
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Mitochondrial Oxidative Stress-Associated Mechanisms in the Development of Metabolic Dysfunction-Associated Steatotic Liver Disease
1 Department of Neonatology, The Affiliated Wuxi People’s Hospital of Nanjing Medical University, Wuxi Children’s Hospital, Wuxi, 214000, China
2 Department of Neonatology, Affiliated Children’s Hospital of Jiangnan University, Wuxi Children's Hospital, Wuxi, 214023, China
3 Department of Pediatric Laboratory, Affiliated Children’s Hospital of Jiangnan University, Wuxi Children’s Hospital, Wuxi, 214023, China
4 Division of Oncology, Department of Clinical Sciences Lund, Lund University, Lund, 22381, Sweden
* Corresponding Authors: Le Zhang. Email: ; Zhenshan Yang. Email:
# These authors contributed equally to this work
(This article belongs to the Special Issue: Mitochondrial Dynamics and Oxidative Stress in Disease: Cellular Mechanisms and Therapeutic Targets)
BIOCELL 2025, 49(3), 399-417. https://doi.org/10.32604/biocell.2025.059908
Received 19 October 2024; Accepted 02 January 2025; Issue published 31 March 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
With the prevalence of obesity, metabolic dysfunction-associated steatotic liver disease (MASLD) has become the most common chronic liver disease worldwide and can cause a series of serious complications. The pathogenesis of MASLD is complex, characterized by oxidative stress, impaired mitochondrial function and lipid metabolism, and cellular inflammation. Mitochondrial biology and function are central to the physiology of the liver. It has been suggested that mitochondrial oxidative stress plays a crucial role in MASLD progression. Excessive oxidative stress response is an important trigger for the occurrence and development of MASLD. In this review, we aim to focus on the recent advances in understanding mitochondrial oxidative stress-related mechanisms in the progression of MASLD. The in-depth elaboration of its related mechanisms is hoped to help find effective methods for treating MASLD.Keywords
Abbreviations
| ACACA | Acetyl CoA Carboxylase 1 |
| AICAR | 5-Aminoimidazole-4-carboxamide ribotide |
| ALCAT1 | Lysocardiolipin acyltransferase 1 |
| AMPK | Adenosine monophosphate-activated protein kinase |
| ATP | Adenosine triphosphate |
| CL | Cardiolipin |
| CPT1A | Carnitine palmitoyltransferase 1α |
| ER | Endoplasmic reticulum |
| ERMCS | ER-mitochondria contact sites |
| FA | Fatty acid |
| GPX | Glutathione peroxidase |
| GSH | Glutathione |
| HCC | Hepatocellular carcinoma |
| 8-OH-dG | 8-Hydroxy-2’-deoxyguanosine |
| 8-OH-G | 8-hydroxyguanine |
| 4-HNE | 4-hydroxy-2-nonenal |
| MDA | Malondialdehyde |
| IP3R1-GRP75-VDAC1 | Inositol 1,4,5-trisphosphate receptor 1-glucose-regulated protein 75-voltage-dependent anion channel 1 |
| Keap1 | Kelch-like ECH-associated protein 1 |
| LD | Lipid droplet |
| LOOH | Lipid hydroperoxide |
| MASLD | Metabolic dysfunction-associated steatotic liver disease |
| MLKL | Mixed lineage kinase domain-like protein |
| mtDNA | Mitochondrial DNA |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Nonalcoholic steatohepatitis |
| NFE2L2 | Nuclear factor erythroid-derived 2-like 2 |
| NF-κB p65 | Nuclear factor kappa B p65 |
| Nrf2 | Nuclear factor-erythroid 2-related factor 2 |
| NOX4 | NAD(P)H oxidase 4 |
| OS | Oxidative stress |
| OXPHOS | Oxidative phosphorylation |
| OxPLs | Oxidized phospholipids |
| PC | Phosphocholine |
| PPARα | Peroxisome proliferator-activated receptor alpha |
| PGC1α | Peroxisome proliferator-activated receptor gamma coactivator-1α |
| PRDX | Peroxiredoxin |
| PTP | Protein tyrosine phosphatases |
| rRNA | Ribosomal RNA |
| tRNA | Transfer RNA |
| RIPK3 | Receptor-interacting protein kinase 3 |
| ROS | Reactive oxygen species |
| SOD | Superoxide dismutase |
| STAT-1 | Signal transducer and activator of transcription-1 |
| SIRT1 | Sirtuin 1 |
| Trx | Thioredoxin |
| TFAM | Transcription factor A |
Non-alcoholic fatty liver disease (NAFLD) is a chronic liver condition. With the improvement of people’s living standards, the prevalence of high-calorie and high-fat diets, as well as sedentary and physically inactive lifestyles, has led to a global increase in obesity and NAFLD [1]. Currently, NAFLD has reached epidemic proportions, affecting 20%–30% of the world’s population [2,3] and becoming the most common cause of chronic liver disease globally [4]. To more accurately reflect its pathogenesis, NAFLD was renamed in 2020 as metabolic dysfunction-associated fatty liver disease (MAFLD) in relation to fatty liver disorders [5,6], which has gained recognition from various global stakeholders [7]. In June 2023, a multi-society Delphi Consensus Statement on the new nomenclature for fatty liver diseases established the term metabolic dysfunction-associated steatotic liver disease (MASLD) [8]. As a result of some patients advancing to liver fibrosis, cirrhosis, and even hepatocellular carcinoma [9], coupled with a propensity for complications such as cardiovascular and cerebrovascular diseases that elevate related mortality and morbidity rates [10], MASLD has emerged as a significant public health issue affecting human health [11].
However, there is currently no definitive safe and effective drug available for the routine treatment of MASLD. Lifestyle modifications, including reduced energy intake and increased physical activity, represent the most effective methods for preventing and treating MASLD [12]. Given the immense societal and economic burdens associated with caring for MASLD patients, efforts are being made to explore effective interventions to alleviate symptoms and even achieve a cure for the disease.
Over the past decade, MASLD has been recognized as a complex metabolic disease [13] arising from the intricate interplay between genetic susceptibility, host metabolic disturbances, and environmental factors. Many factors influence its clinical manifestations and disease progression, including ethnicity, genetic susceptibility, dietary habits, metabolism, immunity, and gut microbiota. Despite the significant expansion of knowledge that has greatly enhanced our understanding of MASLD, the underlying mechanisms of its pathogenesis remain elusive. In hepatocytes, mitochondria account for 18% of the total volume of cells and have an effect on the metabolic functions of the liver and the oxidation of nutrients to generate energy [14]. Liver mitochondria are essential for glucose, lipid, and protein metabolism, as well as energy production. Furthermore, liver mitochondria function as nutrient sensors, with their biogenesis, dynamics, and interactions with other organelles being dynamically regulated to adapt to liver metabolism [15]. To perform these diverse functions, mitochondria interact and dynamically relationships with other organelles, coordinating their stress responses to mitochondrial challenges in both physiological and pathological settings. Therefore, it is unsurprising that mitochondrial dysfunction is intimately tied to the underlying mechanisms of liver metabolic abnormalities.
Numerous studies have demonstrated the presence of widespread mitochondrial abnormalities in patients with MASLD [16], aligning with observations that impaired energy metabolism persists in clinical manifestation. Abnormal mitochondrial functions, such as compromised fatty acid oxidation and oxidative phosphorylation, drive oxidative stress, as depicted in Fig. 1, which serves as an initial and notable manifestation of the MASLD disease [16]. In this context, we shall review the proof of hepatic mitochondrial oxidative stress in human MASLD patients. Additionally, in further sections, discuss the advancements in research on the mechanisms contributing to mitochondrial dysfunction in MASLD, which possibly reveal fresh avenues for therapeutic advancements in the future.
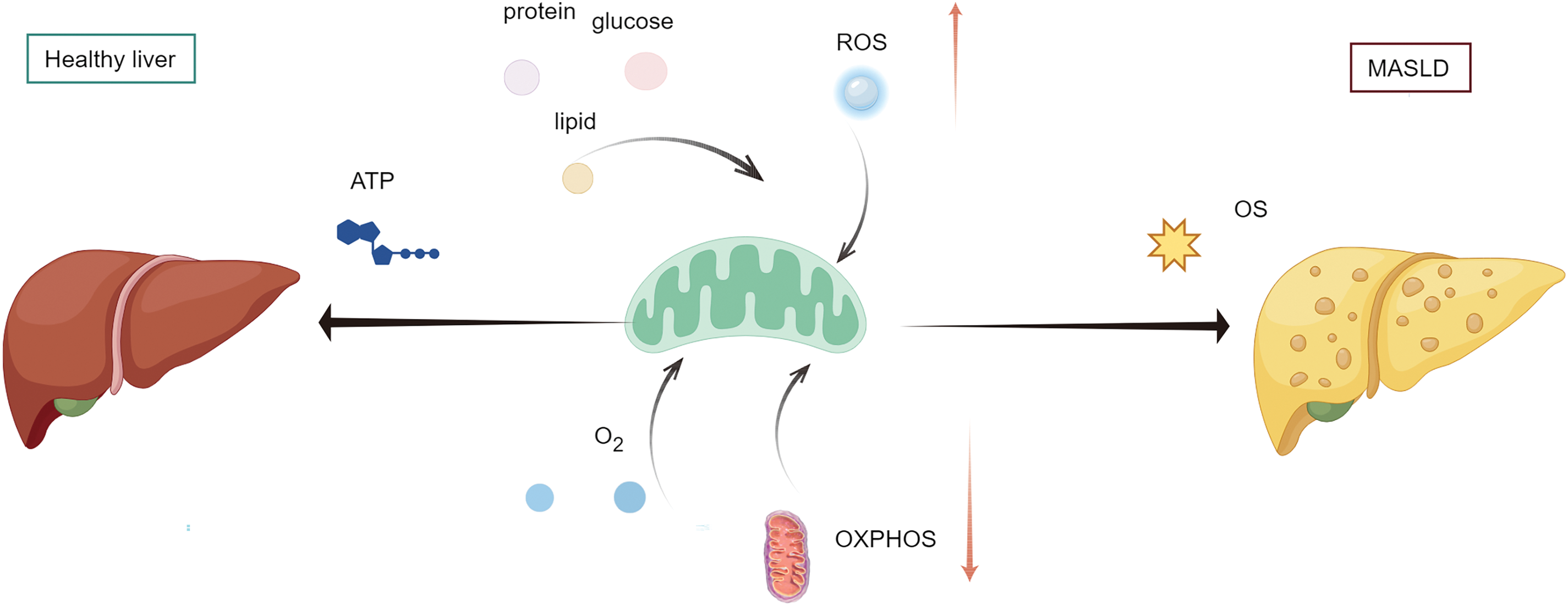
Figure 1: Oxidative stress contributes to MASLD. Under normal conditions, liver mitochondria utilize glucose, lipids, and proteins as substrates for metabolism, generating adenosine triphosphate (ATP) through oxygen consumption. Abnormal mitochondrial function, such as impaired fatty acid oxidation and oxidative phosphorylation (OXPHOS), can drive oxidative stress (OS), leading to metabolic-associated fatty liver disease (MASLD). ROS, reactive oxygen species (By Figdraw)
2 Oxidative Stress Implicates Mitochondrial Dysfunction in MASLD
The liver, which comprises 2%–4% of the total body weight, is nevertheless accountable for 15% of the human body’s oxygen consumption [17]. The significant respiration-to-liver-weight ratio indicates that hepatocytes possess a high abundance of mitochondria, which generate adenosine triphosphate (ATP) by utilizing energy-yielding substrates derived from the catabolism of carbohydrates, lipids, and proteins [14]. Mitochondria are the core power source that performs many vital functions in cells, including oxidative phosphorylation (OXPHOS), reactive oxygen species (ROS) production, nutritional metabolism, intracellular signaling cascades, and apoptosis [18,19]. One of the important functions of mitochondria in the liver is to generate energy in the form of ATP through OXPHOS [15,20]. Mitochondria is the leading site of ROS formation in cells and serves as a critical determinant of maintaining normal energy cellular redox homeostasis in multiple life processes [21]. 90% of cellular ROS in the human body is produced by mitochondria [22,23]. ROS can be divided into free radicals and nonradicals according to their chemical properties. Free radicals mainly include superoxide anion radical (O2•-), hydroxyl radical (HO•), nitric oxide (NO•), nitrogen dioxide (NO2•), carbonate radical anion (CO3•-), and alkoxyl/alkyl peroxyl (RO•/ROO•), and the main nonradical species are H2O2, hypochlorous acid (HOCl), and peroxynitrite (ONOO−)/peroxynitrous acid (ONOOH) [24]. Several major ROS clearance pathways exist to maintain a steady level of ROS. For example, O2•- is disproportionated into H2O2 by superoxide dismutase (SOD), H2O2 is decomposed into H2O and O2 by catalase, H2O2 and lipid hydroperoxide(LOOH) are decomposed by glutathione peroxidase (GPX) using glutathione (GSH) as an electron donor, and H2O2 is reduced to H2O by peroxiredoxin (PRDX) 1-5 using reduced thioredoxin (Trx) as the electron donor [24]. When the production of intracellular ROS exceeds the scavenging capacity of the antioxidant system, it will lead to the accumulation of intracellular oxygen free radicals. Excessive oxygen free radicals can damage the structure and function of cells, which is called oxidative stress (OS) [25]. Excessive OS reaction can affect the mitochondrial respiratory chain, cause mitochondrial energy metabolism disorders, and lead to mitochondrial damage, which in turn can promote the generation of ROS in the body, and then aggravate the liver oxidative stress reaction, and lead to hepatocyte apoptosis. The interplay between mitochondrial dysfunction and oxidative stress forms a vicious cycle.
In fact, OS is an essential feature of MASLD [16]. Through the microscope, researchers can observe different levels of mitochondrial ultrastructural injury, morphological abnormalities, and decreased respiratory chain activity, and other characteristics in liver biopsy of patients with MASLD [26–28], which serves to underscore the pivotal function of OS in MASLD. Numerous pieces of evidence have shown heightened oxidative damage affecting nearly all macromolecules within the livers of individuals suffering from MASLD, encompassing proteins, sugar, lipids, and nucleic acids, which gives rise to the creation of oxidative damage biomarkers [29]. In other words, macromolecules can be damaged by OS, resulting in the formation of toxic substances [30]. For instance, lipids undergo oxidation/peroxidation, producing a range of substances; the primary concern lies with unsaturated fatty acids, particularly those with conjugated double bonds, such as linoleic acid, which are susceptible to attack by free radicals. Their peroxidation leads to the cleavage of these fatty acids, resulting in the production of aldehydes such as malondialdehyde (MDA) and 4-hydroxy-2-nonenal (4-HNE). There are also other products of lipid oxidative damage, such as lipid peroxides and 8-isoprostane. The fatty acyl chains of phospholipids and triglycerides are subject to hydroxyl radical attack, causing the emergence of these molecules. This attack may further lead to the production of reactive aldehydes and/or the disruption of cellular structures and organization. Oxidation of protein can result in modification of amino acids, protein cross-linkage, and formation of protein carbonyls. DNA/RNA oxidation can lead to the fragmentation of single or double-strand nucleic acids or the modification of bases or sugars. Among them, 8-hydroxy-2’-deoxyguanosine (8-OH-dG) and 8-hydroxyguanine (8-OH-G) are the most common DNA/RNA damage products. These biomarkers of OS were determined in clinical samples of MASLD, and their concentrations or activities were increased, although the increase was not significant in a few cases [26,31,32]. The characteristic of MASLD is the accumulation of macrovesicular fat in hepatocytes observed in liver biopsy, which is caused by disruptions in the transport and metabolism of fatty acid (FA) [33]. Fat accumulation can impair fatty acid oxidation, reducing mitochondrial ATP synthesis respiration [34]. Previous studies utilizing mouse models of nonalcoholic steatohepatitis (NASH) and human tissue biopsies have revealed that obesity induces hepatic OS, leading to the oxidation and inactivation of hepatic protein tyrosine phosphatases (PTP). This, in turn, facilitates the activation of signal transducer and activator of transcription-1 (STAT-1) signaling, which promotes the recruitment of cytotoxic T cells and exacerbates the progression of NASH and liver fibrosis [35]. In summary, elevated oxidative oxidative stress contributed to mitochondrial dysfunction and disrupted energy metabolism in MASLD.
3 Mechanisms Underlying Mitochondrial Dysfunction in MASLD
In patients with MASLD, regardless of whether mitochondrial dysfunction serves as the primary or secondary mechanism, heightened oxidative stress within mitochondria, compromised bioenergetic function, and defective mitochondrial quality control mechanisms represent uniform characteristics of mitochondrial abnormalities observed in MASLD. All these interactions contribute to deleterious effects. Although these abnormalities may vary in triggering mitochondrial dysfunction in MASLD patients, as depicted in Fig. 2, depending on each individual’s unique biological, environmental, and genetic factors, any one of these abnormalities has the potential to induce the occurrence of others. Mitochondria serve as the primary site of oxidative metabolism, and OS and lipid peroxidation are major pathogenic factors in MASLD. In MASLD patients, the decline in mitochondrial function leads to the loss of ATP synthesis and reduced consumption of ROS, resulting in the accumulation of ROS. It promotes increased lipid deposition and OS responses in the liver, ultimately leading to hepatocyte apoptosis. Given mitochondrial dysfunction’s pivotal role in the pathogenesis of MASLD, extensive research has been conducted on the underlying mechanisms of mitochondrial dysfunction and related OS in MASLD. Since changes in mitochondrial function often precede clinical symptoms, timely detection of these changes and the implementation of appropriate treatments are significant in preventing or delaying the occurrence and progression of MASLD.
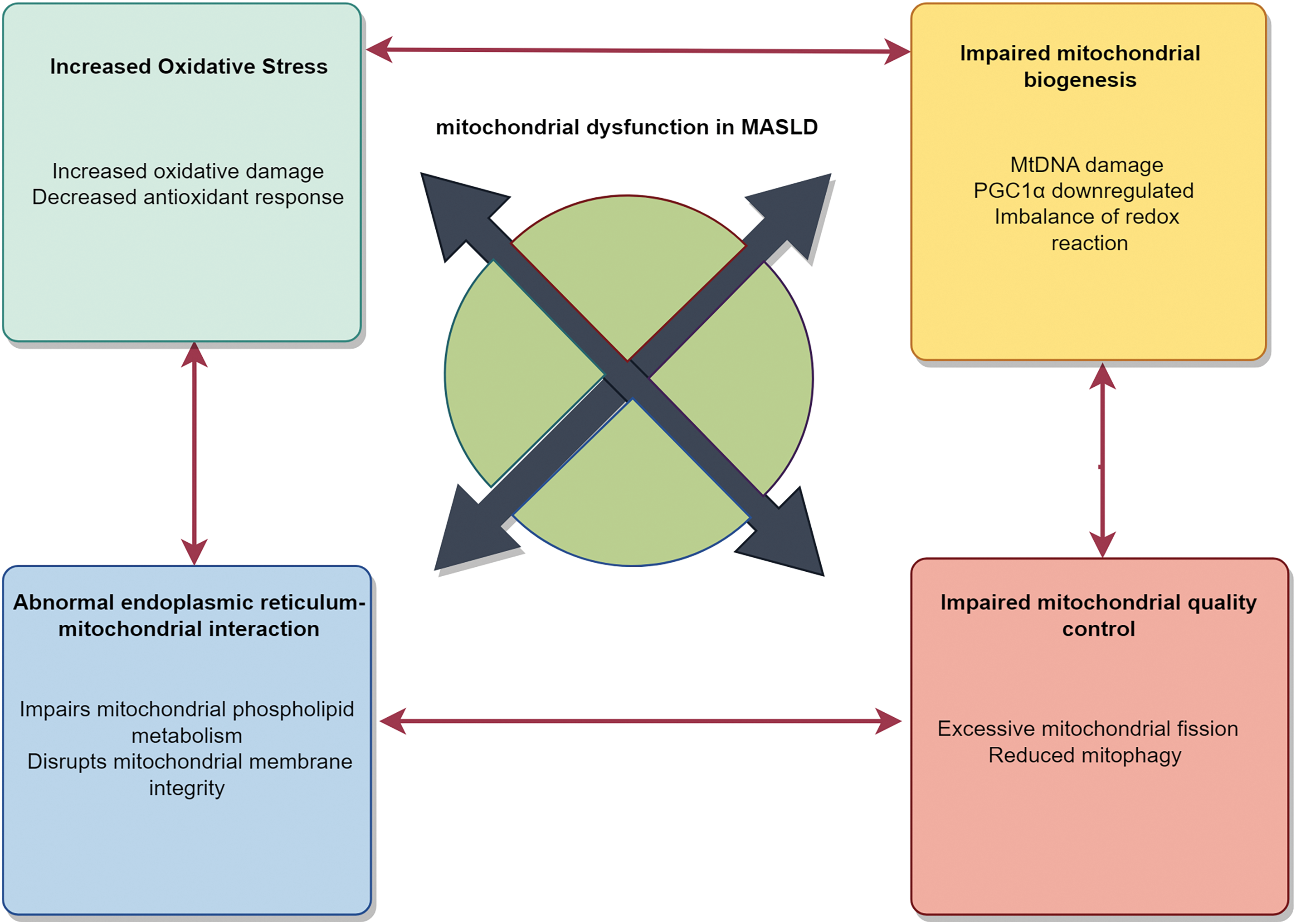
Figure 2: Mechanisms underlying mitochondrial dysfunction in MASLD. Mitochondrial dysfunction induced by oxidative stress plays a pivotal role in metabolic-associated fatty liver disease (MASLD). Oxidative stress, a key driver in MASLD, increases ROS production, damaging macromolecules, impairing mitochondrial function, and disrupting antioxidant defenses. Impaired mitochondrial quality control, especially autophagy dysfunction, hinders damaged mitochondrial clearance. Mitochondrial DNA mutations reduce energy production, affecting cellular physiology. ER-mitochondrial dysfunction, with reduced contact, disrupts material exchange and signaling. While their relative importance in MASLD varies, these abnormalities interact and may induce each other. PGC1α, peroxisome proliferator-activated receptor gamma coactivator-1α; mtDNA, mitochondrial DNA; ER, endoplasmic reticulum (By Figdraw)
3.1 Increased Oxidative Stress in MASLD
In the process of electron transfer in aerobic respiration of mitochondria, the by-product of ROS is inevitable due to the leakage of electrons at complexes I and III [36]. However, ROS play a significant signaling role, whenever there is too much, the rate of intracellular ROS production outweighs the cell’s antioxidant capacity; they result in OS and severe damage despite the presence of antioxidant systems such as catalases, peroxiredoxins, and glutathione/glutathione peroxidase systems [37]. An increase in OS may be the cause of mitochondrial dysfunction.
MASLD is a complex multifactorial disease related to lifestyle, genetic, and environmental factors. In the “multiple-hit” hypothesis about its pathogenesis, OS is considered to be the main factor leading to liver injury and disease progression in MASLD [38] and has also been identified as an essential feature of human steatohepatitis [16]. Given the relationship between mitochondrial oxidative metabolism and the occurrence and development of MASLD, researchers have been continuously exploring over the past two decades to understand better the mechanisms of hepatic mitochondrial oxidative stress in MASLD. This endeavor aims to provide potential therapeutic targets for clinical practice.
Earlier, researchers found that the oxidative stress pathway leads to mitochondrial dysfunction in metabolic diseases. Cardiolipin (CL) is a mitochondrial phospholipid required for mitochondrial bioenergy and membrane structure [39]. Lysocardiolipin acyltransferase 1 (ALCAT1) serves as a catalyst for pathological CL remodeling in several diseases [40]. CL is highly sensitive to ROS oxidative damage due to its double bonds and unique position in mitochondria. At the same time, CL is the only phospholipid in mitochondria that undergo early oxidation during apoptosis. The experiment found that MASLD significantly up-regulated the expression of ALCAT1 protein in the liver of Wild-Type control mice, and ALCAT1 deficiency could dramatically improve insulin sensitivity and reduce liver fat production and fibrosis [41]. In short, the expression of ALCAT1 leads to CL peroxidation, leading to a vicious cycle of oxidative stress and mitochondrial dysfunction and ultimately leading to MASLD [41].
In Europe, MASLD is considered a metabolic predisposition to hepatocellular carcinoma (HCC) [42–44]. As various immunosuppressive mechanisms involving different immune cell subsets have been studied in HCC [45], the role of adaptive immune response in MASLD and HCC has been recognized [46]. Here, researchers found that the imbalance of lipid metabolism in MASLD can lead to the selective loss of CD4 T lymphocytes in the liver, but will not lead to the selective loss of CD8 T lymphocytes, and accelerate the occurrence of liver cancer [47]. It was also found that CD4 T lymphocytes had greater mitochondrial mass than CD8 T lymphocytes and could produce higher levels of ROS. Among the fatty acids accumulated in MASLD, linoleic acid can destroy the function of mitochondria, cause more oxidative damage than other free fatty acids, and mediate the selective loss of CD4 T lymphocytes in the liver. Blocking ROS in vivo can reverse the reduction of CD4 T lymphocytes in the liver induced by MASLD and delay the promotion of HCC by MASLD [47].
In 2018, Baker et al. proposed that the onset of MASLD may originate from the fetal period [48]. Previous human studies have described that infants born to obese mothers have increased intracellular lipid storage and have a higher risk of developing obesity, MASLD, cardiovascular disease, and liver cancer in later life [49–51]. Compared with adult livers, fetal livers normally have fewer mitochondria, lower activity of the fatty acid metabolic enzyme carnitine palmitoyl transferase-1 (CPT1), and little or no gluconeogenesis. However, fetal exposure to excessive maternal fuel can uniquely alter liver fatty acid oxidation, tricarboxylic acid cycle activity, de novo adipogenesis, and mitochondrial health functions. These events can promote the increase of oxidative stress and the excessive storage of this peroxidation leads to the exposure of hydrophilic phosphocholine (PC) head groups and the formation of highly reactive oxidized phospholipids diglycerides. Together with the changes in immune function and epigenetic changes, they make the fetal liver ready for the occurrence of MASLD and may increase the risk of MASLD in the next generation.
Lipid toxic factors produced by lipid peroxidation exist in many inflammatory environments, such as atherosclerosis [52], infectious pulmonary diseases [53], and MASLD [54,55]. Phospholipids containing PC are the main components of low-density lipoprotein, cell membranes, and lipid droplets. They usually contain polyunsaturated fatty acids at the sn-2 position, which are susceptible to free radical-induced lipid peroxidation. This peroxidation leads to the exposure of hydrophilic PC head groups and the formation of highly reactive oxidized phospholipids (OxPLs). OxPLs can induce cell stress and apoptosis by regulating the activity of intracellular signal transduction and gene expression. Studies have shown that OxPLs accumulate in MASLD in humans and mice. In the mouse model, it was found that OxPLs partially inactivate the enzyme manganese superoxide dismutase (MnSOD/SOD2) through covalent modification, which promotes an increase in ROS and induces mitochondrial dysfunction in hepatocytes. Neutralization of OxPLs can significantly reduce steatosis, inflammation, hepatocyte injury, and fibrosis. It is manifested by the reduction in the number of macrophages, the decreased expression of inflammatory genes, and lowered levels of serum cytokines, thereby alleviating liver inflammation [56]. These results suggest that targeting OxPLs may be an effective treatment strategy for MASLD.
It has been discovered that elevated levels of NAD(P)H oxidase 4 (NOX4) and nuclear factor erythroid-derived 2-like 2 (NFE2L2) in hepatocytes during the early stages of disease progression can alleviate mitochondrial oxidative stress [57]. NFE2L2 encodes nuclear factor-erythroid 2-related factor 2 (Nrf2) mediates signaling that plays a pivotal role in maintaining cellular redox homeostasis in hepatocytes [58], which can improve mitochondrial oxidative stress and membrane potential and rescue mitochondrial biogenesis. Previous studies have demonstrated that NFE2L2 drives the expression of NOX4 in muscle, subsequently enhancing NFE2L2 antioxidant defenses, thereby mitigating muscle oxidative damage and insulin resistance [59]. In the majority of patients with MASLD, progression to NASH and fibrosis does not occur, which researchers believe may be due to adaptive mechanisms that mitigate the oxidative damage that might otherwise arise during the progression of MASLD [57]. They have found that the induction and activation of NFE2L2 depend on ROS generated by mitochondria and NOX4. To a certain extent, ROS derived from NOX4 contribute to antioxidant defense, and the deletion or inhibition of NOX4 in hepatocytes decreases ROS and weakens antioxidant defense, leading to mitochondrial oxidative stress and damage to proteins and lipids. Their experimental results indicate that inducing NOX4 in the livers of obese mice triggers the ROS-dependent induction of the NFE2L2 antioxidant defense response, thereby limiting steatosis, mitochondrial oxidative stress, and tissue damage and consequently restraining the progression to NASH and fibrosis [57].
Necrotic apoptosis can be activated in both human and experimental MASLD livers as an immunogenic regulated cell death pathway [60,61], dependent on the activity of mixed lineage kinase domain-like protein (MLKL) and receptor-interacting protein kinase 3 (RIPK3) [62–64]. Research has found that RIPK3-deficient mice with NASH exhibit reduced liver infiltration of inflammatory cells, fibrosis, and precancerous nodules [65]. To investigate the role of RIPK3 in regulating mitochondrial function and the role of lipid droplet (LD) structure in MASLD, researchers conducted functional analyses assessing mitochondrial and LD biology in both NFE2L2 and RIPK3−/− mice. Their results showed that RIPK3 deficiency inhibits mitochondrial dysfunction and oxidative stress [66], indicating that RIPK3 deficiency can restore mitochondrial bioenergetics. This also suggests that RIPK3 inhibition holds promise for improving MASLD.
Relevant studies have shown that improving mitochondrial dysfunction and reducing hepatic cellular oxidative stress contribute to the amelioration of MASLD [67,68]. Acetyl CoA Carboxylase 1 (ACACA) is the key enzyme controlling the speed of fatty acid biosynthesis. It controls the generation of fatty acids via novo lipogenesis, with elevated activity enhancing adipocyte differentiation and fat deposition [69]. Adenosine monophosphate-activated protein kinase (AMPK) acts as an essential sensor of energy in cellular metabolism, which facilitates mitochondrial lipid oxidation and enhances fatty acid uptake, thus leading to reduced lipid accumulation [70]. ACACA can be inactivated by inhibiting lipid synthesis through AMPK phosphorylation. In addition, carnitine palmitoyltransferase 1α (CPT1A), as a downstream target of the nuclear receptor peroxisome proliferator-activated receptor alpha (PPARα), serves as a key regulator of β-oxidation of free fatty acids within mitochondria [71]. Previous studies have shown that the imbalance of PPARα in MASLD may be involved in the pathogenesis of the disease [72]. Recently, researchers have confirmed that suppression of ACACA leads to reduced lipid accumulation by adjusting fatty acid metabolism and influencing the AMPK/PPARα/CPT1A axis. Simultaneously, by enhancing mitochondrial function, it helps to alleviate oxidative stress [73]. These findings indicate that ACACA could be a potential therapeutic target for the treatment of MASLD.
We know that MASLD is multifactorial, with lifestyle, genetic, and environmental factors contributing to its onset and progression. As early as ten years ago, research has shown that the widespread use of plastics and plastic products in our daily lives, their resistance to degradation, and low recycling rates can lead to significant environmental and health issues [74]. In the environment, plastics are continuously fragmented, and particles ≤5 mm and ≤1 µm are defined as microplastics (MPs) and nanoplastics (NPs), respectively. MPs and NPs can contaminate dietary products and be ingested by humans [75]. Previous studies have demonstrated that in various animal models, smaller-sized MPs and NPs preferentially accumulate in the liver [76]. Exogenous substances that induce mitochondrial abnormalities are a fundamental underlying fatty liver disease mechanism. NPs can cause mitochondrial oxidative damage, leading to excessive ROS production and calcium (Ca2+) overload [77]. Jie Wei and Jintao Liu et al. first proved through animal experiments that NPs inhibit proteasome-dependent degradation of the endoplasmic reticulum (ER)-resident protein inositol 1,4,5-trisphosphate receptor 1 (IP3R1), stabilizing and accumulating it at the ER-mitochondrial association membrane (MAM). This leads to the formation of the IP3R1-GRP75-VDAC1 complex, disrupting liver Ca2+homeostasis and redox balance in NP-exposed mice. Furthermore, the reduction of Nrf2, which is sensitive to oxidative stress in the liver of NP-exposed mice, positively regulates miR-26a by directly binding to its promoter region, resulting in downregulation of miR-26a while upregulating 10 genes involved in MAM formation, lipid uptake, inflammation, and fibrosis. Additionally, miR-26a inhibition increases MAM tether VDAC1, promotes nuclear translocation of nuclear factor kappa B p65 (NF-κB p65) and Kelch-like ECH-associated protein 1 (Keap1), and inactivates Nrf2 function, creating a vicious cycle [78]. The liver MAM-tethers/Nrf2/miR-26a feedback loop is a crucial metabolic conversion hub in the development of non-alcoholic steatohepatitis (NASH) from simple steatosis. It represents a promising therapeutic target for oxidative stress-related liver injury and NASH.
3.2 Impaired Mitochondrial Biogenesis in MASLD
Mitochondria have their genetic material, called mitochondrial DNA (mtDNA), which codes for the 13 polypeptides of the OXPHOS system, along with the 2 rRNA (ribosomal RNA) s and 22 tRNA (transfer RNA) s playing a pivotal role in mitochondrial protein synthesis [79]. The normal mitochondrial function depends heavily on mtDNA. Still, owing to its closeness to sites where ROS are generated and the absence of histones that safeguard DNA, along with ineffective DNA repair systems, it is susceptible to oxidative damage, leading to mutations that can impair normal cellular physiological functions [80]. Mitochondrial biosynthesis processes are essential for maintaining stability during the entire mitochondrial lifecycle. In MASLD, an imbalance in redox reactions results in excessive production of ROS, producing oxidative damage to mtDNA. This mtDNA damage impairs mitochondrial function and protein synthesis, subsequently adversely affecting cellular energy metabolism and other mitochondrial functions.
The process of mitochondrial biogenesis necessitates the synchronized expression of both nuclear and mitochondrial genes, and peroxisome proliferator-activated receptor gamma coactivator-1α (PGC1α) acts as a pivotal transcriptional activator and master regulator of this process by activating other transcription factors involved in nuclear and mitochondrial gene expression, such as transcription factor A (TFAM) and sirtuin 1 (SIRT1) [81,82]. Research has shown that PGC1α can associate the ROS defense system with mitochondrial oxidative metabolism, allowing cells to preserve their redox balance when oxidative capacity changes [83]. The expression of PGC-1α may be regulated by mitochondrial ROS, thereby establishing a tight connection with the development of MASLD.
As previous studies have shown that the relative content of mtDNA is affected by various conditions associated with oxidative stress [84,85], Chimienti et al. utilized a rat model of MASLD fed an obese-inducing diet. By examining the mtDNA content in control groups and obese rats without fatty liver disease, they found that the mtDNA content was decreased in obese rats [86], also indicating a significant decrease in PGC-1α. It is well known that PGC-1α regulates the biosynthesis of mtDNA, enhancing mitochondrial biogenesis and oxidative phosphorylation [87,88]. Conversely, it can be inferred that when upon downregulation of PGC-1α, the mtDNA copy count diminishes, resulting in a decreased rate of oxidative phosphorylation, resulting in a reduction in ATP levels. Recent studies showed that compared relative to the control group, the ATP levels were substantially decreased, the mtDNA copy count was markedly decreased, and the expression of PGC-1α decreased in the MASLD model group induced by free fatty acid. However, upregulation of the PGC-1α signaling pathway can enhance mitochondrial biogenesis and ATP production in HepG2 cells [89]. Therefore, mtDNA biogenesis dysfunction may lead to mitochondrial dysfunction, thereby promoting the occurrence and progression of MASLD.
3.3 Abnormal Endoplasmic Reticulum-Mitochondrial Interaction in MASLD
The endoplasmic reticulum (ER) and mitochondria both exhibit continuous tubular membrane network morphologies within the cytoplasm. Notably, approximately 5%–20% of the mitochondrial surface is in close proximity to the ER membrane, with a minute distance of only 10–30 nm separating them. This tight adjacency facilitates the formation of a special structure known as the endoplasmic reticulum-mitochondria contact site (ERMCS). These connection points afford a stable interface for the coordinated performance of the two organelles [90]. With ongoing exploration, the ERMCS have been implicated in an increasing number of crucial physiological functions, including the interchange of calcium ions between the endoplasmic reticulum and mitochondria [91,92], the management of phospholipid production and breakdown, the adjustment of mitochondrial shape and self-eating processes, the activation of the inflammasome complex, and programmed cell death are all critical cellular processes [93,94]. These functions are indispensable for the normal operation of mitochondria. Previous research has found that reducing ER-mitochondrial interaction and calcium exchange is an early event in diet-induced obese mice, which can alter hepatic insulin sensitivity and induce hepatic steatosis. Conversely, enhancing ER-mitochondrial interaction can prevent diet-induced glucose intolerance [95]. Furthermore, they also confirmed that impaired ER-mitochondrial communication occurs in the livers of patients with MASLD [95]. Recent research has indicated that reduced ER-mitochondria contact impairs mitochondrial phospholipid metabolism and disrupts mitochondrial membrane integrity, subsequently leading to weakened mitochondrial respiration, decreased ATP production, and elevated levels of ROS. These alterations collectively contribute to mitochondrial dysfunction. In experimental mice, such dysfunction was observed to result in hepatic lipid accumulation, NASH, and liver fibrosis [96].
3.4 Impaired Mitochondrial Quality Control in MASLD
Mitochondrial quality control refers to the balance between mitochondrial biogenesis and the clearance of damaged mitochondria, encompassing processes such as mitochondrial fusion, fission, and mitophagy [97]. Mitochondrial fusion facilitates the exchange of metabolites and substrates within mitochondria and regulates oxidative metabolism to alleviate organelle stress. Mitochondrial fission involves the mitotic segregation of mitochondria into daughter cells and the separation of damaged or dysfunctional mitochondrial parts, which is essential for subsequent autophagic degradation. The interplay between mitochondrial membrane fusion and fission contributes to regulating mitochondrial morphology and quantity [98]. Mitophagy is the degradation and recycling of dysfunctional or damaged mitochondria, serving as a crucial mechanism for mitochondrial quality control [99,100].
The pathological changes of chronic liver injury are often associated with abnormally enhanced mitochondrial fission [101]. Research has found that excessive mitochondrial fission (such as decreased mitochondrial content, reduced mitochondrial area, and mitochondrial deformation) exists in MASLD experimental models [102]. This suggests a likelihood that alterations in mitochondrial fission play a role in the progression of MASLD. Several studies have demonstrated that mitophagy induction in hepatocytes can maintain an adequate number of functional mitochondria, thereby fulfilling cellular energy demands, preventing hepatocyte injury induced by free fatty acids, and ultimately mitigating the progression of MASLD [103,104]. Previous research findings have suggested that defects in hepatic mitophagy can exacerbate MASLD and contribute to the pathogenesis of nonalcoholic steatohepatitis [105,106]. Recent experiments have demonstrated that reduced mitophagy is an early feature of NAFLD pathogenesis. When the mitophagy process is impaired, damaged mitochondria cannot be cleared in a timely manner, leading to the accumulation of ROS and cellular damage [107]. In summary, the mechanism of mitochondria in MASLD is depicted in Fig. 3.
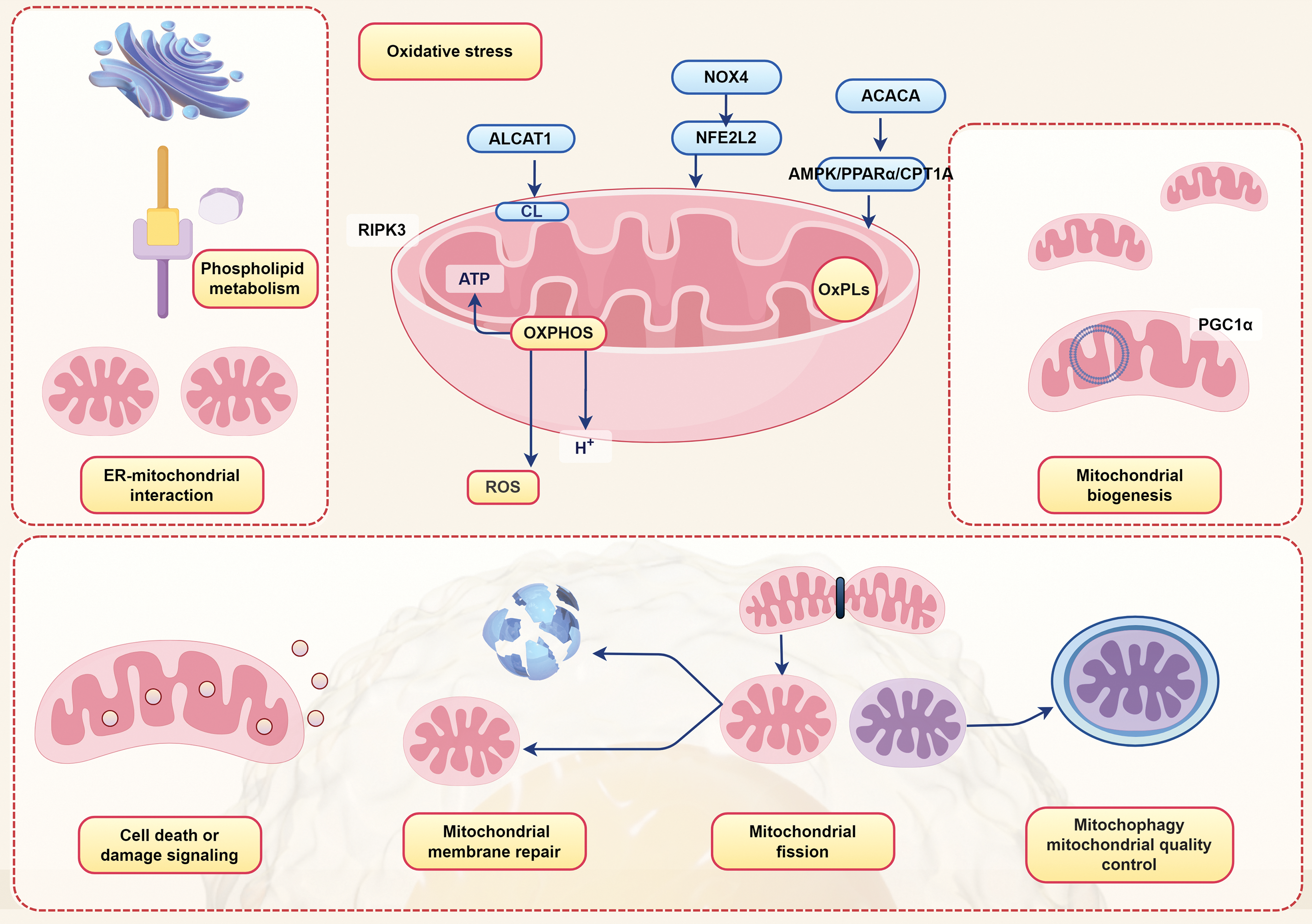
Figure 3: Manifestations of various mechanisms of mitochondria in MASLD. NOX4, NAD(P)H oxidase 4; ACACA, Acetyl CoA Carboxylase 1; ALCAT1, Lysocardiolipin acyltransferase 1; NFE2L2, Nuclear factor erythroid-derived 2-like 2; AMPK, adenosine monophosphate-activated protein kinase; PPARα, peroxisome proliferator-activated receptor alpha; CPT1A, carnitine palmitoyltransferase 1; CL, Cardiolipin; RIPK3, receptor-interacting protein kinase 3; ATP, adenosine triphosphate; OXPHOS, oxidative phosphorylation; OxPLs, oxidized phospholipids; PGC1α, peroxisome proliferator-activated receptor gamma coactivator-1α; ER, endoplasmic reticulum; ROS, reactive oxygen species (By Figdraw)
For humans, assessing various characteristics and quality control of hepatic mitochondrial function remains challenging, and currently, most of the available methods are expensive, time-consuming techniques or invasive tests [108]. Therefore, a detailed examination of hepatic mitochondria necessitates non-invasive methods, which should also allow extended study time and disease progression tracking [109,110]. Existing literature indicates that physical exercise [111,112], bariatric surgery [113], and targeted pharmacological therapies [114] may have the potential to reverse mitochondrial dysfunction. Pharmacological treatments are primarily targeted at the pathogenesis of NASH, which includes oxidative stress, metabolic disorders, apoptosis, and fibrosis. Based on their mechanisms of action, these treatments can be categorized into five types [114]: lipid-lowering, hypoglycemic, anti-apoptotic, antioxidant, and anti-fibrotic. They hold promise for large-scale clinical applications. However, there are still some limitations in understanding the physiological effects of certain drugs. For instance, activators of AMPK, such as metformin, are widely used in treating NAFLD, yet the exact mechanism of action remains unclear mainly [115]. Additionally, although 5-Aminoimidazole-4-carboxamide ribotide (AICAR) exhibits anti-inflammatory and antioxidant properties, the underlying molecular mechanisms preventing mitochondrial dysfunction have yet to be determined [116].
Certain natural compounds [30], like hesperidin and caffeine, are emerging as viable treatment options for MASLD, partly due to their strong antioxidant properties and safe profiles make them attractive options, as many are already incorporated into the typical human diet. Within the body, the vitamin family serves as a significant category of antioxidants [117], controlling oxidative stress and fat accumulation via the catalysis of enzymatic reactions. For instance, the antioxidant activities of vitamins C and E are believed to mitigate hepatocyte damage [118]. Since mitochondria can be easily obtained from cultured cells, and the techniques for mitochondrial isolation and preservation have become increasingly sophisticated, large-scale mitochondrial donation centers can be established. When autologous transplantation is not feasible, a suitable donor can be promptly identified. Currently, mitochondrial transplantation is being investigated as a therapeutic approach for treating MASLD, while the clinical application of mitochondrial transplantation is limited [119]. Certainly, a healthy diet combined with regular exercise remains the first-line non-pharmacological intervention for the management of MASLD [120].
The incidence of MASLD exhibits a notable upward trend [121], and it is also highly prevalent among overweight or obese children and adolescents [122]. Furthermore, the pathogenesis of MASLD is a complex process that involves the interaction of multiple factors. Among them, mitochondrial dysfunction caused by mitochondrial oxidative stress has a pivotal influence on MASLD; according to reports, alleviating mitochondrial dysfunction and decreasing oxidative stress in hepatocytes can contribute to improving MASLD [67], which represents a promising therapeutic target. Over the years, researchers have continued exploring various oxidative stress pathways in MASLD. Whether it is the discovery that the expression of ALCAT1 leads to cardiolipin peroxidation, causing oxidative stress, the finding in MASLD in humans and mice that accumulated OxPLs promote the increase of ROS to induce hepatic mitochondrial dysfunction, or the identification that NOX4 and NFE2L2 can mitigate mitochondrial oxidative stress, any pathway that demonstrates the role of mitochondrial ROS in MASLD contributes to uncovering the pathogenesis of this disease and provides a theoretical basis for developing novel therapeutic strategies. In MASLD, mitochondrial quality control is impaired, particularly with abnormal autophagy function, which hinders the clearance of damaged mitochondria, resulting in ROS accumulation and cellular damage. Additionally, biogenesis dysfunction such as mtDNA damage reduces the efficiency of mitochondrial energy production, impeding their ability to provide sufficient ATP for cellular needs and subsequently affecting normal cellular physiological functions. When mtDNA biofunctional disorders occur, the rate of oxidative phosphorylation slows down, leading to decreased ATP production and compromising cellular energy supply. Furthermore, abnormal endoplasmic reticulum-mitochondrial interplay is also crucial in the pathogenesis of fatty liver disease. The decreased contact between the endoplasmic reticulum and mitochondria disrupts material exchange and signal transmission, resulting in weakened mitochondrial respiration, reduced ATP production, and elevated ROS levels. Although the relative importance of these abnormalities in triggering mitochondrial dysfunction may vary among MASLD patients, any abnormalities can induce other abnormalities, interacting and influencing each other. This implies that restoring one defect may exert an overall beneficial impact on mitochondrial integrity and function. Therefore, enhancing our understanding of how these mitochondrial regulatory mechanisms are involved in MASLD could offer a variety of new therapeutic targets, benefiting all patients with MASLD.
Acknowledgement: None.
Funding Statement: This work was supported by grants from the Top Medical Expert Team of Wuxi Taihu Talent Plan (Grant Nos. DJTD202106, GDTD202105 and YXTD202101), Medical Key Discipline Program of Wuxi Health Commission (Grant Nos. ZDXK2021007 and CXTD2021005), Top Talent Support Program for Young and Middle-Aged People of Wuxi Health Committee (Grant No. BJ2023090), and Scientific Research Program of Wuxi Health Commission (Grant Nos. Z20210 and M202208).
Author Contributions: Juan Yang: Visualization, Writing—original draft, Writing—review & editing. Jiahui Zhang: Writing—original draft, Writing—review & editing. Le Zhang and Zhenshan Yang: Supervision, Writing—original draft, Writing—review & editing. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Younossi Z, Tacke F, Arrese M, Chander Sharma B, Mostafa I, Bugianesi E, et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology. 2019;69(6):2672–82. doi:10.1002/hep.30251. [Google Scholar] [PubMed] [CrossRef]
2. Chan KE, Koh TJL, Tang ASP, Quek J, Yong JN, Tay P, et al. Global prevalence and clinical characteristics of metabolic-associated fatty liver disease: a meta-analysis and systematic review of 10 739 607 individuals. J Clin Endocrinol Metab. 2022;107(9):2691–700. doi:10.1210/clinem/dgac321. [Google Scholar] [PubMed] [CrossRef]
3. Le MH, Le DM, Baez TC, Wu Y, Ito T, Lee EY, et al. Global incidence of non-alcoholic fatty liver disease: a systematic review and meta-analysis of 63 studies and 1,201,807 persons. J Hepatol. 2023;79(2):287–95. doi:10.1016/j.jhep.2023.03.040. [Google Scholar] [PubMed] [CrossRef]
4. Mao T, Sun Y, Xu X, He K. Overview and prospect of NAFLD: significant roles of nutrients and dietary patterns in its progression or prevention. Hepatol Commun. 2023;7(10):e0234. doi:10.1097/HC9.0000000000000234. [Google Scholar] [PubMed] [CrossRef]
5. Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol. 2020;73(1):202–9. doi:10.1016/j.jhep.2020.03.039. [Google Scholar] [PubMed] [CrossRef]
6. Zheng KI, Fan JG, Shi JP, Wong VW, Eslam M, George J, et al. From NAFLD to MAFLD: a “redefining” moment for fatty liver disease. Chin Med J (Engl). 2020;133(19):2271–3. doi:10.1097/CM9.0000000000000981. [Google Scholar] [PubMed] [CrossRef]
7. Méndez-Sánchez N, Bugianesi E, Gish RG, Lammert F, Tilg H, Nguyen MH, et al. Global multi-stakeholder endorsement of the MAFLD definition. Lancet Gastroenterol Hepatol. 2022;7(5):388–90. doi:10.1016/S2468-1253(22)00062-0. [Google Scholar] [PubMed] [CrossRef]
8. Lazarus JV, Newsome PN, Francque SM, Kanwal F, Terrault NA, Rinella ME. Reply: a multi-society Delphi consensus statement on new fatty liver disease nomenclature. Hepatology. 2024;79(3):E93–4. doi:10.1016/j.jhep.2023.06.003. [Google Scholar] [PubMed] [CrossRef]
9. Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. 2021;184(10):2537–64. doi:10.1016/j.cell.2021.04.015. [Google Scholar] [PubMed] [CrossRef]
10. Mantovani A, Csermely A, Petracca G, Beatrice G, Corey KE, Simon TG, et al. Non-alcoholic fatty liver disease and risk of fatal and non-fatal cardiovascular events: an updated systematic review and meta-analysis. Lancet Gastroenterol Hepatol. 2021;6(11):903–13. doi:10.1016/S2468-1253(21)00308-3. [Google Scholar] [PubMed] [CrossRef]
11. Bence KK, Birnbaum MJ. Metabolic drivers of non-alcoholic fatty liver disease. Mol Metab. 2021;50(3):101143. doi:10.1016/j.molmet.2020.101143. [Google Scholar] [PubMed] [CrossRef]
12. Eslam M, Sarin SK, Wong VW, Fan JG, Kawaguchi T, Ahn SH, et al. The Asian Pacific Association for the Study of the Liver clinical practice guidelines for the diagnosis and management of metabolic associated fatty liver disease. Hepatol Int. 2020;14(6):889–919. doi:10.1007/s12072-020-10094-2. [Google Scholar] [PubMed] [CrossRef]
13. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism. 2016;65(8):1038–48. doi:10.1016/j.metabol.2015.12.012. [Google Scholar] [PubMed] [CrossRef]
14. Di Ciaula A, Passarella S, Shanmugam H, Noviello M, Bonfrate L, Wang DQ, et al. Nonalcoholic fatty liver disease (NAFLD). Mitochondria as players and targets of therapies? Int J Mol Sci. 2021;22(10):5375. doi:10.3390/ijms22105375. [Google Scholar] [PubMed] [CrossRef]
15. Morio B, Panthu B, Bassot A, Rieusset J. Role of mitochondria in liver metabolic health and diseases. Cell Calcium. 2021;94(Suppl. 3):102336. doi:10.1016/j.ceca.2020.102336. [Google Scholar] [PubMed] [CrossRef]
16. Fromenty B, Roden M. Mitochondrial alterations in fatty liver diseases. J Hepatol. 2023;78(2):415–29. doi:10.1016/j.jhep.2022.09.020. [Google Scholar] [PubMed] [CrossRef]
17. Shum M, Ngo J, Shirihai OS, Liesa M. Mitochondrial oxidative function in NAFLD: friend or foe? Mol Metab. 2021;50:101134. doi:10.1016/j.molmet.2020.101134. [Google Scholar] [PubMed] [CrossRef]
18. Friedman JR, Nunnari J. Mitochondrial form and function. Nature. 2014;505(7483):335–43. doi:10.1038/nature12985. [Google Scholar] [PubMed] [CrossRef]
19. Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21(2):85–100. doi:10.1038/s41580-019-0173-8. [Google Scholar] [CrossRef]
20. Rui L. Energy metabolism in the liver. Compr Physiol. 2014;4(1):177–97. doi:10.1002/cphy. [Google Scholar] [PubMed] [CrossRef]
21. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94(3):909–50. doi:10.1152/physrev.00026.2013. [Google Scholar] [PubMed] [CrossRef]
22. Janik-Karpinska E, Ceremuga M, Niemcewicz M, Synowiec E, Sliwiński T, Bijak M. Mitochondrial damage induced by T-2 mycotoxin on human skin-fibroblast Hs68 cell line. Molecules. 2023;28(5):2408. doi:10.3390/molecules28052408. [Google Scholar] [PubMed] [CrossRef]
23. Varela CD, Farhana A. Biochemistry, superoxides. In: StatPearls. Treasure Island (FLStatPearls Publishing; 2024. [Google Scholar]
24. Zhang L, Wang X, Cueto R, Effi C, Zhang Y, Tan H, et al. Biochemical basis and metabolic interplay of redox regulation. Redox Biol. 2019;26:101284. doi:10.1016/j.redox.2019.101284. [Google Scholar] [PubMed] [CrossRef]
25. Phull A-R, Arain SQ, Majid A, Fatima H, Ahmed M, Kim S-J. Oxidative stress-mediated epigenetic remodeling, metastatic progression and cell signaling in cancer. Oncologie. 2024;26(4):493–507. doi:10.1515/oncologie-2024-0157. [Google Scholar] [CrossRef]
26. Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015;21(5):739–46. doi:10.1016/j.cmet.2015.04.004. [Google Scholar] [PubMed] [CrossRef]
27. Shami GJ, Cheng D, Verhaegh P, Koek G, Wisse E, Braet F. Three-dimensional ultrastructure of giant mitochondria in human non-alcoholic fatty liver disease. Sci Rep. 2021;11(1):3319. doi:10.1038/s41598-021-82884-z. [Google Scholar] [PubMed] [CrossRef]
28. Moore MP, Cunningham RP, Meers GM, Johnson SA, Wheeler AA, Ganga RR, et al. Compromised hepatic mitochondrial fatty acid oxidation and reduced markers of mitochondrial turnover in human NAFLD. Hepatology. 2022;76(5):1452–65. doi:10.1002/hep.32324. [Google Scholar] [PubMed] [CrossRef]
29. Ore A, Akinloye OA. Oxidative stress and antioxidant biomarkers in clinical and experimental models of non-alcoholic fatty liver disease. Medicina. 2019;55(2):26. doi:10.3390/medicina55020026. [Google Scholar] [PubMed] [CrossRef]
30. Arroyave-Ospina JC, Wu Z, Geng Y, Moshage H. Role of oxidative stress in the pathogenesis of non-alcoholic fatty liver disease: implications for prevention and therapy. Antioxidants. 2021;10(2):174. doi:10.3390/antiox10020174. [Google Scholar] [PubMed] [CrossRef]
31. Stiuso P, Scognamiglio I, Murolo M, Ferranti P, De Simone C, Rizzo MR, et al. Serum oxidative stress markers and lipidomic profile to detect NASH patients responsive to an antioxidant treatment: a pilot study. Oxid Med Cell Longev. 2014;2014:169216. doi:10.1155/2014/169216. [Google Scholar] [PubMed] [CrossRef]
32. Leghi GE, Domenici FA, Vannucchi H. Influence of oxidative stress and obesity in patients with nonalcoholic steatohepatitis. Arq Gastroenterol. 2015;52(3):228–33. doi:10.1590/S0004-28032015000300014. [Google Scholar] [PubMed] [CrossRef]
33. Scorletti E, Carr RM. A new perspective on NAFLD: focusing on lipid droplets. J Hepatol. 2022;76(4):934–45. doi:10.1016/j.jhep.2021.11.009. [Google Scholar] [PubMed] [CrossRef]
34. Talari NK, Mattam U, Meher NK, Paripati AK, Mahadev K, Krishnamoorthy T, et al. Lipid-droplet associated mitochondria promote fatty-acid oxidation through a distinct bioenergetic pattern in male Wistar rats. Nat Commun. 2023;14(1):766. doi:10.1038/s41467-023-36432-0. [Google Scholar] [PubMed] [CrossRef]
35. Grohmann M, Wiede F, Dodd GT, Gurzov EN, Ooi GJ, Butt T, et al. Obesity drives STAT-1-dependent NASH and STAT-3-dependent HCC. Cell. 2018;175(5):1289–306.e20. doi:10.1016/j.cell.2018.09.053. [Google Scholar] [PubMed] [CrossRef]
36. Hernansanz-Agustín P, Enríquez JA. Generation of reactive oxygen species by mitochondria. Antioxidants. 2021;10(3):415. doi:10.3390/antiox10030415. [Google Scholar] [PubMed] [CrossRef]
37. He L, He T, Farrar S, Ji L, Liu T, Ma X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell Physiol Biochem. 2017;44(2):532–53. doi:10.1159/000485089. [Google Scholar] [PubMed] [CrossRef]
38. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24(7):908–22. doi:10.1038/s41591-018-0104-9. [Google Scholar] [PubMed] [CrossRef]
39. Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20(39):14205–18. doi:10.3748/wjg.v20.i39.14205. [Google Scholar] [PubMed] [CrossRef]
40. Li J, Romestaing C, Han X, Li Y, Hao X, Wu Y, et al. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 2010;12(2):154–65. doi:10.1016/j.cmet.2010.07.003. [Google Scholar] [PubMed] [CrossRef]
41. Wang L, Liu X, Nie J, Zhang J, Kimball SR, Zhang H, et al. ALCAT1 controls mitochondrial etiology of fatty liver diseases, linking defective mitophagy to steatosis. Hepatology. 2015;61(2):486–96. doi:10.1002/hep.27420. [Google Scholar] [PubMed] [CrossRef]
42. Sun B, Karin M. Obesity, inflammation, and liver cancer. J Hepatol. 2012;56(3):704–13. doi:10.1016/j.jhep.2011.09.020. [Google Scholar] [PubMed] [CrossRef]
43. Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol. 2013;10(11):656–65. doi:10.1038/nrgastro.2013.183. [Google Scholar] [PubMed] [CrossRef]
44. Wree A, Broderick L, Canbay A, Hoffman HM, Feldstein AE. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol. 2013;10(11):627–36. doi:10.1038/nrgastro.2013.149. [Google Scholar] [PubMed] [CrossRef]
45. Greten TF, Wang XW, Korangy F. Current concepts of immune based treatments for patients with HCC: from basic science to novel treatment approaches. Gut. 2015;64(5):842–8. doi:10.1136/gutjnl-2014-307990. [Google Scholar] [PubMed] [CrossRef]
46. Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell. 2014;26(4):549–64. doi:10.1016/j.ccell.2014.09.003. [Google Scholar] [PubMed] [CrossRef]
47. Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P, et al. NAFLD causes selective CD4+ T lymphocyte loss and promotes hepatocarcinogenesis. Nature. 2016;531(7593):253–7. doi:10.1038/nature16969. [Google Scholar] [PubMed] [CrossRef]
48. Baker PR 2nd, Friedman JE. Mitochondrial role in the neonatal predisposition to developing nonalcoholic fatty liver disease. J Clin Invest. 2018;128(9):3692–703. doi:10.1172/JCI120846. [Google Scholar] [PubMed] [CrossRef]
49. Houghton LC, Ester WA, Lumey LH, Michels KB, Wei Y, Cohn BA, et al. Maternal weight gain in excess of pregnancy guidelines is related to daughters being overweight 40 years later. Am J Obstet Gynecol. 2016;215(2):246.e1–8. doi:10.1016/j.ajog.2016.02.034. [Google Scholar] [PubMed] [CrossRef]
50. Rinella M, Charlton M. The globalization of nonalcoholic fatty liver disease: prevalence and impact on world health. Hepatology. 2016;64(1):19–22. doi:10.1002/hep.28524. [Google Scholar] [PubMed] [CrossRef]
51. Hagström H, Stål P, Hultcrantz R, Hemmingsson T, Andreasson A. Overweight in late adolescence predicts development of severe liver disease later in life: a 39 years follow-up study. J Hepatol. 2016;65(2):363–8. doi:10.1016/j.jhep.2016.03.019. [Google Scholar] [PubMed] [CrossRef]
52. Batty M, Bennett MR, Yu E. The role of oxidative stress in atherosclerosis. Cells. 2022;11(23):3843. doi:10.3390/cells11233843. [Google Scholar] [PubMed] [CrossRef]
53. Baldan A, Gonen A, Choung C, Que X, Marquart TJ, Hernandez I, et al. ABCG1 is required for pulmonary B-1 B cell and natural antibody homeostasis. J Immunol. 2014;193(11):5637–48. doi:10.4049/jimmunol.1400606. [Google Scholar] [PubMed] [CrossRef]
54. Musso G, Cassader M, Paschetta E, Gambino R. Bioactive lipid species and metabolic pathways in progression and resolution of nonalcoholic steatohepatitis. Gastroenterology. 2018;155(2):282–302.e8. doi:10.1053/j.gastro.2018.06.031. [Google Scholar] [PubMed] [CrossRef]
55. Liangpunsakul S, Chalasani N. Lipid mediators of liver injury in nonalcoholic fatty liver disease. Am J Physiol Gastrointest Liver Physiol. 2019;316(1):G75–81. doi:10.1152/ajpgi.00170.2018. [Google Scholar] [PubMed] [CrossRef]
56. Sun X, Seidman JS, Zhao P, Troutman TD, Spann NJ, Que X, et al. Neutralization of oxidized phospholipids ameliorates non-alcoholic steatohepatitis. Cell Metab. 2020;31(1):189–206.e8. doi:10.1016/j.cmet.2019.10.014. [Google Scholar] [PubMed] [CrossRef]
57. Greatorex S, Kaur S, Xirouchaki CE, Goh PK, Wiede F, Genders AJ, et al. Mitochondria- and NOX4-dependent antioxidant defense mitigates progression to nonalcoholic steatohepatitis in obesity. J Clin Invest. 2023;134(3):e162533. doi:10.1172/JCI162533. [Google Scholar] [PubMed] [CrossRef]
58. Upadhyay KK, Jadeja RN, Vyas HS, Pandya B, Joshi A, Vohra A, et al. Carbon monoxide releasing molecule-A1 improves nonalcoholic steatohepatitis via Nrf2 activation mediated improvement in oxidative stress and mitochondrial function. Redox Biol. 2020;28(9):101314. doi:10.1016/j.redox.2019.101314. [Google Scholar] [PubMed] [CrossRef]
59. Xirouchaki CE, Jia Y, McGrath MJ, Greatorex S, Tran M, Merry TL, et al. Skeletal muscle NOX4 is required for adaptive responses that prevent insulin resistance. Sci Adv. 2021;7(51):eabl4988. doi:10.1126/sciadv.abl4988. [Google Scholar] [PubMed] [CrossRef]
60. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25(3):486–541. doi:10.1038/s41418-017-0012-4. [Google Scholar] [PubMed] [CrossRef]
61. Afonso MB, Rodrigues PM, Carvalho T, Caridade M, Borralho P, Cortez-Pinto H, et al. Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin Sci. 2015;129(8):721–39. doi:10.1042/CS20140732. [Google Scholar] [PubMed] [CrossRef]
62. Gautheron J, Vucur M, Schneider AT, Severi I, Roderburg C, Roy S, et al. The necroptosis-inducing kinase RIPK3 dampens adipose tissue inflammation and glucose intolerance. Nat Commun. 2016;7(1):11869. doi:10.1038/ncomms11869. [Google Scholar] [PubMed] [CrossRef]
63. Gautheron J, Gores GJ, Rodrigues CMP. Lytic cell death in metabolic liver disease. J Hepatol. 2020;73(2):394–408. doi:10.1016/j.jhep.2020.04.001. [Google Scholar] [PubMed] [CrossRef]
64. Majdi A, Aoudjehane L, Ratziu V, Islam T, Afonso MB, Conti F, et al. Inhibition of receptor-interacting protein kinase 1 improves experimental non-alcoholic fatty liver disease. J Hepatol. 2020;72(4):627–35. doi:10.1016/j.jhep.2019.11.008. [Google Scholar] [PubMed] [CrossRef]
65. Afonso MB, Rodrigues PM, Mateus-Pinheiro M, Simão AL, Gaspar MM, Majdi A, et al. RIPK3 acts as a lipid metabolism regulator contributing to inflammation and carcinogenesis in non-alcoholic fatty liver disease. Gut. 2021;70(12):2359–72. doi:10.1136/gutjnl-2020-321767. [Google Scholar] [PubMed] [CrossRef]
66. Afonso MB, Islam T, Magusto J, Amorim R, Lenoir V, Simões RF, et al. RIPK3 dampens mitochondrial bioenergetics and lipid droplet dynamics in metabolic liver disease. Hepatology. 2023;77(4):1319–34. doi:10.1002/hep.32756. [Google Scholar] [PubMed] [CrossRef]
67. Li Z, Zhang H, Li Y, Chen H, Wang C, Wong VKW, et al. Phytotherapy using blueberry leaf polyphenols to alleviate non-alcoholic fatty liver disease through improving mitochondrial function and oxidative defense. Phytomedicine. 2020;69:153209. doi:10.1016/j.phymed.2020.153209. [Google Scholar] [PubMed] [CrossRef]
68. Dai X, Kuang Q, Sun Y, Xu M, Zhu L, Ge C, et al. Fisetin represses oxidative stress and mitochondrial dysfunction in NAFLD through suppressing GRP78-mediated endoplasmic reticulum (ER) stress. J Funct Foods. 2022;90(1):104954. doi:10.1016/j.jff.2022.104954. [Google Scholar] [CrossRef]
69. Lally JS, Ghoshal S, DePeralta DK, Moaven O, Wei L, Masia R, et al. Inhibition of acetyl-CoA carboxylase by phosphorylation or the inhibitor ND-654 suppresses lipogenesis and hepatocellular carcinoma. Cell Metabolism. 2019;29(1):174–82.e5. doi:10.1016/j.cmet.2018.08.020. [Google Scholar] [PubMed] [CrossRef]
70. Zhang J, Zhang W, Yang L, Zhao W, Liu Z, Wang E, et al. Phytochemical gallic acid alleviates nonalcoholic fatty liver disease via AMPK-ACC-PPARa axis through dual regulation of lipid metabolism and mitochondrial function. Phytomedicine. 2023;109:154589. doi:10.1016/j.phymed.2022.154589. [Google Scholar] [PubMed] [CrossRef]
71. Liang K. Mitochondrial CPT1A: insights into structure, function, and basis for drug development. Front Pharmacol. 2023;14:1160440. doi:10.3389/fphar.2023.1160440. [Google Scholar] [PubMed] [CrossRef]
72. Montagner A, Polizzi A, Fouché E, Ducheix S, Lippi Y, Lasserre F, et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut. 2016;65(7):1202–14. doi:10.1136/gutjnl-2015-310798. [Google Scholar] [PubMed] [CrossRef]
73. Dong J, Li M, Peng R, Zhang Y, Qiao Z, Sun N. ACACA reduces lipid accumulation through dual regulation of lipid metabolism and mitochondrial function via AMPK- PPARα- CPT1A axis. J Transl Med. 2024;22(1):196. doi:10.1186/s12967-024-04942-0. [Google Scholar] [PubMed] [CrossRef]
74. Rochman CM, Browne MA, Halpern BS, Hentschel BT, Hoh E, Karapanagioti HK, et al. Classify plastic waste as hazardous. Nature. 2013;494(7436):169–71. doi:10.1038/494169a. [Google Scholar] [PubMed] [CrossRef]
75. Jiménez-Arroyo C, Tamargo A, Molinero N, Moreno-Arribas MV. The gut microbiota, a key to understanding the health implications of micro (nano) plastics and their biodegradation. Microbial Biotechnol. 2023;16(1):34–53. doi:10.1111/1751-7915.14182. [Google Scholar] [PubMed] [CrossRef]
76. Yin J, Ju Y, Qian H, Wang J, Miao X, Zhu Y, et al. Nanoplastics and microplastics may be damaging our livers. Toxics. 2022;10(10):586. doi:10.3390/toxics10100586. [Google Scholar] [PubMed] [CrossRef]
77. Li S, Ma Y, Ye S, Tang S, Liang N, Liang Y, et al. Polystyrene microplastics trigger hepatocyte apoptosis and abnormal glycolytic flux via ROS-driven calcium overload. J Hazard Mater. 2021;417(2):126025. doi:10.1016/j.jhazmat.2021.126025. [Google Scholar] [PubMed] [CrossRef]
78. Liu JJ, Wang H, Wen K, Ni X, Lin Y, et al. Nanoplastic propels diet-induced NAFL to NASH via ER-mitochondrial tether-controlled redox switch NASH. J Hazard Mater. 2024;465:133142. doi:10.1016/j.jhazmat.2023.133142. [Google Scholar] [PubMed] [CrossRef]
79. D’Souza AR, Minczuk M. Mitochondrial transcription and translation: overview. Essays Biochem. 2018;62(3):309–20. [Google Scholar] [PubMed]
80. Boczonadi V, Ricci G, Horvath R. Mitochondrial DNA transcription and translation: clinical syndromes. Essays Biochem. 2018;62(3):321–40. [Google Scholar] [PubMed]
81. Dieter F, Esselun C, Eckert GP. Redox active α-lipoic acid differentially improves mitochondrial dysfunction in a cellular model of alzheimer and its control cells. Int J Mol Sci. 2022;23(16):9186. doi:10.3390/ijms23169186. [Google Scholar] [PubMed] [CrossRef]
82. Dos Santos SM, Romeiro CFR, Rodrigues CA, Cerqueira ARL, Monteiro MC. Mitochondrial dysfunction and alpha-lipoic acid: beneficial or harmful in alzheimer’s disease? Oxid Med Cell Longev. 2019;2019:8409329. doi:10.1155/2019/8409329. [Google Scholar] [PubMed] [CrossRef]
83. Choi HI, Kim HJ, Park JS, Kim IJ, Bae EH, Ma SK, et al. PGC-1α attenuates hydrogen peroxide-induced apoptotic cell death by upregulating Nrf-2 via GSK3β inactivation mediated by activated p38 in HK-2 cells. Sci Rep. 2017;7(1):4319. doi:10.1038/s41598-017-04593-w. [Google Scholar] [PubMed] [CrossRef]
84. Orlando A, Chimienti G, Pesce V, Fracasso F, Lezza AMS, Russo F. An in vitro study on mitochondrial compensatory response induced by gliadin peptides in Caco-2 cells. Int J Mol Sci. 2019;20(8):1862. doi:10.3390/ijms20081862. [Google Scholar] [PubMed] [CrossRef]
85. Chimienti G, Picca A, Fracasso F, Russo F, Orlando A, Riezzo G, et al. The age-sensitive efficacy of calorie restriction on mitochondrial biogenesis and mtDNA damage in rat liver. Int J Mol Sci. 2021;22(4):1665. doi:10.3390/ijms22041665. [Google Scholar] [PubMed] [CrossRef]
86. Chimienti G, Orlando A, Russo F, D’Attoma B, Aragno M, Aimaretti E, et al. The mitochondrial trigger in an animal model of nonalcoholic fatty liver disease. Genes. 2021;12(9):1439. doi:10.3390/genes12091439. [Google Scholar] [PubMed] [CrossRef]
87. Coppi L, Ligorio S, Mitro N, Caruso D, De Fabiani E, Crestani M. PGC1s and beyond: disentangling the complex regulation of mitochondrial and cellular metabolism. Int J Mol Sci. 2021;22(13):6913. doi:10.3390/ijms22136913. [Google Scholar] [PubMed] [CrossRef]
88. Jannig PR, Dumesic PA, Spiegelman BM, Ruas JL. SnapShot: regulation and biology of PGC-1α. Cell. 2022;185(8):1444–.e1. doi:10.1016/j.cell.2022.03.027. [Google Scholar] [PubMed] [CrossRef]
89. Wang X, Wang J, Ying C, Xing Y, Su X, Men K. Fenofibrate alleviates NAFLD by enhancing the PPARα/PGC-1α signaling pathway coupling mitochondrial function. BMC Pharmacol Toxicol. 2024;25(1):7. doi:10.1186/s40360-023-00730-6. [Google Scholar] [PubMed] [CrossRef]
90. Csordás G, Weaver D, Hajnóczky G. Endoplasmic reticulum-mitochondrial contactology: structure and signaling functions. Trends Cell Biol. 2018;28(7):523–40. doi:10.1016/j.tcb.2018.02.009. [Google Scholar] [PubMed] [CrossRef]
91. Arruda AP, Pers BM, Parlakgül G, Güney E, Inouye K, Hotamisligil GS. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med. 2014;20(12):1427–35. doi:10.1038/nm.3735. [Google Scholar] [PubMed] [CrossRef]
92. Loncke J, Kaasik A, Bezprozvanny I, Parys JB, Kerkhofs M, Bultynck G. Balancing ER-mitochondrial Ca2+ fluxes in health and disease. Trends Cell Biol. 2021;31(7):598–612. doi:10.1016/j.tcb.2021.02.003. [Google Scholar] [PubMed] [CrossRef]
93. Marchi S, Patergnani S, Pinton P. The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim Biophys Acta. 2014;1837(4):461–9. doi:10.1016/j.bbabio.2013.10.015. [Google Scholar] [PubMed] [CrossRef]
94. Paillusson S, Stoica R, Gomez-Suaga P, Lau DHW, Mueller S, Miller T, et al. There’s something wrong with my MAM; the ER-mitochondria axis and neurodegenerative diseases. Trends Neurosci. 2016;39(3):146–57. doi:10.1016/j.tins.2016.01.008. [Google Scholar] [PubMed] [CrossRef]
95. Beaulant A, Dia M, Pillot B, Chauvin MA, Ji-Cao J, Durand C, et al. Endoplasmic reticulum-mitochondria miscommunication is an early and causal trigger of hepatic insulin resistance and steatosis. J Hepatol. 2022;77(3):710–22. doi:10.1016/j.jhep.2022.03.017. [Google Scholar] [PubMed] [CrossRef]
96. Dong J, Chen L, Ye F, Tang J, Liu B, Lin J, et al. Mic19 depletion impairs endoplasmic reticulum-mitochondrial contacts and mitochondrial lipid metabolism and triggers liver disease. Nat Commun. 2024;15(1):168. doi:10.1038/s41467-023-44057-6. [Google Scholar] [PubMed] [CrossRef]
97. Ghosh R, Vinod V, Symons JD, Boudina S. Protein and mitochondria quality control mechanisms and cardiac aging. Cells. 2020;9(4):933. doi:10.3390/cells9040933. [Google Scholar] [PubMed] [CrossRef]
98. Giacomello M, Pyakurel A, Glytsou C, Scorrano L. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol. 2020;21(4):204–24. doi:10.1038/s41580-020-0210-7. [Google Scholar] [PubMed] [CrossRef]
99. Su L, Zhang J, Gomez H, Kellum JA, Peng Z. Mitochondria ROS and mitophagy in acute kidney injury. Autophagy. 2023;19(2):401–14. doi:10.1080/15548627.2022.2084862. [Google Scholar] [PubMed] [CrossRef]
100. Lin Q, Li S, Jin H, Cai H, Zhu X, Yang Y, et al. Mitophagy alleviates cisplatin-induced renal tubular epithelial cell ferroptosis through ROS/HO-1/GPX4 axis. Int J Biol Sci. 2023;19(4):1192–210. doi:10.7150/ijbs.80775. [Google Scholar] [PubMed] [CrossRef]
101. Zhou H, Zhu P, Wang J, Toan S, Ren J. DNA-PKcs promotes alcohol-related liver disease by activating Drp1-related mitochondrial fission and repressing FUNDC1-required mitophagy. Signal Transduct Target Ther. 2019;4(1):56. doi:10.1038/s41392-019-0094-1. [Google Scholar] [PubMed] [CrossRef]
102. Miyao M, Kawai C, Kotani H, Minami H, Abiru H, Hamayasu H, et al. Mitochondrial fission in hepatocytes as a potential therapeutic target for nonalcoholic steatohepatitis. Hepatol Res. 2022;52(12):1020–33. doi:10.1111/hepr.13832. [Google Scholar] [PubMed] [CrossRef]
103. Li X, Shi Z, Zhu Y, Shen T, Wang H, Shui G, et al. Cyanidin-3-O-glucoside improves non-alcoholic fatty liver disease by promoting PINK1-mediated mitophagy in mice. Br J Pharmacol. 2020;177(15):3591–607. doi:10.1111/bph.15083. [Google Scholar] [PubMed] [CrossRef]
104. Meng Z, Gao M, Wang C, Guan S, Zhang D, Lu J. Apigenin alleviated high-fat-diet-induced hepatic pyroptosis by mitophagy-ROS-CTSB-NLRP3 pathway in mice and AML12 cells. J Agric Food Chem. 2023;71(18):7032–45. doi:10.1021/acs.jafc.2c07581. [Google Scholar] [PubMed] [CrossRef]
105. Zhang NP, Liu XJ, Xie L, Shen XZ, Wu J. Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab Invest. 2019;99(6):749–63. doi:10.1038/s41374-018-0177-6. [Google Scholar] [PubMed] [CrossRef]
106. Li R, Xin T, Li D, Wang C, Zhu H, Zhou H. Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: the role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol. 2018;18:229–43. doi:10.1016/j.redox.2018.07.011. [Google Scholar] [PubMed] [CrossRef]
107. Undamatla R, Fagunloye OG, Chen J, Edmunds LR, Murali A, Mills A, et al. Reduced mitophagy is an early feature of NAFLD and liver-specific PARKIN knockout hastens the onset of steatosis, inflammation and fibrosis. Sci Rep. 2023;13(1):7575. doi:10.1038/s41598-023-34710-x. [Google Scholar] [PubMed] [CrossRef]
108. Georgiev A, Granata C, Roden M. The role of mitochondria in the pathophysiology and treatment of common metabolic diseases in humans. Am J Physiol Cell Physiol. 2022;322(6):C1248–c59. doi:10.1152/ajpcell.00035.2022. [Google Scholar] [PubMed] [CrossRef]
109. Kupriyanova Y, Zaharia OP, Bobrov P, Karusheva Y, Burkart V, Szendroedi J, et al. Early changes in hepatic energy metabolism and lipid content in recent-onset type 1 and 2 diabetes mellitus. J Hepatol. 2021;74(5):1028–37. doi:10.1016/j.jhep.2020.11.030. [Google Scholar] [PubMed] [CrossRef]
110. Pedersen JS, Rygg MO, Chrøis K, Sustarsic EG, Gerhart-Hines Z, Wever Albrechtsen NJ, et al. Influence of NAFLD and bariatric surgery on hepatic and adipose tissue mitochondrial biogenesis and respiration. Nat Commun. 2022;13(1):2931. doi:10.1038/s41467-022-30629-5. [Google Scholar] [PubMed] [CrossRef]
111. Stevanović J, Beleza J, Coxito P, Ascensão A, Magalhães J. Physical exercise and liver “fitness”: role of mitochondrial function and epigenetics-related mechanisms in non-alcoholic fatty liver disease. Mol Metab. 2020;32:1–14. doi:10.1016/j.molmet.2019.11.015. [Google Scholar] [PubMed] [CrossRef]
112. Ignacio DL, Fortunato RS, Silvestre D, Matta L, de Vansconcelos AL, Carvalho DP, et al. Physical exercise improves mitochondrial function in ovariectomized rats. J Endocrinol. 2022;254(2):77–90. doi:10.1530/JOE-22-0057. [Google Scholar] [PubMed] [CrossRef]
113. Lassailly G, Caiazzo R, Ntandja-Wandji LC, Gnemmi V, Baud G, Verkindt H, et al. Bariatric surgery provides long-term resolution of nonalcoholic steatohepatitis and regression of fibrosis. Gastroenterology. 2020;159(4):1290–301.e5. doi:10.1053/j.gastro.2020.06.006. [Google Scholar] [PubMed] [CrossRef]
114. Deng Y, Dong Y, Zhang S, Feng Y. Targeting mitochondrial homeostasis in the treatment of non-alcoholic fatty liver disease: a review. Front Pharmacol. 2024;15:1463187. doi:10.3389/fphar.2024.1463187. [Google Scholar] [PubMed] [CrossRef]
115. Ramanathan R, Ali AH, Ibdah JA. Mitochondrial dysfunction plays central role in nonalcoholic fatty liver disease. Int J Mol Sci. 2022;23(13):7280. doi:10.3390/ijms23137280. [Google Scholar] [PubMed] [CrossRef]
116. Višnjić D, Lalić H, Dembitz V, Tomić B, Smoljo T. AICAr, a widely used AMPK activator with important AMPK-independent effects: a systematic review. Cells. 2021;10(5):1095. doi:10.3390/cells10051095. [Google Scholar] [PubMed] [CrossRef]
117. Abe RAM, Masroor A, Khorochkov A, Prieto J, Singh KB, Nnadozie MC, et al. The role of vitamins in non-alcoholic fatty liver disease: a systematic review. Cureus. 2021;13(8):e16855. doi:10.7759/cureus.16855. [Google Scholar] [PubMed] [CrossRef]
118. Raza S, Tewari A, Rajak S, Sinha RA. Vitamins and non-alcoholic fatty liver disease: a molecular insight. Liver Res. 2021;5(2):62–71. doi:10.1016/j.livres.2021.03.004. [Google Scholar] [PubMed] [CrossRef]
119. Chen Y, Yang F, Chu Y, Yun Z, Yan Y, Jin J. Mitochondrial transplantation: opportunities and challenges in the treatment of obesity, diabetes, and nonalcoholic fatty liver disease. J Transl Med. 2022;20(1):483. doi:10.1186/s12967-022-03693-0. [Google Scholar] [PubMed] [CrossRef]
120. Al Hashmi K, Giglio RV, Pantea Stoian A, Patti AM, Al Waili K, Al Rasadi K, et al. Metabolic dysfunction-associated fatty liver disease: current therapeutic strategies. Front Nutr. 2024;11:1355732. doi:10.3389/fnut.2024.1355732. [Google Scholar] [PubMed] [CrossRef]
121. Le MH, Yeo YH, Li X, Li J, Zou B, Wu Y, et al. 2019 global NAFLD prevalence: a systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2022;20(12):2809–17.e28. doi:10.1016/j.cgh.2021.12.002. [Google Scholar] [PubMed] [CrossRef]
122. Zhang L, El-Shabrawi M, Baur LA, Byrne CD, Targher G, Kehar M, et al. An international multidisciplinary consensus on pediatric metabolic dysfunction-associated fatty liver disease. Med. 2024;5(7):797–815.e2. doi:10.1016/j.medj.2024.03.017. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools