
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

BTG2 interference ameliorates high glucose-caused oxidative stress, cell apoptosis, and lipid deposition in HK-2 cells
1 Department of Blood Transfusion, The First People’s Hospital of Wuhu, Wuhu, 241000, China
2 Department of Clinical Laboratory, The First People’s Hospital of Wuhu, Wuhu, 241000, China
* Corresponding Author: FAN CUI. Email:
BIOCELL 2024, 48(9), 1379-1388. https://doi.org/10.32604/biocell.2024.052205
Received 26 March 2024; Accepted 25 June 2024; Issue published 04 September 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Objective: Diabetic nephropathy (DN) is a deleterious microangiopathy of diabetes, constituting a critical determinant of fatality in diabetic patients. This work is purposed to disclose the effects and modulatory mechanism of BTG anti-proliferation factor 2 (BTG2) during the pathological process of DN. Methods: BTG2 expression in kidney tissues of diabetic mice and high glucose (HG)-exposed human proximal tubular cell line HK-2 was assessed with Western blot and RT-qPCR. The diabetic mice model was constructed by streptozotocin injection and confirmed by the blood glucose level beyond 16.7 mmol/L. Hematoxylin and eosin (H&E) staining and measurement of kidney function hallmarks were conducted to assess kidney injury. Cell counting kit (CCK)-8 method and TUNEL assay appraised cell activity and apoptosis. Oil red O staining assayed lipid accumulation. Relevant commercial kits were used to estimate oxidative stress-related factors. Co-immunoprecipitation (Co-IP) assay testified the binding relationship of BTG2 with protein arginine methyltransferase 1 (PRMT1). Results: BTG2 expression was significantly raised in renal tissues of diabetic mice and HK-2 cells exposed to HG. BTG2 deficiency improved viability and extenuated the apoptosis, lipid deposition as well as oxidative stress in HK-2 cells following HG exposure. In addition, PRMT1 was also overexpressed in HK-2 cells exposed to HG. BTG2 interacted with PRMT1 and positively modulated PRMT1 expression. The effects of BTG2 interference on viability, apoptosis, lipid deposition, and oxidative stress in HG-challenged HK-2 cells were partially abrogated by PRMT1 overexpression. Conclusion: Altogether, BTG2 might aggravate HK-2 cell injury in response to HG by binding with PRMT1, providing a novel target for the therapeutic strategy of DN.Keywords
Diabetes mellitus attributed to the persistently elevated blood glucose remains a complicated metabolic disease with escalating health concerns on the global scale, which may bring about micro and macrovascular complications involving multiple organs, including kidneys [1]. It is reported that the new cases of diabetes may rise to approximately 693 million by 2045 [2]. Considering the global epidemic of diabetes, the prevalence of diabetic nephropathy (DN), a deleterious microangiopathy of diabetes that may develop in 30%–40% of diabetic patients, is also on a drastic upward trend [3]. Despite conventional therapies covering the management of glycemia, blood pressure, and diet, a significant proportion of patients with DN still culminate into end-stage renal disease, contributing to considerable fatality [4,5]. Hence, it is of enormous clinical benefit to seek for effective intervention measures to retard the progression of DN.
The anti-proliferative (APR) BTG/TOB family comprises an emerging gene family implicated in various cellular events such as cell growth, death, differentiation, and survival [6]. Belonging to the BTG/TOB family, BTG anti-proliferation factor 2 (BTG2) is extensively abundant in multiple tissues and features the anti-proliferative property in human malignancies [7–9]. The existing evidence has proposed that BTG2 participates in the regulation of hepatic glucose homeostasis in diabetic mice [10]. Exogenous insulin significantly alters BTG2 gene expression and BTG2 is implicated in the modification of insulin homeostasis and glucose metabolism [11]. Moreover, recent research has identified BTG2 as a novel prospective target of DN therapy [12]. Nonetheless, whether BTG2 functions in the biological processes of DN is unknown.
Based on the GeneMANIA database (https://genemania.org/, accessed on 24 June 2024), BTG2 was preliminarily predicted to interact with protein arginine methyltransferase 1 (PRMT1). As a major member responsible for ~85% of total arginine methylation [13], PRMT1 has been supported by emerging literature to be associated with pathological states, such as tumors [14], cardiovascular diseases [15], brain diseases [16], and so on. Of note, Chen et al. [17] have disclosed that PRMT1 is overexpressed during the process of DN, and its knockdown may mitigate tissue injury and renal fibrosis.
This paper concentrates on delineating the impacts of BTG2 on the biological events in DN and the specific action mechanism related to PRMT1.
A total of 12 male mice (20 ± 2 g; 6 weeks old) on a C57BL/6 background obtained from Cavens Biogle Model Animal Research Co., Ltd. (Changzhou, China) were reared with food and water at liberty under a 12/12 h (light/dark) photoperiod for 7 d of habituation. The standard temperature (23°C ± 1°C) and humidity (55% ± 5%) were maintained. To induce DN, the mice fed with a high-fat diet (HFD) for 8 weeks were given 30 mg/kg streptozotocin (STZ; cat. no. M2082, AbMole Bioscience, Houston, TX, USA) dissolved in sodium citrate buffer via intraperitoneal injection. The mice in the Control group received the same volume of sodium citrate buffer. 72 h post-injection, mice with a casual blood glucose level above 16.7 mmol/L three times indicated a successful diabetic mouse model. Mice were euthanized by intraperitoneally injected with 50 mg/kg sodium pentobarbital. The blood samples and the renal tissues were harvested. All animal experiments were performed by the Regulations for the Administration of Affairs Concerning Experimental Animals approved by the State Council of the People’s Republic of China and were approved by the Animal Ethics Committee of The First People’s Hospital of Wuhu (Approval no. IACUC-20220311-02).
Hematoxylin and Eosin (H&E) staining
Following the separate immobilization, dehydration, and embedment of mice kidney tissues with 4% paraformaldehyde, gradient ethanol, and paraffin, respectively, the tissues were sectioned into 4 µm-thickness slices, followed by a 10 min-staining of hematoxylin and a 5 min-staining of eosin. The histological assessment was implemented by applying a microscope (Leica, DMi8, Wetzlar, Hessen, Germany).
Creatinine (Cre) and blood urea nitrogen (BUN) are hallmarks of kidney damage and compromised renal functions [18]. Serum Cre and BUN levels were examined with corresponding kits (cat. no. C011-2-1 for Cre and cat. no. C013-2-1 for BUN, Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
RPMI-1640 medium harboring 10% fetal bovine serum (FBS; cat. no. BNCC338360, BeNa Culture Collection, Xinyang, China) was adopted for the cultivation of human renal tubular epithelial cells (HK-2) from BeNa Culture Collection (http://www.bncc.com, accessed on 24 June 2024, cat. no. BNCC339833, Xinyang, China) under the circumstance of 5% CO2/95% air at 37°C. For treatment, HK-2 cells exposed to 5.5 mmol/L D-glucose (cat. no. M5567, AbMole Bioscience, Houston, TX, USA) for 48 h served as the Control group; Cells stimulated by 30 mmol/L D-glucose for 48 h served as the high glucose (HG) group; Cells cultivated under 5.5 mmol/L D-glucose plus 24.5 mmol/L D-mannitol (MA; cat. no. M3584, AbMole Bioscience, Houston, TX, USA) for 48 h were assigned as MA group to establish the control osmolarity.
Plasmid construction and transfection
The small interfering RNAs (siRNAs) targeting BTG2 (siRNA-BTG2-1: sense, 5′-GCUCCAUCUGCGUCUUGUACG-3′ and antisense, 5′-UACAAGACGCAGAUGGAGCCG-3′; siRNA-BTG2-2: sense, 5′-GCAUUCGCAUCAACCACAAGA-3′ and antisense, 5′-UUGUGGUUGAUGCGAAUGCAG-3′) and PRMT1 overexpression plasmid (Ov-PRMT1) designed by Obio (Shanghai, China), as well as their negative controls (siRNA-NC (sense, 5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense, 5′-ACGUGACACGUUCGGAGAATT-3′) and Ov-NC) were prepared. Upon 60%–70% confluency, the above plasmids were introduced to HK-2 cells utilizing Lipofectamine® 2000 in conformity with the protocol supplied by Invitrogen (cat. no. 11668027, Thermo Fisher Scientific, Inc., Carlsbad, CA, USA).
Reverse transcription-quantitative PCR (RT-qPCR)
In line with the manual from TRIzol reagent (cat. no. 19201ES60; Yeasen, Shanghai, China), total RNA was extracted from HK-2 cells. Subsequently, after the production of cDNA employing Hifair® II 1st Strand cDNA Synthesis Kit (cat. no. 11119ES60; Yeasen, Shanghai, China), PCR was carried out utilizing Hieff® qPCR SYBR Green Master Mix (cat. no. 11201ES08, Yeasen, Shanghai, China). Based on the 2−ΔΔCq approach, relative BTG2 expression were calibrated, with β-actin as a normalizer. The primer sequences for homo species genes were below BTG2, forward, 5′-CCTACCCCTACCCACCAGAA-3′ and reverse 5′-TTTACCGCTTTGCCTCGTCT-3′; β-actin, forward, 5′-AGCGAGCATCCCCCAAAGTT-3′ and reverse 5′-GGGCACGAAGGCTCATCATT-3′.
After the accomplishment of total protein isolation and quantification by RIPA lysis buffer (cat. no. 20115ES60, Yeasen, Shanghai, China) and BCA Protein Assay Kit (20201ES76, Yeasen, Shanghai, China), the proteins were subjected to separation in 12% SDS-PAGE and then shifted to polyvinylidene fluoride (PVDF) membranes (cat. no. IPVH00010; Millipore, MA, USA) using a constant current of 200 mA. After sealing in 5% bovine serum albumin (BSA; cat. no. 9048-46-8, Solarbio, Beijing, China), and overnight cultivation with primary antibodies recognizing BTG2 (cat. no. PA5-50204, Invitrogen, Thermo Fisher Scientific, Inc., Carlsbad, CA, USA), Bcl-2 (cat. no. #AF6138; Affinity Biosciences, Liyang, China), Bax (cat. no. #AF6614; Affinity Biosciences, Liyang, China), cleaved caspase3 (cat. no. #9661; CST, Boston, MA, USA), caspase3 (cat. no. #9662, CST, Boston, MA, USA), CD36 (cat. no. #DF13262; Affinity Biosciences, Liyang, China), PPARγ (cat. no. #AF6284; Affinity Biosciences, Liyang, China), PRMT1 (cat. no. orb1529734, Biorbyt, Wuhan, China) and β-actin (cat. no. #AF7018; Affinity Biosciences, Liyang, China), the membranes were subsequently labeled by HRP-linked secondary antibody (cat. no. #S0001; Affinity Biosciences, Liyang, China) the next day. Color development and density analysis were severally performed with an Enhanced ECL Chemiluminescent Substrate Kit (cat. no. 36222ES60, Yeasen, Shanghai, China) and ImageJ software version 1.53 (NIH, Bethesda, MD, USA).
5 × 103 HK-2 cells dispensed into 96-well plates were maintained for 48 h with 5.5 or 30 mmol/L of D-glucose. Thereafter, 10 μL of CCK-8 reagent (cat. no. C0037, Beyotime, Shanghai, China) was pipetted to cells. The absorbance at 450 nm was read 2 h later with a microplate reader (DNM9602G, Drawell Scientific, Shanghai, China).
Terminal-deoxynucleotidyl transferase mediated nick end labeling (TUNEL)
HK-2 cells were plated on chamber slides (5 × 104 cells/well) and maintained for 48 h with 5.5 or 30 mmol/L of D-glucose. Following respective immobilization and perforation with 4% paraformaldehyde and 0.1% Triton X-100, HK-2 cells were submerged in a TUNEL reaction mixture (cat. no. 40307ES20, Yeasen, Shanghai, China) for 1 h without light exposure. Nuclear staining was achieved utilizing 10 mg/mL DAPI (cat. no. C1005, Beyotime, Shanghai, China). Finally, TUNEL-positive cells were noticed under a fluorescence microscope (Leica, DMI4000B, Wetzlar, Hessen, Germany).
1 × 104 HK-2 cells were plated on chamber slides and maintained for 48 h with 5.5 or 30 mmol/L of D-glucose. Following 10 min of immobilization with 4% paraformaldehyde, HK-2 cells were subjected to staining with Oil Red O (cat. no. O1391, Sigma-Aldrich, St. Louis, MO, USA) at 4°C for half an hour, before hematoxylin counterstaining. The stained cells were observed under a microscope (Leica, DMi8, Wetzlar, Hessen, Germany).
Dichloro-dihydro-fluorescein diacetate (DCFH-DA) assay
2 × 105 cells were plated in six-well plates and maintained for 48 h with 5.5 or 30 mmol/L D-glucose. HK-2 cells were probed with a DCFH-DA probe (10 μmol/L; cat. no. 50101ES01, Yeasen, Shanghai, China) for half an hour at 37˚C without light exposure. The fluorescencent images were captured, and the fluorescence intensity was quantified to assess the production of intracellular reactive oxygen species (ROS).
Determination of malondialdehyde (MDA) and superoxide Dismutase (SOD) contents
The culture medium of each group was harvested. Subsequently, MDA and SOD concentrations within the medium were measured by MDA Content Assay Kit (cat. no. AKFA013M; Boxbio, Beijing, China) with the absorbance values at 532 nm and SOD Activity Assay Kit (cat. no. AKAO001M; Boxbio, Beijing, China) with the absorbance values at 560 nm with a microplate reader, separately.
Co-immunoprecipitation (Co-IP) assay
With the aid of a Co-IP kit (cat. no. abs955, Absin, Shanghai, China), a Co-IP assay was carried out. Briefly, 2 μg IP-indicated antibody (BTG2 (cat. no. sc-517187, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and PRMT1 (cat. no. sc-166963, Santa Cruz Biotechnology, Santa Cruz, CA, USA)) or IgG (cat. no. sc-515946, Santa Cruz Biotechnology, Santa Cruz, CA, USA) were added to protein lysates of HK-2 cells. followed by incubation with protein A/G beads overnight at 4°C. The untreated proteins served as an input control. The immunoprecipitants were subjected to Western blot analysis with anti-BTG2 antibody (cat. no. PA5-50204, Invitrogen, Thermo Fisher Scientific, Inc., Carlsbad, CA, USA) and anti-PRMT1 antibody (cat. no. orb1529734, Biorbyt, Wuhan, China).
Statistically significant differences were assessed using Student’s t-test between two groups or one-way analysis of variance (ANOVA) as well as Tukey’s post hoc test among >2 groups, which was done with SPSS 22.0 software (SPSS, Inc., Chicago, IL, USA). All data were repeated three times. p < 0.05 were denoted as significant in statistics.
BTG2 exhibits improved expression in DN
To specify the role of BTG2 in DN, a diabetic mice model was successfully established by STZ induction, presented by a casual blood glucose level above 16.7 mmol/L 3 times. As portrayed by H&E staining, the normal renal structure was observed in the Control group, whereas structural derangements of interstitium, glomerular vascular tufts, and tubular epithelia were observed in DN group (Fig. 1A). Also, the concentrations of kidney functional indicators (BUN and Cre) in the serum were both significantly elevated in the DN group relative to the Control group (p < 0.001; Fig. 1B,C), indicating the kidney damage and compromised renal functions. Thus, the above findings confirmed the successful establishment of an in vivo DN model in mice. Further, Western blot analysis determined that BTG2 expression was on an upward trend in the renal tissues of diabetic mice compared to the Control (p < 0.001; Fig. 1D). Concurrently, the in vitro model of DN was also constructed in HK-2 cells exposed to HG. As Fig. 1E illustrated, in comparison to the Control group, the viability of HK-2 cells was robustly eliminated in response to HG (p < 0.01). BTG2 expression was also raised in HK-2 cells challenged with HG as expected (p < 0.001; Fig. 1F,G). All these findings summarized that BTG2 expression was greatly upreagulted following DN both in vitro and in vivo.
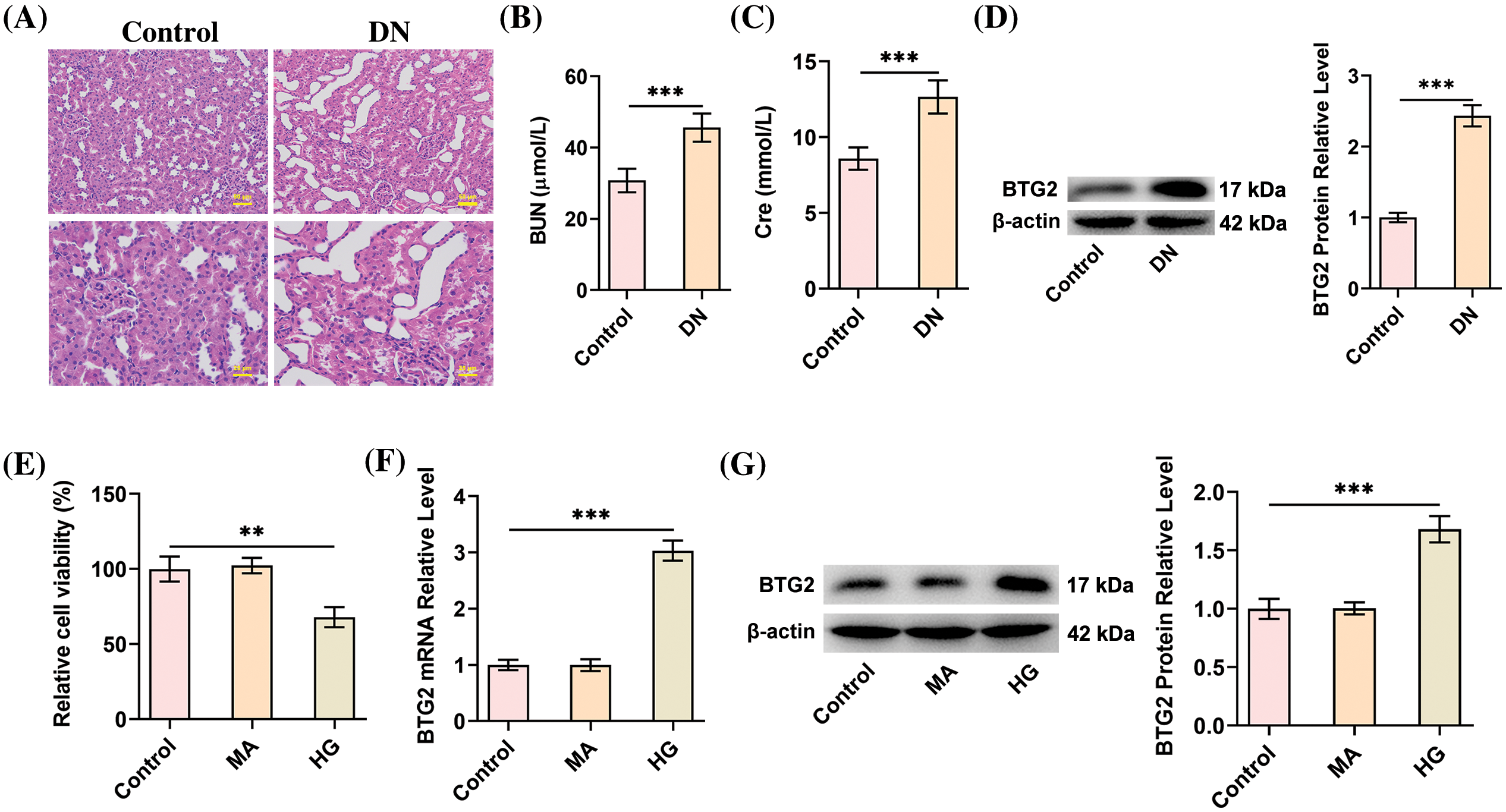
Figure 1: BTG2 exhibits improved expression in DN. (A) Representative histopathological images by H&E staining. (B and C) Biochemical kits evaluated the levels of serum Cre and BUN to assess kidney function. (D) Western blot was utilized for the detection of BTG2 expression in mice kidney tissues. n = 6 mice per group. (E) CCK-8 method was utilized for the assessment of cell activity. (F and G) RT-qPCR and Western blot tested BTG2 expression in HG-cultivated HK-2 cells. n = 3. **p < 0.01 and ***p < 0.001. DN, diabetic nephropathy; Cre, Creatinine; BUN, blood urea nitrogen; BTG2, BTG anti-proliferation factor 2; MA, D-mannitol; HG, high glucose.
BTG2 deletion extenuates HG-triggered viability loss and apoptosis of HK-2 cells
With the aim of identifying the effects of BTG2 on the DN cell model, BTG2 expression was significantly depleted following siRNA-BTG2-1/2 transfection, and the ability of siRNA-BTG2-1 was noted to be more potent in inhibiting BTG2 expression (Fig. 2A,B). Accordingly, we chose siRNA-BTG2-1 for application in the subsequent assays. After BTG2 was silenced, the viability of HG-cultivated HK-2 cells was remarkably boosted (Fig. 2C). Besides, the TUNEL-positive cells were notably elevated by HG exposure and then notably reduced following deficiency of BTG2 (Fig. 2D). Similarly, HG challenging significantly lowered Bcl-2 expression and increased Bax, Cleaved caspase3/caspase3 in HK-2 cells, which was remarkably reversed when BTG2 was down-regulated (Fig. 2E). Anyway, BTG2 interference might improve the viability and repress the apoptosis of HG-cultivated HK-2 cells.
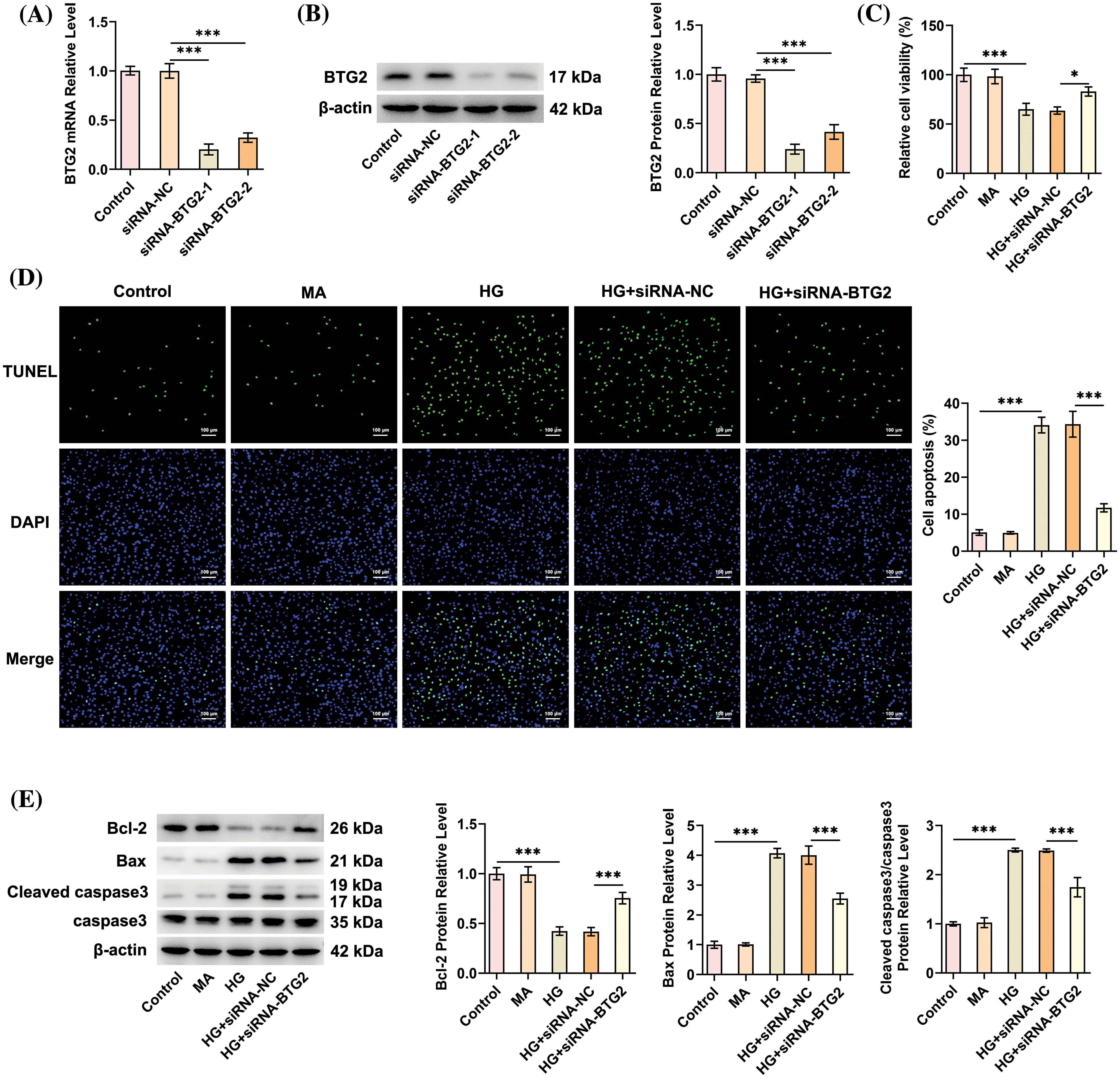
Figure 2: BTG2 deletion extenuates HG-triggered viability loss and apoptosis of HK-2 cells. (A and B) Transfection efficacy of BTG2 interference plasmids by RT-qPCR and Western blot. (C) CCK-8 method assayed cell activity. (D) TUNEL staining appraised cell apoptosis. (E) Western blot tested the expression of proteins implicated in apoptosis. n = 3. *p < 0.05 and ***p < 0.001. BTG2, BTG anti-proliferation factor 2; MA, D-mannitol; HG, high glucose; CCK-8, cell counting kit-8; siRNA-NC, negative control of small interfering RNA; siRNA-BTG2, small interfering RNA targeting BTG2.
BTG2 deletion eases HG-evoked oxidative stress and lipid deposition in HK-2 cells
Moreover, as depicted in Fig. 3A, Oil red O staining demonstrated that BTG2 down-regulation markedly hampered the lipid accumulation which was aggravated in HG-exposed HK-2 cells. In addition, peroxisome proliferator-activated receptor gamma (PPARγ)/CD36 signal is crucial in mediating adipogenesis process, ultimately affecting lipid deposition [19]. In comparison to the Control group, the protein levels of CD36 and PPARγ were up-regulated in HG-challenged HK-2 cells, which were lowered in BTG2-silencing HK-2 cells treated by HG (Fig. 3B). At the same time, upon exposure to HG, ROS and MDA levels were remarkably enhanced whereas SOD level was significantly diminished in HK-2 cells, in comparison to the Control group. Conversely, inhibition of BTG2 contributed to the decrease on ROS and MDA concentrations and the increase on SOD activity in HG-cultured HK-2 cells (Fig. 3C–E). To be concluded, BTG2 knockdown might exert anti-lipid accumulation and anti-oxidative stress activities in HG-elicited cellular model of DN.
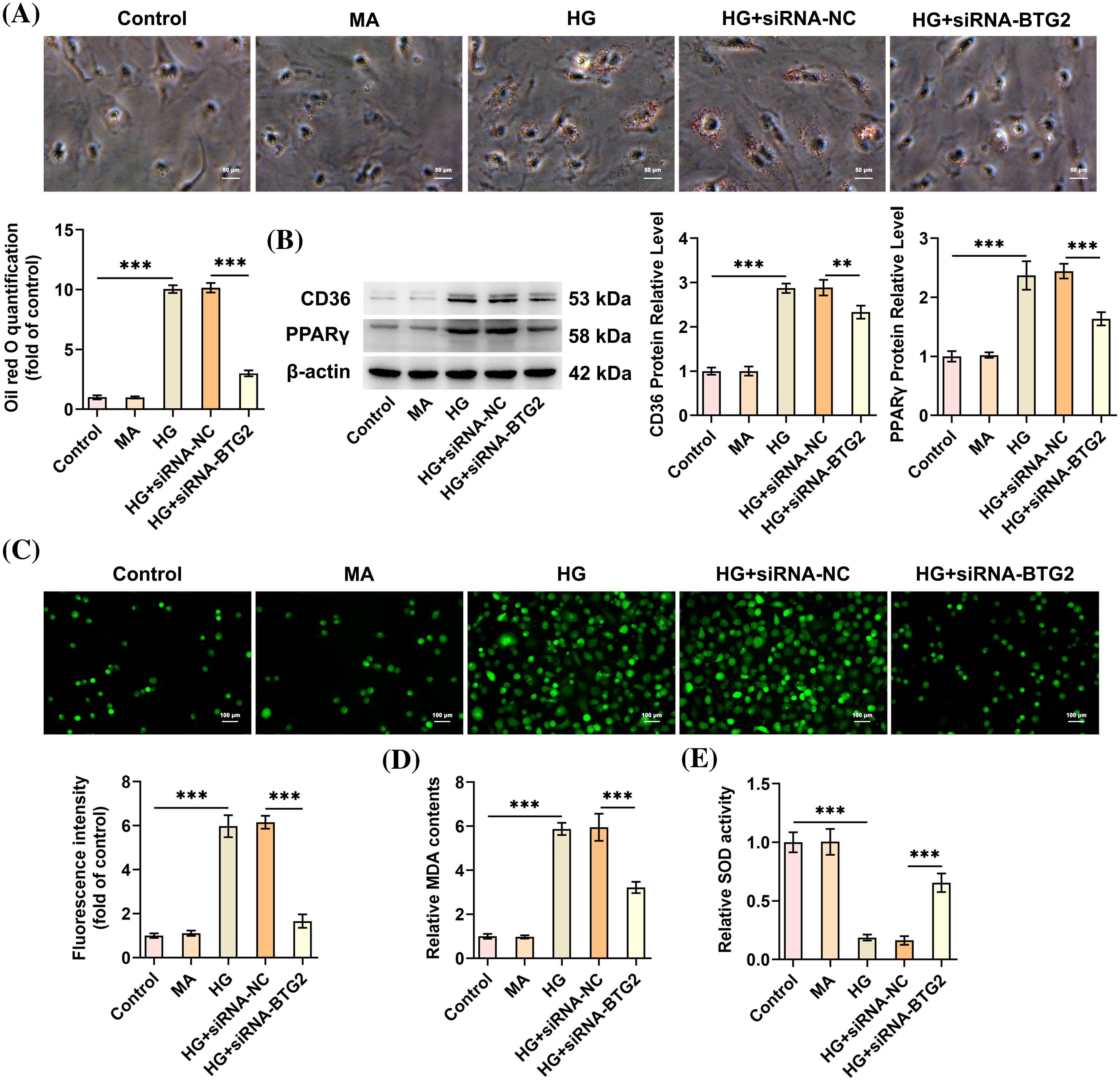
Figure 3: BTG2 deletion eases HG-evoked oxidative stress and lipid deposition in HK-2 cells. (A) Oil red O staining was utilized to assess lipid accumulation. (B) Western blot tested the expression of proteins implicated in lipid deposition. (C) DCFH-DA fluorescence probe assessed ROS production. (D and E) Related assay kits examined MDA and SOD. n = 3. **p < 0.01 and ***p < 0.001. BTG2, BTG anti-proliferation factor 2; MA, D-mannitol; HG, high glucose; CCK-8, cell counting kit-8; siRNA-NC, negative control of small interfering RNA; siRNA-BTG2, small interfering RNA targeting BTG2.
BTG2 interacts with PRMT1 and positively modulates PRMT1 expression
PRMT1 was predicted as one of the potential targets that could bind with BTG2 based on GeneMANIA database. via Western blot, it was discovered that PRMT1 expression was also intensified in HG-cultivated HK-2 cells, which was then declined when BTG2 was depleted (Fig. 4A). Importantly, the strong affinity of BTG2 protein with PRMT1 protein was further proved by Co-IP assay in HG-stimulated HK-2 cells (Fig. 4B). Altogether, BTG2 might have a strong binding with PRMT1 in HG-induced HK-2 cells.
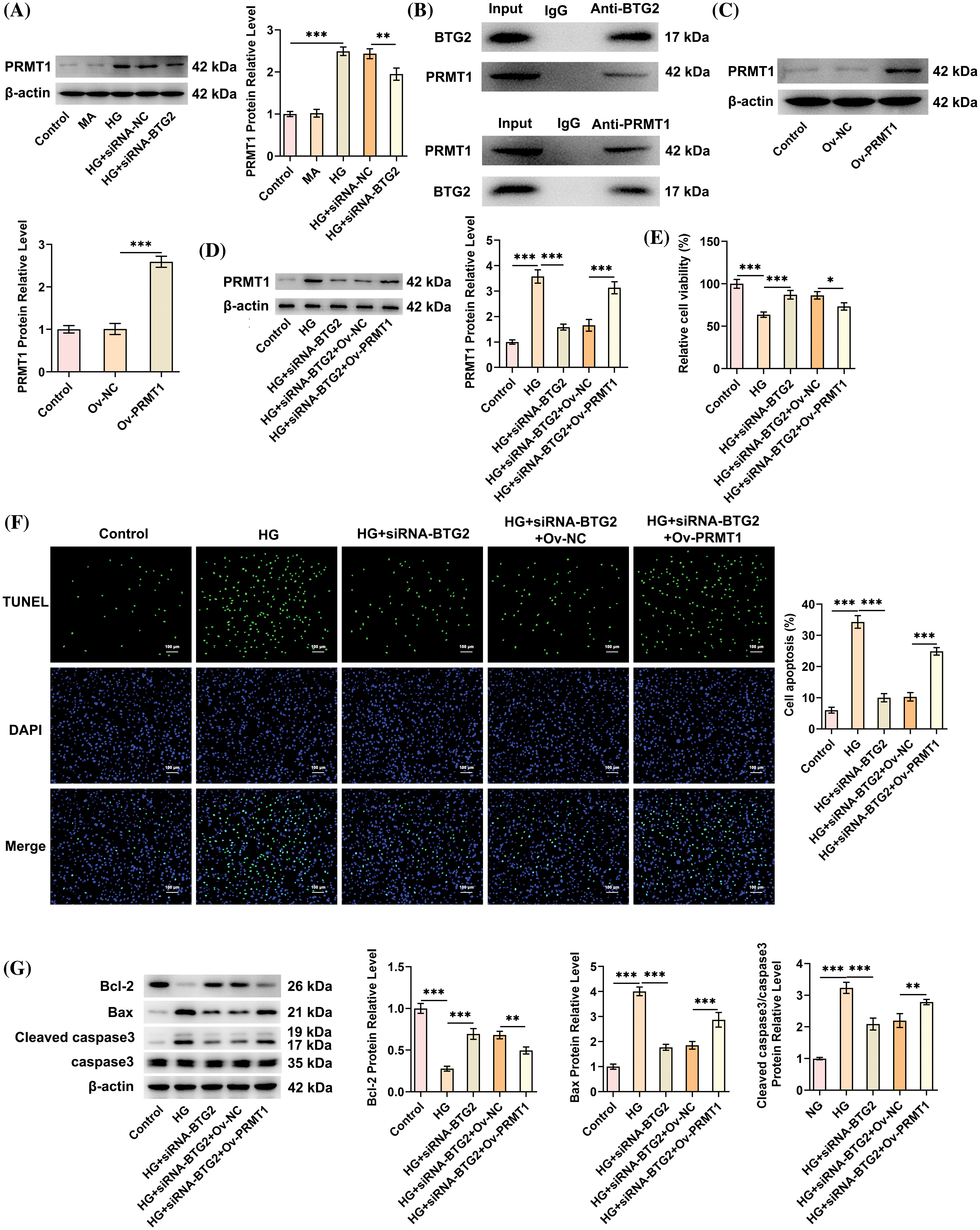
Figure 4: BTG2 interacts with and positively modulates PRMT1 expression, and BTG2 deletion protects against viability loss, apoptosis via suppressing PRMT1. (A) Western blot tested PRMT1 level in HG-treated HK-2 cells with or without BTG2 silencing. (B) Co-IP assay validated the binding of BTG2 with PRMT1. (C) Transfection efficacy of PRMT1 overexpression plasmids by Western blot. (D) Following transfection with siRNA-BTG2 or co-transfection with siRNA-BTG2 and Ov-PRMT1, the PRMT1 level was examined via western blot. (E) CCK-8 method assayed cell activity. (F) TUNEL staining appraised cell apoptosis. (G) Western blot tested apoptosis-related proteins. n = 3. *p < 0.05, **p < 0.01 and ***p < 0.001. BTG2, BTG anti-proliferation factor 2; MA, D-mannitol; HG, high glucose; siRNA-NC, negative control of small interfering RNA; siRNA-BTG2, small interfering RNA targeting BTG2; PRMT1, protein arginine methyltransferase 1; Ov-PRMT1, PRMT1 overexpression plasmid; Ov-NC, negative control of overexpression plasmid; Co-IP, Co-immunoprecipitation.
BTG2 deletion protects against viability loss, apoptosis, lipid accumulation and oxidative stress in HG-cultivated HK-2 cells via suppressing PRMT1
Prior to the implementation of rescue assays with the purpose of corroborating the functional mechanism of BTG2 in HG-stimulated DN cell model involving PRMT1, PRMT1 expression was successfully fortified after transfection of Ov-PRMT1, compared with Ov-NC group (Fig. 4C). The loss- and gain-of functional experiments were conducted via transfection with siRNA-BTG2 alone or co-transfection with siRNA-BTG2 and Ov-PRMT1. Expectedly, the decreased PRMT1 expression in BTG2-silencing HK-2 cells under HG exposure was partly restored following additional PRMT1 overexpression (Fig. 4D). Meanwhile, the strengthened viability of BTG2-silencing HK-2 cells under HG exposure was diminished again when PRMT1 was up-regulated (Fig. 4E). Conversely, PRMT1 elevation improved the weakened apoptotic capacity of HG-cultivated HK-2 cells imposed by BTG2 knockdown, coupled with the descending Bcl-2 expression, the ascending Bax and Cleaved caspase3/caspase3 (Fig. 4F,G). When PRMT1 was up-regulated, the suppressed lipid accumulation, CD36 and PPARγ expression on account of BTG2 silencing was facilitated again in HK-2 cells under HG exposure (Fig. 5A,B). Concurrently, the reduced ROS, MDA contents and the raised SOD content in HG-challenged HK-2 cells attributed to BTG2 shortage were all reversed after PRMT1 was overexpressed (Fig. 5C–E). Overall, PRMT1 elevation might compensate the inhibitory role of BTG2 interference in the viability loss, apoptosis, lipid accumulation as well as oxidative damage in HG-induced cellular model of DN.
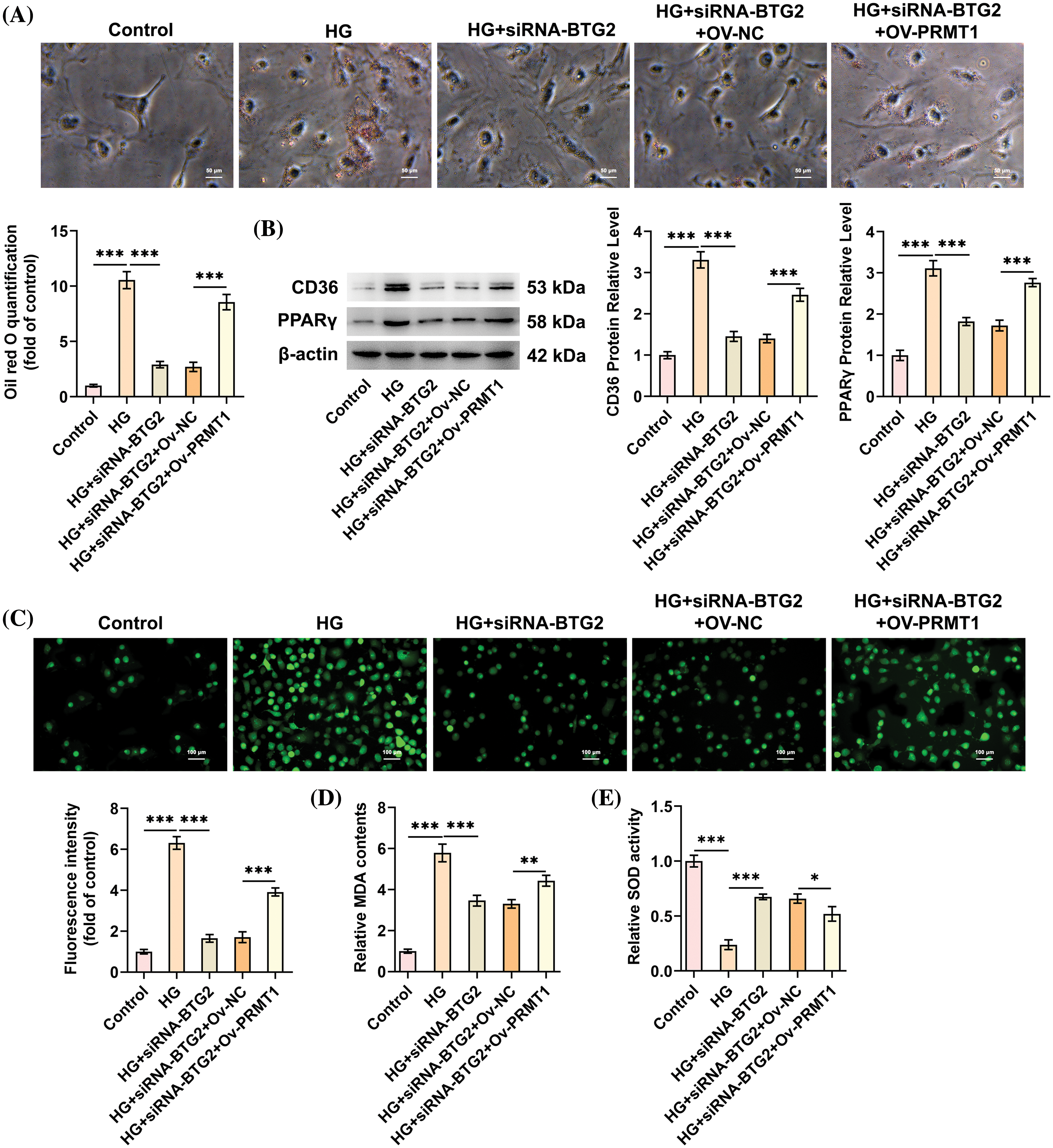
Figure 5: BTG2 deletion protects against oxidative stress and lipid accumulation in HG-cultivated HK-2 cells via suppressing PRMT1. (A) Oil red O staining was utilized to assess lipid accumulation. (B) Western blot tested the levels of proteins implicated in lipid deposition. (C) DCFH-DA fluorescence probe assessed ROS production. (D and E) Related assay kits examined MDA and SOD. n = 3. *p < 0.05, **p < 0.01 and ***p < 0.001. BTG2, BTG anti-proliferation factor 2; HG, high glucose; siRNA-NC, negative control of small interfering RNA; siRNA-BTG2, small interfering RNA targeting BTG2; PRMT1, protein arginine methyltransferase 1; Ov-PRMT1, PRMT1 overexpression plasmid; Ov-NC, negative control of overexpression plasmid.
DN is a frequently-occurring worldwide multifactorial disease where genetic factors are well involved [20]. STZ is commonly viewed as a diabetogenic chemical by selectively disrupting the function of insulin-producing pancreatic β cells in most strains of rodents [21], which was employed here for the construction of in vivo model of DN in mice. It was discovered that the renal tissues were severely injured, and the concentrations of renal indexes including BUN and Cre were markedly elevated in the serum of diabetic mice, suggesting the successful construction of DN mice model. Renal tubular epithelial cells are the major component of the renal parenchyma, and the significance of tubular injury has been emphasized in the etiology of diabetic kidney diseases recently [22]. Our paper established that BTG2 was upregulated in DN tissues and HG-stimulated DN cell model and BTG2 deletion accelerated the viability, eliminated the apoptosis, lipid deposition and oxidative damage in HG-exposed renal tubular epithelial HK-2 cells, as well as proposed a novel mechanism associated with interaction with PRMT1 in mediating the protective role of BTG2 knockdown in DN.
Previous evidence has revealed the up-regulation of BTG2 expression in STZ-treated db/db mice [10]. Consistently, our findings also manifested that BTG2 expression was fortified in renal tissues of diabetic mice and HG-cultivated HK-2 cells. Emerging studies have supported the regulatory role of BTG2 in diabetes. For instance, BTG2 reduces glucose homeostasis impairment in diabetic mice [10]. Inhibition of BTG2 impedes the apoptosis of INS-1 pancreatic beta-cells upon HG circumstances [23]. Moreover, BTG2 has been identified as a possible marker for DN therapy [12]. The experimental results in the current work that when BTG2 was depleted, HG-elicited viability loss and apoptosis of HK-2 cells were both alleviated, accompanied with the promoted expression of anti-apoptotic Bcl-2 and reduced expression of pro-apoptotic Bax and Cleaved caspase3.
Lipids are regarded as potent signaling molecules participating in a multitude of cellular responses [24]. Lipid accumulation is associated with renal dysfunction in human DN and it is currently believed that dysregulated lipid accumulation is the major influencing factor for DN [25,26]. PPARγ, an isotype of peroxisome proliferator-activated receptors (PPARs), is a nuclear hormone receptor serving as a crucial mediator of lipid metabolism [27]. CD36, a common target of PPARγ, is a scavenger receptor that functions in fatty acid uptake and contributes to lipid accumulation [28]. Furthermore, CD36 and PPARγ are both overexpressed in HG-cultured HK-2 cells [29]. Su et al. [30] have reported that BTG2 may regulate lipid accumulation in obesity-associated hepatocellular carcinoma. Our study demonstrated that the accumulated lipid and the ascending CD36, PPARγ expression in HK-2 cells challenged with HG were all repressed by deficiency of BTG2.
Oxidative stress provoked by diabetic conditions such as the persistent hyperglycemic state has been recognized as one of the most crucial underlying processes involved in the advancement of kidney disease in the diabetic population [4,31]. ROS is designated as the highly reactive substances containing oxygen radicals, which has historically been portrayed as an initiating factor of oxidative stress [32]. As a consequence, ROS overabundance is responsible for the up-regulation of MDA, which is the final product of lipid peroxidation [33]. SOD is an important enzyme that scavenges reactive oxygen radicals, constituting the primary enzymatic antioxidant defense mechanism [34]. BTG2 may drive oxidative stress in cardiomyopathy [35] and myocardial infarction [36]. As expected, our data confirmed that silencing of BTG2 prominently lowered ROS and MDA contents whilst improved SOD content in HG-treated HK-2 cells.
PRMT1 represents a chief type I arginine methyltransferase in mammals capable of mediating diversified cellular processes through methylating histone and non-histone proteins [37]. Combined with GeneMANIA database, PRMT1 was predicted as a potential target binding to BTG2. In HG-cultivated HK-2 cells, the strong affinity of BTG2 protein with PRMT1 protein was also corroborated by Co-IP assay. Intriguingly, PRMT1 gene variant is related to the elevated risks of DN in type 2 diabetes mellitus patients in Japan [38]. In particular, PRMT1 is highly expressed in DN [17]. In our work, PRMT1 was overexpressed in HG-cultivated HK-2 cells and then be underexpressed after BTG2 was knocked down. Further, PRMT1 knockdown may delay the process of DN [17,39]. Also, PRMT1 has been clarified to modulate lipid accumulation and oxidative stress in human diseases [40,41]. Through investigation, we demonstrated that when PRMT1 expression was elevated, the protective role of BTG2 depletion against HG-triggered viability loss, apoptosis, lipid accumulation as well as oxidative stress was abolished.
Some limitations still exist in this study. First, this study confirmed the regulatory role of BTG2 in HG-exposed HK-2 cells, an in vitro cell model of DN, but the molecular function of BTG2 in a mouse model of DN remains unclear. Secondly, we preliminarily disclosed the effects of BTG2/PRMT1 on oxidative stress and lipid accumulation in HG-challenged HK-2 cells, while more details of the molecular mechanisms are needed to be investigated.
Taken together, BTG2 down-regulation extenuated apoptosis, lipid deposition as well as oxidative damage in HG-challenged HK-2 cells, which might be attributed to the reduction of the interacting protein PRMT1. Conclusively, targeting BTG2 or PRMT1 might shed new light on therapeutic modalities for DN.
Acknowledgement: None.
Funding Statement: This study was supported by Key Project of Natural Science Research of Anhui Universities (No. KJ2020A0341).
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Fan Cui; data collection: Wenjuan Zhu and Zhengzheng Ju; analysis and interpretation of results: Wenjuan Zhu and Fan Cui; draft manuscript preparation: Wenjuan Zhu and Fan Cui. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: All data generated in this study have been included in this article.
Ethics Approval: All animal experiments were performed in accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals approved by the State Council of People’s Republic of China, and were approved by the Animal Ethics Committee of The First People’s Hospital of Wuhu (Approval no. IACUC-20220311-02).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Gal A, Burchell RK. Diabetes mellitus and the kidneys. Vet Clin North Am Small Anim Pract. 2023;53(3):565–80. doi:10.1016/j.cvsm.2023.01.006. [Google Scholar] [PubMed] [CrossRef]
2. Cho NH, Shaw JE, Karuranga S, Huang Y, da Rocha Fernandes JD, Ohlrogge AW, et al. IDF diabetes atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract. 2018;138:271–81. doi:10.1016/j.diabres.2018.02.023. [Google Scholar] [PubMed] [CrossRef]
3. Papademetriou V, Alataki S, Stavropoulos K, Papadopoulos C, Bakogiannis K, Tsioufis K. Pharmacological management of diabetic nephropathy. Curr Vasc Pharmacol. 2020;18(2):139–47. doi:10.2174/1570161117666190405164749. [Google Scholar] [PubMed] [CrossRef]
4. Samsu N. Diabetic nephropathy: challenges in pathogenesis, diagnosis, and treatment. Biomed Res Int. 2021;2021:1497449. [Google Scholar] [PubMed]
5. Alicic RZ, Rooney MT, Tuttle KR. Diabetic kidney disease: challenges, progress, and possibilities. Clin J Am Soc Nephrol. 2017;12(12):2032–45. doi:10.2215/CJN.11491116. [Google Scholar] [PubMed] [CrossRef]
6. Winkler GS. The mammalian anti-proliferative BTG/Tob protein family. J Cell Physiol. 2010;222(1):66–72. doi:10.1002/jcp.21919. [Google Scholar] [PubMed] [CrossRef]
7. Mao B, Zhang Z, Wang G. BTG2: a rising star of tumor suppressors (review). Int J Oncol. 2015;46(2):459–64. doi:10.3892/ijo.2014.2765. [Google Scholar] [PubMed] [CrossRef]
8. Kim SH, Jung IR, Hwang SS. Emerging role of anti-proliferative protein BTG1 and BTG2. BMB Rep. 2022;55(8):380–8. doi:10.5483/BMBRep.2022.55.8.092. [Google Scholar] [PubMed] [CrossRef]
9. Wang R, Wang R, Tian J, Wang J, Tang H, Wu T, et al. BTG2 as a tumor target for the treatment of luminal a breast cancer. Exp Ther Med. 2022;23(5):339. doi:10.3892/etm.2022.11269. [Google Scholar] [PubMed] [CrossRef]
10. Kim YD, Kim SG, Hwang SL, Choi HS, Bae JH, Song DK, et al. B-cell translocation gene 2 regulates hepatic glucose homeostasis via induction of orphan nuclear receptor Nur77 in diabetic mouse model. Diabetes. 2014;63(6):1870–80. doi:10.2337/db13-1368. [Google Scholar] [PubMed] [CrossRef]
11. Zhao JH, Zhang XL, Gao LG, Guo YJ, Wang ZY, Su CC, et al. Exogenous insulin promotes the expression of B-cell translocation gene 1 and 2 in chicken pectoralis. Poult Sci. 2022;101(7):101875. doi:10.1016/j.psj.2022.101875. [Google Scholar] [PubMed] [CrossRef]
12. Hojjati F, Roointan A, Gholaminejad A, Eshraghi Y, Gheisari Y. Identification of key genes and biological regulatory mechanisms in diabetic nephropathy: meta-analysis of gene expression datasets. Nefrologia. 2023;43(5):575–86. doi:10.1016/j.nefro.2022.06.003. [Google Scholar] [CrossRef]
13. Tang J, Frankel A, Cook RJ, Kim S, Paik WK, Williams KR, et al. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J Biol Chem. 2000;275(11):7723–30. doi:10.1074/jbc.275.11.7723. [Google Scholar] [PubMed] [CrossRef]
14. Yao B, Gui T, Zeng X, Deng Y, Wang Z, Wang Y, et al. PRMT1-mediated H4R3me2a recruits SMARCA4 to promote colorectal cancer progression by enhancing EGFR signaling. Genome Med. 2021;13(1):58. doi:10.1186/s13073-021-00871-5. [Google Scholar] [PubMed] [CrossRef]
15. Zheng S, Zeng C, Huang A, Huang F, Meng A, Wu Z, et al. Relationship between protein arginine methyltransferase and cardiovascular disease (Review). Biomed Rep. 2022;17(5):90. doi:10.3892/br. [Google Scholar] [CrossRef]
16. Hashimoto M, Fukamizu A, Nakagawa T, Kizuka Y. Roles of protein arginine methyltransferase 1 (PRMT1) in brain development and disease. Biochim Biophys Acta Gen Subj. 2021;1865(1):129776. doi:10.1016/j.bbagen.2020.129776. [Google Scholar] [PubMed] [CrossRef]
17. Chen YY, Peng XF, Liu GY, Liu JS, Sun L, Liu H, et al. Protein arginine methyltranferase-1 induces ER stress and epithelial-mesenchymal transition in renal tubular epithelial cells and contributes to diabetic nephropathy. Biochim Biophys Acta Mol Basis Dis. 2019;1865(10):2563–75. doi:10.1016/j.bbadis.2019.06.001. [Google Scholar] [PubMed] [CrossRef]
18. Lan J, Xu B, Shi X, Pan Q, Tao Q. WTAP-mediated N(6)-methyladenosine modification of NLRP3 mRNA in kidney injury of diabetic nephropathy. Cell Mol Biol Lett. 2022;27(1):51. doi:10.1186/s11658-022-00350-8. [Google Scholar] [PubMed] [CrossRef]
19. Zhuang JL, Liu YY, Li ZZ, Zhuang QZ, Tang WZ, Xiong Y, et al. Amentoflavone prevents ox-LDL-induced lipid accumulation by suppressing the PPARgamma/CD36 signal pathway. Toxicol Appl Pharmacol. 2021;431:115733. doi:10.1016/j.taap.2021.115733. [Google Scholar] [PubMed] [CrossRef]
20. Zhang C, Li H, Wang S. Common gene signatures and molecular mechanisms of diabetic nephropathy and metabolic syndrome. Front Public Health. 2023;11:1150122. doi:10.3389/fpubh.2023.1150122. [Google Scholar] [PubMed] [CrossRef]
21. Furman BL. Streptozotocin-induced diabetic models in mice and rats. Curr Protoc. 2021;1(4):e78. doi:10.1002/cpz1.v1.4. [Google Scholar] [CrossRef]
22. Chen SJ, Lv LL, Liu BC, Tang RN. Crosstalk between tubular epithelial cells and glomerular endothelial cells in diabetic kidney disease. Cell Prolif. 2020;53(3):e12763. doi:10.1111/cpr.v53.3. [Google Scholar] [CrossRef]
23. Kooptiwut S, Kaewin S, Semprasert N, Sujjitjoon J, Junking M, Suksri K, et al. Estradiol prevents high glucose-induced β-cell apoptosis by decreased BTG2 expression. Sci Rep. 2018;8(1):12256. doi:10.1038/s41598-018-30698-x. [Google Scholar] [PubMed] [CrossRef]
24. Li-Beisson Y, Nakamura Y, Harwood J. Lipids: from chemical structures, biosynthesis, and analyses to industrial applications. Subcell Biochem. 2016;86:1–18. doi:10.1007/978-3-319-25979-6. [Google Scholar] [CrossRef]
25. Su K, Yi B, Yao BQ, Xia T, Yang YF, Zhang ZH, et al. Liraglutide attenuates renal tubular ectopic lipid deposition in rats with diabetic nephropathy by inhibiting lipid synthesis and promoting lipolysis. Pharmacol Res. 2020;156:104778. doi:10.1016/j.phrs.2020.104778. [Google Scholar] [PubMed] [CrossRef]
26. Han Y, Xiong S, Zhao H, Yang S, Yang M, Zhu X, et al. Lipophagy deficiency exacerbates ectopic lipid accumulation and tubular cells injury in diabetic nephropathy. Cell Death Dis. 2021;12(11):1031. doi:10.1038/s41419-021-04326-y. [Google Scholar] [PubMed] [CrossRef]
27. Janani C, Ranjitha Kumari BD. PPAR gamma gene--a review. Diabetes Metab Syndr. 2015;9(1):46–50. doi:10.1016/j.dsx.2014.09.015. [Google Scholar] [PubMed] [CrossRef]
28. Maréchal L, Laviolette M, Rodrigue-Way A, Sow B, Brochu M, Caron V, et al. The CD36-PPARγ pathway in metabolic disorders. Int J Mol Sci. 2018;19(5):1529. doi:10.3390/ijms19051529. [Google Scholar] [PubMed] [CrossRef]
29. Feng L, Gu C, Li Y, Huang J. High glucose promotes CD36 expression by upregulating peroxisome proliferator-activated receptor γ levels to exacerbate lipid deposition in renal tubular cells. Biomed Res Int. 2017;2017:1414070. doi:10.1155/2017/1414070. [Google Scholar] [PubMed] [CrossRef]
30. Su Q, Xu ZX, Xiong ML, Li HY, Xu MY, Luo SZ. The oncogenic miR-27a/BTG2 axis promotes obesity-associated hepatocellular carcinoma by mediating mitochondrial dysfunction. Neoplasma. 2022;69(4):820–31. doi:10.4149/neo_2022_211227N1837. [Google Scholar] [PubMed] [CrossRef]
31. Darenskaya M, Kolesnikov S, Semenova N, Kolesnikova L. Diabetic nephropathy: significance of determining oxidative stress and opportunities for antioxidant therapies. Int J Mol Sci. 2023;24(15):12378. doi:10.3390/ijms241512378. [Google Scholar] [PubMed] [CrossRef]
32. Jakubczyk K, Dec K, Kaldunska J, Kawczuga D, Kochman J, Janda K. Reactive oxygen species–sources, functions, oxidative damage. Pol Merkur Lekarski. 2020;48(284):124–7. [Google Scholar] [PubMed]
33. Tsikas D. Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: analytical and biological challenges. Anal Biochem. 2017;524:13–30. doi:10.1016/j.ab.2016.10.021. [Google Scholar] [PubMed] [CrossRef]
34. Ali SS, Ahsan H, Zia MK, Siddiqui T, Khan FH. Understanding oxidants and antioxidants: classical team with new players. J Food Biochem. 2020;44(3):e13145. [Google Scholar] [PubMed]
35. Wang Q, Jiang F, Zhao C, Song J, Hu M, Lv Y, et al. miR-21-5p prevents doxorubicin-induced cardiomyopathy by downregulating BTG2. Heliyon. 2023;9(5):e15451. doi:10.1016/j.heliyon.2023.e15451. [Google Scholar] [PubMed] [CrossRef]
36. Hu D, Gu Y, Wu D, Zhang J, Li Q, Luo J, et al. Icariside II protects cardiomyocytes from hypoxia‐induced injury by upregulating the miR‐7‐5p/BTG2 axis and activating the PI3K/Akt signaling pathway. Int J Mol Med. 2020;46(4):1453–65. [Google Scholar] [PubMed]
37. Sudhakar SRN, Khan SN, Clark A, Hendrickson-Rebizant T, Patel S, Lakowski TM, et al. Protein arginine methyltransferase 1, a major regulator of biological processes. Biochem Cell Biol. 2024;102(2):106–26. [Google Scholar] [PubMed]
38. Iwasaki H, Shichiri M. Protein arginine N-methyltransferase 1 gene polymorphism is associated with proliferative diabetic retinopathy in a Japanese population. Acta Diabetol. 2022;59(3):319–27. doi:10.1007/s00592-021-01808-5. [Google Scholar] [PubMed] [CrossRef]
39. Ojima A, Ishibashi Y, Matsui T, Maeda S, Nishino Y, Takeuchi M, et al. Glucagon-like peptide-1 receptor agonist inhibits asymmetric dimethylarginine generation in the kidney of streptozotocin-induced diabetic rats by blocking advanced glycation end product-induced protein arginine methyltranferase-1 expression. Am J Pathol. 2013;182(1):132–41. doi:10.1016/j.ajpath.2012.09.016. [Google Scholar] [PubMed] [CrossRef]
40. Park MJ, Kim DI, Lim SK, Choi JH, Kim JC, Yoon KC, et al. Thioredoxin-interacting protein mediates hepatic lipogenesis and inflammation via PRMT1 and PGC-1α regulation in vitro and in vivo. J Hepatol. 2014;61(5):1151–7. doi:10.1016/j.jhep.2014.06.032. [Google Scholar] [PubMed] [CrossRef]
41. Zhao J, Adams A, Weinman SA, Tikhanovich I. Hepatocyte PRMT1 protects from alcohol induced liver injury by modulating oxidative stress responses. Sci Rep. 2019;9(1):9111. doi:10.1038/s41598-019-45585-2. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools