
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

The Role of Glutamate Receptors in Ischemic Stroke
1 New Drug Screening and Pharmacodynamics Evaluation Center, China Pharmaceutical University, Nanjing, 210009, China
2 Chongqing Innovation Institute of China Pharmaceutical University, Chongqing, 401135, China
* Corresponding Authors: Hao Sun. Email: ; Hong Liao. Email:
(This article belongs to the Special Issue: Cell Death and Inflammation in Signaling and Diseases)
BIOCELL 2025, 49(2), 167-180. https://doi.org/10.32604/biocell.2025.059159
Received 29 September 2024; Accepted 16 December 2024; Issue published 28 February 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Glutamate is an essential excitatory neurotransmitter in the brain, playing a vital role in regulating synaptic activity and maintaining the homeostasis of the cerebral environment but also serves as a central hub for neuronal injury and inflammatory responses. In various pathological conditions, such as ischemic stroke, glutamate is released and accumulates excessively in the brain, leading to heightened stimulation of neurons and excitotoxicity. This phenomenon positions glutamate as a primary inducing factor for neuronal damage following cerebral ischemia. Glutamate exerts its effects primarily through two types of receptors: ionotropic and metabotropic glutamate receptors, both of which are extensively distributed throughout the hippocampus and cortical regions of the brain. Ionotropic receptors mediate rapid excitatory neurotransmission upon activation by glutamate; these are mainly categorized into N-methyl-D-aspartate receptors (NMDARs), α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptors (AMPARs), and kainate receptors (KARs). Conversely, metabotropic receptors function as G-protein-coupled receptors (GPCR) facilitating glutamatergic cellular effects via intracellular second messenger. With the comprehensive investigation of glutamate receptors and their structural characteristics, our understanding of the nerve damage and protective mechanisms associated with glutamate receptors in ischemic stroke is progressively advancing. Consequently, exploring the role of glutamate receptors and their downstream signaling pathways in cerebral ischemia can provide a robust theoretical foundation for targeted therapies aimed at treating cerebral ischemia, stroke, and related disorders. This article reviews the function of glutamate receptors and their mediated downstream signal transduction pathways in the context of ischemic brain injury.Keywords
Stroke is the second leading cause of death and disability worldwide, with the highest incidence occurring in developing countries. It is primarily categorized into ischemic stroke and hemorrhagic stroke, with ischemic stroke accounting for approximately 87% of cases, making it the most prevalent type that poses a significant threat to human health [1]. With the establishment of green channels for emergency care and ongoing advancements in mechanical thrombectomy and thrombolytic therapy in clinical practice, mortality rates have been effectively managed. However, post-treatment rehabilitation remains a critical focus within clinical settings. Clinical data survey results show that the disability rate of stroke after recovery is as high as 70%–80%, which seriously affects the quality of life of patients and increases the economic burden of families [2]. Currently, tissue plasminogen activator (t-PA) is the only drug approved by the FDA for treating acute ischemic stroke [3]. Therefore, clarifying the mechanisms underlying functional rehabilitation after stroke and identifying potential rehabilitation targets hold great significance for drug development and clinical treatment strategies.
Modern medical research indicates that ischemic stroke is a neurological disorder primarily characterized by vascular obstruction. The formation of a thrombus within the brain disrupts blood flow by occluding arteries, leading to the death of numerous neurons and glial cells in the infarction core area due to ischemia. This process is accompanied by synaptic loss and the reduction in synaptic plasticity. One of the most critical mechanisms underlying neuronal injury during acute cerebral ischemia is glutamate-mediated excitotoxicity [4]. Following cerebral ischemia, ischemia and hypoxia in the infarcted region result in significant presynaptic membrane release of glutamate, causing an immediate upregulation of glutamate concentration in the synaptic cleft. This rapid increase acts on glutamate receptors located on the postsynaptic membrane as well as adjacent astrocytes, eliciting various cellular effects.
In recent decades, the structure, classification, distribution, and physiological functions of glutamate and its receptors have been extensively investigated. Specifically, glutamate primarily interacts with two types of receptors: ionotropic glutamate receptors (iGluRs) and metabotropic glutamate receptors (mGluRs), both of which are widely distributed in the hippocampus and cortical regions of the brain. The former is mainly categorized into N-methyl-D-aspartate receptors (NMDARs), α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptors (AMPARs), and kainate receptors (KARs) [5]. In contrast, mGluRs are G-protein-coupled receptors that mediate glutamatergic cellular effects through intracellular second messenger pathways [6]. Research has elucidated the mechanisms underlying neuronal death and survival mediated by glutamate receptors following cerebral ischemia. A significant release of glutamate activates postsynaptic membrane iGluRs particularly NMDA receptors (NMDAR) which can induce various downstream effects to modulate neuronal activity via distinct pathways. The diversity of mGluRs in the brain along with their widespread distribution renders them prime targets for treating a range of neurological and psychiatric disorders. Unlike iGluRs activation responsible for rapid excitatory synaptic transmission, mGluRs play a crucial role in fine-tuning this excitatory transmission as well as regulating circuit-specific activities [7]. Activation of mGluRs is associated with excessive intracellular signaling within the brain; disruptions in one or more of these pathways may significantly impact overall brain function. However, it is noteworthy that glutamate receptors on adjacent astrocytes especially Group II glutamate receptors may mediate certain protective effects [8]. Therefore, understanding the mechanisms governing neuronal death or protection mediated by glutamate receptors holds substantial significance for clinical interventions targeting ischemic stroke. This paper primarily discusses the signal transduction mechanisms involved in neuroprotection and injury mediated by glutamate receptor activity following ischemic stroke.
2 Distribution and Physiological Function of Glutamate Receptors
2.1 Ionic Glutamate Receptors (iGluRs)
Three postsynaptic ionotropic glutamate receptors have been identified: N-methyl-D-aspartate receptors (NMDARs), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (AMPARs), and kainate receptors (KAR). These receptors are responsible for the majority of postsynaptic slow excitatory potential transmission [9]. The function of KAR is not fully understood, although it may modulate presynaptic and postsynaptic excitatory neurotransmission.
NMDARs are one of the subtypes of excitatory iGluRs, which is widely distributed in the hippocampus and cortex and is also the glutamate receptor most directly related to the excitotoxicity of ischemic stroke [10]. It is a heterotetramer composed of two NR1 and NR2 subunits, which have dual ligand-voltage gating characteristics and high Ca2+ permeability [11]. Under physiological conditions, when glutamate is released from the presynaptic membrane to activate glutamate receptors, NMDARs activation mediates inward cationic currents, thereby depolarizing postsynaptic neurons and generating excitatory postsynaptic currents. The GluN1 subunit is essential for all functional NMDARs and has binding sites for glycine and D-serine [12]. In addition, NMDAR also contains an atypical glycine-binding subunit GluN3A, Glycine-binding sites in the GluN1 and GluN3A subunits promote receptor surface transport. Research has demonstrated that mutations within the glycine binding site of the human GluN3A subunit markedly diminish the surface expression of NMDARs [13], indicating that this binding site is crucial for the regulation of NMDARs activity. The GluN2 subunit provides the glutamate binding site and controls the expression and functional properties of NMDARs. Much of the single-channel diversity and pharmacological properties of NMDARs result from the NR2 subunit composition of the receptor [14]. They include competitive antagonists acting at the agonist binding site, sodium and calcium blockers, allosteric sites distinct from the binding site of endogenous agonists, and glycine-site antagonists. These channels regulate Ca2+ permeability and play a role in receptor trafficking, synaptic plasticity, and excitotoxicity [15].
AMPARs are expressed in the postsynaptic membrane and play a crucial role in fast excitatory neurotransmission within the brain [16]. These receptors have heterotetrameric structures consisting of GluA1 to GluA4 (also known as GluRA to GluRD) subunits in various combinations, with GluA2 being a critical site for regulating Ca2+ permeability [17]. The regulation of Ca2+ permeability of GluA2 subunits is influenced by post-transcriptional modifications, and AMPARs lacking the structure of GluA2 are capable of allowing calcium (Ca2+) to pass through. However, most AMPARs in the brain contain edited GluA2 subunits and are therefore impermeable to Ca2+. Growing evidence suggests that Ca2+-permeable AMPARs play significant roles in receptor trafficking, learning, and memory processes, as well as cell death [16].
In addition to AMPARs and NMDARs, KARs are ionic glutamate receptors predominantly expressed in the amygdala, cortex, and hippocampus [18]. Activated by the agonist kainate, KARs play a pivotal role in postsynaptic excitatory neurotransmission. Unlike the aforementioned receptors, KARs are tetrameric structures formed through the assembly of five different subunits of GluK1-GluK5. GluK1, K2, and K3 can undergo editing and splicing to generate variants such as GluK1a, GluK1b, GluK1c, and GluK2a-2c [19]. Upon activation, they elicit a characteristic electrophysiological response characterized by a small amplitude and slowly decaying current attributed to their interaction with auxiliary subunits as well as their biophysical properties.
2.2 Metabotropic Glutamate Receptors (mGluRs)
Metabotropic glutamate receptors (mGluRs) are G-protein-coupled non-ionic glutamate receptors widely distributed throughout the brain, playing a crucial role in regulating both physiological and pathological processes by modulating presynaptic glutamate release and postsynaptic ion receptor activity. mGluRs can be categorized into three main functional subgroups based on differences in amino acid sequences and signal transduction mechanisms of their constituent proteins [20]. The group Ⅰ mGluRs comprises presynaptic mGluR1 and postsynaptic mGluR5, both expressed in astrocytes [21]. The group Ⅱ mGluRs includes mGluR2 and 3 [22], whereas the group Ⅲ encompasses inhibitory presynaptic receptors such as mGluR4, 6, 7, and 8 [23] (Fig. 1).
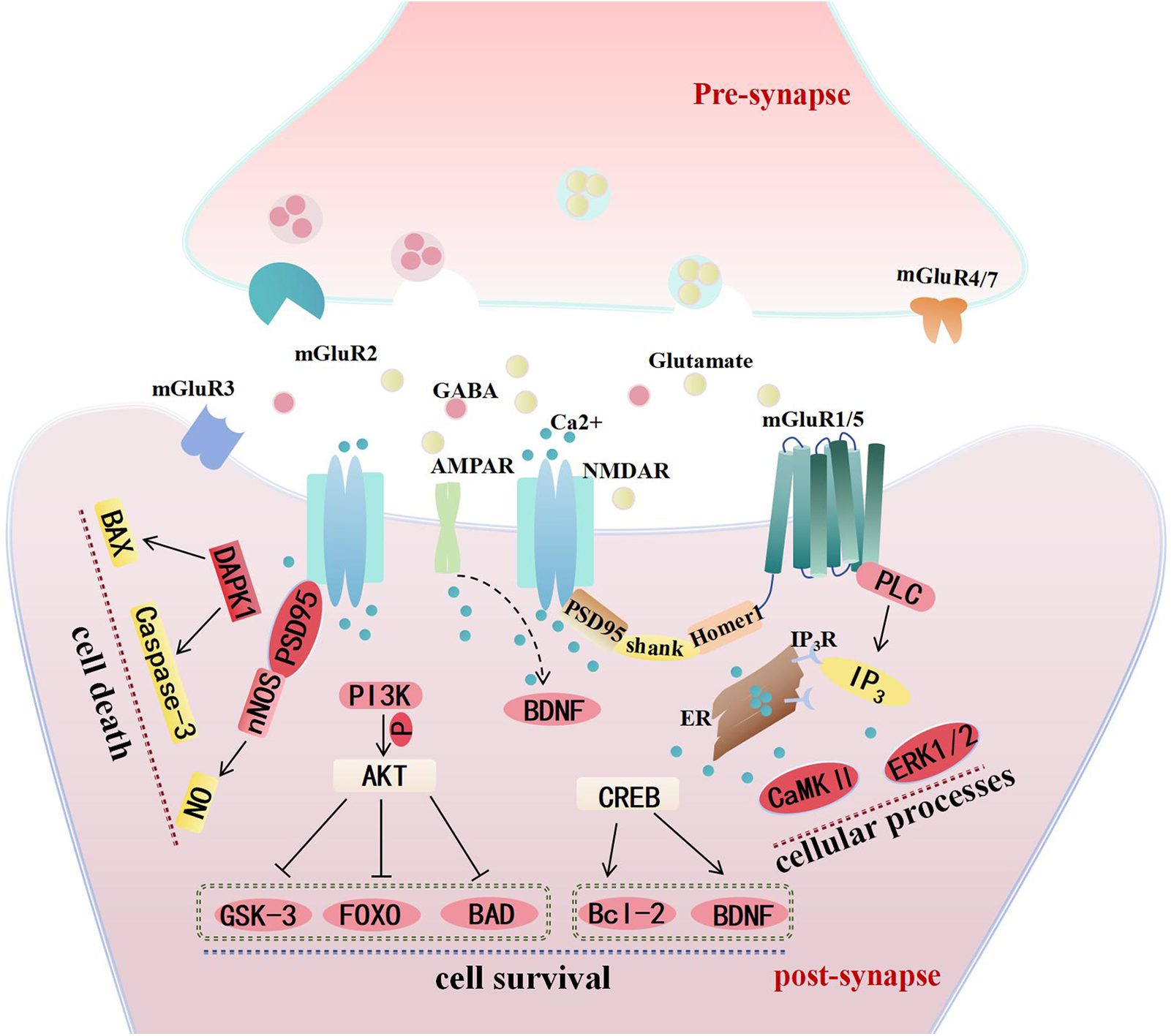
Figure 1: The distribution of glutamate receptors at synapses and the main mechanisms mediating neuronal survival and death in cerebral ischemia. Abbreviations list: N-methyl-D-aspartate receptors (NMDARs), α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptors (AMPARs), Gamma-aminobutyric acid (GABA), metabotropic glutamate receptors (mGluR), phospholipase C (PLC), inositol-1,4,5-triphosphate (IP3), endoplasmic reticulum (ER), extracellular regulated protein kinases 1/2 (ERK1/2), calmodulin-dependent protein kinase Ⅱ (CaMK Ⅱ), postsynaptic density protein 95 (PSD95), brain-derived neurotrophic factor (BDNF), cAMP-response element binding protein (CREB), B-cell lymphoma-2 (Bcl-2), phosphoinositide-3 kinase (PI3K), protein kinase B (AKT), glycogen synthase kinase 3 (GSK-3), Forkhead Box O (FOXO), Bcl-xl/Bcl-2-Associated Death promoter (BAD), death-associated protein kinase 1 (DAPK1), Bcl-2-associated X protein (BAX), neuronal Nitric Oxide Synthase (nNOS). The localization of glutamate receptors at synapses implicated in ischemic stroke and the primary mechanisms underlying ischemic brain injury. It is primarily the NMDAR and the group I mGluRs that mediate neuronal survival or death by activating distinct Ca2+-dependent downstream signaling pathways. AMPAR may enhance neuronal survival by mediating BDNF expression following cerebral ischemia. On one hand, NMDAR initiates apoptotic signaling through DAPK, leading to neuronal death; on the other hand, they inhibit the apoptotic pathway via the PI3K-AKT pathway and upregulate anti-apoptotic mechanisms through the CREB pathway to promote neuronal survival. The interaction between NMDAR and PSD95/nNOS, resulting in neurotoxic NO production, is also a significant contributor to neuronal death. Additionally, the downstream effects of group I mGluRs mediated by Ca2+-dependent protein kinases represent another critical factor influencing neuronal activity.
The group I mGluRs are primarily categorized into presynaptic mGluR1 and postsynaptic mGluR5. Presynaptic mGluR1 predominantly facilitates the vesicular release of glutamate, while postsynaptic mGluR5 is mainly involved in regulating the activity of postsynaptic ionic receptors [24]. Furthermore, mGluR5 is highly expressed in developmental astrocytes and exhibits a rapid decline in adult astrocytes; however, it is re-expressed and plays a role in synaptic plasticity associated with certain central nervous system disorders [25]. Structurally coupled to NMDAR through postsynaptic density protein 95 (PSD95)/Homer on the postsynaptic membrane, mGluR5 mediates the classical GPCR pathway and regulates NMDAR-mediated Ca2+ signaling [26].
The group II mGluRs (mGluR2 and mGluR3) are primarily categorized based on their extensive distribution in the brain, exhibiting similar localization within the cerebral cortex, hippocampus, and cerebellar amygdala. Specifically, mGluR2/3 are equally distributed in microglia and oligodendrocyte; with mGluR2 localized on presynaptic neuronal membranes while mGluR3 is predominantly located on postsynaptic membranes and highly expressed in astrocytes [27]. Their activation inhibits glutamate release from presynaptic nerve terminals and mediates signaling pathways within post-synapses and glial cells. Under physiological conditions, binding of mGluR2/3 to adenylyl cyclase through Gαi/o protein inhibits Cyclic Adenosine Monophosphate (cAMP) and protein kinase A (PKA) signaling ultimately leading to inhibition of neurotransmitter release [28].
The group III mGluRs, including mGluR4, mGluR6, mGluR7, and mGluR8, are primarily located in the presynaptic membrane and terminals of Gamma-aminobutyric acidergic (GABAergic) neurons in the striatum [29]. They play a role in inhibiting neurotransmitter release. However, mGluR6 is predominantly found in the retina with limited research on its involvement in central nervous system diseases. Group III mGluRs are linked to Gi/o proteins to suppress the elevation of cAMP, representing an inhibitory autoreceptor mechanism that regulates excessive glutamate and GABA release from presynaptic terminals [30].
3 Pathophysiological Mechanisms Underlying Cerebral Ischemia and Reperfusion Injury
Contemporary medical research has demonstrated that neuronal damage resulting from cerebral ischemia primarily arises from reduced blood flow to the affected brain regions, leading to embolism and a significant increase in intracranial pressure as well as brain edema [4]. Furthermore, following an ischemia stroke, there is a decrease in oxygen and glucose supply, resulting in insufficient energy availability for neurons within the ischemic area. This leads to the closure of ATP-dependent ion channels and an elevation of intracellular calcium levels, ultimately causing mitochondrial dysfunction. The activation of glial cells along with immune cell infiltration initiates an acute inflammatory response that can directly or indirectly contribute to neuronal death [31]. Conversely, the irreversible damage induced by reperfusion primarily results from an increase in oxygen free radicals and intracellular calcium overload. Ischemia and hypoxia impair the mitochondrial electron transport chain in neurons, leading to the formation of reactive oxygen species from oxygen molecules that enter cells during reperfusion. Intracellular calcium overload predominantly occurs during reperfusion due to enhanced calcium influx [32]. During ischemic conditions, ATP production is diminished, and sodium and potassium pumps become dysfunctional, resulting in a significant accumulation of intracellular Na+. The elevated intracellular Na+ levels during reperfusion not only activate the sodium pump but also facilitate reverse transport via sodium-calcium exchange proteins, thereby exacerbating calcium overload [33].
4 Dual Effects of iGluRs Mediating Neuronal Survival in Stroke
Under physiological conditions, the activation of iGluRs is crucial for neuronal activity, directly or indirectly regulating neuron growth, differentiation, migration, and survival. It also plays a role in dendrite and axon development, as well as synapse and neural circuit formation [34]. Inhibition of iGluRs activation can result in significant neuronal apoptosis, impacting neurogenesis, learning, and memory. Conversely, iGluR activation, particularly NMDAR activation, can lead to neuronal death through various pathways during cerebral ischemia. The main mechanism of damage is triggered by NMDA-mediated Ca2+ influx which initiates a series of cascading damages [35]. Some signaling pathways are pivotal in NMDAR-mediated neuronal protection following ischemic stroke.
4.1 NMDAR-Mediated Neural Protective Pathways
PI3K/AKT pathway In ischemic brain injury diseases, the NMDAR activation strongly activates this pathway. Under the stimulation of ischemia and hypoxia, lipid products phosphatidylinositol diphosphate (IP2) and phosphatidylinositol triphosphate (IP3), produced by Phosphatidylinositide 3-kinases (PI3K) activation, act as second messengers to activate various intracellular target proteins, ultimately regulating cell proliferation, differentiation, survival, and other processes [36]. Serine/threonine-protein kinase (AKT), as a direct downstream target of PI3K, is fully activated by IP3 and interacts with a variety of substrates to catalyze the activation or deactivation of substrates and regulate cell survival or death, which involves many mechanisms. On the one hand, it can inhibit the activity of glycogen synthase kinase 3 (GSK-3) and affect its downstream substrates such as nuclear factor-kappa B (NF-κB) and c-Jun transcription factor, thereby inhibiting the occurrence of neuronal apoptosis. In addition, it catalyzes the phosphorylation of transcription factor Forkhead Box O (FOXO) and reduces its expression, thereby affecting the expression of its downstream apoptotic genes such as FasL and Bim [37]. In addition, it also phosphorylates BAD, a member of the Bcl-2 family of apoptotic genes, thereby inhibiting the blocking effect of BAD on Bcl2/Bcl-xL, a pro-survival family of Bcl-2 [38] (Fig. 1).
CREB pathway cAMP-response element binding protein (CREB) is a member of the DNA-binding transcription factor family, and its transcriptional activation depends on its Ser phosphorylation. NMDAR activation induced by cerebral ischemia with the increase of intracellular Ca2+ concentration can phosphorylate CREB, which directly or indirectly activates the transcription of downstream related genes and regulates the expression of c-fos, c-Jun, Bcl-2, brain-derived neurotrophic factor (BDNF) and other genes [39–41]. Thus, some neurons can survive after cerebral ischemia by inhibiting apoptosis and promoting cell differentiation, regeneration, and repair after injury. The phosphorylation of CREB is regulated by a variety of signal transduction pathways, such as cAMP pathway, Ca2+/calmodulin dependent protein kinase IV (CaMKIV) pathway, Rat sarcoma/Extracellular regulated protein kinases (Ras/ERK) pathway, PI3K/AKT pathway, and stress-induced p38 mitogen-activated protein kinase (p38 MAPK) pathway. The intracellular Ras/ERK pathway and CaMKIV pathway in the nucleus were activated by the Ca2+ influx caused by NMDAR activation [42,43]. CaMKIV rapidly phosphorylates the Ser of CREB, whereas the ERK1/2 pathway promotes long-acting phosphorylation of CREB [44]. In addition to CREB autophosphorylation, several co-activators jointly participate in the initiation and maintenance of transcription of CREB downstream genes. The target of rapamycin complexes (TORCs) is a group of CREB coactivators that promote CREB-dependent gene transcription independent of CREB Ser phosphorylation. The activation of TORCs is mainly regulated by Ca2+ and cAMP pathways. Ca2+ activates calcineurin to dephosphorylate TORC2. cAMP is also required to inhibit the activity of Salt-inducible kinase 2 (SIK2), the kinase of TORC2 [45,46].
4.2 NMDAR-Mediated Neural Injury Pathways
Under pathological conditions, NMDAR activation can also lead to neuronal death through various pathways. The mechanism of injury is primarily initiated by the NMDAR-mediated influx of Ca2+, which in turn triggers a series of cascading damage.
Calpain and calcium-dependent cysteine proteases are activated by Ca2+ influx mediated by NMDAR activation and can be categorized into calpain I and calpain II based on the concentration of Ca2+ they rely on [47]. Under physiological conditions, calpain is involved in regulating neuronal function, including cytoskeleton adjustment, enzyme modification, and Long-term potentiation (LTP) enhancement [48]. In the context of cerebral ischemia and hypoxia, the dysregulation of Ca2+ leads to sustained activation of calpain, resulting in protein and enzyme hydrolysis as well as disruption of cellular homeostasis [49], which is a significant contributor to neuronal death.
The influx of Ca2+ induced by NMDAR activation leads to the upregulation of neuronal Nitric Oxide Synthase (nNOS) and triggers a cascade of downstream toxic reactions, including mitochondrial dysfunction, p38 MAPK activation, and transient receptor potential melastatin (TRPM) channel activity [50,51]. The activation of nNOS results in excessive production of NO and ROS, which can cause cellular damage by inhibiting mitochondrial respiratory chain enzymes and promoting mitochondrial membrane depolarization [52]. Furthermore, their activation subsequently stimulates TRPM7, a recently discovered bifunctional protein with a dual ion channel and protein kinase structure that is permeable to divalent and monovalent cations such as Ca2+, Mg2+, and Na+. During the acute phase of cerebral ischemia, Ca2+ influx through NMDA channels activates nNOS to generate ROS, while ROS in turn activates TRPM7 channels. The resulting Ca2+ influx via TRPM7 further activates nNOS in a positive feedback loop that generates ROS and induces intracellular calcium overload ultimately leading to neuronal death [53]. Another critical signaling pathway implicated in neuronal death is the GluN2B-PSD95-nNOS pathway. During the acute phase of cerebral ischemia, NR2B subunits facilitate cell death by recruiting death-associated protein kinase 1 (DAPK1) and calcium/calmodulin-dependent protein kinase Ⅱ (CaMKⅡ) or by forming NR2B-PSD95-nNOS complexes [54,55]. Following excitotoxicity, PSD95 employs its distinctive molecular architecture to recruit calcium-dependent nNOS from the cytoplasmic matrix to the plasma membrane, thereby catalyzing the production of neurotoxic NO and initiating a cascade of downstream events leading to cell death [56] (Fig. 1).
Considering the correlation between excitotoxic and neuronal death in ischemic stroke, numerous clinical trials have been conducted to enhance cerebral ischemic outcomes through the inhibition of NMDAR. Unfortunately, these efforts have often been hindered by limited treatment windows and adverse side effects [57,58].
AMPAR also plays critical but contradictory roles in the pathophysiology of ischemic stroke. In ischemic injury, AMPAR activation induces the expression of the BDNF, and delayed enhancement of AMPA signaling promotes behavioral recovery after stroke, whereas blocking AMPAR signaling during the same period hinders recovery [59]. Studies have shown that administration of AMPAR agonist on the 5th day after cerebral ischemia can promote the recovery of sensorimotor function in mice, while administration of AMPAR inhibitor during the same period can delay the recovery [60]. This recovery effect is mediated by enhancing BDNF activity around the infarction. In contrast to agonist-mediated protective effects, AMPAR agonist administration in the acute phase after stroke increased the infarct size in the cerebral cortex of mice, which may be related to the anti-inflammatory and anti-apoptotic signals mediated by AMPAR agonist [61] (Fig. 1).
mGluRs are widely distributed throughout the central nervous system and activate various signaling pathways in presynaptic and postsynaptic terminals as well as astrocytes. While not directly involved in rapid excitatory transmission, their activation regulates excitatory neurotransmission and is linked to diverse intracellular signaling. Disruptions in these transduction pathways may significantly impact brain function, offering promising targets for the treatment of central nervous system diseases. The following will discuss their contributions and research progress in ischemic stroke:
Group Ⅰ mGluRs Presynaptic mGluR1 predominantly facilitates the release of glutamate from vesicles, while postsynaptic mGluR5 is involved in modulating the activity of postsynaptic iGluRs. Given the prevalence of excitotoxicity in ischemic injury, the group I mGluRs, which mediates glutamate release and Ca2+ signaling through Gα/q11 coupling, has been extensively researched. Due to the complexity of cell death caused by ischemic brain injury, it is challenging to determine the exact role of these two group I mGluRs in ischemia/reperfusion injury. Existing studies have demonstrated that activation of group I mGluRs can mediate both neuroprotective (AKT/ERK activation) and neurotoxic (intracellular Ca2+ release) cellular signaling pathways. Previous preclinical studies have shown that intraperitoneal injection of a mGluR1 antagonist can reduce cerebral infarct size and neuronal death in two different animal models of middle cerebral artery occlusion (MCAO) induced focal cerebral ischemia and permanent cerebral ischemia in rats [62]. Furthermore, YM202074 and EMQMCM, negative allosteric modulators for mGluR1, reduced infarct size in a dose-dependent manner in a rodent model of MCAO [63]. Based on these observations, it appears plausible to conclude that signaling after mGluR1 activation promotes cell death during the acute phase of cerebral ischemia. However, mice lacking mGluR1 did not show any difference in infarct size compared to control mice after 24 h MCAO. In contrast to mGluR1, both the mGluR5 agonist CHPG and antagonist MTEP demonstrated a dose-dependent reduction in infarct size during ischemia in rats with MCAO [64,65]. In vitro experiments revealed that group I mGluRs agonists could induce neuronal damage. In a mixed culture of cortical neurons, group I mGluRs agonists exacerbated chronic NMDA toxicity and oxygen-glucose deprivation (OGD) induced damage, which was associated with increased intracellular Ca2+ release and extracellular Ca2+ influx, as well as activation of protein kinase C (PKC) [66]. There may be other mechanisms through which group I mGluRs exacerbate neuronal damage, such as enhancing the release of arachidonic acid or the synthesis of nitric oxide [67]. In neonatal animals, the protective effect of mGluRs is mainly mediated by group Ⅰ mGluRs [68]. The addition of group Ⅰ mGluRs agonists to cultured neuronal cells can prevent apoptosis caused by extracellular low K+, which is related to the activation of PKC. Glutamate-induced cell swelling is mediated by mGluR1 activation in cultured astrocytes [69]. The role of arachidonic acid and nitric oxide in nervous system injury has been extensively studied.
Group Ⅱ mGluRs The group Ⅱ mGluRs primarily regulate synaptic plasticity by activating Gαi/o and coupling with cAMP to attenuate the release of presynaptic neurotransmitters [70]. Their activation inhibits glutamate release from presynaptic membrane nerve terminals and mediates signaling pathways in the postsynaptic membrane and glial cells. It has been shown that activation of group II mGluRs reduces the excessive glutamate release associated with neurodegenerative and psychiatric disorders and thus alleviates dysfunction [71], which encourages us to search for effective agonists as potential therapeutic approaches. A compound search identified LY379268 as a potent and systemically available mGlu2/3 receptor agonist. LY379268 is effective in several animal models of stroke, epilepsy, substance abuse, schizophrenia, and pain. Inhibition of motor activity was the main side effect of LY379268. Tolerance exacerbates this side effect with repeated dosing, but the therapeutic effect of LY379268 remains [72]. To date, there are no clinical data for LY379268. Administration of group Ⅱ and Ⅲ mGluRs agonists to the presynaptic membrane in the adult mouse brain can reduce the toxic effects of NMDAR and KA by inhibiting the release of excitatory neurotransmitters [73]. Cell culture experiments also showed that group Ⅱ mGluRs agonists had a protective effect on acute and chronic NMDA toxicity and OGD injury [74]. This protective effect was related to the inhibition of Glutamate release and the reduction of intracellular cAMP levels.
Group Ⅲ mGluRs Group III mGluRs are predominantly localized at the terminals of presynaptic glutamatergic and GABAergic neurons, where they play a critical role in the release of inhibitory neurotransmitters [75]. Research on this receptor subtype in the context of cerebral ischemic injury has been relatively limited, primarily concentrating on the protective roles of mGluR4 and mGluR7 in neuronal survival. Previous studies have demonstrated that knockout of mGluR4 significantly increases cerebral infarction volume in mice subjected to MCAO. Conversely, subcutaneous administration of the selective mGluR4 agonist PHCCC primarily affecting both mGluR4 and mGluR7 substantially reduces cerebral infarction volume after cerebellar ischemia in murine models [76,77]. Furthermore, treatment with ACPT-I, an agonist of group III mGluRs, has been shown to provide significant protection against glutamate-induced excitotoxicity in vitro when applied to primary cortical neurons [77].
It is generally believed that subtypes 1 and 5 of group Ⅰ mGluRs play a neurotoxic role in the pathological stimulation of cerebral ischemia, while subtypes 2 and 3 of group Ⅱ play a neuroprotective role. Based on the understanding of the specific functions of mGluRs in regulating cellular excitability and synaptic transmission, the search for therapeutic effective mGluRs ligands has focused on antagonists of group I mGluRs and agonists of group II and III mGluRs.
During cerebral ischemia, glutamate functions beyond being a mere neurotransmitter; the receptors for glutamate expressed on synapses and glial cells significantly contribute to neuronal death. The excitotoxicity induced by glutamate is a critical factor in neuronal death. This paper primarily explores the mechanisms of neuronal injury and protection mediated by glutamate receptors located on both presynaptic and postsynaptic membranes following cerebral ischemia. The protective effects of NMDAR activation on neurons during ischemic stroke remain a subject of controversy. The molecular mechanisms underlying the diverse signaling pathways mediated by NMDAR may be associated with its composition and localization. Some studies indicate that synaptic NMDAR predominantly activates the PI3K/Akt and CREB signaling pathways, which can inhibit the expression of BCL-2 and other apoptotic genes, thereby exerting protective effects against oxidative stress while promoting neuronal survival. Conversely, extrasynaptic NMDAR inhibits ERK1/2 activation and CREB phosphorylation, activating FOXO and calpain to promote neuronal apoptosis primarily through excitatory amino acid-induced excitotoxicity. Similarly, certain studies have suggested that NR2A-containing NMDAR plays a protective role in ischemic tolerance mechanisms, whereas NR2B-containing NMDAR along with its associated signaling molecules are implicated in excitotoxicity during ischemia. In vitro cultures of mature outer cortical cells reveal that activation of NR2B-containing NMDAR leads to excitotoxic damage; however, activation of NR2A-containing NMDAR promotes neuronal survival while protecting neurons from NMDAR-mediated as well as non-NMDAR-mediated injuries [51].
In addition to glutamate receptors present on neuronal synapses, emerging evidence suggests that mGluRs expressed on glial cells also mediate synaptic function post-cerebral ischemia. Notably, some mGluRs are re-expressed on glial cells following cerebral ischemia. As previously mentioned, mGluR5 is highly expressed in the postsynaptic membrane as well as astrocyte and even somewhat in microglia during developmental stages; however, this expression diminishes sharply three weeks into maturation within glial cells [78]. Subsequent investigations have demonstrated the re-expression of mGluR5 in astrocytes surrounding the infarct area after cerebral ischemia; concurrently mediating secretion of synaptogenic proteins thrombospondin-1/2 (TSP1/2) and Hevin thus facilitating nerve circuit regeneration around the infarction site [79]. Moreover, astrocytes play a crucial role in maintaining the equilibrium between glutamate reuptake and release at the tri-synaptic structure via Excitatory amino acid transporter 2 (EAAT2) located on their membranes, which is implicated in various pathological mechanisms associated with acute brain injury, including those involving GluRs [80]. Furthermore, it has been proposed that mGluRs on glial cells may play an essential role in functional recovery following ischemic stroke beyond their intrinsic roles at neuron synapse structures. Nonetheless, our understanding regarding the role of mGluRs on glial cells across different stages of cerebral ischemic stroke remains limited and requires further clarification.
Despite encouraging findings from both in vitro and in vivo investigations of glutamate receptors in ischemic stroke, clinical trials involving iGluRs antagonists, particularly NMDAR antagonists, have faced significant challenges. These challenges are primarily associated with the emergence of adverse effects, including psychosis, cognitive dysfunctions such as memory impairment and disorientation, as well as dyskinesia [81–83]. In addition, mGluRs have attracted much attention in preclinical studies, but the relationship between different subtypes of mGluRs and iGluRs is poorly understood, and the mechanism of different responses is still unclear, and needs further exploration.
Acknowledgment: None.
Funding Statement: This work was supported by Grants of the National Natural Science Foundation of China (82073831), Natural Science Foundation of Chongqing (CSTB2023NSCQ-MSX0656), and China Postdoctoral Science Foundation (2023M733889).
Author Contributions: The authors confirm their contribution to the paper as follows: study conception and design: Long Qi and Chaoran Wu; draft manuscript preparation: Long Qi; review and editing: Hong Liao and Hao Sun; supervision: Hong Liao and Hao Sun. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Saini V, Guada L, Yavagal DR. Global epidemiology of stroke and access to acute ischemic stroke interventions. Neurology. 2021;97(20 Suppl 2):S6–16. [Google Scholar] [PubMed]
2. Renú A, Millán M, San RL, Blasco J, Martí-Fàbregas J, Terceño M, et al. Effect of intra-arterial alteplase vs placebo following successful thrombectomy on functional outcomes in patients with large vessel occlusion acute ischemic stroke: the CHOICE randomized clinical trial. JAMA. 2022;327(9):826–35. doi:10.1001/jama.2022.1645. [Google Scholar] [PubMed] [CrossRef]
3. Liu Q, Shi K, Wang Y, Shi FD. Neurovascular inflammation and complications of thrombolysis therapy in stroke. Stroke. 2023;54(10):2688–97. doi:10.1161/STROKEAHA.123.044123. [Google Scholar] [PubMed] [CrossRef]
4. Shen Z, Xiang M, Chen C, Ding F, Wang Y, Shang C, et al. Glutamate excitotoxicity: potential therapeutic target for ischemic stroke. Biomed Pharmacother. 2022;151:113125. doi:10.1016/j.biopha.2022.113125. [Google Scholar] [PubMed] [CrossRef]
5. Karakas E, Regan MC, Furukawa H. Emerging structural insights into the function of ionotropic glutamate receptors. Trends Biochem Sci. 2015;40(6):328–37. doi:10.1016/j.tibs.2015.04.002. [Google Scholar] [PubMed] [CrossRef]
6. Lee J, Gonzalez-Hernandez AJ, Kristt M, Abreu N, Roßmann K, Arefin A, et al. Distinct beta-arrestin coupling and intracellular trafficking of metabotropic glutamate receptor homo- and heterodimers. Sci Adv. 2023;9(49):eadi8076. doi:10.1126/sciadv.adi8076. [Google Scholar] [PubMed] [CrossRef]
7. Fernandes G, Mishra PK, Nawaz MS, Donlin-Asp PG, Rahman MM, Hazra A, et al. Correction of amygdalar dysfunction in a rat model of fragile X syndrome. Cell Rep. 2021;37(2):109805. doi:10.1016/j.celrep.2021.109805. [Google Scholar] [PubMed] [CrossRef]
8. Abd-Elrahman KS, Sarasija S, Ferguson SSG. The role of neuroglial metabotropic glutamate receptors in Alzheimer’s disease. Curr Neuropharmacol. 2023;21(2):273–83. doi:10.2174/1570159X19666210916102638. [Google Scholar] [PubMed] [CrossRef]
9. Kalev-Zylinska ML, Morel-Kopp MC, Ward CM, Hearn JI, Hamilton JR, Bogdanova AY. Ionotropic glutamate receptors in platelets: opposing effects and a unifying hypothesis. Platelets. 2021;32(8):998–1008. doi:10.1080/09537104.2020.1852542. [Google Scholar] [PubMed] [CrossRef]
10. Shindo T, Shikanai H, Watarai A, Hiraide S, Iizuka K, Izumi T. D-serine metabolism in the medial prefrontal cortex, but not the hippocampus, is involved in AD/HD-like behaviors in SHRSP/Ezo. Eur J Pharmacol. 2022;923:174930. doi:10.1016/j.ejphar.2022.174930. [Google Scholar] [PubMed] [CrossRef]
11. Hsieh CP, Chang WT, Chen L, Chen HH, Chan MH. Differential inhibitory effects of resveratrol on excitotoxicity and synaptic plasticity: involvement of NMDA receptor subtypes. Nutr Neurosci. 2021;24(6):443–58. doi:10.1080/1028415X.2019.1641995. [Google Scholar] [PubMed] [CrossRef]
12. Bodner O, Radzishevsky I, Foltyn VN, Touitou A, Valenta AC, Rangel IF, et al. D-serine signaling and NMDAR-mediated synaptic plasticity are regulated by system A-type of glutamine/D-serine dual transporters. J Neurosci. 2020;40(34):6489–502. doi:10.1523/JNEUROSCI.0801-20.2020. [Google Scholar] [PubMed] [CrossRef]
13. Mesic I, Madry C, Geider K, Bernhard M, Betz H, Laube B. The N-terminal domain of the GluN3A subunit determines the efficacy of glycine-activated NMDA receptors. Neuropharmacology. 2016;105:133–41. doi:10.1016/j.neuropharm.2016.01.014. [Google Scholar] [PubMed] [CrossRef]
14. Brickley SG, Misra C, Mok MH, Mishina M, Cull-Candy SG. NR2B and NR2D subunits coassemble in cerebellar Golgi cells to form a distinct NMDA receptor subtype restricted to extrasynaptic sites. J Neurosci. 2003;23(12):4958–66. doi:10.1523/JNEUROSCI.23-12-04958.2003. [Google Scholar] [PubMed] [CrossRef]
15. Ma H, Khaled HG, Wang X, Mandelberg NJ, Cohen SM, He X, et al. Excitation-transcription coupling, neuronal gene expression and synaptic plasticity. Nat Rev Neurosci. 2023;24(11):672–92. doi:10.1038/s41583-023-00742-5. [Google Scholar] [PubMed] [CrossRef]
16. Ge Y, Wang YT. GluA1-homomeric AMPA receptor in synaptic plasticity and neurological diseases. Neuropharmacology. 2021;197:108708. doi:10.1016/j.neuropharm.2021.108708. [Google Scholar] [PubMed] [CrossRef]
17. Zheng Z, Keifer J. Sequential delivery of synaptic GluA1- and GluA4-containing AMPA receptors (AMPARs) by SAP97 anchored protein complexes in classical conditioning. J Biol Chem. 2014;289(15):10540–50. doi:10.1074/jbc.M113.535179. [Google Scholar] [PubMed] [CrossRef]
18. Mennesson M, Rydgren E, Lipina T, Sokolowska E, Kulesskaya N, Morello F, et al. Kainate receptor auxiliary subunit NETO2 is required for normal fear expression and extinction. Neuropsychopharmacology. 2019;44(11):1855–66. doi:10.1038/s41386-019-0344-5. [Google Scholar] [PubMed] [CrossRef]
19. Taniguchi S, Stolz JR, Swanson GT. The antiseizure drug perampanel is a subunit-selective negative allosteric modulator of kainate receptors. J Neurosci. 2022;42(28):5499–509. doi:10.1523/JNEUROSCI.2397-21.2022. [Google Scholar] [PubMed] [CrossRef]
20. Goel N, Peng K, Lu Y. Neuromodulation by mGluRs in sound localization circuits in the auditory brainstem. Front Neural Circuits. 2020;14:599600. doi:10.3389/fncir.2020.599600. [Google Scholar] [PubMed] [CrossRef]
21. Mahato PK, Ramsakha N, Ojha P, Gulia R, Sharma R, Bhattacharyya S. Group I metabotropic glutamate receptors (mGluRsins and outs. Adv Exp Med Biol. 2018;1112:163–75. doi:10.1007/978-981-13-3065-0. [Google Scholar] [CrossRef]
22. Frouni I, Kwan C, Bédard D, Hamadjida A, Kang W, Belliveau S, et al. Effect of mGluR2 and mGluR2/3 activators on parkinsonism in the MPTP-lesioned non-human primate. Naunyn Schmiedebergs Arch Pharmacol. 2024 Nov;397(11):9135–47. doi:10.1007/s00210-024-03216-2. [Google Scholar] [PubMed] [CrossRef]
23. Domin H. Group III metabotropic glutamate receptors as promising targets for neuroprotective therapy: particular emphasis on the role of mGlu4 and mGlu7 receptors. Pharmacol Biochem Behav. 2022;219:173452. doi:10.1016/j.pbb.2022.173452. [Google Scholar] [PubMed] [CrossRef]
24. Sanderson TM, Ralph LT, Amici M, Ng AN, Kaang BK, Zhuo M, et al. Selective recruitment of presynaptic and postsynaptic forms of mGluR-LTD. Front Synaptic Neurosci. 2022;14:857675. doi:10.3389/fnsyn.2022.857675. [Google Scholar] [PubMed] [CrossRef]
25. Zehnder T, Petrelli F, Romanos J, De Oliveira Figueiredo EC, Lewis TLJr., Déglon N, et al. Mitochondrial biogenesis in developing astrocytes regulates astrocyte maturation and synapse formation. Cell Rep. 2021;35(2):108952. doi:10.1016/j.celrep.2021.108952. [Google Scholar] [PubMed] [CrossRef]
26. Samojedny S, Czechowska E, Pańczyszyn-Trzewik P, Sowa-Kućma M. Postsynaptic proteins at excitatory synapses in the brain-relationship with depressive disorders. Int J Mol Sci. 2022;23(19):11423. doi:10.3390/ijms231911423. [Google Scholar] [PubMed] [CrossRef]
27. Jiang Y, Zou M, Ren T, Wang Y. Are mGluR2/3 inhibitors potential compounds for novel antidepressants? Cell Mol Neurobiol. 2023;43(5):1931–40. doi:10.1007/s10571-022-01310-8. [Google Scholar] [PubMed] [CrossRef]
28. Jin LE, Wang M, Yang ST, Yang Y, Galvin VC, Lightbourne TC, et al. mGluR2/3 mechanisms in primate dorsolateral prefrontal cortex: evidence for both presynaptic and postsynaptic actions. Mol Psychiat. 2017;22(11):1615–25. doi:10.1038/mp.2016.129. [Google Scholar] [PubMed] [CrossRef]
29. McCullock TW, Cardani LP, Kammermeier PJ. Signaling specificity and kinetics of the human metabotropic glutamate receptors. 2023 Jul 28. doi:10.1101/2023.07.24.550373. [Google Scholar] [PubMed] [CrossRef]
30. Niswender CM, Johnson KA, Luo Q, Ayala JE, Kim C, Conn PJ, et al. A novel assay of Gi/o-linked G protein-coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Mol Pharmacol. 2008;73(4):1213–24. doi:10.1124/mol.107.041053. [Google Scholar] [PubMed] [CrossRef]
31. Yang Q, Huang Q, Hu Z, Tang X. Potential neuroprotective treatment of stroke: targeting excitotoxicity, oxidative stress, and inflammation. Front Neurosci. 2019;13:1036. doi:10.3389/fnins.2019.01036. [Google Scholar] [PubMed] [CrossRef]
32. Amantea D, Greco R, Micieli G, Bagetta G. Paradigm shift to neuroimmunomodulation for translational neuroprotection in stroke. Front Neurosci. 2018;12:241. doi:10.3389/fnins.2018.00241. [Google Scholar] [PubMed] [CrossRef]
33. Verma M, Lizama BN, Chu CT. Excitotoxicity, calcium and mitochondria: a triad in synaptic neurodegeneration. Transl Neurodegener. 2022;11(1):3. doi:10.1186/s40035-021-00278-7. [Google Scholar] [PubMed] [CrossRef]
34. Washburn HR, Chander P, Srikanth KD, Dalva MB. Transsynaptic signaling of ephs in synaptic development, plasticity, and disease. Neuroscience. 2023;508:137–52. doi:10.1016/j.neuroscience.2022.11.030. [Google Scholar] [PubMed] [CrossRef]
35. Li C, Meng L, Li X, Li D, Jiang LH. Non-NMDAR neuronal Ca2+-permeable channels in delayed neuronal death and as potential therapeutic targets for ischemic brain damage. Expert Opin Ther Targets. 2015;19(7):879–92. doi:10.1517/14728222.2015.1021781. [Google Scholar] [PubMed] [CrossRef]
36. Gong Z, Lao D, Wu Y, Li T, Lv S, Mo X, et al. Inhibiting PI3K/Akt-signaling pathway improves neurobehavior changes in anti-NMDAR encephalitis mice by ameliorating blood-brain barrier disruption and neuronal damage. Cell Mol Neurobiol. 2023;43(7):3623–37. doi:10.1007/s10571-023-01371-3. [Google Scholar] [PubMed] [CrossRef]
37. Ding YW, Pan SY, Xie W, Shen HY, Wang HH. Elevated soluble Fas and FasL in cerebrospinal fluid and serum of patients with anti-N-methyl-D-aspartate receptor encephalitis. Front Neurol. 2018;9:904. doi:10.3389/fneur.2018.00904. [Google Scholar] [PubMed] [CrossRef]
38. Liu YJ, Cui ZY, Yang AL, Jallow AW, Huang HL, Shan CL, et al. Anti-apoptotic and pro-survival effect of exercise training on early aged hypertensive rat cerebral cortex. Aging. 2021;13(16):20495–510. doi:10.18632/aging.203431. [Google Scholar] [PubMed] [CrossRef]
39. Xie D, Zhang P, You S, Shen Y, Xu W, Zhan C, et al. Salidroside derivative SHPL-49 attenuates glutamate excitotoxicity in acute ischemic stroke via promoting NR2A-CAMKIIα-Akt/CREB pathway. Phytomedicine. 2024;134(6):155583. doi:10.1016/j.phymed.2024.155583. [Google Scholar] [PubMed] [CrossRef]
40. Yu L, Liu S, Zhou R, Sun H, Su X, Liu Q, et al. Atorvastatin inhibits neuronal apoptosis via activating cAMP/PKA/p-CREB/BDNF pathway in hypoxic-ischemic neonatal rats. FASEB J. 2022;36(4):e22263. doi:10.1096/fj.202101654RR. [Google Scholar] [PubMed] [CrossRef]
41. Li L, Yang JH, Li C, Zhou HF, Yu L, Wu XL, et al. Danhong injection improves neurological function in rats with ischemic stroke by enhancing neurogenesis and activating BDNF/AKT/CREB signaling pathway. Biomed Pharmacother. 2023;163(10204):114887. doi:10.1016/j.biopha.2023.114887. [Google Scholar] [PubMed] [CrossRef]
42. Moriguchi S, Sakagami H, Yabuki Y, Sasaki Y, Izumi H, Zhang C, et al. Stimulation of sigma-1 receptor amelio-rates depressive-like behaviors in CaMKIV null mice. Mol Neurobiol. 2015;52(3):1210–22. doi:10.1007/s12035-014-8923-2. [Google Scholar] [PubMed] [CrossRef]
43. Marsden WN. Synaptic plasticity in depression: molecular, cellular and functional correlates. Prog Neuropsychopharmacol Biol Psychiat. 2013;43(13):168–84. doi:10.1016/j.pnpbp.2012.12.012. [Google Scholar] [PubMed] [CrossRef]
44. Hsu WC, Le HN, Lin YJ, Chen MC, Wang TF, Li CC, et al. Calmodulin/CaMKII-γ mediates prosurvival capability in apicidin-persistent hepatocellular carcinoma cells via ERK1/2/CREB/c-fos signaling pathway. J Cell Biochem. 2021;122(6):612–25. doi:10.1002/jcb.29892. [Google Scholar] [PubMed] [CrossRef]
45. Cai XM, Sun XY, Li R, Wang PJ, Qiu JC, Ge YX, et al. The hippocampal salt-inducible kinase 2-CREB-regulated transcription co-activator 1 system mediates the antidepressant actions of paroxetine in mice. Behav Brain Res. 2024;465:114972. doi:10.1016/j.bbr.2024.114972. [Google Scholar] [PubMed] [CrossRef]
46. Xue K, Chen S, Chai J, Yan W, Zhu X, Ji D, et al. Nitration of cAMP-response element binding protein participates in myocardial infarction-induced myocardial fibrosis via accelerating transcription of Col1a2 and Cxcl12. Antioxid Redox Signal. 2023;38(10–12):709–30. [Google Scholar] [PubMed]
47. Averna M, Pellegrini M, Cervetto C, Pedrazzi M, Bavestrello M, De Tullio R, et al. Physiological roles of calpain 1 associated to multiprotein NMDA receptor complex. PLoS One. 2015;10(10):e0139750. doi:10.1371/journal.pone.0139750. [Google Scholar] [PubMed] [CrossRef]
48. Tang X, Zhang X, Li S, Chi X, Luo A, Zhao Y. NR2B receptor- and calpain-mediated KCC2 cleavage resulted in cognitive deficiency exposure to isoflurane. Neurotoxicology. 2020;76:75–83. doi:10.1016/j.neuro.2019.10.003. [Google Scholar] [PubMed] [CrossRef]
49. Novorolsky RJ, Nichols M, Kim JS, Pavlov EV, Joshua JW, Wilson JJ, et al. The cell-permeable mitochondrial calcium uniporter inhibitor Ru265 preserves cortical neuron respiration after lethal oxygen glucose deprivation and reduces hypoxic/ischemic brain injury. J Cereb Blood Flow Metab. 2020;40(6):1172–81. doi:10.1177/0271678X20908523. [Google Scholar] [PubMed] [CrossRef]
50. Ashraf U, Ding Z, Deng S, Ye J, Cao S, Chen Z. Pathogenicity and virulence of Japanese encephalitis virus: neuroinflammation and neuronal cell damage. Virulence. 2021;12(1):968–80. doi:10.1080/21505594.2021.1899674. [Google Scholar] [PubMed] [CrossRef]
51. Zong P, Feng J, Yue Z, Li Y, Wu G, Sun B, et al. Functional coupling of TRPM2 and extrasynaptic NMDARs exacerbates excitotoxicity in ischemic brain injury. Neuron. 2022;110(12):1944–58. doi:10.1016/j.neuron.2022.03.021. [Google Scholar] [PubMed] [CrossRef]
52. Yu H, Ma L, Liu D, Wang Y, Pei X, Duan Z, et al. Involvement of NMDAR/PSD-95/nNOS-NO-cGMP pathway in embryonic exposure to BPA induced learning and memory dysfunction of rats. Environ Pollut. 2020;266:115055. doi:10.1016/j.envpol.2020.115055. [Google Scholar] [PubMed] [CrossRef]
53. Li WT, Zhang SY, Zhou YF, Zhang BF, Liang ZQ, Liu YH, et al. Carvacrol attenuates traumatic neuronal injury through store-operated Ca2+ entry-independent regulation of intracellular Ca2 homeostasis. Neurochem Int. 2015;90:107–13. doi:10.1016/j.neuint.2015.07.020. [Google Scholar] [PubMed] [CrossRef]
54. Tu W, Xu X, Peng L, Zhong X, Zhang W, Soundarapandian MM, et al. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell. 2010;140(2):222–34. doi:10.1016/j.cell.2009.12.055. [Google Scholar] [PubMed] [CrossRef]
55. Buonarati OR, Cook SG, Goodell DJ, Chalmers NE, Rumian NL, Tullis JE, et al. CaMKII versus DAPK1 binding to GluN2B in ischemic neuronal cell death after resuscitation from cardiac arrest. Cell Rep. 2020;30(1):1–8. doi:10.1016/j.celrep.2019.11.076. [Google Scholar] [PubMed] [CrossRef]
56. Wang F, Xie X, Xing X, Sun X. Excitatory synaptic transmission in ischemic stroke: a new outlet for classical neuroprotective strategies. Int J Mol Sci. 2022;23(16):9381. doi:10.3390/ijms23169381. [Google Scholar] [PubMed] [CrossRef]
57. Zhang X, Peng K, Zhang X. The function of the NMDA receptor in hypoxic-ischemic encephalopathy. Front Neurosci. 2020;14:567665. doi:10.3389/fnins.2020.567665. [Google Scholar] [PubMed] [CrossRef]
58. Choi DW. Excitotoxicity: still hammering the ischemic brain in 2020. Front Neurosci. 2020;14:579953. doi:10.3389/fnins.2020.579953. [Google Scholar] [PubMed] [CrossRef]
59. Clarkson AN, Overman JJ, Zhong S, Mueller R, Lynch G, Carmichael ST. AMPA receptor-induced local brain-derived neurotrophic factor signaling mediates motor recovery after stroke. J Neurosci. 2011;31(10):3766–75. doi:10.1523/JNEUROSCI.5780-10.2011. [Google Scholar] [PubMed] [CrossRef]
60. Achzet LM, Davison CJ, Shea M, Sturgeon I, Jackson DA. Oxidative stress underlies the ischemia/reperfusion-induced internalization and degradation of AMPA receptors. Int J Mol Sci. 2021;22(2):717. doi:10.3390/ijms22020717. [Google Scholar] [PubMed] [CrossRef]
61. Sharma H, Reeta KH, Sharma U, Suri V. Decanoic acid mitigates ischemia reperfusion injury by modulating neuroprotective, inflammatory and oxidative pathways in middle cerebral artery occlusion model of stroke in rats. J Stroke Cerebrovasc Dis. 2023;32(8):107184. doi:10.1016/j.jstrokecerebrovasdis.2023.107184. [Google Scholar] [PubMed] [CrossRef]
62. Li H, Zhang N, Sun G, Ding S. Inhibition of the group I mGluRs reduces acute brain damage and improves long-term histological outcomes after photothrombosis-induced ischaemia. ASN Neuro. 2013;5(3):195–207. doi:10.1042/AN20130002. [Google Scholar] [PubMed] [CrossRef]
63. Kohara A, Takahashi M, Yatsugi S, Tamura S, Shitaka Y, Hayashibe S, et al. Neuroprotective effects of the selective type 1 metabotropic glutamate receptor antagonist YM-202074 in rat stroke models. Brain Res. 2008;1191:168–79. doi:10.1016/j.brainres.2007.11.035. [Google Scholar] [PubMed] [CrossRef]
64. Bao WL, Williams AJ, Faden AI, Tortella FC. Selective mGluR5 receptor antagonist or agonist provides neuroprotection in a rat model of focal cerebral ischemia. Brain Res. 2001;922(2):173–9. doi:10.1016/S0006-8993(01)03062-1. [Google Scholar] [PubMed] [CrossRef]
65. Hakon J, Quattromani MJ, Sjölund C, Talhada D, Kim B, Moyanova S, et al. Inhibiting metabotropic glutamate receptor 5 after stroke restores brain function and connectivity. Brain. 2024;147(1):186–200. doi:10.1093/brain/awad293. [Google Scholar] [PubMed] [CrossRef]
66. Vanzulli I, Butt AM. mGluR5 protect astrocytes from ischemic damage in postnatal CNS white matter. Cell Calcium. 2015;58(5):423–30. doi:10.1016/j.ceca.2015.06.010. [Google Scholar] [PubMed] [CrossRef]
67. Dumuis A, Sebben M, Haynes L, Pin JP, Bockaert J. NMDA receptors activate the arachidonic acid cascade system in striatal neurons. Nature. 1988;336(6194):68–70. doi:10.1038/336068a0. [Google Scholar] [PubMed] [CrossRef]
68. Nasrniya S, Bigdeli MR. Ischemic tolerance induced by normobaric hyperoxia and evaluation of group I and II metabotropic glutamate receptors. Curr Neurovasc Res. 2013;10(1):21–8. doi:10.2174/156720213804805981. [Google Scholar] [PubMed] [CrossRef]
69. Shen KZ, Johnson SW. Group I mGluRs evoke K-ATP current by intracellular Ca2+ mobilization in rat subthalamus neurons. J Pharmacol Exp Ther. 2013;345(1):139–50. doi:10.1124/jpet.112.201566. [Google Scholar] [PubMed] [CrossRef]
70. DiRaddo JO, Miller EJ, Hathaway HA, Grajkowska E, Wroblewska B, Wolfe BB, et al. A real-time method for measuring cAMP production modulated by Gαi/o-coupled metabotropic glutamate receptors. J Pharmacol Exp Ther. 2014;349(3):373–82. doi:10.1124/jpet.113.211532. [Google Scholar] [PubMed] [CrossRef]
71. Bratek-Gerej E, Bronisz A, Ziembowicz A, Salinska E. Pretreatment with mGluR2 or mGluR3 agonists reduces apoptosis induced by hypoxia-ischemia in neonatal rat brains. Oxid Med Cell Longev. 2021;2021:8848015. doi:10.1155/omcl.v2021.1. [Google Scholar] [CrossRef]
72. Imre G. The preclinical properties of a novel group II metabotropic glutamate receptor agonist LY379268. CNS Drug Rev. 2007;13(4):444–64. doi:10.1111/cns.2007.13.issue-4. [Google Scholar] [CrossRef]
73. Bruno V, Battaglia G, Copani A, Casabona G, Storto M, Di Giorgi Gerevini V, et al. Metabotropic glutamate receptors and neurodegeneration. Prog Brain Res. 1998;116:209–21. doi:10.1016/S0079-6123(08)60439-2. [Google Scholar] [PubMed] [CrossRef]
74. Opere CA, Heruye S, Njie-Mbye YF, Ohia SE, Sharif NA. Regulation of excitatory amino acid transmission in the retina: studies on neuroprotection. J Ocul Pharmacol Ther. 2018;34(1–2):107–18. [Google Scholar] [PubMed]
75. Lukacs IP, Francavilla R, Field M, Hunter E, Howarth M, Horie S, et al. Differential effects of group III metabotropic glutamate receptors on spontaneous inhibitory synaptic currents in spine-innervating double bouquet and parvalbumin-expressing dendrite-targeting GABAergic interneurons in human neocortex. Cereb Cortex. 2023;33(5):2101–42. doi:10.1093/cercor/bhac195. [Google Scholar] [PubMed] [CrossRef]
76. Moyanova SG, Mastroiacovo F, Kortenska LV, Mitreva RG, Fardone E, Santolini I, et al. Protective role for type 4 metabotropic glutamate receptors against ischemic brain damage. J Cereb Blood Flow Metab. 2011;31(4):1107–18. doi:10.1038/jcbfm.2010.201. [Google Scholar] [PubMed] [CrossRef]
77. Domin H, Przykaza Ł, Jantas D, Kozniewska E, Boguszewski PM, Śmiałowska M. Neuroprotective potential of the group III mGlu receptor agonist ACPT-I in animal models of ischemic stroke: in vitro and in vivo studies. Neuropharmacology. 2016;102:276–94. doi:10.1016/j.neuropharm.2015.11.025. [Google Scholar] [PubMed] [CrossRef]
78. Sun W, McConnell E, Pare JF, Xu Q, Chen M, Peng W, et al. Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science. 2013;339(6116):197–200. doi:10.1126/science.1226740. [Google Scholar] [PubMed] [CrossRef]
79. Kim SK, Hayashi H, Ishikawa T, Shibata K, Shigetomi E, Shinozaki Y, et al. Cortical astrocytes rewire somatosensory cortical circuits for peripheral neuropathic pain. J Clin Invest. 2016;126(5):1983–97. doi:10.1172/JCI82859. [Google Scholar] [PubMed] [CrossRef]
80. Yang XM, Yu H, Li JX, Li N, Li C, Xu DH, et al. Excitotoxic storms of ischemic stroke: a non-neuronal perspective. Mol Neurobiol. 2024;61(11):9562–81. doi:10.1007/s12035-024-04184-7. [Google Scholar] [PubMed] [CrossRef]
81. Yan J, Bengtson CP, Buchthal B, Hagenston AM, Bading H. Coupling of NMDA receptors and TRPM4 guides discovery of unconventional neuroprotectants. Science. 2020;370(6513):eaay3302. doi:10.1126/science.aay3302. [Google Scholar] [PubMed] [CrossRef]
82. O'Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW. 1,026 experimental treatments in acute stroke. Ann Neurol. 2006;59(3):467–77. doi:10.1002/ana.20741. [Google Scholar] [PubMed] [CrossRef]
83. Lai K, Pritišanac I, Liu ZQ, Liu HW, Gong LN, Li MX, et al. Glutamate acts on acid-sensing ion channels to worsen ischaemic brain injury. Nature. 2024;631(8022):826–34. doi:10.1038/s41586-024-07684-7. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools