
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

The pathogenesis of Parkinson’s disease and crosstalk with other diseases
Institute for Brain Sciences Research, School of Life Sciences, Henan University, Kaifeng, 475004, China
* Corresponding Author: JIANSHE WEI. Email:
(This article belongs to the Special Issue: Exploring the Cellular Mechanisms of Neurodegenerative Diseases)
BIOCELL 2024, 48(8), 1155-1179. https://doi.org/10.32604/biocell.2024.051518
Received 07 March 2024; Accepted 13 May 2024; Issue published 02 August 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
In China, Parkinson’s disease (PD) is the second most prevalent central nervous system (CNS) degenerative illness affecting middle-aged and older persons. Movement disorders including resting tremor, bradykinesia, myotonia, postural instability, and gait instability are the predominant clinical symptoms. The two main types of PD are sporadic and familial, with sporadic PD being the more prevalent of the two. The environment, genetics, mitochondrial dysfunction, oxidative stress, inflammation, protein aggregation and misfolding, loss of trophic factors, cell death, and gut microbiota may all have a role in the etiology of PD. PD is inversely connected with other cancers and positively correlated with COVID-19, diabetes mellitus (DM), melanoma, and ischemic heart disease (IHD) risk. Delaying disease progression, managing motor and non-motor symptoms, and avoiding and controlling dysfunction in the middle and later phases of the disease are the key areas of research and development for its therapy. Presently, the development and progression of PD can be slowed down by using conventional pharmacology, natural items, and innovative technology. This article reviews the pathogenesis of PD, its correlations with other non-genetic diseases, and the research progress of drugs and technologies for alleviating PD.Keywords
Abbreviation
| UA | Uric acid |
| nAChRs | nicotinic acetylcholine receptors |
| OXPHOS | oxidative phosphorylation |
| ETC | electron transport chain |
| TLR | toll-like receptors |
| REDOX | oxidation-reduction reaction |
| ER | endoplasmic reticulum |
| NFs | Neurotrophic factors |
| LPO | lipid peroxidation |
| CMA | chaperone-mediated autophagy |
| SARSCoV-2 | severe acute respiratory syndrome coronavirus 2 |
| ACE2 | angiotensin-converting enzyme 2 |
| DM | Diabetes mellitus |
| HD | Huntington’s chorea |
| PLA2G6 | phospholipase A2, group VI |
| IHD | Ischemic heart disease |
| NRTN | neurturin |
| MNAF | mesencephalic astrocyte-derived neural factor |
| GAD | glutamic acid decarboxylase |
| NPY | neuropeptide Y |
| NRSF | neuron restrictive silencing factor |
| MSCs | mesenchymal stem cells |
| hPSC | pluripotent stem cells |
| ESC | embryonic stem cells |
| iPSC | induced pluripotent stem cells |
| Nurr1 | nuclear receptor related 1 |
| GBA1 | glucosylceramidase beta 1 |
| DMT | disease-modifying therapies |
| LNPs | lipid nanoparticles. |
Parkinson’s disease (PD) is a neurological condition that primarily affects the elderly and is progressive and irreversible on a recessive basis [1]. Despite the fact the disease can strike anyone at any age, the typical age of onset is 60 [2]. After Alzheimer’s disease (AD), PD is the second most common neurodegenerative illness worldwide [3]. PD is the primary cause of disability on a global scale and exhibits the highest rate of growth among neurological conditions. From 1990 to 2016, the total number of patients exceeded 6 million [4]. By 2040, it is predicted that this number will have doubled to over 12 million [5]. Research shows that various factors, such as the extension of life expectancy, the improvement of industrialization, and the decline of the smoking rate, may increase the burden of disease [6], which might reach over 17 million [7]. The increasing incidence and prevalence of this disease worldwide highlight its significant economic, social, and public health impact. According to current theories, there are three types of PD: idiopathic PD, familial PD, and Parkinsonism [8]. According to epidemiological research, only a small percentage of PD patients have the familial form of the disease, whereas the sporadic form affects the vast majority [9]. It is becoming more and more obvious that the development of PD cannot be attributed to a single cause and that, in the majority of instances, it arises from an unpleasant interaction between genetic polymorphisms, epigenetic modifications, and other environmental factors [10].
The disorder shows a wide range of non-motor abnormalities alongside motor symptoms including bradykinesia, stiffness, and tremor. These typical motor symptoms occur due to reduced dopamine (DA) levels in the striatum, caused by the presence of α-synuclein (α-syn) positive inclusions (Lewy bodies (LBs) and Lewy neurites) within neurons and axons, as well as the loss of dopaminergic (DAergic) neurons in the substantia nigra (SN) and other brain regions. The accumulation of α-syn is a pathological hallmark of PD. Despite its primary presence in the central nervous system, α-syn is highly expressed in the peripheral nervous system and the presynaptic terminals connecting synaptic vesicle devices in the brain [11]. The aging of the population was linked to the disease’s rising incidence [12]. Degeneration of DA neurons is not limited to the risk factor of old age but also can be caused by environmental factors and genetic deficiencies [13]. The degeneration of DAergic nigrostriatal neurons in the brain was considered to be the primary cause of PD, followed by complex pathological mechanisms such as mitochondrial dysfunction, oxidative stress, inflammation, protein aggregation, and misfolding, loss of neurotrophic factors, cell death (ferroptosis, apoptotic and autophagy), and gut microbiota [14].
In this review, we present a summary of research studies that provide evidence and contribute to understanding the association in PD. We offer insights into the underlying pathophysiology of PD and other diseases associated with PD, summarize the potential therapeutic drugs and technologies for treating PD, and trials of different ways to interfere (Figs. 1 and 2).
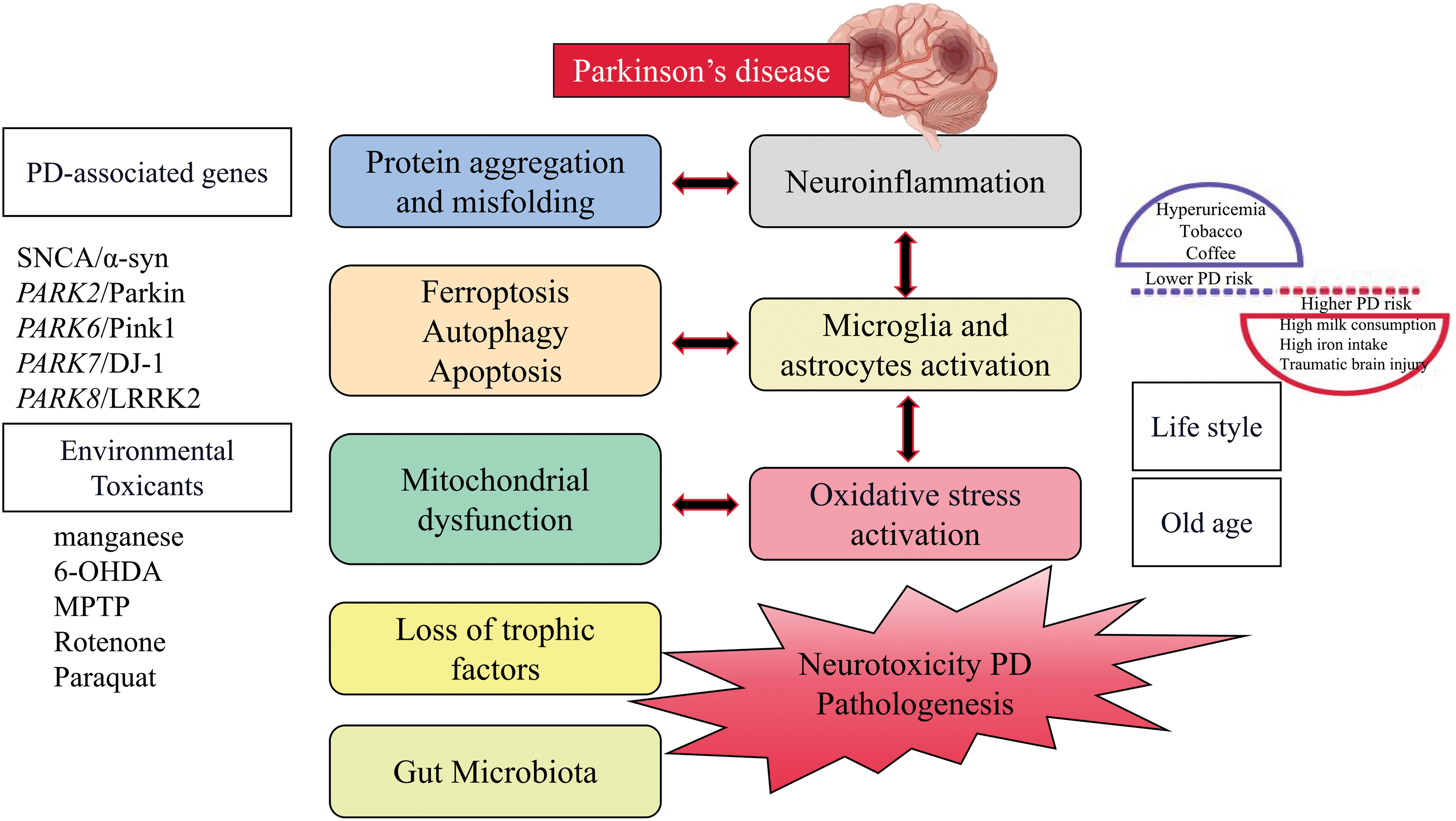
Figure 1: The pathogenic mechanism of Parkinson’s disease.
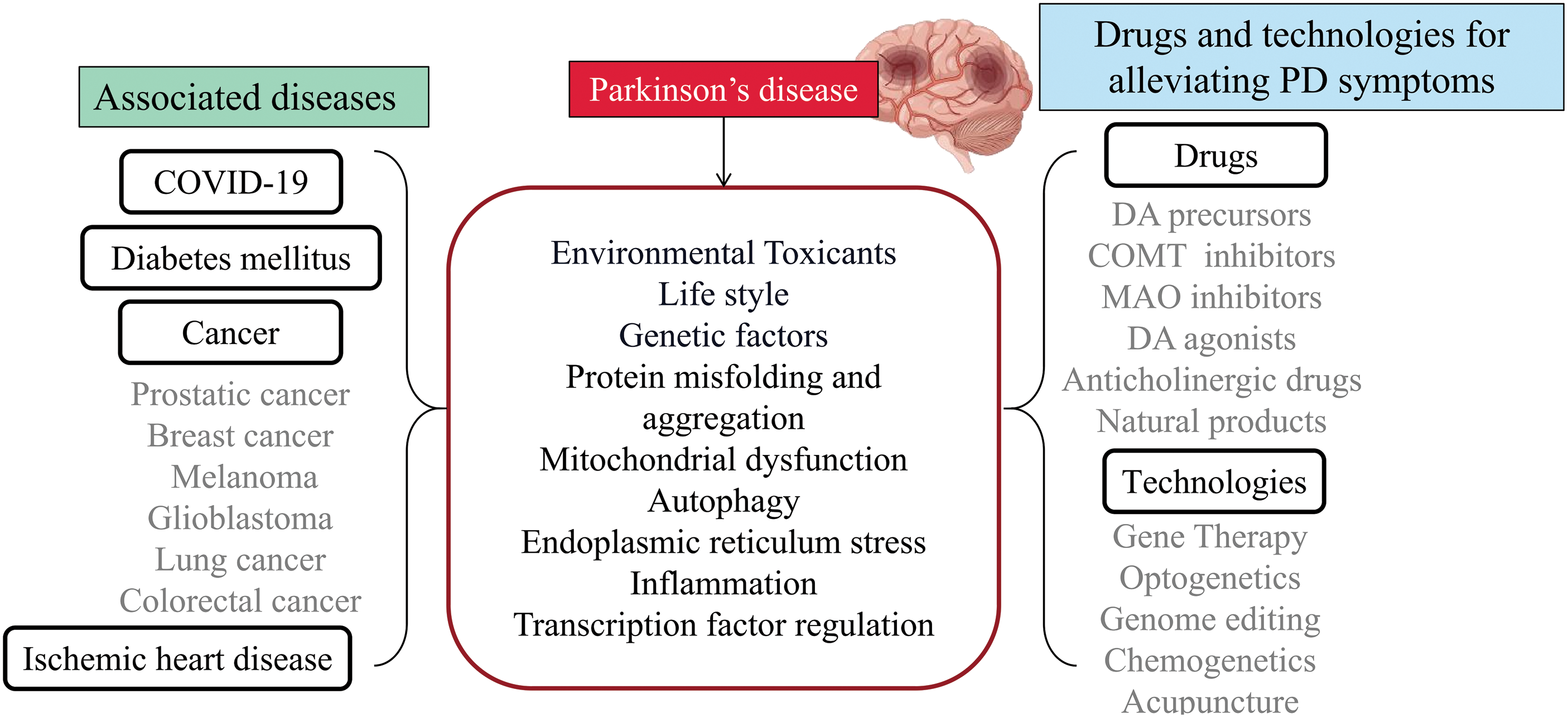
Figure 2: The association between Parkinson’s disease and other diseases, as well as drugs and technologies for alleviating Parkinson’s disease. PD passes through environmental toxicants, lifestyle, genetic factors, protein misfolding and aggregation, mitochondrial dysfunction, autophagy, endoplasmic reticulum stress, inflammation, and transcription factor regulation interacting with other diseases.
Environmental and Genetic Factors & PD
Pesticide exposure and increased risk of PD have been linked to numerous studies conducted over many decades in various populations worldwide [15]. Those who lived in areas with pesticide-treated fields and had occupational exposure were more likely to develop personality disorders. Paraquat, rotenone, 2,4-D, and various dithiocarbamates and organochlorines, among other pesticides associated with PD, have been shown to cause experimental Parkinsonism in laboratory studies, which may indicate causal effects [16,17]. The effect of pesticide exposure on PD risk can be amplified by genetically determined impairment in toxicant handling, which is an example of gene-environment interaction. Conversely, the adoption of healthy eating habits or maintaining a good diet can protect against the negative consequences of pesticide exposure [18,19]. The use of chlorinated solvents such as trichloroethylene, perchloroethylene, and carbon tetrachloride in activities like dry cleaning, degreasing, anesthesia, and viscose rayon manufacturing, as well as the former use of polychlorinated biphenyls as coolants and lubricants, has been associated with an elevated risk of PD and toxicity related to Parkinsonism in animal models [20,21]. It is important to note that organic chemicals and pesticides, including 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), not only directly harm DA neurons but also cause persistent damage to the central nervous system (CNS), specifically affecting DNA methylation and triggering additional inflammatory responses in the brain tissue [22,23]. Therefore, it can be seen that chronic systemic pesticide exposure reproduces the characteristics of PD.
Many lifestyle factors were associated with a reduced risk of PD [24]. These include hyperuricemia and the use of tobacco and coffee, while higher PD risk is associated with high milk consumption, high iron intake, and traumatic brain injury (TBI) in men. Interestingly, some negative risk factors appear to have neuroprotective effects and may be beneficial for patients with early PD, especially in terms of uric acid, smoking (nicotine), and caffeine. Uric acid (UA), a byproduct of purine synthesis, possesses strong antioxidant properties. Animal studies have demonstrated that UA can protect DAergic neurons by regulating neuroinflammation and oxidative stress. It was found that among all non-communicable diseases (cardiovascular and cerebrovascular diseases, diabetes, cancer), the number of smokers was higher than that of non-smokers, except for PD [25].
Epidemiological studies consistently report significantly reduced risks of PD in individuals who have smoked cigarettes at any point [26], possibly due to reduced nicotine responsiveness in PD patients. Nicotine, found in tobacco, shows potential as a treatment for PD. It influences the activity of the striatum and behaviors mediated by the DAergic system through the activation of nicotinic acetylcholine receptors (nAChRs) on DAergic terminals and modulation of DA release [27]. Additionally, nicotine may delay the development of PD by inducing the expression of cytochrome P450 (CYP) metabolic enzymes in the brain, which inactivates neurotoxins [28]. Caffeine, a substance commonly found in coffee and tea, has been the subject of extensive research in the field of neurodegenerative diseases [29]. Its neuroprotective effects are believed to be due to its ability to block adenosine 2A (A2A) receptors in the brain. These receptors are currently being investigated as a potential target for anti-parkinsonian therapy.
These factors hold promise for early diagnosis and intervention to delay PD progression. However, it is not recommended to limit milk consumption based on the currently available evidence for preventing PD, although more research is needed, particularly considering the sex differences observed [30]. Iron, when present in excess, can generate free radicals, participate in lipid peroxidation, and contribute to neurodegeneration. It also appears to enhance the neurotoxicity induced by MPTP, and accelerate α-syn fibrillation and deposition [31]. There are overlapping pathological features between TBI and PD, including neurodegeneration, protein misfolding (α-syn, APP, tau, TDP43), and persistent inflammation [32]. Lifestyle factors can have additive effects on the development of PD, pointing towards a potential approach for disease prevention.
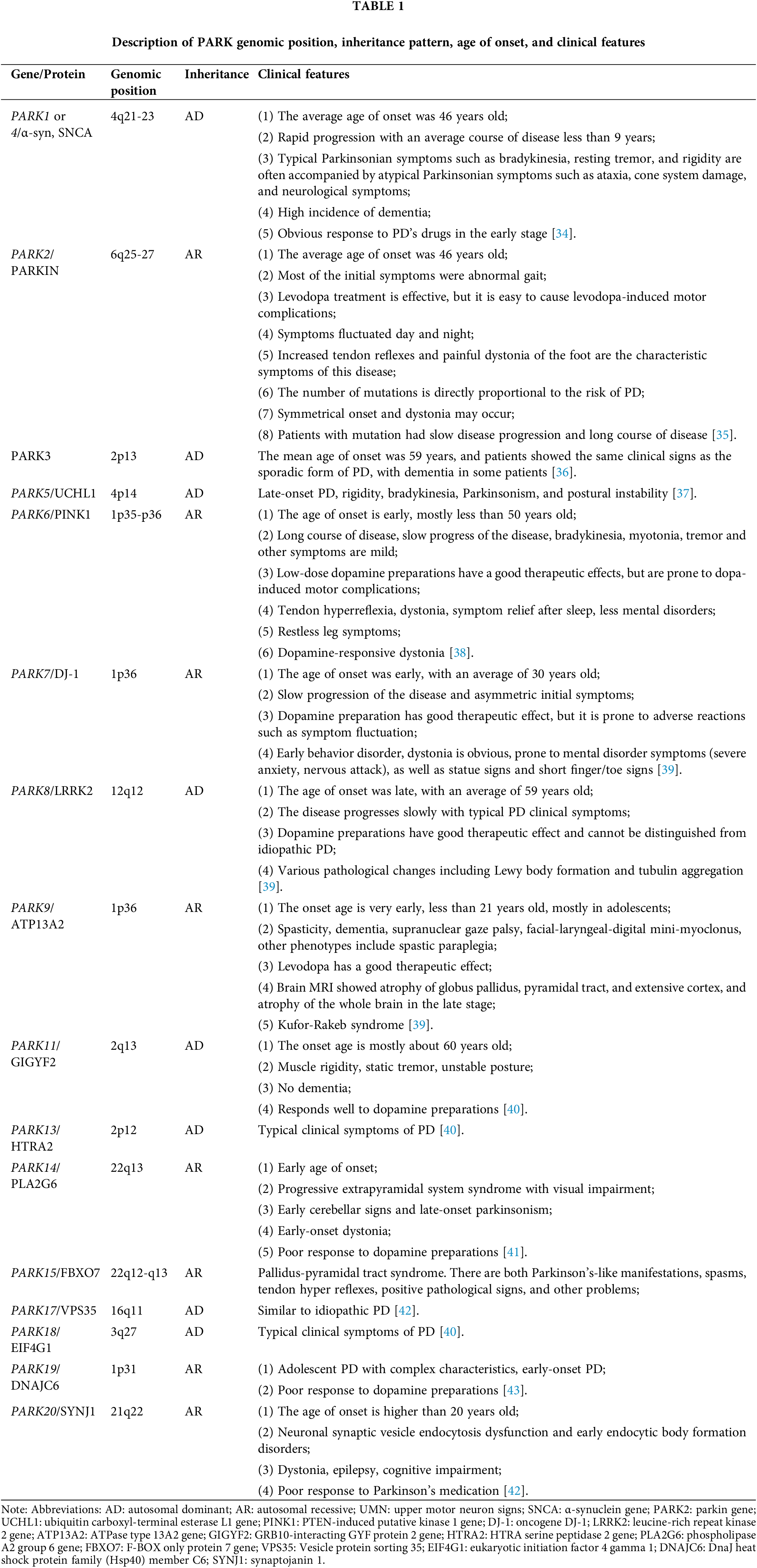
For nearly a century, it was believed that PD had no genetic links. However, a few decades ago, extensive studies on PD patients using linkage analysis revealed mutations in multiple genes [33]. These genetic mutations, as presented in Table 1, are associated with familial PD. Despite the recent progress in our comprehension of the genetic aspects of PD, the exact underlying causes of PD remain incompletely understood. The majority of PD cases (~95%) are considered sporadic, meaning they occur randomly without a clear genetic cause, and are believed to result from a combination of genetic susceptibility and environmental factors. Despite their distinct origins, both sporadic and monogenic (caused by a specific genetic mutation) PD cases display similar biochemistry, pathology, and clinical characteristics. Further information about the PD-associated genes can be found in Table 1.
Mitochondrial Dysfunction and Oxidative Stress & PD
Mitochondrial complex I dysfunction: implications for oxidative stress and PD
Mitochondria play a vital role in cell energy production by producing adenosine triphosphate (ATP) through respiration and oxidative phosphorylation (OXPHOS) [44]. Recent studies suggest that mitochondria are also responsible for generating reactive oxygen species (ROS), which can lead to oxidative stress within the cell. During oxidative phosphorylation, the electrons from nicotinamide adenine dinucleotide (NADH) are transferred through complex I (NADH-quinone oxidoreductase) for oxidation. The administration of rotenone leads to the degeneration of DAergic neurons, however, this toxicity can be mitigated by methylene blue, an alternative electron carrier that circumvents the blockade of complex I/III. This emphasizes the significance of complex I deficiency in the toxic effects induced by rotenone [45]. It is well-known that disruptions in DA homeostasis can impact mitochondrial function. For instance, the modification of DA can inhibit mitochondrial respiratory chain complexes [46]. The SN region of the brain, where DAergic neurons are located, is particularly vulnerable to complex I dysfunction due to the production of ROS during DA metabolism [47]. Therefore, it is possible that impairments in the mitochondrial respiratory chain, particularly deficiencies in complex I, as well as the resulting increase in ROS production, may play a role in the development of sporadic PD, either indirectly or directly.
Many research studies have demonstrated an elevation in oxidative stress levels in PD. The onset of PD is closely linked to an increase in oxidative stress. The inhibition of respiratory chain activity in the SN pars compacta (SNpc) area of PD is associated with the production of ROS and the triggering of apoptosis [48]. Various enzymes, such as monoamine oxidase (MAO), L-amino acid oxidase, and tyrosine hydroxylase, participate in DA metabolism and the generation of ROS [49]. In the normal physiological process, MAO produces hydrogen peroxide (H2O2) in PD, which is then converted to hydroxyl radicals (OH-) by iron through the Fenton reaction. Hence, both H2O2 and OH- contribute to ROS production [50]. The presence of H2O2 and OH- leads to the oxidation of glutathione (GSH) in the cytoplasm, resulting in GSH leakage in PD [51]. The leakage of GSH molecules leads to the formation of glutamyl peptide and cysteine from glutamic acid and cysteine. These peptides are harmful to DAergic cells as they penetrate through the cell membrane and activate ROS in DAergic neurons. Consequently, these peptides downregulate the activity of mitochondrial complex I, leading to increased production of ROS and heightened oxidative stress [52]. Furthermore, oxidative stress promotes protein carbonylation, an irreversible and irreparable modification. Carbonylation contributes to cellular senescence, which leads to the aggregation of proteins. Protein aggregation is a primary pathological characteristic of nigrostriatal DAergic neurons in PD and induces neuroinflammation and oxidative stress [53].
Mitochondrial DNA (mtDNA) is a crucial component in cell structure and function, as it is the only genetic material outside the nucleus with an impact on genetics. Unlike nuclear DNA, mtDNA lacks introns and any mutations occurring in it can affect vital functional regions in the genome [54]. MtDNA is responsible for encoding four enzyme complexes involved in the OXPHOS process. As a result, mutations in mtDNA can directly disrupt the OXPHOS process, leading to decreased mitochondrial respiratory function and energy synthesis. This, in turn, affects brain tissue function and causes degenerative changes [55]. Moreover, mtDNA is located near the inner mitochondrial membrane, where OXPHOS generates ROS. Due to the absence of histones and repair mechanisms, mtDNA is highly susceptible to oxidative damage and mutations. Mutations in mtDNA can lead to defects in its encoded complex and electron transport chain (ETC), resulting in an increased production of ROS, and establishing a harmful cycle [56]. Consequently, mtDNA mutations accumulate over time. The oxidative stress induced by ROS and free radicals causes the degeneration and death of DAergic neurons in the SN, which is closely associated with the development of PD.
Mitochondrial dysfunction and oxidative stress in monogenic PD
The majority of PD cases result from the intricate interplay between environmental and genetic factors. However, it is worth noting that a significant portion of individuals exhibit clear familial inheritance patterns. Research on gene decoding has revealed that mutations in genes such as SNCA, PARKIN or PRKN, PINK1, DJ-1, LRRK2, and others [57] are responsible for familial PD. Additionally, mitochondrial dysfunction and oxidative stress associated with familial PD have been compiled in Table 2.

Inflammation appears to be involved in the pathophysiology of PD, and research has demonstrated its significant impact on the development of the disease [70]. During the progression of PD, inflammation triggers the activation of apoptosis pathways in DAergic cells [71]. This relationship between inflammation and PD is mutual, as inflammation leads to the death of DAergic cells, which in turn stimulates further inflammation through a vicious feedback loop [72]. Additionally, inflammatory factors induce oxidative stress, forcing DAergic cells to activate signals that promote cell death [73]. The CNS consists of neurons and glial cells, including microglia, oligodendrocytes, and astrocytes. When a foreign pathogen enters, glial cells (astrocytes and microglia) are activated, and excessive activation of these cells triggers the release of various neuroinflammatory markers, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), nitric oxide (NO), and cyclooxygenase-2 (COX-2).
Microglia can sense and respond to neural activation, while also providing a negative feedback mechanism to counter excessive neural activity. This unique microglia-mediated neuromodulation mechanism plays a crucial role in protecting the brain from disease invasion. Moreover, inhibiting the conversion of A1 astrocytes by microglia is neuroprotective in PD models [74]. The nuclear factor kappa-B (NF-κB) pathway is mainly responsible to produce proinflammatory enzymes and cytokines in microglia [75]. One of the proinflammatory cytokines, TNF-α, can induce apoptosis through the activation of caspases 1 and 3 via its TNF-R1 receptor death domain [76]. TNF-α, a pro-inflammatory molecule, can decrease the expression of c-Rel–NF-κB, which has a neuroprotective effect on DA neurons by inhibiting apoptosis through the B-cell lymphoma-extra-large pathway. The signaling pathway of interferon-γ (IFN-γ)–interferon-gamma receptor 1 (IFNGR) can phosphorylate the protein LRRK2, which plays a role in various cellular processes in microglia and DA neurons. When LRRK2 is activated, it suppresses the expression of c-Rel–NF-κB. As a result, the insufficiency of c-Rel–NF-κB leads to increased inflammation [77,78]. In PD, high levels of C-X-C motif chemokine receptor 4 (CXCR4) (also known as fusion) and its ligand C-X-C motif chemokine ligand 12 (CXCL12) are found. The interaction between CXCR4 and CXCL12 activates caspase 3, causing neural cell death through apoptosis [79,80].
Additionally, the presence of specific α-syn conformations triggers the activation of microglia, leading to an immune response that involves increased expression of toll-like receptors (TLR) and proinflammatory mediators. This immune response includes excessive microglia activation, changes in the composition and phenotype of T lymphocytes, and an increase in B lymphocyte-produced antibodies. These immune cells from the periphery may actively contribute to the progression of neurodegeneration [81].
Protein aggregation and misfolding
Protein misfolding and subsequent aggregation are common underlying factors in neurodegenerative diseases. Proteins usually fold into a specific three-dimensional structure to carry out their functions effectively. Misfolding can be caused by genetic mutations, mistranslation, environmental factors, ionic strength, pH, metal ions, oxidation-reduction reaction (REDOX) state changes, and protein concentration. In PD, the formation of cytoplasmic LBs and the premature death of DA neurons in the midbrain SN are characteristic features. Familial PD can be caused by mutations in the α-syn and Parkin genes, leading to the deposition of α-syn and Parkin proteins in LBs [82]. Mutations in the α-syn gene increase the number of protein oligomers, promoting LBs formation. Oxidative stress, Aβ, and mitochondrial inhibitors also contribute to the production of α-syn polymers. Additionally, α-syn binds to the S6’ subunit of the 19S CAP on the 26S proteasome, inhibiting proteasome function and hindering synuclein degradation. Parkin mutation affects α-syn metabolism, leading to its accumulation. Furthermore, Parkin aids in removing proteins with abnormal configurations in the endoplasmic reticulum (ER) [83]. Hence, Parkin mutation allows the accumulation of α-syn and other misfolded proteins, leading to intracellular aggregation. DA neurons are especially vulnerable to α-syn and Parkin mutations due to their increased oxidative damage during DA metabolism. It is important to highlight that while protein misfolding and aggregation are key aspects of PD, the disease’s pathogenesis involves a combination of genetic and environmental factors. Here, we discuss the role of protein misfolding and aggregation (α-syn, PARKIN, PINK1, DJ-1, and LRRK2) in PD (Table 3).

Neurotrophic factors (NFs) possess the ability to nourish neurons, stimulate axon regeneration, and regulate microglia. Insufficient NF levels in the SN and striatum can result in the degeneration and necrosis of DA-producing neurons, leading to the clinical symptoms of PD. Various NFs have been identified, including brain-derived neurotrophic factor (BDNF) [90], glial cell line-derived neurotrophic factor (GDNF) [91], ciliary neurotrophic factor (CNTF), nerve growth factor (NGF), fibroblast growth factor (FGF), vascular endothelial growth factor (VEGF), and insulin-like growth factor 1 (IGF-1) [92]. NFs play a crucial role in maintaining normal neuronal functions, participating in nerve cell differentiation, preventing neuronal degeneration and death, regulating calcium balance, enhancing resistance to free radicals, improving the catalase system, and inhibiting the absorption of 6-hydroxydopamine (6-OHDA) by nerve fibers. Notably, neurotrophic factors, especially NGF, interact with local microglia and infiltrating immune cells to modulate immune responses [93].
Ferroptosis is a regulated cell death pathway that is dependent on iron and involves the accumulation of lipid peroxidation (LPO). It shares various characteristics with the pathophysiology of PD. Previous studies have demonstrated that iron accumulation is implicated in the progression of PD. Inhibition of ferroptosis has shown protective effects in PD cells and animal models [94,95]. PD patients also exhibit significant accumulation of LPO, primarily due to iron-mediated cell death caused by prolonged exposure to excessive free radicals from certain neurotoxic substances [96]. Additionally, PD neurons demonstrate excessive iron and LPO accumulation, as well as decreased levels of glutathione (GSH), xCT, DJ-1, and coenzyme Q10, indicating a strong correlation with ferroptosis [97]. Reduced GSH levels render neurons susceptible to oxidative stress and free radical damage. In PD, the downregulation of the xCT gene results in the downregulation of System Xc and subsequently decreases GSH levels. This process ultimately leads to increased oxidative stress, which contributes to the damage of dopaminergic neurons [98]. It is interesting to note that α-syn, a protein closely associated with PD, has been implicated in the regulation of both iron and lipid metabolism. This suggests a potential interaction between dysregulated α-syn and other PD-related pathological features related to ferroptosis [97]. In PD, elevated levels of iron have been particularly observed in glial cells and DAergic neurons in the SNpc, with the severity of the disease correlating to the iron levels [99]. This has been confirmed through iron-sensitive high-field magnetic resonance imaging (MRI) [100], quantitative susceptibility mapping (QSM) analyses, as well as postmortem tissue studies, and is supported by numerous animal models of PD [101]. The abnormal iron accumulation in PD is likely a result of imbalances in the iron homeostatic pathway, which are caused by alterations in iron regulatory proteins. Patients who carry mutations in these iron-related proteins, which are strongly associated with PD, show a higher risk of developing the disease, with connections to macroautophagy (SNCA, LRRRK2, DJ-1, VPS35), chaperone-mediated autophagy (CMA) (SNCA, LRRRK2, DJ-1, VPS35), and mitophagy (SNCA, PARKIN, PINK1, LRRK2, FBXO7, VPS35), and downstream lysosome dysfunction have an increased a risk of developing PD [102]. Indeed, there is growing evidence suggesting a significant association between ferroptosis and the onset and progression of neurodegenerative diseases. Moreover, various ferroptosis inhibitors, including iron chelators, free radical scavengers, and antioxidants, have shown promise in counteracting the pathological mechanisms involved in neurodegenerative diseases to some extent. Consequently, potential therapeutic targets are being investigated, particularly in the realms of iron metabolism, lipid metabolism, and amino acid metabolism, due to their implications in ferroptosis and its role in neurological disorders. These avenues hold promise for developing novel interventions against neurodegenerative diseases. The spread of the 2019 novel coronavirus disease (COVID-19) also accelerates the onset of PD symptoms and increases the activity and mortality rate of PD patients, thereby increasing the complexity of PD. Furthermore, there have been reports indicating that the combined occurrence of ferroptosis, cytokine release, neuroinflammation, and oxidative stress can have a detrimental impact on the prognosis of individuals with co-infections of COVID-19 [103]. This phenomenon presents new challenges in the development of potential therapeutic interventions for neurodegenerative diseases such as PD.
A significant cluster of genes linked to familial and lysosomal function has been identified [104]. In PD, disruptions in autophagy pathways can result in the buildup of proteins and the formation of inclusion bodies in the cytoplasm. Autophagy is thought to play a role in the breakdown of α-syn fibrils, involving both the autophagy-lysosome and ubiquitin-proteasome systems (UPS). Certain proteins associated with PD also contribute to autophagy. For instance, the presence of mutated LRRK2 in neuroblastoma cells leads to axonal shortening and the formation of autophagic vacuoles [105]. Additionally, PARKIN and PINK1, encoded by recessive genes linked to PD, can cause mitochondrial damage, and promote autophagy [106]. Modulating autophagy levels by activating autophagy or enhancing lysosomal activity can reduce the accumulation of α-syn and damaged organelles, thereby inhibiting neuronal degeneration and necrosis. These findings provide valuable insights for the development of potential therapeutic interventions for PD.
Apoptosis refers to a regulated process of cell death which is marked by distinct alterations in cellular morphology, such as cellular shrinkage, nuclear condensation, and DNA degradation. In PD, indications of apoptotic cell death have been documented in both neuronal and non-neuronal cells. Various studies, including postmortem examinations, animal models, and in vitro cultures, have detected DNA fragmentation and characteristic morphological changes in the brains of PD patients, particularly in the nigrostriatal region. These findings suggest the presence of apoptotic cell death and a proapoptotic environment in PD [107]. Oxidative stress, imbalance of Ca2+ homeostasis, mitochondrial damage, and regulation of proteins such as B-cell lymphoma-2 (Bcl-2), Bcl2-associated X (Bax), and caspase 3 are important processes in the apoptosis of DAergic neurons in the SN [108]. Oxidative damage, calcium homeostasis imbalance, and mitochondrial damage can be initiated individually or in combination during the process of DAergic neuron apoptosis in the SN and are a common pathway for many apoptosis-inducing factors. The three mechanisms are often interrelated and mutually causal. The intracellular regulation of apoptosis is an extremely complex process, in which ROS, Bcl-2, Bax, and caspase 3 all play important roles. The core link may be the inhibition of mitochondrial respiratory chain complex I by toxins, oxygen free radicals, and active ions in the internal and external environment, which reduces ATP and GSH in cells, disrupts Ca2+ balance, causes calcium overload, increases the production of free radicals in the respiratory chain, reduces the ratio of Bcl-2 and Bax proteins, and activates caspase 3 to jointly participate in the regulation of cell apoptosis, ultimately leading to the apoptosis of DAergic neurons [109].
The gut microbiota and PD are closely linked. Changes in the composition and abundance of gut microbes, known as dysbiosis, can impact both the enteric nervous system and the CNS. Research has shown that gut microbes can communicate with the CNS and enteric nervous system through various pathways, including metabolites, hormones, the immune system, and afferent nerves. This communication occurs via the “microbial-gut-brain axis” and contributes to the development and progression of PD [110]. Comparisons between PD patients and healthy individuals have revealed differences in the composition of intestinal flora. PD patients tend to have decreased levels of prevotellaceae and lachnosperaceae, while levels of verrucomicrobiaceae (such as akkermansia) and lactobacillaceae are increased. Additionally, markers of intestinal inflammation, namely α1-antitrypsin, and zonulin, are significantly elevated in PD patients. These findings suggest that the gut microbiota may influence the pathogenesis of PD through intestinal inflammation [111]. Furthermore, the activation of colonic mucosal immunity via the toll-like receptor 4 (TLR4) signaling pathway may trigger the spread of α-syn lesions from the gut to the brain along the gut-brain axis, leading to neuroinflammation and neurodegeneration in PD [112].
Association of PD with Other Diseases
The COVID-19 pandemic, caused by the outbreak of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has disrupted all aspects of life in an unprecedented way. The prospect of returning to normalcy feels distant. Extensive discussions have arisen regarding the potential neurological consequences of COVID-19 and its impact on patients with neurodegenerative disorders, such as PD. The virus can potentially affect the CNS through various mechanisms [113]. It can directly invade the brain by spreading through infected neurons, including the olfactory nerve, ocular epithelium, and neurovascular endothelium. Alternatively, it can enter the brain via infected white blood cells crossing the blood-brain barrier during circulation to highly vascularized brain tissues. PD patients are at a higher risk of contracting COVID-19 due to their age and multiple comorbidities. The disease can easily trigger a systemic inflammatory response [114,115]. Moreover, COVID-19 may worsen PD symptoms by interacting with the dopamine-dependent system in the brain, leading to respiratory and cardiac issues. This increases the vulnerability of PD patients to COVID-19 infection.
COVID-19 infections frequently exacerbate symptoms of PD [116]. Although the exact mechanism behind this remains uncertain, potential factors such as alterations in cerebral dopamine metabolism, changes in pharmacodynamics, and direct effects of endotoxins have been proposed [117]. Severe infections like COVID-19 directly impact the motor symptoms associated with PD. In a study, all eight patients infected with COVID-19 experienced a worsening of their Parkinson’s symptoms either before or after infection [118]. A community-based case-control study compared twelve PD patients with COVID-19 to 36 COVID-negative individuals matched for age, sex, and disease, and found that the COVID-19 group experienced a deterioration in levodopa-responsive motor symptoms and an increase in daily OFF-time [119]. Anxiety is the most common neurological and psychiatric complication in PD patients. Quantitative analysis of anxiety levels in Iranian PD patients and age-matched individuals from the general population during the pandemic revealed that 25.5% of cases and 4.8% of the control group experienced severe anxiety [120]. There exists a strong correlation between the severity of anxiety in individuals with PD and their apprehension towards contracting the COVID-19 virus. This correlation is notably higher compared to the control group. Recently, large-scale online studies, such as Fox Insight, have been utilized to evaluate the impact of the pandemic on PD patients. By analyzing data from 5429 PD patients (including 51 individuals who tested positive for SARS-CoV-2), it was discovered that among those infected, 18% reported experiencing new motor symptoms, while 55% reported a worsening of at least one pre-existing motor symptom [121]. Additionally, there were observed occurrences of new or intensified non-motor symptoms, including emotional (71%), cognitive (49%), sleep-related (62%), and autonomic dysfunction (38%). The evidence is therefore substantial in supporting the notion that SARS-CoV-2 directly and indirectly affects both the motor and non-motor symptoms of PD [122]. As a result, these indirect effects of COVID-19 may prove more detrimental to PD patients than the actual virus itself.
There is a hypothesis suggesting that infection with SARS-CoV-2 could contribute to the progression of neurodegeneration. Similar to PD, it is possible that during SARS-CoV-2 infection, there is an increase and buildup of α-syn, which could lead to widespread neurodegeneration. Moreover, α-syn activates TLR4, a receptor presents on microglia and astroglia, resulting in chronic neuroinflammation and proinflammatory activation, thereby further promoting the degeneration of dopamine-producing neurons in PD. The proteins of SARS-CoV-2 interact with host proteins involved in pathways related to aging, causing disruptions in mitochondrial function, autophagy, ER stress, proteostasis, and inflammation. This suggests that SARS-CoV-2 infection may exacerbate the abnormal folding and aggregation of proteins. Additionally, Wu et al. [123] proposed that SARS-CoV-2 RNA has a selective impact on mitochondria, impairing their function and leading to reduced energy production and increased generation of ROS. The entry of SARS-CoV-2 primarily occurs in lung cells through the binding of its spike protein to the angiotensin-converting enzyme 2 (ACE2). Downregulation of ACE2 expression further decreases DA levels in the blood of COVID-19-infected PD patients, resulting in motor symptoms such as tremors and coordination issues [124]. Research has shown that SARS-CoV-2 can directly damage nerve cells. First, high expression of ACE2 can be observed in the neurons of the cerebral cortex. SARS-CoV-2 can regulate ACE2 expression in cerebral cortical neurons to further control the biosynthesis of DA decarboxylase (DDC), thus affecting the progression of PD. DDC is an enzyme that regulates the synthesis of DA, serotonin, and histamine. Therefore, a decrease in serotonin secretion in cerebral blood vessels can be observed in certain ACE2 knockout mice [125]. The inflammatory response caused by SARS-CoV-2 is also considered one of the mechanisms by which COVID-19 exacerbates the symptoms of PD. Previous studies have found that systemic inflammatory responses may be one of the mechanisms that worsen PD symptoms [126], In COVID-19 patients, major immune indicators such as C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), and D-dimer are significantly increased [127]. α-syn is an important component of LBs formed in the brains of PD patients, and inflammation may promote the accumulation of α-syn in the brain [128]. Further accumulation of α-syn promotes the progression of PD and exacerbates existing symptoms. Other pathological mechanisms include oxidative stress, genetic susceptibility, and cellular homeostasis imbalance.
As a result, PD patients are more susceptible to COVID-19 infection and vice versa. Finding effective prevention and treatment methods for both PD and COVID-19 will present a greater challenge. Because PD patients are mostly middle-aged and elderly, and a majority of PD patients have multiple chronic illnesses, this undoubtedly increases their susceptibility to viruses, exacerbating the difficulties in late-stage treatment and prognosis recovery. Therefore, it is of great significance to enhance research on the interaction between COVID-19 and PD, as it relates to the prevention, treatment, and rehabilitation of PD patients with concurrent COVID-19 infection.
Diabetes mellitus (DM) is a common metabolic disease characterized by insulin resistance and impaired pancreatic β-cell function. At present, the incidence of DM is increasing rapidly worldwide [129]. With the change of high-sugar and high-energy diet in contemporary young people, it can be found that there are more and more patients with DM and PD, while the quality of life of Parkinson’s patients was often on the low side, especially after cognitive impairment in the later stage of the disease, patients have a higher risk of depression. Cognitive impairment is a common nonmotor symptom of PD, and as the disease progresses, cognitive abilities such as memory, attention, and executive function may decline. This decline in cognitive ability can affect the quality of life of patients and increase the risk of depression. Depression is a common neurological and psychiatric symptom of PD. Depression symptoms can worsen the motor function of PD patients, while motor symptoms and the life pressure of chronic disease patients can lead to the development of depression [130,131]. DM is a metabolic disorder associated with an increased risk of cognitive impairment and dementia. Chronic high blood sugar levels and insulin resistance can lead to brain inflammation, oxidative stress, and vascular damage, all of which are risk factors for cognitive decline. DM may increase the risk of PD [132].
Managing the blood glucose levels and metabolic health of PD and DM patients may be important for optimizing treatment outcomes and slowing disease progression. Therefore, understanding the potential relationship between DM and PD will be beneficial to clinical disease management. And it has instructive significance for individualized treatment. The pathophysiology of both DM and PD is regulated by genetic and environmental factors. Currently, several potential shared pathological mechanisms are well known, such as protein misfolding, insulin resistance, mitochondrial dysfunction, oxidative stress, chronic inflammation, vitamin D (VD) deficiency, microbial dysbiosis in the gut, and alterations in transcription factors associated with glucose metabolism and insulin regulation.
DM and PD involve the misfolding and deposition of amyloid-like proteins. In diabetes patients, pancreatic β-cells secrete islet amyloid polypeptide (IAPP) to regulate insulin secretion. Fibrous forms of IAPP deposit in the islets of diabetes patients, leading to pancreatic β-cell dysfunction [133]. In PD patients, misfolded α-syn deposits in the cytoplasm of DAergic neurons, form LBs, which are a hallmark pathological feature of PD [82]. Recent studies on IAPP and α-syn have found cross-reactivity between these two proteins, demonstrating that IAPP in type 2 diabetes mellitus (T2DM) can promote α-syn aggregation [134]. Phosphorylated aggregates of α-syn have been detected in pancreatic β-cells of the majority of patients with PD or T2DM. These α-syn deposits show co-localization and interaction with islet amyloid polypeptide (IAPP). Additionally, a separate study suggests that insulin-degrading enzyme (IDE) can impede the aggregation of α-syn by binding to α-syn oligomers. However, in T2DM patients with insulin resistance, IDE is competitively inhibited, resulting in the promotion of α-syn fibril formation. This process may increase vulnerability to PD or exacerbate the progression of the disease [135].
Insulin receptors are present in the basal ganglia [136] and SN [137], which are the brain regions primarily impacted in individuals with PD. Studies conducted using rodent models have demonstrated that insulin resistance can result in diminished expression of DA transporter in the striatum [138], reduced DA turnover [139], and impaired insulin-dependent release of DA in the striatum. Insulin exerts its effects by binding to insulin receptors, not only in the periphery but also in the CNS. Insulin resistance is a pathological state where cells are unable to perceive insulin and respond to it normally. A study in 1996 analyzed the mRNA levels of insulin receptors in the SN of PD brains and found a decrease in insulin receptor mRNA compared to the control group, which may be related to neuronal loss in the SN [140]. Morris et al. also found in their study on cerebral functional imaging in elderly individuals that patients with PD had increased insulin resistance in the cerebral region compared to normal individuals [141]. In individuals with T2DM, insulin resistance can potentially contribute to the creation of advanced glycation end products (AGEs) [136], which can also form in brain areas like the SN. AGEs interact with their receptor (RAGE), triggering downstream pathways that result in oxidative stress, inflammation, and neuronal cell death [142]. Remarkably, AGEs have been found to co-localize with α-syn in LBs) [143]. The glycation of α-syn can potentially worsen the progression of PD through various mechanisms.
Mitochondria play a key role in electron transport and oxidative phosphorylation. In T2DM, some studies have found that the production of ROS in muscle tissue of hyperglycemic mice was related to the damage of muscle mitochondria, while antioxidant therapy and control of blood glucose normalization can restore normal mitochondrial function [144]. When ER is subjected to oxidative stress, it will lead to misfolding or even dysfunction of proteins, and protein accumulation can enhance the oxidative stress of ER at the same time. PARKIN and PINK1 are proteins encoded by two recessive genes associated with PD, which can cause mitochondrial damage and promote autophagy, autophagy disorders lead to protein accumulation. Both ER and mitochondria are involved in lipid and glucose metabolism, and when these organelles are subjected to oxidative stress, they are likely to develop into DM [145,146]. When there is dysfunction in mitochondrial proteins, oxidative stress [147] and cell death [148] tend to increase. In rodent models, MPTP selectively blocks complex I, the initial enzyme in the mitochondrial respiratory chain pathway, which leads to neuronal cell death and neurodegenerative alterations resembling PD [149]. The features of mitochondrial dysfunction may be comparable in both T2DM and PD. Impaired insulin signaling in PD has been shown to increase oxidative stress [150], and a recent study revealed that chronic insulin resistance in diabetic db/db mice can cause mitochondrial damage and degeneration of DAergic neurons [151]. While the precise mechanisms linking mitochondrial dysfunction, oxidative stress, and PD remain uncertain, their involvement is likely significant in the development of PD and may have connections to T2DM [152].
In the CNS, microglial cells are the major cells involved in immune regulation and inflammation. Under various neuroinflammatory stimuli, resting microglial cells can be activated and differentiated into two phenotypes: pro-inflammatory M1 and anti-inflammatory M2. Animal experiments have suggested that in early PD animal models, both M1 and M2 phenotypes coexist, but as the disease progresses, the M1 phenotype starts to dominate [153]. Studies have also indicated that PD patients exhibit more severe imaging manifestations of cranial inflammation compared to healthy individuals [154]. In addition to imaging findings, Liu et al. found that some inflammatory markers, such as IL-2, IL-6, and TNF-α, are significantly elevated in the body fluids of PD patients compared to normal individuals [155]. In DM patients, insulin resistance can lead to the accumulation of glycated end products, which can activate downstream inflammatory reactions by binding to their receptors and also aggregate in the α-syn adjacent to synaptic nuclei, affecting their aggregation and toxicity [143]. Insulin resistance impacts the activation of microglial cells and the generation of neuroinflammation via the NF-κB and phosphoinositide 3-kinase (PI3K)/protein kinase B (PKB or AKT) pathways. The PI3K/AKT pathway regulates microglial cell activation and the expression of inflammatory mediators [156]. Elevated levels of proinflammatory cytokines, such as IL-1β, IL-6, and TNF-α, have been detected in the brain, cerebrospinal fluid, and blood of PD patients. Inflammation also contributes to the development of type 2 diabetes mellitus (T2DM). The levels of immune cells, cytokines, and chemokines are increased in the islets of T2DM patients [157]. Islet inflammation is considered a potential factor responsible for impaired insulin secretion and reduced β-cell function in diabetic patients.
VD deficiency was associated with T2DM, insulin resistance, and metabolic syndrome [158], while some VD receptor polymorphisms are associated with PD. It was found that there was a difference in the expression of this receptor in blood cells of patients with PD compared with the control group and VD intake improves motor symptoms associated with PD. Several possible therapeutic mechanisms have been proposed, in addition to immunomodulatory effects in the brain [159], VD may regulate the expression of GDNF, which can improve motor symptoms in patients with PD. Some metabolic pathways are regulated by regulating transcription factors. For example, recombinant peroxisome proliferator-activated receptor gamma (PPAR-γ) and its coactivator-1α (PGC-1α) regulate gluconeogenesis gene expression and oxidative metabolism [160]. Interestingly, PGC-1α was associated with neuroprotection in several neurodegenerative diseases, including Huntington’s chorea (HD) and PD [161]. In PD, elevated PGC1-α levels protect cultured nerve cells from oxidative stress. A study found that in animals with a high-fat diet, PPAR-γ signals in the brain regulate food intake, energy consumption, and liver insulin activity, suggesting that brain PPAR-γ may regulate liver insulin sensitivity directly through brain-liver axis-mediated pathways [162].
In general, these studies have shown that there was a common pathway between DM and PD, but the factors that mediate metabolic syndrome and neurodegeneration are both complex and similar. it was difficult to determine which factors are the main determinants of neurodegeneration.
Cancer and neurodegenerative disorders collectively impact approximately 41.3 million individuals globally and pose significant health challenges [163,164]. These two groups of diseases appear to be associated with contrasting cellular fate phenotypes, which contribute to their pathological progression. While cancer is characterized by heightened cellular proliferation and resistance to apoptosis, neurodegeneration entails the gradual demise of neuronal cells. PD and cancer share several common risk factors, including aging, DNA damage caused by oxidative stress, metabolic dysregulation, and exposure to environmental chemicals [165]. In biological research, certain interesting phenomena have been discovered. Cancer is a disease characterized by uncontrolled cell proliferation, while the characteristic of PD is abnormal cell apoptosis [166,167] Research suggests that there may be two main connections between neurodegenerative diseases and cancer. On one hand, they share some genes or biological pathways such as the UPS and P53, which are abnormally regulated in different directions, leading to different diseases. On the other hand, they have similar pathological and physiological abnormal states, but they develop into different outcomes, including mitochondrial dysfunction, oxidative stress, and DNA damage [167]. Through these shared genes, biological pathways, and abnormal pathological and physiological states, it is speculated that some biological factors in PD patients may have a protective effect against cancer [166]. PD and overall cancer demonstrated an inverse relationship, particularly with colorectal cancer, lung cancer, prostate cancer, and bladder cancer. This inverse association was held for both cancers related to smoking and those unrelated to smoking. Additionally, PD showed a positive association with melanoma [168]. The dysregulation of glycosphingolipid metabolism, storage, and interaction with α-syn, along with mutations in the phospholipase A2, group VI (PLA2G6) gene that controls the synthesis of the core component ceramide in glycosphingolipids, are linked to both melanoma and PD [169]. Some genes associated with familial PD, such as PARKIN and ubiquitin C-terminal hydrolase L1 (UCHL1), are typically involved in cellular processing and clearance in nerve cells, but they also regulate the cell division cycle [170]. The UPS is the primary pathway for protein degradation in cells, responsible for degrading more than 80% of proteins in the cell. Various cellular physiological processes require the involvement of protein ubiquitination, such as protein transport, organelle generation, ER protein regulation, cell cycle processes, cell proliferation and differentiation, and cell apoptosis [171]. Research suggests that UPS plays a crucial role in PD and cancer, as dysfunction of UPS leads to protein accumulation and the formation of LBs in PD [171]. In contrast, in cancer, UPS function is usually upregulated [172].
A substantial body of evidence suggests shared genetic mechanisms between cancer and neurodegenerative diseases. Mutations in various genes that disrupt cell cycle regulation and protein turnover have been identified in both PD and cancer. Mitophagy, the process of eliminating damaged mitochondria, plays a crucial role in preserving neuronal function, regulating neuronal viability, and potentially reversing neurodegeneration. Mitochondrial dysfunction and autophagy are critical in the pathogenesis of PD, but their role in cancer is still unclear. Disruption of complex I and mitochondrial bioenergetics effectively prevents the proliferative capacity of colon cancer epithelial cells [173]. Mitochondrial metabolism and mitochondrial ROS production are critical for cancer cell proliferation and tumorigenesis [174]. PD and cancer share common pathways associated with mutations in mitochondrial proteins. An increase in DNA damage promotes cells to enter the cell division cycle, similar to what happens in tumor formation and neurodegenerative diseases. The “second-hit” theory in neurodegenerative diseases proposes that oxidative stress and mitotic abnormalities can drive neurons to enter the cell division cycle [175,176]. However, cancer cells can escape cell cycle checkpoints and undergo uncontrolled proliferation, while neurons can undergo mitosis but cannot complete it [177].
There is a growing body of evidence supporting the therapeutic targeting of mitochondria to induce neuroprotective and antitumor effects in PD and cancer. Mitochondrial-targeted drugs have been shown to enhance the production of ROS within mitochondria by inhibiting mitochondrial complexes in cancer cells, thereby activating anti-proliferative REDOX signaling mechanisms. The inhibition of OXPHOS through mitochondrial targeting drugs has been well-established as a key mechanism for antitumorigenesis in cancer. However, the ability of mitochondrial-targeted drugs to provide neuroprotection in PD, where complex I is already compromised, may seem paradoxical. Thus, the neuroprotective potential of mitochondrial targeting depends on the timing of administration and the progression of the disease. Fig. 3 and Table 4 show the expression of PD-related genes in cancer (TIMER database: http://timer.comp-genomics.org/timer/) and their biological functions in different diseases. We found that PD-related genes are low expressed in some tumors, which may be related to genetic mutations, epigenetic regulation, intracellular oxidative stress, mitochondrial dysfunction, and inflammatory response, all of which may lead to a decrease in the expression of these genes.
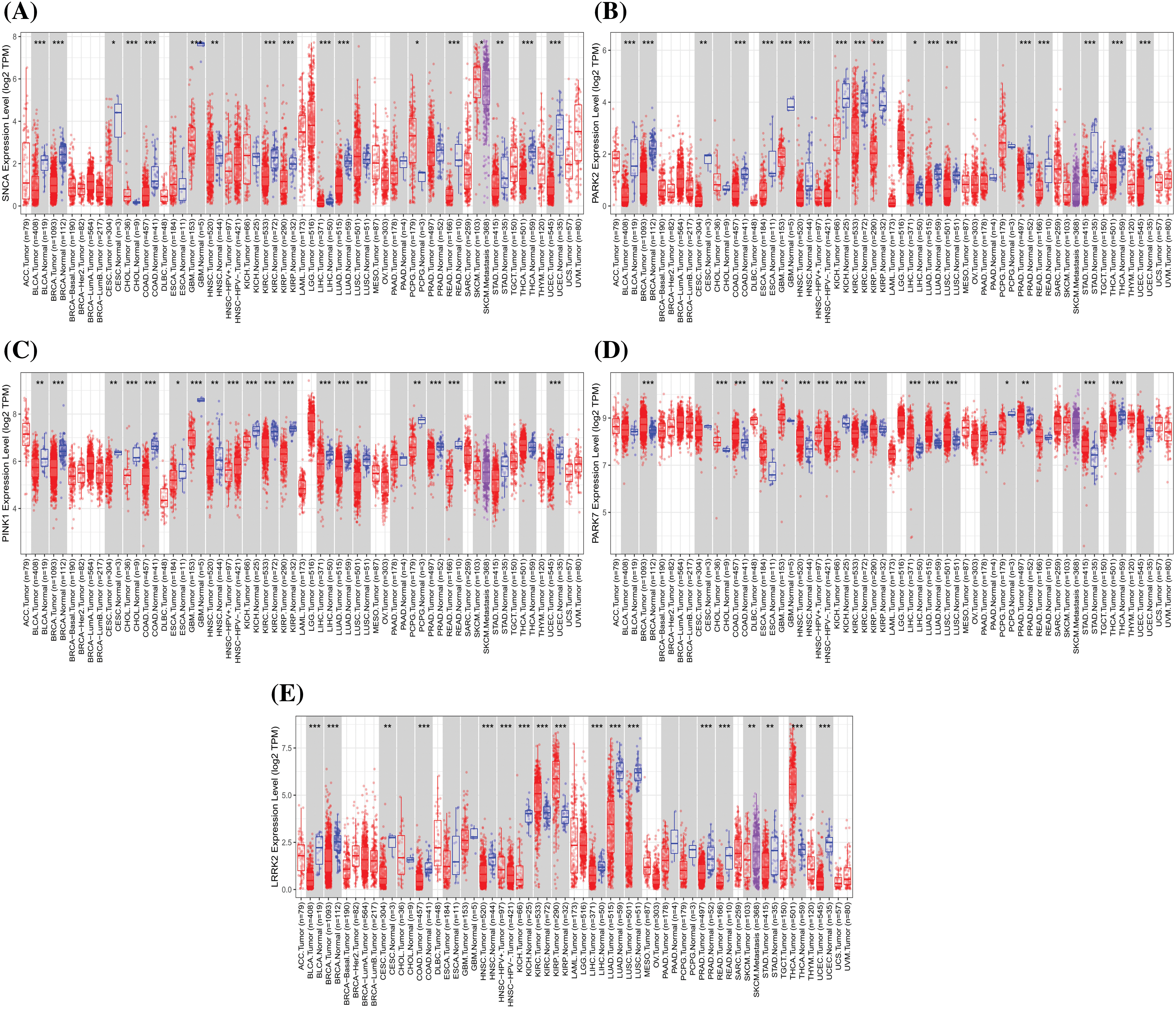
Figure 3: The expression of Parkinson’s disease-related genes in cancer, including SNCA, PARK2, PINK1, PARK7, and LRRK2 (Red represents the tumor group, and blue represents the normal group. Compared with the normal group, *p < 0.05, **p < 0.01, ***p < 0.001).

Ischemic heart disease (IHD) is a leading cause of death and disability worldwide. The negative impacts of acute ischemia/reperfusion injury (IRI) on the heart contribute to the major clinical symptoms of IHD [189]. As a result, there is a need for novel therapeutic targets to protect the heart against IRI, limit the death of cardiomyocytes, preserve myocardial function, and prevent the development of heart failure. An interesting group of mitochondrial proteins associated with familial forms of PD may offer new targets for cardioprotection. The proteins PARKIN, PINK1, DJ-1, LRRK2, and α-syn, are crucial in preventing cell death in DAergic neurons of the SN, as well as being present in the heart. These proteins have various functions, including maintaining normal mitochondrial function, protecting against oxidative stress, mediating mitophagy, and preventing apoptosis [190]. Table 4 provides further details regarding the role of these PD proteins in the heart.
Therapeutic Drugs and Technologies & PD
Conventional pharmacological treatments for PD include DA precursors such as levodopa and l-DOPA (l-3,4 dihydroxyphenylalanine), as well as other symptomatic treatments such as DA agonists (amantadine, apomorphine, bromocriptine, cabergoline, lisuride, pergolide, pramipexole, ropinirole, rotigotine), MAO inhibitors (selegiline, rasagiline), catechol-O-methyltransferase (COMT) inhibitors (entacapone, tolcapone), anticholinergic drugs (phenanthrol), and amantadine [188]. Several classic and novel natural products, such as Vicia faba, Mucuna pruriens, Siegesbeckia pubescens, Sophora flavescens, Carthamus tinctorius, Curcuma longa, Huperzia selago, Diphasiastrum complanatum, Erythrina velutina, Gynostemma pentaphyllum, Echinacoside, Centella Asiatica, Sulforaphane, Oleuropein, flavonoids, Ginsenosides, cyanidin-3-O-glucoside, caffeine, Resveratrol, and other polyphenols, have demonstrated potential efficacy in PD. They selectively protect DAergic neurons from cell death and provide additional benefits such as antioxidant, anti-inflammatory, and neurotrophic effects [191,192]. However, potential neuroprotective drugs such as ubiquinone, creatine, and PYM50028 have not shown any clinical benefits in recent highly anticipated clinical trials [193]. It was exciting to observe the progression of four trials into Phase 3, which included three disease-modifying therapies (DMT) aiming to slow down the progression of PD by targeting the underlying biology of the disease. These trials involved Lactobacillus, BIIB122, and buntanetap. Inflammation is considered to be a crucial factor in the progression of PD, and there are three ongoing trials in this area, all in Phase 1. These trials involve Biovie’s NE-3107 and two trials on sargramostim (leukine) being conducted at the University of Nebraska. It should be noted that some agents categorized under different classes also possess secondary anti-inflammatory properties, such as glucagon-like peptide-1 (GLP-1) receptor agonists. Additionally, there is an ongoing Phase 2 trial at Cambridge University investigating the anti-inflammatory azathioprine [194]. The pharmaceutical company Roche is also testing a novel anti-inflammatory agent, an NOD-like receptor thermal protein domain associated protein 3 (NLRP3) inflammasome inhibitor called selnoflas [195].
The immunotherapy category, previously used for trials involving antibodies to α-syn, has been reclassified as the “targeting α-syn” category, to focus on projects that primarily aim at alpha-synuclein as a target. Within this category, there are five trials for antibody-based projects, with three different agents involved: prasinezumab (Roche), UB-312 (Vaxxinity), and UCB7853 (UCB). Additionally, there is a Phase 3 trial at Wayne State University using memantine to prevent the transmission of α-syn between cells. Other ongoing trials are focused on preventing the aggregation of α-syn using small molecule agents, such as Anle 138b from Modag. A notable highlight in this category is the Phase 3 study of buntanetap by Annovis Bio, which reduces the production of α-syn and consequently mitigates aggregation [196]. While a significant amount of attention has been given to directly targeting the pathology of α-syn, there is also some focus, albeit to a lesser extent, on drugs that target GBA and LRRK2-associated PD. One notable example is the development of BIIB122, an inhibitor of LRRK2, by Biogen and Denali, which is currently undergoing Phase 3 testing. In terms of trials targeting the microbiome/gastrointestinal tract (GIT), there is a focus on both symptomatic treatments (ST-improving or reducing symptoms) and DMT, including the testing of an antibiotic (rifaximin), fecal microbiota transfer, and probiotics. Additionally, there is a study investigating the enhancement of brain small-chain fatty acids through tributyrin supplementation, which mimics the natural production of small-chain fatty acids in the gut [197]. GLP-1 receptor agonists have also garnered increasing attention in the PD clinical pipeline. After showing promising results in early trials [198], multiple clinical trials are now exploring the potential of GLP-1 receptor agonists. This includes repurposing exenatide (currently undergoing Phase 3 trials in the UK) and lixisenatide, as well as research efforts by Neuraly (NLY-01) and Peptron (PT-320). However, the slow progress and limited number of agents advancing from Phase 2 to Phase 3 is a cause for concern. Nonetheless, various stakeholders are making collective efforts to speed up the clinical trial process and facilitate the development of new therapies for the PD community.
Deep brain stimulation (DBS) is a medical technique used to treat nervous system diseases. It involves the implantation of electrodes in specific nerve nuclei within the brain, using a stereotaxic instrument [199]. By applying low or high-frequency electrical stimulation, abnormal electrical activity in neurons can be inhibited, leading to the treatment of various diseases. In the case of patients with PD, the target nuclei for DBS are typically located in the basal ganglia or thalamus. DBS of the subthalamic nucleus (STN), globus pallidus internus (GPi), and ventral intermediate nucleus of the thalamus (VIM) is a distinct symptomatic treatment for PD [200]. This particular approach can enhance DA metabolism in the striatum, resulting in improved motor symptoms related to PD. However, it should be noted that VIM-DBS primarily addresses tremor symptoms and does not provide therapeutic benefits for symptoms like bradykinesia and rigidity. Hence, VIM-DBS is only suitable for PD patients experiencing tremor symptoms, and it cannot be utilized to address other therapeutic targets [201]. Conversely, DBS with STN and GPi is an effective option for improving motor symptoms and controlling long-term complications caused by levodopa therapy, such as end-dose phenomenon and dyskinesia.
PD gene therapy has the potential to revolutionize treatment methods. Currently, there are various approaches such as nutritional factor gene therapy, targeting cerebral DA neurotrophic factor (CDNF), neurturin (NRTN), mesencephalic astrocyte-derived neural factor (MNAF), GNDF, and BDNF. Additionally, there is gene replacement therapy focused on crucial genes related to DA synthesis or key enzymes in the disease metabolic pathway, namely tyrosine hydroxylase (TH), aromatic acid decarboxylase (AADC), guanosine triphosphate cyclohydrolase-1 (GCH1), and glutamic acid decarboxylase (GAD). Furthermore, other genes like glucosylceramidase beta 1 (GBA1), PARKIN, LRRK2, SNCA, PINK1, DJ-1, VPS35, DnaJ heat shock protein family member C13 (DNAJC13), coiled-coil-helix-coiled-coil-helix domain containing 2 (CHCHD2), SLC6A, neuropeptide Y (NPY), and neuron restrictive silencing factor (NRSF) are also being investigated for their potential role in PD gene therapy [202].
Gene-editing technology aims to treat diseases by targeting specific populations of dysfunctional or improved cells. CRISPR-Cas9, a type of genome editing technology, has already reached the clinical trial stage for many diseases caused by a single gene mutation. In the case of PD, editing or regulating the expression of key genes known to be altered in PD, such as GDNF, PINK1, PARKIN, SNCA, or AADC, can help correct dysfunction in related biological pathways [174]. Stem cell-based therapies, including fetal mesencephalic tissue, mesenchymal stem cells (MSCs), and pluripotent stem cells (hPSC), such as embryonic stem cells (ESC) and induced pluripotent stem cells (iPSC), are also being explored [203]. Genome editing holds promise as a new treatment option for both familial and idiopathic PD.
Another emerging technology called optogenetics allows precise control of neural activity in specific cell populations by using light-sensitive ion channels (opsins) or G-protein-coupled receptors. Studies have shown that optogenetic treatment can improve the motor behavior of mice treated with 6-OHDA, a neurotoxin commonly used to model PD [204–206]. As an evolving field, optogenetics may offer viable treatment options for PD.
Designer receptors exclusively activated by designer drugs (DREADDs) are a modified chemical genetics platform based on G-protein-coupled receptors (GPCR). By modifying different GPCRs, synthetic proteins can be transmitted. These modified receptors can only be activated or inhibited by specific synthetic compounds, which then activate the corresponding GPCR signaling pathway, resulting in different changes in cell excitability. Research has shown that temporarily activating cholinergic neurons in the pedunculopontine tegmental nucleus (PPTg) using DREADDs in a rat model of PD, through the peripheral administration of the DREADDs ligand, clozapine-N-oxide (CNO), significantly improves motor symptoms [207]. DREADDs have been extensively used to map neuronal circuits in the CNS, providing control over specific neurons in terms of when and where they are activated or inhibited. This technology is emerging as a promising treatment for PD.
Acupuncture is a cost-effective therapy originating from traditional Chinese medicine in China. It utilizes various techniques to promote health and treat diseases. Multiple clinical trials have shown that acupuncture provides substantial therapeutic benefits for patients with PD, effectively reducing both motor and nonmotor symptoms. Furthermore, acupuncture may help decrease the dosage and frequency of anti-PD medications and mitigate their side effects [208]. It can also reverse the degeneration of dopamine-producing neurons, slow down the progression of PD, and address various underlying factors such as mitochondrial dysfunction, oxidative stress, protein aggregation, impaired autophagy, and neuroinflammation [209].
Nanomedicine refers to the use of nanosized particles in the field of health and medicine. It is an innovative approach that aims to treat diseases with greater accuracy and effectiveness. The main goal is to overcome the limitations of conventional drugs, such as instability, potential immunogenicity, poor solubility, and short plasma half-life [210]. There are some approved treatment methods available for managing PD symptoms, such as transdermal patches. For example, the transdermal patch containing rotigotine (Neupro) is used to relieve restless leg syndrome symptoms associated with PD, while selegiline (Emsam) is prescribed for depression. However, these treatments may also have certain side effects [211]. It is worth noting that lipid nanoparticles (LNPs) are not used in these formulations. One example of LNPs in therapeutics is the intravenous therapeutic patisiran (ONPATTRO), which has been approved for the management of polyneuropathy. LNPs are employed here to deliver a therapeutic short-interfering RNA (siRNA) [212]. However, a recent search of the National Institutes of Health (NIH) library for clinical trials conducted in the past decade showed that LNPs have not yet been extensively utilized as nanocarriers for treating PD. There is currently one Phase 1 study using liposomes, which began in 2021 and is expected to be finished by December 2022. This study aims to assess the safety of Talineuren, a therapeutic that combines GM1 (a monosialotetrahexosylganglioside), with a proprietary liposomal formulation [213]. In contrast, a study utilizing gold nanocrystals has already concluded [214]. Although the results have not been announced yet, they could potentially open up new possibilities for nano-based therapeutics and represent a significant milestone in the field of nanomedicine. However, further research is necessary to investigate the potential of LNPs in developing gene or drug-delivery systems for PD.
Due to their ability to replicate and give rise to various cell types, stem cells have garnered significant interest as a potential solution for repairing damaged tissues. In the context of neurocognitive disorders characterized by dementia, such as AD and PD, stem cell therapy is increasingly being investigated as a potential treatment option. This innovative approach seeks to replace degenerated DAergic neurons with new ones or provide a new cell source capable of developing into DAergic neurons. Different types of stem cells, including induced pluripotent stem cells (iPSCs), mesenchymal stem cells (MSCs), neural stem cells (NSCs), and embryonic stem cells (ESCs), are being explored for transplantation therapies. Recently, disease-modifying strategies have been suggested, such as combining cell replacement therapies with other therapeutic approaches like using natural compounds or biomaterials, to modify the underlying neurodegenerative process [215].
Numerous research studies utilizing various genetic and toxin models of PD have contributed to a better understanding of the disease’s development. This review highlights that exposure to pesticides and familial gene mutations can lead to mitochondrial dysfunction, which is linked to PD in humans and animals. Furthermore, lifestyle factors such as hyperuricemia, as well as tobacco and coffee use, have been found to reduce the risk of PD, possibly due to their impact on the release of DA. These studies consistently indicate that mitochondrial dysfunction is a common factor in both sporadic and monogenic PD, suggesting its significant role in the disease’s pathophysiology. Additionally, complex I deficiency, mtDNA mutations, and oxidative stress are also involved in the development of PD.
In PD patients and animal models, biochemical abnormalities in mitochondria, including complex I damage, decreased ATP synthesis, increased ROS production, mtDNA mutations, and impaired mitochondrial repair pathways, have been observed. Inflammation is another aspect closely associated with PD, with research indicating the involvement of microglia in inflammatory reactions, and their overactivation being a pathophysiological basis for PD. Protein misfolding, specifically α-syn protein, contributes to the formation of LBs and exacerbates PD progression. The loss of neurofilaments (NFs) is an important factor in the decline of brain function and neurodegenerative diseases [216]. NFs improve the nutrition supply to the nervous system, promote neuronal survival, regeneration, and differentiation, alleviate neurodegenerative changes, enhance neuroprotection, improve PD pathology, and reduce the risk of PD [217]. Among the various cell death pathways, including ferroptosis, autophagy, and apoptosis, lipid peroxidation, and protein aggregation play crucial roles. Recent studies have also revealed dysregulation of intestinal flora metabolites in PD patients [111]. The intestinal flora converts into various metabolites and neurotransmitters, such as short-chain fatty acids (SCFA), 5-hydroxytryptamine, nicotinamide, and bile acids, which reach the intestinal submucosa, enter the intestinal-liver circulation, pulmonary circulation, and systemic circulation, ultimately affecting the brain [218]. SCFA or lipopolysaccharides (LPS), which are produced by bacteria in the intestine, can affect the permeability of the intestinal walls and the blood-brain barrier (BBB). When the BBB is damaged, metabolites from the intestinal flora can enter the brain and cause neuroinflammation [219,220].
Patients with PD are at a higher risk of developing COVID-19, DM, melanoma, and IHD. This increased risk is likely due to inflammation, damage to mitochondrial function, oxidative stress, and certain genes such as PARKIN, PINK1, DJ-1, LRRK2, and α-syn [221–224]. Currently, the main treatment for PD involves the use of DAergic drugs, such as levodopa. These drugs supplement the levels of DA in the brain, which helps alleviate symptoms like tremors and muscle stiffness. However, DAergic drugs only provide symptomatic relief and cannot slow down the progression of PD. Additionally, their usage is limited by potential side effects. The research and development strategies for PD therapeutic drugs focus on improving existing DAergic drugs, developing new drugs that target newly discovered molecular targets, and exploring the potential therapeutic uses of existing medications. One area of focus is the investigation of new molecular targets, including nicotinamide adenine dinucleotide phosphate (NADPH) oxidase [225], nuclear receptor-related 1 (Nurr1) [226], A2A [227], nicotine receptors, metabolic glutamate receptors (mGluRs) [228], and glucocerebrosidase (GCase) [229]. These targets provide direction for future research in PD therapeutics. Various therapeutic technologies, such as gene therapy, gene editing, optogenetics, chemogenetics, and physical stimulation therapy, are being explored, but they still have certain limitations.
Nanotechnology presents unique advantages in addressing these issues. Nanomaterials play dual roles as carriers for drug and gene delivery, exhibiting low biological toxicity, controlled drug release, and high drug capacity. They enhance drug stability and pharmacological properties through embedding or surface modification, with certain nanomaterials even displaying anti-inflammatory, anti-oxidant, and neuroprotective effects [230]. Nanotechnology holds great promise in supporting PD stem cell therapy and regulating neural activity. Its application in diagnosing and treating degenerative diseases of the CNS, including PD, is generating considerable interest [231,232]. The pathogenesis of PD is multifaceted, and it will still be a long-term challenge in future clinical treatment.
Pharmacological treatments for PD encompass medications that alleviate both motor and non-motor symptoms. The first type consists of drugs that elevate dopamine levels in the brain, such as levodopa. The second type comprises medications that target other neurotransmitters in the body to alleviate overall symptoms of the disease. For instance, anticholinergic drugs interfere with the creation or absorption of acetylcholine and effectively reduce tremors. The third type includes medications that manage the non-motor symptoms of PD. For instance, PD patients experiencing depression may be prescribed antidepressants [233]. Up to now, different animal models that can be used for PD research have focused on drugs, neurotoxins, pesticides, genetic alterations, α-syn inoculation, viral vector injections, and induced pluripotent stem cells [234,235]. The animal models have demonstrated variable drug responses, which can hinder the reliable evaluation of drug efficacy. This highlights the importance of developing personalized pharmacological approaches to effectively enhance the quality of life for patients with PD [236]. In clinical trials, despite efforts to establish strict eligibility criteria, study participants naturally display more variability compared to preclinical (animal) models, which typically use nearly identical individuals within the animal group. In human studies, apart from the inherent clinical diversity caused by PD itself, there is additional variability within the participant group due to factors unrelated to PD, such as age, genetics, environmental exposures, and other medical conditions. Generally, these various aspects among study subjects are not fully taken into account in human clinical trials, even though most trials exclude independent neurological comorbidities. The impact of this variability on the response of patients to disease-modifying interventions, even when they share a common target relevant to the intervention, remains largely unknown. These considerations are often overlooked when developing preclinical models. Consequently, current preclinical models of PD are likely to fall short in accurately representing the significant aspect of sporadic PD, which is the heterogeneity of the patient population [237]. Studying the potential therapeutic agents to halt or slow down the degenerative process induced by single-neuron neurodegeneration is feasible. However, implementing this model of neurodegeneration in animals is not possible, as it is impossible to inject a single DAergic neuron containing neuromelanin [238]. Unfortunately, there is currently no ideal animal model for PD research. Developing a model that fully replicates the features of human PD, which typically take years to manifest, is a challenging task. Despite its limitations, current animal models do offer a valuable platform to selectively investigate the pathophysiology and the interactions of multiple etiological factors involved in PD, aiding our understanding of disease limitations and treatment options.
Acknowledgement: None.
Funding Statement: This work was supported partly by the National Natural Science Foundation of China (32161143021, 81271410), Henan University Graduate «Talent Program» of Henan Province (SYLYC2023092), and Henan Natural Science Foundation of China (182300410313).
Author Contributions: TL: Conceptualization; DG: Methodology; JW: Data curation; TL: Formal analysis; TL, JW: Funding acquisition; JW: Project administration; TL: Writing-original draft; TL, DG: Writing-review & editing. All authors reviewed the manuscript.
Availability of Data and Materials: All data and material generated or analyzed during this study are included in this published article.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that there is no conflict of interest regarding the publication of this paper.
References
1. Rai SN, Singh P. Advancement in the modelling and therapeutics of Parkinson’s disease. J Chem Neuroanat. 2020;104:101752. doi:10.1016/j.jchemneu.2020.101752. [Google Scholar] [PubMed] [CrossRef]
2. Meléndez-Flores JD, Irabien-Zuñiga M, Cerda-Contreras C, de Los Reyes-Calderón I, Estrada-Bellmann I. Parkinson’s disease phenotype based on age at onset in Latin American patients: a paired-based analysis. Rev Neurol. 2022;74(9):298–302. doi:10.33588/rn.7409.2021518. [Google Scholar] [PubMed] [CrossRef]
3. Almansoub HAMM, Tang H, Wu Y, Wang DQ, Mahaman YAR, Salissou MTM, et al. Oxytocin alleviates MPTP-induced neurotoxicity in mice by targeting MicroRNA-26a/death-associated protein kinase 1 pathway. J Alzheimers Dis. 2020;74(3):883–901. doi:10.3233/JAD-191091. [Google Scholar] [PubMed] [CrossRef]
4. GBD 2016 Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018;17(11):939–53. doi:10.1016/S1474-4422(18)30295-3. [Google Scholar] [PubMed] [CrossRef]
5. Rossi A, Berger K, Chen H, Leslie D, Mailman RB, Huang X. Projection of the prevalence of Parkinson’s disease in the coming decades: revisited. Mov Disord. 2018;33(1):156–9. doi:10.1002/mds.27063. [Google Scholar] [PubMed] [CrossRef]
6. Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007;68(5):384–6. doi:10.1212/01.wnl.0000247740.47667.03. [Google Scholar] [PubMed] [CrossRef]
7. Dorsey ER, Sherer T, Okun MS, Bloem BR. The emerging evidence of the Parkinson pandemic. J Parkinsons Dis. 2018;8(s1):S3–8. doi:10.3233/JPD-181474. [Google Scholar] [PubMed] [CrossRef]
8. Aarsland D, Batzu L, Halliday GM, Geurtsen GJ, Ballard C, Ray Chaudhuri K, et al. Parkinson disease-associated cognitive impairment. Nat Rev Dis Primers. 2021;7(1):47. doi:10.1038/s41572-021-00280-3. [Google Scholar] [PubMed] [CrossRef]
9. Zeng XS, Geng WS, Jia JJ. Neurotoxin-induced animal models of Parkinson disease: pathogenic mechanism and assessment. ASN Neuro. 2018;10:1759091418777438. doi:10.1177/1759091418777438. [Google Scholar] [PubMed] [CrossRef]
10. Elsworth JD. Parkinson’s disease treatment: past, present, and future. J Neural Transm. 2020;127(5):785–91. doi:10.1007/s00702-020-02167-1. [Google Scholar] [PubMed] [CrossRef]
11. Ali R, Alam AA, Rajput S, Ahmad RR. Pharmacotherapeutics and molecular docking studies of alpha-synuclein modulators as promising therapeutics for Parkinson’s disease. BIOCELL. 2022;46(12):2681–94. doi:10.32604/biocell.2022.021224. [Google Scholar] [CrossRef]
12. Zhang K, Zhu S, Li J, Jiang T, Feng L, Pei J, et al. Targeting autophagy using small-molecule compounds to improve potential therapy of Parkinson’s disease. Acta Pharm Sin B. 2021;11(10):3015–34. doi:10.1016/j.apsb.2021.02.016. [Google Scholar] [PubMed] [CrossRef]
13. Kline EM, Houser MC, Herrick MK, Seibler P, Klein C, West A, et al. Genetic and environmental factors in Parkinson’s disease converge on immune function and inflammation. Mov Disord. 2021;36(1):25–36. doi:10.1002/mds.28411. [Google Scholar] [PubMed] [CrossRef]
14. Fujimaki T, Saiki S, Tashiro E, Yamada D, Kitagawa M, Hattori N, et al. Identification of licopyranocoumarin and glycyrurol from herbal medicines as neuroprotective compounds for Parkinson’s disease. PLoS One. 2014;9(6):e100395. doi:10.1371/journal.pone.0100395. [Google Scholar] [PubMed] [CrossRef]
15. Gonzalez-Latapi P, Bayram E, Litvan I, Marras C. Cognitive impairment in Parkinson’s Disease: epidemiology, clinical profile, protective and risk factors. Behav Sci. 2021;11(5):74. doi:10.3390/bs11050074. [Google Scholar] [PubMed] [CrossRef]
16. Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, et al. Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect. 2011;119(6):866–72. doi:10.1289/ehp.1002839. [Google Scholar] [PubMed] [CrossRef]
17. Harrigan J, Brambila DF, Meera P, Krantz DE, Schweizer FE. The environmental toxicant ziram enhances neurotransmitter release and increases neuronal excitability via the EAG family of potassium channels. Neurobiol Dis. 2020;143:104977. doi:10.1016/j.nbd.2020.104977. [Google Scholar] [PubMed] [CrossRef]
18. Bloem BR, Okun MS, Klein C. Parkinson’s disease. Lancet. 2021;397(10291):2284–303. doi:10.1016/S0140-6736(21)00218-X. [Google Scholar] [PubMed] [CrossRef]
19. Marras C, Canning CG, Goldman SM. Environment, lifestyle, and Parkinson’s disease: implications for prevention in the next decade. Mov Disord. 2019;34(6):801–11. doi:10.1002/mds.27720. [Google Scholar] [PubMed] [CrossRef]
20. Nielsen SS, Warden MN, Sallmén M, Sainio M, Uuksulainen S, Checkoway H, et al. Solvent exposed occupations and risk of Parkinson disease in Finland. Clin Park Relat Disord. 2021;4:100092. doi:10.1016/j.prdoa.2021.100092. [Google Scholar] [PubMed] [CrossRef]
21. Dorsey ER, De Miranda BR, Horsager J, Borghammer P. The body, the brain, the environment, and Parkinson’s disease. J Parkinsons Dis. 2024;14(3):363–81. doi:10.3233/JPD-240019. [Google Scholar] [PubMed] [CrossRef]
22. Mustapha M, Mat Taib CN. MPTP-induced mouse model of Parkinson’s disease: a promising direction of therapeutic strategies. Bosn J Basic Med Sci. 2021;21(4):422–33. doi:10.17305/bjbms.2020.5181. [Google Scholar] [PubMed] [CrossRef]
23. Kochmanski J, VanOeveren SE, Patterson JR, Bernstein AI. Developmental dieldrin exposure alters DNA methylation at genes related to dopaminergic neuron development and Parkinson’s Disease in mouse midbrain. Toxicol Sci. 2019;169(2):593–607. doi:10.1093/toxsci/kfz069. [Google Scholar] [PubMed] [CrossRef]
24. Yoon SY, Park YH, Lee HJ, Kang DR, Kim YW. Lifestyle factors and Parkinson disease risk: korean nationwide cohort study with repeated health screening data. Neurology. 2022;98(6):e641–52. doi:10.1212/WNL.0000000000012942. [Google Scholar] [PubMed] [CrossRef]
25. Mappin-Kasirer B, Pan H, Lewington S, Kizza J, Gray R, Clarke R, et al. Tobacco smoking and the risk of Parkinson disease: a 65-year follow-up of 30,000 male British doctors. Neurology. 2020;94(20):e2132–8. doi:10.1212/WNL.0000000000009437. [Google Scholar] [PubMed] [CrossRef]
26. Searles Nielsen S, Gallagher LG, Lundin JI, Longstreth WTJr, Smith-Weller T, Franklin GM, et al. Environmental tobacco smoke and Parkinson’s disease. Mov Disord. 2012;27(2):293–6. doi:10.1002/mds.24012. [Google Scholar] [PubMed] [CrossRef]
27. Ma C, Liu Y, Neumann S, Gao X. Nicotine from cigarette smoking and diet and Parkinson disease: a review. Transl Neurodegener. 2017;6(1):18. doi:10.1186/s40035-017-0090-8. [Google Scholar] [PubMed] [CrossRef]
28. Derkinderen P, Shannon KM, Brundin P. Gut feelings about smoking and coffee in Parkinson’s disease. Mov Disord. 2014;29(8):976–9. doi:10.1002/mds.25882. [Google Scholar] [PubMed] [CrossRef]
29. Kolahdouzan M, Hamadeh MJ. The neuroprotective effects of caffeine in neurodegenerative diseases. CNS Neurosci Ther. 2017;23(4):272–90. doi:10.1111/cns.12684. [Google Scholar] [PubMed] [CrossRef]
30. Ascherio A, Schwarzschild MA. The epidemiology of Parkinson’s disease: risk factors and prevention. Lancet Neurol. 2016;15(12):1257–72. doi:10.1016/S1474-4422(16)30230-7. [Google Scholar] [PubMed] [CrossRef]
31. Logroscino G, Gao X, Chen H, Wing A, Ascherio A. Dietary iron intake and risk of Parkinson’s disease. Am J Epidemiol. 2008;168(12):1381–8. doi:10.1093/aje/kwn273. [Google Scholar] [PubMed] [CrossRef]
32. Delic V, Beck KD, Pang KCH, Citron BA. Biological links between traumatic brain injury and Parkinson’s disease. Acta Neuropathol Commun. 2020;8(1):45. doi:10.1186/s40478-020-00924-7. [Google Scholar] [PubMed] [CrossRef]
33. Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020;19(2):170–8. doi:10.1016/S1474-4422(19)30287-X. [Google Scholar] [PubMed] [CrossRef]
34. Gómez-Benito M, Granado N, García-Sanz P, Michel A, Dumoulin M, Moratalla R. Modeling Parkinson’s disease with the alpha-synuclein protein. Front Pharmacol. 2020;11:356. doi:10.3389/fphar.2020.00356. [Google Scholar] [PubMed] [CrossRef]
35. Wasner K, Grünewald A, Klein C. Parkin-linked Parkinson’s disease: from clinical insights to pathogenic mechanisms and novel therapeutic approaches. Neurosci Res. 2020;159:34–9. doi:10.1016/j.neures.2020.09.001. [Google Scholar] [PubMed] [CrossRef]
36. DeStefano AL, Lew MF, Golbe LI, Mark MH, Lazzarini AM, Guttman M, et al. PARK3 influences age at onset in Parkinson disease: a genome scan in the GenePD study. Am J Hum Genet. 2002;70(5):1089–95. doi:10.1086/339814. [Google Scholar] [PubMed] [CrossRef]
37. Zhang J, Hattori N, Leroy E, Morris HR, Kubo S, Kobayashi T, et al. Association between a polymorphism of ubiquitin carboxy-terminal hydrolase L1 (UCH-L1) gene and sporadic Parkinson’s disease. Parkinsonism Relat Disord. 2000;6(4):195–7. doi:10.1016/S1353-8020(00)00015-8. [Google Scholar] [PubMed] [CrossRef]
38. Abramov AY, Gegg M, Grunewald A, Wood NW, Klein C, Schapira AH. Bioenergetic consequences of PINK1 mutations in Parkinson disease. PLoS One. 2011;6(10):e25622. doi:10.1371/journal.pone.0025622. [Google Scholar] [PubMed] [CrossRef]
39. Crosiers D, Theuns J, Cras P, van Broeckhoven C. Parkinson disease: insights in clinical, genetic and pathological features of monogenic disease subtypes. J Chem Neuroanat. 2011;42(2):131–41. doi:10.1016/j.jchemneu.2011.07.003. [Google Scholar] [PubMed] [CrossRef]
40. Saini P, Rudakou U, Yu E, Ruskey JA, Asayesh F, Laurent SB, et al. Association study of DNAJC13, UCHL1, HTRA2, GIGYF2, and EIF4G1 with Parkinson’s disease. Neurobiol Aging. 2021;100(3):119.e7–13. doi:10.1016/j.neurobiolaging.2020.10.019. [Google Scholar] [PubMed] [CrossRef]
41. Niemann N, Jankovic J. Juvenile parkinsonism: differential diagnosis, genetics, and treatment. Parkinsonism Relat Disord. 2019;67:74–89. doi:10.1016/j.parkreldis.2019.06.025. [Google Scholar] [PubMed] [CrossRef]
42. Wittke C, Petkovic S, Dobricic V, Schaake S. Genotype-phenotype relations for the atypical parkinsonism genes: mDSGene systematic review. Mov Disord. 2021;36(7):1499–510. doi:10.1002/mds.28517. [Google Scholar] [PubMed] [CrossRef]
43. Kurian MA, Abela L, Adam MP, Feldman J, Mirzaa GM, Pagon RA, et al. DNAJC6 Parkinson disease. In: GeneReviews®. Seattle (WAUniversity of Washington; 2021. [Google Scholar]
44. Subramaniam SR, Chesselet MF. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog Neurobiol. 2013;106–107:17–32. doi:10.1016/j.pneurobio.2013.04.004. [Google Scholar] [PubMed] [CrossRef]
45. Wen Y, Li W, Poteet EC, Xie L, Tan C, Yan LJ, et al. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J Biol Chem. 2011;286(18):16504–15. doi:10.1074/jbc.M110.208447. [Google Scholar] [PubMed] [CrossRef]
46. Gautam AH, Zeevalk GD. Characterization of reduced and oxidized dopamine and 3,4-dihydrophenylacetic acid, on brain mitochondrial electron transport chain activities. Biochim Biophys Acta. 2011;1807(7):819–28. doi:10.1016/j.bbabio.2011.03.013. [Google Scholar] [PubMed] [CrossRef]
47. Chinta SJ, Andersen JK. Redox imbalance in Parkinson’s disease. Biochim Biophys Acta. 2008;1780(11):1362–7. doi:10.1016/j.bbagen.2008.02.005. [Google Scholar] [PubMed] [CrossRef]
48. Dionísio PA, Amaral JD, Rodrigues CMP. Oxidative stress and regulated cell death in Parkinson’s disease. Ageing Res Rev. 2021;67:101263. doi:10.1016/j.arr.2021.101263. [Google Scholar] [PubMed] [CrossRef]
49. Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262(5134):689–95. doi:10.1126/science.7901908. [Google Scholar] [PubMed] [CrossRef]
50. Cohen G, Farooqui R, Kesler N. Parkinson disease: a new link between monoamine oxidase and mitochondrial electron flow. Proc Natl Acad Sci U S A. 1997;94(10):4890–4. doi:10.1073/pnas.94.10.4890. [Google Scholar] [PubMed] [CrossRef]
51. Rahimmi A, Khosrobakhsh F, Izadpanah E, Moloudi MR, Hassanzadeh K. N-acetylcysteine prevents rotenone-induced Parkinson’s disease in rat: an investigation into the interaction of parkin and Drp1 proteins. Brain Res Bull. 2015;113:34–40. doi:10.1016/j.brainresbull.2015.02.007. [Google Scholar] [PubMed] [CrossRef]
52. Olanow CW, Schapira AH, LeWitt PA, Kieburtz K, Sauer D, Olivieri G, et al. TCH346 as a neuroprotective drug in Parkinson’s disease: a double-blind, randomised, controlled trial. Lancet Neurol. 2006;5(12):1013–20. doi:10.1016/S1474-4422(06)70602-0. [Google Scholar] [PubMed] [CrossRef]
53. Hassanzadeh K, Rahimmi A. Oxidative stress and neuroinflammation in the story of Parkinson’s disease: could targeting these pathways write a good ending? J Cell Physiol. 2018;234(1):23–32. doi:10.1002/jcp.26865. [Google Scholar] [PubMed] [CrossRef]
54. Giannoccaro MP, La Morgia C, Rizzo G, Carelli V. Mitochondrial DNA and primary mitochondrial dysfunction in Parkinson’s disease. Mov Disord. 2017;32(3):346–63. doi:10.1002/mds.26966. [Google Scholar] [PubMed] [CrossRef]
55. Shang D, Huang M, Wang B, Yan X, Wu Z, Zhang X. mtDNA maintenance and alterations in the pathogenesis of neurodegenerative diseases. Curr Neuropharmacol. 2023;21(3):578–98. doi:10.2174/1570159X20666220810114644. [Google Scholar] [PubMed] [CrossRef]
56. Müller-Nedebock AC, Brennan RR, Venter M, Pienaar IS, van der Westhuizen FH, Elson JL, et al. The unresolved role of mitochondrial DNA in Parkinson’s disease: an overview of published studies, their limitations, and future prospects. Neurochem Int. 2019;129:104495. doi:10.1016/j.neuint.2019.104495. [Google Scholar] [PubMed] [CrossRef]
57. Ryan BJ, Hoek S, Fon EA, Wade-Martins R. Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci. 2015;40(4):200–10. doi:10.1016/j.tibs.2015.02.003. [Google Scholar] [PubMed] [CrossRef]
58. Thomas B, Beal MF. Parkinson’s disease. Hum Mol Genet. 2007;16(R2):R183–94. doi:10.1093/hmg/ddm159. [Google Scholar] [PubMed] [CrossRef]
59. Wang XL, Feng ST, Wang ZZ, Yuan YH, Chen NH, Zhang Y. Parkin, an E3 ubiquitin ligase, plays an essential role in mitochondrial quality control in Parkinson’s disease. Cell Mol Neurobiol. 2021;41(7):1395–411. doi:10.1007/s10571-020-00914-2. [Google Scholar] [PubMed] [CrossRef]
60. Kuroda Y, Mitsui T, Kunishige M, Shono M, Akaike M, Azuma H, et al. Parkin enhances mitochondrial biogenesis in proliferating cells. Hum Mol Genet. 2006;15(6):883–95. doi:10.1093/hmg/ddl006. [Google Scholar] [PubMed] [CrossRef]
61. Darios F, Corti O, Lücking CB, Hampe C, Muriel MP, Abbas N, et al. Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum Mol Genet. 2003;12(5):517–26. doi:10.1093/hmg/ddg044. [Google Scholar] [PubMed] [CrossRef]
62. Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, et al. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279(18):18614–22. doi:10.1074/jbc.M401135200. [Google Scholar] [PubMed] [CrossRef]
63. Wood-Kaczmar A, Gandhi S, Yao Z, Abramov AY, Miljan EA, Keen G, et al. PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS One. 2008;3(6):e2455. doi:10.1371/journal.pone.0002455. [Google Scholar] [PubMed] [CrossRef]
64. Amo T, Sato S, Saiki S, Wolf AM, Toyomizu M, Gautier CA, et al. Mitochondrial membrane potential decrease caused by loss of PINK1 is not due to proton leak, but to respiratory chain defects. Neurobiol Dis. 2011;41(1):111–8. doi:10.1016/j.nbd.2010.08.027. [Google Scholar] [PubMed] [CrossRef]
65. Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441(7097):1162–6. doi:10.1038/nature04779. [Google Scholar] [PubMed] [CrossRef]
66. Heo JY, Park JH, Kim SJ, Seo KS, Han JS, Lee SH, et al. DJ-1 null dopaminergic neuronal cells exhibit defects in mitochondrial function and structure: involvement of mitochondrial complex I assembly. PLoS One. 2012;7(3):e32629. doi:10.1371/journal.pone.0032629. [Google Scholar] [PubMed] [CrossRef]
67. Krebiehl G, Ruckerbauer S, Burbulla LF, Kieper N, Maurer B, Waak J, et al. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLoS One. 2010;5(2):e9367. doi:10.1371/journal.pone.0009367. [Google Scholar] [PubMed] [CrossRef]
68. Mortiboys H, Johansen KK, Aasly JO, Bandmann O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology. 2010;75(22):2017–20. doi:10.1212/WNL.0b013e3181ff9685. [Google Scholar] [PubMed] [CrossRef]
69. Niu J, Yu M, Wang C, Xu Z. Leucine-rich repeat kinase 2 disturbs mitochondrial dynamics via Dynamin-like protein. J Neurochem. 2012;122(3):650–8. doi:10.1111/j.1471-4159.2012.07809.x. [Google Scholar] [PubMed] [CrossRef]
70. Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, et al. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol. 1997;12(1):25–31. [Google Scholar] [PubMed]
71. Gupta A, Dawson VL, Dawson TM. What causes cell death in Parkinson’s disease? Ann Neurol. 2008;64(S2):S3–15. doi:10.1002/ana.21573. [Google Scholar] [PubMed] [CrossRef]
72. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140(6):918–34. doi:10.1016/j.cell.2010.02.016. [Google Scholar] [PubMed] [CrossRef]
73. Ramesh G, MacLean AG, Philipp MT. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediators Inflamm. 2013;2013:480739. doi:10.1155/2013/480739. [Google Scholar] [PubMed] [CrossRef]
74. Rauf A, Badoni H, Abu-Izneid T, Olatunde A, Rahman MM, Painuli S, et al. Neuroinflammatory markers: key indicators in the pathology of neurodegenerative diseases. Molecules. 2022;27(10):3194. doi:10.3390/molecules27103194. [Google Scholar] [PubMed] [CrossRef]
75. Shih RH, Wang CY, Yang CM. NF-kappaB signaling pathways in neurological inflammation: a mini review. Front Mol Neurosci. 2015;8:77. doi:10.3389/fnmol.2015.00077. [Google Scholar] [PubMed] [CrossRef]
76. Mogi M, Togari A, Kondo T, Mizuno Y, Komure O, Kuno S, et al. Caspase activities and tumor necrosis factor receptor R1 (p55) level are elevated in the substantia nigra from parkinsonian brain. J Neural Transm. 2000;107(3):335–41. doi:10.1007/s007020050028. [Google Scholar] [PubMed] [CrossRef]
77. Russo I, Berti G, Plotegher N, Bernardo G, Filograna R, Bubacco L, et al. Leucine-rich repeat kinase 2 positively regulates inflammation and down-regulates NF-κB p50 signaling in cultured microglia cells. J Neuroinflammation. 2015;12(1):230. doi:10.1186/s12974-015-0449-7. [Google Scholar] [PubMed] [CrossRef]
78. López de Maturana R, Lang V, Zubiarrain A, Sousa A, Vázquez N, Gorostidi A, et al. Mutations in LRRK2 impair NF-κB pathway in iPSC-derived neurons. J Neuroinflammation. 2016;13(1):295. doi:10.1186/s12974-016-0761-x. [Google Scholar] [PubMed] [CrossRef]
79. Yacoubian TA, Standaert DG. Targets for neuroprotection in Parkinson’s disease. Biochim Biophys Acta. 2009;1792(7):676–87. doi:10.1016/j.bbadis.2008.09.009. [Google Scholar] [PubMed] [CrossRef]
80. Vallée A, Lecarpentier Y, Guillevin R, Vallée JN. Circadian rhythms, neuroinflammation and oxidative stress in the story of Parkinson’s disease. Cells. 2020;9(2):314. doi:10.3390/cells9020314. [Google Scholar] [PubMed] [CrossRef]
81. Gelders G, Baekelandt V, Van der Perren A. Linking neuroinflammation and neurodegeneration in Parkinson’s disease. J Immunol Res. 2018;2018:4784268. doi:10.1155/2018/4784268. [Google Scholar] [PubMed] [CrossRef]
82. Estaun-Panzano J, Arotcarena ML, Bezard E. Monitoring α-synuclein aggregation. Neurobiol Dis. 2023;176:105966. doi:10.1016/j.nbd.2022.105966. [Google Scholar] [PubMed] [CrossRef]
83. Tan JM, Wong ES, Lim KL. Protein misfolding and aggregation in Parkinson’s disease. Antioxid Redox Signal. 2009;11(9):2119–34. doi:10.1089/ars.2009.2490. [Google Scholar] [PubMed] [CrossRef]
84. Mehra S, Sahay S, Maji SK. α-Synuclein misfolding and aggregation: implications in Parkinson’s disease pathogenesis. Biochim Biophys Acta Proteins Proteom. 2019;1867(10):890–908. doi:10.1016/j.bbapap.2019.03.001. [Google Scholar] [PubMed] [CrossRef]
85. Clark LN, Afridi S, Karlins E, Wang Y, Mejia-Santana H, Harris J, et al. Case-control study of the parkin gene in early-onset Parkinson disease. Arch Neurol. 2006;63(4):548–52. doi:10.1001/archneur.63.4.548. [Google Scholar] [PubMed] [CrossRef]
86. Oliveira SA, Scott WK, Martin ER, Nance MA, Watts RL, Hubble JP, et al. Parkin mutations and susceptibility alleles in late-onset Parkinson’s disease. Ann Neurol. 2003;53(5):624–9. doi:10.1002/ana.10524. [Google Scholar] [PubMed] [CrossRef]
87. Muqit MM, Abou-Sleiman PM, Saurin AT, Harvey K, Gandhi S, Deas E, et al. Altered cleavage and localization of PINK1 to aggresomes in the presence of proteasomal stress. J Neurochem. 2006;98(1):156–69. doi:10.1111/j.1471-4159.2006.03845.x. [Google Scholar] [PubMed] [CrossRef]
88. Olzmann JA, Brown K, Wilkinson KD, Rees HD, Huai Q, Ke H, et al. Familial Parkinson’s disease-associated L166P mutation disrupts DJ-1 protein folding and function. J Biol Chem. 2004;279(9):8506–15. doi:10.1074/jbc.M311017200. [Google Scholar] [PubMed] [CrossRef]
89. Greggio E, Lewis PA, van der Brug MP, Ahmad R, Kaganovich A, Ding J, et al. Mutations in LRRK2/dardarin associated with Parkinson disease are more toxic than equivalent mutations in the homologous kinase LRRK1. J Neurochem. 2007;102(1):93–102. doi:10.1111/j.1471-4159.2007.04523.x. [Google Scholar] [PubMed] [CrossRef]
90. Palasz E, Wysocka A, Gasiorowska A, Chalimoniuk M, Niewiadomski W, Niewiadomska G. BDNF as a promising therapeutic agent in Parkinson’s disease. Int J Mol Sci. 2020;21(3):1170. doi:10.3390/ijms21031170. [Google Scholar] [PubMed] [CrossRef]
91. d’Anglemont de Tassigny X, Pascual A, López-Barneo J. GDNF-based therapies, GDNF-producing interneurons, and trophic support of the dopaminergic nigrostriatal pathway. Implications for Parkinson’s disease. Front Neuroanat. 2015;9:10. doi:10.3389/fnana.2015.00010. [Google Scholar] [PubMed] [CrossRef]
92. Mufson EJ, Kroin JS, Sendera TJ, Sobreviela T. Distribution and retrograde transport of trophic factors in the central nervous system: functional implications for the treatment of neurodegenerative diseases. Prog Neurobiol. 1999;57(4):451–84. doi:10.1016/s0301-0082(98)00059-8. [Google Scholar] [PubMed] [CrossRef]
93. Tiberi A, Capsoni S, Cattaneo A. A microglial function for the nerve growth factor: predictions of the unpredictable. Cells. 2022;11(11):1835. doi:10.3390/cells11111835. [Google Scholar] [PubMed] [CrossRef]
94. Wang Y, Lv MN, Zhao WJ. Research on ferroptosis as a therapeutic target for the treatment of neurodegenerative diseases. Ageing Res Rev. 2023;91:102035. doi:10.1016/j.arr.2023.102035. [Google Scholar] [PubMed] [CrossRef]
95. Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi AA, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. 2021;6(1):49. doi:10.1038/s41392-020-00428-9. [Google Scholar] [PubMed] [CrossRef]
96. Dexter DT, Carter CJ, Wells FR, Javoy-Agid F, Agid Y, Lees A, et al. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J Neurochem. 1989;52(2):381–9. doi:10.1111/j.1471-4159.1989.tb09133.x. [Google Scholar] [PubMed] [CrossRef]
97. Mahoney-Sánchez L, Bouchaoui H, Ayton S, Devos D, Duce JA, Devedjian JC. Ferroptosis and its potential role in the physiopathology of Parkinson’s disease. Prog Neurobiol. 2021;196:101890. doi:10.1016/j.pneurobio.2020.101890. [Google Scholar] [PubMed] [CrossRef]
98. Vallerga CL, Zhang F, Fowdar J, McRae AF, Qi T, Nabais MF, et al. Analysis of DNA methylation associates the cystine-glutamate antiporter SLC7A11 with risk of Parkinson’s disease. Nat Commun. 2020;11(1):1238. doi:10.1038/s41467-020-15065-7. [Google Scholar] [PubMed] [CrossRef]
99. Hirsch EC, Brandel JP, Galle P, Javoy-Agid F, Agid Y. Iron and aluminum increase in the substantia nigra of patients with Parkinson’s disease: an X-ray microanalysis. J Neurochem. 1991;56(2):446–51. doi:10.1111/j.1471-4159.1991.tb08170.x. [Google Scholar] [PubMed] [CrossRef]
100. Pyatigorskaya N, Sharman M, Corvol JC, Valabregue R, Yahia-Cherif L, Poupon F, et al. High nigral iron deposition in LRRK2 and Parkin mutation carriers using R2* relaxometry. Mov Disord. 2015;30(8):1077–84. doi:10.1002/mds.26218. [Google Scholar] [PubMed] [CrossRef]
101. Wang Y, Spincemaille P, Liu Z, Dimov A, Deh K, Li J, et al. Clinical quantitative susceptibility mapping (QSMbiometal imaging and its emerging roles in patient care. J Magn Reson Imaging. 2017;46(4):951–71. doi:10.1002/jmri.25693. [Google Scholar] [PubMed] [CrossRef]
102. Masaldan S, Bush AI, Devos D, Rolland AS, Moreau C. Striking while the iron is hot: iron metabolism and ferroptosis in neurodegeneration. Free Radic Biol Med. 2019;133:221–33. doi:10.1016/j.freeradbiomed.2018.09.033. [Google Scholar] [PubMed] [CrossRef]
103. Huang P, Zhang LY, Tan YY, Chen SD. Links between COVID-19 and Parkinson’s disease/Alzheimer’s disease: reciprocal impacts, medical care strategies and underlying mechanisms. Transl Neurodegener. 2023;12(1):5. doi:10.1186/s40035-023-00337-1. [Google Scholar] [CrossRef]
104. Hou X, Watzlawik JO, Fiesel FC, Springer W. Autophagy in Parkinson’s disease. J Mol Biol. 2020;432(8):2651–72. doi:10.1016/j.jmb.2020.01.037. [Google Scholar] [PubMed] [CrossRef]
105. Plowey ED, Cherra SJ3rd, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem. 2008;105(3):1048–56. doi:10.1111/j.1471-4159.2008.05217.x. [Google Scholar] [PubMed] [CrossRef]
106. Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85(2):257–73. doi:10.1016/j.neuron.2014.12.007. [Google Scholar] [PubMed] [CrossRef]
107. Lev N, Melamed E, Offen D. Apoptosis and Parkinson’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27(2):245–50. doi:10.1016/S0278-5846(03)00019-8. [Google Scholar] [PubMed] [CrossRef]
108. Elfawy HA, Das B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: etiologies and therapeutic strategies. Life Sci. 2019;218(9):165–84. doi:10.1016/j.lfs.2018.12.029. [Google Scholar] [PubMed] [CrossRef]
109. Nicotra A, Parvez S. Apoptotic molecules and MPTP-induced cell death. Neurotoxicol Teratol. 2002;24(5):599–605. doi:10.1016/s0892-0362(02)00213-1. [Google Scholar] [PubMed] [CrossRef]
110. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell. 2016;167(6):1469–80.e12. doi:10.1016/j.cell.2016.11.018. [Google Scholar] [PubMed] [CrossRef]
111. Wang Q, Luo Y, Ray Chaudhuri K, Reynolds R, Tan EK, Pettersson S. The role of gut dysbiosis in Parkinson’s disease: mechanistic insights and therapeutic options. Brain. 2021;144(9):2571–93. doi:10.1093/brain/awab156. [Google Scholar] [PubMed] [CrossRef]
112. Mulak A, Bonaz B. Brain-gut-microbiota axis in Parkinson’s disease. World J Gastroenterol. 2015;21(37):10609–20. doi:10.3748/wjg.v21.i37.10609. [Google Scholar] [PubMed] [CrossRef]
113. Sepehrinezhad A, Shahbazi A, Negah SS. COVID-19 virus may have neuroinvasive potential and cause neurological complications: a perspective review. J Neurovirol. 2020;26(3):324–9. doi:10.1007/s13365-020-00851-2. [Google Scholar] [PubMed] [CrossRef]
114. Sulzer D, Antonini A, Leta V, Nordvig A, Smeyne RJ, Goldman JE, et al. COVID-19 and possible links with Parkinson’s disease and parkinsonism: from bench to bedside. NPJ Parkinsons Dis. 2020;6:18. doi:10.1038/s41531-020-00123-0. [Google Scholar] [PubMed] [CrossRef]
115. McLean G, Hindle JV, Guthrie B, Mercer SW. Co-morbidity and polypharmacy in Parkinson’s disease: insights from a large Scottish primary care database. BMC Neurol. 2017;17(1):126. doi:10.1186/s12883-017-0904-4. [Google Scholar] [PubMed] [CrossRef]
116. Dulski J, Sławek J. Incidence and characteristics of post-COVID-19 parkinsonism and dyskinesia related to COVID-19 vaccines. Neurol Neurochir Pol. 2023;57(1):53–62. doi:10.5603/PJNNS.a2023.0011. [Google Scholar] [PubMed] [CrossRef]
117. Brugger F, Erro R, Balint B, Kägi G, Barone P, Bhatia KP. Why is there motor deterioration in Parkinson’s disease during systemic infections-a hypothetical view. npj Parkinsons Dis. 2015;1:15014. doi:10.1038/npjparkd.2015.14. [Google Scholar] [PubMed] [CrossRef]
118. Artusi CA, Romagnolo A, Imbalzano G, Marchet A, Zibetti M, Rizzone MG, et al. COVID-19 in Parkinson’s disease: report on prevalence and outcome. Parkinsonism Relat Disord. 2020;80:7–9. doi:10.1016/j.parkreldis.2020.09.008. [Google Scholar] [PubMed] [CrossRef]
119. Cilia R, Bonvegna S, Straccia G, Andreasi NG, Elia AE, Romito LM, et al. Effects of COVID-19 on Parkinson’s disease clinical features: a community-based case-control study. Mov Disord. 2020;35(8):1287–92. doi:10.1002/mds.28170. [Google Scholar] [PubMed] [CrossRef]
120. Salari M, Zali A, Ashrafi F, Etemadifar M, Sharma S, Hajizadeh N, et al. Incidence of anxiety in Parkinson’s disease during the coronavirus disease (COVID-19) pandemic. Mov Disord. 2020;35(7):1095–6. doi:10.1002/mds.28116. [Google Scholar] [PubMed] [CrossRef]
121. Brown EG, Chahine LM, Goldman SM, Korell M, Mann E, Kinel DR, et al. The effect of the COVID-19 pandemic on people with Parkinson’s disease. J Parkinsons Dis. 2020;10(4):1365–77. doi:10.3233/JPD-202249. [Google Scholar] [PubMed] [CrossRef]
122. Fearon C, Fasano A. Parkinson’s Disease and the COVID-19 pandemic. J Parkinsons Dis. 2021;11(2):431–44. doi:10.3233/JPD-202320. [Google Scholar] [PubMed] [CrossRef]
123. Wu KE, Fazal FM, Parker KR, Zou J, Chang HY. RNA-GPS predicts SARS-CoV-2 RNA residency to host mitochondria and nucleolus. Cell Syst. 2020;11(1):102–8.E3. doi:10.1016/j.cels.2020.06.008. [Google Scholar] [PubMed] [CrossRef]
124. Nataf S. An alteration of the dopamine synthetic pathway is possibly involved in the pathophysiology of COVID-19. J Med Virol. 2020;92(10):1743–4. doi:10.1002/jmv.25826. [Google Scholar] [PubMed] [CrossRef]
125. Tiwari S, Yadav N, Singh S. COVID-19 and Parkinson’s disease: possible links in pathology and therapeutics. Neurotox Res. 2022;40(5):1586–96. doi:10.1007/s12640-022-00540-4. [Google Scholar] [PubMed] [CrossRef]
126. Al-Kuraishy HM, Al-Gareeb AI, Kaushik A, Kujawska M, Ahmed EA, Batiha GE. SARS-COV-2 infection and Parkinson’s disease: possible links and perspectives. J Neurosci Res. 2023;101(6):952–75. doi:10.1002/jnr.25171. [Google Scholar] [PubMed] [CrossRef]
127. Xi X, Han L. Exploring the relationship between novel Coronavirus pneumonia and Parkinson’s disease. Medicine. 2022;101(46):e31813. doi:10.1097/MD.0000000000031813. [Google Scholar] [PubMed] [CrossRef]
128. Dehay B, Bourdenx M, Gorry P, Przedborski S, Vila M, Hunot S, et al. Targeting α-synuclein for treatment of Parkinson’s disease: mechanistic and therapeutic considerations. Lancet Neurol. 2015;14(8):855–66. doi:10.1016/S1474-4422(15)00006-X. [Google Scholar] [PubMed] [CrossRef]
129. Khan MAB, Hashim MJ, King JK, Govender RD, Mustafa H, Al Kaabi J. Epidemiology of Type 2 diabetes—global burden of disease and forecasted trends. J Epidemiol Glob Health. 2020;10(1):107–11. doi:10.2991/jegh.k.191028.001. [Google Scholar] [PubMed] [CrossRef]
130. Harrison PJ, Luciano S. Incidence of Parkinson’s disease, dementia, cerebrovascular disease and stroke in bipolar disorder compared to other psychiatric disorders: an electronic health records network study of 66 million people. Bipolar Disord. 2021;23(5):454–62. doi:10.1111/bdi.13022. [Google Scholar] [PubMed] [CrossRef]
131. Iijima M, Okuma Y, Suzuki K, Yoshii F, Nogawa S, Osada T, et al. Associations between probable REM sleep behavior disorder, olfactory disturbance, and clinical symptoms in Parkinson’s disease: a multicenter cross-sectional study. PLoS One. 2021;16(2):e0247443. doi:10.1371/journal.pone.0247443. [Google Scholar] [PubMed] [CrossRef]
132. Aune D, Schlesinger S, Mahamat-Saleh Y, Zheng B, Udeh-Momoh CT, Middleton LT. Diabetes mellitus, prediabetes and the risk of Parkinson’s disease: a systematic review and meta-analysis of 15 cohort studies with 29.9 million participants and 86,345 cases. Eur J Epidemiol. 2023;38(6):591–604. doi:10.1007/s10654-023-00970-0. [Google Scholar] [PubMed] [CrossRef]
133. Wang H, Raleigh DP. The ability of insulin to inhibit the formation of amyloid by pro-islet amyloid polypeptide processing intermediates is significantly reduced in the presence of sulfated glycosaminoglycans. Biochemistry. 2014;53(16):2605–14. doi:10.1021/bi4015488. [Google Scholar] [PubMed] [CrossRef]
134. Horvath I, Wittung-Stafshede P. Cross-talk between amyloidogenic proteins in type-2 diabetes and Parkinson’s disease. Proc Natl Acad Sci U S A. 2016;113(44):12473–77. doi:10.1073/pnas.1610371113. [Google Scholar] [PubMed] [CrossRef]
135. Lv YQ, Yuan L, Sun Y, Dou HW, Su JH, Hou ZP, et al. Long-term hyperglycemia aggravates α-synuclein aggregation and dopaminergic neuronal loss in a Parkinson’s disease mouse model. Transl Neurodegener. 2022;11(1):14. doi:10.1186/s40035-022-00288-z. [Google Scholar] [PubMed] [CrossRef]
136. Athauda D, Foltynie T. Insulin resistance and Parkinson’s disease: a new target for disease modification? Prog Neurobiol. 2016;145–146:98–120. doi:10.1016/j.pneurobio. [Google Scholar] [CrossRef]
137. Qeva E, Sollazzo C, Bilotta F. Insulin signaling in the central nervous system, a possible pathophysiological mechanism of anesthesia-induced delayed neurocognitive recovery/postoperative neurocognitive disorder: a narrative review. Expert Rev Neurother. 2022;22(10):839–47. doi:10.1080/14737175.2022.2144234. [Google Scholar] [PubMed] [CrossRef]
138. Jones KT, Woods C, Zhen J, Antonio T, Carr KD, Reith ME. Effects of diet and insulin on dopamine transporter activity and expression in rat caudate-putamen, nucleus accumbens, and midbrain. J Neurochem. 2017;140(5):728–40. doi:10.1111/jnc.13930. [Google Scholar] [PubMed] [CrossRef]
139. Baladi MG, Horton RE, Owens WA, Daws LC, France CP. Eating high fat chow decreases dopamine clearance in adolescent and adult male rats but selectively enhances the locomotor stimulating effects of cocaine in adolescents. Int J Neuropsychopharmacol. 2015;18(7):pyv024. doi:10.1093/ijnp/pyv024. [Google Scholar] [PubMed] [CrossRef]
140. Takahashi M, Yamada T, Tooyama I, Moroo I, Kimura H, Yamamoto T, et al. Insulin receptor mRNA in the substantia nigra in Parkinson’s disease. Neurosci Lett. 1996;204(3):201–4. doi:10.1016/0304-3940(96)12357-0. [Google Scholar] [PubMed] [CrossRef]
141. Morris JK, Vidoni ED, Perea RD, Rada R, Johnson DK, Lyons K, et al. Insulin resistance and gray matter volume in neurodegenerative disease. Neuroscience. 2014;270:139–47. doi:10.1016/j.neuroscience.2014.04.006. [Google Scholar] [PubMed] [CrossRef]
142. Vicente Miranda H, El-Agnaf OM, Outeiro TF. Glycation in Parkinson’s disease and Alzheimer’s disease. Mov Disord. 2016;31(6):782–90. doi:10.1002/mds.26566. [Google Scholar] [PubMed] [CrossRef]
143. König A, Vicente Miranda H, Outeiro TF. Alpha-synuclein glycation and the action of anti-diabetic agents in Parkinson’s disease. J Parkinsons Dis. 2018;8(1):33–43. doi:10.3233/JPD-171285. [Google Scholar] [PubMed] [CrossRef]
144. Bonnard C, Durand A, Peyrol S, Chanseaume E, Chauvin MA, Morio B, et al. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest. 2008;118(2):789–800. doi:10.1172/JCI32601. [Google Scholar] [PubMed] [CrossRef]
145. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin was recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183(5):795–803. doi:10.1083/jcb.200809125. [Google Scholar] [PubMed] [CrossRef]
146. Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107(1):378–83. doi:10.1073/pnas.0911187107. [Google Scholar] [PubMed] [CrossRef]
147. Choi ML, Chappard A, Singh BP, Maclachlan C, Rodrigues M, Fedotova EI, et al. Pathological structural conversion of α-synuclein at the mitochondria induces neuronal toxicity. Nat Neurosci. 2022;25(9):1134–48. doi:10.1038/s41593-022-01140-3. [Google Scholar] [PubMed] [CrossRef]
148. Kleinridders A, Cai W, Cappellucci L, Ghazarian A, Collins WR, Vienberg SG, et al. Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. Proc Natl Acad Sci U S A. 2015;112(11):3463–8. doi:10.1073/pnas.1500877112. [Google Scholar] [PubMed] [CrossRef]
149. Aviles-Olmos I, Limousin P, Lees A, Foltynie T. Parkinson’s disease, insulin resistance and novel agents of neuroprotection. Brain. 2013;136(2):374–84. doi:10.1093/brain/aws009. [Google Scholar] [PubMed] [CrossRef]
150. Wang S, Zhang C, Sheng X, Zhang X, Wang B, Zhang G. Peripheral expression of MAPK pathways in Alzheimer’s and Parkinson’s diseases. J Clin Neurosci. 2014;21(5):810–4. doi:10.1016/j.jocn.2013.08.017. [Google Scholar] [PubMed] [CrossRef]
151. Khang R, Park C, Shin JH. Dysregulation of parkin in the substantia nigra of db/db and high-fat diet mice. Neuroscience. 2015;294:182–92. doi:10.1016/j.neuroscience.2015.03.017. [Google Scholar] [PubMed] [CrossRef]
152. Hassan A, Sharma Kandel R, Mishra R, Gautam J, Alaref A, Jahan N. Diabetes mellitus and Parkinson’s disease: shared pathophysiological links and possible therapeutic implications. Cureus. 2020;12(8):e9853. doi:10.7759/cureus.9853. [Google Scholar] [PubMed] [CrossRef]
153. Joers V, Tansey MG, Mulas G, Carta AR. Microglial phenotypes in Parkinson’s disease and animal models of the disease. Prog Neurobiol. 2017;155:57–75. doi:10.1016/j.pneurobio.2016.04.006. [Google Scholar] [PubMed] [CrossRef]
154. Zhang PF, Gao F. Neuroinflammation in Parkinson’s disease: a meta-analysis of PET imaging studies. J Neurol. 2022;269(5):2304–14. doi:10.1007/s00415-021-10877-z. [Google Scholar] [PubMed] [CrossRef]
155. Liu TW, Chen CM, Chang KH. Biomarker of neuroinflammation in Parkinson’s disease. Int J Mol Sci. 2022;23(8):4148. doi:10.3390/ijms23084148. [Google Scholar] [PubMed] [CrossRef]
156. Qi G, Mi Y, Fan R, Li R, Liu Z, Liu X. Nobiletin protects against systemic inflammation-stimulated memory impairment via MAPK and NF-κB signaling pathways. J Agric Food Chem. 2019;67(18):5122–34. doi:10.1021/acs.jafc.9b00133. [Google Scholar] [PubMed] [CrossRef]
157. Lisco G, Giagulli VA, De Pergola G, Guastamacchia E, Jirillo E, Triggiani V. Hyperglycemia-induced immune system disorders in diabetes mellitus and the concept of hyperglycemic memory of innate immune cells: a perspective. Endocr Metab Immune Disord Drug Targets. 2022;22(4):367–70. doi:10.2174/1871530321666210924124336. [Google Scholar] [PubMed] [CrossRef]
158. Nakashima A, Yokoyama K, Yokoo T, Urashima M. Role of vitamin D in diabetes mellitus and chronic kidney disease. World J Diabetes. 2016;7(5):89–100. doi:10.4239/wjd.v7.i5.89. [Google Scholar] [PubMed] [CrossRef]
159. Bivona G, Gambino CM, Iacolino G, Ciaccio M. Vitamin D and the nervous system. Neurol Res. 2019;41(9):827–35. doi:10.1080/01616412.2019.1622872. [Google Scholar] [PubMed] [CrossRef]
160. Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1α interaction. Nature. 2003;423(6939):550–5. doi:10.1038/nature01667. [Google Scholar] [PubMed] [CrossRef]
161. Pasinetti GM, Wang J, Marambaud P, Ferruzzi M, Gregor P, Knable LA, et al. Neuroprotective and metabolic effects of resveratrol: therapeutic implications for Huntington’s disease and other neurodegenerative disorders. Exp Neurol. 2011;232(1):1–6. doi:10.1016/j.expneurol.2011.08.014. [Google Scholar] [PubMed] [CrossRef]
162. Mullur R, Liu YY, Brent GA. Thyroid hormone regulation of metabolism. Physiol Rev. 2014;94(2):355–82. doi:10.1152/physrev.00030.2013. [Google Scholar] [PubMed] [CrossRef]
163. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49. doi:10.3322/caac.21660. [Google Scholar] [PubMed] [CrossRef]
164. Deuschl G, Beghi E, Fazekas F, Varga T, Christoforidi KA, Sipido E, et al. The burden of neurological diseases in Europe: an analysis for the Global Burden of Disease Study 2017. Lancet Public Health. 2020;5(10):e551–67. doi:10.1016/S2468-2667(20)30190-0. [Google Scholar] [PubMed] [CrossRef]
165. Ganguli M. Cancer and dementia: it’s complicated. Alzheimer Dis Assoc Disord. 2015;29(2):177–82. doi:10.1097/WAD.0000000000000086. [Google Scholar] [PubMed] [CrossRef]
166. Bajaj A, Driver JA, Schernhammer ES. Parkinson’s disease and cancer risk: a systematic review and meta-analysis. Cancer Causes Control. 2010;21(5):697–707. doi:10.1007/s10552-009-9497-6. [Google Scholar] [PubMed] [CrossRef]
167. Driver JA. Inverse association between cancer and neurodegenerative disease: review of the epidemiologic and biological evidence. Biogerontology. 2014;15(6):547–57. doi:10.1007/s10522-014-9523-2. [Google Scholar] [PubMed] [CrossRef]
168. Zhang X, Guarin D, Mohammadzadehhonarvar N, Chen X, Gao X. Parkinson’s disease and cancer: a systematic review and meta-analysis of over 17 million participants. BMJ Open. 2021;11(7):e046329. doi:10.1136/bmjopen-2020-046329. [Google Scholar] [PubMed] [CrossRef]
169. Feng DD, Cai W, Chen X. The associations between Parkinson’s disease and cancer: the plot thickens. Transl Neurodegener. 2015;4(1):20. doi:10.1186/s40035-015-0043-z. [Google Scholar] [PubMed] [CrossRef]
170. Sturchio A, Dwivedi AK, Vizcarra JA, Chirra M, Keeling EG, Mata IF, et al. Genetic parkinsonisms and cancer: a systematic review and meta-analysis. Rev Neurosci. 2020;32(2):159–67. doi:10.1515/revneuro-2020-0083. [Google Scholar] [PubMed] [CrossRef]
171. Sherman MY, Goldberg AL. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29(1):15–32. doi:10.1016/S0896-6273(01)00177-5. [Google Scholar] [PubMed] [CrossRef]
172. Manasanch EE, Orlowski RZ. Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol. 2017;14(7):417–33. doi:10.1038/nrclinonc.2016.206. [Google Scholar] [PubMed] [CrossRef]
173. Boyle KA, Van Wickle J, Hill RB, Marchese A, Kalyanaraman B, Dwinell MB. Mitochondria-targeted drugs stimulate mitophagy and abrogate colon cancer cell proliferation. J Biol Chem. 2018;293(38):14891–904. doi:10.1074/jbc.RA117.001469. [Google Scholar] [PubMed] [CrossRef]
174. Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010;107(19):8788–93. doi:10.1073/pnas.1003428107. [Google Scholar] [PubMed] [CrossRef]
175. Zhu X, Lee HG, Perry G, Smith MA. Alzheimer disease, the two-hit hypothesis: an update. Biochim Biophys Acta. 2007;1772(4):494–502. doi:10.1016/j.bbadis.2006.10.014. [Google Scholar] [PubMed] [CrossRef]
176. Lombardi VR, García M, Rey L, Cacabelos R. Characterization of cytokine production, screening of lymphocyte subset patterns and in vitro apoptosis in healthy and Alzheimer’s Disease (AD) individuals. J Neuroimmunol. 1999;97(1–2):163–71. doi:10.1016/S0165-5728(99)00046-6. [Google Scholar] [PubMed] [CrossRef]
177. Folch J, Junyent F, Verdaguer E, Auladell C, Pizarro JG, Beas-Zarate C, et al. Role of cell cycle re-entry in neurons: a common apoptotic mechanism of neuronal cell death. Neurotox Res. 2012;22(3):195–207. doi:10.1007/s12640-011-9277-4. [Google Scholar] [PubMed] [CrossRef]
178. Rockenstein E, Nuber S, Overk CR, Ubhi K, Mante M, Patrick C, et al. Accumulation of oligomer-prone α-synuclein exacerbates synaptic and neuronal degeneration in vivo. Brain. 2014;137(5):1496–513. doi:10.1093/brain/awu057. [Google Scholar] [PubMed] [CrossRef]
179. Kam TI, Mao X, Park H, Chou SC, Karuppagounder SS, Umanah GE, et al. Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson’s disease. Science. 2018;362(6414):eaat8407. doi:10.1126/science.aat8407. [Google Scholar] [PubMed] [CrossRef]
180. Navarro-Otano J, Gelpi E, Mestres CA, Quintana E, Rauek S, Ribalta T, et al. Alpha-synuclein aggregates in epicardial fat tissue in living subjects without parkinsonism. Parkinsonism Relat Disord. 2013;19(1):27. doi:10.1016/j.parkreldis.2012.07.005. [Google Scholar] [PubMed] [CrossRef]
181. Panicker N, Dawson VL, Dawson TM. Activation mechanisms of the E3 ubiquitin ligase parkin. Biochem J. 2017;474(18):3075–86. doi:10.1042/BCJ20170476. [Google Scholar] [PubMed] [CrossRef]
182. Quinn PMJ, Moreira PI, Ambrósio AF, Alves CH. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol Commun. 2020;8(1):189. doi:10.1186/s40478-020-01062-w. [Google Scholar] [PubMed] [CrossRef]
183. Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, et al. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. 2013;288(2):915–26. doi:10.1074/jbc.M112.411363. [Google Scholar] [PubMed] [CrossRef]
184. Liu X, Ye B, Miller S, Yuan H, Zhang H, Tian L, et al. Ablation of ALCAT1 mitigates hypertrophic cardiomyopathy through effects on oxidative stress and mitophagy. Mol Cell Biol. 2012;32(21):4493–504. doi:10.1128/MCB.01092-12. [Google Scholar] [PubMed] [CrossRef]
185. Repici M, Giorgini F. DJ-1 in Parkinson’s disease: clinical insights and therapeutic perspectives. J Clin Med. 2019;8(9):1377. doi:10.3390/jcm8091377. [Google Scholar] [PubMed] [CrossRef]
186. Billia F, Hauck L, Grothe D, Konecny F, Rao V, Kim RH, et al. Parkinson-susceptibility gene DJ-1/PARK7 protects the murine heart from oxidative damage in vivo. Proc Natl Acad Sci U S A. 2013;110(15):6085–90. doi:10.1073/pnas.1303444110. [Google Scholar] [PubMed] [CrossRef]
187. Webber PJ, West AB. LRRK2 in Parkinson’s disease: function in cells and neurodegeneration. FEBS J. 2009;276(22):6436–44. doi:10.1111/j.1742-4658.2009.07342.x. [Google Scholar] [PubMed] [CrossRef]
188. Gehrke S, Imai Y, Sokol N, Lu B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature. 2010;466(7306):637–41. doi:10.1038/nature09191. [Google Scholar] [PubMed] [CrossRef]
189. Gagno G, Ferro F, Fluca AL, Janjusevic M, Rossi M, Sinagra G, et al. From brain to heart: possible role of Amyloid-β in ischemic heart disease and ischemia-reperfusion injury. Int J Mol Sci. 2020;21(24):9655. doi:10.3390/ijms21249655. [Google Scholar] [PubMed] [CrossRef]
190. Mukherjee UA, Ong SB, Ong SG, Hausenloy DJ. Parkinson’s disease proteins: novel mitochondrial targets for cardioprotection. Pharmacol Ther. 2015;156(3):34–43. doi:10.1016/j.pharmthera.2015.10.005. [Google Scholar] [PubMed] [CrossRef]
191. Cacabelos R. Parkinson’s disease: from pathogenesis to pharmacogenomics. Int J Mol Sci. 2017;18(3):551. doi:10.3390/ijms18030551. [Google Scholar] [PubMed] [CrossRef]
192. Kim HJ, Jeon B, Chung SJ. Professional ethics in complementary and alternative medicines in management of Parkinson’s disease. J Parkinsons Dis. 2016;6(4):675–83. doi:10.3233/JPD-160890. [Google Scholar] [PubMed] [CrossRef]
193. Athauda D, Foltynie T. The ongoing pursuit of neuroprotective therapies in Parkinson disease. Nat Rev Neurol. 2015;11(1):25–40. doi:10.1038/nrneurol.2014.226. [Google Scholar] [PubMed] [CrossRef]
194. Cutting E, Williams-Gray G. A trial investigating whether suppressing the immune system with azathioprine slows the progression of Parkinson’s disease. 2020. Available from: https://www.isrctn.com/ISRCTN14616801. [Accessed 2022]. [Google Scholar]
195. Trials C. A study to investigate the safety, tolerability, pharmacokinetics, and pharmacodynamics of RO7486967 in participants with early idiopathic Parkinson’s disease. 2022. Available from: https://www.isrctn.com/ISRCTN85338453. [Accessed 2022]. [Google Scholar]
196. Fang C, Hernandez P, Liow K, Damiano E, Zetterberg H, Blennow K, et al. Buntanetap, a novel translational inhibitor of multiple neurotoxic proteins, proves to be safe and promising in both Alzheimer’s and Parkinson’s patients. J Prev Alzheimers Dis. 2023;10(1):25–33. doi:10.14283/jpad.2022.84. [Google Scholar] [PubMed] [CrossRef]
197. Belén Sanz-Martos A, Fernández-Felipe J, Merino B, Cano V, Ruiz-Gayo M, Del Olmo N. Butyric acid precursor tributyrin modulates hippocampal synaptic plasticity and prevents spatial memory deficits: role of PPARγ and AMPK. Int J Neuropsychopharmacol. 2022;25(6):498–511. doi:10.1093/ijnp/pyac015. [Google Scholar] [PubMed] [CrossRef]
198. Athauda D, Maclagan K, Skene SS, Bajwa-Joseph M, Letchford D, Chowdhury K, et al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390(10103):1664–75. doi:10.1016/S0140-6736(17)31585-4. [Google Scholar] [PubMed] [CrossRef]
199. França C, Carra RB, Diniz JM, Munhoz RP, Cury RG. Deep brain stimulation in Parkinson’s disease: state of the art and future perspectives. Arq Neuropsiquiatr. 2022;80(5 Suppl 1):105–15. doi:10.1590/0004-282X-ANP-2022-S133. [Google Scholar] [PubMed] [CrossRef]
200. Pollak P, Fraix V, Krack P, Moro E, Mendes A, Chabardes S, et al. Treatment results: parkinson’s disease. Mov Disord. 2002;17(S3):S75–83. doi:10.1002/mds.10146. [Google Scholar] [PubMed] [CrossRef]
201. Fukaya C, Yamamoto T. Deep brain stimulation for Parkinson’s disease: recent trends and future direction. Neurol Med Chir. 2015;55(5):422–31. doi:10.2176/nmc.ra.2014-0446. [Google Scholar] [PubMed] [CrossRef]
202. Axelsen TM, Woldbye DPD. Gene therapy for Parkinson’s disease, an update. J Parkinsons Dis. 2018;8(2):195–215. doi:10.3233/JPD-181331. [Google Scholar] [PubMed] [CrossRef]
203. Liu Z, Cheung HH. Stem cell-based therapies for Parkinson disease. Int J Mol Sci. 2020;21(21):8060. doi:10.3390/ijms21218060. [Google Scholar] [PubMed] [CrossRef]
204. Sanders TH, Jaeger D. Optogenetic stimulation of cortico-subthalamic projections is sufficient to ameliorate bradykinesia in 6-ohda lesioned mice. Neurobiol Dis. 2016;95:225–37. doi:10.1016/j.nbd.2016.07.021. [Google Scholar] [PubMed] [CrossRef]
205. Yoon HH, Park JH, Kim YH, Min J, Hwang E, Lee CJ, et al. Optogenetic inactivation of the subthalamic nucleus improves forelimb akinesia in a rat model of Parkinson disease. Neurosurgery. 2014;74(5):533–41. doi:10.1227/NEU.0000000000000297. [Google Scholar] [PubMed] [CrossRef]
206. Yoon HH, Min J, Hwang E, Lee CJ, Suh JK, Hwang O, et al. Optogenetic inhibition of the subthalamic nucleus reduces levodopa-induced dyskinesias in a rat model of Parkinson’s disease. Stereotact Funct Neurosurg. 2016;94(1):41–53. doi:10.1159/000442891. [Google Scholar] [PubMed] [CrossRef]
207. Pienaar IS, Gartside SE, Sharma P, De Paola V, Gretenkord S, Withers D, et al. Pharmacogenetic stimulation of cholinergic pedunculopontine neurons reverses motor deficits in a rat model of Parkinson’s disease. Mol Neurodegener. 2015;10:47. doi:10.1186/s13024-015-0044-5. [Google Scholar] [PubMed] [CrossRef]
208. Huang J, Qin X, Cai X, Huang Y. Effectiveness of acupuncture in the treatment of Parkinson’s disease: an overview of systematic reviews. Front Neurol. 2020;11:917. doi:10.3389/fneur.2020.00917. [Google Scholar] [PubMed] [CrossRef]
209. Ko JH, Lee H, Kim SN, Park HJ. Does acupuncture protect dopamine neurons in Parkinson’s disease rodent model?: a systematic review and meta-analysis. Front Aging Neurosci. 2019;11:102. doi:10.3389/fnagi.2019.00102. [Google Scholar] [PubMed] [CrossRef]
210. Saidi T, Fortuin J, Douglas TS. Nanomedicine for drug delivery in South Africa: a protocol for systematic review. Syst Rev. 2018;7(1):154. doi:10.1186/s13643-018-0823-5. [Google Scholar] [PubMed] [CrossRef]
211. Silva S, Almeida AJ, Vale N. Importance of nanoparticles for the delivery of antiparkinsonian drugs. Pharmaceutics. 2021;13(4):508. doi:10.3390/pharmaceutics13040508. [Google Scholar] [PubMed] [CrossRef]
212. Urits I, Swanson D, Swett MC, Patel A, Berardino K, Amgalan A, et al. A review of patisiran (ONPATTRO®) for the treatment of polyneuropathy in people with hereditary transthyretin amyloidosis. Neurol Ther. 2020;9(2):301–15. doi:10.1007/s40120-020-00208-1. [Google Scholar] [PubMed] [CrossRef]
213. ClinicalTrialsgov2022. Available from: https://clinicaltrials.gov/ct2/results?cond=Parkinson+Disease&term=liposomes. [Accessed 2022]. [Google Scholar]
214. ClinicalTrialsgov2022. Available from: https://clinicaltrials.gov/ct2/results?recrs=&cond=Parkinson+Disease&term=nanoparticles. [Accessed 2022]. [Google Scholar]
215. Nasrolahi A, Shabani Z, Sadigh-Eteghad S, Salehi-Pourmehr H, Mahmoudi J. Stem cell therapy for the treatment of Parkinson’s disease: what promise does it hold? Curr Stem Cell Res Ther. 2024;19(2):185–99. doi:10.2174/1574888X18666230222144116. [Google Scholar] [PubMed] [CrossRef]
216. Diekämper E, Brix B, Stöcker W, Vielhaber S, Galazky I, Kreissl MC, et al. Neurofilament levels are reflecting the loss of presynaptic dopamine receptors in movement disorders. Front Neurosci. 2021;15:690013. doi:10.3389/fnins.2021.690013. [Google Scholar] [PubMed] [CrossRef]
217. Buhmann C, Magnus T, Choe CU. Blood neurofilament light chain in Parkinson’s disease. J Neural Transm. 2023;130(6):755–62. doi:10.1007/s00702-023-02632-7. [Google Scholar] [PubMed] [CrossRef]
218. Raj K, Singh S, Chib S, Mallan S. Microbiota- brain-gut-axis relevance to Parkinson’s disease: potential therapeutic effects of probiotics. Curr Pharm Des. 2022;28(37):3049–67. doi:10.2174/1381612828666221003112300. [Google Scholar] [PubMed] [CrossRef]
219. Sun MF, Shen YQ. Dysbiosis of gut microbiota and microbial metabolites in Parkinson’s disease. Ageing Res Rev. 2018;45:53–61. doi:10.1016/j.arr.2018.04.004. [Google Scholar] [PubMed] [CrossRef]
220. Liang Y, Cui L, Gao J, Zhu M, Zhang Y, Zhang HL. Gut microbial metabolites in Parkinson’s disease: implications of mitochondrial dysfunction in the pathogenesis and treatment. Mol Neurobiol. 2021;58(8):3745–58. doi:10.1007/s12035-021-02375-0. [Google Scholar] [PubMed] [CrossRef]
221. Cartella SM, Terranova C, Rizzo V, Quartarone A, Girlanda P. Covid-19 and Parkinson’s disease: an overview. J Neurol. 2021;268(12):4415–21. doi:10.1007/s00415-021-10721-4. [Google Scholar] [PubMed] [CrossRef]
222. Cullinane PW, de Pablo Fernandez E, König A, Outeiro TF, Jaunmuktane Z, Warner TT. Type 2 diabetes and Parkinson’s disease: a focused review of current concepts. Mov Disord. 2023;38(2):162–77. doi:10.1002/mds.29298. [Google Scholar] [PubMed] [CrossRef]
223. Filippou PS, Outeiro TF. Cancer and Parkinson’s disease: common targets, emerging hopes. Mov Disord. 2021;36(2):340–6. doi:10.1002/mds.28425. [Google Scholar] [PubMed] [CrossRef]
224. Grosu L, Grosu AI, Crisan D, Zlibut A, Perju-Dumbrava L. Parkinson’s disease and cardiovascular involvement: edifying insights (Review). Biomed Rep. 2023;18(3):25. doi:10.3892/br.2023.1607. [Google Scholar] [PubMed] [CrossRef]
225. Belarbi K, Cuvelier E, Destée A, Gressier B, Chartier-Harlin MC. NADPH oxidases in Parkinson’s disease: a systematic review. Mol Neurodegener. 2017;12(1):84. doi:10.1186/s13024-017-0225-5. [Google Scholar] [PubMed] [CrossRef]
226. Spathis AD, Asvos X, Ziavra D, Karampelas T, Topouzis S, Cournia Z, et al. Nurr1: RXRα heterodimer activation as monotherapy for Parkinson’s disease. Proc Natl Acad Sci U S A. 2017;114(15):3999–4004. doi:10.1073/pnas.1616874114. [Google Scholar] [PubMed] [CrossRef]
227. Kulisevsky J, Poyurovsky M. Adenosine A2A-receptor antagonism and pathophysiology of Parkinson’s disease and drug-induced movement disorders. Eur Neurol. 2012;67(1):4–11. doi:10.1159/000331768. [Google Scholar] [PubMed] [CrossRef]
228. Amalric M. Targeting metabotropic glutamate receptors (mGluRs) in Parkinson’s disease. Curr Opin Pharmacol. 2015;20:29–34. doi:10.1016/j.coph.2014.11.001. [Google Scholar] [PubMed] [CrossRef]
229. Esfandiary A, Finkelstein DI, Voelcker NH, Rudd D. Clinical sphingolipids pathway in Parkinson’s disease: from GCase to integrated-biomarker discovery. Cells. 2022;11(8):1353. doi:10.3390/cells11081353. [Google Scholar] [PubMed] [CrossRef]
230. Chen ZJ, Liang CY, Yang LQ, Ren SM, Xia YM, Cui L, et al. Association of Parkinson’s disease with microbes and microbiological therapy. Front Cell Infect Microbiol. 2021;11:619354. doi:10.3389/fcimb.2021.619354. [Google Scholar] [PubMed] [CrossRef]
231. Li A, Tyson J, Patel S, Patel M, Katakam S, Mao X, et al. Emerging nanotechnology for treatment of Alzheimer’s and Parkinson’s disease. Front Bioeng Biotechnol. 2021;9:672594. doi:10.3389/fbioe.2021.672594. [Google Scholar] [PubMed] [CrossRef]
232. Zoey FL, Palanivel M, Padmanabhan P, Gulyás B. Parkinson’s disease: a nanotheranostic approach targeting alpha-synuclein aggregation. Front Cell Dev Biol. 2021;9:707441. doi:10.3389/fcell.2021.707441. [Google Scholar] [PubMed] [CrossRef]
233. Nwabufo CK, Aigbogun OP. Diagnostic and therapeutic agents that target alpha-synuclein in Parkinson’s disease. J Neurol. 2022;269(11):5762–86. doi:10.1007/s00415-022-11267-9. [Google Scholar] [PubMed] [CrossRef]
234. Dovonou A, Bolduc C, Soto Linan V, Gora C, Peralta MRIII, Lévesque M. Animal models of Parkinson’s disease: bridging the gap between disease hallmarks and research questions. Transl Neurodegener. 2023;12(1):36. doi:10.1186/s40035-023-00368-8. [Google Scholar] [PubMed] [CrossRef]
235. Valadez-Barba V, Juárez-Navarro K, Padilla-Camberos E, Díaz NF, Guerra-Mora JR, Díaz-Martínez NE. Parkinson’s disease: an update on preclinical studies of induced pluripotent stem cells. Neurología (Engl Ed). 2023;38(9):681–94. doi:10.1016/j.nrleng.2023.10.004. [Google Scholar] [PubMed] [CrossRef]
236. Cesaroni V, Blandini F, Cerri S. Dyskinesia and Parkinson’s disease: animal model, drug targets, and agents in preclinical testing. Expert Opin Ther Targets. 2022;26(10):837–51. doi:10.1080/14728222.2022.2153036. [Google Scholar] [PubMed] [CrossRef]
237. Mari Z, Mestre TA. The disease modification conundrum in Parkinson’s disease: failures and hopes. Front Aging Neurosci. 2022;14:810860. doi:10.3389/fnagi.2022.810860. [Google Scholar] [PubMed] [CrossRef]
238. Huenchuguala S, Segura-Aguilar J. Single-neuron neurodegeneration as a degenerative model for Parkinson’s disease. Neural Regen Res. 2024;19(3):529–35. doi:10.4103/1673-5374.380878. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools