
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

The role of tazarotene-induced gene 1 in carcinogenesis: is it a tumor suppressor gene or an oncogene?
1 Department of Dermatology, Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, New Taipei City, 231, Taiwan
2 School of Medicine, Tzu Chi University, Hualien, 970, Taiwan
3 Department of Life Sciences, Ministry of Science and Technology, Taipei, 106, Taiwan
4 Department of Internal Medicine, Taipei Tzu Chi Hospital, The Buddhist Tzu Chi Medical Foundation, New Taipei City, 231, Taiwan
5 Department of Research, Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, New Taipei City, 231, Taiwan
* Corresponding Author: FU-MING TSAI. Email:
BIOCELL 2024, 48(9), 1285-1297. https://doi.org/10.32604/biocell.2024.053746
Received 09 May 2024; Accepted 21 June 2024; Issue published 04 September 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Tazarotene-induced gene 1 (TIG1) is induced by a derivative of vitamin A and is known to regulate many important biological processes and control the development of cancer. TIG1 is widely expressed in various tissues; yet in many cancer tissues, it is not expressed because of the methylation of its promoter. Additionally, the expression of TIG1 in cancer cells inhibits their growth and invasion, suggesting that TIG1 acts as a tumor suppressor gene. However, in some cancers, poor prognosis is associated with TIG1 expression, indicating its protumor growth characteristics, especially in promoting the invasion of inflammatory breast cancer cells. This review comprehensively summarizes the roles of the TIG1 gene in cancer development and details the mechanisms through which TIG1 regulates cancer development, with the aim of understanding its various roles in cancer development.Keywords
Abbreviations
| 5-aza-dC | 5-aza-2’-deoxycytidine |
| AGBL2 | ATP/GTP binding protein-like 2 |
| AMPK | AMP-activated protein kinase |
| CCP2 | Cytoplasmic carboxypeptidase 2 |
| CCPs | Cytosolic carboxypeptidases |
| CDK | Cyclin-dependent kinase |
| CRC | Colorectal cancer |
| CTCs | Circulating tumor cells |
| CXCR4 | C-X-C chemokine receptor type 4 |
| DNAJC8 | DnaJ heat shock protein family member C8 |
| EBV | Epstein-Barr virus |
| ECM | Extracellular matrix |
| EMT | Epithelial-to-mesenchymal transition |
| ER | Endoplasmic reticulum |
| FBXW7 | F-box/WD repeat-containing protein 7 |
| Glut | Glucose transporter |
| GPCRs | G protein coupled receptors |
| GRK5 | G protein-coupled receptor kinase 5 |
| HCC | Hepatocellular carcinoma |
| HDAC | Histone deacetylase |
| HOX | Homeobox |
| IBC | Inflammatory breast cancer |
| ICAM | Intercellular adhesion molecule |
| IL | Interleukin |
| ITGB3 | Integrin subunit beta 3 |
| KLKs | Kallikrein-related peptidases |
| MAT | Mesenchymal-to-amoeboid transition |
| MMPs | Matrix metalloproteinases |
| mTOR | Mammalian target of rapamycin |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NPC | Nasopharyngeal carcinoma |
| PDAC | Pancreatic ductal adenocarcinoma |
| PGE2 | Prostaglandin E2 |
| PKM2 | Pyruvate kinase M2 |
| PLK | Polo-like kinase |
| RA | Retinoic acid |
| RAR | Retinoic acid receptor |
| RARRES1 | Retinoic acid receptor responder protein 1 |
| RCC | Renal cell carcinoma |
| ROCK | Rho-associated kinase |
| SIRT1 | Sirtuin 1 |
| SOX1 | SRY-box transcription factor 1 |
| SPINK | Serine protease inhibitor Kazal-type protein |
| TAM | Tyro3: Axl: and Mer |
| TCPs | Tubulin carboxypeptidases |
| TIG1 | Tazarotene-induced gene 1 |
| TKIs | Tyrosine kinase inhibitors |
| TMEM192 | Transmembrane protein 192 |
| TNF | Tumor necrosis factor |
| TTL | Tubulin tyrosine ligase |
| uPA | Urokinase plasminogen activator |
In 1996, Tazarotene-induced gene 1 (TIG1), also known as retinoic acid receptor responder protein 1 (RARRES1), was discovered in human skin tissues and was shown to be induced by a retinoic acid receptor (RAR)β/γ-selective retinoid [1]. The TIG1 gene, which is located on chromosome 3q25.32, is translated into TIG1 isoform 1, consisting of 294 amino acids (GenBank: NP_996846.1), or TIG1 isoform 2, consisting of 228 amino acids (GenBank: NP_002879.2). Amino acid sequence alignment showed a high similarity of TIG1 to a protein of the Latexin family, which possesses inhibitory activities against rat carboxypeptidase A1 (CPA1) and CPA2 [2].
In addition to its expression in bone marrow and various subsequent cell types, skeletal muscle, and the brain, the TIG1 gene is widely expressed in various other organs and tissues [3,4]. Analysis of data from The Human Protein Atlas database (https://www.proteinatlas.org/ENSG00000118849-RARRES1/tissue, accessed on 12/04/2024) also revealed that in addition to its expression in ocular tissues, the TIG1 gene is broadly expressed across various tissues. Further analysis of its expression patterns in different tissues revealed that TIG1 was predominantly expressed in terminally differentiated tissues, such as acinar epithelial cells of normal prostate tissues [4], kidney tubules [5], and mucosal tissues of the colon [6]. Mice lacking the TIG1 gene develop normally but are more prone to follicular lymphoma, even though they exhibit increased B-cell survival and inhibited B-cell differentiation [7]. Based on the results from gene knockout mice and the distribution of TIG1 in normal tissues, the expression of TIG1 may be associated with tissue or cell differentiation.
The progression of cancer involves various genetic variations, including oncogenes produced by the activation of proto-oncogenes and the loss of function of tumor suppressor genes, which act as brakes and help monitor and maintain controlled cell cycles. Classical tumor suppressor genes are inherently recessive and anti-proliferative, and they often lose their activity due to mutations found in cancer tissues. However, recent research has elucidated that certain tumor suppressor genes do not conform to these standard definitions and exhibit dual roles, potentially having opposite effects within the cell. These dual-role tumor suppressor genes may include transcription factors and kinases that can initiate transcription or activate target molecules in a carcinogenic manner depending on specific cellular contexts, without involving any genetic modifications. Alternatively, these tumor suppressor genes may maintain fundamental cellular functions, such as promoting cell survival, through protein-protein interactions. Consequently, these tumor suppressor genes may perform pro-cancer functions during specific stages of carcinogenesis or in specific tissues through unique protein binding partners [8,9].
Reports indicate chromosomal 3q allelic loss in cancer cells [10–12]; however, instances of TIG1 gene mutations in cancer cells are rare. The results from numerous in vitro experiments suggest that TIG1 can influence cell metabolism, regulate mitochondrial activity, and inhibit cell growth [13], suggesting its role as a tumor suppressor gene in tumorigenesis. However, some studies have reported that TIG1 can promote cancer cell metastasis [14] or that the expression of TIG1 in cancer tissues is negatively correlated with patient survival [15]. These findings elucidate the “dual-agent” role of TIG1. Moreover, integrated results from the literature and database analyses indicate that TIG1 may play different roles in cancer cell development across different tissues. This review summarizes the expression patterns of the TIG1 gene in various cancer tissues and systematically explores signaling pathways that are targeted by TIG1 in the process of cancer cell evolution, with the aim of understanding the role of the TIG1 gene in cancer development.
Expression of TIG1 in Various Cancers
TIG1 is a transmembrane protein involved in regulating cell growth, differentiation, and apoptosis. It has been found to be dysregulated in multiple types of cancer, although its role as an oncogene or a tumor suppressor gene remains unclear. Table 1 summarizes the literature data on the expression of TIG1 in different cancer tissues and cells and explores the findings in the context of various cancer types.
Prostate cancer affects middle-aged men and is one of the malignant tumors contributing significantly to male mortality worldwide [30,31]. After hormone therapy fails, appropriate treatment options for advanced prostate cancer become scarce. Derivatives of vitamin A have shown significant effects in combating prostate cancer and are worth developing [32]. Jing et al. reported that TIG1 gene expression was detectable in all normal prostate tissues and benign prostatic hyperplasia tissues; however, among 51 malignant prostate tissues, only four exhibited TIG1 expression [4]. Research by Zhang et al. revealed that among 50 prostate cancer specimens, 26 had the TIG1 gene promoter methylated, while in eight adjacent normal tissue samples, no TIG1 promoter methylation was detected [16]. Additionally, treating prostate cancer cells that did not express TIG1 with a DNA methyltransferase inhibitor (5-aza-2’-deoxycytidine; 5-aza-dC) induced intracellular TIG1 expression, suggesting that the low expression of TIG1 in prostate cancer is associated with the methylation of its promoter.
In prostate epithelial cells, retinoic acid (RA) induces the expression of TIG1 and its homolog gene, Latexin. Compared to epithelial cells, TIG1 and Latexin have lower expression levels in mouse hematopoietic stem cells. In primary prostate cell cultures, inhibiting the expression of TIG1 or its homolog Latexin increased the cells’ stemness and invasive capabilities. Conversely, overexpressing TIG1 or Latexin had the opposite effects, suppressing cell colony formation and invasion [2]. In human prostate cells, TIG1 expression regulates autophagy-related and endoplasmic reticulum (ER) stress-related proteins, promoting cellular autophagy and inhibiting angiogenesis [17]. These findings collectively indicate that TIG1 acts as a tumor suppressor gene, restraining cancer cell growth in the prostate. They suggest that RA could be developed as a treatment for prostate cancer, potentially through the expression of TIG1 in the prostate, which may inhibit cancer cells from transforming into tumor stem cells and suppress angiogenesis.
TIG1 in kidney cancer (Renal cell carcinoma: RCC)
Kidney cancer is one of the most common cancers in the United States. Approximately 75% of RCC patients are diagnosed with clear cell renal cell carcinoma, which typically originates from the renal tubular epithelium and can metastasize to other organs, leading to most cancer-related deaths. Somatic mutations in the Von Hippel–Lindau gene have been known to lead to overexpression of growth factors, but further genomic studies have identified mutations in genes involved in epigenetic regulation, highlighting tumor heterogeneity [33,34]. Although RA can be used to treat kidney diseases or renal cancer, it is more concerning that it causes severe side effects on the kidneys, including tubular necrosis, granulomatous interstitial nephritis, and cortical necrosis [35].
Zimpfer et al. analyzed 903 cases of renal cell carcinoma and reported that TIG1 could be distributed in the cytoplasm or in the cell nucleus, with approximately 72.5% of cancer tissues showing cytoplasmic TIG1 expression. Additionally, higher expression of cytoplasmic TIG1 was positively correlated with a higher cancer grade, and patients with high cytoplasmic TIG1 expression had a shorter overall survival [18]. However, in a study by Peterfi et al., TIG1 expression was categorized as either membranous or cytoplasmic/negative. The study indicated that the absence of TIG1 expression (negative expression) or cytoplasmic TIG1 expression was a significant risk factor for tumor progression, whereas membranous TIG1 expression was associated with better disease outcomes [5]. Additionally, when TIG1’s binding partner AGBL2 co-expressed with membranous TIG1, it further enhanced the positive effects of membranous TIG1 and increased the patient’s survival rate.
The potential mechanisms underlying RA-mediated side effects in kidney diseases include the release of proinflammatory cytokines such as interleukin (IL)-1β or tumor necrosis factor (TNF)-α causing cell necrosis, upregulation of thrombomodulin leading to a hypercoagulable state, and infiltration of immune cells [36]. However, TIG1 may also play a significant role in RA-mediated side effects. High expression of TIG1 is associated with poorer kidney survival rates, suggesting that TIG1 is a risk factor for RA-induced kidney disease [37]. Additionally, analysis of data from The Cancer Genome Atlas showed that TIG1 expression was negatively correlated with the survival rate of patients with renal cell carcinoma. Further research by Geng et al. indicated that TIG1 could upregulate intercellular adhesion molecule (ICAM)-1 expression, thereby facilitating macrophage infiltration and activation and leading to cancer cell apoptosis [15]. These findings suggest that while RA can inhibit cancer cell growth as a treatment for kidney cancer, its related side effects can impact its efficacy. The side effects mediated by RA may be related to TIG1 expression, leading to immune cell infiltration and kidney tissue damage.
TIG1 in nasopharyngeal carcinoma (NPC)
NPC is cancer associated with Epstein‒Barr Virus (EBV) infection, characterized by a high tendency for lymphatic spread and distant metastasis [38–40]. Undifferentiation is a primary characteristic of NPC, and promoting cellular differentiation offers a unique therapeutic opportunity. The activation of the RA metabolism pathway, which regulates SRY-box transcription factor 1(SOX1), plays a key role in the differentiation of NPC cells, providing a potential target for differentiation therapy in NPC [41]. Additionally, early studies have shown that retinoids can effectively inhibit EBV proliferation [42]. Kwong et al. reported that TIG1 expression was absent in 80% of nasopharyngeal carcinoma cell lines and 33% of nasopharyngeal carcinoma xenografts [19]. Moreover, methylation of the TIG1 gene promoter was associated with the absence of TIG1 expression, and treating cancer cells lacking TIG1 with 5-aza-2’-deoxycytidine restored TIG1 expression. Normal epithelial cells did not exhibit TIG1 gene promoter methylation. These findings suggest that the epigenetic silencing of TIG1 in primary nasopharyngeal tumors may indicate its potential role in the pathogenesis of nasopharyngeal carcinoma.
Kwok et al. demonstrated that overexpressing or knocking down TIG1 in HK1 NPC cells significantly reduced or enhanced cell proliferation, respectively [20]. However, in EBV-infected HK1 cells, while the expression of TIG1 did not affect the proliferation of these cells, the knockdown of TIG1 enhanced the invasive ability of EBV-infected HK1 cells. These results suggest that TIG1 may play a role in the proliferation and invasion of NPC cells, with its function potentially dependent on EBV infection. Additionally, the study did not address whether TIG1 expression affects the proliferation of EBV within the cells. Therefore, the importance of TIG1 in RA regulation of NPC requires further investigation.
TIG1 in inflammatory breast cancer (IBC)
In studies involving either breast cancer cell lines or mouse models of breast cancer, RA has been shown to inhibit the growth of mammary tumor cells. In addition to directly regulating cancer cell growth, RA can suppress tumor angiogenesis and modulate the immune status of the tumor microenvironment to achieve inhibition of cancer cell proliferation [43]. IBC involves cancer cells that block lymphatic vessels in the skin of the breast, making it a rare, highly lethal, and extremely aggressive disease [44]. In a study by Wang et al., TIG1 was found to be highly expressed in IBC cell lines but not in non-IBC breast cancer cell lines or normal breast tissue. Moreover, TIG1 expression in cancerous tissues was negatively correlated with the median survival time of IBC patients [14]. Knockdown of TIG1 expression inhibited cell proliferation and invasion of IBC cell lines and reduced the ability of cancer cells to grow in xenografts. Currently, there are no literature reports on whether RA regulates the growth of IBC, so the importance of the RA activation pathway in IBC invasion cannot be determined. Nonetheless, these study results suggest that TIG1 may act as an oncogene, positively regulating the malignant properties of IBC cells.
TIG1 in pancreatic ductal adenocarcinoma (PDAC)
PDAC is a relatively rare cancer. Owing to its often-nonspecific symptoms, high invasiveness, and late-stage diagnosis, PDAC is expected to become the second leading cause of cancer-related deaths in the United States by 2030 [45–47]. Despite the availability of new chemotherapy drugs, such as FOLFIRINOX, for PDAC patients, the effectiveness of these cytotoxic agents is significantly limited by the frequent development of chemotherapy resistance [48,49]. In addition to chemotherapy, the antitumor activity of RA in the treatment of PDAC has begun to gain attention [50], and this treatment has now entered clinical trials [51].
Nitschke et al. used stable isotope labeling by amino acids in cell culture to identify differentially expressed genes between chemoresistant and chemosensitive PDAC cells and reported that TIG1 was highly expressed in chemoresistant PDAC cells but not in normal pancreas cells or other PDAC cell lines [21]. The study also revealed that patients with PDAC who had circulating tumor cells (CTCs) had a poorer prognosis, and if these CTCs expressed TIG1, they could represent a more aggressive CTC subtype. These findings suggest that TIG1 could be a marker for a specific subtype of CTCs in PDAC patients. They also imply that during the combined treatment of PDAC with RA and chemotherapy, inducing TIG1 expression may become a risk factor, potentially transforming cancer cells into a more invasive state.
TIG1 in colorectal cancer (CRC)
CRC is the second most lethal cancer, and its global incidence and mortality rates are expected to further increase over the next few decades. CRC displays considerable genetic diversity, with numerous somatic clonal mutations found in many cancer cells, arising from variations in gene expression. This makes CRC one of the most mutation-rich malignancies [52–54]. Furthermore, disruptions in RA signaling pathways are associated with the occurrence and progression of CRC cancer. Reduced expression of RA target genes correlates with poorer prognosis in CRC, suggesting that RA signaling pathways could serve as therapeutic targets for CRC [55]. A study by Wang et al. revealed that TIG1 was highly expressed in normal colon tissues, but its expression tended to decrease in cancerous tissues [22,23]. Analysis of the distribution of the TIG1 protein in colorectal cancer tissues revealed that TIG1 was located primarily in normal colon tissues and in highly differentiated colorectal adenocarcinomas, while its expression was low in poorly differentiated colorectal adenocarcinomas [5]. However, the expression levels of TIG1 in colorectal cancer tissues were not significantly correlated with the survival rates of CRC patients [6], suggesting that TIG1 alone may not be sufficient to correct the disrupted RA signaling in CRC.
Nonetheless, overexpression of TIG1 in the CRC cell lines HCT116 and SW620 was shown to inhibit cell growth [3,22,24]. Additionally, the overexpression of TIG1 suppressed the invasion of cancer cells, while the knockdown of TIG1 promoted cell invasiveness [23], suggesting that TIG1 plays a tumor-suppressing role in CRC.
Choriocarcinoma is a relatively rare and invasive form of trophoblastic disease. Choriocarcinoma is primarily categorized into two types, namely, gestational and nongestational choriocarcinoma. In western countries, the likelihood of choriocarcinoma occurrence in pregnant women is approximately one in 40,000, while that in patients with molar pregnancy is approximately one in 40 [56]. Both alone and in combination with chemotherapy drugs, RA effectively inhibits the growth of choriocarcinoma cells [57].
A study by Huebner et al. revealed that the expression of TIG1 in choriocarcinoma cells was lower than that in healthy placental tissue, and this downregulation was associated with a high level of TIG1 gene promoter methylation [25]. Furthermore, as cell density increased during the growth of Swan71 trophoblast cells, TIG1 expression was induced, with the upregulation of E-cadherin and downregulation of N-cadherin. These results suggest that TIG1 may play a tumor-suppressive role in the development of choriocarcinoma.
TIG1 in hepatocellular carcinoma (HCC)
Liver cancer is one of the most common cancers worldwide, with HCC accounting for 90% of cases. Chemotherapy drugs have limited efficacy in treating HCC; however, combining treatment with RA has been shown to effectively increase patients’ two-year survival rates [58]. Additionally, although tyrosine kinase inhibitors (TKIs) are currently used for treatment, the survival rates of patients with advanced HCC have not significantly improved [59].
Guo et al. used a high-throughput approach to screen liver cancer cells for differentially expressed genes associated with the sensitivity to the first-line TKI lenvatinib. They found that cells sensitive to lenvatinib showed significantly increased TIG1 expression [26]. Overexpression of TIG1 in the HCC cell lines Huh7 and SKHEP1 inhibited their cell growth and in vivo tumor growth and increased the sensitivity to lenvatinib. Conversely, silencing TIG1 enhanced HCC cell proliferation and migration. These findings suggest that TIG1 can suppress HCC progression and modulate its sensitivity to lenvatinib. Although current multicenter clinical trials primarily involve combining TKIs with immune checkpoint inhibitors for treating HCC [60], the increased sensitivity of liver cancer cells to TKIs due to TIG1 expression suggests that combining RA with TKIs could also be considered as a candidate approach for treating HCC.
Testicular cancer is one of the most common malignancies, accounting for approximately 1% of all male cancers and 5% of genitourinary malignancies. The causes of testicular cancer are multifactorial, with testicular dysgenesis syndrome identified as a common risk factor for testicular cancer [61]. The survival rate for testicular germ cell tumors is high, and chemotherapy remains the primary treatment modality. However, due to the long-term side effects of platinum-based chemotherapy leading to overtreatment, alternative drugs are being continuously studied. Among them, RA treatment can alter the morphology of testicular cancer cells and induce differentiation [62], suggesting its potential as an adjunct therapy for treating testicular germ cell tumors.
Shyu et al. reported that the TIG1 gene was highly expressed in normal testicular tissues but was significantly downregulated in testicular carcinoma and testicular seminoma tissues [27]. The expression of TIG1 in NT2/D1 testicular cancer cells inhibited their invasion and influenced the expression of proteins related to epithelial-mesenchymal transition, indicating that TIG1 may have a role in suppressing the progression of testicular carcinoma.
Melanoma arises from melanocytes and is the most aggressive and deadly form of skin cancer [63]. Early-stage melanoma can be treated surgically, but once it metastasizes to distant organs, the efficacy of available drug therapies is limited, and treatment does not significantly improve patient survival rates [64]. The use of RA has been shown to effectively prevent the formation of more than 40% of melanomas, but the widespread use of RA is limited because of its side effects [65,66]. RA induces the expression of TIG1, suggesting that TIG1 may play a crucial role in RA’s ability to suppress melanoma formation.
A study by Wang et al. revealed that TIG1 gene expression was increased in normal skin tissues but was significantly decreased in melanoma tissues [28,29]. In A2058 and A375 melanoma cells, TIG1 expression inhibited insulin-induced cell proliferation and regulated the expression of cell cycle-related proteins, suggesting that TIG1 may play an important role in the RA-mediated suppression of melanoma development.
Molecular Pathways Involved in the Regulation of Tumor Progression by TIG1
Cancer invasion and metastasis of malignant tumor cells are among the leading causes of cancer-related deaths worldwide and have been a focus of research. Distant metastasis of malignant tumor cells is a complex process, involving local infiltration of tumor cells into neighboring tissues, transmigration of cancer cells across the endothelium into blood vessels, survival in the circulatory system, extravasation, and subsequent proliferation and colonization in distant organs [67,68]. During the progression of cancer as described above, epithelial tumor cells not only retain their ability for local cell proliferation but also exhibit plasticity through morphological and phenotypic transitions, such as epithelial-to-mesenchymal transition (EMT) and mesenchymal-to-amoeboid transition (MAT). Morphological changes in cancer cells and tissue invasion involve various genes; for instance, a hallmark of EMT is the downregulation or loss of E-cadherin, facilitated by transcriptional repressors such as Snail/SNAI1, Slug/SNAI2, or Twist, which leads to the disassembly of adherens junctions and nuclear translocation of membrane-bound β-catenin, thereby regulating the transcription of genes like c-myc or cyclin D1, followed by upregulation of mesenchymal markers such as vimentin and N-cadherin. MAT relies on Rac and Rho/Rho-associated kinase (ROCK) signaling pathways, influencing the expression state of cortical actin filaments in leading-edge cells and causing cell deformation. Whether EMT or MAT, the transformation process of cells involves reduced cell-cell or cell-extracellular matrix (ECM) adhesion, relying on protease activities such as matrix metalloproteinases (MMPs), serine proteases, and cathepsins [69–71].
Deletion of the TIG1 gene increases B cell survival but inhibits B cell differentiation in mice [7]. Overexpression of TIG1 causes defects in limb distal element regeneration in axolotls and induces proximal displacement of embryonic cells [72]. Moreover, excessive activation of TIG1 expression inhibits RIO kinase 1, leading to p53 activation and subsequent induction of apoptosis in mouse podocytes, whereas podocyte-specific TIG1 gene deletion alleviates proteinuria and glomerular injury in adriamycin-induced nephropathy in mice [37]. These in vivo animal study results highlight the importance of TIG1 in organ development and disease. Currently, there are no literature reports on the regulation of tumor cell growth and invasion by TIG1 in vivo. Nevertheless, through cell experiments and clinical data, numerous studies have explored the mechanisms by which TIG1 regulates cell growth and death, autophagy, cancer cell invasion, and cell differentiation. By examining protein-protein interactions or indirectly regulating downstream gene expression, the potential role of TIG1 in tumorigenesis can be revealed. In the following sections, an outline of the roles of TIG1 based on different cellular functional activities is provided.
The role of TIG1 in posttranslational modifications of tubulin
Microtubules are the largest component of the cytoskeleton and are involved in regulating various cellular activities, such as intracellular transport, chromatid separation during mitosis, cell migration, and the establishment of cell polarity. Microtubules are typically composed of 13 laterally associated protofilaments of α-tubulin and β-tubulin heterodimers, which form a dynamic hollow tubular structure. Both α-tubulin and β-tubulin can undergo multiple posttranslational modifications, which are linked to the regulation of microtubule properties and functionality. Known posttranslational modifications include acetylation, detyrosination, polyglutamylation, and polyglycylation. Acetylation of α-tubulin and β-tubulin is mediated by the acetyltransferase MEC-17/TAT1 or San; deacetylation is catalyzed by histone deacetylase 6 (HDAC6) and sirtuin 2. Detyrosination and retyrosination of α-tubulin are catalyzed by tubulin carboxypeptidases (TCPs) and tubulin tyrosine ligase (TTL), respectively. Polyglutamylation and deglutamylation are mediated by various TTL-like (poly) glutamylases and cytosolic carboxypeptidases (CCPs), respectively [73].
Sahab et al. reported that cytoplasmic carboxypeptidase 2 (CCP2/AGBL2) was involved in the detyrosination of α-tubulin, with TIG1 binding to and inhibiting the activity of AGBL2. Knockdown of AGBL2 prevented α-tubulin from undergoing detyrosination, whereas knockdown of TIG1 increased the levels of detyrosinated α-tubulin, suggesting that TIG1 regulates tubulin modification via AGBL2 [74]. The same team also reported that the interaction between TIG1 and AGBL2 inhibited tubulin deglutamylation, thereby regulating mitochondrial voltage-dependent anion channel 1, the mitochondrial membrane potential, and the activation of the AMP-activated protein kinas (AMPK) signaling pathway. Knockdown of TIG1 increased the expression of stem cell markers and promoted anoikis-and anchorage-independent growth, indicating that the interaction between TIG1 and AGBL2 is crucial for the metabolic regulation of cancer cells [13]. Furthermore, Peterfi et al. noted that the expression levels of TIG1 and AGBL2 were significant for assessing the outcomes of renal cell carcinoma [5], indicating that TIG1 and AGBL2 could indeed play a role in cancer cell growth in vivo.
The role of TIG1 in the regulation of the cell cycle
The cell cycle is regulated by various protein kinases, with cyclin-dependent kinase (CDK), polo-like kinase (PLK), and aurora kinase being among the most critical regulators [75–77]. PLKs are a family of serine/threonine kinases that regulate cell division, are highly conserved in eukaryotes, and are involved in controlling centrosome maturation, DNA checkpoint activation, mitotic entry, spindle assembly, and cytokinesis. In mammals, there are four types of PLKs, namely, PLK1, PLK2/Snk, PLK3/Fnk/Prk, and PLK4/Sak [78].
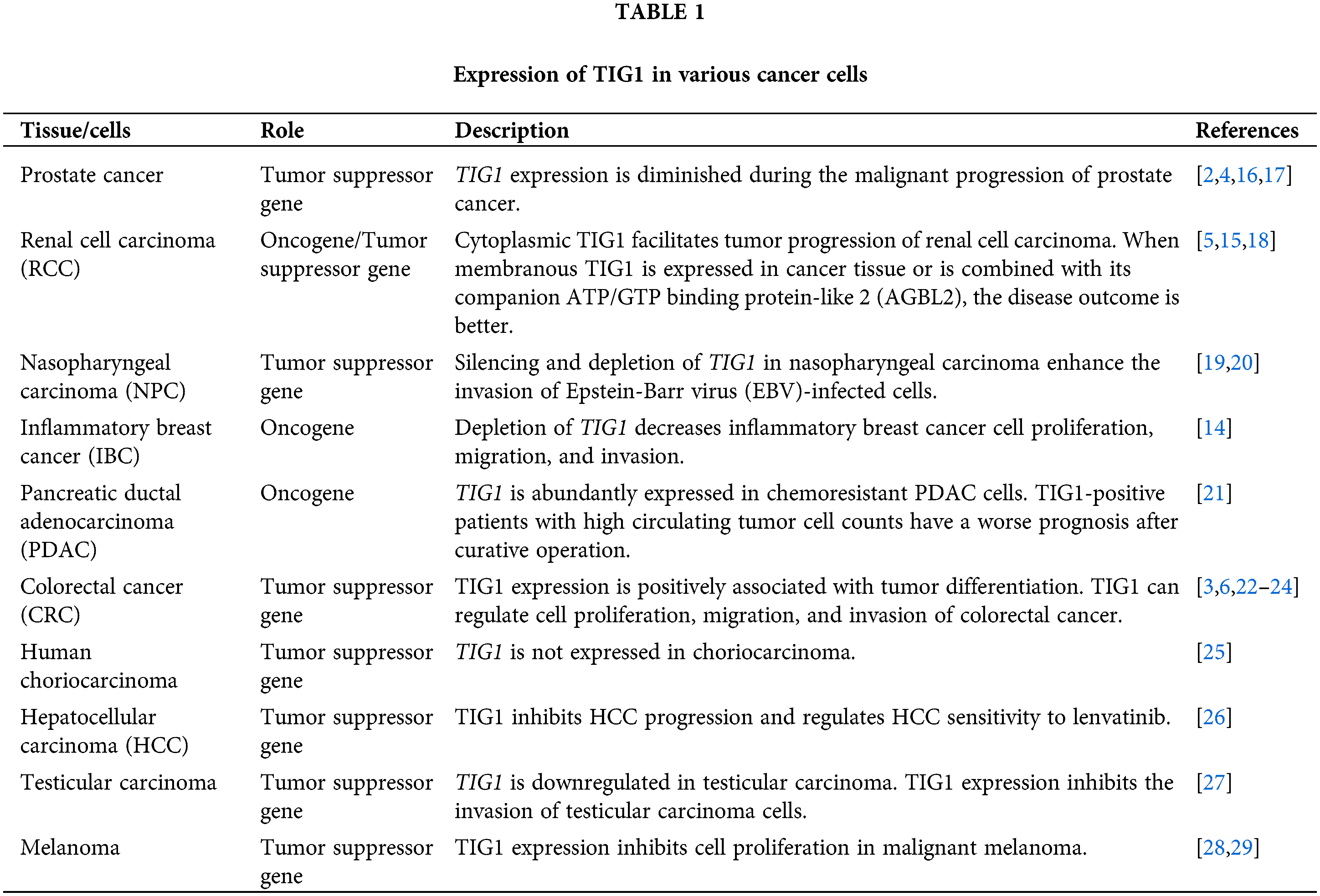
PLK2 is overexpressed in colorectal cancer tissues [22]. Activation of PLK2 inhibits the expression of the tumor suppressor protein F-box/WD repeat-containing protein 7 (FBXW7) and upregulates its downstream protein cyclin E1, which is regulated by FBXW7 to promote cell proliferation [79,80]. The interaction between TIG1 and PLK2 can prevent PLK2 from promoting CRC cell proliferation, which suggests that TIG1 can regulate the CRC cell cycle by modulating PLK2 activity, thereby inhibiting tumorigenesis [22], as illustrated in Fig. 1.
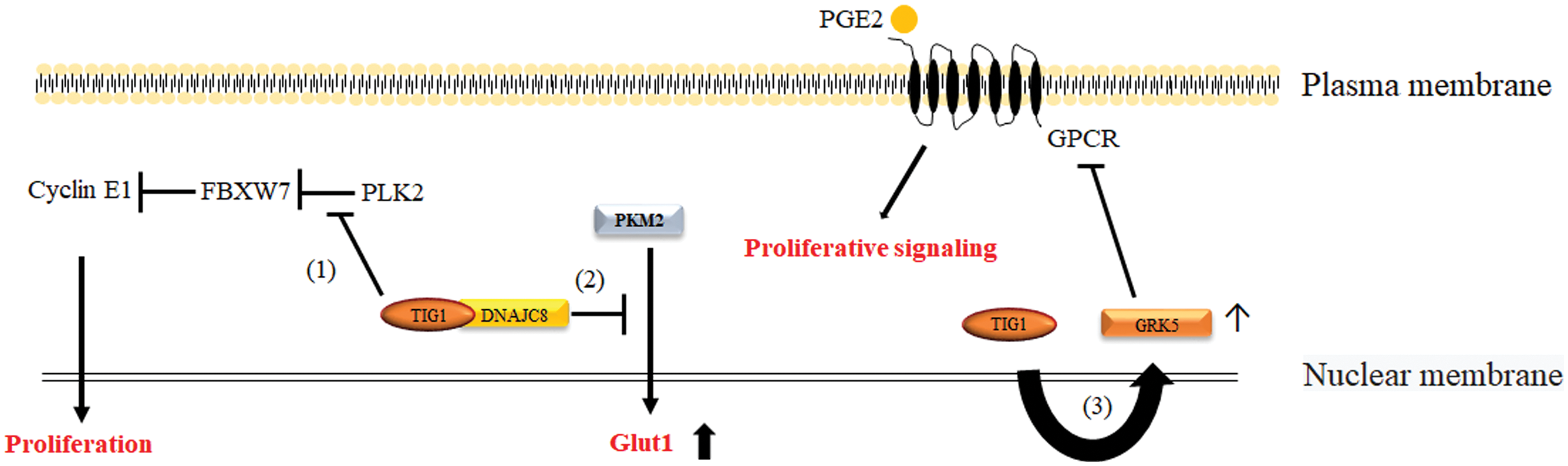
Figure 1: Mechanisms by which TIG1 inhibits cell proliferation. The pathways through which TIG1 suppresses cell proliferation include TIG1 binding to PLK2 to inhibit the production of the cell cycle regulator cyclin E1 (Path 1); TIG1 binding to DNAJC8 to prevent PKM2 from entering the nucleus and inducing Glut1 transcription (Path 2); and TIG1 inducing GRK5 expression to block PGE2-mediated signaling via GPCRs involved in cell proliferation (Path 3).
The role of TIG1 in the regulation of glucose uptake
Glucose metabolism involves transporting glucose from the bloodstream into cells and processing it by various enzymes to generate energy for cellular activities. Glucose is a hydrophilic molecule that requires the sodium-independent facilitative glucose transporter (Glut)1-Glut4 for crossing cell membranes. Among these proteins, Glut1 is critical for glucose uptake by cancer cells and is often upregulated in cancer cells [81].
Pyruvate kinase M2 (PKM2) primarily regulates cellular metabolism by converting phosphoenolpyruvate to pyruvate [82]. Additionally, PKM2 can enter the cell nucleus as a transcriptional cofactor, contributing to the transcription of Glut1 [83–85]. TIG1, by binding to DnaJ heat shock protein family member C8 (DNAJC8), sequesters PKM2 in the cytoplasm, thereby inhibiting Glut1 transcription, which in turn reduces glucose uptake and prevents cell proliferation [86]. These findings suggest that TIG1 indirectly regulates Glut1 gene expression, thereby reducing glucose uptake and inhibiting cancer cell growth (Fig. 1).
The role of TIG1 in the prostaglandin E2 (PGE2) pathway
PGE2 is a key mediator involved in chronic inflammation and directly contributes to tumor development by regulating cancer cell growth and migration, apoptosis, epithelial-mesenchymal transition, and angiogenesis [87]. PGE2 activates EP1-EP4 G protein-coupled receptors, leading to the activation of intracellular second messengers such as cyclic AMP, Ca2+, and inositol phosphates, which promote cancer progression [88].
Gene expression array analysis showed that TIG1 induced the expression of G protein-coupled receptor kinase 5 (GRK5) in CRC cells [3]. GRK5 is known to participate in the phosphorylation of G protein-coupled receptors (GPCRs), thereby reducing their signaling sensitivity [89,90]. Studies have shown that TIG1 induces GRK5 gene expression, preventing external factors, such as prostaglandin E2, from activating intracellular signaling, such as cyclic AMP production and nuclear entry of β-catenin, thus blocking PGE2-driven proliferation of CRC cells [3,24] (Fig. 1).
The role of TIG1 in regulating autophagy
Autophagy is an evolutionarily conserved process in which cells use lysosomes to degrade and recycle damaged organelles, misfolded proteins, and other large molecules [91]. Autophagy can be classified into general (non-selective) or selective types. General autophagy involves packaging portions of the cytoplasm into autophagosomes, which are then delivered to lysosomes for degradation. Selective autophagy, on the other hand, functions by recognizing specific targets such as damaged mitochondria and the ER. Generally, targeted selective autophagy may be a more effective cancer treatment strategy compared to non-selective autophagy because it selectively degrades damaged mitochondria, thereby precisely maintaining cellular homeostasis. Autophagy regulation plays a dual role in many cancers: activating autophagy under stress conditions can suppress tumor growth, yet it can also allow certain tumor cells to adapt to stress and thrive opportunistically. Hence, further research is needed to clarify the precise mechanisms of autophagy in cancer for the development of promising therapeutic targets [92].
Research on intracellular signaling has revealed that tumor suppressors are negatively regulated by mammalian targets of rapamycin (mTOR) and AMPK, leading to induction of autophagy and inhibition of cancer development [93]. Conversely, oncogenes may be activated by mammalian targets of mTOR and AKT pathways, suppressing autophagy and enhancing cancer formation [94,95]. These findings indicate that autophagy can suppress the generation of cancer cells. However, in cells with activated RAS mutations, high levels of autophagy can aid cancer cell survival. Inhibiting autophagy-related proteins can increase the accumulation of damaged mitochondria and reduce cancer cell growth. These results highlight the significant role of autophagy in the survival of tumors dependent on RAS activation [96]. Roy et al. reported that TIG1 upregulated the expression of the autophagy regulator sirtuin 1 (SIRT1) in prostate cancer cells and downregulated mTOR and the p38 MAPK, which inhibit autophagy activation, thereby activating autophagy [17]. Additionally, through its interaction with transmembrane protein 192 (TMEM192), TIG1 can activate the expression of autophagy-related proteins such as Beclin-1 and LC3B to induce autophagy [97] (Fig. 2). Although the ultimate effect of TIG1-induced cellular autophagy on cell growth in RAS-mutant cancer cells remains unknown, it is inferred that TIG1 can regulate selective autophagy through the mTOR pathway, thereby potentially inhibiting cancer cell growth through cellular autophagy mechanisms.
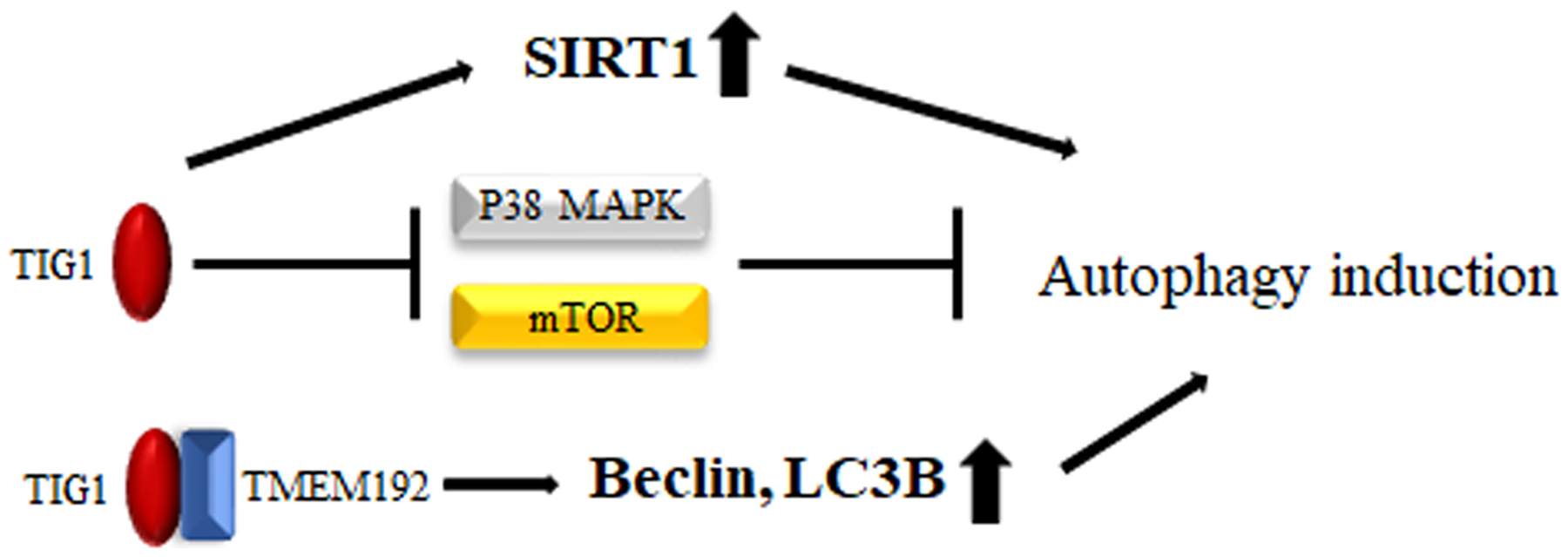
Figure 2: Mechanisms by which TIG1 induces autophagy. TIG1 can induce SIRT1 expression, inhibit the expression of mTOR and the p38 MAPK, or bind to TMEM192 to induce the production of Beclin-1 and LC3B, leading to the activation of autophagy.
The role of TIG1 in the activation of Axl receptor tyrosine kinase
AXL is a member of the Tyro3, Axl, and Mer (TAM) family of receptor tyrosine kinases and is considered a key contributor to tumor progression and metastasis. Its expression is associated with lower survival rates in various cancers. The role of Axl in promoting cancer progression is largely attributed to its ability to induce EMT. EMT provides tumor cells with various advantages, such as enhanced cell invasion, apoptosis resistance, and stem cell-like characteristics, thereby promoting tumor progression and contributing to cancer cell drug resistance [98].
A study by Wang et al. revealed that in inflammatory breast cancer, TIG1 was bound to and stabilized AXL to prevent its proteasomal degradation. Stabilized AXL continually activates downstream AKT and nuclear factor kappa-B (NF-κB) signaling and promotes MMP-9 gene transcription, which leads to increased cancer cell invasiveness [14] (Fig. 3).
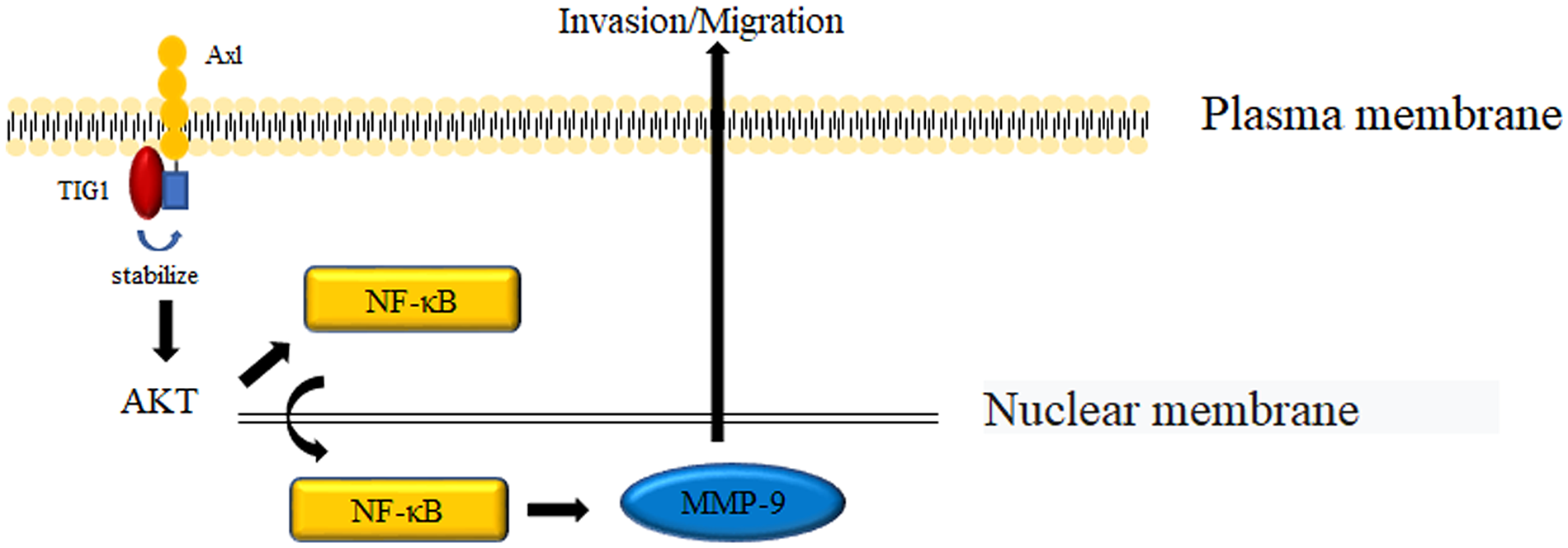
Figure 3: Mechanisms by which TIG1 promotes cell invasion. TIG1 stabilizes the Axl protein, which activates the AKT and NF-κB pathways to induce MMP-9 expression and promote cell invasion.
The role of TIG1 in the activation of the urokinase plasminogen activator (uPA) signaling pathway
uPA is a serine protease with various activities; when bound to its receptor uPAR, uPA can degrade most substrates in the extracellular matrix [99,100]. Thus, activation of the uPA signaling pathway is crucial for tumor cells to migrate from the primary site to distant organs. Inactive single-chain pro-uPA can be cleaved into active two-chain uPA by a number of cathepsins [101–103]. TIG1 binds to cathepsin V and leads to its rapid degradation, thereby inhibiting cathepsin V-induced EMT and suppressing cancer cell invasion in CRC [23] (Fig. 4).
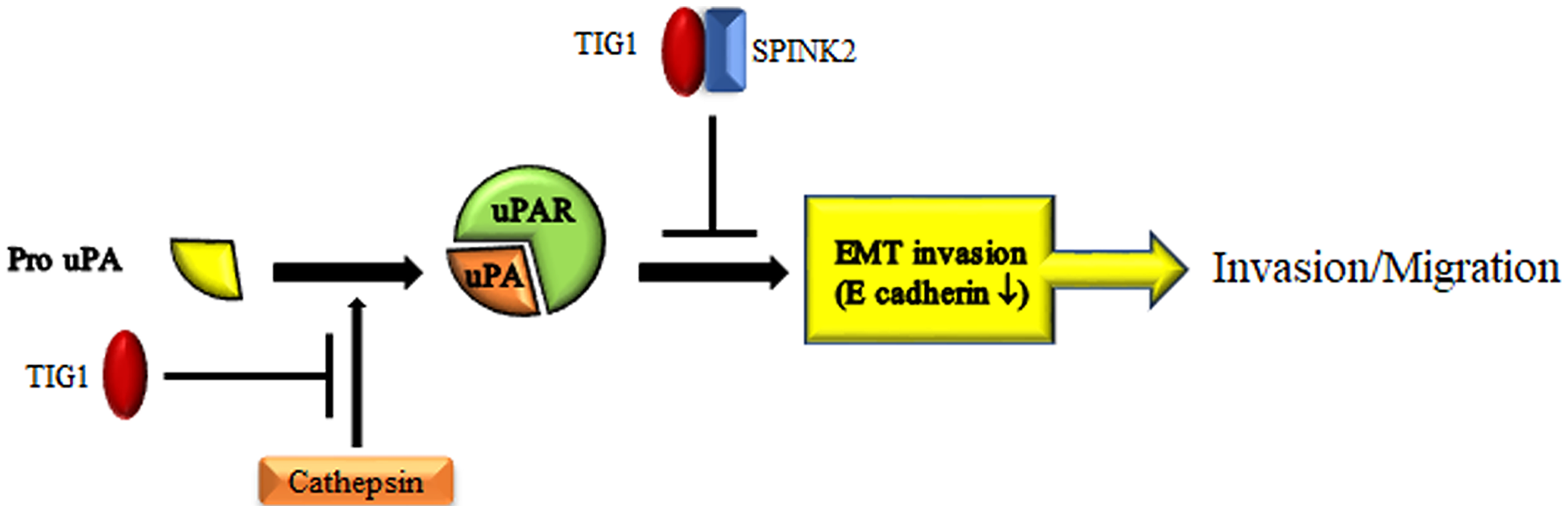
Figure 4: Mechanisms by which TIG1 inhibits cell invasion. TIG1 blocks cathepsin activation or collaborates with SPINK2 to interrupt uPA/uPAR signaling, thereby suppressing cell invasion.
Additionally, the activation of the uPA/uPAR pathway activates kallikrein-related peptidases (KLKs), and the KLK family is involved in the degradation of various interstitial substrates [104]. KLKs are targeted by the serine protease inhibitor Kazal-type protein (SPINK) family, and the synergy between SPINK and KLK is involved in the activation of the uPA/uPAR pathway [105,106]. TIG1 interacts with SPINK2, reducing the activity of uPA in testicular carcinoma cells and inhibiting their invasiveness [27]. These findings indicate that TIG1 can block the activation of uPA/uPAR signaling through two different pathways, which leads to the suppression of cancer cell invasion (Fig. 4).
While TIG1 appears to be involved in cell differentiation, organ development, and the regulation of cancer cell growth, there are currently no specific drugs developed to induce or inhibit TIG1 activity. RA upregulates TIG1 expression, and in carcinogenesis, promoter methylation often leads to TIG1 silencing, suggesting the potential importance of RA or drugs inhibiting DNA methylation like 5-aza-dC in cancer treatment assessment. Oral RA is the preferred treatment for adult acute promyelocytic leukemia and neuroblastoma [107,108]. Unfortunately, due to cancer cells developing resistance to RA or ineffective transport of RA to its designated site in the body, its clinical application is limited. Additionally, RA has relevant literature reports for treating gastric cancer, liver cancer, lung cancer, melanoma, and PDAC. Despite most evidence being from in vitro experiments, clinical trials have shown poor drug efficacy due to inefficient drug delivery or rapid clearance in the body [109]. 5-aza-dC has been approved by the European Medicines Agency for treating acute myeloid leukemia, and preclinical studies suggest its potential effectiveness against solid tumors. However, varying results from clinical trials have yet to confirm its efficacy [110].
Apart from being targeted for clinical treatment to achieve tumor therapy, TIG1 is more commonly used in clinical practice as a biomarker for diagnostic purposes. Database findings indicate that TIG1 is considered an adverse prognostic marker in renal cancer and glioma (https://www.proteinatlas.org/ENSG00000118849-RARRES1/pathology, accessed on 12/04/2024), although some concerns remain to be verified. For example, TIG1 is predominantly expressed in peripheral or highly differentiated colorectal adenoma tissue compared to most colorectal tissues that do not express TIG1 [6], making it difficult to assess detailed tumor conditions based on TIG1 expression levels in tumor blocks. Furthermore, the location of TIG1 expression may influence its protein function. Membranous TIG1 and cytoplasmic TIG1 appear to play different roles in cancer cells [18]. While TIG1 is considered an adverse prognostic marker in renal cancer and glioma, we further observe that cytoplasmic TIG1 is predominantly present in cancer tissues. Therefore, using TIG1 expression for clinical diagnosis of tumor conditions should consider the appropriate tissue distribution of TIG1.
Cancer is a group of diseases characterized by abnormal cell growth and the potential to spread to other parts of the body. Both external environmental factors and endogenous genetic factors play crucial roles in the development of cancer. While oncogenes can lead to uncontrolled cell growth and potentially cause cancer, tumor suppressor genes can prevent cells from becoming malignant, suggesting that their roles within the cell are seemingly opposite. These dual-function genes are mostly transcription factors or kinases, capable of both positively and negatively regulating transcription within cells to execute various biological activities. Take Homeobox (HOX) genes as an example: HOX genes encode a series of transcription factors that regulate embryogenesis. Furthermore, dysregulation of HOX genes is closely associated with many human malignancies [111]. For instance, HOXA5 can induce TP53 expression and inhibit β-catenin expression, thereby performing tumor suppressor functions in cervical cancer cells [112]. However, HOXB5 in CRC upregulates the expression of C-X-C chemokine receptor type 4 (CXCR4) and integrin subunit beta 3 (ITGB3), promoting cell invasion [113].
In general, cancer results from the accumulation of successive somatic mutations. Numerous studies have indicated that both gaining oncogene function and losing tumor suppressor gene function are necessary for the ability to transform normal cells into cancer cells. From this perspective, the lack of TIG1 expression in many cancer tissues due to the methylation of its promoter suggests that TIG1 functions as a tumor suppressor gene. However, it promotes cell survival and assists in cancer cell metastasis in IBC and PDAC. Studies indicate that TIG1 is not a transcription factor, unlike common dual-function tumor suppressor genes, but it may indirectly regulate the activity of specific transcription factors through protein-protein interactions. Given the unique roles of the TIG1 gene in tumorigenesis, cancer biologists need to understand its distribution across different cancer tissues, its potential targets, and its biological functions to better comprehend the contribution of TIG1 to tumor development. We hope that our review of TIG1 research will provide cancer biologists with a comprehensive understanding of the TIG1 gene functions from multiple perspectives.
Acknowledgement: None.
Funding Statement: This study was supported by the Taipei Tzu Chi Hospital through grants from the Buddhist Tzu Chi Medical Foundation under the Numbers TCRD-TPE-111-23 (2/3) and TCRD-TPE-113-20, Taipei, Taiwan.
Author Contributions: The authors confirm their contribution to the paper as follows: study conception and design: Chun-Hua Wang, Lu-Kai Wang; draft manuscript preparation: Chun-Hua Wang; review and editing: Chun-Hua Wang, Lu-Kai Wang, Fu-Ming Tsai; visualization: Rong-Yaun Shyu; supervision: Fu-Ming Tsai. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Nagpal S, Patel S, Asano AT, Johnson AT, Duvic M, Chandraratna RA. Tazarotene-induced gene 1 (TIG1a novel retinoic acid receptor-responsive gene in skin. J Invest Dermatol. 1996;106(2):269–74. doi:10.1111/1523-1747.ep12340668. [Google Scholar] [PubMed] [CrossRef]
2. Oldridge EE, Walker HF, Stower MJ, Simms MS, Mann VM, Collins AT, et al. Retinoic acid represses invasion and stem cell phenotype by induction of the metastasis suppressors RARRES1 and LXN. Oncogenesis. 2013;2(4):e45. doi:10.1038/oncsis.2013.6. [Google Scholar] [PubMed] [CrossRef]
3. Wu CC, Tsai FM, Shyu RY, Tsai YM, Wang CH, Jiang SY. G protein-coupled receptor kinase 5 mediates Tazarotene-induced gene 1-induced growth suppression of human colon cancer cells. BMC Cancer. 2011;11:175. doi:10.1186/1471-2407-11-175. [Google Scholar] [PubMed] [CrossRef]
4. Jing C, El-Ghany MA, Beesley C, Foster CS, Rudland PS, Smith P, et al. Tazarotene-induced gene 1 (TIG1) expression in prostate carcinomas and its relationship to tumorigenicity. J Natl Cancer Inst. 2002;94(7):482–90. doi:10.1093/jnci/94.7.482. [Google Scholar] [PubMed] [CrossRef]
5. Peterfi L, Banyai D, Yusenko MV, Bjercke T, Kovacs G. Expression of RARRES1 and AGBL2 and progression of conventional renal cell carcinoma. Br J Cancer. 2020;122(12):1818–24. doi:10.1038/s41416-020-0798-6. [Google Scholar] [PubMed] [CrossRef]
6. Wu CC, Shyu RY, Chou JM, Jao SW, Chao PC, Kang JC, et al. RARRES1 expression is significantly related to tumour differentiation and staging in colorectal adenocarcinoma. Eur J Cancer. 2006;42(4):557–65. [Google Scholar] [PubMed]
7. Patel J, Xun D, Creswell K, Kim DK, Wu M, Hwang JW, et al. Loss of RARRES1 function promotes follicular lymphomagenesis and inhibits B cell differentiation in mice. Int J Biol Sci. 2022;18(7):2670–82. [Google Scholar] [PubMed]
8. Datta N, Chakraborty S, Basu M, Ghosh MK. Tumor suppressors having oncogenic functions: the double agents. Cells. 2020;10(1):46. [Google Scholar] [PubMed]
9. Kim JW, Mo HY, Son HJ, Yoo NJ, Ann CH, Lee SH. Genes with dual proto-oncogene and tumor suppressor gene activities are frequently altered by protein losses in colon cancers. Pathol Res Pract. 2023;248:154659. [Google Scholar] [PubMed]
10. Yamaguchi T, Toguchida J, Yamamuro T, Kotoura Y, Takada N, Kawaguchi N, et al. Allelotype analysis in osteosarcomas: frequent allele loss on 3q, 13q, 17p, and 18q. Cancer Res. 1992;52(9):2419–23. [Google Scholar] [PubMed]
11. Dannenberg H, Speel EJ, Zhao J, Saremaslani P, Van Der Harst E, Roth J, et al. Losses of chromosomes 1p and 3q are early genetic events in the development of sporadic pheochromocytomas. Am J Pathol. 2000;157(2):353–9. doi:10.1016/S0002-9440(10)64547-6. [Google Scholar] [PubMed] [CrossRef]
12. Field J, Tsiriyotis C, Zoumpourlis V, Howard P, Spandidos D, Jones A. Allele loss on chromosome-3 in squamous-cell carcinoma of the head and neck correlates with poor clinical prognostic indicators. Int J Oncol. 1994;4(3):543–9. doi:10.3892/ijo.4.3.543. [Google Scholar] [PubMed] [CrossRef]
13. Maimouni S, Lee MH, Sung YM, Hall M, Roy A, Ouaari C, et al. Tumor suppressor RARRES1 links tubulin deglutamylation to mitochondrial metabolism and cell survival. Oncotarget. 2019;10(17):1606–24. doi:10.18632/oncotarget.26600. [Google Scholar] [PubMed] [CrossRef]
14. Wang X, Saso H, Iwamoto T, Xia W, Gong Y, Pusztai L, et al. TIG1 promotes the development and progression of inflammatory breast cancer through activation of Axl kinase. Cancer Res. 2013;73(21):6516–25. doi:10.1158/0008-5472.CAN-13-0967. [Google Scholar] [PubMed] [CrossRef]
15. Geng X, Chi K, Liu C, Fu Z, Wang X, Meng L, et al. Interaction of RARRES1 with ICAM1 modulates macrophages to suppress the progression of kidney renal clear cell carcinoma. Front Immunol. 2022;13:982045. doi:10.3389/fimmu.2022.982045. [Google Scholar] [PubMed] [CrossRef]
16. Zhang J, Liu L, Pfeifer GP. Methylation of the retinoid response gene TIG1 in prostate cancer correlates with methylation of the retinoic acid receptor beta gene. Oncogene. 2004;23(12):2241–9. [Google Scholar] [PubMed]
17. Roy A, Ramalinga M, Kim OJ, Chijioke J, Lynch S, Byers S, et al. Multiple roles of RARRES1 in prostate cancer: autophagy induction and angiogenesis inhibition. PLoS One. 2017;12(7):e0180344. [Google Scholar] [PubMed]
18. Zimpfer A, Dammert F, Glass A, Zettl H, Kilic E, Maruschke M, et al. Expression and clinicopathological correlations of retinoid acid receptor responder protein 1 in renal cell carcinomas. Biomark Med. 2016;10(7):721–32. [Google Scholar] [PubMed]
19. Kwong J, Lo KW, Chow LS, Chan FL, To KF, Huang DP. Silencing of the retinoid response gene TIG1 by promoter hypermethylation in nasopharyngeal carcinoma. Int J Cancer. 2005;113(3):386–92. [Google Scholar] [PubMed]
20. Kwok WK, Pang JC, Lo KW, Ng HK. Role of the RARRES1 gene in nasopharyngeal carcinoma. Cancer Genet Cytogenet. 2009;194(1):58–64. [Google Scholar] [PubMed]
21. Nitschke C, Markmann B, Tolle M, Kropidlowski J, Belloum Y, Goetz MR, et al. Characterization of RARRES1 expression on circulating tumor cells as unfavorable prognostic marker in resected pancreatic ductal adenocarcinoma patients. Cancers. 2022;14(18):4405. doi:10.3390/cancers14184405. [Google Scholar] [PubMed] [CrossRef]
22. Wang CH, Lu TJ, Wang LK, Wu CC, Chen ML, Kuo CY, et al. Tazarotene-induced gene 1 interacts with Polo-like kinase 2 and inhibits cell proliferation in HCT116 colorectal cancer cells. Cell Biol Int. 2021;45(11):2347–56. doi:10.1002/cbin.11681. [Google Scholar] [PubMed] [CrossRef]
23. Wang CH, Wang LK, Wu CC, Chen ML, Kuo CY, Shyu RY, et al. Cathepsin V mediates the Tazarotene-induced Gene 1-induced reduction in invasion in colorectal cancer cells. Cell Biochem Biophys. 2020;78(4):483–94. doi:10.1007/s12013-020-00940-3. [Google Scholar] [PubMed] [CrossRef]
24. Tsai FM, Wu CC, Shyu RY, Wang CH, Jiang SY. Tazarotene-induced gene 1 inhibits prostaglandin E2-stimulated HCT116 colon cancer cell growth. J Biomed Sci. 2011;18:88. doi:10.1186/1423-0127-18-88. [Google Scholar] [PubMed] [CrossRef]
25. Huebner H, Strick R, Wachter DL, Kehl S, Strissel PL, Schneider-Stock R, et al. Hypermethylation and loss of retinoic acid receptor responder 1 expression in human choriocarcinoma. J Exp Clin Cancer Res. 2017;36(1):165. doi:10.1186/s13046-017-0634-x. [Google Scholar] [PubMed] [CrossRef]
26. Guo Y, Chai B, Zhang H, Chai X, Chen Y, Xu J, et al. RARRES1 inhibits hepatocellular carcinoma progression and increases its sensitivity to lenvatinib through interaction with SPINK2. Biol Direct. 2024;19(1):15. [Google Scholar] [PubMed]
27. Shyu RY, Wang CH, Wu CC, Wang LK, Chen ML, Kuo CY, et al. Tazarotene-induced gene 1 (TIG1) interacts with serine protease inhibitor kazal-Type 2 (SPINK2) to inhibit cellular invasion of testicular carcinoma cells. Biomed Res Int. 2019;2019:6171065. [Google Scholar] [PubMed]
28. Wang CH, Wang LK, Wu CC, Chen ML, Kuo CY, Shyu RY, et al. TIG1 inhibits the mTOR signaling pathway in malignant melanoma through the VAC14 protein. Anticancer Res. 2023;43(6):2635–43. [Google Scholar] [PubMed]
29. Wang CH, Tzeng IS, Wang LK, Wu CC, Chen ML, Kuo CY, et al. Tazarotene-induced Gene 1 induces melanoma cell death by triggering endoplasmic reticulum stress response. Front Biosci. 2024;29(6):233. [Google Scholar]
30. Sekhoacha M, Riet K, Motloung P, Gumenku L, Adegoke A, Mashele S. Prostate cancer review: genetics, diagnosis, treatment options, and alternative approaches. Molecules. 2022;27(17):5730. [Google Scholar] [PubMed]
31. Wasim S, Park J, Nam S, Kim J. Review of current treatment intensification strategies for prostate cancer patients. Cancers. 2023;15(23):5615. [Google Scholar] [PubMed]
32. Halubiec P, Lazarczyk A, Szafranski O, Bohn T, Dulinska-Litewka J. Synthetic retinoids as potential therapeutics in prostate cancer—An update of the last decade of research: a review. Int J Mol Sci. 2021;22(19):10537. [Google Scholar] [PubMed]
33. Tran J, Ornstein MC. Clinical review on the management of metastatic renal cell carcinoma. JCO Oncol Pract. 2022;18(3):187–96. [Google Scholar] [PubMed]
34. Kase AM, George DJ, Ramalingam S. Clear cell renal cell carcinoma: from biology to treatment. Cancers. 2023;15(3):665. [Google Scholar] [PubMed]
35. DiKun KM, Gudas LJ. Vitamin A and retinoid signaling in the kidneys. Pharmacol Ther. 2023;248:108481. [Google Scholar] [PubMed]
36. Chen A, Liu Y, Lu Y, Lee K, He JC. Disparate roles of retinoid acid signaling molecules in kidney disease. Am J Physiol Renal Physiol. 2021;320(5):F683–92. [Google Scholar] [PubMed]
37. Chen A, Feng Y, Lai H, Ju W, Li Z, Li Y, et al. Soluble RARRES1 induces podocyte apoptosis to promote glomerular disease progression. J Clin Invest. 2020;130(10):5523–35. [Google Scholar] [PubMed]
38. Siak PY, Heng WS, Teoh SSH, Lwin YY, Cheah SC. Precision medicine in nasopharyngeal carcinoma: comprehensive review of past, present, and future prospect. J Transl Med. 2023;21(1):786. [Google Scholar] [PubMed]
39. Wong KCW, Hui EP, Lo KW, Lam WKJ, Johnson D, Li L, et al. Nasopharyngeal carcinoma: an evolving paradigm. Nat Rev Clin Oncol. 2021;18(11):679–95. [Google Scholar] [PubMed]
40. Jicman Stan D, Niculet E, Lungu M, Onisor C, Rebegea L, Vesa D, et al. Nasopharyngeal carcinoma: a new synthesis of literature data (Review). Exp Ther Med. 2022;23(2):136. [Google Scholar] [PubMed]
41. Lei XX, Liu Y, Wang JX, Cai Q, Yan M, He HP, et al. SOX1 promotes differentiation of nasopharyngeal carcinoma cells by activating retinoid metabolic pathway. Cell Death Dis. 2020;11(5):331. [Google Scholar] [PubMed]
42. Zeng Y, Zhou HM, Xu SP. Inhibitory effect of retinoids on Epstein-Barr virus induction in Raji cells. Intervirology. 1981;16(1):29–32. [Google Scholar] [PubMed]
43. Caricasulo MA, Zanetti A, Terao M, Garattini E, Paroni G. Cellular and micro-environmental responses influencing the antitumor activity of all-trans retinoic acid in breast cancer. Cell Commun Signal. 2024;22(1):127. [Google Scholar] [PubMed]
44. De Schepper M, Nguyen HL, Richard F, Rosias L, Lerebours F, Vion R, et al. Treatment response, tumor infiltrating lymphocytes and clinical outcomes in inflammatory breast cancer-treated with neoadjuvant systemic therapy. Cancer Res Commun. 2024;4(1):186–99. [Google Scholar] [PubMed]
45. Park W, Chawla A, O’Reilly EM. Pancreatic cancer: a review. JAMA. 2021;326(9):851–62. [Google Scholar] [PubMed]
46. Sarfraz H, Saha A, Jhaveri K, Kim DW. Review of current systemic therapy and novel systemic therapy for pancreatic ductal adenocarcinoma. Curr Oncol. 2023;30(6):5322–36. [Google Scholar] [PubMed]
47. Jiang Y, Sohal DPS. Pancreatic adenocarcinoma management. JCO Oncol Pract. 2023;19(1):19–32. [Google Scholar] [PubMed]
48. Janssen QP, Van Dam JL, Doppenberg D, Prakash LR, Van Eijck CHJ, Jarnagin WR, et al. FOLFIRINOX as initial treatment for localized pancreatic adenocarcinoma: a retrospective analysis by the trans-atlantic pancreatic surgery consortium. J Natl Cancer Inst. 2022;114(5):695–703. [Google Scholar] [PubMed]
49. Conroy T, Castan F, Lopez A, Turpin A, Ben Abdelghani M, Wei AC, et al. Five-year outcomes of FOLFIRINOX vs gemcitabine as adjuvant therapy for pancreatic cancer: a randomized clinical trial. JAMA Oncol. 2022;8(11):1571–8. [Google Scholar] [PubMed]
50. Mere Del Aguila E, Tang XH, Gudas LJ. Pancreatic ductal adenocarcinoma: new insights into the actions of vitamin A. Oncol Res Treat. 2022;45(5):291–8. [Google Scholar] [PubMed]
51. Kocher HM, Basu B, Froeling FEM, Sarker D, Slater S, Carlin D, et al. Phase I clinical trial repurposing all-trans retinoic acid as a stromal targeting agent for pancreatic cancer. Nat Commun. 2020;11(1):4841. [Google Scholar] [PubMed]
52. Hossain MS, Karuniawati H, Jairoun AA, Urbi Z, Ooi J, John A, et al. Colorectal cancer: a review of carcinogenesis, global epidemiology, current challenges, risk factors, preventive and treatment strategies. Cancers. 2022;14(7):1732. [Google Scholar] [PubMed]
53. Alzahrani SM, Al Doghaither HA, Al-Ghafari AB. General insight into cancer: an overview of colorectal cancer (Review). Mol Clin Oncol. 2021;15(6):271. [Google Scholar] [PubMed]
54. Biller LH, Schrag D. Diagnosis and treatment of metastatic colorectal cancer: a review. JAMA. 2021;325(7):669–85. [Google Scholar] [PubMed]
55. Wester RA, Van Voorthuijsen L, Neikes HK, Dijkstra JJ, Lamers LA, Frolich S, et al. Retinoic acid signaling drives differentiation toward the absorptive lineage in colorectal cancer. iScience. 2021;24(12):103444. [Google Scholar] [PubMed]
56. Wang Y, Wang Z, Zhu X, Wan Q, Han P, Ying J, et al. Intestinal metastasis from choriocarcinoma: a case series and literature review. World J Surg Oncol. 2022;20(1):173. [Google Scholar] [PubMed]
57. G. SEL, Harma M, Harma MI, Tekin IO. Comparison of the effects of all-trans retinoic acid, methotrexate, actinomycin D, and combined chemotherapy on different choriocarcinoma cell culture models. Med Sci. 2019;23(95):19–23. [Google Scholar]
58. Sun J, Liu C, Wang N, Jiang D, Zhang F, Shi J, et al. All-trans-retinoic acid plus oxaliplatin/fluorouracil/leucovorin for advanced hepatocellular carcinoma with pulmonary metastasis: a multicenter retrospective study. Cancer Manage Res. 2022;14:1663–70. [Google Scholar]
59. Mou L, Tian X, Zhou B, Zhan Y, Chen J, Lu Y, et al. Improving outcomes of tyrosine kinase inhibitors in hepatocellular carcinoma: new data and ongoing trials. Front Oncol. 2021;11:752725. [Google Scholar] [PubMed]
60. Wu JY, Wu JY, Li YN, Qiu FN, Zhou SQ, Yin ZY, et al. Lenvatinib combined with anti-PD-1 antibodies plus transcatheter arterial chemoembolization for neoadjuvant treatment of resectable hepatocellular carcinoma with high risk of recurrence: a multicenter retrospective study. Front Oncol. 2022;12:985380. [Google Scholar] [PubMed]
61. Cassell A, Jalloh M, Ndoye M, Yunusa B, Mbodji M, Diallo A, et al. Review of testicular tumor: diagnostic approach and management outcome in Africa. Res Rep Urol. 2020;12:35–42. [Google Scholar] [PubMed]
62. Lobo J, Cardoso AR, Miranda-Goncalves V, Looijenga LHJ, Lopez M, Arimondo PB, et al. Targeting germ cell tumors with the newly synthesized flavanone-derived compound MLo1302 efficiently reduces tumor cell viability and induces apoptosis and cell cycle arrest. Pharmaceutics. 2021;13(1):73. [Google Scholar] [PubMed]
63. Zakhem GA, Pulavarty AN, Lester JC, Stevenson ML. Skin cancer in people of color: a systematic review. Am J Clin Dermatol. 2022;23(2):137–51. [Google Scholar] [PubMed]
64. Schadendorf D, Van Akkooi ACJ, Berking C, Griewank KG, Gutzmer R, Hauschild A, et al. Melanoma. Lancet. 2018;392(10151):971–84. [Google Scholar] [PubMed]
65. Asgari MM, Brasky TM, White E. Association of vitamin A and carotenoid intake with melanoma risk in a large prospective cohort. J Invest Dermatol. 2012;132(6):1573–82. [Google Scholar] [PubMed]
66. Zhang YP, Chu RX, Liu H. Vitamin A intake and risk of melanoma: a meta-analysis. PLoS One. 2014;9(7):e102527. [Google Scholar] [PubMed]
67. Fares J, Fares MY, Khachfe HH, Salhab HA, Fares Y. Molecular principles of metastasis: a hallmark of cancer revisited. Signal Transduct Target Ther. 2020;5(1):28. [Google Scholar] [PubMed]
68. Gerstberger S, Jiang Q, Ganesh K. Metastasis. Cell. 2023;186(8):1564–79. [Google Scholar] [PubMed]
69. Wu JS, Jiang J, Chen BJ, Wang K, Tang YL, Liang XH. Plasticity of cancer cell invasion: patterns and mechanisms. Transl Oncol. 2021;14(1):100899. [Google Scholar] [PubMed]
70. Bakir B, Chiarella AM, Pitarresi JR, Rustgi AK. EMT, MET, plasticity, and tumor metastasis. Trends Cell Biol. 2020;30(10):764–76. [Google Scholar] [PubMed]
71. Aouad P, Quinn HM, Berger A, Brisken C. Tumor dormancy: EMT beyond invasion and metastasis. Genesis. 2024;62(1):e23552. [Google Scholar] [PubMed]
72. Oliveira CR, Knapp D, Elewa A, Gerber T, Gonzalez Malagon SG, Gates PB, et al. Tig1 regulates proximo-distal identity during salamander limb regeneration. Nat Commun. 2022;13(1):1141. [Google Scholar] [PubMed]
73. Lopes D, Maiato H. The tubulin code in mitosis and cancer. Cells. 2020;9(11):2356. [Google Scholar] [PubMed]
74. Sahab ZJ, Hall MD, Me Sung Y, Dakshanamurthy S, Ji Y, Kumar D, et al. Tumor suppressor RARRES1 interacts with cytoplasmic carboxypeptidase AGBL2 to regulate the alpha-tubulin tyrosination cycle. Cancer Res. 2011;71(4):1219–28. [Google Scholar] [PubMed]
75. Lee SY, Jang C, Lee KA. Polo-like kinases (Plksa key regulator of cell cycle and new potential target for cancer therapy. Dev Reprod. 2014;18(1):65–71. [Google Scholar] [PubMed]
76. Ding L, Cao J, Lin W, Chen H, Xiong X, Ao H, et al. The roles of cyclin-dependent kinases in cell-cycle progression and therapeutic strategies in human breast cancer. Int J Mol Sci. 2020;21(6):1960. doi:10.3390/ijms21061960. [Google Scholar] [PubMed] [CrossRef]
77. Willems E, Dedobbeleer M, Digregorio M, Lombard A, Lumapat PN, Rogister B. The functional diversity of Aurora kinases: a comprehensive review. Cell Div. 2018;13:7. doi:10.1186/s13008-018-0040-6. [Google Scholar] [PubMed] [CrossRef]
78. Llamazares S, Moreira A, Tavares A, Girdham C, Spruce BA, Gonzalez C, et al. polo encodes a protein kinase homolog required for mitosis in Drosophila. Genes Dev. 1991;5(12A):2153–65. doi:10.1101/gad.5.12a.2153. [Google Scholar] [PubMed] [CrossRef]
79. Yeh CH, Bellon M, Nicot C. FBXW7: a critical tumor suppressor of human cancers. Mol Cancer. 2018;17(1):115. doi:10.1186/s12943-018-0857-2. [Google Scholar] [PubMed] [CrossRef]
80. Cizmecioglu O, Krause A, Bahtz R, Ehret L, Malek N, Hoffmann I. Plk2 regulates centriole duplication through phosphorylation-mediated degradation of Fbxw7 (human Cdc4). J Cell Sci. 2012;125:981–92. doi:10.1242/jcs.095075. [Google Scholar] [PubMed] [CrossRef]
81. Zambrano A, Molt M, Uribe E, Salas M. Glut 1 in cancer cells and the inhibitory action of resveratrol as a potential therapeutic strategy. Int J Mol Sci. 2019;20(13):3374. [Google Scholar] [PubMed]
82. Zahra K, Dey T, Ashish, Mishra SP, Pandey U. Pyruvate kinase M2 and cancer: the role of PKM2 in promoting tumorigenesis. Front Oncol. 2020;10:159. [Google Scholar] [PubMed]
83. Salani B, Ravera S, Amaro A, Salis A, Passalacqua M, Millo E, et al. IGF1 regulates PKM2 function through Akt phosphorylation. Cell Cycle. 2015;14(10):1559–67. [Google Scholar] [PubMed]
84. Yang W, Lu Z. Regulation and function of pyruvate kinase M2 in cancer. Cancer Lett. 2013;339(2):153–8. [Google Scholar] [PubMed]
85. Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, et al. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol. 2012;14(12):1295–304. [Google Scholar] [PubMed]
86. Wang CH, Shyu RY, Wu CC, Chen ML, Lee MC, Lin YY, et al. Tazarotene-induced gene 1 interacts with DNAJC8 and regulates glycolysis in cervical cancer cells. Mol Cells. 2018;41(6):562–74. [Google Scholar] [PubMed]
87. Finetti F, Paradisi L, Bernardi C, Pannini M, Trabalzini L. Cooperation between prostaglandin E2 and epidermal growth factor receptor in cancer progression: a dual target for cancer therapy. Cancers. 2023;15(8):2374. [Google Scholar] [PubMed]
88. Finetti F, Travelli C, Ercoli J, Colombo G, Buoso E, Trabalzini L. Prostaglandin E2 and cancer: insight into tumor progression and immunity. Biol. 2020;9(12):434. [Google Scholar]
89. Gambardella J, Franco A, Giudice CD, Fiordelisi A, Cipolletta E, Ciccarelli M, et al. Dual role of GRK5 in cancer development and progression. Transl Med UniSa. 2016;14:28–37. [Google Scholar] [PubMed]
90. Harris DM, Cohn HI, Pesant S, Eckhart AD. GPCR signalling in hypertension: role of GRKs. Clin Sci. 2008;115(3):79–89. [Google Scholar]
91. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–41. doi:10.1016/j.cell.2011.10.026. [Google Scholar] [PubMed] [CrossRef]
92. Liu J, Wu Y, Meng S, Xu P, Li S, Li Y, et al. Selective autophagy in cancer: mechanisms, therapeutic implications, and future perspectives. Mol Cancer. 2024;23(1):22. doi:10.1186/s12943-024-01934-y. [Google Scholar] [PubMed] [CrossRef]
93. Park JM, Lee DH, Kim DH. Redefining the role of AMPK in autophagy and the energy stress response. Nat Commun. 2023;14(1):2994. doi:10.1038/s41467-023-38401-z. [Google Scholar] [PubMed] [CrossRef]
94. Panwar V, Singh A, Bhatt M, Tonk RK, Azizov S, Raza AS, et al. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct Target Ther. 2023;8(1):375. doi:10.1038/s41392-023-01608-z. [Google Scholar] [PubMed] [CrossRef]
95. Xu Z, Han X, Ou D, Liu T, Li Z, Jiang G, et al. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl Microbiol Biotechnol. 2020;104(2):575–87. doi:10.1007/s00253-019-10257-8. [Google Scholar] [PubMed] [CrossRef]
96. Debnath J, Gammoh N, Ryan KM. Autophagy and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol. 2023;24(8):560–75. [Google Scholar] [PubMed]
97. Shyu RY, Wang CH, Wu CC, Chen ML, Lee MC, Wang LK, et al. Tazarotene-induced gene 1 enhanced cervical cell autophagy through transmembrane protein 192. Mol Cells. 2016;39(12):877–87. [Google Scholar] [PubMed]
98. Goyette MA, Cote JF. AXL receptor tyrosine kinase as a promising therapeutic target directing multiple aspects of cancer progression and metastasis. Cancers. 2022;14(3):466. [Google Scholar] [PubMed]
99. Duffy MJ, Maguire TM, McDermott EW, O’Higgins N. Urokinase plasminogen activator: a prognostic marker in multiple types of cancer. J Surg Oncol. 1999;71(2):130–5. [Google Scholar] [PubMed]
100. Mahmood N, Mihalcioiu C, Rabbani SA. Multifaceted role of the urokinase-type plasminogen activator (uPA) and its receptor (uPARdiagnostic, prognostic, and therapeutic applications. Front Oncol. 2018;8:24. [Google Scholar] [PubMed]
101. Goretzki L, Schmitt M, Mann K, Calvete J, Chucholowski N, Kramer M, et al. Effective activation of the proenzyme form of the urokinase-type plasminogen activator (Pro-uPA) by the cysteine protease cathepsin L. FEBS Lett. 1992;297(1–2):112–8. doi:10.1016/0014-5793(92)80339-I. [Google Scholar] [PubMed] [CrossRef]
102. Kobayashi H, Schmitt M, Goretzki L, Chucholowski N, Calvete J, Kramer M, et al. Cathepsin B efficiently activates the soluble and the tumor cell receptor-bound form of the proenzyme urokinase-type plasminogen activator (Pro-uPA). J Biol Chem. 1991;266(8):5147–52. doi:10.1016/S0021-9258(19)67767-7. [Google Scholar] [CrossRef]
103. Maynadier M, Farnoud R, Lamy PJ, Laurent-Matha V, Garcia M, Rochefort H. Cathepsin D stimulates the activities of secreted plasminogen activators in the breast cancer acidic environment. Int J Oncol. 2013;43(5):1683–90. doi:10.3892/ijo.2013.2095. [Google Scholar] [PubMed] [CrossRef]
104. Emami N, Diamandis EP. New insights into the functional mechanisms and clinical applications of the kallikrein-related peptidase family. Mol Oncol. 2007;1(3):269–87. doi:10.1016/j.molonc.2007.09.003. [Google Scholar] [PubMed] [CrossRef]
105. Azouz NP, Ynga-Durand MA, Caldwell JM, Jain A, Rochman M, Fischesser DM, et al. The antiprotease SPINK7 serves as an inhibitory checkpoint for esophageal epithelial inflammatory responses. Sci Transl Med. 2018;10(444):895. doi:10.1126/scitranslmed.aap9736. [Google Scholar] [PubMed] [CrossRef]
106. Cai S, Zhang P, Dong S, Li L, Cai J, Xu M. Downregulation of SPINK13 promotes metastasis by regulating uPA in ovarian cancer cells. Cell Physiol Biochem. 2018;45(3):1061–71. doi:10.1159/000487348. [Google Scholar] [PubMed] [CrossRef]
107. Yilmaz M, Kantarjian H, Ravandi F. Acute promyelocytic leukemia current treatment algorithms. Blood Cancer J. 2021;11(6):123. doi:10.1038/s41408-021-00514-3. [Google Scholar] [PubMed] [CrossRef]
108. Zimmerman MW, Durbin AD, He S, Oppel F, Shi H, Tao T, et al. Retinoic acid rewires the adrenergic core regulatory circuitry of childhood neuroblastoma. Sci Adv. 2021;7(43):eabe0834. doi:10.1126/sciadv.abe0834. [Google Scholar] [PubMed] [CrossRef]
109. Giuli MV, Hanieh PN, Giuliani E, Rinaldi F, Marianecci C, Screpanti I, et al. Current trends in ATRA delivery for cancer therapy. Pharmaceutics. 2020;12(8):707. doi:10.3390/pharmaceutics12080707. [Google Scholar] [PubMed] [CrossRef]
110. Gleneadie HJ, Baker AH, Batis N, Bryant J, Jiang Y, Clokie SJH, et al. The anti-tumour activity of DNA methylation inhibitor 5-aza-2’-deoxycytidine is enhanced by the common analgesic paracetamol through induction of oxidative stress. Cancer Lett. 2021;501:172–86. doi:10.1016/j.canlet.2020.12.029. [Google Scholar] [PubMed] [CrossRef]
111. Feng Y, Zhang T, Wang Y, Xie M, Ji X, Luo X, et al. Homeobox genes in cancers: from carcinogenesis to recent therapeutic intervention. Front Oncol. 2021;11:770428. [Google Scholar] [PubMed]
112. Ma HM, Cui N, Zheng PS. HOXA5 inhibits the proliferation and neoplasia of cervical cancer cells via downregulating the activity of the Wnt/β-catenin pathway and transactivating TP53. Cell Death Dis. 2020;11(6):420. [Google Scholar] [PubMed]
113. Feng W, Huang W, Chen J, Qiao C, Liu D, Ji X, et al. CXCL12-mediated HOXB5 overexpression facilitates colorectal cancer metastasis through transactivating CXCR4 and ITGB3. Theranostics. 2021;11(6):2612–33. [Google Scholar] [PubMed]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools