
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
COMMENTARY

A Commentary on RIPK2: The Hidden Driver of MHC-I Related Immune Suppression in Cancer
Faculty of Medicine, Department of Experimental Medicine 1, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, 91054, Germany
* Corresponding Author: Lydia Meder. Email:
(This article belongs to the Special Issue: Genetic Biomarkers of Cancer: Insights into Molecular and Cellular Mechanisms)
BIOCELL 2025, 49(2), 161-165. https://doi.org/10.32604/biocell.2025.060227
Received 27 October 2024; Accepted 06 January 2025; Issue published 28 February 2025
This article is a commentary on:
Receptor-interacting Protein Kinase 2 is an immunotherapy target in pancreatic cancer
Read original article
 View Full Text
View Full Text Download PDF
Download PDFAbstract
This article has no abstract.Keywords
1 MHC-I Loss in the Immune Evasion of Cancer Cells
Pancreatic ductal adenocarcinoma (PDAC) is a lethal cancer with a poor prognosis due to its aggressive nature and late detection. Recently, new discoveries in PDAC demonstrated receptor-interacting protein kinase 2 (RIPK2) triggering immune evasion. Mechanistically, RIPK2 drives the desmoplastic tumor microenvironment (TME) and restricts the activation and density of tumor-infiltrating effector T cells by impairing the expression of major histocompatibility complex class I (MHC-I) [1]. This process might be relevant in different solid cancer entities as illustrated by analyzing publicly available databases.
MHC-I plays a crucial role in immune recognition, particularly in activating CD8+ T cells and the immune system’s ability to identify and eliminate infected or abnormal cells. In melanoma, truncating mutations in Janus kinase 1 (JAK1) or Janus kinase 2 (JAK2) causes a lack of responsiveness to interferon-γ, rendering cancer cells insensitive to its anti-proliferative and immune-stimulatory effects, and beta-2-microglobulin (B2M) truncating mutations results in the loss of surface expression of MHC-I [2]. In PDAC, a decrease in MHC-I expression is often observed. Still, mutations leading to the loss of MHC-I are very rare, indicating that MHC-I expression may have other regulatory mechanisms [3].
In lung cancer, primary tumors acquire immune checkpoint blockade resistance by MHC-I loss but show a simultaneous increase in programmed-death receptor ligand-1 (PD-L1) [4] already suggesting an interferon-independent mechanism. Nguyen et al. demonstrated that selective inhibition of lysine-specific demethylase 1 (LSD1) can restore MHC-I surface expression and activate the antigen presentation pathway epigenetically [5]. However, in PDAC, Sang and colleagues explored a novel mechanism in cancer cells regulating MHC-I protein stability dependent on RIPK2 [1].
2 RIPK2 and the Autophagolysosomal Degradation of MHC-I
RIPK2 is a downstream adaptor kinase of the nucleotide-binding oligomerization domain containing 1 and 2 (NOD1/2) signaling pathway and is initially identified as a key player in bacterial sensing and intestinal immunity. NOD1/2 recruits RIPK2 and then induces RIPK2 phosphorylation, which promotes RIPK2 ubiquitination by E3 ubiquitin ligases [6]. A previous study on Crohn’s disease showed that RIPK2 is essential in the NOD2-mediated autophagy pathway, and RIPK2 may directly interact with proteins responsible for regulating autophagosome formation [6]. In the context of cancer, RIPK2 has been shown to participate in cell growth, migration, and invasion in pancreatic cancer by promoting phosphorylation in the nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells (NF-κB), c-Jun N-terminal kinases (JNK) and extracellular signal-regulated kinase (ERK) pathway. Moreover, high expression of RIPK2 predicts poor prognosis in pancreatic cancer patients [7]. In their recent work, Sang and colleagues [1] furthermore implicated RIPK2 in the context of immunosuppression in the TME, by showing that RIPK2 ablation in PDAC promotes intra-tumoral CD8+ T-cell proliferation and augments the production of cytolytic granules and cytokines in tumor-infiltrating CD8+ T cells. They demonstrated that the knockdown of RIPK2 increases MHC-I expression in human PDAC cell lines by affecting MHC-I protein stability and increasing its degradation [1].
The autophagolysosomal degradation of MHC-I via selective autophagy receptor NBR1 in PDAC was discovered earlier [3]. However, the authors validated these prior findings with their functional screening approach, and advanced the understanding of the molecular mechanism behind this process by establishing RIPK2 as a critical link between NBR1 and MHC-I [1] (Fig. 1A). Yamamoto et al. tied the NBR1-dependent degradation of MHC-I to canonical selective macro-autophagy instead of non-canonical autophagy-related pathways LC3-Associated Phagocytosis (LAP) and LC3-associated endocytosis (LANDO) [3]. Herein, autophagy is initiated by the heavily regulated formation of the phagophore, a nascent reactive double membrane. During this process, microtubule-associated proteins 1A/1B light chain 3 (MAP1LC3), also known as LC3-I, are tethered to the phagophore by conjugation to phosphatidylethanolamine, forming LC3-II. NBR1 is a selective autophagy receptor (SAR) that associates with MHC-I in a RIPK2-dependent manner [1]. NBR1 recruits the phagophore by binding LC3-II. The phagophore then engulfs and closes around the LC3-II bound cargo-SAR-complex to form the autophagosome [3,8].
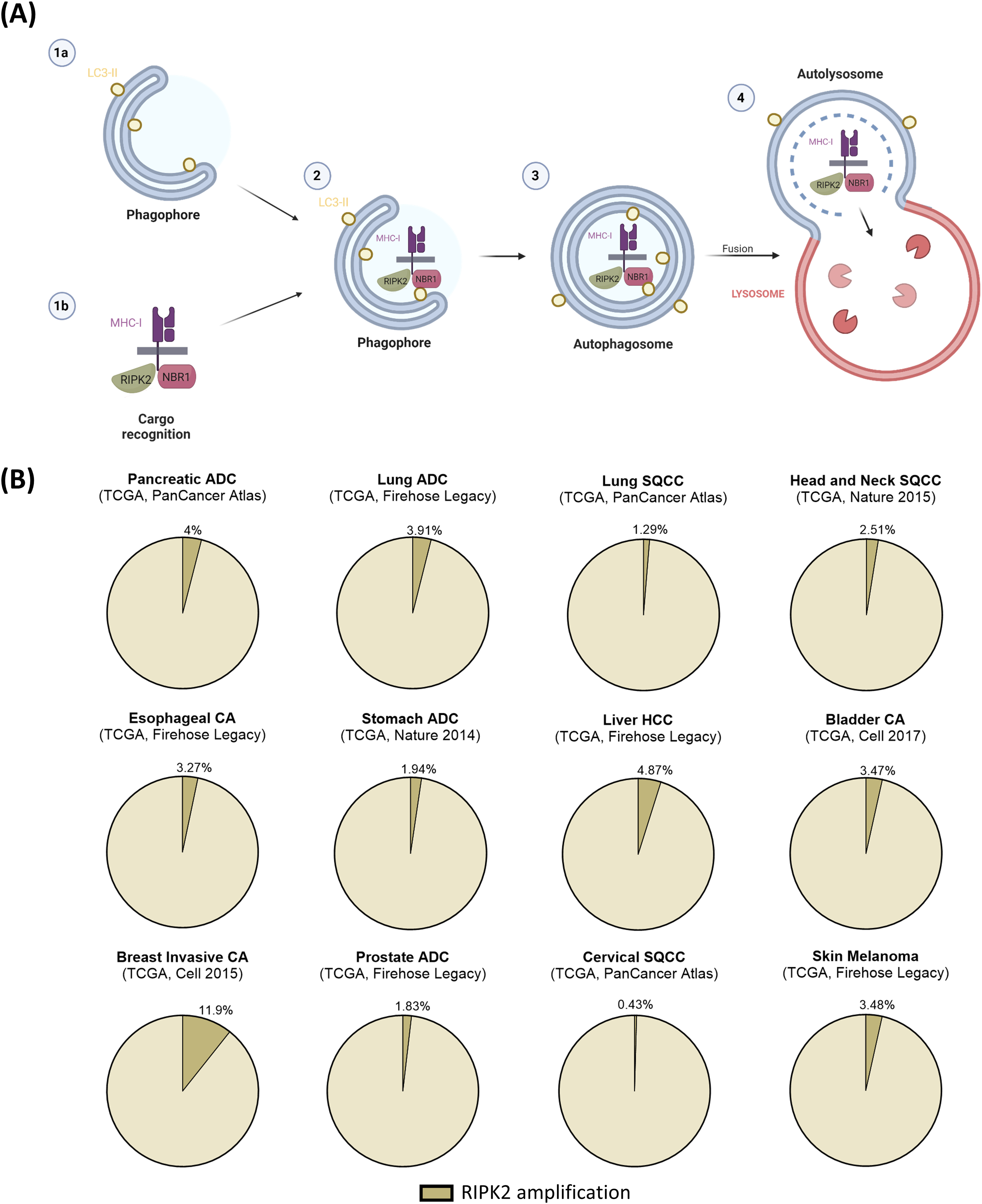
Figure 1: Potential RIPK2 function in immune evasion in solid cancers. (A) Scheme of RIPK2 function in the context of selective macroautophagy in cancer cells. LC3-II is anchored to the nascent phagophore (1a). At the same time, MHC-I is recognized as cargo for autophagosomal degradation by NBR1 in dependence on colocalization with RIPK2 (1b). The cargo complex is recruited to the phagophore by binding NBR1 to LC3-II (2) and subsequently engulfed forming the autophagosome (3). Lastly, fusion of the outer autophagosomal membrane with a lysosome forms the autolysosome, facilitating the degradation of first, the inner autophagosomal membrane, and second, the enclosed cargo by lysosomal enzymes (4). (B) Analysis of publically available data sets of The Cancer Genome Atlas (TCGA) obtained from cBioportal revealed RIPK2 amplification frequencies in different solid cancer entities. Amplifications are annotated as extracted from cBioportal. ADC—Adenocarcinoma, SQCC—Squamous cell carcinoma, HCC—Hepatocellular carcinoma, CA—carcinoma/cancer
Autophagy in cancer, especially in anti-tumor immunity, has been shown to possess a dual role. On the one hand, tumor cell autophagy can promote anti-tumor immunity by supplying tumor antigens to be cross-presented by antigen-presenting cells, thus enhancing CD8+ anti-tumor immunity. On the other hand, mechanisms like the degradation of MHC-I via autophagy, as described by both Yamamoto et al. [3] and Sang et al. [1] exist. Taken together, the role of autophagy is changing throughout tumor development. During initiation and early tumor stages, autophagy can assist tumor suppression but switches towards tumor maintenance in advanced stages [8].
3 Pharmacological Targeting of RIPK2
Considering these findings, it is not surprising that targeting autophagy unselectively for cancer treatment has been difficult, as autophagy is involved in an abundance of cellular processes and across cell types [8]. However, in this context, RIPK2 is a link in this process that, according to current knowledge, is not a direct part of the autophagy machinery itself, but rather selective for the recruitment of MHC-I to the autophagosome. In turn, targeting RIPK2 might impair one of the tumor-progressing functions of autophagy while leaving the rest of the autophagy machinery intact. Targeting RIPK2 as a stand-in for MHC-I autophagolysosomal degradation and combining it with immunotherapy as illustrated by Sang and colleagues, has the potential to enhance anti-tumor immunity while impairing the mechanism of acquired therapy resistance by reduction of tumor cell MHC-I.
While no RIPK2 inhibitors are currently developed to clinical stages, recent efforts of pharmacological development showed promising results in the preclinical field, focusing on cancer therapy and treatment of inflammatory diseases. Notable substances include Boehringer Ingelheim’s BI 706039 and Novartis’s GSK derivative 17, hopefully making pharmacological RIPK2 inhibition clinically feasible in the near future [9].
According to selected data sets of The Cancer Genome Atlas (TCGA) obtained from cBioportal [10], RIPK2 is amplified in various tumor entities, including pancreatic, lung, liver, breast, and other solid cancers (Fig. 1B), classifying RIPK2 a considerable target for cancer therapy.
Sang and colleagues’ [1] research sheds light on the complex protein dynamics of MHC-I in tumor immunology, specifically in PDAC. Our cBioportal analyses suggest that the RIPK2 mechanism to regulate MHC-I may be significant across cancer entities and provide a strategy to further boost the anti-tumor immune response systemically, by combining validated immunotherapy with RIPK2 inhibition. If the results on RIPK2 in the setting of MHC-I autophagolysosomal degradation can be reproduced in other tumor entities, this approach may be appropriate not only for the infamously immunologically cold PDAC but possibly for other cancer types, as well.
Acknowledgement: None.
Funding Statement: QY was funded by the Chinese Scholarship Council, grant number 202308420032, https://www.chinesescholarshipcouncil.com/, accessed on 05 January 2025.
Author Contributions: The authors confirm their contribution to the paper as follows: study conception and design: Lydia Meder; data collection: Qian Yang, Alina Leonie Ruff; analysis and interpretation of results: Qian Yang, Alina Leonie Ruff, Lydia Meder; draft manuscript preparation: Qian Yang, Alina Leonie Ruff, Lydia Meder. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: All data analyzed in this study are publicly available at https://www.cbioportal.org/, accessed on 05 January 2025.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Sang W, Zhou Y, Chen H, Yu C, Dai L, Liu Z, et al. Receptor-interacting Protein Kinase 2 is an immunotherapy target in pancreatic cancer. Cancer Discov. 2024;14(2):326–47. doi:10.1158/2159-8290.CD-23-0584. [Google Scholar] [PubMed] [CrossRef]
2. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375(9):819–29. doi:10.1056/NEJMoa1604958. [Google Scholar] [PubMed] [CrossRef]
3. Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature. 2020;581(7806):100–5. doi:10.1038/s41586-020-2229-5. [Google Scholar] [PubMed] [CrossRef]
4. Meder L, Orschel CI, Otto CJ, Koker M, Bragelmann J, Ercanoglu MS, et al. Blocking the angiopoietin-2-dependent integrin β-1 signaling axis abrogates small cell lung cancer invasion and metastasis. JCI Insight. 2024;9(10):e166402. doi:10.1172/jci.insight.166402. [Google Scholar] [PubMed] [CrossRef]
5. Nguyen EM, Taniguchi H, Chan JM, Zhan YA, Chen X, Qiu J, et al. Targeting lysine-specific demethylase 1 rescues major histocompatibility complex class I antigen presentation and overcomes programmed death-ligand 1 blockade resistance in SCLC. J Thorac Oncol. 2022;17(8):1014–31. doi:10.1016/j.jtho.2022.05.014. [Google Scholar] [PubMed] [CrossRef]
6. Topal Y, Gyrd-Hansen M. RIPK2 NODs to XIAP and IBD. Semin Cell Dev Biol. 2021;109:144–50. doi:10.1016/j.semcdb.2020.07.001. [Google Scholar] [PubMed] [CrossRef]
7. Jiao J, Ruan L, Cheng CS, Wang F, Yang P, Chen Z. Paired protein kinases PRKCI-RIPK2 promote pancreatic cancer growth and metastasis via enhancing NF-κB/JNK/ERK phosphorylation. Mol Med. 2023;29(1):47. doi:10.1186/s10020-023-00648-z. [Google Scholar] [PubMed] [CrossRef]
8. Debnath J, Gammoh N, Ryan KM. Autophagy and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol. 2023;24(8):560–75. doi:10.1038/s41580-023-00585-z. [Google Scholar] [PubMed] [CrossRef]
9. You J, Wang Y, Chen H, Jin F. RIPK2: a promising target for cancer treatment. Front Pharmacol. 2023;14:1192970. doi:10.3389/fphar.2023.1192970. [Google Scholar] [PubMed] [CrossRef]
10. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4. doi:10.1158/2159-8290.CD-12-0095. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools