
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

A Review of Mitochondrial Involvement in Cell Death Pathways Induced by Oncogenic Viruses
1 Basic and Molecular Epidemiology of Gastrointestinal Disorders Research Center, Research Institute for Gastroenterology and Liver Diseases, Shahid Beheshti University of Medical Sciences, Tehran, 1985717413, Iran
2 Department of Microbiology and Microbial Biotechnology, Faculty of Life Sciences and Biotechnology, Shahid Beheshti University, Tehran, 1983969411, Iran
3 Gastroenterology and Liver Diseases Research Center, Research Institute for Gastroenterology and Liver Diseases, Shahid Beheshti University of Medical Sciences, Tehran, 1985717413, Iran
* Corresponding Author: Seyed Reza Mohebbi. Email:
(This article belongs to the Special Issue: Exploring Mitochondria: Unraveling Structure, Function, and Implications in Health and Disease)
BIOCELL 2025, 49(2), 221-251. https://doi.org/10.32604/biocell.2025.059787
Received 17 October 2024; Accepted 16 January 2025; Issue published 28 February 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Oncogenic viruses include both DNA and RNA viruses which contribute to cancer development by disrupting cellular regulation and interfering in the immune responses. These viruses do not directly cause cancer but instead integrate their genetic material into the host genome thus, affecting cell cycle and tumor suppression. This deregulation also leads to impaired immune function and promotes tumor progression by disrupting the removal of infected cells. Generally, innate immunity consists of two important members, including mitochondria and cell deaths, which impact each other as well. Due to the close correlation between viruses, cell death pathways (apoptosis, necroptosis, and pyroptosis), and mitochondria (mitochondrial antiviral-signaling protein and reactive oxygen species generation), targeting these immune system representatives may offer therapeutic strategies to control the progression of oncogenic viral infections. Some previous studies have covered the association of oncogenic viruses with mitochondria and cell death pathways, respectively, but mitochondria and cell death interact with each other, separately, and this interaction may play a role in the progression of cancer induced by oncogenic viruses. Hence, the purpose of this review is to discuss the relationship between cell death, mitochondria, and viral oncogenesis, focusing on the most surveyed oncogenic viruses’ mechanisms of action.Keywords
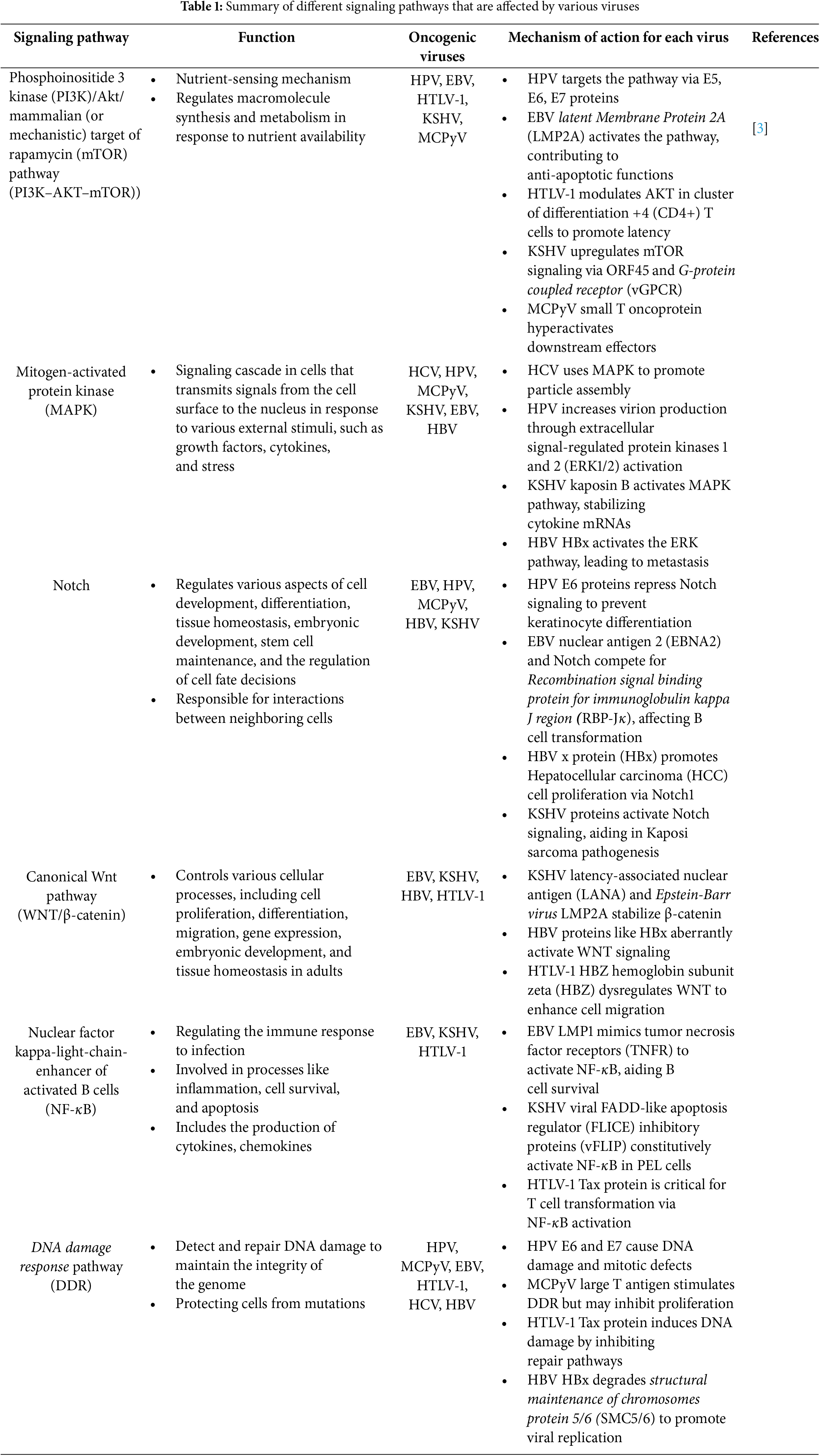
Cancer is a biological term referring to a situation when cells undergo uncontrolled changes in their metabolism and cell cycle. Cells’ life cycle is strictly controlled and suppressed to maintain their normal tissue-related function and mortality by molecules such as p53 and retinoblastoma protein (pRb). When a cell cannot be controlled and undergoes severe alteration, the immune system tries to eliminate it to inhibit further damage, but sometimes the cell becomes immortal, which means the cell death pathways are not sufficient to remove it. When faulty cells are accumulated a structure called a tumor is formed, which might be able to perform metastatic behavior [1–3]. Various factors are capable of disrupting cells’ standard functions including genetic mutations, physical and chemical damage, and pathogenic invasions, especially by viruses. Detection of viral genome in tumor cells started the hypothesis that viruses can be the reason behind the inflammations that lead to cancer. These viruses are known as oncogenic viruses. It’s estimated that oncogenic viruses are behind about 15%–20% of cancers all over the world [4]. It’s noteworthy to know that as a result of cancer development in viral infection, neoplasm can be observed which is not even desirable to the virus itself and they can be as deadly to the virus as they are to the host’s cells because the viruses are non-permissive for replication in the tumor cells [5]. Technically, viruses begin to prepare the essential factors and manipulate specific metabolic pathways such as pathways regulating glucose metabolism, fatty acid metabolism, and some oncogene expressions to ensure their persistence, which leads to disruption of the cell cycle machinery and finally alters the cell’s proliferation and division, because viruses are not able to replicate independently. Signaling pathways which are manipulated by oncogenic viruses to disrupt the normal function of their host cells and the immune system include Phosphoinositide 3 kinase (PI3K)/Akt/mammalian (or mechanistic) target of rapamycin (mTOR) pathway (PI3K–AKT–mTOR, Mitogen-activated protein kinase (MAPK), Notch, Canonical Wingless-related integration site (Wnt) pathway (WNT/β-catenin), Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), DNA damage response pathway (DDR).
If these changes are not controlled and inhibited, they can lead to severe side effects or even death [6]. Hence, oncogenic viruses are not the only reason behind cancer development but their presence can accelerate this process. Generally, viruses have some major mechanisms to contribute to oncogenesis: I) encoding viral oncogenic proteins, called proto-oncogenes, II) beginning a chronic inflammation and suppressing immune system function [7,8], III) creating genomic instability or alterations in the microenvironment of host’s cells. Oncogenic viruses can be divided into two groups DNA viruses and RNA viruses based on their genetic materials. Insertional mutagenesis is referred to as viral DNA integration which leads to changes in the host’s genomic structure or transcription levels of the genes. DNA viruses are able to directly integrate their genome into the host’s genome but RNA viruses have to go through a reverse transcription mechanism [4,7]. Hence, long-term interaction between oncogenic viruses and their host cells is essential to lead to their tumorigenesis ability [1,8,9]. Seven viruses have been recognized to be involved in cancer so far. Among RNA viruses Hepatitis C virus (HCV) and Human T-cell lymphotropic virus type 1 (HTLV-1) and among DNA viruses Kaposi’s sarcoma-associated herpesvirus (KSHV), Epstein–Barr virus (EBV), Human papillomavirus (HPV), Hepatitis B virus (HBV), and Merkel cell polyomavirus (MCV or MCPyV) are known to be involved in cancer development and pathogenicity of the oncogenic viruses by some specific proteins encoded by their own genome in order to defeat immune system and ensure viral replication and cells transition including trans-activating transcriptional regulatory protein (Tax), HBZ hemoglobin subunit zeta (HBZ), and P12 encoded by HTLV-1, Epstein-Barr nuclear antigen A2 (EBNA2), Epstein-Barr nuclear antigen leader protein (EBNA-LP), latent membrane protein 1/2A/2B (LMP1, LMP2A, LMP2B), BamHI Epstein-Barr virus replication activator 1 (BZLF1), Epstein-Barr nuclear antigen (EBNA3A, EBNA3B, EBNA3C, EBNA1), BRLF1 encoded by EBV, E6, E7, E2, E5, L1, and L2 encoded by HPV, Hepatitis B virus core protein (HBc) and Hepatitis B virus x protein (HBx) encoded by HBV, non-structural protein 3/4A/5A/5B/s2 (NS3, NS4A, NS5A, NS5B, NS2), E1, E2, and P7 encoded by HCV, Latency-associated nuclear antigen (LANA), viral cyclin (v-Cyclin), VIRF2, Kaposin, Open reading frames 45/52/64 (ORF45, ORF52, ORF64), MIR1, MIR2, K1, and K2 encoded by KSHV, and Large antigen (LT) and small antigen (ST) encoded by MCPyV. The range of host tissue that can be infected by each virus is closely related to its tropism and is exclusive to each of them. For example, HBV is restricted to lead to primary hepatocellular carcinoma, and HTLV-1 causes adult T-cell leukemia/lymphoma. Instead, HPV can develop cervical, head and neck, and anal cancers in addition to squamous epithelial cancer. However, KSHV and EBV can infect a wide range of tissues and cause tumor formation. KSHV is the reason behind endothelial Kaposi’s sarcoma, B-cell primary effusion lymphoma as well as a B-cell lymphoproliferative syndrome and EBV causes B-cell lymphomas, nasopharyngeal carcinoma, stomach cancer, T-cell lymphomas, and a rare form of leiomyosarcoma [6,9]. Fig. 1 mentions these seven oncogenic viruses and the various cancers related to their infection.
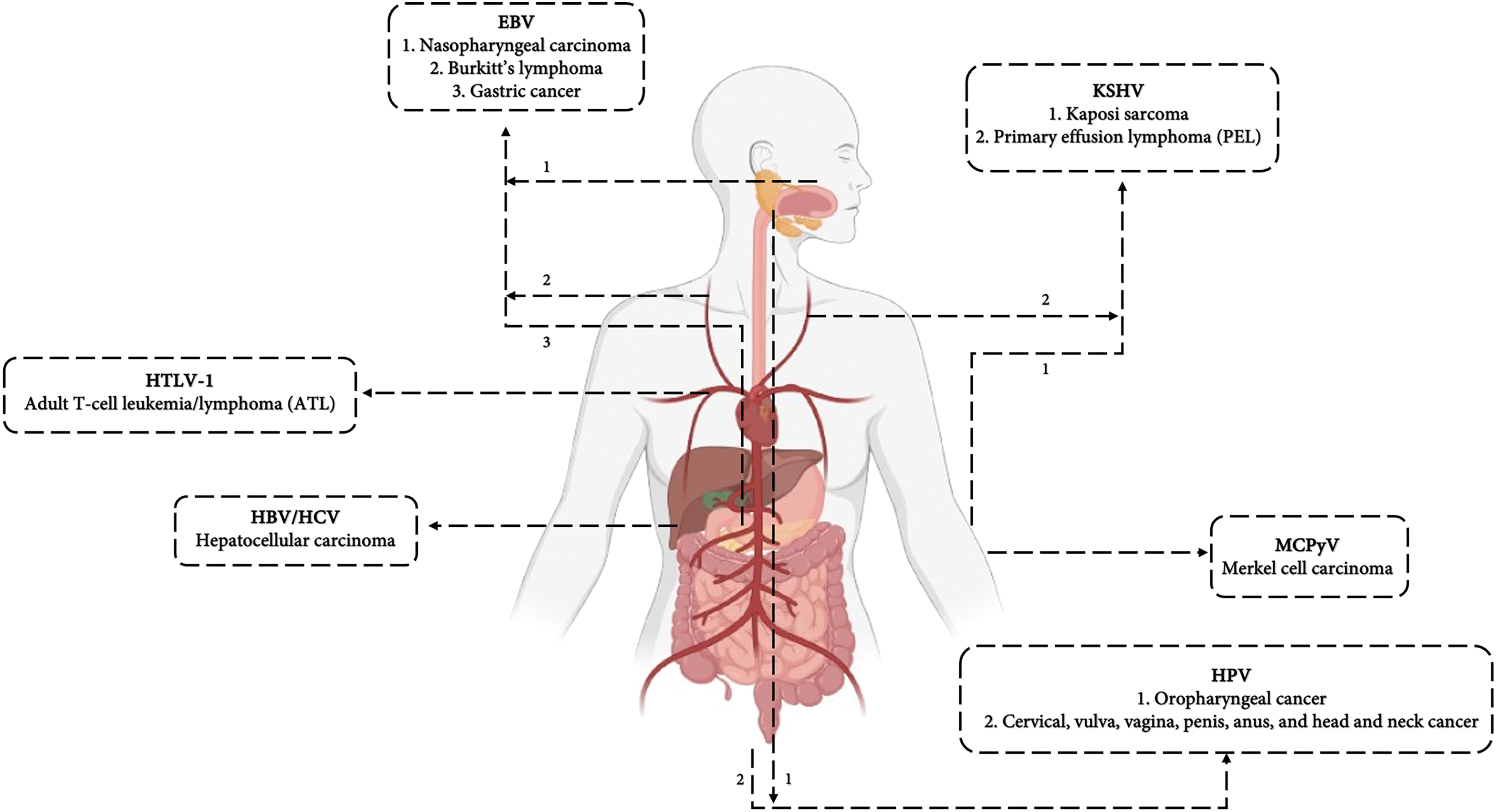
Figure 1: HPV, HCV, HBV, EBV, KSHV, MCPyV, and HTLV-1 have been recognized as viruses involved in cancer development. Their ability to cause cancer in particular target tissues is related to the virus attachment potential. This figure displays the cancers related to each oncogenic virus, pointing to their tissue targets. For instance, the Hepatitis B virus or Hepatitis C virus (HBV/HCV) are both responsible for hepatocellular carcinoma in liver, Human papilloma virus (HPV) is the reason behind cancer in the genital area in males and females, Human T-cell lymphotropic virus type 1 (HTLV-1) infects blood cells causing T-cell leukemia and lymphoma, Epstein–Barr virus (EBV) infection leads to cancer in lymphatic system and nasopharyngeal carcinoma, and KSHV and MCPyV have been identified to cause specific types of skin cancers such as Kaposi sarcoma (KS) and Merkel cell carcinoma (MCC), respectively
On one hand, the immune system has adopted mechanisms to control and defeat the invasion of pathogenic factors such as viruses, which lead to the normal function and proliferation of the cells to maintain homeostasis. Tumor viruses interact with the immune system members to downregulate the function of immune sensors such as Toll-like receptors (TLRs) which are responsible for detecting viral proteins, negatively influence the DDR which recognizes the viral genetic materials, and disrupt the immune signaling pathway including interferon regulatory factor 3/7 (IRF3/7) and NF-kB, and interrupt the cell death pathways to prevent the efficient immune responses [2,6,8]. The metabolic and immune signaling manipulated by oncoviruses through their oncoproteins are discussed in more detail in Table 1. On the other hand, oncogenic viruses target the cell cycle checkpoints and interact with p53 and pRb molecules by encoding oncoproteins. Oncoproteins induce the S phase of the life cycle to have access to the cellular replication machinery and enhance DNA synthesis in order to accelerate their replication [2,3]. Based on the previous studies, the interaction between oncogenic viruses, mitochondria (Mitochondrial antiviral-signaling protein (MAVS) and mitochondrial reactive oxygen species (mROS) production), and cell death pathways, respectively, has been discovered. Still, the associations of mitochondria and cell death pathways remain inconclusive and need more attention because they might be an appropriate therapeutic target to control cancer development. Here we discuss the interactions between the mentioned oncogenic viruses, mitochondria, and cell death pathways to propose that the cell death pathways and mitochondria cooperation might be a promising target in the process of tumor progression.
• Hepatitis C virus (HCV): Hepatitis C virus is identified as a member of Flaviviridae, and consists of a positive single-stranded RNA genome engulfed within an envelope. This virus establishes a chronic infection leading to liver damage and hepatocellular carcinoma (HCC) [7,8]. The genome of HCV is approximately 9.6 kilobases (kb) and encodes various viral proteins such as core protein, NS3, NS4A, NS4B, NS5A, NS5B, NS2, E1, E2, and P7, which associate in a wide range of cell signaling pathways and the mechanism of tumorigenesis.
• Human T cell leukemia virus 1 (HTLV-1): Human T cell leukemia virus 1 is a member of the Retroviridea family. HTLV-1 is recognized as a single-stranded positive-sense RNA virus which is 8.5 kb in length. This virus can form a persistent infection by reverse transcribing its RNA genome and integrating the resulting double-stranded DNA copy of its genome into the host cellular chromatin and forming a provirus. The infection later leads to CD4+ T cell malignancy, called adult T cell leukemia/lymphoma, or HTLV-1-associated myelopathy, which is known as a progressive inflammatory disease of the spinal cord [4,10]. HTLV-1 infection is mostly latent which refers to the ability of the virus to cause asymptomatic infection and production of undetectable proteins [8]. Its genome encodes regulatory factors such as Tax protein, HBZ, p12, p13, p8, p30, and Rex protein which are involved in the pathogenicity and oncogenicity of the virus.
• Epstein-Barr virus (EBV): Epstein-Barr virus is a 184 kb double-stranded DNA member of herpesviruses (human herpesvirus 4 or HHV-4) and the first virus that was detected to associate with cancer development [2,7]. EBV’s infection pattern is classified into two forms known as latent and lytic infection and the latent phase is divided into three groups recognized as latency I, II, and III based on the expression pattern of the latent phase genes. In latent infection the genome of the virus is partially expressed but, in lytic infection, the EBV genome is completely expressed causing a high expression level of the viral oncogenes [4,7]. Both B lymphocytes and epithelial cells can be infected by the lytic infection of EBV, but in latent infection, the influenced cells are restricted to the B cells. EBV genome encodes six lytic-related antigens (EBNA1, EBNA2, EBNA3A, EBNA3B, EBNAC, and EBNA-LP) and two latent-related proteins (LMP1 and LMP2) [5,7].
• Human papillomavirus (HPV): Human papillomavirus is known as an 8 Kb non-enveloped DNA virus that primarily infects basal keratinocytes of the skin or mucous membranes belonging to the Papillomaviridae family [2]. HPV’s genome comprises three regions including I) the early region (E), which encodes E1 to E7 proteins necessary for viral replication and cellular transformation, II) the late region (L), which encodes capsid proteins L1 and L2, III) the long control region, which contains the origin of replication and transcription factor binding sites [5]. HPV is further classified into low- and high-risk groups based on their ability to cause malignant lesions and the high-risk HPVs (types 16 and 18) are the main causes of cervical cancer [4]. HPVs must synchronize their life cycle with that of their host’s squamous epithelial cells. Two key signaling pathways govern this process: integrins on basal epithelial cells that promote proliferation and repress differentiation and NOTCH signaling that induces differentiation in detached cells. Persistent and long-term HPV infection can lead to squamous proliferative lesions, such as warts, papillomas, and condylomas by encoding different oncoproteins, most importantly E6 and E7 [11]. The E6 and E7 oncoproteins lead to the immortalization and transformation of host cells by interacting with key cellular proteins such as pRb and p53, promoting their degradation.
• Hepatitis B virus (HBV): Hepatitis B virus is a member of the Hepadnaviridae family with a small double-stranded DNA with 3.2 kb in length. HBV genome includes four overlapping reading frames that encode a diverse range of proteins involved in its pathogenesis and oncogenesis (S which encodes the viral surface proteins (HBs), and P which encode the viral polymerase, X that encodes the regulatory X protein (HBx), and pre-C that encodes ‘e’ and ‘c’ antigens) [2,5,7]. HBx and HBV core proteins are two of the most important proteins encoded by the virus which play an essential role in the oncogenesis of HBV. HBV forms chronic inflammation that leads to a multi-step process to cause hepatocellular carcinoma (HCC) [12]. Environmental carcinogens including Aflatoxin B, cigarette smoking, and alcohol consumption may promote the tumorigenic properties of HBV [10]. Myeloid differentiation primary response 88 (MyD88), Toll/interleukin-1 receptor (TRIF), and MAVS are recognized to play an important role in controlling HBV replication. Hence, HBV has adopted mechanisms by encoding specific proteins to disrupt immune responses such as Toll-like receptor (TLR) signaling pathways and Retinoic acid-inducible gene 1 (RIG-I) signaling, as well as a stimulator of interferon genes (STING)-mediated interferons (IFN) induction [11].
• Kaposi’s sarcoma herpesvirus (KSHV): KSHV also known as human herpesvirus 8 (HHV-8) is recognized as the most commonly identified cancer in untreated acquired immunodeficiency syndrome (AIDS) patients [1,4]. KSHV consists of a double-stranded DNA which is 165 kb in length, encoding oncoproteins such as LANA, v-Cyclin, vFLIP, and K12. As a member of gamma herpesviruses, this virus can establish either latent or lytic infections. Initially, KSHV exists in a latent state, making the tumor cells resistant to current antiviral treatments targeting lytic replication. In the latency phase, KSHV expresses some specific proteins such as LANA, v-cyclin, and vFLIP, which contribute to cell survival, proliferation, and immune evasion [13].
• Merkel cell polyomavirus (MCV): Merkel cell polyomavirus (MCV), a small non-enveloped virus with a double-stranded DNA genome, belongs to a polyomavirus family. MCC is identified as the reason behind a rare and aggressive skin cancer which can be seen mostly in elderly and immunosuppressed individuals. The genome of this virus, which is 5.4 in length, encodes several proteins involved in its pathogenesis and tumorigenesis, such as large T antigen (LT) and small T antigen (ST), which are involved in the disruption of tumor suppressors such as pRb and p53, facilitating the transformation of normal cells into tumor cells [2,4,5].
Table 2 provides information on the proteins of oncogenic viruses, highlighting their oncoprotein’s roles in pathogenicity and oncogenicity.

4 Apoptosis, Necroptosis, and Pyroptosis Act as a Weapon for Innate Immunity
• Apoptosis: Apoptosis is known as a caspase-dependent cell death pathway. Caspases are proteolytic enzymes that drive the morphological changes associated with some specific cell deaths [31]. During this type of cell death, cell integrity is preserved, and the release of intracellular components such as cytokines out of the cell cannot be seen thus, apoptosis is immunologically silent [31–33]. To initiate apoptosis a specific signal must be detected by the cells. While the integrity of the plasma membrane is maintained, the phosphatidylserine (PS) moves from the inner leaflet of the plasma membrane to the surface of the cell membrane. Then macrophages detect PS as an “eat me” signal [34]. Phagocytes encapsulate the dying cell and form vesicles known as “apoptotic bodies”. These vesicles act as traps consisting of microbial products which should remain engulfed [35]. Apoptosis is classified into two pathways called extrinsic and intrinsic apoptosis. The intrinsic pathway relies on sensing internal stress such as disruption in mitochondrial membrane integrity and the release of some components such as cytokine C (cytC), DNA damage, and oxidative stress [36]. During apoptosis, the BH3 interacting-domain death agonist (BID) protein translocates to mitochondria and activates the B-cell leukemia/lymphoma 2 protein (Bcl2) family protein and BAK protein. After the loss of mitochondrial outer membrane (MOM) integrity, the release of mitochondrial proapoptotic contents cytC can be observed, which assists the assembly of apoptotic protease activating factor (APAF) and cooperates in the formation of a platform named apoptosome. Apoptosomes induce the activation of initiator caspase 9 which activates the executioner caspases (caspase 3,6,7), especially caspase 3 [37]. The extrinsic pathway is a classical ligand-cell surface receptor interaction dependent on death receptors (DR) and extracellular stresses, which can be sensed by extracellular receptors. When the specific ligands bind to their receptors, apoptosis is induced by the activation of caspases similar to the intrinsic pathway [37,38]. (Fig. 2)
• Necroptosis: Necroptotic cell death is controlled by receptor-interacting protein 1 and 3 (RIP1, RIP3), and mixed lineage kinase domain-like pseudokinase (MLKL). Tumor necrosis factors (TNFs) can activate both apoptosis and necroptosis. When TNF binds to TNF receptor 1, it initiates the trimerization of these receptors leading to the formation of complex 1. Complex 1 undergoes reshuffle and forms complex 2. In complex 2, fas-associated death domain (FADD) protein and RIP3 are recruited leading to MLKL activation [39]. After the formation of complex II, procaspase 8 is recruited by FADD and RIP3. Then RIP3 hires MLKLs and they go through self-oligomerization [40]. MLKLs interact with the N-terminal of phosphatidylinositol phosphate (PIP) in the plasma membrane of the dying cell. This interaction inserts MLKLs into the plasma membrane and forms pores to disrupt plasma membrane integrity [35,36] (Fig. 2B).
• Pyroptosis: Pyroptosis is a lytic cell death that includes some common features of both necroptosis and apoptosis [38,41–43]. Pyroptosis leads to inflammatory responses because of the release of cell components [44]. Cysteine-dependent aspartate specific protease (caspase) and gasdermin (GSDM) are two protein families that have the most essential roles in the pyroptosis pathway [38,45,46]. Caspases 1, 4, and 5 are identified to be involved in pyroptotic cell death in humans. GSDM family protein consists of diverse members that each have a particular function in different molecular patterns. The most important member of this family involved in pyroptosis is recognized to be gasdermin D (GSDMD) [33,35,41,42,47]. GSDMD protein contains an N-terminal domain (NTD) responsible for pore-forming activity in the plasma membrane of the dying cell to induce cytolysis [35,44,48]. Activation of pyroptosis is dependent on stimulation of a protein complex known as “Inflammasome”. Inflammasome consists of sensor proteins and adaptor proteins. Sensor proteins are known as pattern recognition receptors (PRRS) such as NOD-like receptors, the DNA receptor Absent in Melanoma 2 (AIM2), and pyrin receptor and adaptor proteins known as apoptosis-associated speck-like protein (ASC) [35,45,49]. PRRs in inflammasome sense the damage-associated molecular patterns (DAMPs) (such as adenosine triphosphate (ATP) and pathogen-associated molecular pattern molecules (PAMPs) (including viral RNAs) and initiate the cell death process. Once the PRRs are triggered, caspase activity leads to GSDMD cleavage [50]. GSDMDs form structures in the plasma membrane of the dying cell that act as pores to disrupt the integrity of the cell membrane. Mitochondria are also able to trigger NLRP3 inflammasome by its reactive oxygen species (ROS) production and mitochondrial DNA release which act as DAMP for the cell. The release of mitochondrial DNA can be the result of viral infection, especially after sensing the viral RNAs in the host cell [51]. Based on the kind of caspase involved in cell death, pyroptosis can be divided into several groups. The most important pathways are known as “canonical and non-canonical pathways”. Caspase 1 is responsible for the canonical pathway and caspase 4 and 5 in humans initiate the non-canonical pathway [35,44,45,52]. In the non-canonical pathway, caspases 4 and 5 are not able to cleave GSDMD directly, so they initially stimulate the activation of caspase 1. Then, Caspase 1 cleaves GSDMDs which leads to pore-forming activity of GSDMDs [33]. GSDMD-N typically forms large oligomeric ring-shaped structures minutes after the initiation of full-length GSDMD cleavage by caspases [35,53]. The pore formation function of GSDMD causes non-selective ion flux and the release of cytosolic content such as interleukins (IL-1b and IL-18), ATP, high mobility group box 1 (HMGB1) [54], and immunogenic organelles contents such as mitochondrial DNA and cytC into intracellular space. These cytosolic contents act as DAMPs to other cells in order to give them a signal about the infection or unexpected condition [33]. Each oncogenic virus has adopted a specific mechanism by encoding oncoproteins in order to interfere with or disrupt the cell death pathways to control cell signaling (Fig. 2C). Here we have a quick recap on the most studied viruses including HPV, EBV, HCV, HBV, and KSHV, and their impacts on these three mentioned cell death pathways. The information related to cell death pathways differences and the oncoproteins activities are illustrated in Fig. 1 and more discussed in Table 3.
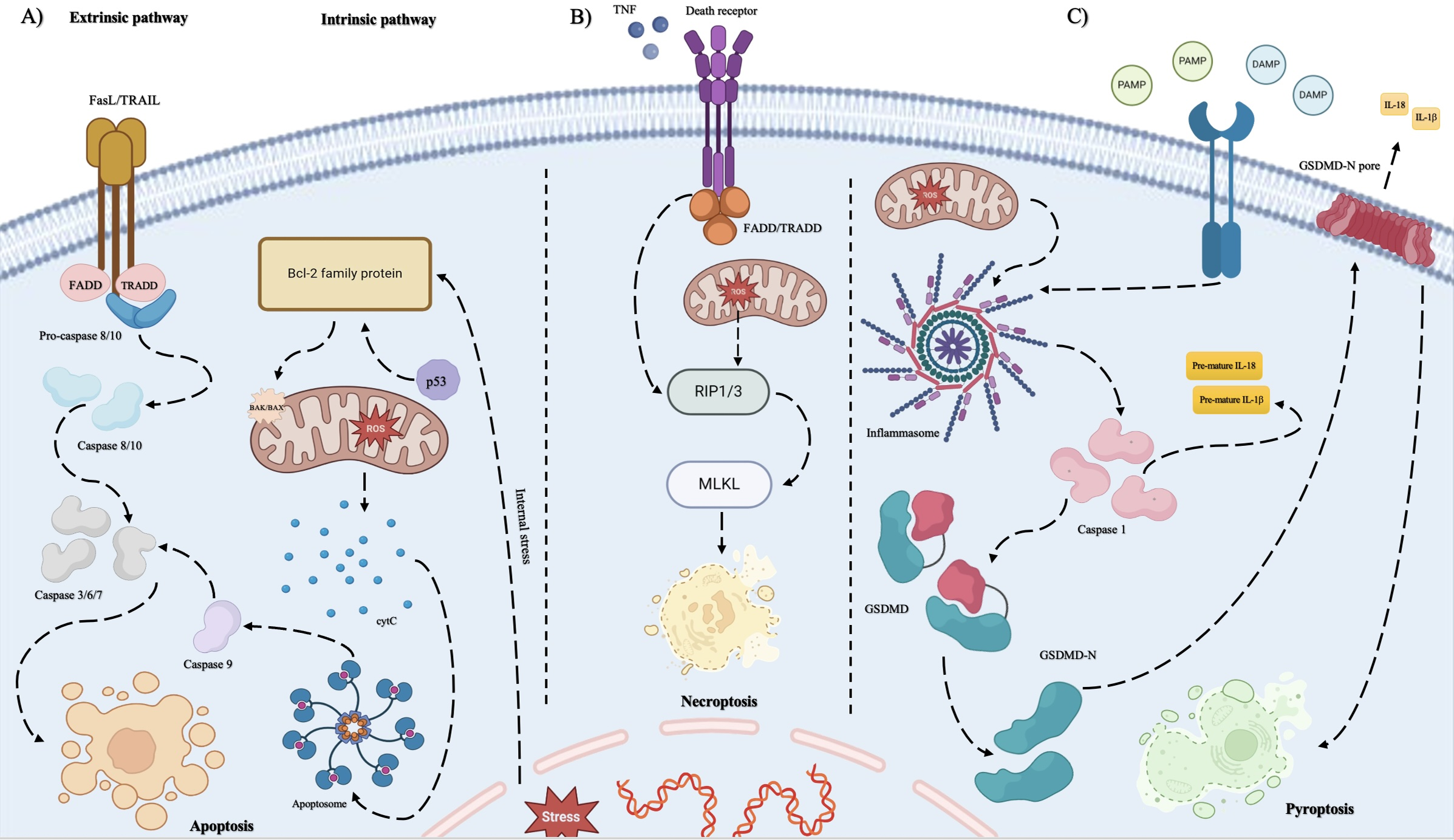
Figure 2: The three discussed cell death pathways including apoptosis, necroptosis, and pyroptosis differ at some points in their mechanisms and the molecules involved in their pathways. A) Apoptosis includes two pathways known as the intrinsic pathway and the extrinsic pathway. When the integrity of the mitochondrial outer membrane (MOM) is disrupted by internal stresses such as viral DNA integration into the nuclear DNA of the host cell, the release of proapoptotic factors into cytosol happens. This event is controlled by BID, Bcl-2 family protein. At first, the BID protein translocates to mitochondria. Then, BAK and BAX are activated, and the release of mitochondrial cytC can be observed. cytC cooperates in the formation of a platform named apoptosome by its caspase recruitment domain (CARD). Apoptosome induces the activation of initiator caspase 9 then, they activate the executioner caspases, especially caspase 3. This pathway is dependent on extracellular receptors known as death receptors (DR) such as tumor necrosis factor (TNF) superfamily (TNF receptor 1), apoptosis antigen 1, and death receptors 3, 4, and 5. When the specific ligands bind to their receptors, apoptosis is induced by the activation of caspases like the intrinsic pathway. In addition, increased levels of ROS lead to triggering mitochondrial dysfunction and initiating apoptosis by causing an imbalance between pro- and anti-apoptotic factors leads to apoptosis. B) Necroptosis is initiated by cytokines such as TNF when they bind to death receptors such as tumor necrosis factor receptor type 1-associated death domain protein (TRADD). This activation leads to receptor-interacting protein kinase 1/3 (RIP 1/3) activation. A high level of RIP3 level prevents apoptotic cell death. Then, MLKL is activated by RIPs which results in necroptosis. Excessive ROS production drives necroptosis in cancer cells. Metformin enhances this process by increasing endogenous ROS and inhibiting antioxidant defenses, amplifying oxidative stress. Phosphorylation of RIPK1, RIPK3, and MLKL further promotes necroptosis, releasing DAMPs that activate the immune system against cancer cells. C) Pyroptotic cell death is primarily activated when DAMPs and PAMPs trigger the cells. Then, inflammasome formation occurs which is a protein complex and leads to caspase1 activation. As the result of caspase 1 activation, GSDMD molecules are cleaved to have their N-terminal free and the maturation of interleukins is seen, respectively. The free GSDMD-N is responsible for the pore formation activity in the plasma membrane which leads to mature interleukins release and the inflammatory signs of pyroptosis. Moreover, reactive oxygen species (ROS) function as signaling molecules that trigger pyroptosis in endothelial cells. Elevated ROS levels can stimulate multiple intracellular pathways, resulting in inflammation and cell death. One primary mechanism involves the activation of the NLRP3 inflammasome, a crucial regulator of pyroptosis. The oncogenic viruses’ impacts on each cell death pathway are mentioned in Table 3

5 Oncogenic Viruses Association with Cell Death Modes
• HPV: Oncogenic human papillomaviruses (HPVs) use E6 and E7 proteins in order to disrupt cellular regulation, resulting in cell proliferation. The E6 oncoprotein promotes p53 degradation through the ubiquitin-proteasome pathway, thus blocking apoptosis. E6 has also the ability to degrade the Bak protein independently of p53 [55–59]. Moreover, E7 targets the pRB protein and other apoptosis pathways like tumor necrosis factor (TNF-α) and tumor-necrosis factor-related apoptosis-inducing ligand (TRAIL) [60]. The E5 protein encoded by HPV downregulates Fas receptors, further inhibiting apoptosis [57,61]. On the contrary, the E2 protein can induce apoptosis via p53-dependent manner, but E6 and E7 counteract this signaling to promote viral persistence [62–66]. High-risk HPV (hrHPV) enables keratinocytes to resist necroptosis by reducing RIPK3 expression, allowing the virus to evade immune responses and facilitating lesion progression [66–68]. Proteins like sirtuin (SIRT1) also inhibit pyroptosis, contributing to persistence of HPV’s and cervical cancer progression [69].
• EBV: EBV is capable of modulating apoptosis in order to ensure viral persistence and cell transformation. EBNA2 protein promotes cell survival by upregulating pro-survival proteins and downregulating pro-apoptotic members of apoptosis [70]. In addition, EBNA3A and EBNA3C work together to silence tumor suppressors resulting in inhibition of apoptosis [71,72]. LMP1 protein also activates NF-κB, upregulates anti-apoptotic proteins, and modulates caspases to enhance cell survival [73–75]. LMP2A inhibits transforming growth factor (TGF-β)-related apoptosis, while viral BCL-2 homologs protect against apoptosis by binding pro-apoptotic proteins [76,77]. When the virus is in its lytic state, proteins such as BZLF1 and BRLF1 balance apoptosis to support viral replication [78]. EBV suppresses necroptosis as well via LMP1, which modulates RIPK3 to prevent necrosome formation [79–81]. It also evades pyroptosis, utilizing BRLF1 which blocks the inflammasome activation [82–84].
• HBV: As discussed earlier, the Hepatitis B virus encodes a protein known as HBx which has the most interference in cell death pathways. HBx blocks apoptosis in a p53-dependent and -independent manner, activating survival signals including NF-κB [85–88]. Moreover, elevated levels of RIPK3 protein in HBV-associated infection suggest a role in promoting necroptosis, although it needs to be studied more [89]. HBx also triggers pyroptosis through NLRP3 inflammasome activation, increasing inflammation and contributing to liver disease [90,91].
• HCV: Hepatitis C virus core protein is recognized to have dual roles in promoting and inhibiting apoptosis. This protein primarily activates the NF-κB pathway in order to inhibit apoptotic cell death, but also sensitizes hepatocytes to TNF and Fas ligand-induced apoptosis. Some other HCV proteins such as NS4A and NS3 are able to promote apoptosis through mitochondrial pathways, and cytotoxic T-cell responses further drive the death of infected hepatocytes [92]. It is identified that HCV also induces pyroptotic cell death, which is recognized to contribute to inflammation, liver fibrosis, and HCC, driven by NF-κB and ROS production [93,94].
• KSHV: Kaposi’s Sarcoma-Associated Herpesvirus is capable of preventing apoptosis mainly via its vFlip protein encoded by its genome, which activates anti-apoptotic NF-κB pathways [95,96]. KSHV also suppresses pyroptosis through the SOX protein, which inhibits AIM2 inflammasome activation, and other viral proteins like PAN RNA and ORF63, preventing inflammasome activity [96].
6 Mitochondria, Cells’ Power House Which Is Involved in Antiviral Responses
Mitochondria are an organelle in eukaryotic cells with two membranes and their own DNA (mtDNA) which are not dependent on the nucleolus in order to replicate [104,105]. Primarily, this organelle is recognized as the powerhouse of the cell which produces ATP, but also plays important roles in generating reactive oxygen species (ROS), cell signaling, and regulating innate immune responses [106]. However, mitochondria can play a dual role in viral tumorigenesis. They may either aid the viral invasion and tumor development by integrating viral proteins and affecting retrograde pathways, or they might help cease the infection by triggering appropriate immune responses and cell deaths [107]. Mitochondrial dysfunction, caused by factors like oxidative stress or inflammations, can disrupt processes such as cell metabolism, and cell proliferation [108–110]. Recently mitochondria have dragged too much attention and many studies have focused on mitochondria’s role in different human diseases. Thus, researches lead to targeting mitochondrial proteins and pathways for drug discovery. Cancer cells rely on mitochondrial adaptation to their changing environment, which affects bioenergetics, susceptibility to cell death, regulation of oxidative stress, and signaling pathways. This correlation has been linked to cancer metabolism since the Warburg effect was discovered in 1956, representing that cancer cells prefer aerobic glycolysis, breaking down glucose into lactic acid, even in the presence of oxygen, rather than relying on full mitochondrial respiration [111].
ROS production is another important function of mitochondria which is associated with viral infections. The production of mitochondrial reactive oxygen species (mROS) primarily results from electron leakage in the electron transport chain (ETC). This process is strictly under control by some pathways such as the ROS scavenging system, comprising both enzymatic and non-enzymatic components. This system efficiently eliminates ROS, including mROS, and is collectively known as the protective enzyme system. Notably, when intracellular ROS levels rise, the mitochondrial antioxidant defense system works to reduce the cytotoxic effects induced by ROS. At low levels, mROS serve as signaling molecules involved in processes like immune response, apoptosis, and adaptation to hypoxia. Thus, controlled ROS levels can help combat pathogens and modulate immune responses and overproduction of mROS can overwhelm the antioxidant defense systems, leading to damage of mitochondrial DNA (mtDNA), proteins, and lipids [112]. For example, high ROS levels can lead to the oxidation of lipids, proteins, and DNA, contributing to genomic instability, which influences the condition of the viruses. So, it’s pivotal for tumor cells to maintain ROS at levels that promote proliferation without causing toxicity, highlighting mitochondria’s role in cellular adaptability and cancer development. ROS influences tyrosine phosphatases, which regulate carcinogenic signaling pathways involved in cancer cell migration and metastasis [113]. In addition to metabolic pathways and ROS production, mitochondria constantly undergo biogenesis including fission and fusion. Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) and PTEN-induced kinase 1 (PINK1) are two of the identified members of the signalling pathways involved in mitochondrial biogenesis, while proteins such as SIRT1 and SIRT3 play critical roles in energy production and oxidative phosphorylation which are some of the targets of viruses [114,115]. Hence, viruses must be able to manipulate mitochondrial dynamics, metabolism, and ROS production in order to control innate immune signaling, affecting early immune responses. Mitochondria is involved in regulating innate immune responses, especially in viral infections with a specific pathway known as MAVS, which was discovered in 2005 [116].
MAVS is located on the outer mitochondrial membrane. Research in 2011 revealed that mitofusin proteins, including Mfn1, and Mfn2 which are involved in mitochondrial fusion and membrane potential interfere in the MAVS function and other innate immune responses such as interferon production [117–119]. Based on the type of viruses (RNA or DNA viruses), different pathways of detection and immune responses are initiated. For instance, the cGAS-STING pathway primarily senses DNA viruses but can also respond to RNA viruses, but the RIG-I-MDA5-MAVS pathway only detects RNA viruses. Finally, both pathways lead to on NF-κB and IRF3, which regulate the production of type I interferon [120,121]. Then, interferons α and β trigger the expression of interferon-stimulated genes (ISGs) through the JAK-STAT pathway, inhibiting viral replication in the host cells.
HTLV-1, EBV, HBV, HCV, and HPV are examples of viruses whose interactions with the mitochondria of their host cells have been under investigation. These viruses can manipulate and disrupt mitochondrial functions such as energy supply, calcium homeostasis, ROS production, and cell death to support their survival in the host cell and maintain their oncogenic potentials [120]. Here we discuss the most well-known oncogenic viruses that are involved in the disruption of mitochondrial function which also might interfere in the cell death pathways. Fig. 3 illustrates the involvement of oncogenic viruses in disrupting the normal function of mitochondria.
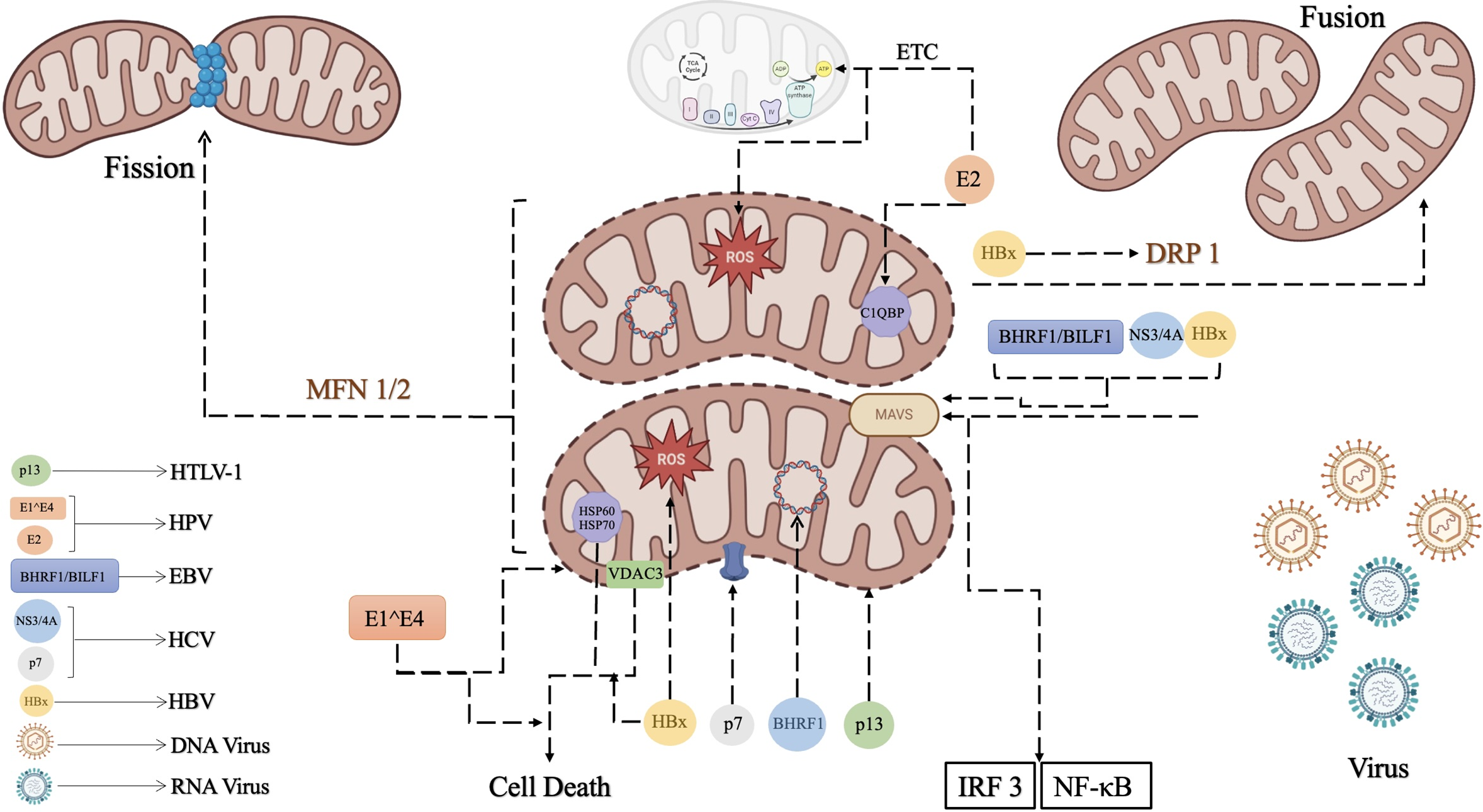
Figure 3: Mitochondria is known as the producer of the main source of energy in cells but is involved in other cellular signaling too. A) Mitochondria interferes with the antiviral activities by the MAVS pathway. When the viral RNA is sensed its receptors such as RIG-1 like receptors (RLRs), activate MAVS, located in the mitochondrial membrane. Then, MAVS triggers the activation of TANK binding kinase 1 (TBK1) and IRF3/7. This signaling pathway results in the stimulation of NF-κB to activate interferon production to respond to the viral invasion. B) Another important function of mitochondria is its biogenesis which includes fusion and fission. Mitochondrial fusion and fission are strictly controlled by proteins such as DRP1 and MFN 1/2, respectively. Oncogenic viruses have adopted some strategies to disrupt normal mitochondrial functions. Oncogenic viruses’ mechanisms by which they interfere with balanced mitochondrial pathways are shown in this Fig. 3. 1) HTLV-1 impacts mitochondria with p13, a protein encoded by its genome. The p13 protein localizes to the inner mitochondrial membrane and functions as a viroporin, which disrupts mitochondrial morphology and membrane permeability by forming ion channels. 2) BHRF1 and BILF1 are oncoproteins encoded by EBV. On one hand, BHRF1 prevents antiviral responses by inhibiting MAVS protein function, helping EBV survival and replication. Moreover, BHRF1 blocks inflammatory responses and enhances viral persistence. On the other hand, BHRF1 interferes with mtDNA replication by relocating mtDNA replication proteins to the nucleus, which disrupts mtDNA processes. 3) HCV interferes with mitochondrial integrity with p7 protein. p7 forms ion channels in the membrane and induces mitochondrial damage, leading to a high level of ROS production, which results in enhanced viral replication. Additionally, its NS3/4A protease targets MAVS, impairing the host’s antiviral response and weakening IFN signaling, which is crucial for the immune response. 4) HBx is a protein encoded by HBV which is recognized to disrupt MAVS, blocking antiviral signaling and finally, reducing IFN-β production. It also induces mitochondrial fragmentation, promotes ROS production, and alters mitochondrial dynamics. 5) HPV associates with mitochondria with its E2 and E1^E4 proteins. The E2 protein is responsible for stimulating mitochondrial biogenesis by enhancing the expression of the protein P32/gC1qR, which normally regulates its morphology. It also interacts with the mitochondrial respiratory chain, increasing ROS production. Furthermore, the E1^E4 protein encoded by HPV induces mitochondrial clustering near the nucleus, reduces mitochondrial membrane potential, and leads to apoptosis. On the other hand, it is noteworthy to consider that in some cases elevated levels of mtROS can cause cellular oxidative stress, leading to mitochondrial dysfunction and impaired viral replication, especially HBV and HCV.
7 Oncogenic Viruses Association with Mitochondria
• HTLV-1: As discussed earlier, the genome of HTLV-1 encodes several nonstructural proteins, including Tax, Rex, p12, p13, p21Rex, p30/Tof, and HBZ [122]. For instance, Tax is responsible for driving transcription from the viral 5′-LTR (long terminal repeat) promoter critical for the expression of viral genes, Rex promotes the expression of incompletely spliced viral transcripts, including those coding for structural proteins of the virion [123,124]. The protein p13 is a smaller, 87-amino acid protein localized mainly to the inner mitochondrial membrane. Investigation indicates that when this protein is expressed in the HeLa-derived cell line Hltat, it alters mitochondrial morphology, leading to the formation of isolated clusters of rounded, fragmented mitochondria. So p13 might function as a viroporin, a class of small, hydrophobic viral proteins that can alter membrane permeability by forming ion channels or pores in membranes [122].
• EBV: EBV encodes several proteins to enhance its viral persistence such as BHRF1, BZLF1, BALF1, and LMP2A. BHRF1 disrupts mitochondrial antiviral signaling protein (MAVS) which inhibits IFN activation and innate immune responses [125]. BHRF1 achieves this by binding to Bcl-2 proteins and preventing the formation of Bax/Bak oligomers on the outer mitochondrial membrane (OMM). This action helps EBV-infected cells survive and supports viral persistence and replication [126–128]. BILF1 is also involved in MAVS and NLRP3 inflammasome activation to block inflammatory responses during reactivation. This allows the virus to suppress immune pathways and promote replication [129,130]. BHRF1 also disrupts mtDNA replication by moving mitochondrial single-stranded DNA-binding protein (mtSSB), which initiates mtDNA replication by a transcription-priming process, from the mitochondria to the nucleus, inhibiting mtDNA replication [131].
• HCV: The p7 protein encoded by HCV forms membrane- membrane depolarization, and interacts with interferon-inducible protein 6–16 (IFI6-16). HCV and mitochondria interaction can result in increased mitochondrial ROS production and viral replication [120]. The Hepatitis C virus employs NS3/4A protease and targets the key components of the host’s antiviral defense. It specifically cleaves and dislocates MAVS and impairs IFN induction of the antiviral signaling in the host cell. MAVS localization is crucial for its antiviral activity and the disruption in its function by HCV’s NS3/4A proves the importance of MAVS in antiviral immune responses [92,132].
• HBV: HBx is a multifunctional protein that can localize to various mitochondrial compartments, including the outer mitochondrial membrane (OMM), inner mitochondrial membrane (IMM), and matrix. It has roles in cellular transcription, cell cycle regulation, calcium signaling, DNA repair, ROS regulation, and apoptosis. Generally, HBV can interact with mitochondria by some of its encoded specific proteins which result in different functions such as HBV polymerase (pol) that has a mitochondrial targeting signal and is localized to mitochondria during HBV replication. It inhibits innate immune responses and interacts with cellular proteins involved in mitochondrial functions. HBx also interacts with MAVS disrupting MAVS-mediated antiviral signaling and attenuating IFN-β induction [133]. Higher levels of mtDNA have also been observed in the peripheral blood of chronic HBV patients and are associated with liver cirrhosis and tumor size in HCC patients. Elevated mitochondrial superoxide and altered mitochondria are also reported. HBx-induced mitochondrial fragmentation and mitophagy are linked to viral persistence and cell survival. These processes also influence ROS generation and carcinogenesis. HBx induces mitochondrial ROS production and alters mtDNA, which can be linked to chronic HBV infection and HCC. Dynamin-related protein 1 (Drp1) is another protein in mitochondria which is essential for mitochondrial fission. HBx interacts with Drp1 and interfere with normal mitochondrial biogenesis [130]. HBx also interacts with various mitochondrial proteins such as Membrane Associated Ring-CH-Type Finger 5 (MARCH 5) which can regulate mitochondrial dynamics and decreases HBx-induced NF-κB and COX-2 activity, AIM2 protein reduction by HBx contributes to HCC metastasis through epithelial-mesenchymal transition (EMT), p53 and Bax, two pro-apoptotic proteins, which HBx interacts with and enhances apoptosis, voltage-dependent anion-selective channel protein 3 (VDAC3) which is an outer membrane mitochondrial protein that HBx binds to it and affects mitochondrial membrane potential and contributes to cell death [133,134], HSP60 and HSP70 (Heat Shock Proteins) that HBx interacts with and enhances apoptosis. Parvulins (Par14 and Par17) also interact with HBx, influencing its nuclear and mitochondrial localization and HBV replication [133]. HBx also induces mitochondrial accumulation of p53 and loss of mitochondrial membrane potential, which contributes to apoptotic cell death [135].
• HPV: The E2 protein encoded by HPV enhances mitochondrial biogenesis by up-regulating mitochondrial transcription factor A (TFAM)-interacting protein known as P32/gC1qR. C1QBP is a mitochondrial protein involved in regulating mitochondria and endoplasmic reticulum (ER) morphology and cell metabolism. HPV-18 E2 protein also can directly interact with the respiratory chain, leading to an increase in mitochondrial ROS production. The increase in ROS level is associated with the stabilization of hypoxia-inducible factor 1-alpha (HIF-1α) and enhanced glycolysis [136–138]. In addition, in cells lacking cytokeratin, HPV16 E1^E4 protein binds to mitochondria and induces detachment of mitochondria from microtubules, which results in the formation of a large mitochondrial cluster near the nucleus and leads to reduced mitochondrial membrane potential (MMP) and induces apoptotic cell death. E1^E4 also causes the collapse of the cytokeratin network in keratinocytes, which leads to the accumulation of E1^E4 in mitochondria. This clustering is associated with a severe reduction in mitochondrial membrane potential and increased apoptosis [122,139].
Based on the conducted research, the complex interactions between oncogenic viruses, mitochondria, and cell death, highlight the unneglectable role of mitochondria and cell death in viral pathogenesis and tumor development, conversely. Oncogenic viruses, such as HPV, EBV, HBV, and HCV can manipulate mitochondrial functions, including energy metabolism, ROS production, and cell death regulation, to establish persistent infections and evade immune responses. These viruses utilize their encoded oncoproteins to interfere with mitochondrial pathways, often suppressing apoptotic signals while promoting alternative cell death pathways like pyroptosis and necroptosis, depending on their needs and the favorable environment in their host cell. This ability to alter mitochondrial dynamics and functions contributes not only to viral survival but also to its oncogenesis potentials, as these processes lead to immune evasion, chronic inflammation, and genomic instability. Thus, targeting the mitochondria-cell death axis represents a promising therapeutic strategy against virus-induced cancers. Further research is crucial to uncover the detailed mechanisms of how specific viral proteins modulate mitochondrial function, which could lead to new treatments aimed at restoring normal cell death processes and preventing cancer progression. As discussed earlier in this review article, previous research has not directly focused on the possible influences that mitochondria and cell death pathways association might have on the oncogenicity of oncogenic viruses. Here we tried to mention and discuss the discovered associations between mitochondria, cell death pathways, and oncogenic viruses to propose a new hypothesis that these interactions might play a pivotal role in the progression of cancer induced by oncogenic viruses. Hence, this criterion might act as a promising target for personalized medicine and more advanced therapeutic targets to restrict cancer development.
Acknowledgement: The authors thank all members of the medical virology lab for their help.
Funding Statement: No funding was received for conducting this study.
Author Contributions: The authors confirm their contribution to the paper as follows: Study conception and design: Sahel Abyar, Seyed Reza Mohebbi; Draft manuscript preparation: Sahel Abyar, Shahrzad Shoraka; Review and editing: Seyed Masoud Hosseini, Mohammad Reza Zali, Seyed Reza Mohebbi; Visualization: Sahel Abyar, Seyed Reza Mohebbi, Shahrzad Shoraka; Supervision: Seyed Masoud Hosseini, Mohammad Reza Zali. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Moles R, Sarkis S, Galli V, Omsland M, Artesi M, Bissa M, et al. NK cells and monocytes modulate primary HTLV-1 infection. PLoS Pathog. 2022;18(4):e1010416. doi:10.1371/journal.ppat.1010416. [Google Scholar] [PubMed] [CrossRef]
2. Tornesello ML, Annunziata C, Tornesello AL, Buonaguro L, Buonaguro FM. Human oncoviruses and p53 tumor suppressor pathway deregulation at the origin of human cancers. Cancers. 2018;10(7):213. doi:10.3390/cancers10070213. [Google Scholar] [PubMed] [CrossRef]
3. Krump NA, You J. Molecular mechanisms of viral oncogenesis in humans. Nat Rev Microbiol. 2018;16(11):684–98. doi:10.1038/s41579-018-0064-6. [Google Scholar] [PubMed] [CrossRef]
4. Hatano Y, Ideta T, Hirata A, Hatano K, Tomita H, Okada H, et al. Virus-driven carcinogenesis. Cancers. 2021;13(11):2625. doi:10.3390/cancers13112625. [Google Scholar] [PubMed] [CrossRef]
5. Tempera I, Lieberman PM. Oncogenic viruses as entropic drivers of cancer evolution. Front Virol. 2021;1:753366. doi:10.3389/fviro.2021.753366. [Google Scholar] [PubMed] [CrossRef]
6. Charostad J, Azaran A, Nakhaei M, Astani A, Kaydani GA, Motamedfar A, et al. Upregulation of interleukin-6 in HPV-positive breast cancer patients. Iran J Immunol. 2021;18(4):315–30. doi:10.22034/IJI.2021.89107.1930. [Google Scholar] [PubMed] [CrossRef]
7. Gaballah A, Bartosch B. An update on the metabolic landscape of oncogenic viruses. Cancers. 2022;14(23):5742. doi:10.3390/cancers14235742. [Google Scholar] [PubMed] [CrossRef]
8. Bruni D, Angell HK, Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer. 2020;20(11):662–80. doi:10.1038/s41568-020-0285-7. [Google Scholar] [PubMed] [CrossRef]
9. Zapatka M, Borozan I, Brewer DS, Iskar M, Grundhoff A, Alawi M, et al. The landscape of viral associations in human cancers. Nat Genet. 2020;52(3):320–30. doi:10.1038/s41588-019-0558-9. [Google Scholar] [PubMed] [CrossRef]
10. Mesri EA, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host Microbe. 2014;15(3):266–82. doi:10.1016/j.chom.2014.02.011. [Google Scholar] [PubMed] [CrossRef]
11. Fang X, Tong W, Wu S, Zhu Z, Zhu J. The role of intratumoral microorganisms in the progression and immunotherapeutic efficacy of head and neck cancer. Oncologie. 2024;26(3):349–60. doi:10.1515/oncologie-2023-0511. [Google Scholar] [CrossRef]
12. Shen C, Jiang X, Li M, Luo Y. Hepatitis virus and hepatocellular carcinoma: recent advances. Cancers. 2023;15(2):533. doi:10.3390/cancers15020533. [Google Scholar] [PubMed] [CrossRef]
13. Naik AQ, Zafar T, Shrivastava VK, Shaik B, Ucar S. The microbiome and cancer: understanding the role of microorganisms in tumor development and treatment. In: The microbiome and cancer: understanding the role of microorganisms in tumor development and treatment. Singapore: Springer Nature Singapore; 2024. p. 1–25. [Google Scholar]
14. Hopcraft SE, Damania B. Tumour viruses and innate immunity. Philos Trans R Soc Lond B Biol Sci. 2017;372(1732):20160267. doi:10.1098/rstb.2016.0267. [Google Scholar] [PubMed] [CrossRef]
15. Mannová P, Beretta L. Activation of the N-Ras-PI3K-Akt-mTOR pathway by hepatitis C virus: control of cell survival and viral replication. J Virol. 2005;79(14):8742–9. doi:10.1128/JVI.79.14.8742-8749.2005. [Google Scholar] [PubMed] [CrossRef]
16. McLaughlin-Drubin ME, Munger K. Viruses associated with human cancer. Biochim Biophys Acta. 2008;1782(3):127–50. doi:10.1016/j.bbadis.2007.12.005. [Google Scholar] [PubMed] [CrossRef]
17. Wei S, Liu C, Du L, Wu B, Zhong J, Tong Y, et al. Identification of a novel class of cyclic penta-peptides against hepatitis C virus as p7 channel blockers. Comput Struct Biotechnol J. 2022;20(10):5902–10. doi:10.1016/j.csbj.2022.10.035. [Google Scholar] [PubMed] [CrossRef]
18. Mahgoub M, Yasunaga JI, Iwami S, Nakaoka S, Koizumi Y, Shimura K, et al. Sporadic on/off switching of HTLV-1 Tax expression is crucial to maintain the whole population of virus-induced leukemic cells. Proc Natl Acad Sci U S A. 2018;115(6):E1269–78. doi:10.1073/pnas.1715724115. [Google Scholar] [PubMed] [CrossRef]
19. Van Prooyen N, Gold H, Andresen V, Schwartz O, Jones K, Ruscetti F, et al. Human T-cell leukemia virus type 1 p8 protein increases cellular conduits and virus transmission. Proc Natl Acad Sci U S A. 2010;107(48):20738–43. doi:10.1073/pnas.1009635107. [Google Scholar] [PubMed] [CrossRef]
20. Rowe M, Raithatha S, Shannon-Lowe C. Counteracting effects of cellular Notch and Epstein-Barr virus EBNA2: implications for stromal effects on virus-host interactions. J Virol. 2014;88(20):12065–76. doi:10.1128/JVI.01431-14. [Google Scholar] [PubMed] [CrossRef]
21. Adamson AL, Le BT, Siedenburg BD. Inhibition of mTORC1 inhibits lytic replication of Epstein-Barr virus in a cell-type specific manner. Virol J. 2014;11(1):110. doi:10.1186/1743-422X-11-110. [Google Scholar] [PubMed] [CrossRef]
22. Brimer N, Lyons C, Wallberg AE, Vande Pol SB. Cutaneous papillomavirus E6 oncoproteins associate with MAML1 to repress transactivation and NOTCH signaling. Oncogene. 2012;31(43):4639–46. doi:10.1038/onc.2011.589. [Google Scholar] [PubMed] [CrossRef]
23. Cruz-Gregorio A, Aranda-Rivera AK, Aparicio-Trejo OE, Coronado-Martínez I, Pedraza-Chaverri J, Lizano M. E6 Oncoproteins from high-risk human papillomavirus induce mitochondrial metabolism in a head and neck squamous cell carcinoma model. Biomolecules. 2019;9(8):351. doi:10.3390/biom9080351. [Google Scholar] [PubMed] [CrossRef]
24. Xie Q, Fan F, Wei W, Liu Y, Xu Z, Zhai L, et al. Multi-omics analyses reveal metabolic alterations regulated by hepatitis B virus core protein in hepatocellular carcinoma cells. Sci Rep. 2017;7(1):41089. doi:10.1038/srep41089. [Google Scholar] [PubMed] [CrossRef]
25. Shin HJ, Park YH, Kim SU, Moon HB, Park DS, Han YH, et al. Hepatitis B virus X protein regulates hepatic glucose homeostasis via activation of inducible nitric oxide synthase. J Biol Chem. 2011;286(34):29872–81. doi:10.1074/jbc.M111.259978. [Google Scholar] [PubMed] [CrossRef]
26. Zhang G, Chan B, Samarina N, Abere B, Weidner-Glunde M, Buch A, et al. Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc Natl Acad Sci U S A. 2016;113(8):E1034–43. doi:10.1073/pnas.1516812113. [Google Scholar] [PubMed] [CrossRef]
27. Kajikawa M, Li PC, Goto E, Miyashita N, Aoki-Kawasumi M, Mito-Yoshida M, et al. The intertransmembrane region of Kaposi’s sarcoma-associated herpesvirus modulator of immune recognition 2 contributes to B7-2 downregulation. J Virol. 2012;86(9):5288–96. doi:10.1128/JVI.00219-12. [Google Scholar] [PubMed] [CrossRef]
28. Chen L, Ding L, Wang X, Huang Y, Gao S-J. Activation of glucocorticoid receptor signaling inhibits KSHV-induced inflammation and tumorigenesis. mBio. 2024;15(1):e0301123. doi:10.1128/mbio.03011-23. [Google Scholar] [PubMed] [CrossRef]
29. Charostad J, Nakhaie M, Dehghani A, Faghihloo E. The interplay between EBV and KSHV viral products and NF-κB pathway in oncogenesis. Infect Agent Cancer. 2020;15(1):62. doi:10.1186/s13027-020-00317-4. [Google Scholar] [PubMed] [CrossRef]
30. Houben R, Shuda M, Weinkam R, Schrama D, Feng H, Chang Y, et al. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J Virol. 2010;84(14):7064–72. doi:10.1128/JVI.02400-09. [Google Scholar] [PubMed] [CrossRef]
31. Yuan J, Ofengeim D. A guide to cell death pathways. Nat Rev Mol Cell Biol. 2024;25(5):379–95. doi:10.1038/s41580-023-00689-6. [Google Scholar] [PubMed] [CrossRef]
32. Sanz AB, Sanchez-Niño MD, Ramos AM, Ortiz A. Regulated cell death pathways in kidney disease. Nat Rev Nephrol. 2023;19(5):281–99. doi:10.1038/s41581-023-00694-0. [Google Scholar] [PubMed] [CrossRef]
33. Tsuchiya K. Switching from apoptosis to pyroptosis: gasdermin-elicited inflammation and antitumor immunity. Int J Mol Sci. 2021;22(1):426. doi:10.3390/ijms22010426. [Google Scholar] [PubMed] [CrossRef]
34. Wang Q, Imamura R, Motani K, Kushiyama H, Nagata S, Suda T. Pyroptotic cells externalize eat-me and release find-me signals and are efficiently engulfed by macrophages. Int Immunol. 2013;25(6):363–72. doi:10.1093/intimm/dxs161. [Google Scholar] [PubMed] [CrossRef]
35. Zhang Y, Chen X, Gueydan C, Han J. Plasma membrane changes during programmed cell deaths. Cell Res. 2018;28(1):9–21. doi:10.1038/cr.2017.133.Epub Epub 2017 Oct 27. [Google Scholar] [CrossRef]
36. Yan GE, Elbadawi M, Efferth T. Multiple cell death modalities and their key features. Acad Sci J. 2020;2(2):39–48. [Google Scholar]
37. Kesavardhana S, Malireddi RKS, Kanneganti TD. Caspases in cell death, inflammation, and pyroptosis. Annu Rev Immunol. 2020;38(1):567–95. doi:10.1146/annurev-immunol-073119-095439. [Google Scholar] [PubMed] [CrossRef]
38. Herr DR, Yam TYA, Tan WSD, Koh SS, Wong WSF, Ong WY, et al. Ultrastructural characteristics of DHA-induced pyroptosis. Neuromolecular Med. 2020;22(2):293–303. doi:10.1007/s12017-019-08586-y. [Google Scholar] [PubMed] [CrossRef]
39. Christgen S, Zheng M, Kesavardhana S, Karki R, Malireddi RKS, Banoth B, et al. Identification of the PANoptosome: a molecular platform triggering pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol. 2020;10:237. doi:10.3389/fcimb.2020.00237. [Google Scholar] [PubMed] [CrossRef]
40. Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol. 2021;18(5):1106–21. doi:10.1038/s41423-020-00630-3. [Google Scholar] [PubMed] [CrossRef]
41. Guo R, Wang H, Cui N. Autophagy regulation on pyroptosis: mechanism and medical implication in sepsis. Mediators Inflamm. 2021;2021:9925059. doi:10.1155/2021/9925059. [Google Scholar] [PubMed] [CrossRef]
42. Song H, Yang B, Li Y, Qian A, Kang Y, Shan X. Focus on the mechanisms and functions of pyroptosis, inflammasomes, and inflammatory caspases in infectious diseases. Oxid Med Cell Longev. 2022;2022(15):2501279. doi:10.1155/2022/2501279. [Google Scholar] [PubMed] [CrossRef]
43. Mulvihill E, Sborgi L, Mari SA, Pfreundschuh M, Hiller S, Müller DJ. Mechanism of membrane pore formation by human gasdermin-D. EMBO J. 2018;37(14):e98321. doi:10.15252/embj.201798321. [Google Scholar] [PubMed] [CrossRef]
44. Li J, Chen L, Zhou C, Bai Y, Zhao R, Zhang J, et al. Insight to pyroptosis in viral infectious diseases. Health. 2021;13(5):574–90. doi:10.4236/health.2021.135043. [Google Scholar] [CrossRef]
45. Wei X, Xie F, Zhou X, Wu Y, Yan H, Liu T, et al. Role of pyroptosis in inflammation and cancer. Cell Mol Immunol. 2022;19(9):971–92. doi:10.1038/s41423-022-00905-x. [Google Scholar] [PubMed] [CrossRef]
46. Liu J, Hong M, Li Y, Chen D, Wu Y, Hu Y. Programmed cell death tunes tumor immunity. Front Immunol. 2022;13:847345. doi:10.3389/fimmu.2022.847345. [Google Scholar] [PubMed] [CrossRef]
47. Chen J, Jiang R, Guan W, Cao Q, Tian Y, Dong K, et al. Novel model of pyroptosis-related molecular signatures for prognosis prediction of clear cell renal cell carcinoma patients. Int J Med Sci. 2024;21(3):496–507. doi:10.7150/ijms.88301. [Google Scholar] [PubMed] [CrossRef]
48. Liu Z, Wang C, Yang J, Zhou B, Yang R, Ramachandran R, et al. Crystal structures of the full-length murine and human Gasdermin D reveal mechanisms of autoinhibition, lipid binding, and oligomerization. Immunity. 2019;51(1):43–9. doi:10.1016/j.immuni.2019.04.017. [Google Scholar] [PubMed] [CrossRef]
49. Li S, Sun Y, Song M, Song Y, Fang Y, Zhang Q, et al. NLRP3/caspase-1/GSDMD-mediated pyroptosis exerts a crucial role in astrocyte pathological injury in mouse model of depression. JCI Insight. 2021;6(23):e146852. doi:10.1172/jci.insight.146852. [Google Scholar] [PubMed] [CrossRef]
50. Wang Y, Liu Y, Liu Q, Zheng Q, Dong X, Liu X, et al. Caspase-1-dependent pyroptosis of peripheral blood mononuclear cells is associated with the severity and mortality of septic patients. Biomed Res Int. 2020;2020(1):9152140. doi:10.1155/2020/9152140. [Google Scholar] [PubMed] [CrossRef]
51. Choudhury SM, Ma X, Abdullah SW, Zheng H. Activation and Inhibition of the NLRP3 inflammasome by RNA viruses. J Inflamm Res. 2021;14:1145–63. doi:10.2147/JIR.S295706. [Google Scholar] [PubMed] [CrossRef]
52. Zhou C, Shen Y, Jin Y, Shen Z, Ye D, Shen Y, et al. A novel Pyroptosis-related long non-coding RNA signature for predicting the prognosis and immune landscape of head and neck squamous cell carcinoma. Cancer Med. 2022;11(24):5097–112. doi:10.1002/cam4.4819. [Google Scholar] [PubMed] [CrossRef]
53. Chen X, He WT, Hu L, Li J, Fang Y, Wang X, et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 2016;26(9):1007–20. doi:10.1038/cr.2016.100. [Google Scholar] [PubMed] [CrossRef]
54. Zhou D, Zhu Y, Jiang P, Zhang T, Zhuang J, Li T, et al. Identifying pyroptosis-and inflammation-related genes in intracranial aneurysms based on bioinformatics analysis. Biol Res. 2023;56(1):50. doi:10.1186/s40659-023-00464-z. [Google Scholar] [PubMed] [CrossRef]
55. Akash S, Bayıl I, Hossain MS, Islam MR, Hosen ME, Mekonnen AB, et al. Novel computational and drug design strategies for inhibition of human papillomavirus-associated cervical cancer and DNA polymerase theta receptor by Apigenin derivatives. Sci Rep. 2023;13(1):16565. doi:10.1038/s41598-023-43175-x. [Google Scholar] [PubMed] [CrossRef]
56. Araldi RP, Sant’Ana TA, Módolo DG, de Melo TC, Spadacci-Morena DD, de Cassia Stocco R, et al. The human papillomavirus (HPV)-related cancer biology: an overview. Biomed Pharmacother. 2018;106(593):1537–56. doi:10.1016/j.biopha.2018.06.149. [Google Scholar] [PubMed] [CrossRef]
57. Cai Q, Lv L, Shao Q, Li X, Dian A. Human papillomavirus early proteins and apoptosis. Arch Gynecol Obstet. 2013;287(3):541–8. doi:10.1007/s00404-012-2665-z. [Google Scholar] [PubMed] [CrossRef]
58. Haghighi ZMS, Tabatabaei T, Rafigh M, Karampour R, Babaei F, Amjad ZS, et al. Human papillomavirus maybe is a critical player in the regulation of chemoresistance related factors (P53, Rb, TWIST, Bcl-2, Bcl-XL, c-IAP2, cytochrome C, and caspase 3) in breast cancer. Pathol Res Pract. 2023;248(2012):154653. doi:10.1016/j.prp.2023.154653. [Google Scholar] [PubMed] [CrossRef]
59. Vats A, Trejo-Cerro O, Thomas M, Banks L. Human papillomavirus E6 and E7: what remains? Tumour Virus Res. 2021;11(2):200213. doi:10.1016/j.tvr.2021.200213. [Google Scholar] [PubMed] [CrossRef]
60. Skelin J, Sabol I, Tomaić V. Do or die: hPV E5, E6 and E7 in cell death evasion. Pathogens. 2022;11(9):1027. doi:10.3390/pathogens11091027. [Google Scholar] [PubMed] [CrossRef]
61. Basukala O, Banks L. The not-so-good, the bad and the ugly: hPV E5, E6 and E7 oncoproteins in the orchestration of carcinogenesis. Viruses. 2021;13(10):1892. doi:10.3390/v13101892. [Google Scholar] [PubMed] [CrossRef]
62. Wells SI, Francis DA, Karpova AY, Dowhanick JJ, Benson JD, Howley PM. Papillomavirus E2 induces senescence in HPV-positive cells via pRB- and p21CIP-dependent pathways. EMBO J. 2000;19(21):5762–71. doi:10.1093/emboj/19.21.5762. [Google Scholar] [PubMed] [CrossRef]
63. Brown C, Kowalczyk AM, Taylor ER, Morgan IM, Gaston K. P53 represses human papillomavirus type 16 DNA replication via the viral E2 protein. Virol J. 2008;5(1):5. doi:10.1186/1743-422X-5-5. [Google Scholar] [PubMed] [CrossRef]
64. Jamal DF, Rozaimee QA, Osman NH, Mohd Sukor A, Elias MH, Shamaan NA, et al. Human papillomavirus 16 E2 as an apoptosis-inducing protein for cancer treatment: a systematic review. Int J Mol Sci. 2022;23(20):12554. doi:10.3390/ijms232012554. [Google Scholar] [PubMed] [CrossRef]
65. Fontan CT, James CD, Prabhakar AT, Bristol ML, Otoa R, Wang X, et al. A critical role for p53 during the HPV16 life cycle. Microbiol Spectr. 2022;10(3):e0068122. doi:10.1128/spectrum.00681-22. [Google Scholar] [PubMed] [CrossRef]
66. Teng Y, Xu D, Yang X, Tang H, Tao X, Fan Y, et al. The emerging roles of pyroptosis, necroptosis, and ferroptosis in non-malignant dermatoses: a review. J Inflamm Res. 2023;16:1967–77. doi:10.2147/JIR.S409699. [Google Scholar] [PubMed] [CrossRef]
67. Balachandran S, Adams GP. Interferon-γ-induced necrosis: an antitumor biotherapeutic perspective. J Interferon Cytokine Res. 2013;33(4):171–80. doi:10.1089/jir.2012.0087. [Google Scholar] [PubMed] [CrossRef]
68. Thapa RJ, Nogusa S, Chen P, Maki JL, Lerro A, Andrake M, et al. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci U S A. 2013;110(33):E3109–18. doi:10.1073/pnas.1301218110. [Google Scholar] [PubMed] [CrossRef]
69. Fang Y, Tian S, Pan Y, Li W, Wang Q, Tang Y, et al. Pyroptosis: a new frontier in cancer. Biomed Pharmacother. 2020;121(2):109595. doi:10.1016/j.biopha.2019.109595. [Google Scholar] [PubMed] [CrossRef]
70. Fitzsimmons L, Kelly GL. EBV and apoptosis: the viral master regulator of cell fate? Viruses. 2017;9(11):339. doi:10.3390/v9110339. [Google Scholar] [PubMed] [CrossRef]
71. Anderton E, Yee J, Smith P, Crook T, White RE, Allday MJ. Two Epstein-Barr virus (EBV) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor Bim: clues to the pathogenesis of Burkitt’s lymphoma. Oncogene. 2008;27(4):421–33. doi:10.1038/sj.onc.1210668. [Google Scholar] [PubMed] [CrossRef]
72. Lurain KA, Ramaswami R, Krug LT, Whitby D, Ziegelbauer JM, Wang H-W, et al. HIV-associated cancers and lymphoproliferative disorders caused by Kaposi sarcoma herpesvirus and Epstein-Barr virus. Clin Microbiol Rev. 2024;37(3):e0002223. doi:10.1128/cmr.00022-23. [Google Scholar] [PubMed] [CrossRef]
73. Fierti AO, Yakass MB, Okertchiri EA, Adadey SM, Quaye O. The role of epstein-barr virus in modulating key tumor suppressor genes in associated malignancies: epigenetics, transcriptional, and post-translational modifications. Biomolecules. 2022;12(1):127. doi:10.3390/biom12010127. [Google Scholar] [PubMed] [CrossRef]
74. Huang J, Zhang X, Nie X, Zhang X, Wang Y, Huang L, et al. Assembly and activation of EBV latent membrane protein 1. Cell. 2024;187(18):4996–5009. doi:10.1016/j.cell.2024.06.021. [Google Scholar] [PubMed] [CrossRef]
75. Mitra B, Beri NR, Guo R, Burton EM, Murray-Nerger LA, Gewurz BE. Characterization of target gene regulation by the two Epstein-Barr virus oncogene LMP1 domains essential for B-cell transformation. mBio. 2023;14(6):e0233823. doi:10.1128/mbio.02338-23. [Google Scholar] [PubMed] [CrossRef]
76. Fukuda M, Longnecker R. Epstein-Barr virus latent membrane protein 2A mediates transformation through constitutive activation of the Ras/PI3-K/Akt Pathway. J Virol. 2007;81(17):9299–306. doi:10.1128/JVI.00537-07. [Google Scholar] [PubMed] [CrossRef]
77. Sun Y, Shi D, Sun J, Zhang Y, Liu W, Luo B. Regulation mechanism of EBV-encoded EBER1 and LMP2A on YAP1 and the impact of YAP1 on the EBV infection status in EBV-associated gastric carcinoma. Virus Res. 2024;343(10):199352. doi:10.1016/j.virusres.2024.199352. [Google Scholar] [PubMed] [CrossRef]
78. Kalla M, Schmeinck A, Bergbauer M, Pich D, Hammerschmidt W. AP-1 homolog BZLF1 of Epstein-Barr virus has two essential functions dependent on the epigenetic state of the viral genome. Proc Natl Acad Sci U S A. 2010;107(2):850–5. doi:10.1073/pnas.0911948107. [Google Scholar] [PubMed] [CrossRef]
79. Xiao L, Hu ZY, Dong X, Tan Z, Li W, Tang M, et al. Targeting Epstein-Barr virus oncoprotein LMP1-mediated glycolysis sensitizes nasopharyngeal carcinoma to radiation therapy. Oncogene. 2014;33(37):4568–78. doi:10.1038/onc.2014.32. [Google Scholar] [PubMed] [CrossRef]
80. Lu J, Tang M, Li H, Xu Z, Weng X, Li J, et al. EBV-LMP1 suppresses the DNA damage response through DNA-PK/AMPK signaling to promote radioresistance in nasopharyngeal carcinoma. Cancer Lett. 2016;380(1):191–200. doi:10.1016/j.canlet.2016.05.032. [Google Scholar] [PubMed] [CrossRef]
81. Liu X, Li Y, Peng S, Yu X, Li W, Shi F, et al. Epstein-Barr virus encoded latent membrane protein 1 suppresses necroptosis through targeting RIPK1/3 ubiquitination. Cell Death Dis. 2018;9(2):53. doi:10.1038/s41419-017-0081-9. [Google Scholar] [PubMed] [CrossRef]
82. Long X, Yang J, Zhang X, Yang Z, Li Y, Wang F, et al. BRLF1 suppresses RNA Pol III-mediated RIG-I inflammasome activation in the early EBV lytic lifecycle. EMBO Rep. 2021;22(1):e50714. doi:10.15252/embr.202050714. [Google Scholar] [PubMed] [CrossRef]
83. Ansari MA, Singh VV, Dutta S, Veettil MV, Chikoti D, Lu L, et al. Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I, II, and III in B and epithelial cells. J Virol. 2013;87(15):8606–23. doi:10.1128/JVI.00805-13. [Google Scholar] [PubMed] [CrossRef]
84. Skinner CM, Ivanov NS, Barr SA, Chen Y, Skalsky RL. An epstein-barr virus MicroRNA blocks interleukin-1 (IL-1) signaling by targeting IL-1 receptor 1. J Virol. 2017;91(21):e00530–17. doi:10.1128/JVI.00530-17. [Google Scholar] [PubMed] [CrossRef]
85. Zhang HT, Chen GG, Hu BG, Zhang ZY, Yun JP, He ML, et al. Hepatitis B virus x protein induces autophagy via activating death-associated protein kinase. J Viral Hepat. 2014;21(9):642–9. doi:10.1111/jvh.12191. [Google Scholar] [PubMed] [CrossRef]
86. Jiang Y, Han Q, Zhao H, Zhang J. The mechanisms of HBV-induced hepatocellular carcinoma. J Hepatocell Carcinoma. 2021;8:435–50. doi:10.2147/JHC.S307962. [Google Scholar] [PubMed] [CrossRef]
87. Lei Y, Xu X, Liu H, Chen L, Zhou H, Jiang J, et al. HBx induces hepatocellular carcinogenesis through ARRB1-mediated autophagy to drive the G1/S cycle. Autophagy. 2021;17(12):4423–41. doi:10.1080/15548627.2021.1917948. [Google Scholar] [PubMed] [CrossRef]
88. Kim SJ, Khan M, Quan J, Till A, Subramani S, Siddiqui A. Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013;9(12):e1003722. doi:10.1371/journal.ppat.1003722.Epub Epub 2013 Dec 5. [Google Scholar] [CrossRef]
89. Han LY, Yang JR, Zhao ZH, Gao S, Fan YC, Wang K. RIPK3 mRNA level acts as a diagnostic biomarker in hepatitis B virus-associated hepatocellular carcinoma. Pathol Res Pract. 2020;216(10):153147. doi:10.1016/j.prp.2020.153147. [Google Scholar] [PubMed] [CrossRef]
90. Huang XY, Li D, Chen ZX, Huang YH, Gao WY, Zheng BY, et al. Hepatitis B Virus X protein elevates Parkin-mediated mitophagy through Lon Peptidase in starvation. Exp Cell Res. 2018;368(1):75–83. doi:10.1016/j.yexcr.2018.04.016. [Google Scholar] [PubMed] [CrossRef]
91. Schollmeier A, Glitscher M, Hildt E. Relevance of HBx for hepatitis B virus-associated pathogenesis. Int J Mol Sci. 2023;24(5):4964. doi:10.3390/ijms24054964. [Google Scholar] [PubMed] [CrossRef]
92. Lee J, Ou JJ. Hepatitis C virus and intracellular antiviral response. Curr Opin Virol. 2022;52:244–9. doi:10.1016/j.coviro.2021.12.010. [Google Scholar] [PubMed] [CrossRef]
93. Kofahi HM, Taylor NG, Hirasawa K, Grant MD, Russell RS. Hepatitis C virus infection of cultured human hepatoma cells causes apoptosis and pyroptosis in both infected and bystander cells. Sci Rep. 2016;6(1):37433. doi:10.1038/srep37433. [Google Scholar] [PubMed] [CrossRef]
94. Wallace HL, Wang L, Gardner CL, Corkum CP, Grant MD, Hirasawa K, et al. Crosstalk between pyroptosis and apoptosis in hepatitis C virus-induced cell death. Front Immunol. 2022;13:788138. doi:10.3389/fimmu.2022.788138. [Google Scholar] [PubMed] [CrossRef]
95. Wang HW, Sharp TV, Koumi A, Koentges G, Boshoff C. Characterization of an anti-apoptotic glycoprotein encoded by Kaposi’s sarcoma-associated herpesvirus which resembles a spliced variant of human survivin. EMBO J. 2002;21(11):2602–15. doi:10.1093/emboj/21.11.2602. [Google Scholar] [PubMed] [CrossRef]
96. Zhang X, Lan Q, Zhang M, Wang F, Shi K, Li X, et al. Inhibition of AIM2 inflammasome activation by SOX/ORF37 promotes lytic replication of Kaposi’s sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A. 2023;120(27):e2300204120. doi:10.1073/pnas.2300204120. [Google Scholar] [PubMed] [CrossRef]
97. De Re V, Caggiari L, De Zorzi M, Fanotto V, Miolo G, Puglisi F, et al. Epstein-Barr virus BART microRNAs in EBV-associated Hodgkin lymphoma and gastric cancer. Infect Agent Cancer. 2020;15(1):42. doi:10.1186/s13027-020-00307-6. [Google Scholar] [PubMed] [CrossRef]
98. Wyatt S, Glover K, Dasanna S, Lewison M, González-García M, Colbert CL, et al. Epstein-barr virus encoded BCL2, BHRF1, downregulates autophagy by noncanonical binding of BECN1. Biochemistry. 2023;62(20):2934–51. doi:10.1021/acs.biochem.3c00225. [Google Scholar] [PubMed] [CrossRef]
99. Wyżewski Z, Mielcarska MB, Gregorczyk-Zboroch KP, Myszka A. Virus-mediated inhibition of apoptosis in the context of EBV-associated diseases: molecular mechanisms and therapeutic perspectives. Int J Mol Sci. 2022;23(13):7265. doi:10.3390/ijms23137265. [Google Scholar] [PubMed] [CrossRef]
100. Marusawa H, Hijikata M, Chiba T, Shimotohno K. Hepatitis C virus core protein inhibits Fas-and tumor necrosis factor alpha-mediated apoptosis via NF-κB activation. J Virol. 1999;73(6):4713–20. doi:10.1128/JVI.73.6.4713-4720.1999. [Google Scholar] [PubMed] [CrossRef]
101. Park J, Kang W, Ryu SW, Kim WI, Chang DY, Lee DH, et al. Hepatitis C virus infection enhances TNFκ-induced cell death via suppression of NF-κB. Hepatology. 2012;56(3):831–40. doi:10.1002/hep.25726. [Google Scholar] [PubMed] [CrossRef]
102. Punj V, Matta H, Schamus S, Yang T, Chang Y, Chaudhary PM. Induction of CCL20 production by Kaposi sarcoma-associated herpesvirus: role of viral FLICE inhibitory protein K13-induced NF-κB activation. Blood. 2009;113(22):5660–8. doi:10.1182/blood-2008-10-186403. [Google Scholar] [PubMed] [CrossRef]
103. Moses AV, Jarvis MA, Raggo C, Bell YC, Ruhl R, Luukkonen BG, et al. Kaposi’s sarcoma-associated herpesvirus-induced upregulation of the c-kit proto-oncogene, as identified by gene expression profiling, is essential for the transformation of endothelial cells. J Virol. 2002;76(16):8383–99. doi:10.1128/JVI.76.16.8383-8399.2002. [Google Scholar] [PubMed] [CrossRef]
104. Koshiba T. Mitochondrial-mediated antiviral immunity. Biochim Biophys Acta. 2013;1833(1):225–32. doi:10.1016/j.bbamcr.2012.03.005. [Google Scholar] [PubMed] [CrossRef]
105. Rosenberg RN, Pascual JM. Rosenberg’s molecular and genetic basis of neurological and psychiatric disease. In: Rosenberg’s Molecular and genetic basis of neurological and psychiatric disease. Waltham, MA, USA: Academic Press; 2020. Vol. 2, p. 389–400. [Google Scholar]
106. Zhang X, Hu Y. Mitochondrial thermogenesis in cancer cells. Oncologie. 2023;25(6):591–603. doi:10.1515/oncologie-2023-0298. [Google Scholar] [CrossRef]
107. Foo J, Bellot G, Pervaiz S, Alonso S. Mitochondria-mediated oxidative stress during viral infection. Trends Microbiol. 2022;30(7):679–92. doi:10.1016/j.tim.2021.12.011. [Google Scholar] [PubMed] [CrossRef]
108. Murphy MP, Hartley RC. Mitochondria as a therapeutic target for common pathologies. Nat Rev Drug Discov. 2018;17(12):865–86. doi:10.1038/nrd.2018.174. [Google Scholar] [PubMed] [CrossRef]
109. Eldeeb MA, Thomas RA, Ragheb MA, Fallahi A, Fon EA. Mitochondrial quality control in health and in Parkinson’s disease. Physiol Rev. 2022;102(4):1721–55. doi:10.1152/physrev.00041.2021. [Google Scholar] [PubMed] [CrossRef]
110. Osellame LD, Blacker TS, Duchen MR. Cellular and molecular mechanisms of mitochondrial function. Best Pract Res Clin Endocrinol Metab. 2012;26(6):711–23. doi:10.1016/j.beem.2012.05.003. [Google Scholar] [PubMed] [CrossRef]
111. Devic S. Warburg effect—a consequence or the cause of carcinogenesis? J Cancer. 2016;7(7):817–22. doi:10.7150/jca.14274. [Google Scholar] [PubMed] [CrossRef]
112. Zhang B, Pan C, Feng C, Yan C, Yu Y, Chen Z, et al. Role of mitochondrial reactive oxygen species in homeostasis regulation. Redox Rep. 2022;27(1):45–52. doi:10.1080/13510002.2022.2046423. [Google Scholar] [PubMed] [CrossRef]
113. Sarmiento-Salinas FL, Perez-Gonzalez A, Acosta-Casique A, Ix-Ballote A, Diaz A, Treviño S, et al. Reactive oxygen species: role in carcinogenesis, cancer cell signaling and tumor progression. Life Sci. 2021;284(6):119942. doi:10.1016/j.lfs.2021.119942. [Google Scholar] [PubMed] [CrossRef]
114. Yeo D. Control of mitochondrial homeostasis in disused and aging skeletal muscle: role of Pgc-1Alpha and sirtuins., [Ph.D. dissertation]. Twin Cities, MN, USA: University of Minnesota; 2020. [Google Scholar]
115. Abu Shelbayeh O, Arroum T, Morris S, Busch KB. PGC-1α is a master regulator of mitochondrial lifecycle and ROS stress response. Antioxidants. 2023;12(5):1075. doi:10.3390/antiox12051075. [Google Scholar] [PubMed] [CrossRef]
116. Elesela S, Lukacs NW. Role of mitochondria in viral infections. Life. 2021;11(3):232. doi:10.3390/life11030232. [Google Scholar] [PubMed] [CrossRef]
117. Kim IS, Silwal P, Jo EK. Mitofusin 2, a key coordinator between mitochondrial dynamics and innate immunity. Virulence. 2021;12(1):2273–84. doi:10.1080/21505594.2021.1965829. [Google Scholar] [PubMed] [CrossRef]
118. Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146(3):448–61. doi:10.1016/j.cell.2011.06.041. [Google Scholar] [PubMed] [CrossRef]
119. Koshiba T, Yasukawa K, Yanagi Y, Kawabata S. Mitochondrial membrane potential is required for MAVS-mediated antiviral signaling. Sci Signal. 2011;4(158):ra7. doi:10.1126/scisignal.2001147. [Google Scholar] [PubMed] [CrossRef]
120. Moreno-Altamirano MMB, Kolstoe SE, Sánchez-García FJ. Virus control of cell metabolism for replication and evasion of host immune responses. Front Cell Infect Microbiol. 2019;9:95. doi:10.3389/fcimb.2019.00095. [Google Scholar] [PubMed] [CrossRef]
121. Sharma A, Kontodimas K, Bosmann M. The MAVS immune recognition pathway in viral infection and sepsis. Antioxid Redox Signal. 2021;35(16):1376–92. doi:10.1089/ars.2021.0167. [Google Scholar] [PubMed] [CrossRef]
122. Cavallari I, Scattolin G, Silic-Benussi M, Raimondi V, D’Agostino DM, Ciminale V. Mitochondrial proteins coded by human tumor viruses. Front Microbiol. 2018;9:81. doi:10.3389/fmicb.2018.00081. [Google Scholar] [PubMed] [CrossRef]
123. Pluta A, Jaworski JP, Douville RN. Regulation of expression and latency in BLV and HTLV. Viruses. 2020;12(10):1079. doi:10.3390/v12101079. [Google Scholar] [PubMed] [CrossRef]
124. Nakano K, Watanabe T. Tuning Rex rules HTLV-1 pathogenesis. Front Immunol. 2022;13:959962. doi:10.3389/fimmu.2022.959962. [Google Scholar] [PubMed] [CrossRef]
125. Nash A, Ryan EJ. The oncogenic gamma herpesviruses Epstein-Barr virus (EBV) and Kaposi’s sarcoma-associated herpesvirus (KSHV) hijack retinoic acid-inducible gene I (RIG-I) facilitating both viral and tumour immune evasion. Tumour Virus Res. 2022;14:200246. doi:10.1016/j.tvr.2022.200246. [Google Scholar] [PubMed] [CrossRef]
126. Fitzsimmons L, Cartlidge R, Chang C, Sejic N, Galbraith LCA, Suraweera CD, et al. EBV BCL-2 homologue BHRF1 drives chemoresistance and lymphomagenesis by inhibiting multiple cellular pro-apoptotic proteins. Cell Death Differ. 2020;27(5):1554–68. doi:10.1038/s41418-019-0435-1. [Google Scholar] [PubMed] [CrossRef]
127. Rosemarie Q, Sugden B. Epstein-barr virus: how its lytic phase contributes to oncogenesis. Microorganisms. 2020;8(11):1824. doi:10.3390/microorganisms8111824. [Google Scholar] [PubMed] [CrossRef]
128. Henderson S, Rowe M, Gregory C, Croom-Carter D, Wang F, Longnecker R, et al. Induction of bcl-2 expression by Epstein-Barr virus latent membrane protein 1 protects infected B cells from programmed cell death. Cell. 1991;65(7):1107–15. doi:10.1016/0092-8674(91)90007-l. [Google Scholar] [PubMed] [CrossRef]
129. Yiu SPT, Zerbe C, Vanderwall D, Huttlin EL, Weekes MP, Gewurz BE. An Epstein-Barr virus protein interaction map reveals NLRP3 inflammasome evasion via MAVS UFMylation. Mol Cell. 2023;83(13):2367–86. doi:10.1016/j.molcel.2023.05.018. [Google Scholar] [PubMed] [CrossRef]
130. Yiu SPT, Guo R, Zerbe C, Weekes MP, Gewurz BE. Epstein-Barr virus BNRF1 destabilizes SMC5/6 cohesin complexes to evade its restriction of replication compartments. Cell Rep. 2022;38(10):110411. doi:10.1016/j.celrep.2022.110411. [Google Scholar] [PubMed] [CrossRef]
131. Miralles Fusté J, Shi Y, Wanrooij S, Zhu X, Jemt E, Persson Ö, et al. In vivo occupancy of mitochondrial single-stranded DNA binding protein supports the strand displacement mode of DNA replication. PLoS Genet. 2014;10(12):e1004832. doi:10.1371/journal.pgen.1004832. [Google Scholar] [PubMed] [CrossRef]
132. Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci U S A. 2005;102(49):17717–22. doi:10.1073/pnas.0508531102. [Google Scholar] [PubMed] [CrossRef]
133. Hossain MG, Akter S, Ohsaki E, Ueda K. Impact of the interaction of hepatitis B virus with mitochondria and associated proteins. Viruses. 2020;12(2):175. doi:10.3390/v12020175. [Google Scholar] [PubMed] [CrossRef]
134. Schollmeier A, Basic M, Glitscher M, Hildt E. The impact of HBx protein on mitochondrial dynamics and associated signaling pathways strongly depends on the hepatitis B virus genotype. J Virol. 2024;98(5):e0042424. doi:10.1128/jvi.00424-24. [Google Scholar] [PubMed] [CrossRef]
135. Sivasudhan E, Blake N, Lu Z, Meng J, Rong R. Hepatitis B viral protein HBx and the molecular mechanisms modulating the hallmarks of hepatocellular carcinoma: a comprehensive review. Cells. 2022;11(4):741. doi:10.3390/cells11040741. [Google Scholar] [PubMed] [CrossRef]
136. Cruz-Gregorio A, Aranda-Rivera AK, Roviello GN, Pedraza-Chaverri J. Targeting mitochondrial therapy in the regulation of HPV infection and HPV-related cancers. Pathogens. 2023;12(3):402. doi:10.3390/pathogens12030402. [Google Scholar] [PubMed] [CrossRef]
137. Kierans SJ, Taylor CT. Regulation of glycolysis by the hypoxia-inducible factor (HIFimplications for cellular physiology. J Physiol. 2021;599(1):23–37. doi:10.1113/JP280572. [Google Scholar] [PubMed] [CrossRef]
138. Liang S, Dong S, Liu W, Wang M, Tian S, Ai Y, et al. Accumulated ROS activates HIF-1α-induced glycolysis and exerts a protective effect on sensory hair cells against noise-induced damage. Front Mol Biosci. 2022;8:806650. doi:10.3389/fmolb.2021.806650. [Google Scholar] [PubMed] [CrossRef]
139. Raj K, Berguerand S, Southern S, Doorbar J, Beard P. E1 empty set E4 protein of human papillomavirus type 16 associates with mitochondria. J Virol. 2004;78(13):7199–207. doi:10.1128/JVI.78.13.7199-7207.2004. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools