Res. 2022;12(5):2102–17. [Google Scholar] [PubMed]
72. Groen K, Steffens Reinhardt L, Bourdon JC, Avery-Kiejda KA. It is not all about the alpha: elevated expression of p53β variants is associated with lower probability of survival in a retrospective melanoma cohort. Cancer Cell Int. 2023;23(1):228. doi:10.1186/s12935-023-03083-6. [Google Scholar] [PubMed] [CrossRef]
73. Chana JS, Wilson GD, Cree IA, Alexander RA, Myatt N, Neale M, et al. c-myc, p53, and Bcl-2 expression and clinical outcome in uveal melanoma. Brit J Ophthalm. 1999;83(1):110–4. doi:10.1136/bjo.83.1.110. [Google Scholar] [PubMed] [CrossRef]
74. Togashi K, Suzuki S, Mitobe Y, Nakagawa-Saito Y, Sugai A, Takenouchi S, et al. CEP-1347 dually targets MDM4 and PKC to activate p53 and inhibit the growth of uveal melanoma cells. Cancers. 2023;16(1):118. doi:10.3390/cancers16010118. [Google Scholar] [PubMed] [CrossRef]
75. Vuong L, Conley SM, Al-Ubaidi MR. Expression and role of p53 in the retina. Invest Ophthalmol Vis Sci. 2012;53(3):1362. doi:10.1167/iovs.11-8909. [Google Scholar] [PubMed] [CrossRef]
76. Cepko CL, Austin CP, Yang X, Alexiades M, Ezzeddine D. Cell fate determination in the vertebrate retina. Proc Natl Acad Sci U S A. 1996;93(2):589–95. doi:10.1073/pnas.93.2.589. [Google Scholar] [PubMed] [CrossRef]
77. Holzer P, Masuya K, Furet P, Kallen J, Valat-Stachyra T, Ferretti S, et al. Discovery of a dihydroisoquinolinone derivative (NVP-CGM097a highly potent and selective MDM2 inhibitor undergoing phase 1 clinical trials in p53wt tumors. J Med Chem. 2015;58(16):6348–58. doi:10.1021/acs.jmedchem.5b00810. [Google Scholar] [PubMed] [CrossRef]
78. Heijkants RC, Nieveen M, Hart KC‘T, Teunisse AFAS, Jochemsen AG. Targeting MDMX and PKCδ to improve current uveal melanoma therapeutic strategies. Oncogenesis. 2018;7(3):33. doi:10.1038/s41389-018-0041-y. [Google Scholar] [PubMed] [CrossRef]
79. Matin RN, Chikh A, Law Pak Chong S, Mesher D, Graf M, Sanza’ P, et al. p63 is an alternative p53 repressor in melanoma that confers chemoresistance and a poor prognosis. J Exp Med. 2013;210(3):581–603. doi:10.1084/jem.20121439. [Google Scholar] [PubMed] [CrossRef]
80. Kilic E, Brüggenwirth HT, Meier M, Naus NC, Beverloo HB, Meijerink JP, et al. Increased expression of p73Δex2 transcript in uveal melanoma with loss of chromosome 1p. Melanoma Res. 2008;18(3):208–13. doi:10.1097/CMR.0b013e3283036aa1. [Google Scholar] [PubMed] [CrossRef]
81. Zhilnikova MV, Troitskaya OS, Novak DD, Atamanov VV, Koval OA. Uveal melanoma: molecular and genetic mechanisms of development and therapeutic approaches. Mol Biol. 2024;58(2):165–77. doi:10.1134/S0026893324020183. [Google Scholar] [CrossRef]
82. Onken MD, Noda SE, Kaltenbronn KM, Frankfater C, Makepeace CM, Fettig N, et al. Oncogenic Gq/11 signaling acutely drives and chronically sustains metabolic reprogramming in uveal melanoma. J Biol Chem. 2022;298(1):101495. doi:10.1016/j.jbc.2021.101495. [Google Scholar] [PubMed] [CrossRef]
83. Shi Y. Mechanisms of caspase activation and inhibition during apoptosis. Mol Cell. 2002;9(3):459–70. doi:10.1016/S1097-2765(02)00482-3. [Google Scholar] [PubMed] [CrossRef]
84. Yan B. Research progress on Livin protein: an inhibitor of apoptosis. Mol Cell Biochem. 2011;357(1–2):39–45. doi:10.1007/s11010-011-0873-7. [Google Scholar] [PubMed] [CrossRef]
85. Vucic D, Stennicke HR, Pisabarro MT, Salvesen GS, Dixit VM. ML-IAP, a novel inhibitor of apoptosis that is preferentially expressed in human melanomas. Curr Biol. 2000;10(21):1359–66. doi:10.1016/S0960-9822(00)00781-8. [Google Scholar] [PubMed] [CrossRef]
86. Yan H, Brouha B, Liu T, Raj D, Biddle D, Lee R, et al. Proteolytic cleavage of Livin (ML-IAP) in apoptotic melanoma cells potentially mediated by a non-canonical caspase. J Dermatol Sci. 2006;43(3):189–200. doi:10.1016/j.jdermsci.2006.05.007. [Google Scholar] [PubMed] [CrossRef]
87. Vucic D, Franklin MC, Wallweber HJA, Das K, Eckelman BP, Shin H, et al. Engineering ML-IAP to produce an extraordinarily potent caspase 9 inhibitor: implications for Smac-dependent anti-apoptotic activity of ML-IAP. Biochem J. 2005;385(1):11–20. doi:10.1042/BJ20041108. [Google Scholar] [PubMed] [CrossRef]
88. De Maria A, Bassnett S. Birc7: a late fiber gene of the crystalline lens. Invest Ophthalmol Vis Sci. 2015;56(8):4823. doi:10.1167/iovs.15-16968. [Google Scholar] [PubMed] [CrossRef]
89. Wang H, Yang Y, Wang W, Guan B, Xun M, Zhang H, et al. Single-chain antibody-delivered Livin siRNA inhibits human malignant melanoma growth in vitro and in vivo. Tumour Biol. 2017;39(5):101042831770164. doi:10.1177/1010428317701645. [Google Scholar] [PubMed] [CrossRef]
90. Xu H, Liu H, Li Z, Xu Q, Lin N, Li X. BIRC7 is beneficial for melanoma progression and hypoxic response. CCID. 2022;15:1109–17. doi:10.2147/CCID.S370969. [Google Scholar] [PubMed] [CrossRef]
91. Lederman M, Meir T, Zeschnigk M, Pe’er J, Chowers I. Inhibitor of apoptosis proteins gene expression and its correlation with prognostic factors in primary and metastatic uveal melanoma. Curr Eye Res. 2008;33(10):876–84. doi:10.1080/02713680802382989. [Google Scholar] [PubMed] [CrossRef]
92. Lederman M, Meir T, Pe’er J, Chowers I. Inhibitor of apoptosis proteins (IAP) expression in primary and metastatic uveal melanoma. ARVO Annual Meet Abst. 2006;47(13):2241. [Google Scholar]
93. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266–82. doi:10.1038/s41580-020-00324-8. [Google Scholar] [PubMed] [CrossRef]
94. Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature. 2019;572(7769):402–6. doi:10.1038/s41586-019-1426-6. [Google Scholar] [PubMed] [CrossRef]
95. Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes tophospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16(3):302–9. doi:10.1038/s41589-020-0472-6. [Google Scholar] [PubMed] [CrossRef]
96. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688–92. doi:10.1038/s41586-019-1705-2. [Google Scholar] [PubMed] [CrossRef]
97. Talty R, Bosenberg M. The role of ferroptosis in melanoma. Pigment Cell Melanoma Res. 2022;35(1):18–25. doi:10.1111/pcmr.13009. [Google Scholar] [PubMed] [CrossRef]
98. Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2021;12(8):599–620. doi:10.1007/s13238-020-00789-5. [Google Scholar] [PubMed] [CrossRef]
99. Probst L, Dächert J, Schenk B, Fulda S. Lipoxygenase inhibitors protect acute lymphoblastic leukemia cells from ferroptotic cell death. Biochem Pharmacol. 2017;140(15):41–52. doi:10.1016/j.bcp.2017.06.112. [Google Scholar] [PubMed] [CrossRef]
100. Nguyen NT, Kim J, Le XT, Lee WT, Lee ES, Oh KT, et al. Amplified fenton-based oxidative stress utilizing ultraviolet upconversion luminescence-fueled nanoreactors for apoptosis-strengthened ferroptosis anticancer therapy. ACS Nano. 2023;17(1):382–401. doi:10.1021/acsnano.2c08706. [Google Scholar] [PubMed] [CrossRef]
101. Ta N, Jiang X, Zhang Y, Wang H. Ferroptosis as a promising therapeutic strategy for melanoma. Front Pharmacol. 2023;14:1252567. doi:10.3389/fphar.2023.1252567. [Google Scholar] [PubMed] [CrossRef]
102. Chattopadhyay C, Roszik J, Bhattacharya R, Alauddin M, Mahmud I, Yadugiri S, et al. Imipridones inhibit tumor growth and improve survival in an orthotopic liver metastasis mouse model of human uveal melanoma. Br J Cancer. 2024;131(11):1846–57. doi:10.1038/s41416-024-02866-6. [Google Scholar] [PubMed] [CrossRef]
103. Han A, Purwin TJ, Bechtel N, Liao C, Chua V, Seifert E, et al. BAP1 mutant uveal melanoma is stratified by metabolic phenotypes with distinct vulnerability to metabolic inhibitors. Oncogene. 2021;40(3):618–32. doi:10.1038/s41388-020-01554-y. [Google Scholar] [PubMed] [CrossRef]
104. Del Carpio LP, Algarra MA, Sabaté-Llobera A, Rodriguez-Vida A, Rossi-Seoane S, Ruiz S, et al. Differences in glucose metabolic activity in liver metastasis separates two groups of metastatic uveal melanoma patients with different prognosis. Cancer Med. 2023;12(13):14062–71. doi:10.1002/cam4.6058. [Google Scholar] [PubMed] [CrossRef]
105. Jaklitsch E, Vats K, Kruglov O, Bunimovich Y. 43385 Melanogenesis regulates susceptibility of melanoma cells to ferroptosis. J Am Acad Dermatol. 2023;89(3):AB63. doi:10.1016/j.jaad.2023.07.255. [Google Scholar] [CrossRef]
106. Massi D, Mihic-Probst D, Schadendorf D, Dummer R, Mandalà M. Dedifferentiated melanomas: morpho-phenotypic profile, genetic reprogramming and clinical implications. Cancer Treat Rev. 2020;88:102060. doi:10.1016/j.ctrv.2020.102060. [Google Scholar] [PubMed] [CrossRef]
107. Hu DN, Wakamatsu K, Ito S, McCormick SA. Comparison of eumelanin and pheomelanin content between cultured uveal melanoma cells and normal uveal melanocytes. Melanoma Res. 2009;19(2):75–9. doi:10.1097/CMR.0b013e328329ae49. [Google Scholar] [PubMed] [CrossRef]
108. Zhao L, Zhou X, Xie F, Zhang L, Yan H, Huang J, et al. Ferroptosis in cancer and cancer immunotherapy. Cancer Commun. 2022;42(2):88–116. doi:10.1002/cac2.12250. [Google Scholar] [PubMed] [CrossRef]
109. Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7(7):e2307. doi:10.1038/cddis.2016.208. [Google Scholar] [PubMed] [CrossRef]
110. Chakraborty S, John R, Nag A. Cytoglobin in tumor hypoxia: novel insights into cancer suppression. Tumor Biol. 2014;35(7):6207–19. doi:10.1007/s13277-014-1992-z. [Google Scholar] [PubMed] [CrossRef]
111. De Backer J, Maric D, Zuhra K, Bogaerts A, Szabo C, Vanden Berghe W, et al. Cytoglobin silencing promotes melanoma malignancy but sensitizes for ferroptosis and pyroptosis therapy response. Antioxidants. 2022;11(8):1548. doi:10.3390/antiox11081548. [Google Scholar] [PubMed] [CrossRef]
112. Yao F, Cui X, Zhang Y, Bei Z, Wang H, Zhao D, et al. Iron regulatory protein 1 promotes ferroptosis by sustaining cellular iron homeostasis in melanoma. Oncol Lett. 2021;22(3):657. doi:10.3892/ol.2021.12918. [Google Scholar] [PubMed] [CrossRef]
113. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270–4. doi:10.1038/s41586-019-1170-y. [Google Scholar] [PubMed] [CrossRef]
114. Kong R, Wang N, Han W, Bao W, Lu J. IFNγ-mediated repression of system xc−drives vulnerability to induced ferroptosis in hepatocellular carcinoma cells. J Leukoc Biol. 2021;110(2):301–14. doi:10.1002/JLB.3MA1220-815RRR. [Google Scholar] [PubMed] [CrossRef]
115. Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. 2018;20(10):1181–92. doi:10.1038/s41556-018-0178-0. [Google Scholar] [PubMed] [CrossRef]
116. Zou Z, Yu Q, Yang Y, Wang F, Zhu P, Zhang X, et al. Cytoglobin attenuates melanoma malignancy but protects melanoma cells from ferroptosis. Mol Med Rep. 2024;30(6):219. doi:10.3892/mmr.2024.13343. [Google Scholar] [PubMed] [CrossRef]
117. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15(3):234–45. doi:10.1016/j.chembiol.2008.02.010. [Google Scholar] [PubMed] [CrossRef]
118. Cheff DM, Huang C, Scholzen KC, Gencheva R, Ronzetti MH, Cheng Q, et al. The ferroptosis inducing compounds RSL3 and ML162 are not direct inhibitors of GPX4 but of TXNRD1. Redox Biol. 2023;62(13):102703. doi:10.1016/j.redox.2023.102703. [Google Scholar] [PubMed] [CrossRef]
119. Bonadonna M, Altamura S, Tybl E, Palais G, Qatato M, Polycarpou-Schwarz M, et al. Iron regulatory protein (IRP)–mediated iron homeostasis is critical for neutrophil development and differentiation in the bone marrow. Sci Adv. 2022;8(40):eabq4469. doi:10.1126/sciadv.abq4469. [Google Scholar] [PubMed] [CrossRef]
120. Zeng H, Jiang Q, Zhang R, Zhuang Z, Wu J, Li Y, et al. Immunogenic cell death signatures from on-treatment tumor specimens predict immune checkpoint therapy response in metastatic melanoma. Sci Rep. 2024;14(1):22872. doi:10.1038/s41598-024-74636-6. [Google Scholar] [PubMed] [CrossRef]
121. Wang L, Tong L, Xiong Z, Chen Y, Zhang P, Gao Y, et al. Ferroptosis-inducing nanomedicine and targeted short peptide for synergistic treatment of hepatocellular carcinoma. J Nanobiotechnol. 2024;22(1):533. doi:10.1186/s12951-024-02808-7. [Google Scholar] [PubMed] [CrossRef]
122. Luo H, Ma C. A Novel ferroptosis-associated gene signature to predict prognosis in patients with uveal melanoma. Diagnostics. 2021;11(2):219. doi:10.3390/diagnostics11020219. [Google Scholar] [PubMed] [CrossRef]
123. Slater K, Heeran AB, Garcia-Mulero S, Kalirai H, Sanz-Pamplona R, Rahman A, et al. High cysteinyl leukotriene receptor 1 expression correlates with poor survival of uveal melanoma patients and cognate antagonist drugs modulate the growth, cancer secretome, and metabolism of uveal melanoma cells. Cancers. 2020;12(10):2950. doi:10.3390/cancers12102950. [Google Scholar] [PubMed] [CrossRef]
124. Slater K, Bosch R, Smith KF, Jahangir CA, Garcia-Mulero S, Rahman A, et al. 1,4-dihydroxy quininib modulates the secretome of uveal melanoma tumour explants and a marker of oxidative phosphorylation in a metastatic xenograft model. Front Med. 2023;9:1036322. doi:10.3389/fmed.2022.1036322. [Google Scholar] [PubMed] [CrossRef]
125. Tonelotto V, Costa-Garcia M, O’Reilly E, Smith KF, Slater K, Dillon ET, et al. 1,4-dihydroxy quininib activates ferroptosis pathways in metastatic uveal melanoma and reveals a novel prognostic biomarker signature. Cell Death Discov. 2024;10(1):70. doi:10.1038/s41420-023-01773-8. [Google Scholar] [PubMed] [CrossRef]
126. Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375(6586):1254–61. doi:10.1126/science.abf0529. [Google Scholar] [PubMed] [CrossRef]
127. Li SR, Bu LL, Cai L. Cuproptosis: lipoylated TCA cycle proteins-mediated novel cell death pathway. Sig Transduct Target Ther. 2022;7(1):158. doi:10.1038/s41392-022-01014-x. [Google Scholar] [PubMed] [CrossRef]
128. Yang Y, Li Q, Chen J, Guo Y, Cai Y, Zhao W, et al. A cuproptosis-related prognostic signature for guiding clinical diagnosis and treatment in uveal melanoma patients. Heliyon. 2024;10(16):e36324. doi:10.1016/j.heliyon.2024.e36324. [Google Scholar] [PubMed] [CrossRef]
129. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. doi:10.1056/NEJMoa1504030. [Google Scholar] [PubMed] [CrossRef]
130. Heppt MV, Amaral T, Kähler KC, Heinzerling L, Hassel JC, Meissner M, et al. Combined immune checkpoint blockade for metastatic uveal melanoma: a retrospective, multi-center study. J Immunother Cancer. 2019;7(1):299. doi:10.1186/s40425-019-0800-0. [Google Scholar] [PubMed] [CrossRef]
131. Piulats JM, Espinosa E, De La Cruz Merino L, Varela M, Alonso Carrión L, Martín-Algarra S, et al. Nivolumab plus Ipilimumab for treatment-naïve metastatic uveal melanoma: an open-label, multicenter, phase II trial by the spanish multidisciplinary melanoma group (GEM-1402). J Clin Oncol. 2021;39(6):586–98. doi:10.1200/JCO.20.00550. [Google Scholar] [PubMed] [CrossRef]
132. Pelster MS, Gruschkus SK, Bassett R, Gombos DS, Shephard M, Posada L, et al. Nivolumab and Ipilimumab in metastatic uveal melanoma: results from a single-arm phase II study. J Clin Oncol. 2021;39(6):599–607. doi:10.1200/JCO.20.00605. [Google Scholar] [PubMed] [CrossRef]
133. Ny L, Jespersen H, Karlsson J, Alsén S, Filges S, All-Eriksson C, et al. The PEMDAC phase 2 study of pembrolizumab and entinostat in patients with metastatic uveal melanoma. Nat Commun. 2021;12(1):5155. doi:10.1038/s41467-021-25332-w. [Google Scholar] [PubMed] [CrossRef]
134. Spreafico A, Couselo EM, Irmisch A, Bessa J, Au-Yeung G, Bechter O, et al. Phase 1, first-in-human study of TYRP1-TCB (RO7293583a novel TYRP1-targeting CD3 T-cell engager, in metastatic melanoma: active drug monitoring to assess the impact of immune response on drug exposure. Front Oncol. 2024;14:1346502. doi:10.3389/fonc.2024.1346502. [Google Scholar] [PubMed] [CrossRef]
135. Le Naour J, Kroemer G. Trial watch: toll-like receptor ligands in cancer therapy. Oncoimmunology. 2023;12(1):2180237. doi:10.1080/2162402X.2023.2180237. [Google Scholar] [PubMed] [CrossRef]
136. George TJ, Lee JH, DeRemer DL, Hosein PJ, Staal S, Markham MJ, et al. Phase II trial of the PARP inhibitor, Niraparib, in BAP1 and other DNA damage response pathway-deficient neoplasms. JCO Precis Oncol. 2024;8(8):e2400406. doi:10.1200/PO-24-00406. [Google Scholar] [PubMed] [CrossRef]
137. Cennamo G, Montorio D, Andrea D’, Farella L, Matano A, Giuliano E, et al. Long-term outcomes in uveal melanoma after Ruthenium-106 brachytherapy. Front Oncol. 2022;11:754108. doi:10.3389/fonc.2021.754108. [Google Scholar] [PubMed] [CrossRef]
138. Skalet AH, Liu L, Binder C, Miller AK, Crilly R, Hung AY, et al. Longitudinal detection of radiation-induced peripapillary and macular retinal capillary ischemia using OCT angiography. Ophthalmol Retina. 2020;4(3):320–6. doi:10.1016/j.oret.2019.10.001. [Google Scholar] [PubMed] [CrossRef]
139. Toro MD, Gozzo L, Tracia L, Cicciù M, Drago F, Bucolo C, et al. New Therapeutic perspectives in the treatment of uveal melanoma: a systematic review. Biomedicines. 2021;9(10):1311. doi:10.3390/biomedicines9101311. [Google Scholar] [PubMed] [CrossRef]
140. Olofsson BR, Nelson A, Shafazand A, All-Eriksson C, Cahlin C, Elander N, et al. Isolated hepatic perfusion with melphalan for patients with isolated uveal melanoma liver metastases: a multicenter, randomized, open-label, phase III trial (the SCANDIUM Trial). J Clin Oncol. 2023;41(16):3042–50. doi:10.1200/JCO.22.01705. [Google Scholar] [PubMed] [CrossRef]
141. Wang S, Guo Q, Zhou L, Xia X. Ferroptosis: a double-edged sword. Cell Death Discov. 2024;10(1):265. doi:10.1038/s41420-024-02037-9. [Google Scholar] [PubMed] [CrossRef]
142. Liu J, Wang X, Chen AT, Gao X, Himes BT, Zhang H, et al. ZNF117 regulates glioblastoma stem cell differentiation towards oligodendroglial lineage. Nat Commun. 2022;13(1):2196. doi:10.1038/s41467-022-29884-3. [Google Scholar] [PubMed] [CrossRef]
143. Kolesnikova V, Revishchin A, Fab L, Alekseeva A, Ryabova A, Pronin I, et al. GQIcombi application to subdue glioma via differentiation therapy. Front Oncol. 2024;14:1322795. doi:10.3389/fonc.2024.1322795. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools