
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

ABHD17C represses apoptosis and pyroptosis in hepatocellular carcinoma cells
1 Division of Hepatobiliary and Pancreatic Surgery, Department of Surgery, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, 310030, China
2 Department of Hepatobiliary and Pancreatic Surgery, The Second Affiliated Hospital of Fujian Medical University, Quanzhou, 362000, China
* Corresponding Authors: WEI WANG. Email: ; JIAN WU. Email:
(This article belongs to the Special Issue: Navigating the Interplay of Cancer, Autophagy, ER Stress, Cell Cycle and Apoptosis: Mechanisms, Therapies, and Future Directions)
BIOCELL 2024, 48(9), 1299-1310. https://doi.org/10.32604/biocell.2024.051756
Received 14 March 2024; Accepted 22 May 2024; Issue published 04 September 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: Alpha/beta hydrolase domain-containing protein 17C (ABHD17C) is a depalmitoylation enzyme that removes the S-palmitoylation of targeted proteins. The hepatocellular carcinoma (HCC) cells SNU449 and Hep3B use ABHD17C as an oncogene; however, the exact mechanism of this action is yet unknown. Methods: The expression of ABHD17C in liver cancer tissues was analyzed by bioinformatics, and the expression of ABHD17C in clinical liver cancer tissues and adjacent normal tissues was detected. Then, the proliferative viability of HCC cells after overexpression or knockdown of ABHD17C was examined, and pyroptosis and apoptosis proteins were detected. Results: ABHD17C was overexpressed in human HCC tissues as well as numerous HCC cell lines. Depletion of ABHD17C caused reduced viability, cell cycle arrest, and defective invasion and migration in HCC cells, while overexpression of ABHD17C exhibited the opposite effect. Moreover, we discovered that the knockdown of ABHD17C resulted in enhanced apoptotic and pyroptotic phenotypes of HCC cells, whereas overexpression of ABHD17C attenuated such phenotypes. Conclusions: It suggests that ABHD17C contributes to HCC carcinogenesis, making it a promising target for medication treatment.Keywords
Globally, liver cancer is one of the most common cancers and causes a high death rate [1,2]. Currently, the most common type of liver cancer is hepatocellular carcinoma (HCC), which makes up around 80% of all primary liver cancers [2–5]. In addition, the risk of HCC is increased by exposure to Hepatitis C viruses, heavy alcohol consumption, metabolic syndromes, nonalcoholic fatty liver disease, and other factors [6–9]. Despite considerable efforts have been made to combat HCC, and novel drugs and therapies have been developed and clinically administrated to treat HCC [10,11], the outcome of these treatments is limited by drug delivery efficiency and drug resistance [12,13], which results in a poor prognosis for patients with HCC. In order to combat this deadly disease, new therapeutic options are urgently needed.
The Alpha/beta hydrolase domain-containing protein 17 (ABHD17) family consists of three members: ABHD17A, ABHD17B, and ABHD17C, which have highly similar sequences and are broadly expressed in various cell types, removing S-palmitoylation from their substrates [14,15].
An essential posttranslational change known as S-palmitoylation connects the targeted protein’s cysteine residues to the 16-carbon saturated fatty acid [16], thereby regulating their intracellular distribution and function [17–20]. Given the essential role of protein palmitoylation in cancers [21,22], it is not surprising that the ABHD17 proteins influence cancer development. For instance, ABHD17 proteins are able to facilitate cancer growth by depalmitoylating the oncogenic rat sarcoma (RAS) proteins, leading to the release of RAS from the cell membrane to activate the oncogenic signaling pathway [14,15]. Our recent research indicates that ABHD17C overexpression in HCC tissues is associated with poor cancer outcomes [23]. Moreover, ABHD17C acts downstream of USP35 in promoting HCC development by activating phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) signaling [24]. However, cancer-related ABHD17 expression and the mechanism by which they control cancer development remains elusive.
Apoptosis, pyroptosis, ferroptosis, necroptosis, and other forms of regulated cell death (RCD) are key regulators of physiological processes, and dysregulated RCD contributes to cancer development [25]. Drugs targeting genes controlling RCD hold great potential in cancer treatment [26]. Among different types of RCD, pyroptosis is distinct from other types of RCD by its close relationship with inflammation [27]. Caspase 1 or Caspase 11 could initiate pyroptosis, which is capable of cleaving Gasdermin D (GSDMD), and the resulting N-terminal of GSDMD oligomerizes to induce the creation of pores in the cellular membrane, therefore facilitating membrane rupture and release of interleukin (IL)-18 and IL-1 [28,29]. Recognition of the involvement of pyroptosis in HCC is gaining momentum [30,31], but the mechanisms regulating pyroptosis in HCC are not well characterized. Furthermore, unknown is ABHD17C’s function in controlling pyroptosis in HCC.
In this work, HCC tissues and HCC cell lines were examined for ABHD17C expression. In the next step, we investigated the role of regulating ABHD17C expression on HCC viability, invasion, cell cycle, and migration. Finally, an assessment of the role of ABHD17C in HCC apoptosis and pyroptosis was conducted. Examining how ABHD17C affects SNU-182 and PLC/PRF/5 cell activity and partially elucidating its potential processes were the goals of this endeavor.
The Second Affiliated Hospital of Fujian Medical University provided fresh, paraffin-embedded HCC and adjacent NT liver tissues, which were obtained with permission from the hospital’s Ethics Committee (Approval number: No. 27(2023)). The patients provided informed consent in compliance with the Declaration of Helsinki’s principles. Tissues fixed in paraffin from five HCC patients were sectioned in preparation for immunohistochemistry (IHC) analysis.
The HCC patients’ RNA profiling information was acquired from Gene Expression Omnibus. (GSE25097, GSE45436, GSE112790, and GSE76297, https://www.ncbi.nlm.nih.gov/geo/, accessed on 15/11/2023) and subject to gene expression analysis using the R package “GSEA” (v4.2.1) in R (v4.3.0). Statistical significance was defined as p < 0.05. The IHC results of ABHD17C in control liver and HCC tissues can be found on the Human Protein Atlas (HPA) website (https://www.proteinatlas.org/, accessed on 15/11/2023).
Immocell Biotechnology (Xiamen, China) provided human HCC cell lines SNU449, SNU-182, MHCC-97H, Hep3B, PLC/PRF/5, SK-HEP-1, and HepG2 cells. MHCC-97H, Hep3B, SK-HEP-1, and HepG2 cell lines were kept in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, 11995065, CA, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco, 10270106, CA, USA). SNU449 and SNU-182 cell lines were kept in RPMI-1640 (IMMOCELL, IMC-202, Xiamen, China) supplemented with 10% FBS. The PLC/PRF/5 cell line was kept in MEM (IMMOCELL, IMC-204, Xiamen, China) supplemented with 10% FBS. 0.1 mg/mL penicillin-streptomycin (Gibco, 15140-122, CA, USA) was supplemented to cell lines and cultured at 37°C with 5% CO2 in a humidified incubator.
The coding sequence of ABHD17C and shRNAs targeting ABHD17C were respectively inserted into the pLVX-CMV-pGK-puro vector and pLKO.1 vector and the resulting plasmids were named ABHD17C OE and shABHD17Cs. All the plasmids were subject to sequencing to confirm the accuracy of the inserts. Table 1 shows the primer sequences needed to construct plasmids.

Following the manufacturer’s recommendations, Lipofectamine 3000 (Invitrogen, L3000150, Carlsbad, CA, USA) was used for all transfections in cells that were 70% confluent. The cells were transfected for 48 h before being used in subsequent assays.
Quantitative polymerase chain reaction (qPCR)
To perform qPCR experiments, total cellular RNA needs to be extracted first using the Trizol reagent (Invitrogen, 15596026CN, CA, USA) in compliance with the manufacturer’s instructions. HiScript Reverse Transcriptase (Vazyme, R201-01, Nanjing, China) was used to synthesise cDNA. Using the ChamQ SYBR qPCR Master Mix (Vazyme, Q311-02, Nanjing, China), qPCR was performed on a LightCycler 96 device (Roche, Basel, Canton Basel, Switzerland). The gene expression was calculated using the 2−ΔΔCt algorithm, and the results were normalized using 18S rRNA. The qPCR primers designed based on the sequences of human ABHD17C (GenBank: NM_021214) and RNA18S (GenBank: NR_003286) are as follows: ABHD17C-qF: 5′-GCAGCGGTAATTCTCCATTCCC-3′, ABHD17C-qR: 5′-GACCAACACAGGAGAGGTGACT-3′; RNA18S-qF: 5′-ACCCGTTGAACCCCATTCGTGA-3′, RNA18S-qR: 5′-GCCTCACTAAACCATCCAATCGG-3′.
In compliance with the manufacturer’s instructions, the RIPA lysis solution that contained protease inhibitors (Beyotime Biotechnology, P0013B, Shanghai, China) was used to extract the total protein that was present in the cells. The protein of each sample was collected, and a BCA kit (Beyotime Biotechnology, P0009, Shanghai, China) was used to measure the protein content. The samples were then denatured, and 10% SDS-PAGE gels were loaded with 10 μg of protein from each sample for separation, followed by transfer onto polyvinylidene fluoride (PVDF) membranes (Millipore, IPVH10100, Massachusetts, USA), which were blocked for 1 h at 25°C in five percent nonfat milk (Beyotime Biotechnology, P0216-300 g, Shanghai, China) and then incubated with the primary antibody at a temperature of 4°C for an entire night. After that, it was incubated for 2 h at 25°C with the addition of a secondary antibody. In order to see the signal, the membrane was lastly incubated with an ECL working solution (Beyotime Biotechnology, P0018FM, Shanghai, China), which was recorded using a ChemiDoc device (BioRad, ChemiDoc, Hercules, CA, USA). Image J (NIH, Bethesda, MD, USA) was utilised to quantify the band intensity. Table 2 lists the primary antibodies that were employed.
The patients’ liver tissues were fixed and then embedded using paraffin. The tissue was then sectioned which was rehydrated after being dewaxed. After increasing the permeability of tissue cells, a 1% bovine serum albumin block (Solarbio, SW3015, Beijing, China) was applied to the section. The ABHD17C antibody (Invitrogen, PA5-61831, CA, USA), diluted according to the instructions, was used to incubate the sections overnight at 4°C. Then, the sections were washed three more times and incubated with horse radish peroxidase (HRP)-conjugated goat anti-rabbit IgG (Proteintech, SA00001-1, Wuhan, China) for 1 h at 25°C. Subsequently, the sections were stained with diaminobenzidine chromogenic solution (Solarbio, DA1010, Beijing, China), and the nuclei were further restained with hematoxylin solution (Solarbio, H8070, Beijing, China) before mounting. Finally, the different liver tissue sections were observed with an X-73 inverted microscope (OLYMPUS, IX73, Tokyo, Japan) and photographed for documentation.
3-(4,5-dimethylthiazol)-2,5-diphenyl-tetrazolium bromide (MTT) assay
In compliance with the manufacturer’s instructions, the MTT test was carried out to assess cell viability using a detection kit (Beyotime Biotechnology, C0009S, Shanghai, China). In short, transfected cells were cultured for a whole night after being seeded at 3000 cells per well onto 96-well plates. After that, a 10 μL MTT (5 mg/mL) was put into the plates. The cells were then left to incubate for 6 h at 37°C. Subsequently, 100 μL of DMSO was pipetted into every plate well in order to address crystallisation. After 3 h of incubation, the plates were agitated for 10 min at 300 rpm using an oscillator. Finally, a microplate reader (MOLECULAR, SpectraMax 190, Sunnyvale, CA, USA) determined the OD570 values of the plates.
Each well of 6-well plates seeded 700 cells, which were then cultivated for 12 days. The cells were subsequently treated with 4% paraformaldehyde (Solarbio, P1110, Beijing, China) for a duration of 30 min. Following this, the cells were subjected to a 15-min staining process using 0.5% crystal violet (Solarbio, G1062, Beijing, China). Subsequently, Phosphate Buffered Saline (PBS) (Solarbio, P1010, Beijing, China) was used to wash the cells once again and then examined and photographed using an inverted microscope (OLYMPUS, IX73, Tokyo, Japan). The colonies were quantified using Image J (NIH, Bethesda, MD, USA).
Cell cycle and apoptosis assay
The cell cycle was determined by a 7-amino actinomycin D (7-AAD) staining assay. Briefly, 5 × 106 cells were collected and allowed to fix overnight at −20°C in ethanol. Afterward, the cells were placed in PBS and treated with RNase A (Solarbio, 9001-99-4, Beijing, China) at 37°C for 30 min to remove the interference of double-stranded RNA. Following the removal of RNAse A, the cells were incubated at 4°C in a light-free environment for a duration of 30 min. During this time, the cells were exposed to a PBS solution that contained 5 μL of the 7-AAD mixture (eBioscience, 00-6993-50, MA, USA). Finally, flow cytometry analysis (ACEA, D2060R, San Diego, CA, USA) was performed on the cells, and Flowjo (BD Bioscience, Franklin, NY, USA) was used to analyze the results.
To test apoptosis, 5 × 105 cells were collected, cleaned with PBS, and then reconstituted using 100 μL binding buffer of apoptosis detection kit (Vazyme, A211-01, Nanjing, China). Subsequently, the kit is intended for staining cells, as specified by the manufacturer’s instructions. The cells were then subjected to a 10-min incubation at 25°C in the dark. Finally, after adding 400 μL of the kit’s binding buffer to the cells, the cell suspension underwent flow cytometry analysis (ACEA, D2060R, San Diego, CA, USA), and Flowjo (BD Bioscience, Franklin, NY, USA) was used to analyze the results.
Plates with small chambers (Corning, #3422, Corning, New York, NY, USA) were used for transwell assay. Each top chamber received 100 μL of Matrigel (BD, 356235, NJ, USA), and at a temperature of 37°C, the plates were put through an incubation process of 2 h with the purpose of assessing the invasion potential. The cells were then collected and resuspended in the serum-free media. Each upper compartment contained 60,000 cells, while the lower chamber contained 700 μL of full medium. After a 48-h period of incubation, the cells were exposed to methanol at 25°C for 30 min to fix them. Afterward, the cell was subjected for 5 min using a 0.05% concentration of crystal violet (Solarbio, G1062, Beijing, China). Ultimately, an inverted microscope (OLYMPUS, IX73, Tokyo, Japan) was utilized to observe and capture images of the outcomes. The same experimental techniques used for the invasion assay were used to determine the migratory capacity, with the exception that matrix glue was not applied to the upper chambers. Image J (NIH, Bethesda, MD, USA) was used for quantification.
6-well plate wells were seeded with 1 × 106 HCC cells, which were then transfected using the designated plasmids. Following a 24-h culture period, a pipette tip was used to make a wound in each well, and the cells were thoroughly cleaned with PBS to remove any remaining cell debris. Subsequently, each well added 4 mL of serum-free media and the cells were imaged immediately and after another 24 h of culture under an IX73 inverted microscope (OLYMPUS, IX73, Tokyo, Japan). The size of wounds was measured by Image J (NIH, Bethesda, MD, USA).
Dichloro-dihydro-fluorescein diacetate (DCFH-DA) staining
The level of reactive oxygen species (ROS) was assessed by DCFH-DA staining. Briefly, the cells were subjected to the PBS wash and subsequently exposed to 1 mL of a 10 μM solution of DCFH-DA (Sigma-Aldrich, 4091-99-0, MO, USA)/PBS for 15 min in the dark. Then, the cells were treated with 0.25% trypsin (Sigma-Aldrich, T4049, MO, USA) for digestion. Finally, the cell suspension underwent flow cytometry examination (ACEA, D2060R, San Diego, CA, USA), and Flowjo (BD Bioscience, Franklin, NY, USA) was used to analyze the results.
Enzyme-linked immunosorbent assay (ELISA)
The concentrations of IL-1β, IL-18, and tumor necrosis factor-alpha (TNF-α) in the conditioned medium were assessed using the following ELISA kits: Human TNF-α ELISA Kit (R&D systems, DTA00D, MN, USA), Human IL-18 ELISA Kit (R&D systems, DL180, MN, USA), and Human IL-1β ELISA Kit (R&D systems, DLB50, MN, USA). The procedures were followed with the manufacturer’s instructions.
Transmission electronic microscopy (TEM)
To prepare TEM samples, the cells were subjected to a fixation treatment with glutaraldehyde at a concentration of 2.5%. Subsequently, the cells were harvested, centrifuged, and fixed with 2.5% glutaraldehyde again for 2 h. TEM was carried out by Pinuofei Biological Technology (Wuhan, Hubei, China).
Statistical analysis and chart production were carried out using Prism 8.0 (GraphPad, San Diego, CA, USA). Unpaired two-tailed Student t-tests were utilized in order to determine the deviation that existed between the two sets of data. In order to compare a number of different groups, a one-way analysis of variance was utilized. The sample size of each group was n = 3. The mean value of the data was displayed, either plus or minus the standard deviation. To be considered statistically significant, a p-value has to be less than 0.05.
ABHD17C exhibited significant upregulation in human HCC samples and multiple HCC cell lines
In our prior study, we demonstrated that ABHD17C mRNA level is elevated in two data sets and our HCC cohort [24]. To verify this observation in a larger number of HCC patients, we analyzed more HCC RNA profiling data sets. The results confirmed that ABHD17C mRNA is overexpressed in all four additional HCC cohorts (Fig. 1A). Based on the HPA database, we observed that ABHD17C protein expression is also increased in HCC samples compared with controls (Fig. 1B). The IHC results demonstrated that the protein level of ABHD17C was higher in HCC tissues as well (Fig. 1C). To determine whether ABHD17C is overexpressed in HCC cell lines, we performed qPCR and Western blot. qPCR data revealed that compared to SNU449 cells, ABHD17C mRNA expression was significantly elevated in Hep3B, SNU-182, MHCC-97H, HepG2, and PLC/PRF/5, but not in SK-HEP-1 cells (Fig. 1D). Western blot results confirmed the overexpression of ABHD17C in SNU-182, MHCC-97H, Hep3B, HepG2, and PLC/PRF/5 cells compared to SNU449 and SK-HEP-1 cells (Fig. 1E,F). Among these cell lines, PLC/PRF/5 cells expressed the highest level of ABHD17C, while SNU-182 cells exhibited the least elevation of ABHD17C expression (Figs. 1E and 1F). These findings suggest that ABHD17C is overexpressed in human HCC samples and most HCC cell lines investigated in this work.
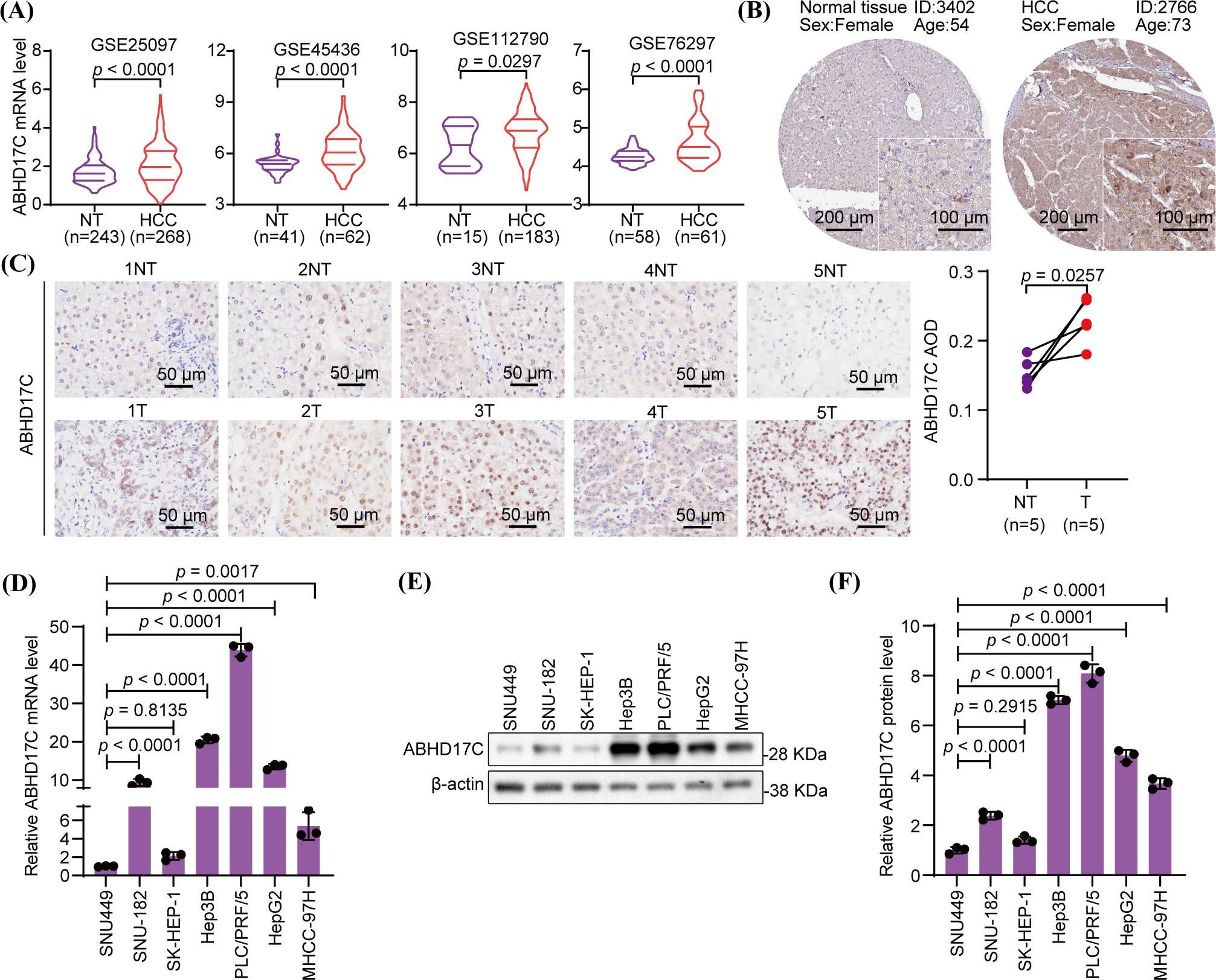
Figure 1: ABHD17C is overexpressed in human HCC tissues and cell lines (n = 3). (A) The bioinformatic investigation shows that HCC tissues have higher mRNA expression of ABHD17C than NT liver tissues. (B) IHC data from the HPA database show that the ABHD17C protein level is higher in an HCC sample than in normal liver tissue. (C) IHC data from our HCC cohort reveal that ABHD17C protein levels are elevated in HCC tumor (T) tissues compared to adjacent NT liver tissues. (D) qPCR outcomes uncover that ABHD17C mRNA levels are upregulated in multiple HCC cell lines. (E) Western blot results confirmed the upregulation of ABHD17C protein levels in these HCC cell lines. (F) Quantification of the Western blot data present in (E).
ABHD17C facilitates the viability and cell cycle of HCC cells
Hep3B cells with ABHD17C deficiency are less viable and exhibit cell cycle arrest [23]. To ascertain if ABHD17C performs similar roles in other cell lines with HCC, we depleted ABHD17C in PLC/PRF/5 cells and overexpressed ABHD17C in SNU-182 cells. qPCR and Western blot data verified the efficient depletion and overexpression of ABHD17C in PLC/PRF/5 and SNU-182 cells, respectively (Fig. A1A–D). Next, we evaluated the influence of ABHD17C on the survival of these cells. MTT assay results indicated that ABHD17C knockdown significantly decreased PLC/PRF/5 cell viability, while overexpression of ABHD17C remarkably increased SNU-182 cell viability (Fig. 2A,B). In line with this, PLC/PRF/5 and SNU-182 cells were respectively inhibited and strengthened by ABHD17C knockdown and overexpression (Fig. 2C,D). In addition, ABHD17C deficiency arrested the cell cycle, while excessive ABHD17C facilitated the cell cycle of SNU-182, as demonstrated by 7-AAD staining assays (Fig. 2E,F).
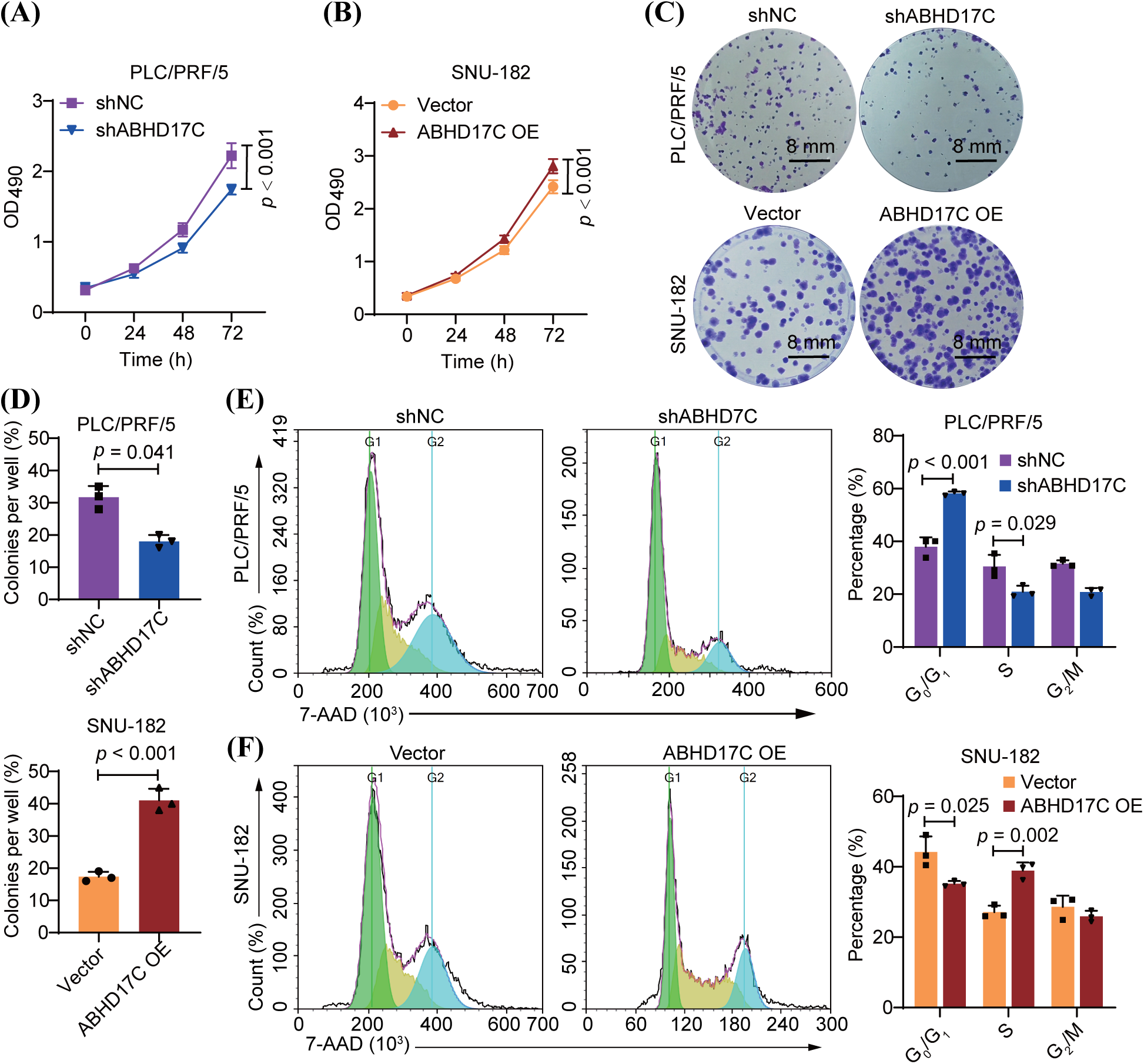
Figure 2: ABHD17C positively controls the viability and cell cycle (n = 3). (A) MTT assay data show that the survival of PLC/PRF/5 cells was impaired by ABHD17C knockdown. (B) MTT assay data indicate that overexpression of ABHD17C enhanced the viability of SNU-182 cells. (C) Colony formation assay results suggest that ABHD17C deficiency attenuated the colony formation capacity of PLC/PRF/5 cells, while excessive ABHD17C strengthened colony formation by SNU-182 cells. (D) Statistical results of the data from the colony formation assay in (C). (E) 7-AAD staining outcomes reveal that ABHD17C depletion resulted in cell cycle arrest in PLC/PRF/5 cells. (F) 7-AAD staining outcomes illustrate that excessive ABHD17C promoted cell cycle progression of SNU-182 cells.
ABHD17C increases the invasion and migration capacity of HCC cells
Next, PLC/PRF/5 and SNU-182 cells were analyzed for their invasion and migration in response to ABHD17C. Wound healing assay results uncovered that depletion of ABHD17C notably attenuated the migration ability, while overexpression of ABHD17C had an opposite function in SNU-182 cells (Fig. 3A and 3B). In support of this finding, Transwell assay outcomes confirmed the positive regulation of ABHD17C on the migration of PLC/PRF/5 and SNU-182 cells (Fig. 3C,D). Moreover, ABHD17C-deficient PLC/PRF/5 cells had impaired invasion capacity compared to control cells, while ABHD17C-overexpressing SNU-182 cells exhibited a stronger invasion capacity than control cells, as indicated by Transwell assay data (Fig. 3E,F). The combination of these results indicates that HCC invasion and migration are positively correlated with the expression of ABHD17C.
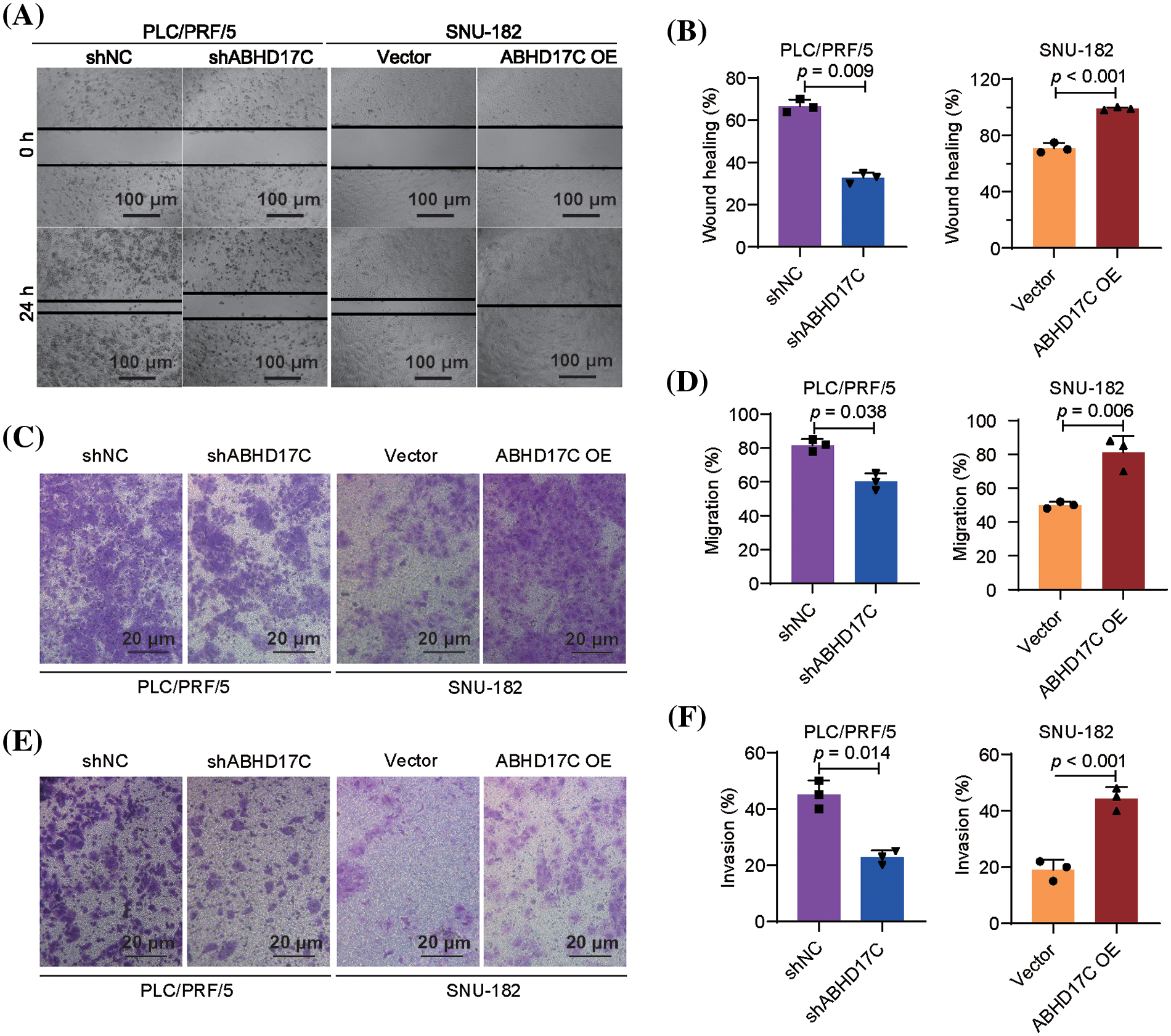
Figure 3: ABHD17C facilitates the invasion and migration (n = 3). (A) Wound healing assay data show that the migration is impaired upon ABHD17C knockdown, while overexpression of ABHD17C enhanced SNU-182 cells’ migration. (B) Statistical results of the data from the wound healing assay in (A). (C) Transwell assay data verified the positive effect of ABHD17C on the migration. (D) Statistical results of data from the transwell assay to detect cell migration in (C). (E) Transwell assay results demonstrate that ABHD17C depletion caused the mitigated invasion of PLC/PRF/5 cells, while excessive ABHD17C promoted the invasion of SNU-182 cells. (F) Statistical results of data from the transwell assay to detect cell invasion in (E).
ABHD17C represses the apoptosis and pyroptosis of HCC cells
To investigate how ABHD17C controls HCC cell survival, we examined two types of RCD including apoptosis and pyroptosis. As indicated by Western blot data, Cleaved-Caspase 3 was dramatically elevated as well as pro-apoptotic BAX, while the expression of anti-apoptotic Bcl2 was reduced in PLC/PRF/5 cells upon ABHD17C deficiency. By contrast, as a result of ABHD17C overexpression in SNU-182 cells, the levels of Cleaved-Caspase 3 and BAX were downregulated, while the level of Bcl2 increased (Fig. 4A–D), indicating ABHD17C represses apoptosis in these HCC cells. Simultaneously, the levels of apoptosis associated speck-like protein containing a CARD (ASC), Cleaved-GSDMD, NOD-like receptor protein 3 (NLRP3), and Cleaved-Caspase 1, which constitute a signaling cascade triggering pyroptosis [32], were substantially upregulated in ABHD17C-deficient PLC/PRF/5 cells but were decreased in ABHD17C-overexpressing SNU-182 cells (Fig. 4A–D), implicating that ABHD17C may inhibit pyroptosis in HCC cells.
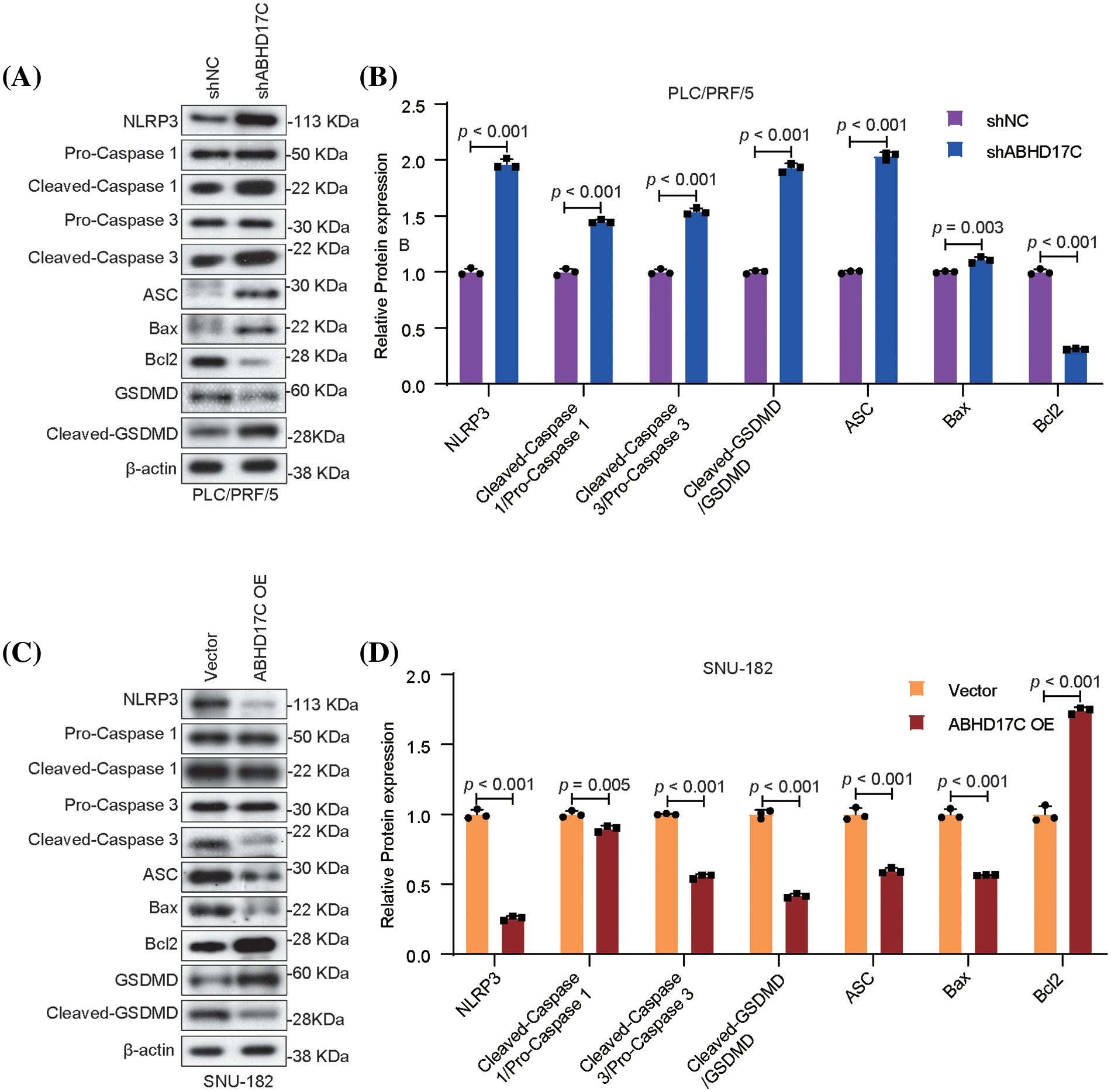
Figure 4: In HCC cells, ABHD17C controls the expression of markers linked to pyroptosis and apoptosis (n = 3). (A) Western blot data show that ABHD17C deficiency caused increased levels of BAX, Cleaved-Caspase, ASC, NLRP3, Cleaved-Caspase 1, and Cleaved-GSDMD, but led to a reduced level of Bcl2 in PLC/PRF/5 cells. (B) Statistical bar graph of Western blot data in (A). (C) Western blot results revealed that excessive ABHD17C repressed the expression of BAX, Cleaved-Caspase, ASC, NLRP3, Cleaved-Caspase 1, and Cleaved-GSDMD, but induced the level of Bcl2 in SNU-182 cells. (D) Statistical bar graph of Western blot data in (C).
Subsequently, we evaluated the ROS level in these HCC cells as it is an inducer of pyroptosis in multiple contexts [33,34]. DCF-DA staining results revealed that ABHD17C-deficient PLC/PRF/5 cells had a higher level of ROS than control cells. However, ABHD17C-overexpressing SNU-182 cells exhibited a comparable ROS level to control cells (Fig. 5A,B). Correspondingly, pyroptosis-induced production of TNF-α, IL-1β, and IL-18 was enhanced in ABHD17C-deficient PLC/PRF/5 cells. However, there was no change in ABHD17C-overexpressing SNU-182 cells (Fig. 5C,D). Additionally, the mitochondria exhibited defects in ABHD17C-deficient PLC/PRF/5 cells and were more intact in ABHD17C-overexpressing SNU-182 cells (Fig. 5E,F). These observations illustrate that ABHD17C negatively controls the apoptosis and pyroptosis in HCC cells.
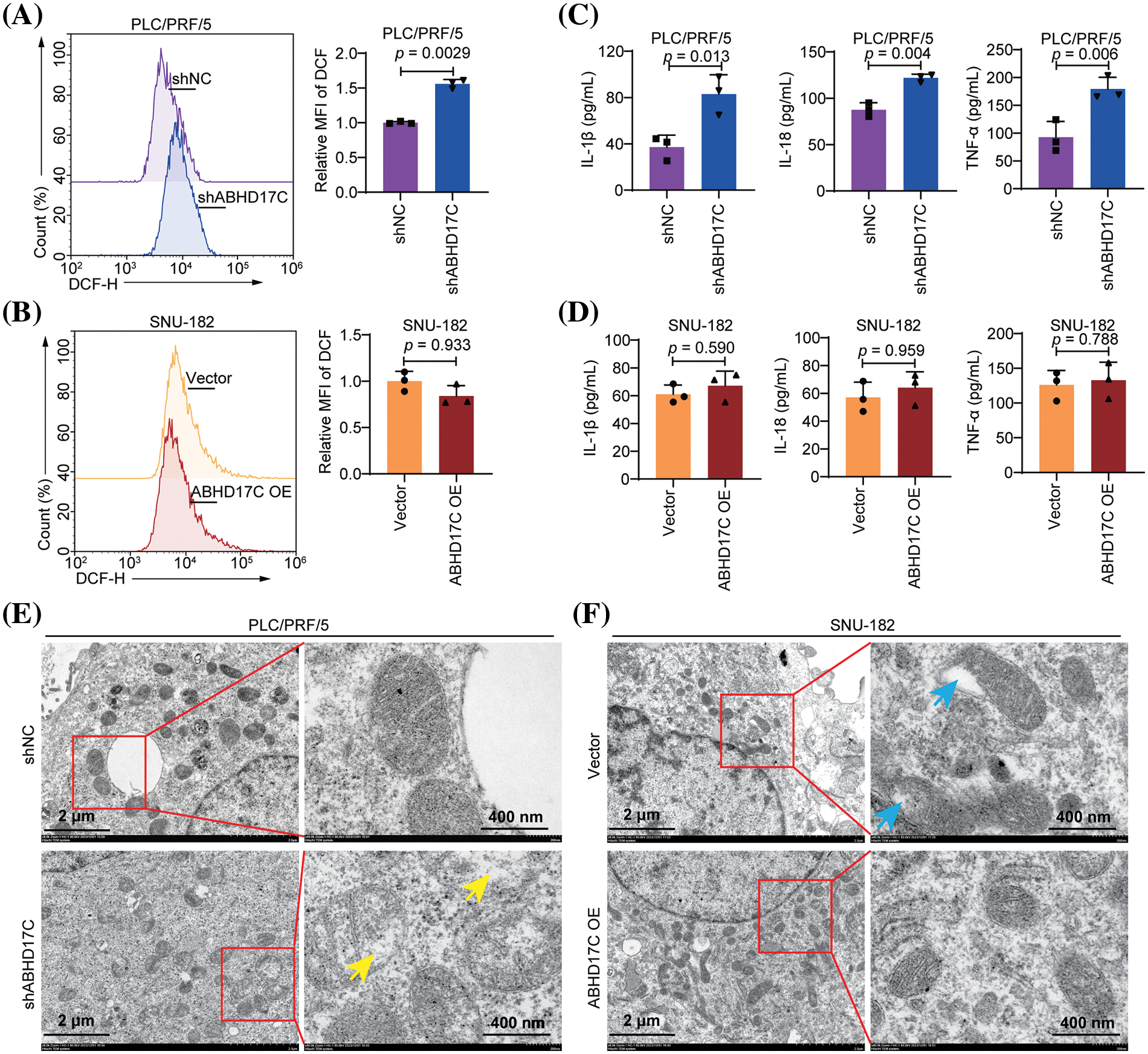
Figure 5: ABHD17C controls the ROS level, pro-cytokine secretion, and mitochondrial function in HCC cells (n = 3). (A) DCF-DA staining results indicate that ABHD17C-deficient PLC/PRF/5 cells had an increased ROS level compared with control cells. (B) DCF-DA staining results suggest that ABHD17C overexpression had no significant impact on the ROS level of SNU-182 cells. (C) The ELISA data demonstrated that the lack of ABHD17C resulted in the increased production of TNF-α, IL-18, and IL-1β by PLC/PRF/5 cells. (D) ELISA data uncover that excessive ABHD17C exhibited a minimal effect on the production of pro-inflammatory cytokines by SNU-182 cells. (E) TEM data show that the structure of mitochondria was disrupted in ABHD17-deficient PLC/PRF/5 cells (yellow arrows). (F) TEM data reveal the presence of defective mitochondria (blue arrows) in control SNU-182 cells, but such mitochondria were not observed in ABHD17-overexpressing SNU-182 cells.
ABHD17 proteins are novel depalmitoylases and their function, particularly in various diseases, is far from clear. Aside from its role as an oncogene in cancer cells, ABHD17C is also implicated in neuronal and cardiovascular diseases [35,36]. Consistently, our current data consolidate that ABHD17C has an oncogene role in HCC cells. Since ABHD17C mRNA is overexpressed in human HCC tissue, we examined its expression in more human HCC tissues as well as in multiple HCC cell lines. As expected, HCC tissues, as well as HCC cells with different extents show significantly increased ABHD17C protein and mRNA levels compared with SNU449 cells. Notably, the ABHD17C expression is unchanged in SNU449 and SK-HEP-1 HCC cells. The genetic heterogeneity or cancer stages of the original donors may be attributed to this disparity.
The mechanism regulating ABHD17C expression in HCC cells remains incompletely understood. In our previous study, miR-145-5p was identified as a negative regulator of ABHD17C mRNA, binding to the 3’UTR of the molecule and significantly increasing ABHD17C expression when miR-145-5p was knocked down. Furthermore, the expression of miR-145-5p is significantly reduced in HCC tissues, whereas ABHD17C expression is increased [23]. These findings reflect the control of ABHD17C at the translational level. Interestingly, we have recently discovered that multiple ubiquitin-specific proteases, which stabilize their target proteins by removing the ubiquitination [37], such as ubiquitin-specific-processing proteases 35 (USP35), are involved in degrading the ABHD17C protein [24]. This finding underscores the importance of posttranscriptional regulation of ABHD17C in HCC cells. It would be interesting to test whether the levels of other USPs in addition to USP35, are differentially expressed between HCC cells expressing high levels and normal levels of ABHD17C.
Given their depalmitoylase properties, ABHD17C can promote HCC development by regulating the S-palmitoylation of other oncogenic proteins like N-RAS, which contributes to the emergence of diverse forms of malignancies [24,38]. Through mitogen-activated protein kinases (MAPK) and PI3K/AKT signaling, overactivated RAS promotes HCC progression [39–41]. Furthermore, excessive N-RAS contributes to the induction of spontaneous HCC in animal models [42–44]. Therefore, it is not surprising that ABHD17C regulates the localization and function of N-RAS in HCC cells as it does in acute myeloid leukemia cells [15,24]. Considering the broad range of substrates by depalmitoylases, whether ABHD17C exerts oncogenic function in HCC cells by additional mechanisms warrants further investigation.
While the oncogenic function of ABHD17C in HCC cells could be mediated by increased RAS signaling, which is essential for cell proliferation, apoptosis, and migration [45,46], the anti-pyroptotic ABHD17C function may be mediated by additional substrates and mechanisms, as the connection between the RAS signaling and pyroptosis is not well established. A comprehensive screen of ABHD17C substrates would be essential to identify more substrates regulated by ABHD17C in modulating its anti-pyroptotic effect on HCC.
Notably, while the depletion of ABHD17C is sufficient to cause increased pyroptotic protein expression, ROS level, secretion of pro-inflammatory cytokines, and interrupted mitochondrial structure in PLC/PRF/5 cells, excessive ABHD17C had minimal effect on the ROS level and cytokine secretion in SNU-182 cells, despite of the reduced expression of pyroptosis-related markers and improved mitochondrial structure in these cells. This obvious difference may be explained by the differential requirement of ABHD17C by different HCC cells, i.e., PLC/PRF/5 cells need more ABHD17C to function properly whereas a low level of ABHD17C in SNU-182 cells is enough to support the survival of these cells. Alternatively, the substrate responsible for mediating the anti-pyroptotic role of ABHD17C in HCC cells is not sensitive enough to overdosed ABHD17C. It is also possible that SNU-182 cells have a low level of pyroptosis, making it difficult to detect any significant change in pyroptosis even with the presence of a pyroptosis repressor like ABHD17C. Further investigations are required to test the possibility of these assumptions.
In summary, this study fully evaluates ABHD17C’s role in promoting the malignant phenotype of HCC cells and demonstrates the cancer-causing function of ABHD17C in the formation of HCC, thereby highlighting the potential therapeutic value of ABHD17C inhibitors as an option for HCC treatment.
Although this study showed that ABHD17C can affect the malignant phenotype of HCC cells, there are still many shortcomings in this study. Human normal hepatocyte LO-2 cells have been contaminated, and there is a lack of human normal hepatocyte cell lines. The control cells we used in this study, SNU499, are HCC cells, which are different from normal hepatocytes. The experimental results would have been more scientific and reliable if we could have used human normal hepatocytes as the control cells to study the changes in ABHD17C expression in HCC cells. In addition, this study did not clarify whether ABHD17C and NLRP3 proteins bind to each other and whether ABHD17C acts directly on NLRP3 or regulates cellular pyroptosis by affecting the expression of other genes and proteins still needs to be further elucidated. Lastly, this study lacks animal experiments; if the results of cellular experiments in vitro can be further verified in animal experiments in vivo, it will make the experimental results more comprehensive and effectively illustrate that a possible treatment target for HCC may be ABHD17C.
Acknowledgement: None.
Funding Statement: The study was supported by the Quanzhou High-Level Talents Project (2021C048R).
Author Contributions: Study conception and design: Linpei Wang, Wei Wang, Jian Wu; data collection and experimental operation: Linpei Wang, Jiawei Wang, Chunfeng Shi; analysis and interpretation of results: Linpei Wang, Jiawei Wang, Chunfeng Shi, Wei Wang, Jian Wu; draft manuscript preparation: Linpei Wang; review and revise manuscript: Jiawei Wang, Chunfeng Shi, Wei Wang, Jian Wu. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The data that support the findings of this study are available on request from the corresponding authors upon reasonable requests and reasons.
Ethics Approval: Ethics approval was obtained from The Second Affiliated Hospital of Fujian Medical University Hospital with the permission of the Hospital’s Ethics Committee (Approval number: No. 27(2023)). Written informed consent was obtained from the patients, adhering to the principles of the Declaration of Helsinki.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Singal AG, Lampertico P, Nahon P. Epidemiology and surveillance for hepatocellular carcinoma: new trends. J Hepatol. 2020;72(2):250–61. doi:10.1016/j.jhep.2019.08.025. [Google Scholar] [PubMed] [CrossRef]
2. Cao G, Liu J, Liu M. Global, regional, and national trends in incidence and mortality of primary liver cancer and its underlying etiologies from 1990 to 2019: results from the global burden of disease study 2019. J Epidemiol Glob Health. 2023;13(2):344–60. doi:10.1007/s44197-023-00109-0. [Google Scholar] [PubMed] [CrossRef]
3. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492. [Google Scholar] [PubMed] [CrossRef]
4. Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6. doi:10.1038/s41572-020-00240-3. [Google Scholar] [PubMed] [CrossRef]
5. Rumgay H, Ferlay J, de Martel C, Georges D, Ibrahim AS, Zheng R, et al. Global, regional and national burden of primary liver cancer by subtype. Eur J Cancer. 2022;161:108–18. doi:10.1016/j.ejca.2021.11.023. [Google Scholar] [PubMed] [CrossRef]
6. McGlynn KA, Petrick JL, El-Serag HB. Epidemiology of hepatocellular carcinoma. Hepatology. 2021;73(Suppl 1):4–13. [Google Scholar] [PubMed]
7. Review T, Labrecque DR, Abbas Z, Anania F, Ferenci P, Khan AG, et al. World gastroenterology organisation global guidelines: nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. J Clin Gastroenterol. 2014;48(6):467–73. doi:10.1097/MCG.0000000000000116. [Google Scholar] [PubMed] [CrossRef]
8. McKillop IH, Schrum LW. Role of alcohol in liver carcinogenesis. Semin Liver Dis. 2009;29(2):222–32. doi:10.1055/s-0029-1214377. [Google Scholar] [PubMed] [CrossRef]
9. Janevska D, Chaloska-Ivanova V, Janevski V. Hepatocellular carcinoma: risk factors, diagnosis and treatment. Open Access Maced J Med Sci. 2015;3(4):732–6. doi:10.3889/oamjms.2015.111. [Google Scholar] [PubMed] [CrossRef]
10. Zhang H, Zhang W, Jiang L, Chen Y. Recent advances in systemic therapy for hepatocellular carcinoma. Biomark Res. 2022;10(1):3. doi:10.1186/s40364-021-00350-4. [Google Scholar] [PubMed] [CrossRef]
11. Vogel A, Saborowski A. Medical therapy of HCC. J Hepatol. 2022;76(1):208–10. doi:10.1016/j.jhep.2021.05.017. [Google Scholar] [PubMed] [CrossRef]
12. Zhong C, Li Y, Yang J, Jin S, Chen G, Li D, et al. Immunotherapy for hepatocellular carcinoma: current limits and prospects. Front Oncol. 2021;11:589680. doi:10.3389/fonc.2021.589680. [Google Scholar] [PubMed] [CrossRef]
13. Kumari R, Sahu MK, Tripathy A, Uthansingh K, Behera M. Hepatocellular carcinoma treatment: hurdles, advances and prospects. Hepat Oncol. 2018;5(2):HEP08. doi:10.2217/hep-2018-0002. [Google Scholar] [PubMed] [CrossRef]
14. Lin DT, Conibear E. ABHD17 proteins are novel protein depalmitoylases that regulate N-Ras palmitate turnover and subcellular localization. eLife. 2015;4:e11306. doi:10.7554/eLife.11306. [Google Scholar] [PubMed] [CrossRef]
15. Remsberg JR, Suciu RM, Zambetti NA, Hanigan TW, Firestone AJ, Inguva A, et al. ABHD17 regulation of plasma membrane palmitoylation and N-Ras-dependent cancer growth. Nat Chem Biol. 2021;17(8):856–64. doi:10.1038/s41589-021-00785-8. [Google Scholar] [PubMed] [CrossRef]
16. Sobocinska J, Roszczenko-Jasinska P, Ciesielska A, Kwiatkowska K. Protein palmitoylation and its role in bacterial and viral infections. Front Immunol. 2017;8:2003. [Google Scholar] [PubMed]
17. Rocks O, Gerauer M, Vartak N, Koch S, Huang ZP, Pechlivanis M, et al. The palmitoylation machinery is a spatially organizing system for peripheral membrane proteins. Cell. 2010;141(3):458–71. doi:10.1016/j.cell.2010.04.007. [Google Scholar] [PubMed] [CrossRef]
18. Won SJ, Martin BR. Temporal profiling establishes a dynamic S-palmitoylation cycle. ACS Chem Biol. 2018;13(6):1560–8. doi:10.1021/acschembio.8b00157. [Google Scholar] [PubMed] [CrossRef]
19. Martin BR, Wang C, Adibekian A, Tully SE, Cravatt BF. Global profiling of dynamic protein palmitoylation. Nat Methods. 2011;9(1):84–9. doi:10.1038/nmeth.1769. [Google Scholar] [PubMed] [CrossRef]
20. Main A, Fuller W. Protein S-palmitoylation: advances and challenges in studying a therapeutically important lipid modification. FEBS J. 2022;289(4):861–82. doi:10.1111/febs.15781. [Google Scholar] [PubMed] [CrossRef]
21. Zhou B, Hao Q, Liang Y, Kong E. Protein palmitoylation in cancer: molecular functions and therapeutic potential. Mol Oncol. 2023;17(1):3–26. doi:10.1002/1878-0261.13308. [Google Scholar] [PubMed] [CrossRef]
22. Ko PJ, Dixon SJ. Protein palmitoylation and cancer. EMBO Rep. 2018;19(10):5437. doi:10.15252/embr.201846666. [Google Scholar] [PubMed] [CrossRef]
23. Wang L, Ma X, Chen Y, Zhang J, Zhang J, Wang W, et al. MiR-145-5p suppresses hepatocellular carcinoma progression by targeting ABHD17C. Oncologie. 2022;24(4):897–912. doi:10.32604/oncologie.2022.025693. [Google Scholar] [CrossRef]
24. Wang L, Wang J, Ma X, Ju G, Shi C, Wang W, et al. USP35 promotes HCC development by stabilizing ABHD17C and activating the PI3K/AKT signaling pathway. Cell Death Discov. 2023;9(1):421. doi:10.1038/s41420-023-01714-5. [Google Scholar] [PubMed] [CrossRef]
25. Peng F, Liao M, Qin R, Zhu S, Peng C, Fu L, et al. Regulated cell death (RCD) in cancer: key pathways and targeted therapies. Signal Transduct Target Ther. 2022;7(1):286. doi:10.1038/s41392-022-01110-y. [Google Scholar] [PubMed] [CrossRef]
26. Tong X, Tang R, Xiao M, Xu J, Wang W, Zhang B, et al. Targeting cell death pathways for cancer therapy: recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research. J Hematol Oncol. 2022;15(1):174. doi:10.1186/s13045-022-01392-3. [Google Scholar] [PubMed] [CrossRef]
27. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99–109. doi:10.1038/nrmicro2070. [Google Scholar] [PubMed] [CrossRef]
28. Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol. 2017;27(9):673–84. doi:10.1016/j.tcb.2017.05.005. [Google Scholar] [PubMed] [CrossRef]
29. Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006;8(11):1812–25. doi:10.1111/cmi.2006.8.issue-11. [Google Scholar] [CrossRef]
30. Zou Z, Zhao M, Yang Y, Xie Y, Li Z, Zhou L, et al. The role of pyroptosis in hepatocellular carcinoma. Cell Oncol. 2023;46(4):811–23. doi:10.1007/s13402-023-00787-9. [Google Scholar] [PubMed] [CrossRef]
31. Chu Q, Jiang Y, Zhang W, Xu C, Du W, Tuguzbaeva G, et al. Pyroptosis is involved in the pathogenesis of human hepatocellular carcinoma. Oncotarget. 2016;7(51):84658–65. doi:10.18632/oncotarget.v7i51. [Google Scholar] [CrossRef]
32. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. 2021;6(1):128. doi:10.1038/s41392-021-00507-5. [Google Scholar] [PubMed] [CrossRef]
33. Wang Y, Shi P, Chen Q, Huang Z, Zou D, Zhang J, et al. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J Mol Cell Biol. 2019;11(12):1069–82. doi:10.1093/jmcb/mjz020. [Google Scholar] [PubMed] [CrossRef]
34. Tian K, Yang Y, Zhou K, Deng N, Tian Z, Wu Z, et al. The role of ROS-induced pyroptosis in CVD. Front Cardiovasc Med. 2023;10:1116509. doi:10.3389/fcvm.2023.1116509. [Google Scholar] [PubMed] [CrossRef]
35. Kolifarhood G, Sabour S, Akbarzadeh M, Sedaghati-Khayat B, Guity K, Rasekhi Dehkordi S, et al. Genome-wide association study on blood pressure traits in the Iranian population suggests ZBED9 as a new locus for hypertension. Sci Rep. 2021;11(1):11699. doi:10.1038/s41598-021-90925-w. [Google Scholar] [PubMed] [CrossRef]
36. Zhang L, Zhang W, He L, Cui H, Wang Y, Wu X, et al. Impact of gallstone disease on the risk of stroke and coronary artery disease: evidence from prospective observational studies and genetic analyses. BMC Med. 2023;21(1):353. doi:10.1186/s12916-023-03072-6. [Google Scholar] [PubMed] [CrossRef]
37. Young MJ, Hsu KC, Lin TE, Chang WC, Hung JJ. The role of ubiquitin-specific peptidases in cancer progression. J Biomed Sci. 2019;26(1):42. doi:10.1186/s12929-019-0522-0. [Google Scholar] [PubMed] [CrossRef]
38. Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7(4):295–308. doi:10.1038/nrc2109. [Google Scholar] [PubMed] [CrossRef]
39. Fan RH, Li J, Wu N, Chen PS. Late SV40 factor: a key mediator of Notch signaling in human hepatocarcinogenesis. World J Gastroenterol. 2011;17(29):3420–30. doi:10.3748/wjg.v17.i29.3420. [Google Scholar] [PubMed] [CrossRef]
40. Fan R, Chen P, Zhao D, Tong JL, Li J, Liu F. Cooperation of deregulated Notch signaling and Ras pathway in human hepatocarcinogenesis. J Mol Histol. 2011;42(5):473–81. doi:10.1007/s10735-011-9353-3. [Google Scholar] [PubMed] [CrossRef]
41. Delire B, Starkel P. The Ras/MAPK pathway and hepatocarcinoma: pathogenesis and therapeutic implications. Eur J Clin Investig. 2015;45(6):609–23. doi:10.1111/eci.12441. [Google Scholar] [PubMed] [CrossRef]
42. Gao M, Liu D. CRISPR/Cas9-based Pten knock-out and sleeping beauty transposon-mediated Nras knock-in induces hepatocellular carcinoma and hepatic lipid accumulation in mice. Cancer Biol Ther. 2017;18(7):505–12. doi:10.1080/15384047.2017.1323597. [Google Scholar] [PubMed] [CrossRef]
43. Leung HW, Leung CON, Lau EY, Chung KPS, Mok EH, Lei MML, et al. EPHB2 activates β-catenin to enhance cancer stem cell properties and drive sorafenib resistance in hepatocellular carcinoma. Cancer Res. 2021;81(12):3229–40. doi:10.1158/0008-5472.CAN-21-0184. [Google Scholar] [PubMed] [CrossRef]
44. Wu M, Xia X, Hu J, Fowlkes NW, Li S. WSX1 act as a tumor suppressor in hepatocellular carcinoma by downregulating neoplastic PD-L1 expression. Nat Commun. 2021;12(1):3500. doi:10.1038/s41467-021-23864-9. [Google Scholar] [PubMed] [CrossRef]
45. Drosten M, Dhawahir A, Sum EY, Urosevic J, Lechuga CG, Esteban LM, et al. Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J. 2010;29(6):1091–104. doi:10.1038/emboj.2010.7. [Google Scholar] [PubMed] [CrossRef]
46. Ney GM, Yang KB, Ng V, Liu L, Zhao M, Kuk W, et al. Oncogenic N-Ras mitigates oxidative stress-induced apoptosis of hematopoietic stem cells. Cancer Res. 2021;81(5):1240–51. doi:10.1158/0008-5472.CAN-20-0118. [Google Scholar] [PubMed] [CrossRef]
Appendix
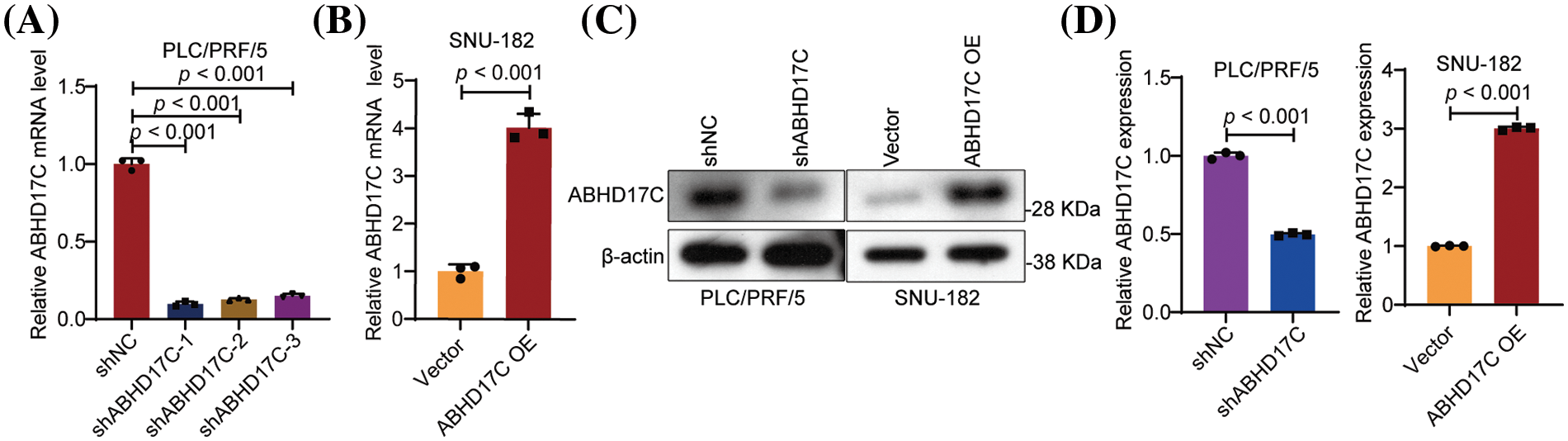
Figure A1: Verification of ABHD17C expression in ABHD17C-deficient and ABHD17-overexpressing cells (n = 3). (A) qPCR data show that all three shRNAs targeting ABHD17C effectively repressed the ABHD17C mRNA level in PLC/PRF/5 cells. shABHD17C-1 was selected for subsequent experiments due to its best knockdown efficiency. (B) qPCR results indicated that the ABHD17C overexpression plasmid substantially increased the mRNA level of ABHD17C in SNU-182 cells. (C) Western blot results validated the reduction of ABHD17C expression in PLC/PRF/5 cells and the upregulation of ABHD17C in SNU-182 cells. (D) Statistical bar graph of Western blot data in (C).
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools