
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Paclitaxel induces human KOSC3 oral cancer cell apoptosis through caspase pathways
1 School of Medicine, College of Medicine, I-Shou University, Kaohsiung, 82445, Taiwan
2 Department of Otolaryngology, An Nan Hospital, China Medical University, Tainan, 70965, Taiwan
3 Department of Cell Biology and Anatomy, College of Medicine, National Cheng Kung University, Tainan, 70101, Taiwan
4 Department of Medical Research, China Medical University Hospital, China Medical University, Taichung, 40402, Taiwan
* Corresponding Authors: CHIA-YIH WANG. Email: ; BU-MIIN HUANG. Email:
(This article belongs to the Special Issue: Navigating the Interplay of Cancer, Autophagy, ER Stress, Cell Cycle and Apoptosis: Mechanisms, Therapies, and Future Directions)
BIOCELL 2024, 48(7), 1047-1054. https://doi.org/10.32604/biocell.2024.050701
Received 14 February 2024; Accepted 25 March 2024; Issue published 03 July 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
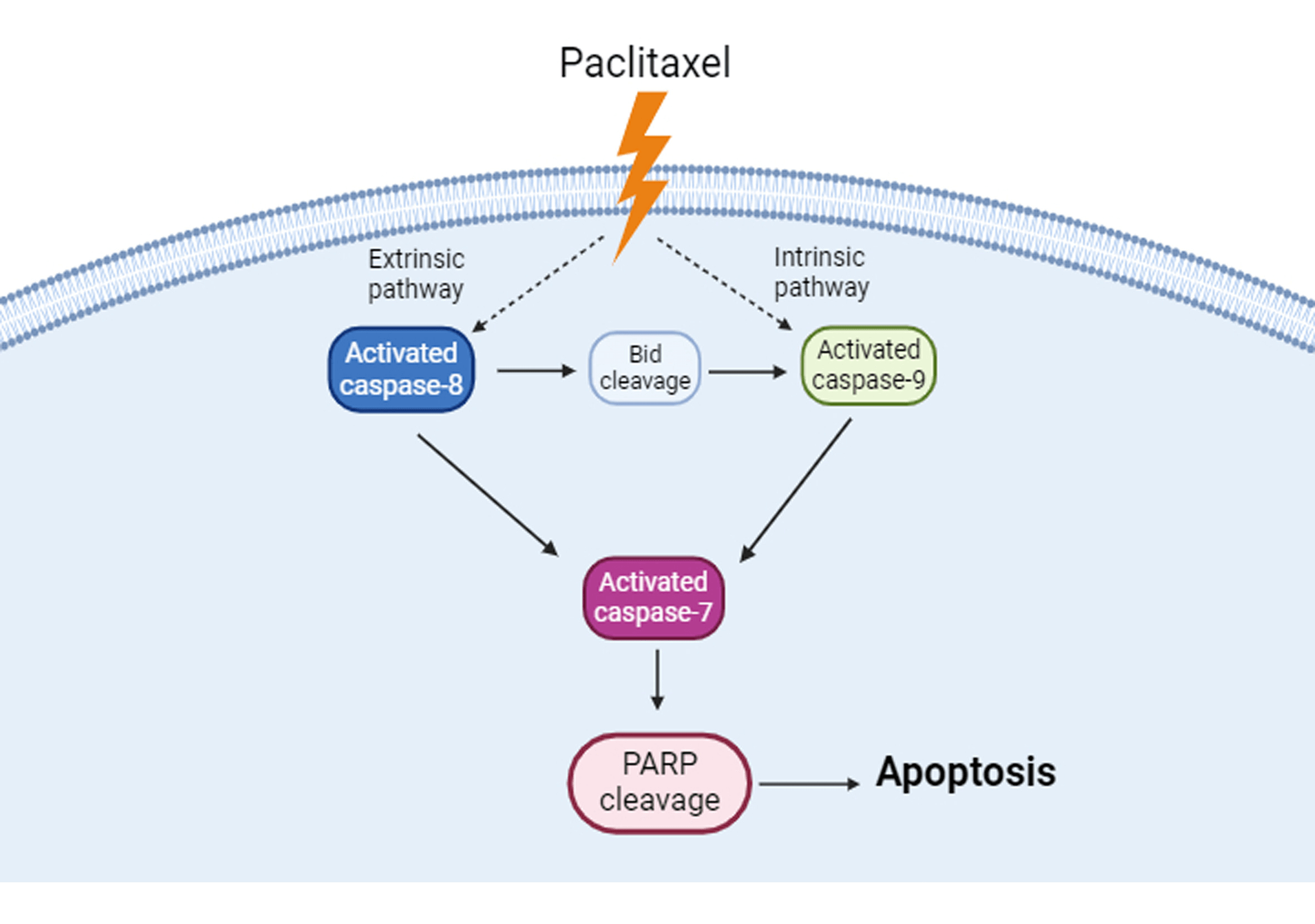
Background: Paclitaxel is a compound derived from Pacific yew bark that induces various cancer cell apoptosis. However, whether it also has anticancer activities in KOSC3 cells, an oral cancer cell line, is unclear. Methods: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, flow cytometry, and western blotting assays were carried out to assess cell viability, subG1 phase of the cell cycle, and apoptosis-related protein expression, respectively. Results: Our findings indicate that paclitaxel could inhibit cell viability and increase the expression of apoptotic markers, including plasma membrane blebbing and the cleavage of poly ADP-ribose polymerase in KOSC3 cells. Also, the treatment with paclitaxel remarkably elevated the percentage of the subG1 phase in KOSC3 cells. In addition, treatment with a pan-caspase inhibitor could recover paclitaxel-inhibited cell viability. Moreover, caspase-8, caspase-9, caspase-7, and BH3 interacting domain death agonist (Bid) were activated in paclitaxel-treated KOSC3 cells. Conclusions: Paclitaxel induced apoptosis through caspase cascade in KOSC3 cells.Graphic Abstract
Keywords
Abbreviations
| Bak | Bcl-2 homologous antagonist/killer |
| Bax | Bcl-2-associated X protein |
| Bcl-2 | B cell lymphoma 2 |
| Bid | BH3 interacting domain death agonist |
| DMSO | Dimethyl sulfoxide |
| ELISA | Enzyme-linked immunosorbent assay |
| lncRNA | Long non-coding RNA |
| HRP | Horseradish peroxidase |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PARP | ADP-ribose polymerase |
| PBS | Phosphate buffered saline |
| PI | Propidium iodide |
| RT | Room temperature |
Oral cancer is a type of head and neck squamous cancer [1] that typically arises from the lips, gums, tongue, mouth, or palate. Squamous cell carcinomas account for more than 90% of all cases [2]. Worldwide, over 400,000 people are diagnosed with this public health problem annually [3]. It has been reported that several areas and countries have a high incidence rate of oral cancers, such as India, Sri Lanka, Southern China, and the Taiwan region [4]. Chronic oral inflammation, alcohol consumption, tobacco smoking, and betel quid chewing are the relevant risk factors for oral cancers [5,6]. Despite various treatment modalities available, the 5-year survival rate of oral cancer patients is only approximately 50%, meaning that most patients did not benefit from the treatments [7]. Therefore, we must seek more effective methods or improve current treatment modalities to treat oral cancer.
Recently, some natural products extracted from plants have been reported to have anticancer activity. For example, the extract of Caesalpinia sappan L. can trigger cell death in lung and breast cancer cells [8]. In addition, the extract of Eclipta alba L. can inhibit viability, colony formation, and migration in colon cancer cells [9]. Also, the extract of Hibiscus sabdariffa leaf induces apoptosis in melanoma cancer cells [10]. Paclitaxel, a naturally occurring substance, was obtained from the Pacific yew bar (Taxus brevifolia), which also has anticancer activity and induces apoptosis in various cancer cell lines, such as lung, colon, nasopharyngeal, and oral cancer cells [11–14]. It can trigger apoptosis through multiple pathways. For instance, paclitaxel can induce apoptosis in the prostate cancer cells via reactive oxygen species-c-Jun N-terminal kinase-caspase-3 pathway [15]. Also, paclitaxel can activate B cell lymphoma 2 (Bcl-2) homologous antagonist/killer (Bak), a pro-apoptotic protein, to trigger apoptosis in breast cancer cells [16].
Apoptosis and necrosis are forms of cell death. When necrosis occurs, cells release cellular components into the extracellular space and initiate an inflammatory response, while apoptosis does not harm the host and causes inflammatory reactions [17,18]. Apoptosis has several biological features, including plasma membrane blebbing, cell shrinkage, formation of apoptotic body, and chromosomal DNA fragmentation [19,20]. Two major signaling pathways mediate apoptosis, the extrinsic (death receptor) and intrinsic (mitochondrial) pathways, and caspases are the vital proteins involved in executing both pathways. Caspases are a type of endoproteases that are classified into initiator caspases and effector caspases [19]. Caspase-8 and caspase-9 are the important initiator caspases, contributing to the intrinsic and extrinsic apoptosis pathways, respectively [19–21]. When initiator caspases are activated, they can cleave and activate their downstream effector caspases, such as caspase-7, resulting in poly ADP-ribose polymerase (PARP) cleavage that causes DNA fragmentation [19,22].
Previous studies have shown that paclitaxel could induce apoptosis in multiple oral cancer cell lines, such as OEC-M1, OC3, and CAL27 [11,23,24]. However, whether it also triggers KOSC3 oral cancer cell line apoptosis is still unknown. This study aims to evaluate whether paclitaxel can induce apoptosis in KOSC3 cells and investigate the underlying mechanism to provide a basic mechanism for designing and improving more effective paclitaxel-combining regimens to treat oral cancers.
Cell culture and drug treatment
KOSC3 oral cancer cell line was purchased from Health Science Research Resources Bank (Osaka, Japan) and cultured in RPMI-1640 medium (SH30096.02, Hyclone, UT, USA) supplemented with 10% fetal bovine serum (SH30070.03, Hyclone, UT, USA) at 37°C with 5% CO2. Paclitaxel was purchased from Sigma-Aldrich, Inc. (T7191, MO, USA), dissolved in Dimethyl sulfoxide (DMSO; D8418, Sigma-Aldrich, Inc., MO, USA) to make the 1 μM paclitaxel stock, and stored at −20°C. To explore the role of caspases in paclitaxel-induced cell death, KOSC3 cells were treated with a 10 μM pan-caspase inhibitor, Z-VAD-FMK (FMK001, R&D Systems, MN, USA), for 48 h.
The MTT assay was utilized to evaluate cell viability in accordance with a previous study protocol [25]. KOSC3 cells were seeded at a density of 1 × 104 cells per well in a 96-well plate. Following a 24 h incubation period at 37°C with 5% CO2, the cells were exposed to varying concentrations of paclitaxel for 24 and 48 h, respectively. The supernatants were aspirated at the indicated time points, and MTT reagent (M2128, Sigma-Aldrich, MO, USA; 0.5 mg/mL in phosphate buffered saline (PBS)) was added to each well. After a subsequent 4 h incubation at 37°C with 5% CO2, the supernatants were aspirated, and 100 μLof dimethyl sulfoxide (DMSO) was added to dissolve the formed crystals. The optical density readings were obtained at 570 nm using an enzyme-linked immunosorbent assay (ELISA) reader from BMG LABTECH, Ortenberg, Germany. The experiments were repeated independently a minimum of three times.
The alterations in the subG1 phase of the cell cycle, indicative of DNA fragmentation, were evaluated through flow cytometry analysis with propidium iodide (PI) staining (P4170, Sigma-Aldrich, Inc., MO, USA) staining [24]. After exposure to paclitaxel at specified time intervals, the cells were detached using 0.05% trypsin (25300054, Thermo Fisher Scientific Inc., MA, USA). Subsequently, they were rinsed with PBS thrice and then fixed with 70% ethanol at RT for 20 min. After 20 min, the fixed cells were rewashed with PBS thrice and then stained with PI (P4170, Sigma-Aldrich, Inc., MO, USA) staining solution (contained 40 µg/ml and RNase A (EN0531, Thermo Fisher Scientific Inc., MA, USA) 100 µg/mL in PBS) at RT for 1h. A FACScan flow cytometer (Becton, Dickinson and Company, NJ, USA) was carried out to analyze the cell cycle distribution of KOSC3 cells. The CellQuest software (version 5.1, Becton, Dickinson and Company, NJ, USA) was utilized to assess the proportion of cells in the subG1 phase of KOSC3 cells. The experiments were repeated independently a minimum of three times.
Total cell lysate preparation and Western blotting were performed as previously described [24]. Primary antibodies used in this study, including anti-cleaved caspase-8 (catalog number 9496; 1:1000), anti-cleaved caspase-9 (catalog number 7237; 1:1000), cleaved caspase-7 (catalog number 8438; 1:1000), anti-PARP (catalog number 9535; 1:1000), anti-BH3 interacting domain death agonist (Bid) (catalog number 2003; 1:1000), and anti-β-actin antibody (catalog number 58169; 1:50000) were purchased from Cell Signaling Technology (Cell Signaling Technology, MA, USA). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (catalog number 111-035-144; 1:5,000) and HRP-conjugated goat anti-mouse IgG (catalog number 111-035-146; 1:5,000) were purchased from Jackson ImmunoResearch, Inc. (PA, USA). The optical density of protein bands was quantified using Quantity One (PDI, Huntington Station, NY, USA), a computer-assisted image analysis system. The experiments were independently repeated a minimum of three times.
Data were presented as mean ± SEM. Statistical analyses were conducted using the SPSS software version 17.0. To compare differences between the two groups, the unpaired Student t-test was used. To compare differences among multiple groups the One-way ANOVA and Tukey’s post-hoc tests were utilized. p-value < 0.05 was defined as statistical significance.
Paclitaxel inhibits cell viability in KOSC3 cells
In a previous study, we investigated the apoptotic effects of paclitaxel at concentrations of 5, 50, and 500 nM on OEC-M1 and OC3 oral cancer cell lines [11]. To minimize the difference in experimental results caused by changes in paclitaxel concentration, we used the same three concentrations in this study to explore the mechanism of paclitaxel-induced apoptosis in KOSC3 oral cancer cells. We treated KOSC3 cells with different paclitaxel concentrations (0, 5, 50, and 500 nM) at 24 and 48 h, respectively, to evaluate whether paclitaxel affected the viability of KOSC3 cells. Our MTT data showed that treatment with paclitaxel at 50 and 500 nM for 24 h (Fig. 1A, p < 0.05) and treatment with paclitaxel at 50 and 500 nM for 48 h, respectively, could significantly reduce the cell viability of KOSC3 cells (Fig. 1B, p < 0.05).
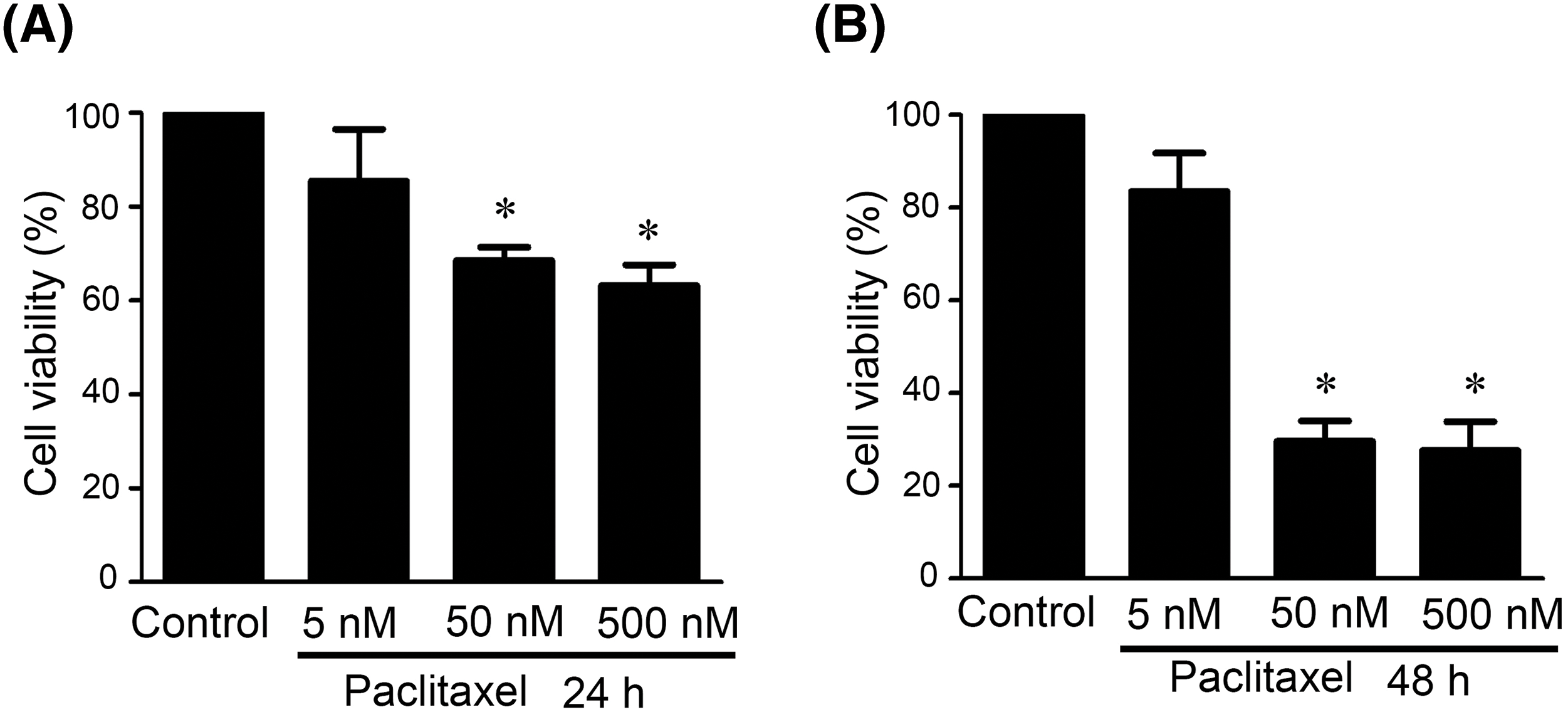
Figure 1: Paclitaxel inhibited cell viability in KOSC3 cells. KOSC3 cells were treated without (control) or with various concentrations of paclitaxel (0, 5, 50 and 500 nM) for 24 (A) and 48 h (B), respectively. The MTT assay was carried out to analyze the cell viability. n = 3, One-way ANOVA, and Tukey’s post-hoc test was performed, *p < 0.05 vs. control.
Paclitaxel induces apoptosis in KOSC3 cells
Next, we evaluated whether paclitaxel could induce apoptosis in KOSC3 cells. From observing the morphological changes between control and paclitaxel-treated cells, we observed that paclitaxel-treated cells exhibited more membrane blebbing (Fig. 2A), a feature of apoptosis, than control cells. To further confirm that paclitaxel can induce apoptosis, we treated KOSC3 with paclitaxel at 8, 16, 20, 24, and 48 h to evaluate the changes in the subG1 phase of the cell cycle. The quantitative analysis showed that the percentage of paclitaxel-treated KOSC3 cells in the subG1 phase was increased in each treatment time compared to control (Fig. 2B). We also investigated whether the cleavage of PARP, an apoptotic marker [20], was also affected by paclitaxel treatment. Thus, we treated KOSC3 cells with paclitaxel for 16, 24, and 48 h and performed Western blotting to detect cleaved PARP expression (Fig. 2C). Quantitative evaluation of the Western blots demonstrated a significant elevation in cleaved PARP expression levels compared to the control group at 16, 24, and 48 h post-paclitaxel treatment (Fig. 2D, p < 0.05). Our results suggest that paclitaxel induced KOSC3 cell death through apoptosis.
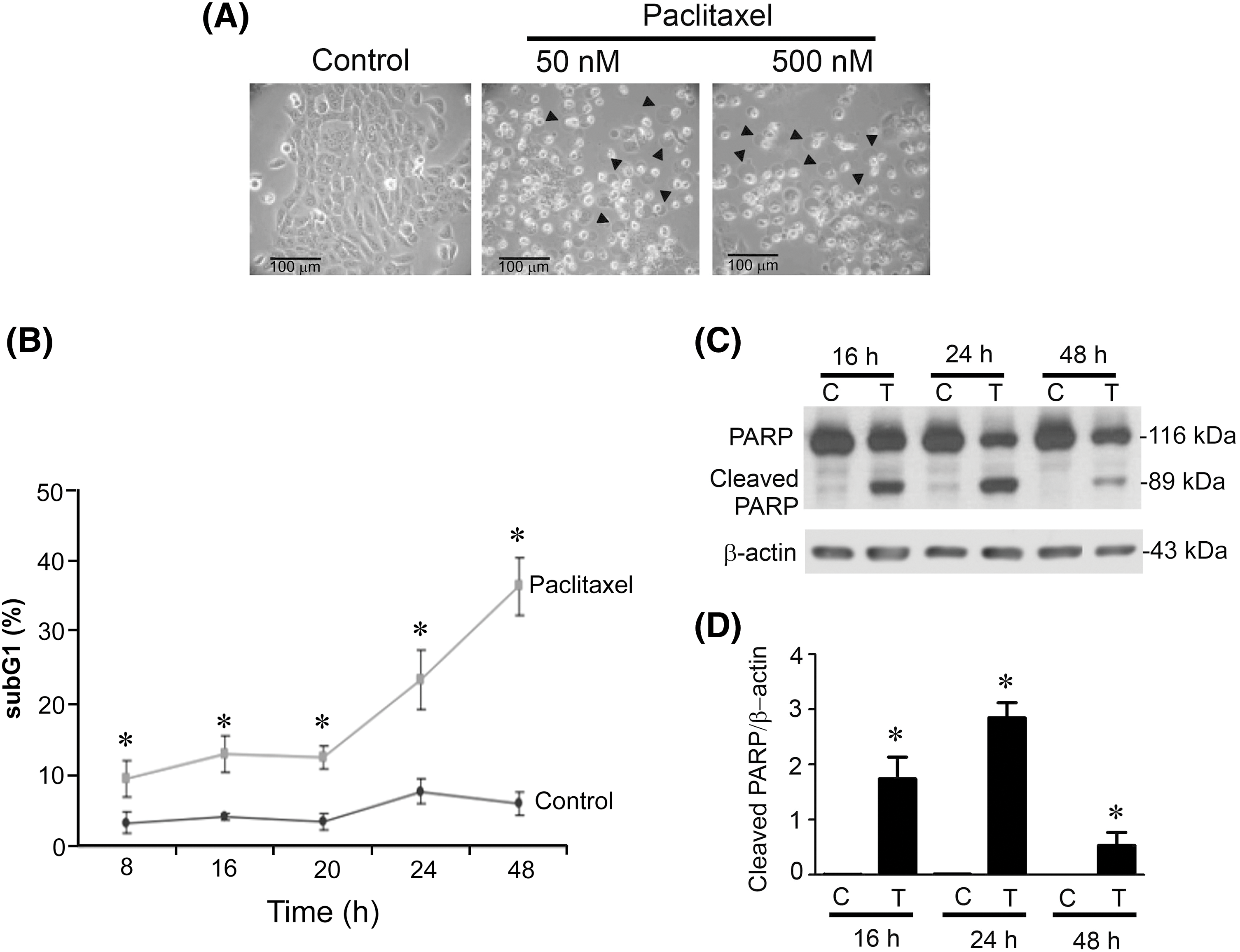
Figure 2: Paclitaxel induced apoptosis in KOSC3 cells. (A) KOSC3 cells were treated without (control) or with 50 nM paclitaxel for 48 h. The morphological changes of cells were observed using light microscopy (scale bar: 100 μm; arrowheads: membrane-blebbed cells). (B) KOSC3 cells were treated without (control) or with 50 nM paclitaxel for 8, 20, 24, and 48 h. A FACScan flow cytometer was used to analyze the changes in the subG1 phase of the cell cycle, and the percentage of KOSC3 cells in the subG1 phase was analyzed using CellQuest software. (C) KOSC3 cells were treated without (C) or with 50 nM paclitaxel (T) for 16, 24, and 48 h. Western blotting was used to analyze cleaved PARP and β-actin protein expression. (D) The Quantity One image analysis system was used to quantify the cleaved PARP expression. n = 3, unpaired Student’s t-test was performed, *p < 0.05 vs. control.
Caspases are involved in paclitaxel-induced KOSC3 cell death
Since caspases are the mediators of apoptosis in both extrinsic and intrinsic pathways [19,21], we treated KOSC3 cells with a pan-caspase inhibitor, Z-VAD-FMK, to investigate whether caspases participated in paclitaxel-induced cell death. Fig. 3 (p < 0.05) shows that treatment with Z-VAD-FMK could significantly recover the cell viability inhibited by paclitaxel, indicating caspases were involved in paclitaxel-induced cell death.
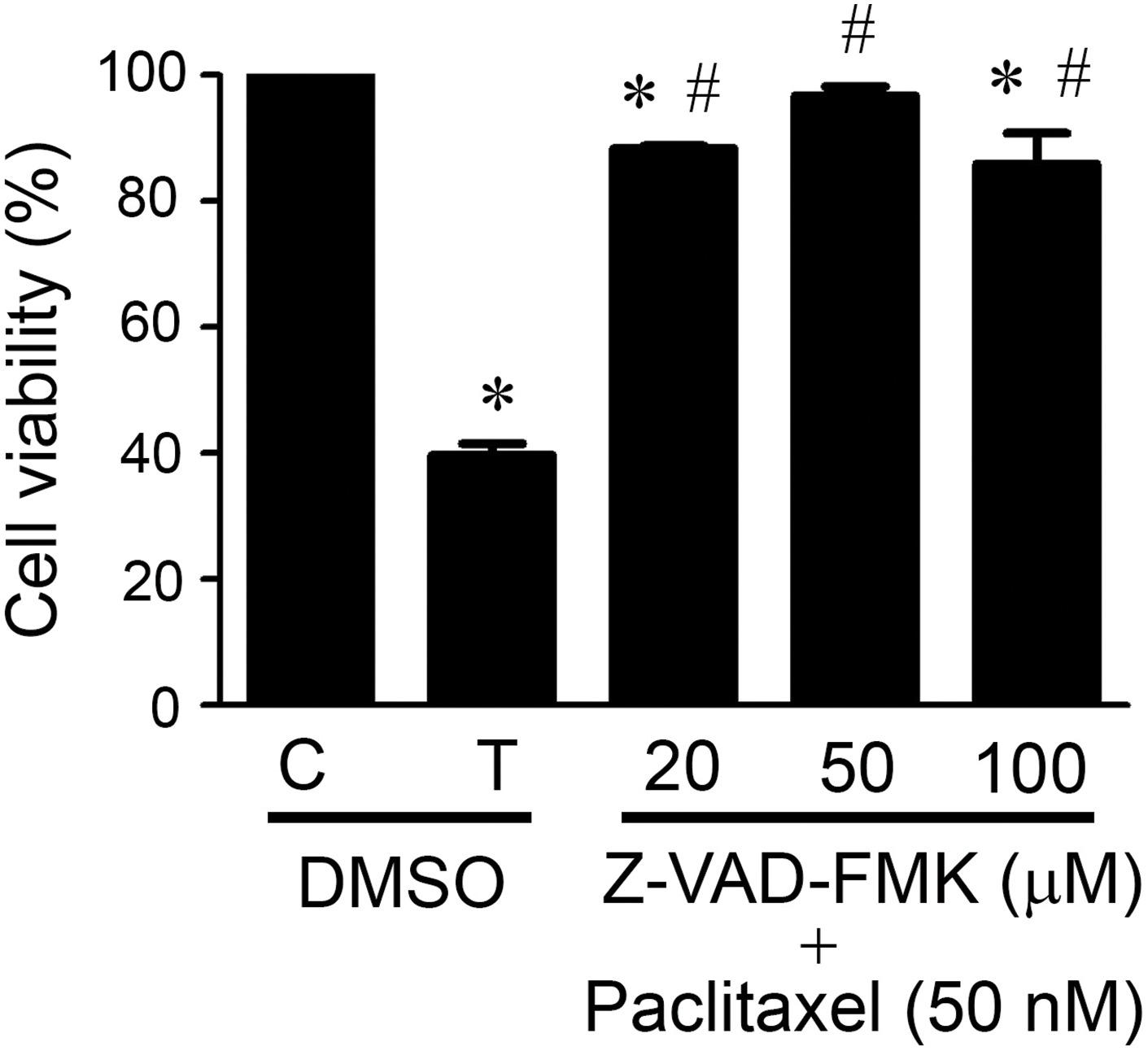
Figure 3: Caspases were involved in paclitaxel-induced KOSC3 cell death. KOSC3 cells were treated without (C) or with 50 nM paclitaxel (T) and concurrently treated with the solvent control DMSO or various concentrations of Z-VAD-FMK for 48 h. The MTT assay was carried out to analyze the cell viability. n = 3, One-way ANOVA, and Tukey’s post-hoc test was performed, *p < 0.05 vs. C + DMSO group. #p < 0.05 vs. T+ DMSO group.
Caspase-8, caspase-9, and caspase-7 are activated in paclitaxel-treated KOSC3 cells
It is well known that caspase-8 mediates the extrinsic pathway, while caspase-9 initiates the intrinsic pathway [19,21]. Therefore, the activation of caspase-8 or caspase-9 represents the initiation of the intrinsic or extrinsic pathway of apoptosis. We performed Western blot analysis to determine whether paclitaxel-induced apoptosis occurred through the extrinsic or intrinsic pathways by detecting the expression of cleaved (activated) initiator caspase-8 and initiator caspase-9 (Fig. 4A). The quantitative results showed that cleaved caspase-8 expression levels were more significantly increased than the control at 16, 24, and 48 h after paclitaxel treatment (Fig. 4B, p < 0.05). Similarly, after paclitaxel treatment, the cleaved caspase-9 protein expression levels were also significantly increased at 16, 24, and 48 h than the control (Fig. 4C, p < 0.05), suggesting that both pathways were activated in paclitaxel-treated KOSC3 cells. In addition, caspase-7 is a downstream effector caspase of caspase-8 and caspase-9. We also investigated whether caspase-7 was cleaved in paclitaxel-treated KOSC3 cells (Fig. 4D). The quantitative analysis of the blots showed that the expression levels of cleaved caspase-7 were significantly increased than the control at 16, 24, and 48 h after paclitaxel treatment (Fig. 4D, p < 0.05). Our results suggest that both initiator and effector caspases were activated in paclitaxel-treated KOSC3 cells.
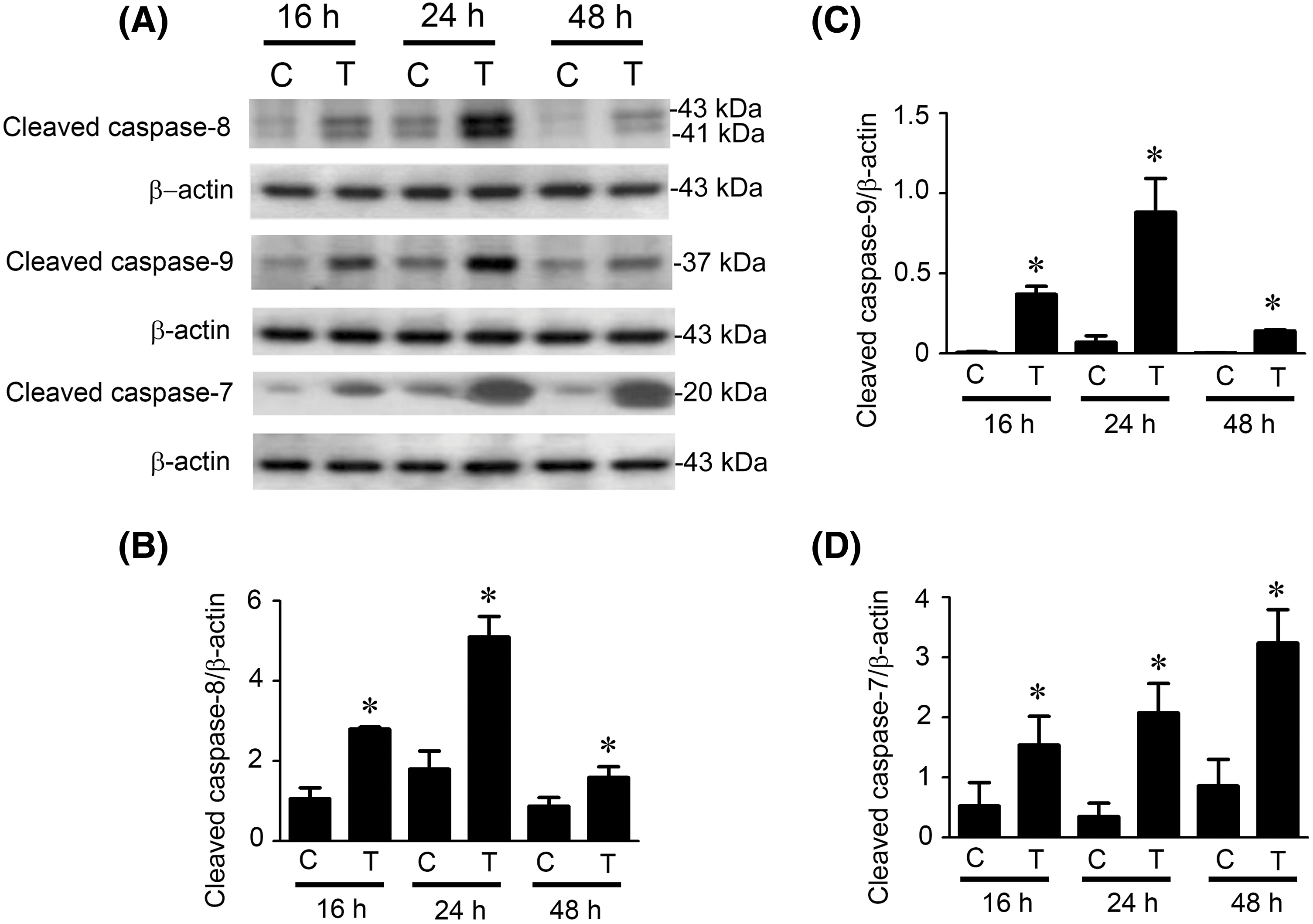
Figure 4: Caspase-8, caspase-9, and caspase-7 were activated in paclitaxel-treated KOSC3 cells. (A) KOSC3 cells were treated without (C) or with 50 nM paclitaxel (T) for 16, 24, and 48 h. Western blotting was performed to detect the cleaved caspase-8, cleaved caspase-9, cleaved caspase-7, and β-actin expression. The Quantity One image analysis system quantified protein expression levels of (B) cleaved caspase-8, (C) cleaved caspase-9, and (D) cleaved caspase-7. n = 3, unpaired Student’s t-test was performed, *p < 0.05 vs. control.
Paclitaxel induces bid cleavage in KOSC3 cells
Bid, a pro-apoptotic Bcl-2 protein, is downstream of caspase-8 [19,21]. Caspase-8 could cleave Bid into t-Bid, which causes Bid activation, resulting in mitochondrial pathway initiation [19,21]. We performed the Western blotting to detect whether Bid was cleaved after paclitaxel treatment (Fig. 5A). The blots were quantitatively analyzed, which showed that truncation of Bid (Bid activation) remarkably occurred at 24, 48, and 72 h post-paclitaxel treatment (Fig. 5B, p < 0.05).
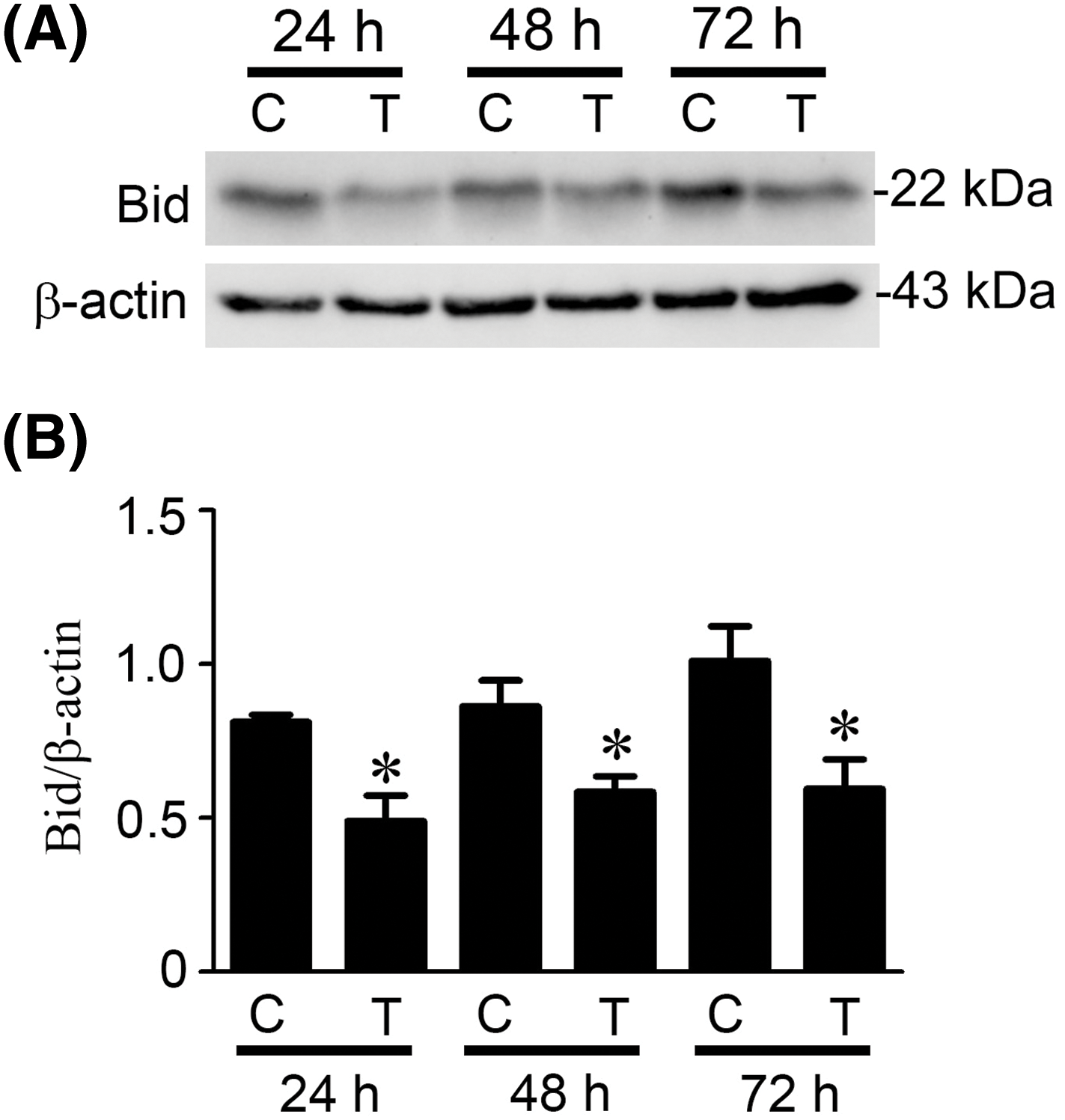
Figure 5: Paclitaxel induced Bid cleavage in KOSC3 cells. KOSC3 cells were treated without (C) or with 50 nM paclitaxel (T) for 24, 48, and 72 h. (A) Protein expression of Bid and β-actin was examined using Western blotting. (B) The Quantity One image analysis system was used to quantify the cleavage of Bid. n = 3, unpaired Student’s t-test was performed, *p < 0.05 vs. control.
Paclitaxel is an extracted substance from the bark of Pacific yew (Taxus brevifolia). Although previous studies have demonstrated it has anticancer activity on several oral cancer cell lines, such as OEC-M1, OC3, and CAL27 cells [11,23,24], it is still unclear whether paclitaxel also has anticancer effects on KOSC3 cells, a cancer cell line derived from gingival squamous cell carcinoma. In this study, data from MTT assay showed that paclitaxel could inhibit cell viability in KOSC3 cells (Fig. 1). Our previous study showed that after a 24 h paclitaxel treatment, cell viability decreased by approximately 20% in OEC-M1 cells and 60% in OC3 cells [11]. The present study also found that cell viability only reduced by approximately 25% in KOSC3 cells after paclitaxel treatment for 24 h (Fig. 1A). We observed that OC3 is more sensitive to paclitaxel than OEC-M1 or KOSC3 cells. It has been shown that p53 plays a critical role in promoting apoptosis under paclitaxel treatment in cancer cells [26]. As OEC-M1 and KOSC3 are p53 mutation cell lines [27,28], the mutation might be the reason for their insensitivity to paclitaxel. Our findings may provide related evidence supporting that p53 plays a vital role in cell viability inhibition under paclitaxel treatment in oral cancer cells.
The flow cytometry and Western blotting results further revealed that the subG1 phase percentage (Fig. 2B) and the cleaved of PARP (Fig. 2C) were increased in paclitaxel-treated KOSC3, indicating paclitaxel induced KOSC3 cell death through apoptosis. We observed that the protein expression level of cleaved PARP was remarkably increased at 24 h (Fig. 2C), but the protein was reduced substantially at 48 h (Fig. 2C) after paclitaxel treatment. This is consistent with our previous findings in two oral cancer cell lines, OEC-M1 and OC3 [11]. The N-terminus of proteins is involved in regulating protein stability [29]. It is known that PARP contains a cleavage site for effector caspases on its N-terminus [30]. Therefore, the cleaved PARP may lose its N-terminus, accelerating the protein degradation.
Also, treatment with a pan-caspase inhibitor (Z-VAD-FMK) could significantly restore paclitaxel-induced KOSC3 viability inhibition (Fig. 3). These results found in the present study are consistent with the results of paclitaxel treatment in OC3 and OEC-M1 oral cancer cell lines [11], suggesting that paclitaxel induces apoptosis in the three oral cancer cell lines through the caspase pathway.
Since treatment with Z-VAD-FMK significantly blocked paclitaxel-induced cell death, suggesting that caspase activation is involved in cell death induced by paclitaxel in KOSC3 cells (Fig. 3). We used a Western blotting assay to examine whether caspases were cleaved (activated) in paclitaxel-treated KOSC3 cells. Our results demonstrated that the cleaved of initiator caspases, caspase-8 and caspase-9 (Fig. 4A), plus the effector caspase, caspase-7 (Fig. 4A), were increased by paclitaxel treatment, indicating the caspase cascade is activated under paclitaxel treatment in KOSC3 cells.
Activation of initiator caspase-8 or initiator caspase-9 represents the induction of the intrinsic or extrinsic apoptotic pathways, respectively [19,21], our results showed that the activities of caspase-8 (Fig. 4B) and caspase-9 (Fig. 4C) were initiated at 16 h, peaked at 20 h, and degraded at 48 h after paclitaxel treatment. This suggests that paclitaxel may activate the extrinsic and intrinsic pathways simultaneously in KOSC3 cells. In addition, Bid, a pro-apoptotic Bcl-2 protein cleaved by caspase-8, can contribute to the link between intrinsic and extrinsic apoptotic pathways [19,21]. Our study found that Bid was cleaved (activated) by paclitaxel treatment (Fig. 5). Therefore, the extrinsic may connect with the intrinsic pathway to induce apoptosis in paclitaxel-treated KOSC3 cells.
This study also found that either treatment with caspase-8 or caspase-9 inhibitors alone did not inhibit paclitaxel-induced KOSC3 cell death (data not shown). Furthermore, combined treatment with both inhibitors did not recover paclitaxel-induced KOSC3 cell death (data not shown). In addition to caspase-8, caspase-9, caspase-2, and caspase-10 are also initiator caspases [19,31], and they can cause Bid cleavage, contributing to apoptosis [19,32,33]. The present study discovered that paclitaxel also induced Bid cleavage. Thus, it is speculated that other initiator caspases, such as caspase-2 and caspase-10, may play a role in paclitaxel-induced apoptosis. Further studies could be investigated for confirmation.
It is still unknown how paclitaxel activated extrinsic and intrinsic pathways in KOSC3 cells. Paclitaxel is known to target the tubulin and suppress the dynamics of microtubules, which causes mitotic arrest [34]. This leads to DNA damage and p53 activation [35], ultimately contributing to apoptosis [36,37]. However, as KOSC3 is a p53 mutation cell line [28], other cellular signaling pathways may be involved in paclitaxel-induced apoptosis in these cells. For instance, DNA damage can activate the c-Jun N-terminal kinase and extracellular signal-regulated kinase pathways [38,39], which promote the expression of death receptor ligands such as Fas ligand and tumor necrosis factor-α [40–42]. The binding of these death receptor ligands to their death receptors will initiate the extrinsic pathway [19,21]. It is known that the expression of pro-apoptotic Bcl-2 protein, such as Bax and Bak, can activate the intrinsic pathway [19,21]. In this study, we found that paclitaxel can activate Bid. In addition, previous studies also report that paclitaxel can trigger the expression of Bax and Bak [43,44]. According to these studies, paclitaxel could induce death receptor ligands or pro-apoptotic Bcl-2 proteins to initiate extrinsic and intrinsic apoptotic pathways in KOSC3 cells. It should be interesting to examine the expression of the related apoptotic proteins, such as death receptor ligands and pro-apoptotic Bcl-2 proteins, in paclitaxel-treated KOSC3 cells further in the future.
In addition, long non-coding RNA (lncRNA) is involved in regulating caspase expression and activation [45]. Due to budget and technological limitations, we could not investigate the involvement of lncRNA in paclitaxel-induced KOSC3 apoptosis in this study. However, in the future, we plan to collaborate with lncRNA-related laboratories to understand better whether lncRNA is involved in regulating paclitaxel-induced apoptosis.
The side effects of paclitaxel treatment include hypersensitivity reactions, peripheral neuropathy, myalgias, neutropenia, and arthralgia [46], which may cause significant discomfort and complications for paclitaxel-treated patients. It has been found that certain natural extracts, such as resveratrol [47] and curcumin [48], can increase the effectiveness of paclitaxel-induced apoptosis. Our study shows that paclitaxel could induce the apoptosis of KOSC3 oral cancer cells. In the future, we will also investigate whether other natural extracts with anti-cancer properties, such as fucoidan [49] and oligonol [50], can enhance paclitaxel-induced apoptosis to reduce the side effects of paclitaxel in oral cancer treatment.
This study demonstrates that paclitaxel is able to induce apoptosis in KOSC3 cells through the intrinsic and extrinsic apoptotic pathways. Our findings may serve as a reference for designing more effective paclitaxel-based therapeutics to enhance apoptotic cell death in oral cancer cells.
Acknowledgement: None.
Funding Statement: The present study was supported by the National Science and Technology Council, Taiwan (MOST-107-2320-B-471-001 to YYL and MOST-110-2320-B-006-025-MY3 to BMH) and by An Nan Hospital (ANHRF111-55 to TCC and BMH).
Author Contributions: YYL, TCC, and BMH designed the present study and analyzed the data. YYL, TCC, and YPL performed all the experiments. YYL, TCC, and BMH wrote the initial manuscript. CYW and BMH revised the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Paduano FAE, Marrelli B, Dattilo V, Hussaini HM, Cooper PR, Tatullo M. Translational aspects of the modern genetics in head and neck cancers. BIOCELL. 2022;46(8):1827–35. doi:10.32604/biocell.2022.020462. [Google Scholar] [CrossRef]
2. Chen SH, Hsiao SY, Chang KY, Chang JY. New insights into oral squamous cell carcinoma: from clinical aspects to molecular tumorigenesis. Int J Mol Sci. 2021;22(5):2252. doi:10.3390/ijms22052252. [Google Scholar] [PubMed] [CrossRef]
3. Abati S, Bramati C, Bondi S, Lissoni A, Trimarchi M. Oral cancer and precancer: a narrative review on the relevance of early diagnosis. Int J Environ Res Public Health. 2020;17(24):9160. doi:10.3390/ijerph17249160. [Google Scholar] [PubMed] [CrossRef]
4. Wang M, Xiao C, Ni P, Yu JJ, Wang XW, Sun H. Correlation of betel quid with oral cancer from 1998 to 2017: a study based on bibliometric analysis. Chin Med J. 2018;131(16):1975–82. doi:10.4103/0366-6999.238140. [Google Scholar] [PubMed] [CrossRef]
5. Irani S, Barati I, Badiei M. Periodontitis and oral cancer—current concepts of the etiopathogenesis. Oncol Rev. 2020;14(1):465. [Google Scholar] [PubMed]
6. Liu H, Yu Z, Xu Z, Liu T, Liu W. A scientometric study of tobacco and alcohol use as risk factors for oral cavity health. J Dent Sci. 2023;18(4):1883–8. doi:10.1016/j.jds.2023.05.016. [Google Scholar] [PubMed] [CrossRef]
7. Pignon JP, le Maitre A, Maillard E, Bourhis J, Group MNC. Meta-analysis of chemotherapy in head and neck cancer (MACH-NCan update on 93 randomised trials and 17,346 patients. Radiother Oncol. 2009;92(1):4–14. doi:10.1016/j.radonc.2009.04.014. [Google Scholar] [PubMed] [CrossRef]
8. Naik Bukke A, Nazneen Hadi F, Babu KS, Shankar PC. In vitro studies data on anticancer activity of Caesalpinia sappan L. heartwood and leaf extracts on MCF7 and A549 cell lines. Data Brief. 2018;19:868–77. doi:10.1016/j.dib.2018.05.050. [Google Scholar] [PubMed] [CrossRef]
9. Nelson VK, Sahoo NK, Sahu M, Sudhan HH, Pullaiah CP, Muralikrishna KS. In vitro anticancer activity of Eclipta alba whole plant extract on colon cancer cell HCT-116. BMC Complement Med Ther. 2020;20(1):355. doi:10.1186/s12906-020-03118-9. [Google Scholar] [PubMed] [CrossRef]
10. Chiu CT, Hsuan SW, Lin HH, Hsu CC, Chou FP, Chen JH. Hibiscus sabdariffa leaf polyphenolic extract induces human melanoma cell death, apoptosis, and autophagy. J Food Sci. 2015;80(3):H649–58. [Google Scholar] [PubMed]
11. Hsiao JR, Leu SF, Huang BM. Apoptotic mechanism of paclitaxel-induced cell death in human head and neck tumor cell lines. J Oral Pathol Med. 2009;38(2):188–97. doi:10.1111/jop.2009.38.issue-2. [Google Scholar] [CrossRef]
12. von Eiff D, Bozorgmehr F, Chung I, Bernhardt D, Rieken S, Liersch S, et al. Paclitaxel for treatment of advanced small cell lung cancer (SCLCa retrospective study of 185 patients. J Thorac Dis. 2020;12(3):782–93. doi:10.21037/jtd. [Google Scholar] [CrossRef]
13. Hao Y, Li Z, Chang M, Zhang X. Effects of salidroside combined with paclitaxel on proliferation, migration, and epithelial mesenchyme of colorectal cancer cells. Drug Des Devel Ther. 2022;16:4079–89. doi:10.2147/DDDT.S384151. [Google Scholar] [PubMed] [CrossRef]
14. Zhao C, Chen HY, Zhao F, Feng HJ, Su JP. Acylglycerol kinase promotes paclitaxel resistance in nasopharyngeal carcinoma cells by regulating FOXM1 via the JAK2/STAT3 pathway. Cytokine. 2021;148:155595. doi:10.1016/j.cyto.2021.155595. [Google Scholar] [PubMed] [CrossRef]
15. Zhang Y, Tang Y, Tang X, Wang Y, Zhang Z, Yang H. Paclitaxel induces the apoptosis of prostate cancer cells via ROS-mediated HIF-1α expression. Molecules. 2022;27(21):7183. doi:10.3390/molecules27217183. [Google Scholar] [PubMed] [CrossRef]
16. Miller AV, Hicks MA, Nakajima W, Richardson AC, Windle JJ, Harada H. Paclitaxel-induced apoptosis is BAK-dependent, but BAX and BIM-independent in breast tumor. PLoS One. 2013;8(4):e60685. doi:10.1371/journal.pone.0060685. [Google Scholar] [PubMed] [CrossRef]
17. Yang Y, Jiang G, Zhang P, Fan J. Programmed cell death and its role in inflammation. Mil Med Res. 2015;2:12. [Google Scholar] [PubMed]
18. Proskuryakov SY, Konoplyannikov AG, Gabai VL. Necrosis: a specific form of programmed cell death? Exp Cell Res. 2003;283(1):1–16. doi:10.1016/S0014-4827(02)00027-7. [Google Scholar] [PubMed] [CrossRef]
19. Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17(7):395–417. doi:10.1038/s41571-020-0341-y. [Google Scholar] [PubMed] [CrossRef]
20. Hajibabaie F, Abedpoor N, Mohamadynejad P. Types of cell death from a molecular perspective. Biology. 2023;12(11):1426. doi:10.3390/biology12111426. [Google Scholar] [PubMed] [CrossRef]
21. Vitale I, Pietrocola F, Guilbaud E, Aaronson SA, Abrams JM, Adam D, et al. Apoptotic cell death in disease-current understanding of the NCCD 2023. Cell Death Differ. 2023;30(5):1097–154. doi:10.1038/s41418-023-01153-w. [Google Scholar] [PubMed] [CrossRef]
22. Mashimo M, Onishi M, Uno A, Tanimichi A, Nobeyama A, Mori M, et al. The 89-kDa PARP1 cleavage fragment serves as a cytoplasmic PAR carrier to induce AIF-mediated apoptosis. J Biol Chem. 2021;296:100046. doi:10.1074/jbc.RA120.014479. [Google Scholar] [PubMed] [CrossRef]
23. Liu M, Zhang J, Li JF, Wang XX. Roles of curcumin combined with paclitaxel on growth inhibition and apoptosis of oral squamous cell carcinoma cell line CAL27 in vitro. Shanghai Kou Qiang Yi Xue. 2016;25(5):538–41. [Google Scholar] [PubMed]
24. Lan YY, Chen YH, Liu C, Tung KL, Wu YT, Lin SC, et al. Role of JNK activation in paclitaxel-induced apoptosis in human head and neck squamous cell carcinoma. Oncol Lett. 2021;22(4):705. doi:10.3892/ol. [Google Scholar] [CrossRef]
25. Nonaka M, Ikeda H, Fujisawa A, Uehara M, Inokuchi T. Induction of apoptosis by paclitaxel in human oral carcinoma cells. Int J Oral Maxillofac Surg. 2006;35(7):649–52. doi:10.1016/j.ijom.2006.01.011. [Google Scholar] [PubMed] [CrossRef]
26. Wu W, Wei T, Li Z, Zhu J. p53-dependent apoptosis is essential for the antitumor effect of paclitaxel response to DNA damage in papillary thyroid carcinoma. Int J Med Sci. 2021;18(14):3197–205. doi:10.7150/ijms.61944. [Google Scholar] [PubMed] [CrossRef]
27. Lin YS, Chen SF, Liu CL, Nieh S. The chemoadjuvant potential of grape seed procyanidins on p53-related cell death in oral cancer cells. J Oral Pathol Med. 2012;41(4):322–31. doi:10.1111/jop.2012.41.issue-4. [Google Scholar] [CrossRef]
28. Inagaki T, Matsuwari S, Takahashi R, Shimada K, Fujie K, Maeda S. Establishment of human oral-cancer cell lines (KOSC-2 and -3) carrying p53 and c-myc abnormalities by geneticin treatment. Int J Cancer. 1994;56(2):301–8. doi:10.1002/ijc.v56:2. [Google Scholar] [CrossRef]
29. Xu S, Xu X, Wang Z, Wu R. A systematic investigation of proteoforms with N-terminal glycine and their dynamics reveals its impacts on protein stability. Angew Chem Int Ed Engl. 2024;63(6):e202315286. doi:10.1002/anie.v63.6. [Google Scholar] [CrossRef]
30. Kamaletdinova T, Fanaei-Kahrani Z, Wang ZQ. The enigmatic function of PARP1: from PARylation activity to PAR readers. Cells. 2019;8(12):1625. doi:10.3390/cells8121625. [Google Scholar] [PubMed] [CrossRef]
31. Sahoo G, Samal D, Khandayataray P, Murthy MK. A review on caspases: key regulators of biological activities and apoptosis. Mol Neurobiol. 2023;60(10):5805–37. doi:10.1007/s12035-023-03433-5. [Google Scholar] [PubMed] [CrossRef]
32. Vigneswara V, Ahmed Z. The role of caspase-2 in regulating cell fate. Cells. 2020;9(5):1259. doi:10.3390/cells9051259. [Google Scholar] [PubMed] [CrossRef]
33. Milhas D, Cuvillier O, Therville N, Clave P, Thomsen M, Levade T, et al. Caspase-10 triggers Bid cleavage and caspase cascade activation in FasL-induced apoptosis. J Biol Chem. 2005;280(20):19836–42. doi:10.1074/jbc.M414358200. [Google Scholar] [PubMed] [CrossRef]
34. Weaver BA. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell. 2014;25(18):2677–81. doi:10.1091/mbc.e14-04-0916. [Google Scholar] [PubMed] [CrossRef]
35. Orth JD, Loewer A, Lahav G, Mitchison TJ. Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and p53 induction. Mol Biol Cell. 2012;23(4):567–76. doi:10.1091/mbc.e11-09-0781. [Google Scholar] [PubMed] [CrossRef]
36. Roos WP, Kaina B. DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013;332(2):237–48. doi:10.1016/j.canlet.2012.01.007. [Google Scholar] [PubMed] [CrossRef]
37. Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018;25(1):104–13. doi:10.1038/cdd.2017.169. [Google Scholar] [PubMed] [CrossRef]
38. Hayakawa J, Depatie C, Ohmichi M, Mercola D. The activation of c-Jun NH2-terminal kinase (JNK) by DNA-damaging agents serves to promote drug resistance via activating transcription factor 2 (ATF2)-dependent enhanced DNA repair. J Biol Chem. 2003;278(23):20582–92. doi:10.1074/jbc.M210992200. [Google Scholar] [PubMed] [CrossRef]
39. Lu Y, Liu B, Liu Y, Yu X, Cheng G. Dual effects of active ERK in cancer: a potential target for enhancing radiosensitivity. Oncol Lett. 2020;20(2):993–1000. doi:10.3892/ol. [Google Scholar] [CrossRef]
40. Dong Y, Shen X, He M, Wu Z, Zheng Q, Wang Y, et al. Activation of the JNK-c-Jun pathway in response to irradiation facilitates Fas ligand secretion in hepatoma cells and increases hepatocyte injury. J Exp Clin Cancer Res. 2016;35(1):114. doi:10.1186/s13046-016-0394-z. [Google Scholar] [PubMed] [CrossRef]
41. Wu MH, Tsai CH, Huang YL, Fong YC, Tang CH. Visfatin Promotes IL-6 and TNF-α Production in Human Synovial Fibroblasts by Repressing miR-199a-5p through ERK, p38 and JNK Signaling Pathways. Int J Mol Sci. 2018;19(1):190. doi:10.3390/ijms19010190. [Google Scholar] [PubMed] [CrossRef]
42. Tanimoto Y, Kizaki H. Proteasome inhibitors block Ras/ERK signaling pathway resulting in the downregulation of Fas ligand expression during activation-induced cell death in T cells. J Biochem. 2002;131(3):319–26. doi:10.1093/oxfordjournals.jbchem.a003106. [Google Scholar] [PubMed] [CrossRef]
43. Wang YM, Xu X, Tang J, Sun ZY, Fu YJ, Zhao XJ, et al. Apatinib induces endoplasmic reticulum stress-mediated apoptosis and autophagy and potentiates cell sensitivity to paclitaxel via the IRE-1α-AKT-mTOR pathway in esophageal squamous cell carcinoma. Cell Biosci. 2021;11(1):124. doi:10.1186/s13578-021-00640-2. [Google Scholar] [PubMed] [CrossRef]
44. Mohiuddin M, Kasahara K. Paclitaxel Impedes EGFR-mutated PC9 cell growth via reactive oxygen species-mediated DNA damage and EGFR/PI3K/AKT/mTOR signaling pathway suppression. Cancer Genom Proteom. 2021;18(5):645–59. doi:10.21873/cgp.20287. [Google Scholar] [PubMed] [CrossRef]
45. Javed Z, Khan K, Iqbal MZ, Ahmad T, Raza Q, Sadia H, et al. Long non-coding RNA regulation of TRAIL in breast cancer: a tangle of non-coding threads. Oncol Lett. 2020;20(4):37. [Google Scholar] [PubMed]
46. Sousa-Pimenta M, Estevinho LM, Szopa A, Basit M, Khan K, Armaghan M, et al. Chemotherapeutic properties and side-effects associated with the clinical practice of terpene alkaloids: paclitaxel, docetaxel, and cabazitaxel. Front Pharmacol. 2023;14:1157306. doi:10.3389/fphar.2023.1157306. [Google Scholar] [PubMed] [CrossRef]
47. Jiang Q, Yang M, Qu Z, Zhou J, Zhang Q. Resveratrol enhances anticancer effects of paclitaxel in HepG2 human liver cancer cells. BMC Complement Altern Med. 2017;17(1):477. doi:10.1186/s12906-017-1956-0. [Google Scholar] [PubMed] [CrossRef]
48. Liu Y, Shen Z, Zhu T, Lu W, Fu Y. Curcumin enhances the anti-cancer efficacy of paclitaxel in ovarian cancer by regulating the miR-9-5p/BRCA1 axis. Front Pharmacol. 2022;13:1014933. [Google Scholar] [PubMed]
49. Chantree P, Na-Bangchang K, Martviset P. Anticancer activity of fucoidan via apoptosis and cell cycle arrest on cholangiocarcinoma cell. Asian Pac J Cancer Prev. 2021;22(1):209–17. doi:10.31557/APJCP.2021.22.1.209. [Google Scholar] [PubMed] [CrossRef]
50. Jo EH, Lee SJ, Ahn NS, Park JS, Hwang JW, Kim SH, et al. Induction of apoptosis in MCF-7 and MDA-MB-231 breast cancer cells by Oligonol is mediated by Bcl-2 family regulation and MEK/ERK signaling. Eur J Cancer Prev. 2007;16(4):342–7. doi:10.1097/01.cej.0000236247.86360.db. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools