
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Influence of Liriodendrin on NLRP3-Mediated Pyroptosis and Proinflammatory Pathways in Mice Experiencing Acute Respiratory Distress Syndrome Induced by Lipopolysaccharide
1 Department of Internal Medicine, Division of Chest Medicine, Changhua Christian Hospital, Changhua, 500, Taiwan
2 Department of Pharmacology, School of Medicine, Chung Shan Medical University, Taichung, 402, Taiwan
3 Department of Pharmacy, Chung Shan Medical University Hospital, Taichung, 402, Taiwan
4 Department of Internal Medicine, School of Medicine, Chung Shan Medical University, Taichung, 402, Taiwan
5 Department of Internal Medicine, School of Medicine, Chung Shan Medical University Hospital, Taichung, 402, Taiwan
6 Institute of Medicine, Chung Shan Medical University, Taichung, 402, Taiwan
7 Department of Internal Medicine, Division of Nephrology, Chung Shan Medical University Hospital, Taichung, 402, Taiwan
8 Department of Dermatology, The Wilshire Lab and Aesthetic Clinic, Shenzhen, 518054, China
9 Department of Dermatology, Shiso Municipal Hospital, Hyogo, 671-2576, Japan
10 Department of Physics, R. O. C. Military Academy, Fengshan, Kaohsiung, 830, Taiwan
* Corresponding Author: Yu-Hsiang Kuan. Email:
# These two authors shared equal contributions to the present study
(This article belongs to the Special Issue: Cellular and Molecular Mechanisms Underlying Inflammation and Immune Regulation: From Genotoxicity to Apoptosis)
BIOCELL 2025, 49(2), 315-334. https://doi.org/10.32604/biocell.2025.061073
Received 16 November 2024; Accepted 10 February 2025; Issue published 28 February 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
ABSTRACT: Background: Acute respiratory distress syndrome (ARDS) is the major therapeutic dilemma associated with significant inflammation and severe pulmonary dysfunction. Liriodendrin is a bioactive compound extract from traditional Chinese medicine, historically utilized for modulating inflammatory responses and alleviating symptoms in multiple disease models. Methods: At present, BALB/c mice were used to explore the effects of liriodendrin on lipopolysaccharide (LPS)-induced ARDS. Before LPS was administered, the mice were treated with either liriodendrin or dexamethasone. Leukocyte infiltration, lung edema, and alveolar-capillary barrier integrity were evaluated in the bronchoalveolar lavage fluid (BALF) and pulmonary parenchyma. The expression of adhesion molecules and proinflammatory cytokines in BALF was evaluated by enzyme-linked immunosorbent assay. Western blotting assay facilitated the analysis of the expression or phosphorylation of inducible nitric oxide synthase (iNOS), cyclooxygenase- 2 (COX-2), NOD-like receptor family pyrin domain-containing 3 (NLRP3), apoptosis-associated speck-like protein containing a CARD (ASC), cleaved caspase-1 (CL-caspase-1), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), inhibitor of kappa B (IκB), mitogen-activated protein kinase (MAPK), and protein kinase B (Akt) in the lungs. In addition, the anti-inflammatory effects of liriodendrin were evaluated in LPS-stimulated RAW264.7 macrophages. Before LPS was administered, the RAW264.7 macrophages were treated with either liriodendrin or dexamethasone. Nitric Oxide (NO) production was measured using the Griess reaction assay, while ELISA assessed IL-1β, IL-6, and TNF-α levels. Western blot analysis evaluated NF-κB phosphorylation and the expression of NLRP3, ASC, and CL-caspase-1. Results: These outcomes revealed that liriodendrin intervention markedly ameliorated the pathological features of LPS-induced ARDS, including leukocyte infiltration, lung edema, and alveolar-capillary barrier disruption. Liriodendrin also reduced the LPS-induced secretion of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), expression of iNOS and COX2, and production of proinflammatory cytokines. Finally, we further discovered that the concentration trend of liriodendrin amelioration of ARDS was similar to those of NLRP3 formation, NF-κB pathway activation, and p38 MAPK, c-Jun N-terminal kinase (JNK), and Akt phosphorylation but not to that of extracellular signal-regulated kinase (ERK) phosphorylation. Liriodendrin inhibited LPS-induced inflammatory responses in RAW264.7 macrophages. It markedly reduced NO production, propro-inflammatorytokines, NF-κB phosphorylation, and NLRP3 formation. Conclusions: In summary, liriodendrin effectively ameliorated the pathological features of LPS-induced ARDS in mice, demonstrating significant antiinflammatory properties attributed to NLRP3 formation through NF-κB pathway activation by p38 MAPK, JNK, and Akt phosphorylation. In LPS-treated RAW264.7 macrophages, liriodendrin reduced NO production, pro-inflammatory cytokines, and NLRP3 formation, suggesting its potential as an agent for ARDS and relative inflammation.Keywords
Acute respiratory distress syndrome (ARDS) could be a severe pulmonary disorder that substantially affects patient survival, leading to high morbidity and mortality [1–3]. Since its discovery in the late 1960s, ARDS has posed a major challenge in intensive care units worldwide because of its complex pathophysiology and clinical management requirements [2,4,5]. Endotoxins, specifically lipopolysaccharide (LPS) originating from the cell walls of Gram-negative bacteria, initiate a cascade of reactions that activate nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [5–7]. This activation process triggers the production of several proinflammatory mediators (including tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6), adhesion molecules (including intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1)), and proinflammatory mediator proteins (including inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2)), all of which contribute to extensive lung damage and severe pulmonary distress [5–7]. In LPS-induced ARDS, the mitogen-activated protein kinase (MAPK) family, which includes extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 MAPK, along with Akt, plays a key role in the activation and phosphorylation of NF-κB, which further exacerbates ARDS [6–8]. In addition, NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasomes activate pyroptotic death pathways, which in turn exacerbate the inflammatory response, neutrophil infiltration, and pulmonary damage through apoptosis-associated speck-like protein containing a CARD (ASC) in mice with LPS-induced ARDS [9,10].
Liriodendrin is a potent bioactive compound found in certain plants, including Sargentodoxa cuneata, Liriodendron tulipifera, and Kalopanax pictus [11–13]. These plants are typically used in traditional Chinese medicine because of their efficacy in treating abdominal pain, ulcers, and menstrual discomfort and in ameliorating swelling, arthritis, inflammation, and pain [13–16]. Multiple studies have indicated the various biological and pharmacological benefits of liriodendrin, including its strong antioxidative capacity, anti-inflammatory effect, and ability to reduce myocardial infarction [15,17]. According to the literature, liriodendrin inhibits the phosphorylation of NF-κB and modulates the Src/signal transducer and activator of transcription 3 (STAT3)/MAPK signaling pathways, thereby ameliorating acute lung injuries (ALI) indirectly caused by cecal ligation and puncture (CLP) and IgG immune complexes (IgG-IC) [18,19]. However, despite these findings, no studies have yet examined whether liriodendrin directly mitigates the inflammatory response of ARDS induced by intratracheal LPS administration. In this study, we examined the effects of liriodendrin on NLRP3-mediated pyroptosis and proinflammatory pathways in LPS-induced ARDS.
2.1 Animals and Animal Preparation
Fifty-four male BALB/c mice, each weighing approximately 25 ± 2 g and aged between 9 and 12 weeks, were procured from the BioLASCO Experimental Animal Center (Taipei, Taiwan). The mice were housed in a controlled laboratory environment to ensure their health and acclimatization. They experienced a 12-h light/dark cycle and a consistent temperature of 21°C ± 2°C. Relative humidity was maintained at 60% ± 5%. These conditions were in place for one week prior to any experimental procedures to help them adjust. All animal studies were approved by the Institutional Animal Ethics Committee of Chung Shan Medical University (IACUC no. 112006). The mice were assigned to six separate groups at random that were treated with normal saline + normal saline in the normal group, saline + 5 mg/kg LPS (Sigma-Aldrich/Merck Millipore, L3024, St. Louis, MO, USA) in the LPS group, 10 μM liriodendrin (ChemFaces, CFN98964, Wuhan, China) + 5 mg/kg LPS, 30 μM liriodendrin + 5 mg/kg LPS, 50 μM liriodendrin + 5 mg/kg LPS, 1 mg/kg dexamethasone (Sigma-Aldrich/Merck Millipore, D4902) + 5 mg/kg LPS, and 50 μM liriodendrin + saline. After saline, liriodendrin, or dexamethasone was intraperitoneally administered for 30 min, the saline and LPS at the concentration of 5 mg/kg were subsequently administered via the intratracheal route. After 6 h, the mice were humanely euthanized, and both their lungs and bronchoalveolar lavage fluid (BALF) were harvested for subsequent assessment. Lung tissues were fixed in 10% formalin, processed for paraffin embedding, and sectioned at 5 µm. Hematoxylin and eosin (H&E) Stain Kit (Vectorlabs, H-3502, New York, NY, USA) included hematoxylin for nuclei and eosin for cytoplasm and extracellular structures. Sections were deparaffinized, rehydrated, stained, and mounted for analysis [6,7,20].
2.2 Bronchoalveolar Lavage Fluid (BALF) and Leukocyte Count
Following euthanasia, bronchoalveolar lavage was performed to collect BALF, as described in previous studies [6,20]. After endotracheal intubation, sterile saline was instilled into the lungs and subsequently aspirated to collect BALF. Subsequently, the BALF samples were centrifuged, and the cells were collected in a pellet and resuspended within phosphate-buffered saline. Flow cytometry (BD Biosciences, FACS Accuri C6, San Jose, CA, USA) was utilized to determine the total leukocyte count. PE/Cyanine7 anti-mouse CD45 antibody (1:100, BioLegend, 103114, San Diego, CA, USA) was used to detect the proportion of leukocytes in BALF. PE anti-mouse Ly-6G Antibody (1:100, BioLegend, 1164504, San Diego, CA, USA) was used to detect the proportion of neutrophils in BALF.
2.3 Alveolar-Capillary Membrane Dysfunction
Increased protein concentration in BALF is a primary biomarker of alveolar-capillary membrane dysfunction, which is the key indicator of compromised pulmonary barrier integrity. Such dysfunction typically occurs when the permeability of the alveolar-capillary barrier increases, triggered by ARDS. In this study, the levels of proteins in BALF were measured using a Quick Start™ Bradford Protein Assay (Bio-Rad Laboratories Inc., 5000201, Hercules, CA, USA). The Bradford protein assay is used to measure protein concentration in a sample. It can assess the extravasation of proteins from the bloodstream into the lung tissue, which reflects increased alveolar-capillary membrane dysfunction [6,7]. Absorbance was measured at 560 nm using the microplate reader (Agilent Technologies, BioTek Synergy HT Multi-Mode, Winooski, VT, USA) [6,7].
The lung wet-to-dry weight ratio is a key indicator of lung water content. An elevated ratio signifies pulmonary edema, indicating increased water retention in lung tissue [20,21]. To assess the severity of lung edema, the lungs of the mice were removed and weighed immediately to obtain the wet weight. They were then placed into the incubator at 80°C for 72 h before being weighed again to determine the dry weight. The comparison of wet and dry weights provides critical insight into the extent of fluid accumulation in the lungs, called lung edema [20].
2.5 Measurements of Adhesion Molecules and Proinflammatory Cytokines with Enzyme-Linked Immunosorbent Assay
Enzyme-linked immunosorbent assay (ELISA) was used to examine the expression of adhesion molecules, including mouse ICAM-1/CD54 ELISA kits (BioTechne/R&D Systems, MIC100, Minneapolis, MN, USA) and mouse VCAM-1/CD106 ELISA kit (BioTechne/R&D Systems, MVC100) and proinflammatory cytokines including Mouse IL-1 beta/IL-1F2 Quantikine ELISA Kit (BioTechne/R&D Systems, MLB00C), Mouse IL-6 ELISA Quantikine Kit (BioTechne/R&D Systems, MB6000B), and Mouse TNF-α Quantikine ELISA Kit (BioTechne/R&D Systems, MTA00B) in BALF. These experiments were conducted with strict adherence to the manufacturer’s guidelines and were informed by previous studies [22].
2.6 Western Blot Analysis of Protein Phosphorylation and Expression
Western blotting was used to determine the levels of protein expression and phosphorylation in the lungs of mice with LPS-induced ARDS. After lung tissues were collected, they were homogenized in T-PER Tissue Protein Extraction Reagent (Thermo Fisher Scientific, 78510, Waltham, MA, USA) supplemented with a protease inhibitor cocktail (Sigma-Aldrich/Merck Millipore, P1860) and phosphatase inhibitor cocktail 2 (Sigma-Aldrich/Merck Millipore, P5726). After centrifugation, the supernatants of the lung homogenates were collected. To analyze the protein samples, an equivalent amount of protein from each sample was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to polyvinylidene difluoride membranes (Thermo Fisher Scientific, 88520). This technique facilitated the separation of proteins depending on their molecular weight and enabled their subsequent membrane transfer for analysis. After incubating the membranes with 5% skim milk for one hour at room temperature for blocking, they were treating for 16 h at 4°C with anti-iNOS (1:500, Santa Cruz Biotechnology, SC-7271, St Louis, MO, USA), anti-COX2 (1:1000, Santa Cruz Biotechnology, SC-19999), anti-NLRP3 (1:500, Santa Cruz Biotechnology, SC-134306), anti-ASC (1:500, Santa Cruz Biotechnology, SC-514414), anti-caspase-1 (1:1000, Santa Cruz Biotechnology, sc-56036), anti-IκB (1:1000, Santa Cruz Biotechnology, SC-514414), anti-p-p65 (1:1000, Santa Cruz Biotechnology, SC-166748), anti-p65 (1:1000, Santa Cruz Biotechnology, SC-8008), anti-p-Akt (1:1000, Santa Cruz Biotechnology, SC-514032), anti-Akt (1:1000, Santa Cruz Biotechnology, SC-81434), anti-p-p38 MAPK (1:1000, Santa Cruz Biotechnology, SC-7973), anti-p38 MAPK (1:1000, Santa Cruz Biotechnology, SC-7972), anti-p-JNK (1:1000, Santa Cruz Biotechnology, SC-6254), anti-JNK (1:1000, Santa Cruz Biotechnology, SC-571), anti-p-ERK (1:1000; Santa Cruz Biotechnology, SC-7383), anti-ERK (1:1000, Santa Cruz Biotechnology, SC-514302), and anti-β-actin (1:5000, Santa Cruz Biotechnology, SC-47778) antibodies. Subsequently, horseradish peroxidase-conjugated AffiniPure™ Donkey Anti-Mouse IgG (1:10000, Jackson ImmunoResearch Labs, 715-545-151, West Grove, PA, USA) secondary antibodies were applied and allowed to incubate at room temperature for a duration of one hour. Finally, the expression and phosphorylation of proteins were evaluated using enhanced chemiluminescence on a FUSIN Solo Vision apparatus (Vilber, Lourmat, Collégien, France) and analyzed using densitometry [20,23,24].
2.7 Cell Culture and Treatment
RAW 264.7 macrophage cells were obtained from a reliable cell bank, Bioresource Collection and Research Center (BCRC, 60001, Hsinchu, Taiwan) and cultured in Dulbecco’s Modified Eagle Medium (DMEM; Cytiva HycloneTM, SH30243.FS, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS; Cytiva HycloneTM, SH30088.03) and 1% penicillin-streptomycin (Cytiva HycloneTM, SV30010) at 37°C in a humidified atmosphere containing 5% CO2. All reagents used in cell culture and treatment, including FBS, DMEM, and antibiotics, were sourced from certified suppliers and verified to be mycoplasma-free to minimize contamination risks. Cells were seeded into culture plates at an appropriate density and allowed to adhere overnight. Treatments included the solvent of liriodendrin which is the dimethyl sulfoxide (DMSO; Sigma-Aldrich/Merck Millipore, D8418), liriodendrin at concentrations of 10, 30 μM, or 50 μM, and dexamethasone (1 mg/L) as a reference anti-inflammatory agent. Treatments were administered 30 min before stimulation with either saline or LPS (500 ng/mL).
The level of nitric oxide (NO) in the culture supernatant was determined using the Nitrite Assay Kit (Griess Reagent) (Sigma-Aldrich/Merck Millipore, MAK367). Briefly, an equal volume of culture supernatant and Griess reagent was mixed and incubated at room temperature for 10 min. The absorbance was measured at 540 nm using a microplate reader (Synergy HT, Multi-mode microplate reader, BioTek Instruments, Inc., Winooski, VT, USA), and NO levels were calculated based on a standard curve prepared with sodium nitrite.
Statistical analyses were conducted using SPSS Statistics (version 21.0, IBM SPSS Statistics, IBM Corporation, Armonk, NY, USA), with results presented as means ± standard deviation (SD). To evaluate the significance of our findings, a one-way analysis of variance (ANOVA) was employed. All experiments were conducted with four replicates (n = 4). In instances requiring multiple comparisons, Bonferroni’s post hoc test was applied, and a p value of less than 0.05 was deemed indicative of statistically significant results.
3.1 Liriodendrin Ameliorated the Pathological Features of Mice with LPS-Induced ARDS
To determine the effect of liriodendrin on LPS-induced ARDS, we examined multiple pathological features, including leukocyte infiltration, neutrophil infiltration, lung edema, and alveolar-capillary membrane dysfunction. After the intratracheal administration of LPS, significant increases were observed in histopathological exchange, leukocyte infiltration, neutrophil infiltration, lung edema, and protein leakage into BALF as a result of alveolar-capillary membrane dysfunction (p < 0.05, Fig. 1). The findings indicate that liriodendrin and dexamethasone treatment significantly mitigated LPS-induced lung injury, as shown by reduced alveolar collapse and improved structural integrity. Additionally, liriodendrin did not cause adverse histopathological changes in lung tissues under normal conditions (Fig. 1A). In addition, liriodendrin inhibited LPS-induced leukocyte infiltration, neutrophil infiltration, and lung edema in a dose-dependent manner, with its inhibitory effect initially observed at a concentration of 30 μM (p < 0.05, Fig. 1B–D). Liriodendrin also inhibited LPS-induced alveolar-capillary membrane dysfunction in a dose-dependent manner, with its inhibitory effect initially observed at a concentration of 10 μM (p < 0.05, Fig. 1E). In the positive control group, 1 mg/kg dexamethasone ameliorated the pathological features of mice with LPS-induced ARDS (Fig. 1). Moreover, we further found that pathological features of mice with ARDS could not induced by liriodendrin at the concentration of 500 μM (Fig. 1).
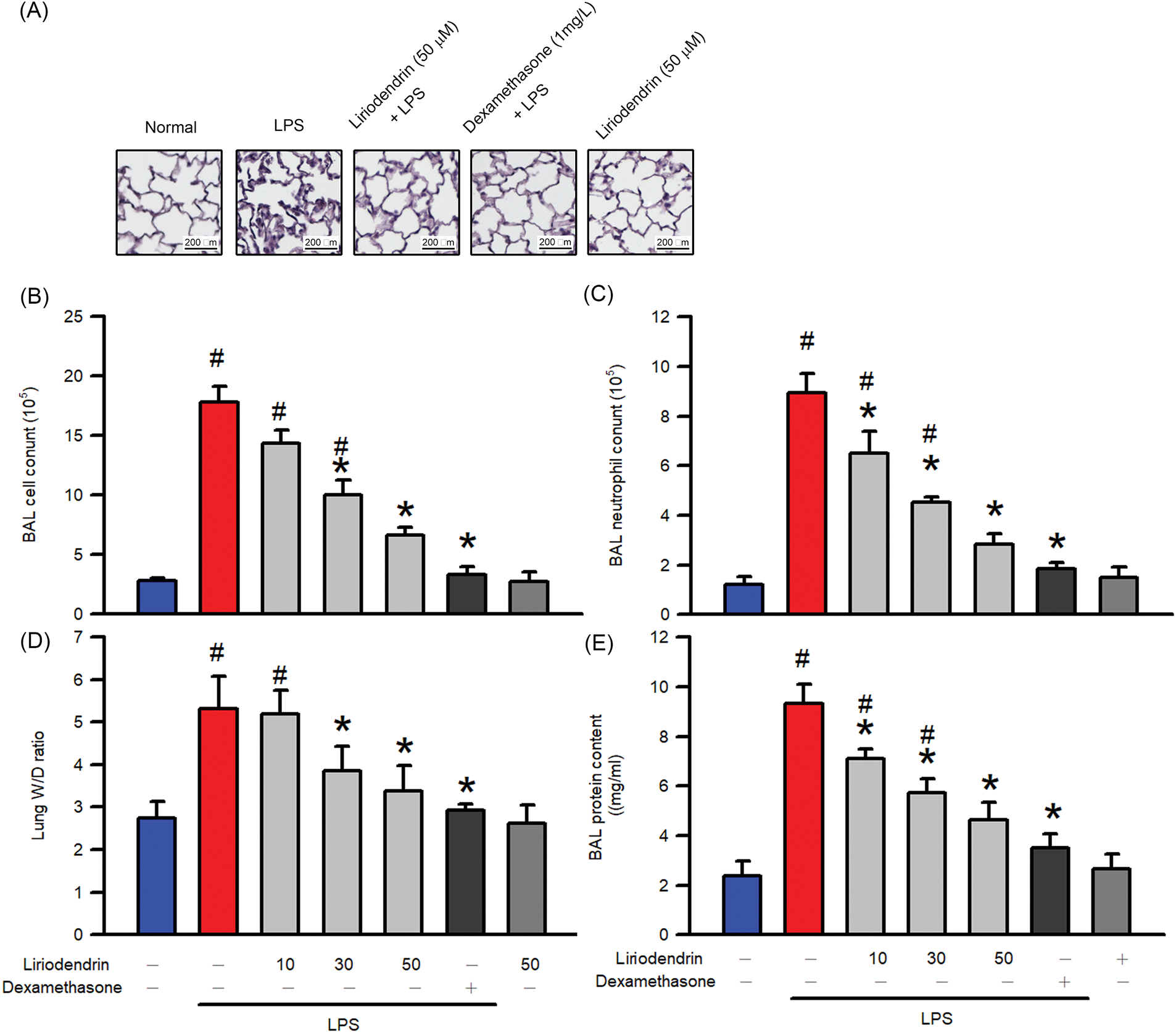
Figure 1: Liriodendrin ameliorated the multiple pathological features, including histopathological exchange, leukocyte infiltration, neutrophil infiltration, lung edema, and alveolar-capillary membrane dysfunction in LPS-induced ARDS mice. Saline, liriodendrin (10, 30, or 50 μM), or dexamethasone (1 mg/kg) was administered intraperitoneally at specified concentrations 30 min prior to the intratracheal administration of saline or LPS (5 mg/kg). (A) The histopathological examination in each treatment was performed using H&E staining. (B) The counts of leukocyte and (C) neutrophil infiltration into BALF in each treatment. (D) Lung edema was evaluated by the ratio of wet and dry weight of lungs in each treatment. (E) The alveolar-capillary membrane dysfunction was evaluated using the Bradford protein assay in each treatment. All data were presented as mean ± SD and derived from experiments performed on groups with four replicates (n = 4). #p < 0.05 significantly differed from the normal group, as shown in blue bars. *p < 0.05 significantly differed from the LPS group, as shown in red bars. LPS: lipopolysaccharide; BALF: bronchoalveolar lavage fluid; ARDS: acute respiratory distress syndrome
3.2 Liriodendrin Downregulated the Expression of Adhesion Molecules in Mice with LPS-Induced ARDS
ELISA was used to evaluate protein expression and to determine whether liriodendrin modulated the secretion of adhesion molecules, including ICAM-1 and VCAM-1, in the BALF of mice with ARDS. After the intratracheal administration of LPS, a significant increase in the secretion of adhesion molecules into BALF was observed (p < 0.05, Fig. 2). By contrast, liriodendrin inhibited this effect in a dose-dependent manner, with its inhibitory effect initially observed at a concentration of 30 μM (p < 0.05, Fig. 2). Dexamethasone also inhibited the secretion of adhesion molecules in mice with LPS-induced ARDS (p < 0.05, Fig. 2).
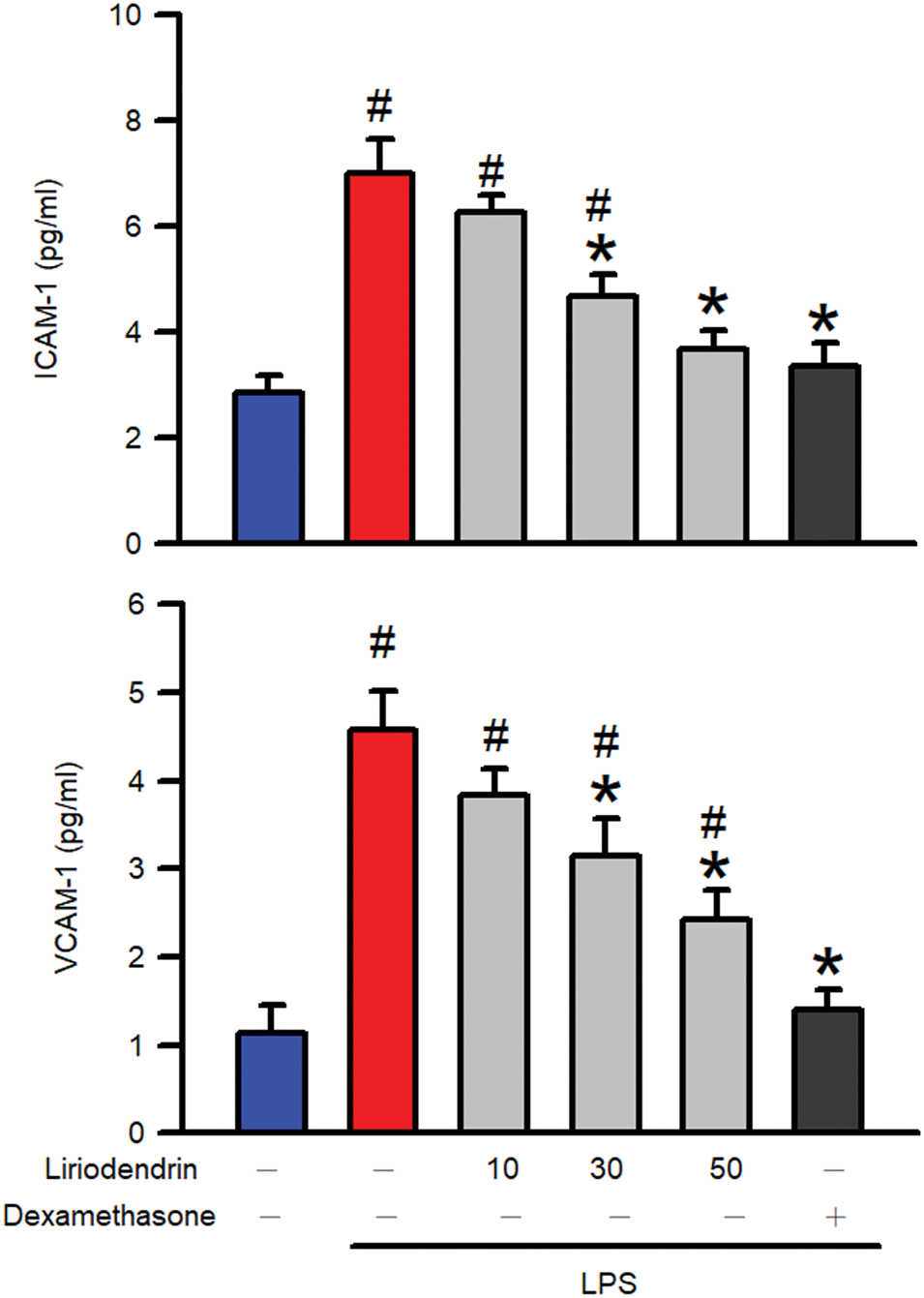
Figure 2: Liriodendrin downregulated the expression of adhesion molecules, including ICAM-1 and VCAM-1, in LPS-induced ARDS mice. Saline, liriodendrin (10, 30, or 50 μM), or dexamethasone (1 mg/kg) was administered intraperitoneally at specified concentrations 30 min before the intratracheal administration of saline or LPS (5 mg/kg). The expression of adhesion molecules, including ICAM-1 and VCAM-1, was evaluated using the ELISA assay in each treatment. All data were presented as mean ± SD and derived from experiments performed on groups with four replicates (n = 4). #p < 0.05 significantly differed from the normal group, as shown in blue bars. *p < 0.05 significantly differed from the LPS group, as shown in red bars. ICAM-1: intercellular adhesion molecule-1; VCAM-1: vascular cell adhesion molecule-1; ELISA: enzyme-linked immunosorbent assay
3.3 Liriodendrin Downregulated the Expression of iNOS and COX2 in Mice with LPS-Induced ARDS
Western blotting was used to evaluate protein expression and to determine whether liriodendrin modulated the expression of iNOS and COX2 in the lung tissues of mice with ARDS. After the intratracheal administration of LPS, significant increases in the expression of iNOS and COX2 were observed in the lung tissues of the mice (p < 0.05, Fig. 3). By contrast, liriodendrin inhibited this effect in a dose-dependent manner, with its inhibitory effect initially observed at a concentration of 30 μM (p < 0.05, Fig. 3). Dexamethasone also inhibited the expression of iNOS and COX2 in mice with LPS-induced ARDS (p < 0.05, Fig. 3).
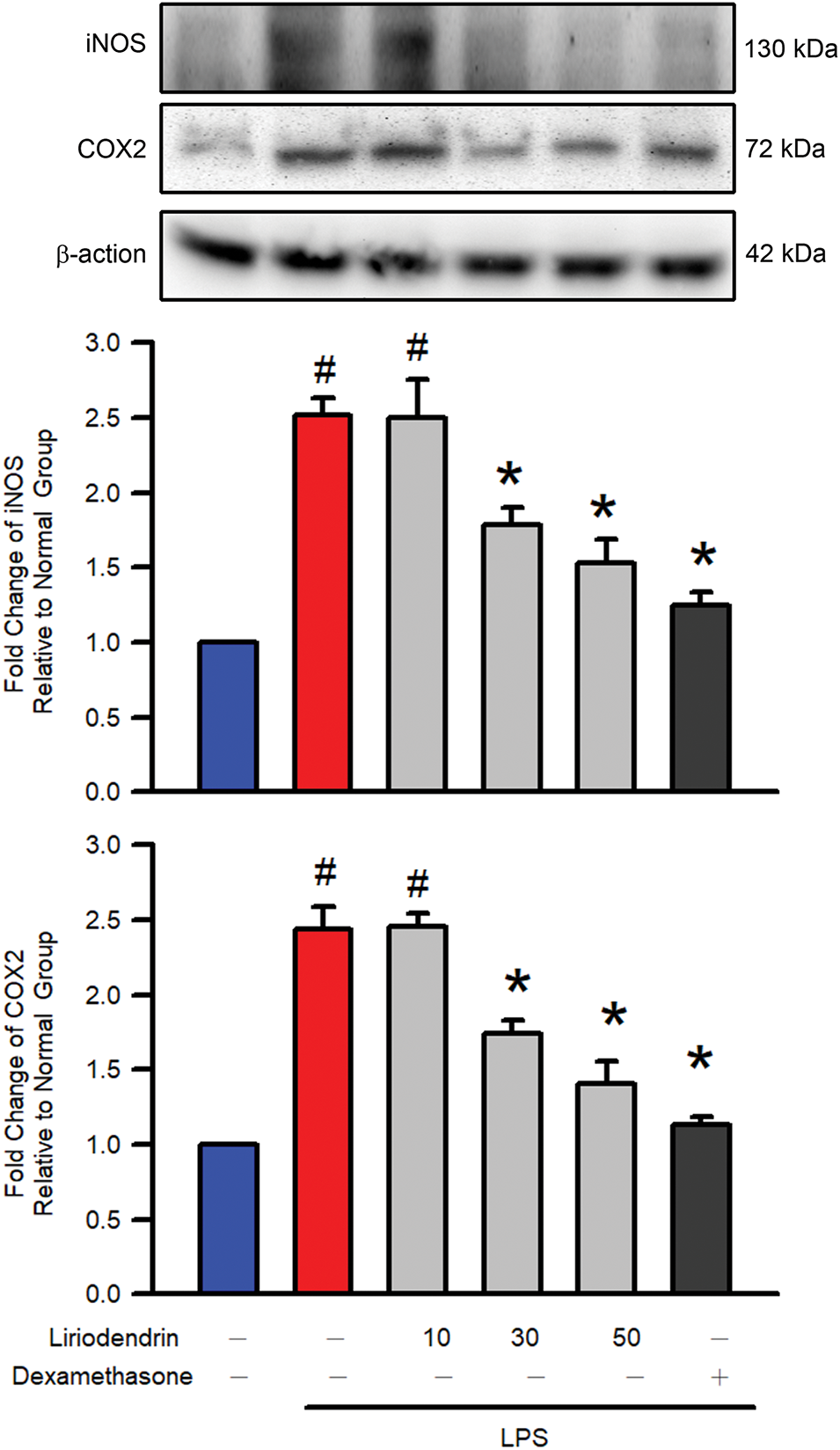
Figure 3: Liriodendrin downregulated the expression of iNOS and COX2 in LPS-induced ARDS mice. Saline, liriodendrin (10, 30, or 50 μM), or dexamethasone (1 mg/kg) was administered intraperitoneally at specified concentrations 30 min before the intratracheal administration of saline or LPS (5 mg/kg). The expression of iNOS and COX2 was evaluated using the Western blotting assay in each treatment. All quantitative data were presented as mean ± SD and derived from experiments performed on groups with four replicates (n = 4). #p < 0.05 significantly differed from the normal group, as shown in blue bars. *p < 0.05 significantly differed from the LPS group, as shown in red bars
3.4 Liriodendrin Hindered the Production of Proinflammatory Cytokines in Mice with LPS-Induced ARDS
ELISA was used to measure the production and release of proinflammatory cytokines including IL-1β, IL-6, and TNFα, into BALF and to determine whether liriodendrin modulated this process in mice with ARDS. After the intratracheal administration of LPS, a significant increase in the production of proinflammatory cytokines was observed in the lung tissues of the mice (p < 0.05, Fig. 4). By contrast, liriodendrin inhibited this effect in a dose-dependent manner, with its inhibitory effect initially observed at a concentration of 30 μM (p < 0.05, Fig. 4). Dexamethasone also inhibited the secretion of proinflammatory cytokines in mice with LPS-induced ARDS (p < 0.05, Fig. 4).
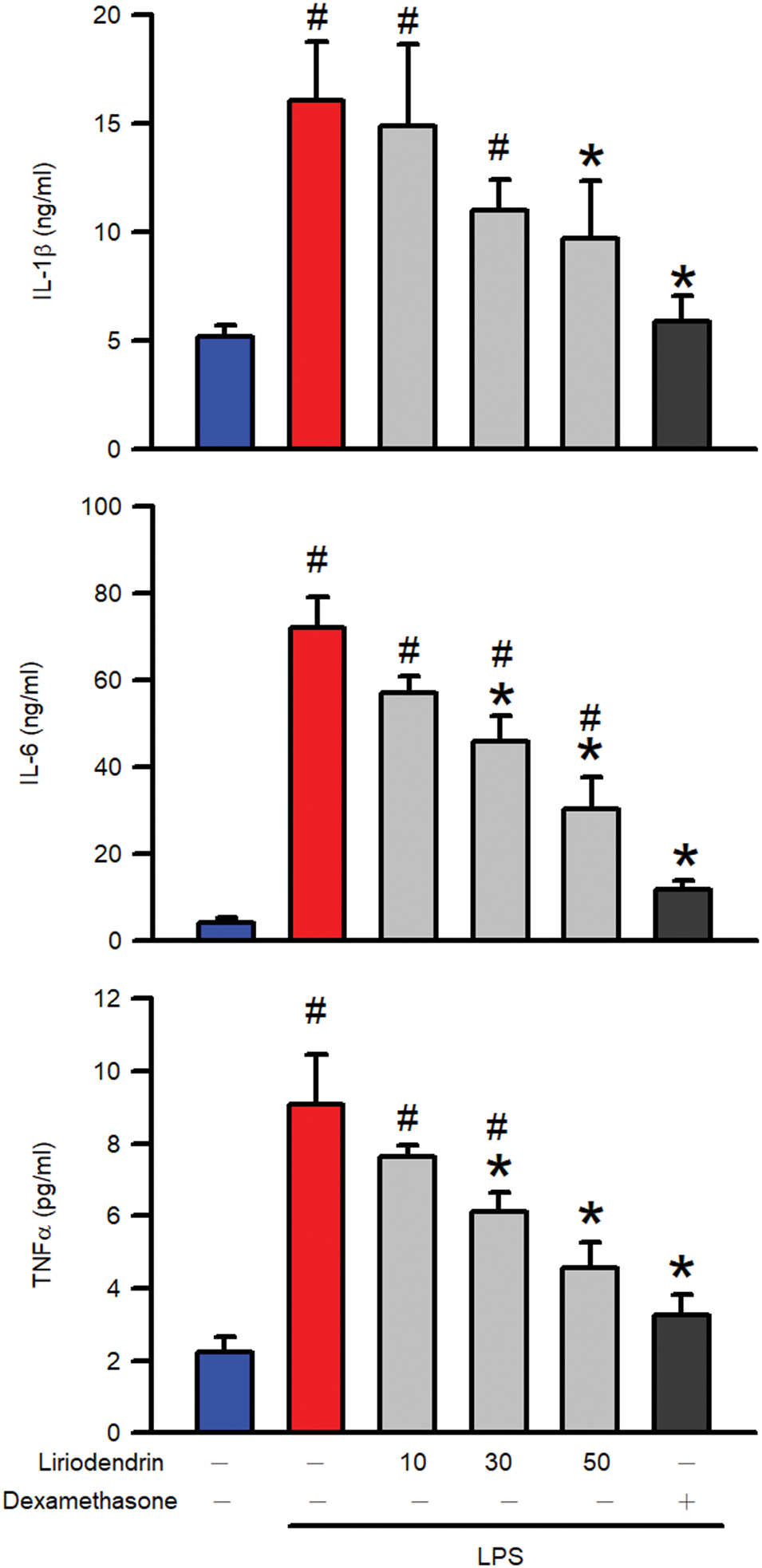
Figure 4: Liriodendrin downregulated the production of proinflammatory cytokines in LPS-induced ARDS mice. Saline, liriodendrin (10, 30, or 50 μM), or dexamethasone (1 mg/kg) was administered intraperitoneally at specified concentrations 30 min prior to the intratracheal administration of saline or LPS (5 mg/kg). The production of proinflammatory cytokines, including IL-1β, IL-6, and TNFα, was evaluated using the ELISA assay in each treatment. All data were presented as mean ± SD and derived from experiments performed on groups with four replicates (n = 4). #p < 0.05 significantly differed from the normal group, as shown in blue bars. *p < 0.05 significantly differed from the LPS group, as shown in red bars. IL: interleukin; TNF: tumor necrosis factor
3.5 Liriodendrin Downregulated the Expression of NLRP3, ASC, and Cleaved Caspase-1 in Mice with LPS-Induced ARDS
Western blotting was used to evaluate protein expression and to determine whether liriodendrin modulated the expression of NLRP-3, ASC, and cleaved caspase-1 in the lung tissues of mice with ARDS. After the intratracheal administration of LPS, significant increases in the expression of NLRP-3, ASC, and cleaved caspase-1 were observed in the lung tissues of the mice (p < 0.05, Fig. 5). By contrast, liriodendrin inhibited this effect in a dose-dependent manner, with its inhibitory effect initially observed at a concentration of 30 μM (p < 0.05, Fig. 5). Dexamethasone also inhibited the expression of NLRP-3, ASC, and cleaved caspase-1 in mice with LPS-induced ARDS (p < 0.05, Fig. 5).
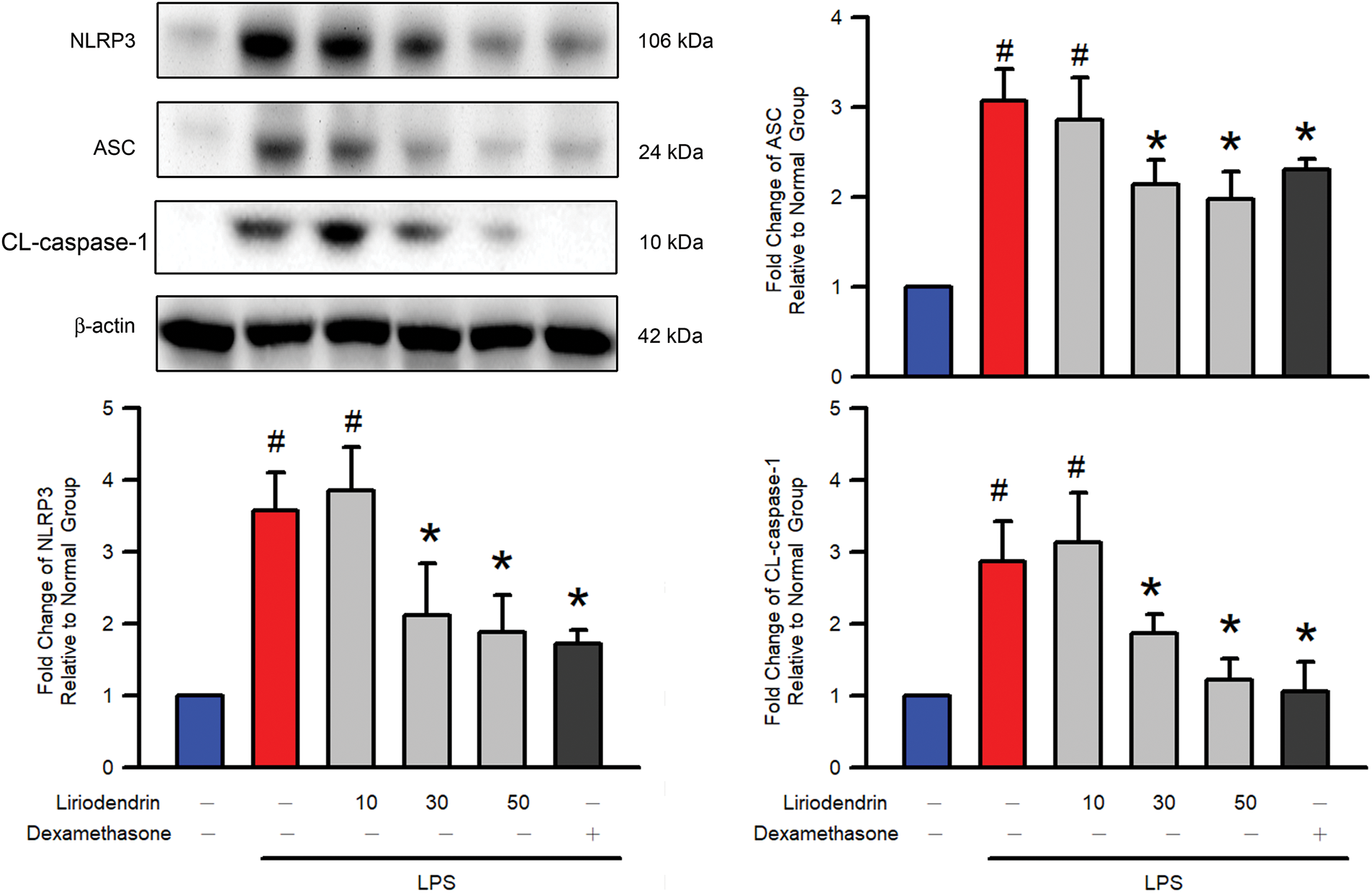
Figure 5: Liriodendrin downregulated the expression of NLRP3, ASC, and cleaved caspase-1 in LPS-induced ARDS mice. Saline, liriodendrin (10, 30, or 50 μM), or dexamethasone (1 mg/kg) was administered intraperitoneally at specified concentrations 30 min before the intratracheal administration of saline or LPS (5 mg/kg). The expression of NLRP3, ASC, and CL-caspase-1 was evaluated using the Western blotting assay in each treatment. All quantitative data were presented as mean ± SD and derived from experiments performed on groups with four replicates (n = 4). #p < 0.05 significantly differed from the normal group, as shown in blue bars. *p < 0.05 significantly differed from the LPS group, as shown in red bars. NLRP3: NOD-like receptor family pyrin domain-containing 3; ASC: apoptosis-associated speck-like protein containing a CARD; CL-csapase-1: cleaved caspase-1
3.6 Liriodendrin Reduced IκB Degradation and NF-κB Phosphorylation in Mice with LPS-Induced ARDS
Western blotting was used to determine the effect of liriodendrin on the degradation of IκB and the phosphorylation of NF-κB in mice with LPS-induced ARDS. After the intratracheal administration of LPS, significant increases in the degradation of IκB and the phosphorylation of NF-κB were observed (p < 0.05, Fig. 6). By contrast, liriodendrin inhibited this effect in a dose-dependent manner, with its inhibitory effect initially observed at a concentration of 30 μM (p < 0.05, Fig. 6). Dexamethasone also inhibited the degradation of IκB and the phosphorylation of NF-κB in mice with LPS-induced ARDS (p < 0.05, Fig. 6).
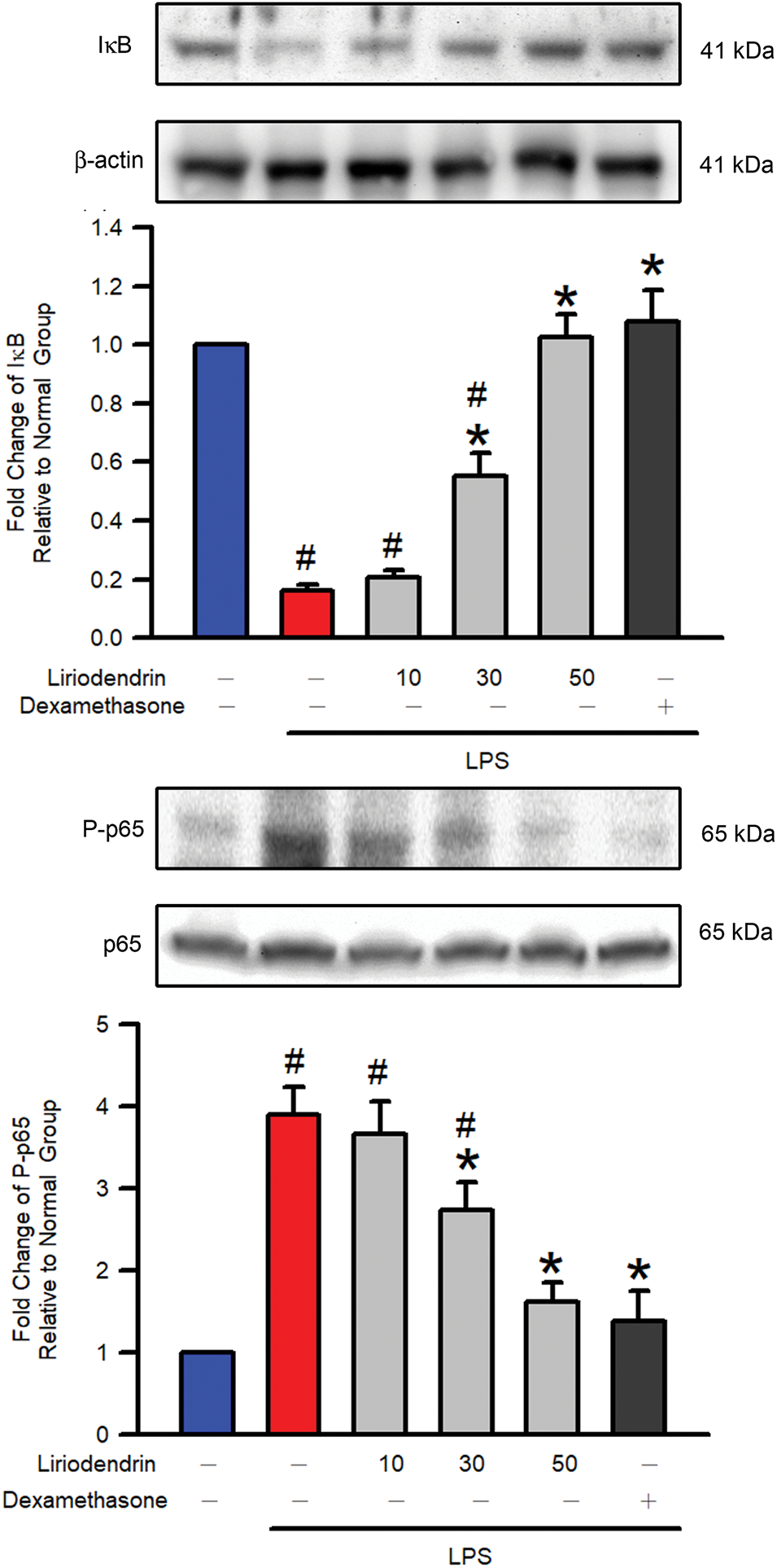
Figure 6: Liriodendrin downregulated the degradation of IκB and the phosphorylation of NF-κB p65 in LPS-induced ARDS mice. Saline, liriodendrin (10, 30, or 50 μM), or dexamethasone (1 mg/kg) was administered intraperitoneally at specified concentrations 30 min prior to the intratracheal administration of saline or LPS (5 mg/kg). The degradation of IκB and the phosphorylation of NF-κB p65 were evaluated using the Western blotting assay in each treatment. The levels of IκB expression and p65 phosphorylation were normalized to the level of actin and p65, respectively. All quantitative data were presented as mean ± SD and derived from experiments performed on groups with four replicates (n = 4). #p < 0.05 significantly differed from the normal group, as shown in blue bars. *p < 0.05 significantly differed from the LPS group, as shown in red bars. NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; IκB: inhibitor of NF-κB
3.7 Liriodendrin Reduced the Phosphorylation of Akt, p38 MAPK, and JNK in Mice with LPS-Induced ARDS
Western blotting was used to determine the effect of liriodendrin on the phosphorylation of Akt and the MAPK family, including p38 MAPK, JNK, and ERK, in mice with LPS-induced ARDS. After the intratracheal administration of LPS, a significant increase was observed in the phosphorylation of Akt, p38 MAPK, JNK, and ERK (p < 0.05, Fig. 7). By contrast, liriodendrin inhibited this effect in a dose-dependent manner, with its inhibitory effect initially observed at a concentration of 30 μM (p < 0.05, Fig. 7). Despite these findings, liriodendrin had no effect on the phosphorylation of ERK in mice with LPS-induced ARDS. Dexamethasone inhibited the phosphorylation of Akt, p38 MAPK, JNK, and ERK in mice with LPS-induced ARDS (p < 0.05, Fig. 7).
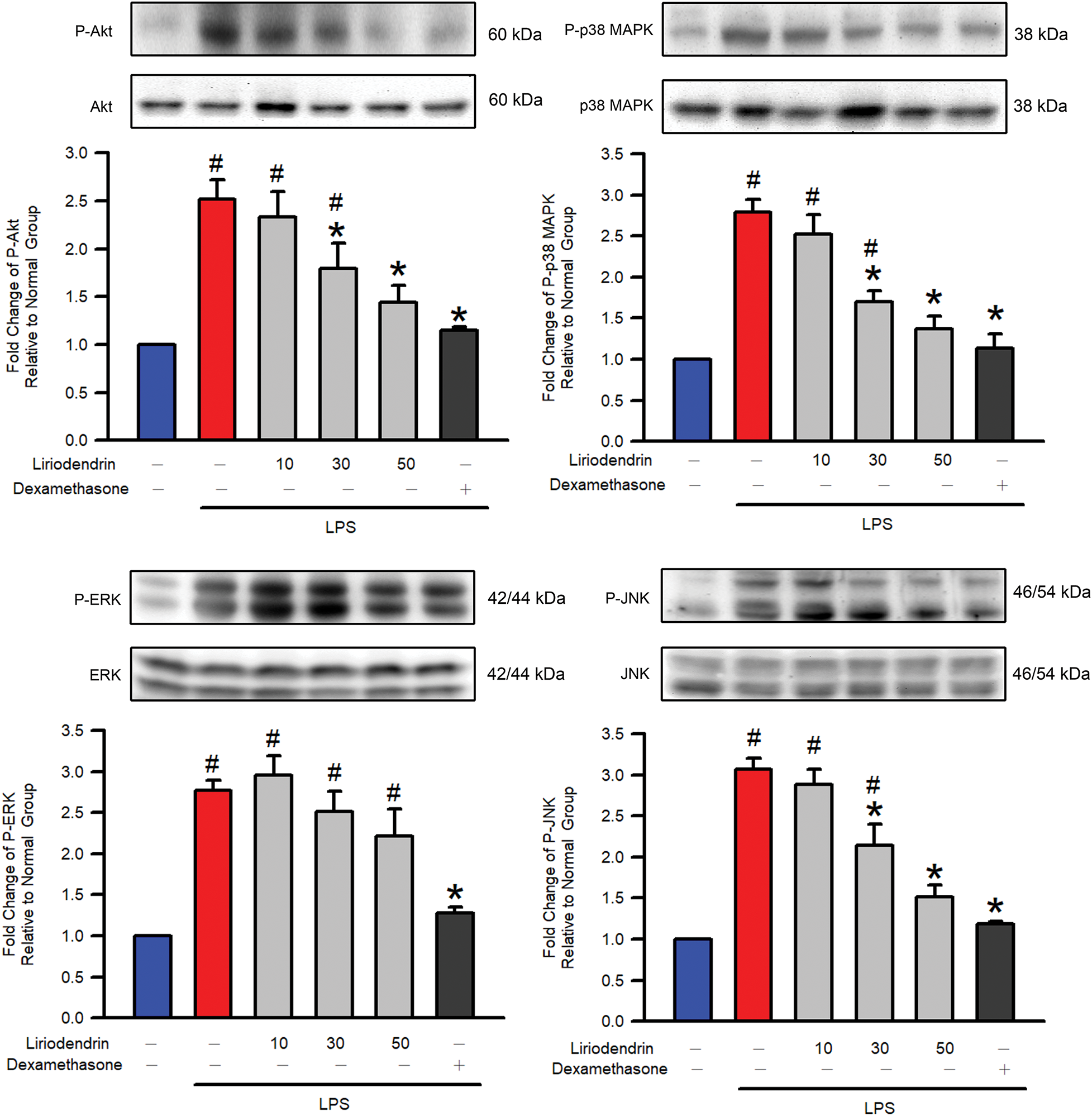
Figure 7: Liriodendrin downregulated the phosphorylation of Akt, p38 MAPK, and JNK in LPS-induced ARDS mice. The phosphorylation of Akt and the MAPK family, including p38 MAPK, JNK, and ERK, were evaluated using the Western blotting assay in each treatment. The phosphorylated levels of Akt, p38 MAPK, and JNK were normalized to the non-phosphorylated levels of Akt, p38 MAPK, and JNK, respectively. Saline, liriodendrin (10, 30, or 50 μM), or dexamethasone (1 mg/kg) was administered intraperitoneally at specified concentrations 30 min prior to the intratracheal administration of saline or LPS (5 mg/kg). All quantitative data were presented as mean ± SD and derived from experiments performed on groups with four replicates (n = 4). #p < 0.05 significantly differed from the normal group, as shown in blue bars. *p < 0.05 significantly differed from the LPS group, as shown in red bars. MAPK: mitogen-activated protein kinase; JNK: c-Jun N-terminal kinase; ERK: extracellular signal-regulated kinase
3.8 Liriodendrin Reduced the NO Generation, Proinflammatory Cytokines Expression, NF-κB p65 Phosphorylation, and NLRP3, ASC, and Cleaved Caspase-1 Expression in LPS-Stimulated Macrophage Activation
Griess reaction assay was used to determine the effect of liriodendrin on NO generation in LPS-stimulated macrophage activation. After the administration of LPS, a significant increase was observed in the NO generation (p < 0.05, Fig. 8A). By contrast, liriodendrin inhibited this effect in a dose-dependent manner, with its inhibitory effect initially observed at a concentration of 30 μM (p < 0.05, Fig. 8A). On the other hand, we further found that proinflammatory cytokines, including IL-1β, IL-6, and TNFα, were significantly increased by LPS (p < 0.05, Fig. 8B). Liriodendrin inhibited this effect in a dose-dependent manner, with its inhibitory effect initially observed at a concentration of 30 μM (p < 0.05, Fig. 8B). Finally, NF-κB p65 phosphorylation, NLRP3, ASC, and cleaved caspase-1 expression were significantly increased by LPS (p < 0.05, Fig. 8C). Liriodendrin inhibited this effect in a dose-dependent manner, with its inhibitory effect initially observed at a concentration of 30 μM (p < 0.05, Fig. 8C). Dexamethasone inhibited the NO generation, proinflammatory cytokines expression, NF-κB p65 phosphorylation, and NLRP3, ASC, and cleaved caspase-1 expression in LPS-stimulated RAW264.7 macrophage (p < 0.05, Fig. 8).
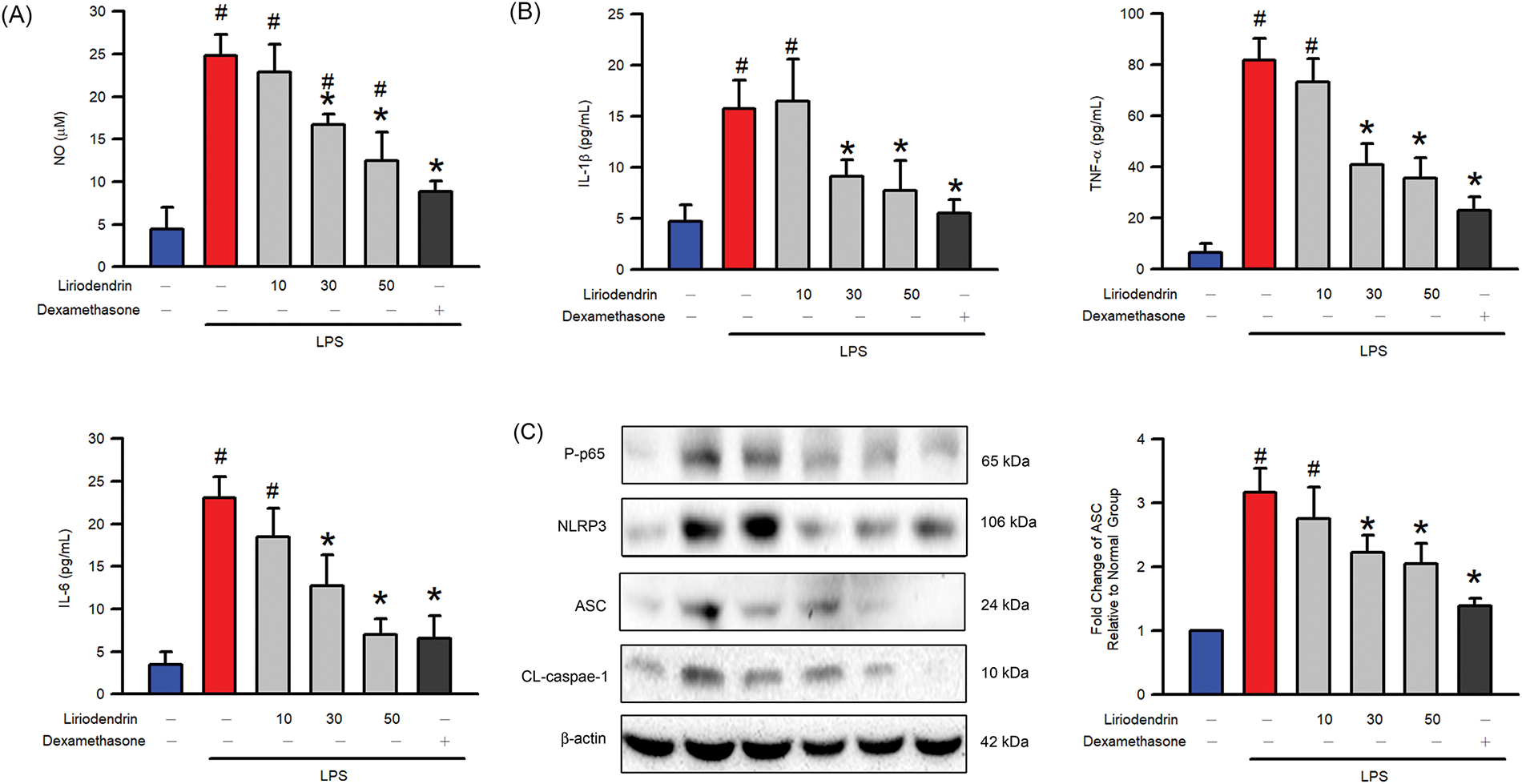
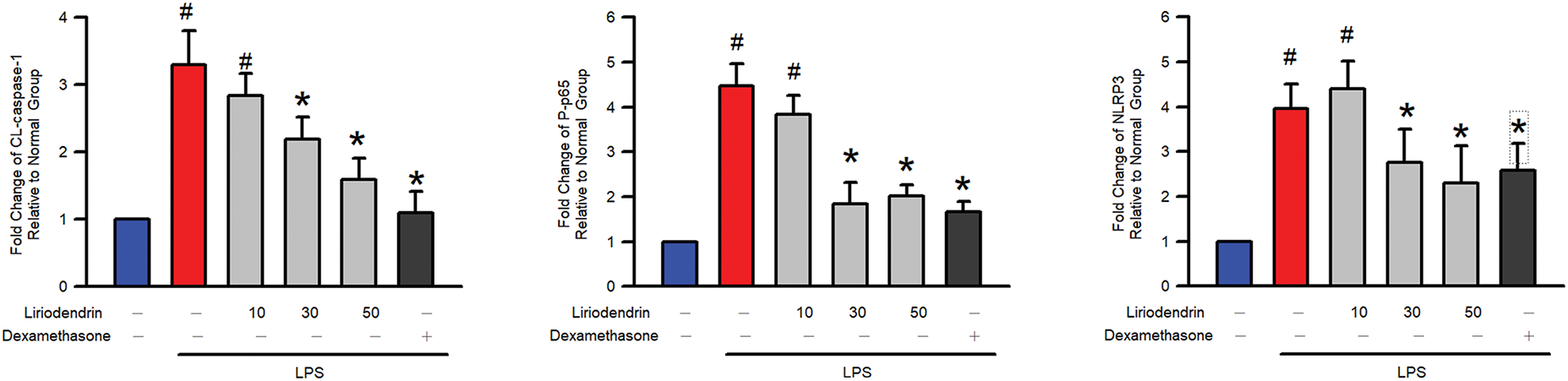
Figure 8: Liriodendrin downregulated the NO generation, proinflammatory cytokines expression, phosphorylation of NF-κB p65, and expression of NLRP3, ASC, and cleaved caspase-1 in LPS-stimulated macrophage activation. (A) The level of NO was evaluated using the Griess reaction assay in each treatment. (B) The production of proinflammatory cytokines, including IL-1β, IL-6, and TNFα, was evaluated using the ELISA assay in each treatment. (C) The phosphorylation of NF-κB p65 and expression of NLRP3, ASC, and cleaved caspase-1 were evaluated using the Western blotting assay in each treatment. DMSO, liriodendrin (10, 30, or 50 μM), or dexamethasone (1 mg/L) was treated at specified concentrations 30 min prior to the administration of saline or LPS (500 ng/mL). All quantitative data were presented as mean ± SD and derived from experiments performed on groups with four replicates (n = 4). #p < 0.05 significantly differed from the normal group, as shown in blue bars. *p < 0.05 significantly differed from the LPS group, as shown in red bars. NO: nitric oxide; IL: interleukin; TNF: tumor necrosis factor; NLRP3: NOD-like receptor family pyrin domain-containing 3; ASC: apoptosis-associated speck-like protein containing a CARD; CL-csapase-1: cleaved caspase-1
ARDS is a severe pathological condition characterized by inflammation, alveolar epithelial damage, and increased capillary permeability, leading to compromised gas exchange. This critical condition manifests as severe hypoxemia and bilateral pulmonary leukocyte infiltration [2,4,5]. It may result from indirect injuries related to systemic conditions, such as sepsis induced by CLP or IgG-IC [18,19]. Direct injuries such as bacterial pneumonia can affect lung tissues, eliciting a strong local inflammatory response [25,26]. To simulate this pathology in experimental models, LPS is typically intratracheally administered to directly induce ARDS [27,28]. This technique mimics bacterial lung infections and triggers a robust local immune response, leading to acute inflammation, alveolar-capillary barrier disruption, and extensive edema. These pathological features are similar to the clinical presentation and progression of ARDS in humans [2,27,28]. In previous study, it has discovered that 20 mg/kg liriodendrin reduced vascular permeability and neutrophil infiltration in the lung tissues of mice with CLP-induced septic ALIs [19]. We further discovered that liriodendrin significantly mitigated the degree of pulmonary edema in mice with LPS-induced ARDS and reduced vascular permeability and leukocyte infiltration. These ameliorative ARDS effects of liriodendrin were found to be dose-dependent. Taken together, these findings indicate that liriodendrin has the potential to ameliorate the pathological features of ARDS in mice treated with LPS.
Leukocyte infiltration and activation are key drivers of ARDS pathogenesis, primarily because of their disruptive effects on the alveolar-capillary barrier [4,29,30]. After LPS stimulation, adhesion molecules such as ICAM-1 and VCAM-1 are significantly upregulated and play a pivotal role in the pathogenesis of ARDS by recruiting leukocytes [5,31,32]. This upregulation facilitates the rolling, adhesion, and migration of leukocytes to the pulmonary endothelium, which further exacerbates the condition [4,33–35]. Both the accumulation and infiltration of leukocytes into the pulmonary parenchyma play a key role in tissue edema and injury by releasing proinflammatory mediators such as iNOS and COX2 and proinflammatory cytokines such as IL-6 and TNF-α [2,5,35]. In the uterine tissues of rats with chronic endometritis (CE), liriodendrin suppresses the expression of iNOS. In addition, pretreatment with liriodendrin substantially downregulates the expression of proinflammatory cytokines such as IL-1β, IL-6, and TNF-α in the lung tissues of mice with IgG-IC or sepsis-induced ALIs, the uterine tissues of mice with CE, the liver tissues of mice with hepatic ischemia-reperfusion injury, the intestinal tissues of mice with radiation enteritis, the colon tissues of mice with colitis induced by dextran sulfate sodium (DSS), and the myocardial tissues of mice with myocardial infarction, as well as LPS-induced RAW 264.7 macrophages [15,36,37]. In the present study, we discovered that liriodendrin inhibits the expression of iNOS and the secretion of proinflammatory cytokines in mice with LPS-induced ARDS. We also discovered that liriodendrin reduces the expression of COX2 and adhesion molecules such as ICAM-1 and VCAM-1. Taken together, these findings indicate that liriodendrin has the potential to alleviate the symptoms of ARDS by targeting adhesion molecules, proinflammatory mediators, and proinflammatory cytokines.
In the molecular pathogenesis of ARDS, pyroptosis contributes to lung damage by enhancing leukocyte recruitment and perpetuating inflammatory cycles [9,10]. Pyroptosis is an inflammatory form of cell death mediated by the formation of NLRP3 inflammasomes [38,39]. NLRP3 functions as a sensor that detects various danger signals through its leucine-rich repeat domain and initiates oligomerization through its nucleotide-binding domain. This structural transformation is essential because it facilitates the recruitment and activation of the adaptor protein ASC and caspase-1. This recruitment of ASC through pyrin-pyrin domain interactions forms a platform for pro-caspase-1 engagement. Once assembled, pro-caspase-1 undergoes autocleavage into its active form, which subsequently processes proinflammatory cytokines such as IL-1β. In the pathogenesis of ARDS, LPS can induce the formation of NLRP3 inflammasomes, leading to the production of IL-1β in the lungs [9,10,40]. According to the literature, Sargentodoxa cuneata extracts contain liriodendrin and downregulate the expression of IL-1β and NLRP3 in the colons of mice with DSS-induced ulcerative colitis [13]. To the best of our knowledge, the present study was the first to examine the protective role of liriodendrin against lung damage in mouse models of LPS-induced ARDS through the reduction of NLRP3 inflammasome formation. We discovered that liriodendrin considerably reduced the production of IL-1β and the expression of NLRP3 and ASC in a murine model of LPS-induced ARDS. Taken together, our findings indicate that liriodendrin has the potential to ameliorate the proinflammatory features associated with ARDS by inhibiting the assembly of NLRP3 inflammasomes and the subsequent pyroptotic response.
In the pathogenesis of ARDS inflammation, the priming phase starts with the activation of NF-κB through molecular signaling cascades after the activation of Toll-like receptors induced by LPS [41,42]. Upon exposure to LPS, IκB kinase phosphorylates IκB proteins, targeting them for degradation and thus enabling the NF-κB complex to translocate into the nucleus and bind to DNA at κB sites. NF-κB activation and phosphorylation play a key role in initiating the transcription of various proinflammatory genes associated with proinflammatory mediators, adhesion molecules, cytokines, NLRP3, and pro-IL-1β [9,10,43,44]. In the colon tissues of mice with DSS-induced colitis, pretreatment with liriodendrin significantly inhibits the phosphorylation of IκB and NF-κB [15]. Similarly, in the myocardial tissues of mice with myocardial infarction, pretreatment with liriodendrin significantly inhibits LPS-induced RAW 264.7 macrophages. Liriodendrin also significantly inhibits the phosphorylation of NF-κB in the uterine tissues of mice with CE, the intestinal tissues of mice with radiation enteritis, and the lung tissues of mice with IgG-IC or sepsis-induced ALI [17–19,30,37]. Similarly, liriodendrin significantly inhibits the expression of NF-κB in the liver tissues of mice with hepatic ischemia-reperfusion injury and effectively inhibits the phosphorylation of NF-κB in the lung tissues of mice with LPS-induced ARDS [45]. We’ve also found that liriodendrin can effectively inhibit the phosphorylation of NF-κB in the lungs of LPS-induced ARDS mice. Moreover, liriodendrin effectively mitigates the degradation of IκB. Taken together, these findings indicate that liriodendrin hinders the formation of NLRP3 inflammasomes and inhibits the associated pyroptotic pathway through NF-κB pathway activation, including NF-κB phosphorylation and IκB degradation in mice with LPS-induced ARDS.
In this study, the activation of NF-κB was influenced by specific upstream kinases, including Akt and the MAPK family, in LPS-induced ARDS [46–49]. In LPS-induced ARDS, abnormal activation of AKT and MAPK signaling pathways occurs in key effector cells, including alveolar epithelial cells, macrophages, and endothelial cells, driving inflammation and lung injury. Alveolar epithelial cells are essential for maintaining the integrity of the alveolar-capillary barrier and for surfactant production. When stimulated by LPS, these cells activate the AKT and MAPK pathways, leading to apoptosis and a compromised barrier function. Macrophages respond to LPS by releasing pro-inflammatory cytokines. The activation of the AKT and MAPK pathways further amplifies this inflammatory response, exacerbating lung injury. In endothelial cells, similar activation increases vascular permeability and promotes leukocyte adhesion, contributing to pulmonary edema and systemic inflammation. This evidence also discovered that the phosphorylation of the MAPK family, which includes p38 MAPK, JNK, ERK, and Akt, indicated the activation of these proteins [47,50,51]. In the colon tissues of mice with DSS-induced colitis, pretreatment with liriodendrin significantly inhibited the phosphorylation of Akt and LPS-induced RAW 264.7 macrophages [15]. In the lung tissues of mice with IgG-IC-induced ALIs, liriodendrin significantly inhibited the phosphorylation of p38 MAPK and JNK [18]. We also discovered that liriodendrin inhibited the phosphorylation of Akt, p38 MAPK, and JNK in the lung tissues of mice with LPS-induced ARDS. Taken together, these findings indicate that liriodendrin hinders NF-κB pathway activation through the activation of NF-κB, including the phosphorylation of Akt, p38 MAPK, and JNK in mice with LPS-induced ARDS.
The suppression of NO and pro-inflammatory cytokines indicates a potential reduction in lung damage associated with oxidative stress, vascular permeability, and leukocyte recruitment, which are essential factors in the progression of ARDS. Additionally, the downregulation of NF-κB activity suggests that liriodendrin interferes with critical transcriptional pathways that enhance inflammatory responses. Moreover, the inhibition of NLRP3 inflammasome activation illustrates the capacity of liriodendrin to prevent pyroptotic cell death, effectively disrupting the cycle of inflammation and tissue damage. These findings derived from macrophage models, in conjunction with results from animal studies, provide valuable molecular insights and point to the significant therapeutic potential of liriodendrin for ARDS treatment.
Dexamethasone demonstrated effective anti-inflammatory properties in the study, showing results comparable to 50 µM Liriodendrin in reducing LPS-induced lung injury. However, its long-term use is limited due to significant side effects, including immunosuppression, hyperglycemia, and osteoporosis, particularly in patients requiring ongoing treatment for ARDS. In contrast, Liriodendrin, a natural plant compound, exhibits similar efficacy with potentially fewer side effects, making it a promising and safer alternative for treating ARDS. Further research is essential to confirm its safety and effectiveness.
This study has several limitations. First, while the LPS-induced murine ARDS model replicates key features of ARDS, it cannot fully capture the complexity of human cases involving various causes and patient-specific factors. Second, the pharmacokinetics, bioavailability, and metabolic pathways of liriodendrin remain unexamined, limiting clinical application. Additionally, the research focused mainly on anti-inflammatory and pyroptosis-modulating effects, neglecting other potentially relevant mechanisms such as oxidative stress regulation and tissue repair. Finally, the evaluation was restricted to short-term effects, leaving long-term safety and efficacy unaddressed. Future studies should address these limitations by including pharmacokinetic analyses, evaluating long-term outcomes, and exploring additional molecular mechanisms. Clinical trials are essential to validate the potential of liriodendrin as a therapeutic option for ARDS and related inflammatory conditions.
In conclusion, liriodendrin ameliorates the pathological features of mice with LPS-induced ARDS, including leukocyte infiltration, lung edema, and alveolar–capillary membrane dysfunction. Liriodendrin also mitigates proinflammatory responses, including the expression of adhesion molecules including ICAM-1 and VCAM-1, expression of iNOS and COX2, and production of proinflammatory cytokines in mice with ARDS. In the lung tissues of mice with LPS-induced ARDS, liriodendrin plays a protective role by downregulating the expression of NLRP3 and ASC; degrading IκB; and phosphorylation of NF-κB through the phosphorylation of p38 MAPK, JNK, and Akt. In a dose-dependent manner, liriodendrin ameliorates the pathological features of mice with LPS-induced ARDS and plays a protective role for them. Taken together, these findings indicate that liriodendrin prevents the development of ALIs associated with LPS-induced ARDS by ameliorating the pathological features and proinflammatory responses associated with NLRP3 formation through NF-κB pathway activation and its upstream factor containing p38 MAPK, JNK, and Akt phosphorylation (Fig. 9). These findings demonstrate the dose-dependent protective effects of liriodendrin in the LPS-induced ARDS model and suggest its potential as a therapeutic candidate for other inflammation-related pulmonary and systemic disorders.
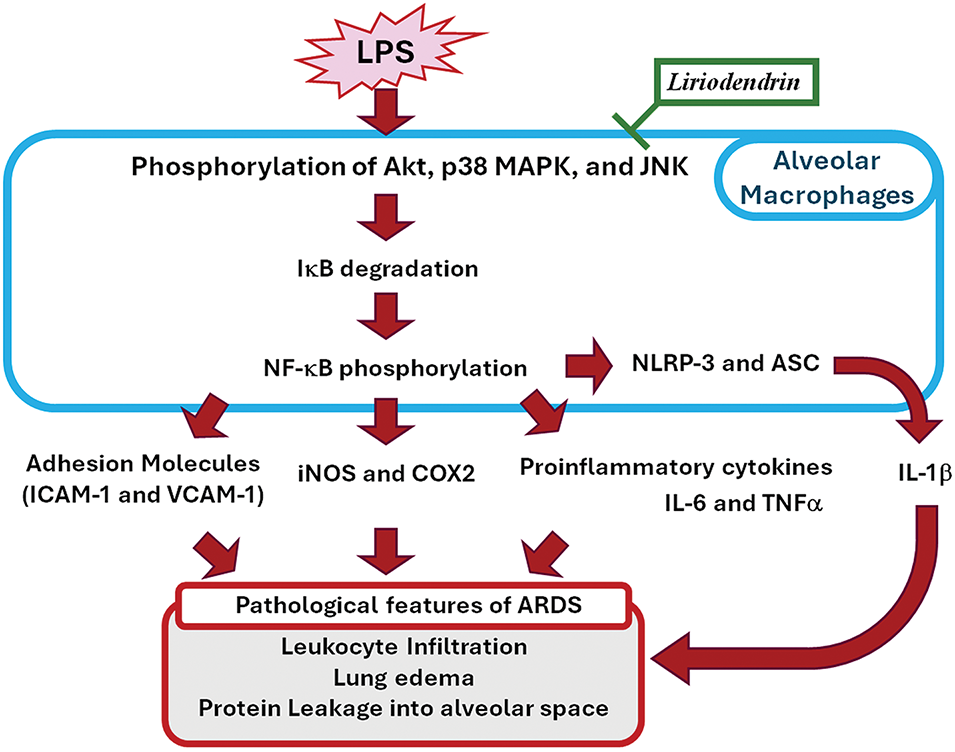
Figure 9: Schemes of the mechanism of the ameliorated effects of liriodendrin against NLRP3-Mediated pyroptosis and proinflammatory pathways in LPS-induced ARDS
Acknowledgement: None.
Funding Statement: This study was supported by Chung Shan Medical University and Changhua Christian Hospital (CSMU-CCH-111-08). This study was supported by research grants from the Chung Shan Medical University Hospital, Taichung, Taiwan (CSH-2023-C-023). The authors would like to thank the National Science and Technology Council, Taiwan, for financially supporting this research under Contract Nos. NSTC 112-2320-B-040-017, NSTC112-2314-B-040-009, and NSTC 112-2320-B-040-011.
Author Contributions: The authors confirm contribution to the paper as follows: Study conception and design: Kuo-Yang Huang, Yu-Hsiang Kuan; Investigation and methodology: Kuo-Yang Huang, Sheng-Chien Lin, Chun-Hung Su, Sheng-Wen Wu, Ching-Chi Tseng, Wei-Chin Hung, Shih-Pin Chen; Visualization; Kuo-Yang Huang, Sheng-Chien Lin, Chun-Hung Su; writing—original draft: Kuo-Yang Huang, Sheng-Wen Wu; Funding acquisition: Kuo-Yang Huang, Yu-Hsiang Kuan; Shih-Pin Chen; Data curation and formal analysis: Sheng-Chien Lin, Chun-Hung Su, Shih-Pin Chen; writing—review and editing: Yu-Hsiang Kuan, Shih-Pin Chen. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics Approval: All animal studies were approved by the Institutional Animal Ethics Committee of Chung Shan Medical University (IACUC no. 112006).
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers. 2019;5(1):18. doi:10.1038/s41572-019-0069-0. [Google Scholar] [PubMed] [CrossRef]
2. Matthay MA, Arabi Y, Arroliga AC, Bernard G, Bersten AD, Brochard LJ, et al. A new global definition of acute respiratory distress syndrome. Am J Respir Crit Care Med. 2024;209(1):37–47. doi:10.1164/rccm.202303-0558WS. [Google Scholar] [PubMed] [CrossRef]
3. Palakshappa JA, Krall JTW, Belfield LT, Files DC. Long-term outcomes in acute respiratory distress syndrome: epidemiology, mechanisms, and patient evaluation. Crit Care Clin. 2021;37(4):895–911. doi:10.1016/j.ccc.2021.05.010. [Google Scholar] [PubMed] [CrossRef]
4. Rizzo AN, Aggarwal NR, Thompson BT, Schmidt EP. Advancing precision medicine for the diagnosis and treatment of acute respiratory distress syndrome. J Clin Med. 2023;12(4):1563. doi:10.3390/jcm12041563. [Google Scholar] [PubMed] [CrossRef]
5. Wu W, He Y, Lin D, Zhang G, Zhang X, Zhang N. Dexmedetomidine mitigates lipopolysaccharide-induced acute lung injury by modulating heat shock protein A12B to inhibit the toll-like receptor 4/nuclear factor-kappa B signaling pathway. Chem Biol Interact. 2024;398:111112. doi:10.1016/j.cbi.2024.111112. [Google Scholar] [PubMed] [CrossRef]
6. Lee CY, Chen SP, Huang-Liu R, Gau SY, Li YC, Chen CJ, et al. Fucoxanthin decreases lipopolysaccharide-induced acute lung injury through the inhibition of RhoA activation and the NF-κB pathway. Environ Toxicol. 2022;37(9):2214–22. doi:10.1002/tox.23587. [Google Scholar] [PubMed] [CrossRef]
7. Ng YY, Ho YC, Yen CH, Lee SS, Tseng CC, Wu SW et al. Protective effect of hibifolin on lipopolysaccharide-induced acute lung injury through akt phosphorylation and NFκB pathway. Environ Toxicol. 2024. doi: 10.1002/tox.24383. [Google Scholar] [PubMed] [CrossRef]
8. Li W, Li D, Chen Y, Abudou H, Wang H, Cai J et al. Classic signaling pathways in alveolar injury and repair involved in sepsis-induced ALI/ARDS: new research progress and prospect. Dis Markers. 2022;2022:6362344. doi:10.1155/2022/6362344. [Google Scholar] [PubMed] [CrossRef]
9. Gu W, Zeng Q, Wang X, Jasem H, Ma L. Acute lung injury and the NLRP3 inflammasome. J Inflamm Res. 2024;17:3801–13. doi:10.2147/JIR.S464838. [Google Scholar] [PubMed] [CrossRef]
10. Zhou S, Yang X, Mo K, Ning Z. Pyroptosis and polarization of macrophages in septic acute lung injury induced by lipopolysaccharide in mice. Immun Inflamm Dis. 2024;12(3):e1197. doi:10.1002/iid3.1197. [Google Scholar] [PubMed] [CrossRef]
11. Li DH, Wang Y, Lv YS, Liu JH, Yang L, Zhang SK et al. Preparative purification of liriodendrin from sargentodoxa cuneata by macroporous resin. Biomed Res Int. 2015;2015:861256. doi:10.1155/2015/861256. [Google Scholar] [PubMed] [CrossRef]
12. Sohn YA, Hwang SA, Lee SY, Hwang IY, Kim SW, Kim SY, et al. Protective effect of liriodendrin isolated from kalopanax pictus against gastric injury. Biomol Ther. 2015;23(1):53–9. doi:10.4062/biomolther.2014.103. [Google Scholar] [PubMed] [CrossRef]
13. Yu P, Xu F, Wu H, Wang X, Ding Q, Zhang M, et al. Anti-ulcerative colitis effects and active ingredients in ethyl acetate extract from decoction of Sargentodoxa cuneata. Molecules. 2023;28(22):7663. doi:10.3390/molecules28227663. [Google Scholar] [PubMed] [CrossRef]
14. Zhang W, Sun CP, Peng YL, Zhao WY, Wang ZY, Ning J, et al. Isolation and identification of two new sargentodoxosides from Sargentodoxa cuneata and their agonistic effects against FXR. Nat Prod Res. 2022;36(14):3665–72. doi:10.1080/14786419.2021. [Google Scholar] [CrossRef]
15. Zhang Z, Yang L, Wang B, Zhang L, Zhang Q, Li D, et al. Protective role of liriodendrin in mice with dextran sulphate sodium-induced ulcerative colitis. Int Immunopharmacol. 2017;52:203–10. doi:10.1016/j.intimp.2017.09.012. [Google Scholar] [PubMed] [CrossRef]
16. Lee HY, Park YM, Hwang HM, Shin DY, Jeong HN, Kim JG, et al. The effect of the mixed extract of Kalopanax pictus Nakai and Achyranthes japonica Nakai on the improvement of degenerative osteoarthritis through inflammation inhibition in the monosodium iodoacetate-induced mouse model. Curr Issues Mol Biol. 2023;45(8):6395–414. doi:10.3390/cimb45080404. [Google Scholar] [PubMed] [CrossRef]
17. Cheng F, Li D, Ma X, Wang Y, Lu L, Hu B, et al. Liriodendrin exerts protective effects against chronic endometritis in rats by modulating gut microbiota composition and the arginine/nitric oxide metabolic pathway. Int Immunopharmacol. 2024;126:111235. doi:10.1016/j.intimp.2023.111235. [Google Scholar] [PubMed] [CrossRef]
18. Zhang S, Hu D, Zhuo Y, Cui L, Li D, Zhang L, et al. Protective effect of liriodendrin on IgG immune complex-induced acute lung injury via inhibiting SRC/STAT3/MAPK signaling pathway: a network pharmacology research. Naunyn Schmiedebergs Arch Pharmacol. 2023;396(11):3269–83. doi:10.1007/s00210-023-02534-1. [Google Scholar] [PubMed] [CrossRef]
19. Yang L, Li D, Zhuo Y, Zhang S, Wang X, Gao H. Protective role of liriodendrin in sepsis-induced acute lung injury. Inflammation. 2016;39(5):1805–13. doi:10.1007/s10753-016-0416-1. [Google Scholar] [PubMed] [CrossRef]
20. Ni YL, Shen HT, Ng YY, Chen SP, Lee SS, Tseng CC, et al. Hibifolin protected pro-inflammatory response and oxidative stress in LPS-induced acute lung injury through antioxidative enzymes and the AMPK2/Nrf-2 pathway. Environ Toxicol. 2024;39(7):3799–807. doi:10.1002/tox.24233. [Google Scholar] [PubMed] [CrossRef]
21. Zhao X, Mago E, Weng D. Role of RIPK1 in the pathogenesis of acute respiratory distress syndrome. BioCell. 2023;47(10):2151–62. doi:10.32604/biocell.2023.030570. [Google Scholar] [CrossRef]
22. Lin FC, Lee SS, Li YC, Ho YC, Chen WY, Chen CJ, et al. Protective effects of Kirenol against lipopolysaccharide-induced acute lung injury through the modulation of the proinflammatory NFκB pathway and the AMPK2-/Nrf2-mediated HO-1/AOE pathway. Antioxidants. 2021;10(2):204. doi:10.3390/antiox10020204. [Google Scholar] [PubMed] [CrossRef]
23. Lu YC, Chiang CY, Chen SP, Hsu YW, Chen WY, Chen CJ, et al. Chlorpyrifos-induced suppression of the antioxidative defense system leads to cytotoxicity and genotoxicity in macrophages. Environ Toxicol Pharmacol. 2024;108:104468. doi:10.1016/j.etap.2024.104468. [Google Scholar] [PubMed] [CrossRef]
24. Lee CY, Wu SW, Yang JJ, Chen WY, Chen CJ, Chen HH, et al. Vascular endothelial dysfunction induced by 3-bromofluoranthene via MAPK-mediated-NFκB pro-inflammatory pathway and intracellular ROS generation. Arch Toxicol. 2024;98(7):2247–59. doi:10.1007/s00204-024-03751-0. [Google Scholar] [PubMed] [CrossRef]
25. Long ME, Mallampalli RK, Horowitz JC. Pathogenesis of pneumonia and acute lung injury. Clin Sci. 2022;136(10):747–69. doi:10.1042/CS20210879. [Google Scholar] [PubMed] [CrossRef]
26. Lee KY. Pneumonia, acute respiratory distress syndrome, and early immune-modulator therapy. Int J Mol Sci. 2017;18(2):388. doi:10.3390/ijms18020388. [Google Scholar] [PubMed] [CrossRef]
27. Bastarache JA, Blackwell TS. Development of animal models for the acute respiratory distress syndrome. Dis Model Mech. 2009;2(5–6):218–23. doi:10.1242/dmm.001677. [Google Scholar] [PubMed] [CrossRef]
28. Ballard-Croft C, Wang D, Sumpter LR, Zhou X, Zwischenberger JB. Large-animal models of acute respiratory distress syndrome. Ann Thorac Surg. 2012;93(4):1331–9. doi:10.1016/j.athoracsur.2011.06.107. [Google Scholar] [PubMed] [CrossRef]
29. Wong JJM, Leong JY, Lee JH, Albani S, Yeo JG. Insights into the immuno-pathogenesis of acute respiratory distress syndrome. Ann Transl Med. 2019;7(19):504. doi:10.21037/atm.2019.09.28. [Google Scholar] [PubMed] [CrossRef]
30. Rebetz J, Semple JW, Kapur R. The pathogenic involvement of neutrophils in acute respiratory distress syndrome and transfusion-related acute lung injury. Transfus Med Hemother. 2018;45(5):290–8. doi:10.1159/000492950. [Google Scholar] [PubMed] [CrossRef]
31. Ren J, Deng G, Li R, Jin X, Liu J, Li J, et al. Possible pharmacological targets and mechanisms of sivelestat in protecting acute lung injury. Comput Biol Med. 2024;170:108080. doi:10.1016/j.compbiomed.2024.108080. [Google Scholar] [PubMed] [CrossRef]
32. Kim J, Baalachandran R, Li Y, Zhang CO, Ke Y, Karki P, et al. Circulating extracellular histones exacerbate acute lung injury by augmenting pulmonary endothelial dysfunction via TLR4-dependent mechanism. Am J Physiol Lung Cell Mol Physiol. 2022;323(3):L223–39. doi:10.1152/ajplung.00072.2022. [Google Scholar] [PubMed] [CrossRef]
33. Grönloh MLB, Arts JJG, Van Buul JD. Neutrophil transendothelial migration hotspots—mechanisms and implications. J Cell Sci. 2021;134(7):jcs255653. doi:10.1242/jcs.255653. [Google Scholar] [PubMed] [CrossRef]
34. Su Y, Lucas R, Fulton DJR, Verin AD. Mechanisms of pulmonary endothelial barrier dysfunction in acute lung injury and acute respiratory distress syndrome. Chin Med J Pulm Crit Care Med. 2024;2(2):80–7. doi:10.1016/j.pccm.2024.04.002. [Google Scholar] [PubMed] [CrossRef]
35. Bhatia M, Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol. 2004;202(2):145–56. doi:10.1002/path.1491. [Google Scholar] [PubMed] [CrossRef]
36. Li B, Yao BC, Chen QL, Song YQ, Jiang N, Zhao LL, et al. The protective role and mechanism of liriodendrin in rats with myocardial infarction. J Thorac Dis. 2022;14(1):135–46. doi:10.21037/jtd-21-1998. [Google Scholar] [PubMed] [CrossRef]
37. Li J, Zheng X, Li X, Yang J, Liu W, Yang L, et al. Study on the protective effect and mechanism of Liriodendrin on radiation enteritis in mice. J Radiat Res. 2022;63(2):213–20. doi:10.1093/jrr/rrab128. [Google Scholar] [PubMed] [CrossRef]
38. Huang Y, Xu W, Zhou R. NLRP3 inflammasome activation and cell death. Cell Mol Immunol. 2021;18(9):2114–27. doi:10.1038/s41423-021-00740-6. [Google Scholar] [PubMed] [CrossRef]
39. Wu Y, Zhang J, Yu S, Li Y, Zhu J, Zhang K, et al. Cell pyroptosis in health and inflammatory diseases. Cell Death Discov. 2022;8(1):191. doi:10.1038/s41420-022-00998-3. [Google Scholar] [PubMed] [CrossRef]
40. Wei T, Zhang C, Song Y. Molecular mechanisms and roles of pyroptosis in acute lung injury. Chin Med J. 2022;135(20):2417–26. doi:10.1097/CM9.0000000000002425. [Google Scholar] [PubMed] [CrossRef]
41. Anwar HM, Salem GEM, El-Latief AHM, Osman AAE, Ghanem SK, Khan H et al. Therapeutic potential of proteases in acute lung injury and respiratory distress syndrome via TLR4/Nrf2/NF-kB signaling modulation. Int J Biol Macromol. 2024;267:131153. doi:10.1016/j.ijbiomac.2024.131153. [Google Scholar] [PubMed] [CrossRef]
42. Lee SA, Lee SH, Kim JY, Lee WS. Effects of glycyrrhizin on lipopolysaccharide-induced acute lung injury in a mouse model. J Thorac Dis. 2019;11(4):1287–302. doi:10.21037/jtd.2019.04.14. [Google Scholar] [PubMed] [CrossRef]
43. Shan M, Wan H, Ran L, Ye J, Xie W, Lu J, et al. Dynasore alleviates LPS-induced acute lung injury by inhibiting NLRP3 inflammasome-mediated pyroptosis. Drug Des Devel Ther. 2024;18:1369–84. doi:10.2147/DDDT.S444408. [Google Scholar] [PubMed] [CrossRef]
44. Jin H, Zhao Y, Yao Y, Zhao J, Luo R, Fan S, et al. Therapeutic effects of tea polyphenol-loaded nanoparticles coated with platelet membranes on LPS-induced lung injury. Biomater Sci. 2023;11(18):6223–35. doi:10.1039/D3BM00802A. [Google Scholar] [PubMed] [CrossRef]
45. Yu ZY, Cheng G. Protective effect of liriodendrin against liver ischaemia/reperfusion injury in mice via modulating oxidative stress, inflammation and nuclear factor kappa B/toll-like receptor 4 pathway. Folia Morphol. 2023;82(3):668–76. doi:10.5603/FM.a2022.0049. [Google Scholar] [PubMed] [CrossRef]
46. Chen J, Ding W, Zhang Z, Li Q, Wang M, Feng J, et al. Shenfu injection targets the PI3K-AKT pathway to regulate autophagy and apoptosis in acute respiratory distress syndrome caused by sepsis. Phytomedicine. 2024;129:155627. doi:10.1016/j.phymed.2024.155627. [Google Scholar] [PubMed] [CrossRef]
47. Yan C, Chen J, Wang B, Wang J, Luo M, Tong J, et al. PD-L1 expression is increased in LPS-induced acute respiratory distress syndrome by PI3K-AKT-Egr-1/C/EBPδ signaling pathway. Inflammation. 2024;47(4):1459–78. doi:10.1007/s10753-024-01988-6. [Google Scholar] [PubMed] [CrossRef]
48. Zhou M, Meng L, He Q, Ren C, Li C. Valsartan attenuates LPS-induced ALI by modulating NF-κB and MAPK pathways. Front Pharmacol. 2024;15:1321095. doi:10.3389/fphar.2024.1321095. [Google Scholar] [PubMed] [CrossRef]
49. Jangam A, Tirunavalli SK, Adimoolam BM, Kasireddy B, Patnaik SS, Erukkambattu J, et al. Anti-inflammatory and antioxidant activities of Gymnema Sylvestre extract rescue acute respiratory distress syndrome in rats via modulating the NF-κB/MAPK pathway. Inflammopharmacology. 2023;31(2):823–44. doi:10.1007/s10787-022-01133-5. [Google Scholar] [PubMed] [CrossRef]
50. Jung F, Liu JS, Yang SH, Tseng HY, Chou SP, Lin JC, et al. FJU-C28 inhibits the endotoxin-induced pro-inflammatory cytokines expression via suppressing JNK, p38 MAPK and NF-κB signaling pathways. Pharmacol Res Perspect. 2021;9(6):e00876. doi:10.1002/prp2.876. [Google Scholar] [PubMed] [CrossRef]
51. Huang X, Zeng Y, Jiang Y, Qin Y, Luo W, Xiang S et al. Lipopolysaccharide-binding protein downregulates fractalkine through activation of p38 MAPK and NF-κB. Mediators Inflamm. 2017;2017:9734837. doi:10.1155/2017/9734837. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools