
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Sciadopitysin exerts anticancer effects on HepG2 hepatocellular carcinoma cells by regulating reactive oxygen species-mediated signaling pathways
1 Department of Biochemistry and Molecular Biology, Heilongjiang Bayi Agricultural University, Daqing, China
2 Hemodialysis Center, Daqing Oilfield General Hospital, Daqing, China
3 Department of Food Science and Engineering, Heilongjiang Bayi Agricultural University, Daqing, China
4 Department of Molecular Biology, Chifeng University, Chifeng, China
5 National Coarse Cereals Engineering Research Center, Daqing, China
* Corresponding Authors: TIAN-ZHU LI. Email: ; CHENG-HAO JIN. Email:
(This article belongs to the Special Issue: Navigating the Interplay of Cancer, Autophagy, ER Stress, Cell Cycle and Apoptosis: Mechanisms, Therapies, and Future Directions)
BIOCELL 2024, 48(7), 1055-1069. https://doi.org/10.32604/biocell.2024.050515
Received 08 February 2024; Accepted 09 April 2024; Issue published 03 July 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Objectives: Sciadopitysin (SP) is a flavonoid in Ginkgo biloba that exhibits various pharmacological activities. This study aimed to investigate its antitumor effects and the underlying molecular mechanism of SP in hepatocellular carcinoma (HCC) cells. Methods: Network pharmacology was used for target prediction analysis. Cell Counting Kit-8 (CCK-8) assay was used to test the cell viability. Flow cytometry was used to test the cell cycle distribution, apoptosis status, and reactive oxygen species (ROS) levels. Transwell and wound-healing assay was used to test the migration effect of SP on HepG2 cells. Western Blot assay was used to test the expression levels of proteins. Results: Network pharmacology analysis results showed that the mitogen-activated protein kinase (MAPK) and other signaling pathways are involved in the SP anti-HCC biological process. CCK-8 assay results demonstrated that SP showed an obvious killing effect on three types of HCC cells and low cytotoxic effect on normal cells. Western Blot and flow cytometry results showed that SP regulated MAPK/signal transducer and activator of transcription 3 (STAT3)/nuclear factor kappa-B (NF-κB) signaling pathway to induce mitochondrion-dependent apoptosis in HepG2 cells. Additionally, SP can arrest the G0/G1 phase cell cycle via the protein kinase B (AKT)/p21/p27/cyclin-dependent kinase (CDK)/Cyclin signaling pathway. Wound healing and transwell assays showed that SP inhibited cell motility and invasion through the AKT/glycogen synthase kinase3β (GSK-3β)/vimentin/β-catenin signaling pathway. Conclusion: These findings demonstrated that SP induced mitochondrion-dependent apoptosis, arrested cell cycle in the G0/G1 phase, and inhibited cell migration by regulating the ROS-mediated signaling pathway in HepG2 cells. Thus, SP could serve as a therapeutic agent for the treatment of human HCC.Graphic Abstract
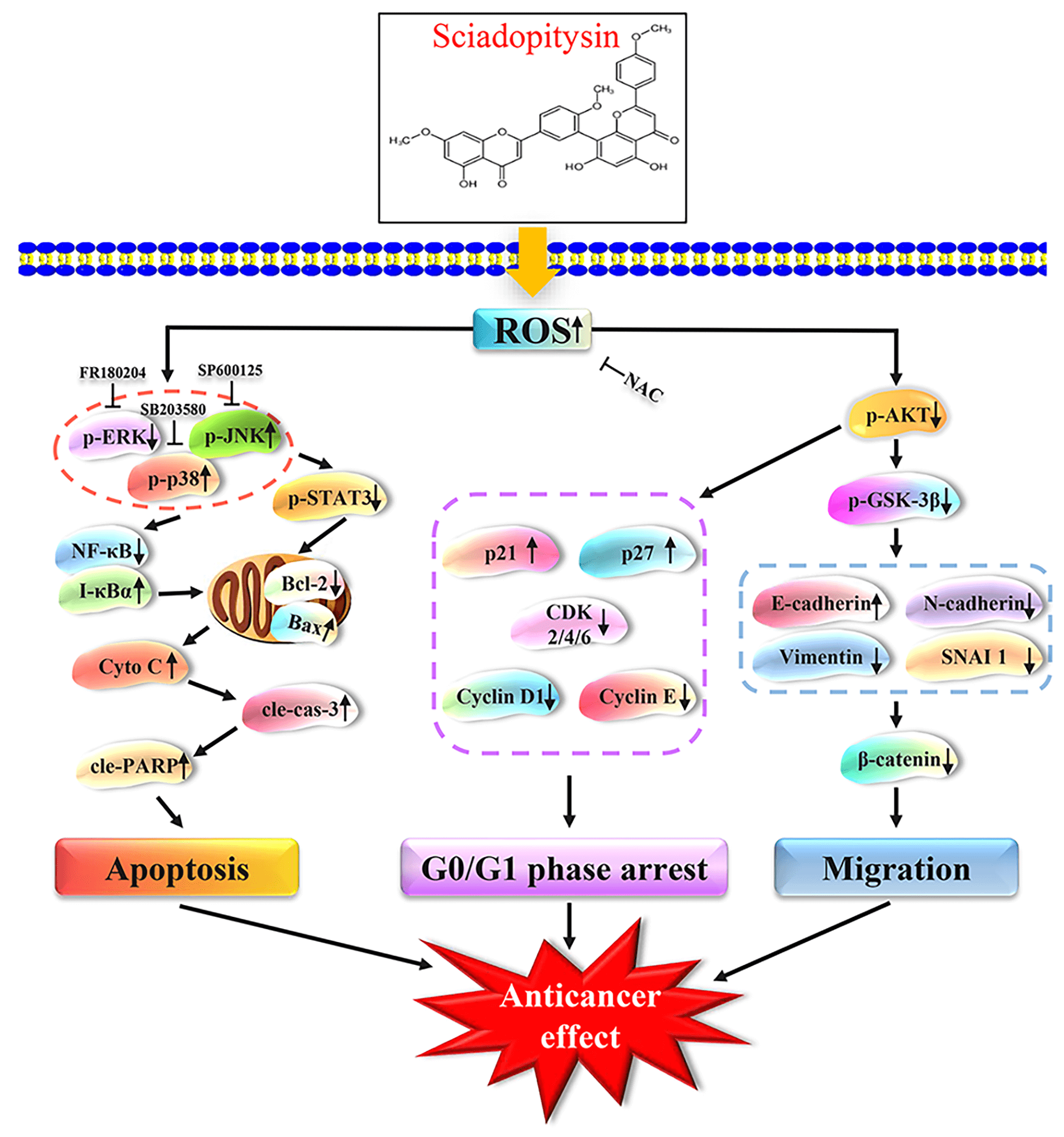
Keywords
Abbreviations
| SP | Sciadopitysin |
| 5-FU | 5-Fluorouracil |
| PPI | Protein–Protein Interaction |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| BP | Biological Processes |
| MF | Molecular Functions |
| CC | Cellular Components |
| PTGS | Post-transcriptional GeneSilencing |
The incidence of liver cancer has risen sharply and is within the top three leading factors contributing to cancer mortality worldwide, according to the Global Cancer Statistics 2020 study [1,2]. Hepatocellular carcinoma (HCC) has serious consequences for human health, which is a dangerous form of liver cancer [3,4]. Each treatment for liver cancer has its disadvantages. For example, the risk of recurrence after hepatectomy is high, and transcatheter arterial chemoembolization often has no noticeable therapeutic effect on patients with advanced liver cancer [5]. In addition, the drug resistance caused by chemotherapy seriously alters normal liver function in patients [6]. Therefore, an efficient and safe drug for liver cancer needs to be developed [7]. Apoptosis, a spontaneous process regulating cell death, plays a vital role in maintaining an organism’s stable internal environment by triggering a series of complex signaling pathways and molecular events [8]. Cell apoptosis as a therapeutic approach for cancer has garnered considerable attention in recent years [9]. For example, Quercetin can induce apoptosis in HepG2 cells through ataxia telangiectasia mutated (ATM)/c-Jun N-terminal kinase (JNK)/signal transducer and activator of transcription 3 (STAT3) signaling pathway [10]. Mitochondria are essential components of eukaryotic cells. In certain types of cancer cells, they are involved in the initiation, progression, and metastasis of tumors. They also have a vital function in the biological procedure of apoptosis. Possible therapeutic and preventative targets for tumors include reactive oxygen species (ROS) generated through mitochondrial metabolism [11]. Several investigations have demonstrated that ROS are important for cancer treatment. ROS can dynamically affect the internal surroundings and activate signaling cascades in tumors, such as the signaling route for mitogen-activated protein kinase (MAPK) [12,13]. Crucial participants in the MAPK protein subfamily include extracellular regulated kinase 1/2 (ERK1/2), c-Jun N-terminal kinase (JNK), and p38, which may be involved in regulating the biological processes of a variety of tumors [14]. The STAT3 is carcinogenic, which is the central hub for multiple signaling pathways in tumors. It possesses the ability to nuclear factor kappa-B (NF-κB) and demonstrates an intricate interplay with NF-κB. This interaction between the two proteins can have significant implications for various tumors.
Small-molecule drugs can achieve a therapeutic effect by promoting apoptosis of cancer cells [15,16]. In the past few years, growing numbers of traditional Chinese herbal remedies have shown promise as small-molecule pharmaceuticals with potent anti-cancer effects [17]. Compared with conventional chemotherapy drugs, traditional Chinese herbal remedies have the characteristics of having multiple targets, low toxicity, and high efficiency, which can considerably increase the lifespan and quality of life of people with cancer [18]. Sciadopitysin (SP) is a flavonoid compound that can be isolated from Ginkgo biloba. In recent decades, researchers have found SP to have promising pharmacological actions. The application and efficacy of SP have been demonstrated in numerous studies. It inhibits osteoclast differentiation and bone resorption, protects cells from damage, and upregulates mitochondrial biosynthesis [19–21]. SP also has antioxidant properties, which can protect SK-N-MC cells from methylglyoxal-induced oxidative damage, thus playing a protective role in nerve [22]. In addition, recent scientific studies have shown that SP can interact with caspase-3 in a silico approach in breast cancer to induce the initiation of apoptosis [23]. However, the precise mechanism by which SP contributes to apoptosis in HCC remains largely elusive. To clarify this point, we explored the intrinsic mechanism of SP-induced apoptosis in HepG2 cells.
This research explored the impact of the killing effect, apoptosis induction, ROS accumulation, cycle arrest, and the migration of SP in HCC cells.
We used the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) and the SwissTargetPrediction platform (http://www.swisstargetprediction.ch/) to choose Homo sapiens to predict the target collection for SP. Next, using the GeneCards disease database (https://www.genecards.org/) associated with HCC screening targets and using the Venny platform (http://www.liuxiaoyuyuan.cn/) to take the intersection, we obtained potential anti-HCC targets for SP. Furthermore, the protein-protein interaction (PPI) network was constructed by the Search Tool for Recurring Instances of Neighbouring Genes (STRING) platform (https://cn.string-db.org/) and analyzed the relationship among protein targets. The SP–HCC target-pathway network diagram was prepared using Cytoscape software (Cytoscape 3.8.2, USA) to analyze the interaction. Then, using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) database (https://david.ncifcrf.gov/) and bioinformatics platform (http://www.bioinformatics.com.cn/) extraction and biological information for the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of the targets.
Three HCC cell lines, HepG2, Huh-7, and Hep3B, human normal hepatocyte THLE-2 cells, human normal renal epithelial 293T cells, and human normal gastric mucous GES-1 were acquired from the American Type Culture Collection (Manassas, VA, USA) or OWTO Biotech Inc. (Shenzhen, China), and Shanghai Qingqi Biotechnology Development Co., Ltd. (China). Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM, C11995500BT, Gibco, Waltham, MA, USA), which also contained 10% fetal bovine serum (FBS, 11011-8611, Gibco) and 1% penicillin/streptomycin (PS) (15140122, Gibco), next placed in an incubator with full of carbon dioxide at 37°C.
Cell viability was measured by Cell Counting Kit-8 (CCK-8) obtained from Solarbio (CA1210-100, Beijing, China). HepG2, Huh-7, Hep3B, THLE-2, 293T, and GES-1 cells were seeded into 96-well plates (1 × 104 cells/well). SP (J-041, Herbpurify, Chengdu, China) and 5-Fluorouracil (5-FU, IF0170, Solarbio, Beijing, China) were used to treat the cultured cells at different concentrations from 20 to 100 μM for 24 h or at different durations from 6 to 30 h. 5-FU, a common chemotherapeutic agent, was also used as a positive control for cell apoptosis. Each well was treated with CCK-8 solution, and the cells were detected by an automatic enzyme-labeling instrument (INFINITE E PLEX, Tecan, Shanghai, China) at 490 nm. The half-maximal inhibitory concentration (IC50) values of the above cells were calculated using GraphPad Prism (GraphPad Prism 5.0, GraphPad Software, San Diego, CA, USA).
Cell apoptosis was detected using an Annexin V-FITC/PI Apoptosis Kit (FXP018, 4 A Biotech, Beijing, China). HepG2 cells were seeded into 6-well plates (1 × 105 cells/well). This was followed by 38.02 µM SP (the IC50 value of HepG2 cells) treatment of each well. The Annexin V-FITC and propidium iodide (PI) were used for staining and subsequent observation and recording of HepG2 cell apoptosis were conducted using a fluorescent microscope (MF52-N, Mshot, Guangzhou, China). Subsequently, the apoptotic rate of SP-treated HepG2 cells was determined using flow cytometry (CyFlow Cube8, Sysmex Co, Kobe, Japan).
Mitochondrial membrane potential assay
Mitochondrial membrane potential (MMP) was detected by an MMP detection kit (M8650, Solarbio, Beijing, China). HepG2 cells were seeded into 6-well plates (1 × 105 cells/well), followed by 38.02 µM SP treatment for 3, 6, 12, and 24 h. The JC-1 staining working solution and JC-1 binding buffer were dispensed accurately in appropriate proportions. Subsequently, MMP in HepG2 cells that were treated with SP were used the flow cytometry to analyze.
Western Blot was carried out following accepted practices. HepG2 cells were treated with 38.02 µM SP or JNK inhibitor (SP600125), p38 inhibitor (SB203580), and ERK inhibitor (FR180204) (MedChemExpress, Shanghai, China) or N-Acetylcysteine (NAC, Co., 3050, SIGMA-ALDRICH, St. Louis, USA). The proteins were extracted after cell collection and subjected to electrophoresis. Subsequently, transferred proteins to the nitrocellulose filter membrane at 320 mA. Then, combined the nitrocellulose filter membranes with primary antibodies, which included Bcl-2-associated X protein (Bax) (Cat#sc-493, 1:1500), B-cell lymphoma-2 (Bcl-2) (Cat#sc-7382, 1:1500), Cytochrome C (cyto C) (Cat#sc-13156, 1:2000), Caspase-3 (Cat#sc-373730, 1:1500), poly ADP-ribose polymerase poly ADP-ribose polymerase (PARP) (Cat#sc-8007, 1:1500), ERK (Cat#sc-154, 1:1000), p-ERK (Cat#sc-7383, 1:1000), JNK (Cat#sc-7345, 1:1500), p-JNK (Cat#sc-6254, 1:1500), p38 (Cat#sc-7149, 1:1500), p-p38 (Cat#sc-7973, 1:1500), STAT3 (Cat#sc-8019, 1:1500), p-STAT3 (Cat#sc-8059, 1:1500), NF-κB (Cat#sc-8008, 1:1500), I-κBα (Cat#sc-1643, 1:1500), protein kinase B (AKT) (Cat#sc-8312, 1:1000), p-AKT(Cat#sc-7985-R, 1:1000), p21 (Cat#sc-397, 1:1500), p27 (Cat#sc-528, 1:1500), CDK 2 (Cat#sc-163, 1:1500), CDK 4 (Cat#sc-260, 1:1500), CDK 6 (Cat#sc-177, 1:1500), cyclin D1 (Cat#sc-753, 1:1500), cyclin E (Cat#sc-182, 1:1500), glycogen synthase kinase3β (GSK-3β) (Cat#sc-377213, 1:1500), p-GSK-3β (Cat#sc-373800, 1:1000), E-cadherin (Cat#sc-8426, 1:1000), N-cadherin (Cat#sc-59987, 1:1000), vimentin (Cat# sc-6260, 1:1000), SNAI 1 (Cat#sc-271977, 1:1000), β-catenin (Cat#sc-7963, 1:1000), and α-tubulin (Cat#sc-47778, 1:2500) (Santa Cruz Biotechnology, Dallas, TX, USA), and stored in a 4°C refrigerator overnight. Then, they were treated with goat anti-mouse IgG (ZB-2305, 1:50,000, ZSGB-bio, Beijing, China) or goat anti-rabbit IgG (ZB-2306, 1:50,000, ZSGB-bio, Beijing, China) secondary antibody and allowed to rest. The use of enhanced chemiluminescence (180-501, Tanon, Shanghai, China) in combination with proteins facilitated the detection and analysis of protein expression levels in HepG2 cells treated with SP via ImageJ software (lmageJ 1.46r, National Institutes of Health, USA).
The ROS levels were detected by a ROS detection kit (S0033S, Beyotime, Shanghai, China). HepG2 cells were seeded into a 3.5-cm plate (1 × 105 cells/plate), followed by treatment with 38.02 µM SP for 3, 6, 12, and 24 h. The collected HepG2 cells were added with a 10 μmol/L probe. Subsequently, the ROS level of the SP-treated HepG2 cells was detected via flow cytometry and fluorescent microscope.
The cell cycle was determined using DNA quantification kits (CA1510, Solarbio, Beijing, China). HepG2 cells were seeded a 3.5-cm cell plates (1 × 105 cells/plate), and 38.02 µM SP was treated for 3, 6, 12, and 24 h. The collected HepG2 cells were incubated in a 4°C refrigerator. Then, 100 μL RNase and 500 μL PI were dispensed accurately following the manufacturer’s instructions. Subsequently, DNA quantification of the SP-treated HepG2 cells was determined using flow cytometry.
Wound healing assay was used to degree cell migration. HepG2 cells were seeded into 6-well plates (1 × 105 cells/well). Scratch the HepG2 cells in the horizontal direction in the 6-well plates. DMEM medium with 10% FBS and 38.02 µM SP were applied to the cells. The degree of cell migration healing was then assessed and quantified using an inverted microscope (MF52-N, Mshot, Guangzhou, China).
The cell invasion degree was determined by transwell assay. HepG2 cells were seeded at a density of 1 × 105 cells/well in the upper chamber of a 6-well transwell plate containing DMEM serum-free medium, and the lower chamber with 15% FBS. Then, 38.02 µM SP treatment was performed for 3, 6, 12, and 24 h. Then use the 0.1% crystal violet staining solution (Cat#1064, Solarbio, Beijing, China) for staining. Subsequently, we observed and counted the number of cell migrations of SP-treated HepG2 cells using the inverted microscope.
Molecular docking experiments assay
The structural formula of target proteins was downloaded from the Protein Data Bank (PDB) database (https://www.rcsb.org/) and the protein was preprocessed by Pymol software (Pymol software 3.0, DeLano Scientific LLC, Palo Alto, CA, USA). The 3D structure of SP downloaded from PubChem was transformed into mol2 by Open Babel GUI software 3.1.1, and then the protein structure of target proteins and the 3D structure of SP were docked by AutoDockTools software (AutoDockTools software 1.5.6, Scripps Research Institute, La Jolla, CA, USA). The optimal docking location was determined and subsequently visualized using the Pymol software.
The data were analyzed using SPSS version 21.0. All data are reported as the mean ± standard deviation (SD) of at least three replicates. Multiple comparisons across the groups were conducted using one-way analysis of variance. A p-value of <0.05 was regarded as significant.
Network pharmacological analysis of SP treatment for HCC
The potential biological effects of SP on HepG2 cells were predicted by network pharmacology, and the therapeutic HCC targets were screened. The 2D structure of SP obtained from PubChem was imported into the SwissTargetPrediction platform, resulting in 100 targets. After removing 56 targets with a confidence score of “0,” we identified 44 effective targets. Relevant targets for HCC were searched using the GeneCards platform, leading to the identification of 25 intersection targets between SP and HCC (Fig. 1A). These intersection targets were then used to construct a protein-protein interaction (PPI) network diagram using Cytoscape 3.8.2 software, which revealed the target–pathway interactions associated with SP–HCC treatment (Fig. 1B). Notably, the glycogen synthase kinase3β (GSK-3β) protein exhibited a high degree value in this network analysis, suggesting its potential importance in SP treatment of HCC. The main anti-HCC targets influenced by SP were further enriched, and resulting in the identification of 119 items and involvement in 20 signaling pathways. These included various biological processes (BP), cellular components (CC), and molecular functions (MF), such as positive regulation of the NF-κB signaling pathway, the MAP kinase activity, the PI3K-AKT signaling pathway, the chemical carcinogenesis–ROS pathway, pathways in cancer, and the MAPK signaling pathway (Figs. 1C, 1D).
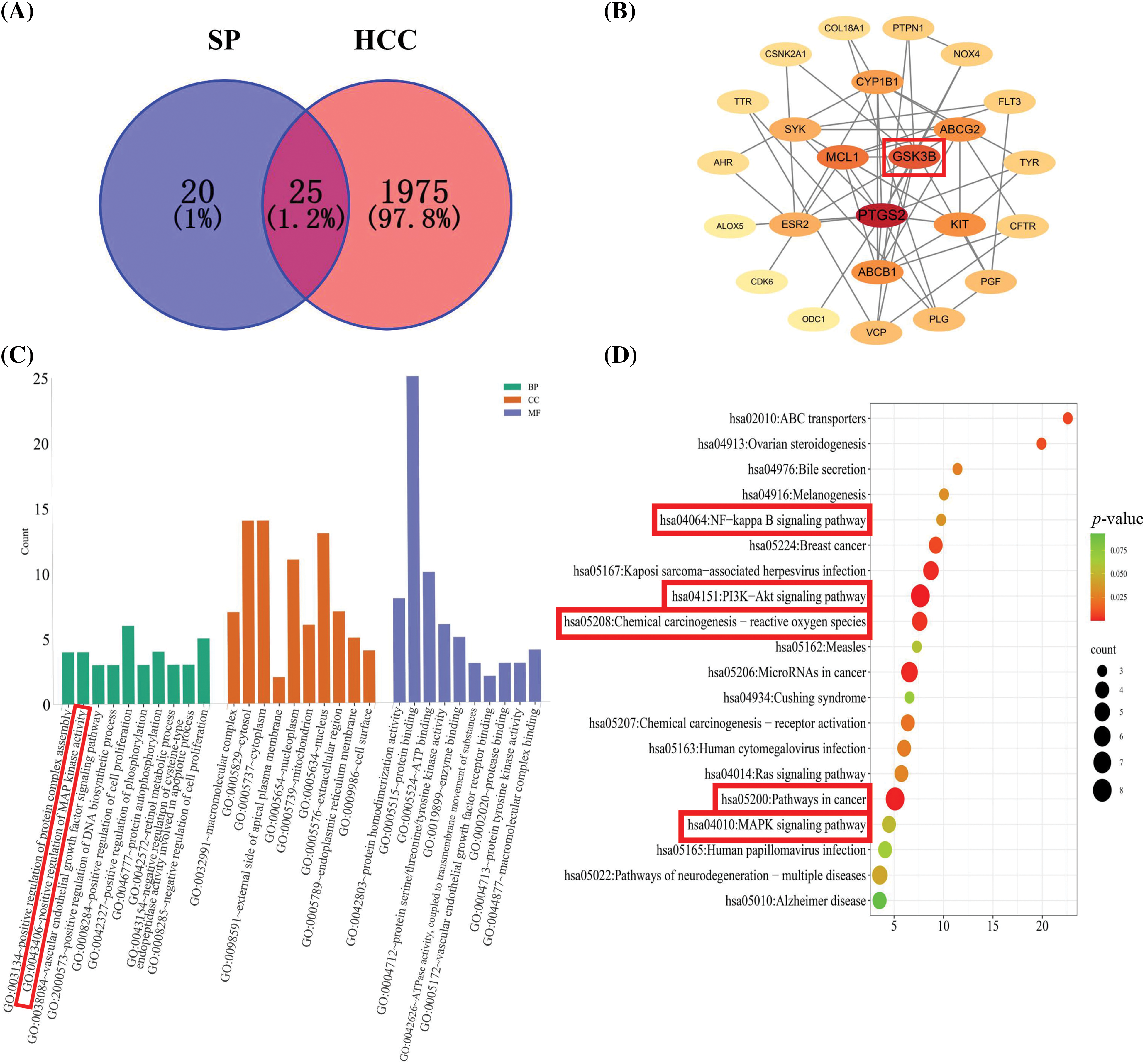
Figure 1: Network pharmacology prediction of SP treatment on HepG2 cells. (A) Venn diagram of SP and HCC targets. (B) Hub gene network of the target proteins was analyzed using Cytoscape. (C) GO enrichment of the anti-HCC effect of SP. (D) KEGG pathway enrichment of the anti-HCC effect of SP.
SP reduced the survival rate of HCC cells
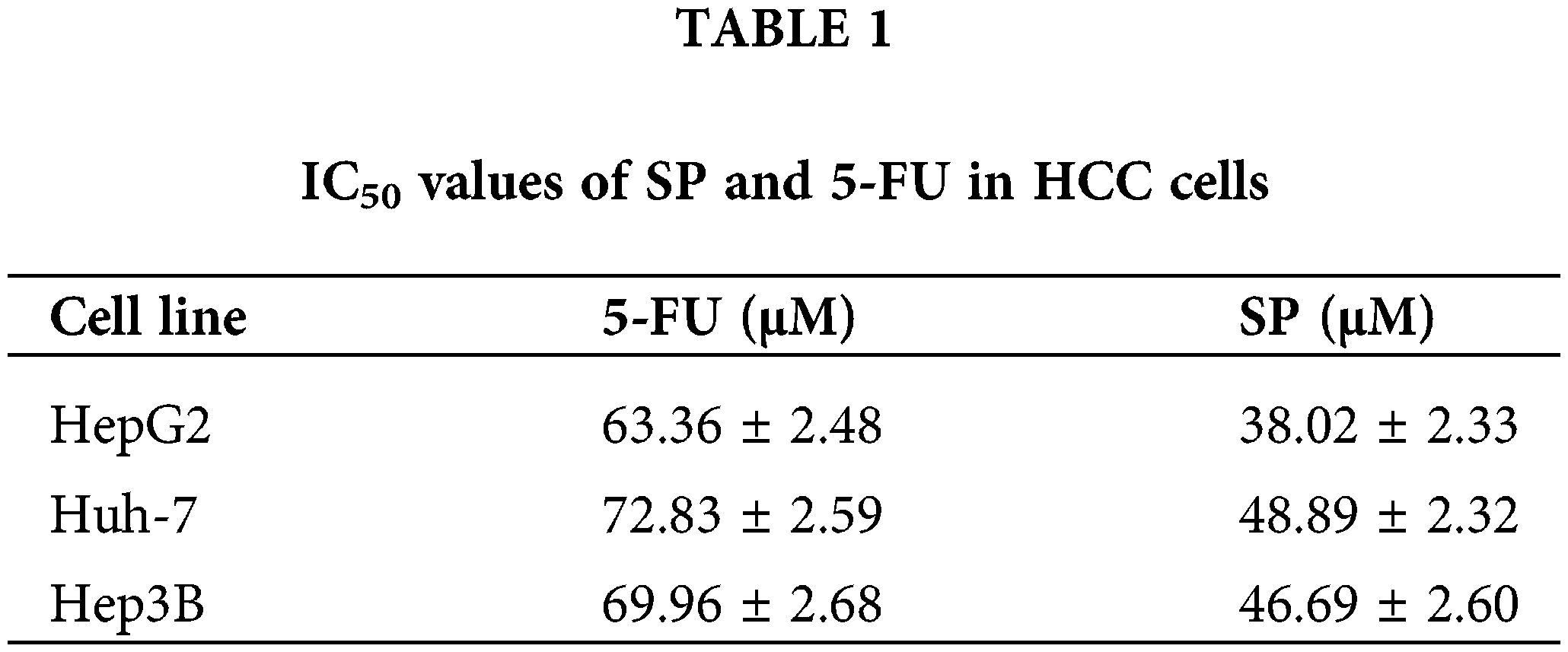
The cell viability was evaluated by the CCK-8 assay. As the SP treatment concentration and time were increased, the cell viability decreased sharply in HepG2, Huh-7, and Hep3B cells (Figs. 2A, 2B), whereas it did not decrease substantially in THLE-2, 293T, and GES-1 cells (Figs. 2C, 2D). The effect of the SP group was considerably better compared to the 5-FU treatment group, indicating that SP effectively reduced the survival rate of the three HCC cells. The IC50 values of HepG2, Huh-7, and Hep3B cells after SP treatment were 38.02, 48.89, and 46.69 μΜ (Table 1). Then, 6, 12, 18, 24, and 30 h were selected in subsequent analysis. In addition, among the three HCC cancer cells tested, the IC50 values of HepG2 cells treated with SP were the lowest, suggesting they were the most susceptible to SP. In subsequent analyses, HepG2 cells were selected as the cell model.
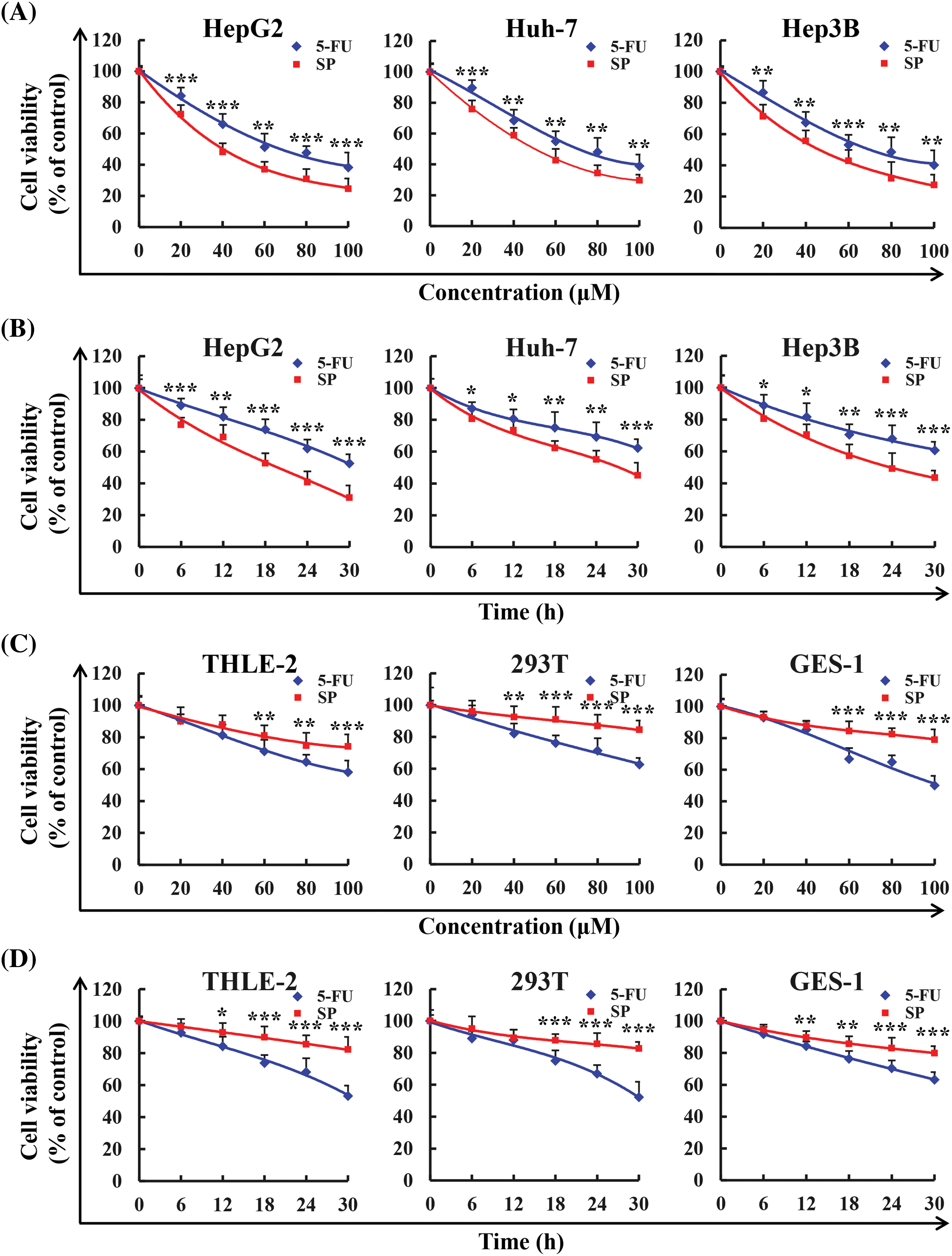
Figure 2: Cytotoxic effect of SP. (A) Three human hepatocellular carcinoma cell lines (HepG2, Huh-7, and Hep3B) were treated with 20, 40, 60, 80, and 100 μM SP or 5-FU for 24 h, and the CCK-8 assay detected the effects on cell activity. (B) Three human hepatocellular carcinoma cell lines (HepG2, Huh-7, and Hep3B) were treated with 38.02 μM (IC50 value) SP or 5-FU for 6, 12, 18, 24, and 30 h, and the CCK-8 assay detected the effects on cell activity. (C) Three human normal cell lines (THLE-2, 293T, and GES-1) were treated with 20, 40, 60, 80, and 100 μM SP or 5-FU for 24 h, and the CCK-8 assay detected the effects on cell activity. (D) Three human normal cell lines (THLE-2, 293T, and GES-1) were treated with 38.02 μM SP or 5-FU for 6, 12, 18, 24, and 30 h, and the effect of cell viability was detected using the CCK-8 assay (*p < 0.05, **p < 0.01, ***p < 0.001 vs. 5-FU), n = 3.
SP-induced apoptosis of HepG2 cells
The apoptotic effect of SP on HepG2 cells was evaluated using Annexin V-FITC/PI assay. As the SP treatment duration was increased, HepG2 cells exhibited a rounder and smaller morphology, accompanied by a considerable increase in fluorescence intensity, indicating extensive apoptosis (Fig. 3A), and this effect was most pronounced at 24 h. Flow cytometry analysis further assessed the apoptosis of HepG2 cells following SP treatment. The results showed that with increased SP treatment time, the degree of apoptosis of HepG2 cells gradually increased from 1.26% to 37.29% (Fig. 3B). Additionally, with increased SP treatment time, the MMP decreased from 90.59% to 71.96% (Fig. 3C). Western Blot analysis revealed SP treatment increased Bax, cyto C, cle-cas-3, and cle-PARP expression, and decreased Bcl-2 expression (Fig. 3D).
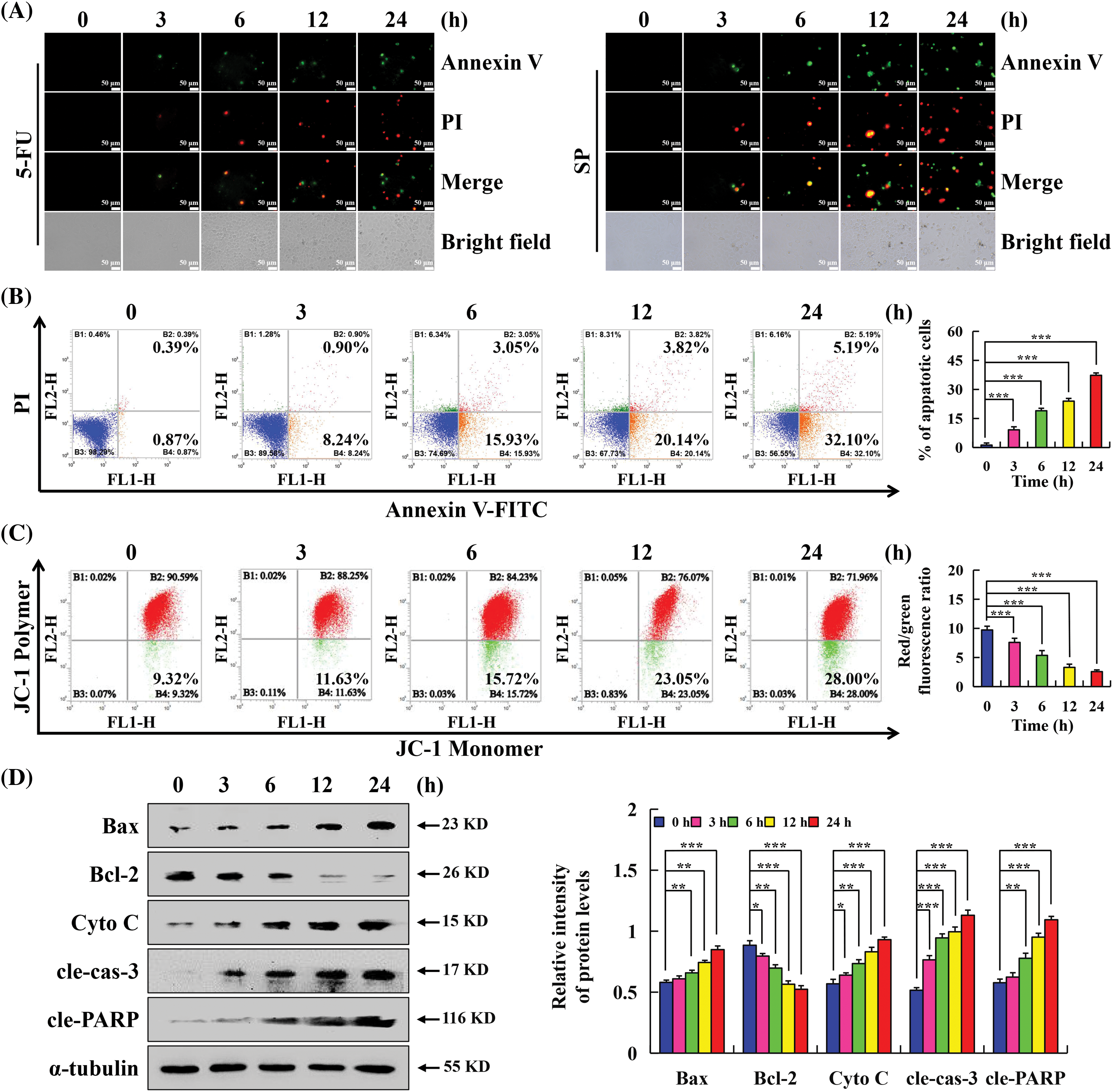
Figure 3: Apoptotic effects of SP on HepG2 cells. (A) Annexin V-FITC/PI staining was used to observe the morphology and fluorescence staining of HepG2 cells under a fluorescence microscope. (Original magnification, 200×). (B) Rate of HepG2 cells after SP treatment was determined via flow cytometry. (C) Mitochondrial membrane potential of HepG2 cells determined using flow cytometry. (D) Western Blot analysis of the expression levels of Bax, Bcl-2, cyto C, cle-cas-3, and cle-PARP of HepG2 cells, with α-tubulin as the control. The protein changes were analyzed using ImageJ software (*p < 0.05, **p < 0.01, ***p < 0.001 vs. 0 h), n = 3.
SP-induced apoptosis of HepG2 cells via the MAPK/STAT3/NF-κB signaling pathway
Apoptosis is a tightly regulated process involving the interaction of multiple signaling pathways. MAPK, as a signal transduction molecule, can regulate STAT3 and NF-κB signaling pathways as downstream molecules to induce apoptosis. Western Blot was used to detect the expression level of MAPK/STAT3/NF-κB signaling pathway-related proteins, to explore the molecular mechanism of SP-induced apoptosis in HepG2 cells. Fig. 4A showed that with increasing treatment time of SP, the p-JNK, p-p38, and I-κBα protein expression levels gradually increased. In contrast, p-ERK, NF-κB, and p-STAT3 protein expression levels gradually decreased. To further verify the interaction and influence between MAPK and STAT3 signaling pathways, HepG2 cells were pretreated with MAPK inhibitors for detection. The SP + JNK inhibitor and SP + p38 inhibitor groups exhibited considerably lower expression levels of p-JNK, p-p38, cle-cas-3, and cle-PARP but higher expression levels of p-STAT3 in these groups than in the SP group. In contrast, in the SP + ERK inhibitor group, the protein expression level of cle-cas-3 and cle-PARP increased, and the protein expression level of p-ERK and p-STAT3 decreased (Figs. 4B–4D).
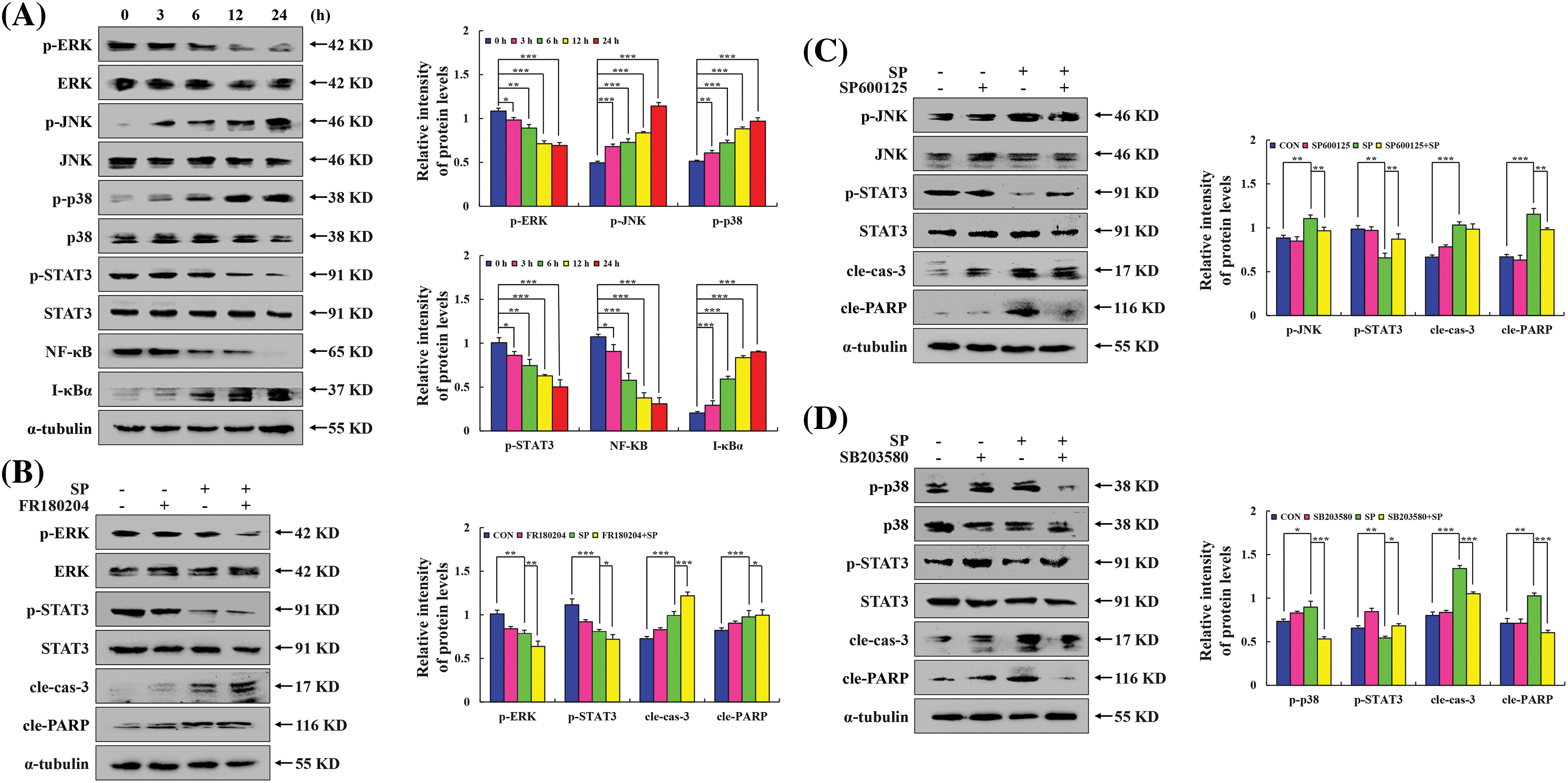
Figure 4: Effect of SP on the MAPK/STAT3/NF-κB signaling pathway in HepG2 cells. (A) HepG2 cells were treated with SP for 3, 6, 12, and 24 h, and the expression level of MAPK/STAT3/NF-κB was analyzed via Western Blot, with α-tubulin as the control. (B) ERK inhibitor treatment for detecting the expression levels of p-ERK, p-STAT3, cle-cas-3, and cle-PARP. (C) JNK inhibitor treatment for detecting the expression levels of p-JNK, p-STAT3, cle-cas-3, and cle-PARP. (D) p38 inhibitor treatment for detecting the expression levels of p-p38, p-STAT3, cle-cas-3, and cle-PARP (*p < 0.05, **p < 0.01, ***p < 0.001 vs. CON or SP + MAPK inhibition), n = 3.
SP-induced apoptosis of HepG2 cells via ROS-mediated signaling pathways
Used the ROS kit to detect whether SP-induced apoptosis was related to ROS. Flow cytometry showed that the accumulation of ROS in HepG2 cells increased with the duration of SP treatment (Fig. 5A). The results of inverted fluorescence microscopy showed that with the increase of SP treatment time, the number of stained HepG2 cells gradually increased, and the fluorescence intensity became stronger (Fig. 5B). Moreover, the combined of SP and the ROS scavenger NAC, significantly attenuated the proportion of apoptotic cells (Fig. 5C). We performed Western Blot and found that the protein expression level in the SP + NAC group were reversed (Fig. 5D).
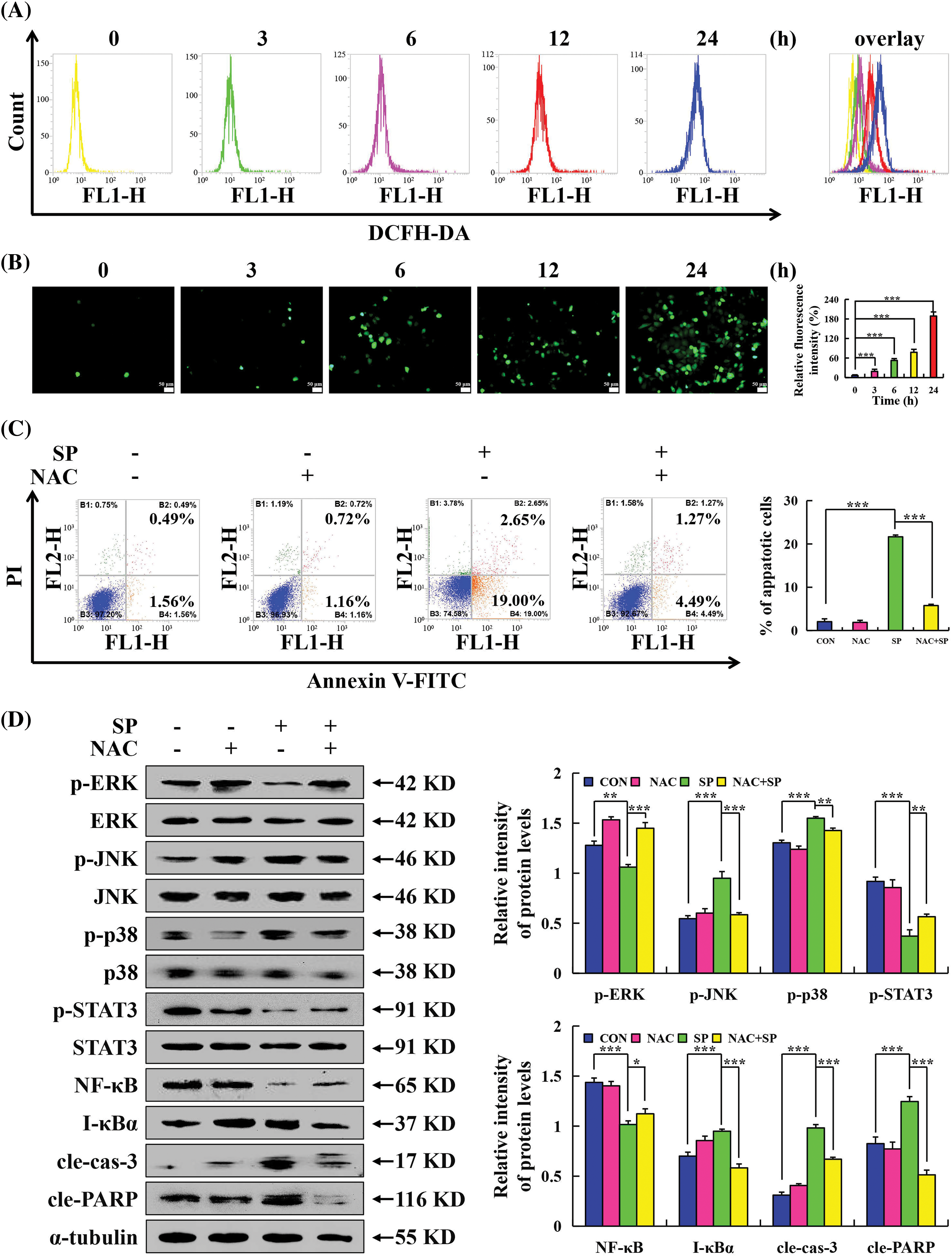
Figure 5: Promoting effects of SP on ROS levels in HepG2 cells. (A) Accumulation of ROS in HepG2 cells after SP treatment detected using flow cytometry. (B) Accumulation of ROS in HepG2 cells after SP treatment detected using fluorescence microscopy. (C) HepG2 cells were treated with SP and/or NAC for 24 h, and the number of apoptotic cells was determined using flow cytometry. (D) Western Blot analysis of the expression levels of MAPK, STAT3, and NF-κB signaling pathway-related proteins, cle-cas-3, and cle-PARP, in HepG2 cells, with α-tubulin as the control (*p < 0.05, **p < 0.01, ***p < 0.001 vs. CON or SP + NAC), n = 3.
SP induced G0/G1 phase arrest of HepG2 cells
The cell cycle arrest effect of SP on HepG2 cells was investigated using flow cytometry. Fig. 6A showed that as the SP treatment time was increased, a noticeable shift in the distribution of cell cycle phases was observed. Specifically, the number of HepG2 cells in the G0/G1 phase increase from 71.74% to 81.87%. Then pretreatment with NAC showed that the SP + NAC group had considerably fewer cells in the G0/G1 phase than the SP treatment alone group (Fig. 6B). These findings indicated that SP could arrest HepG2 cells in the G0/G1 phase. Furthermore, Western Blot analysis revealed decreased expression levels of p-AKT, CDK 2, CDK 4, and CDK 6, cyclin D1, and cyclin E. Conversely, we observed increased expression levels of p21 and p27 (Fig. 6C), indicating their involvement in mediating SP-induced cell cycle arrest. Importantly, when NAC and SP were combined, the expression levels of the proteins reversed compared to those treated with SP alone (Fig. 6D).
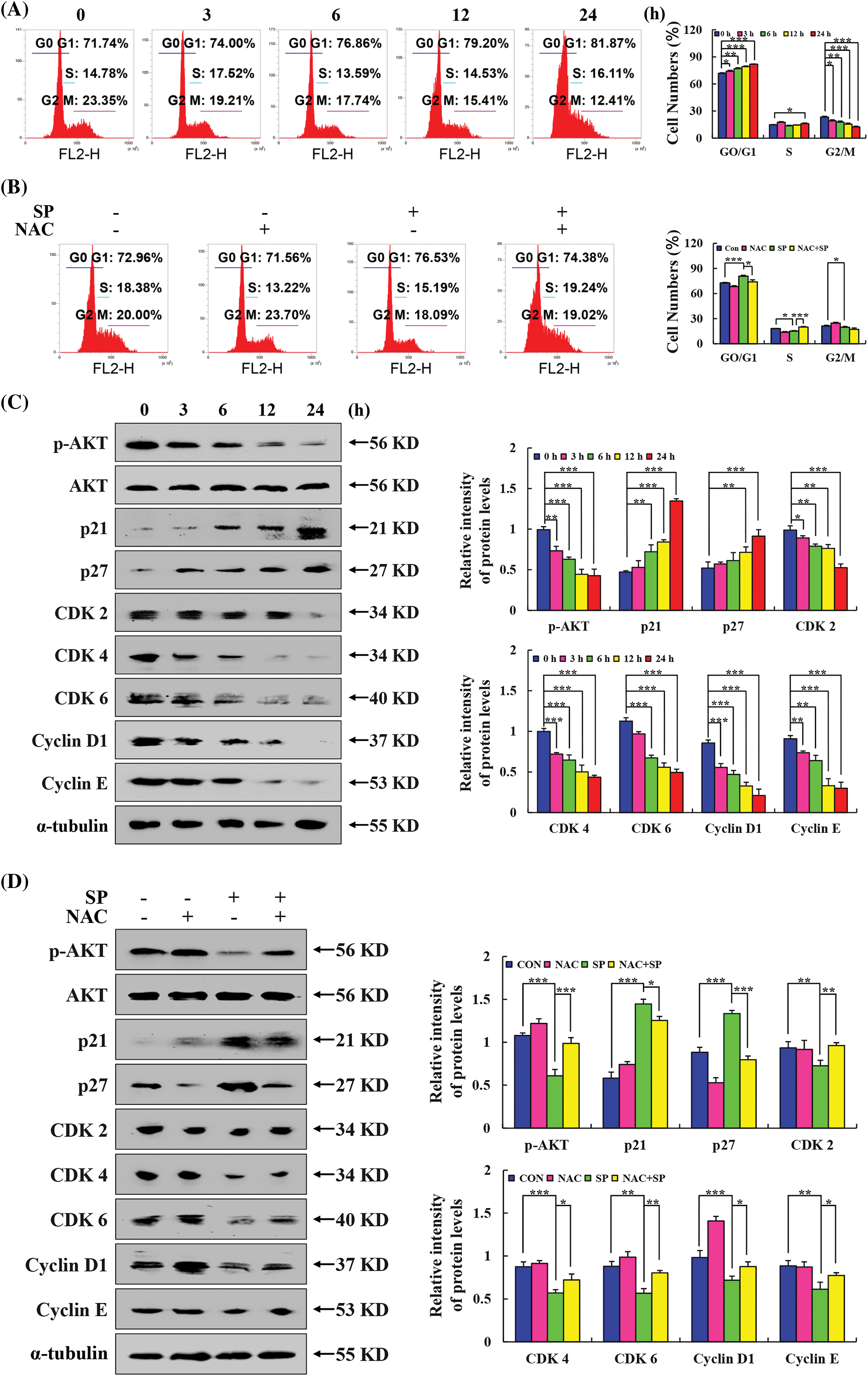
Figure 6: Arresting effects of SP on the cell cycle in HepG2 cells. (A) Arresting of the cell cycle of HepG2 cells after SP treatment was determined using flow cytometry. (B) HepG2 cells were treated with SP and/or NAC for 24 h, and the arrest of the cell cycle in HepG2 cells was determined by flow cytometry. (C) Western Blot analysis of the expression levels of p21, p27, CDK 2, CDK 4, CDK 6, cyclin D1, and cyclin E in HepG2 cells, with α-tubulin as the control. (D) Western Blot analysis was used to detect the expression levels of p21, p27, CDK 2, CDK 4, CDK 6, cyclin D1, and cyclin E in HepG2 cells after they were treated with SP and/or NAC for 24 h, with α-tubulin as the control (*p < 0.05, **p < 0.01, ***p < 0.001 vs. CON or SP + NAC), n = 3.
SP inhibited the migration of HepG2 cells
Wound healing and transwell assays were employed to evaluate the migration and invasion effect on HepG2 cells treatment SP. Cellular changes are shown in Figs. 7A and 7B. Compared with the control group, cell migration in the SP treatment group was significantly inhibited, with a more pronounced effect at 24 h. To examine whether ROS affected the capacity of SP to inhibit HepG2 cell migration, pretreated cells with NAC. The SP + NAC treatment group exhibited faster cell migration and a larger area of cell wound healing than the SP treatment group (Fig. 7C). SP treatment increased E-cadherin expression level, and decreased p-AKT, p-GSK-3β, N-cadherin, vimentin, SNAI 1, and β-catenin expression level significantly decreased in SP-treated HepG2 cells (Fig. 7D). However, these protein expression levels were reversed with NAC + SP treatment (Fig. 7E).
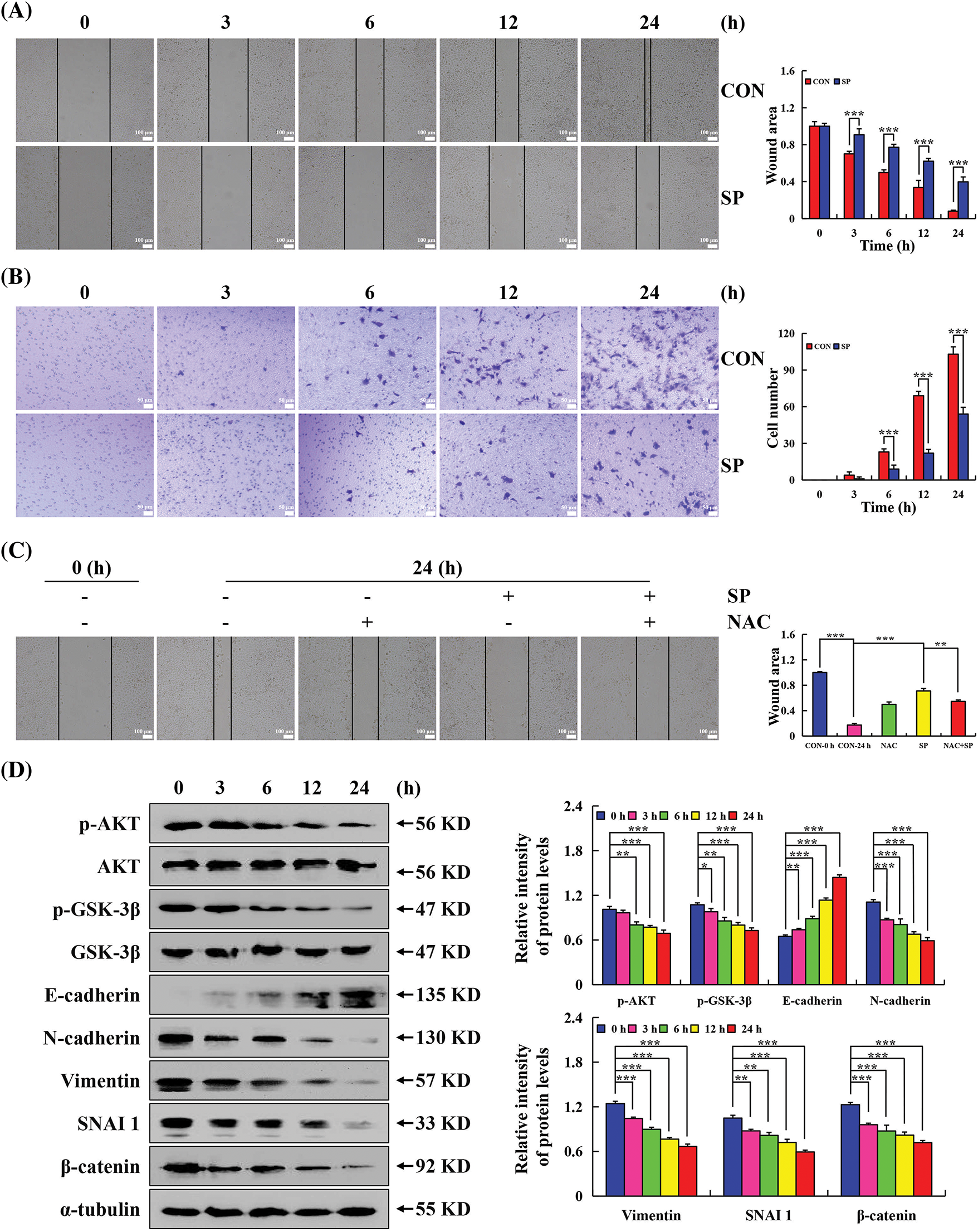
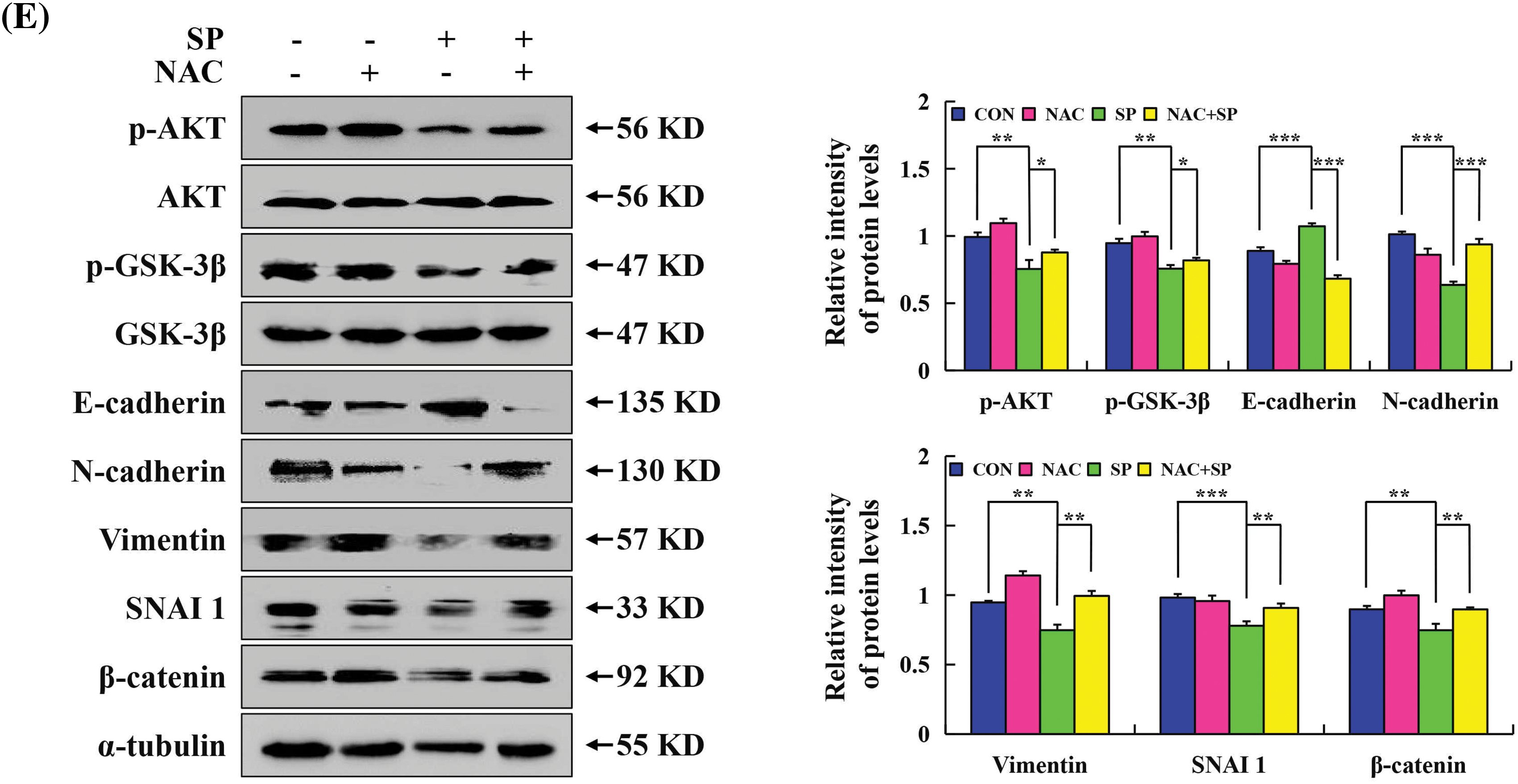
Figure 7: Inhibiting effects of SP on the migration in HepG2 cells. (A) Cell migration was used to analyze the migration ability of HepG2 cells. (B) Migration of HepG2 cells after SP treatment was detected using the transwell assay. (C) Cell migration assay was used to analyze the migration ability of HepG2 cells after they were treated with SP and/or NAC for 24 h. (D) Western Blot analysis was used to determine the expression level of p-AKT, p-GSK-3β, E-cadherin, N-cadherin, vimentin, SNAI 1, and β-catenin in HepG2 cells, with α-tubulin as the control. (E) Western Blot analysis used to determine the expression level of p-AKT, p-GSK-3β, E-cadherin, N-cadherin, vimentin, SNAI 1, and β-catenin in HepG2 cells after they were treated with SP and/or NAC for 24 h, with α-tubulin as the control (*p < 0.05, **p < 0.01, ***p < 0.001 vs. CON or SP + NAC), n = 3.
Molecular docking of SP to target proteins
The results of molecular docking showed that SP was mainly connected with AKT protein by hydrogen bonds, with docking sites including ASP-32, THR-34 and LYS-268 (Fig. 8A), and with GSK-3β protein by hydrogen bonds, with docking sites including GLN-206 and ARG-96 (Fig. 8B). SP can also connect to ERK protein by hydrogen bonds, and the docking site is ARG-277 (Fig. 8C), and with p38 protein by hydrogen bonds, and docking sites include PRO-242, ASN-269, LYS-267, ASN-272, and THR-241 (Fig. 8D).
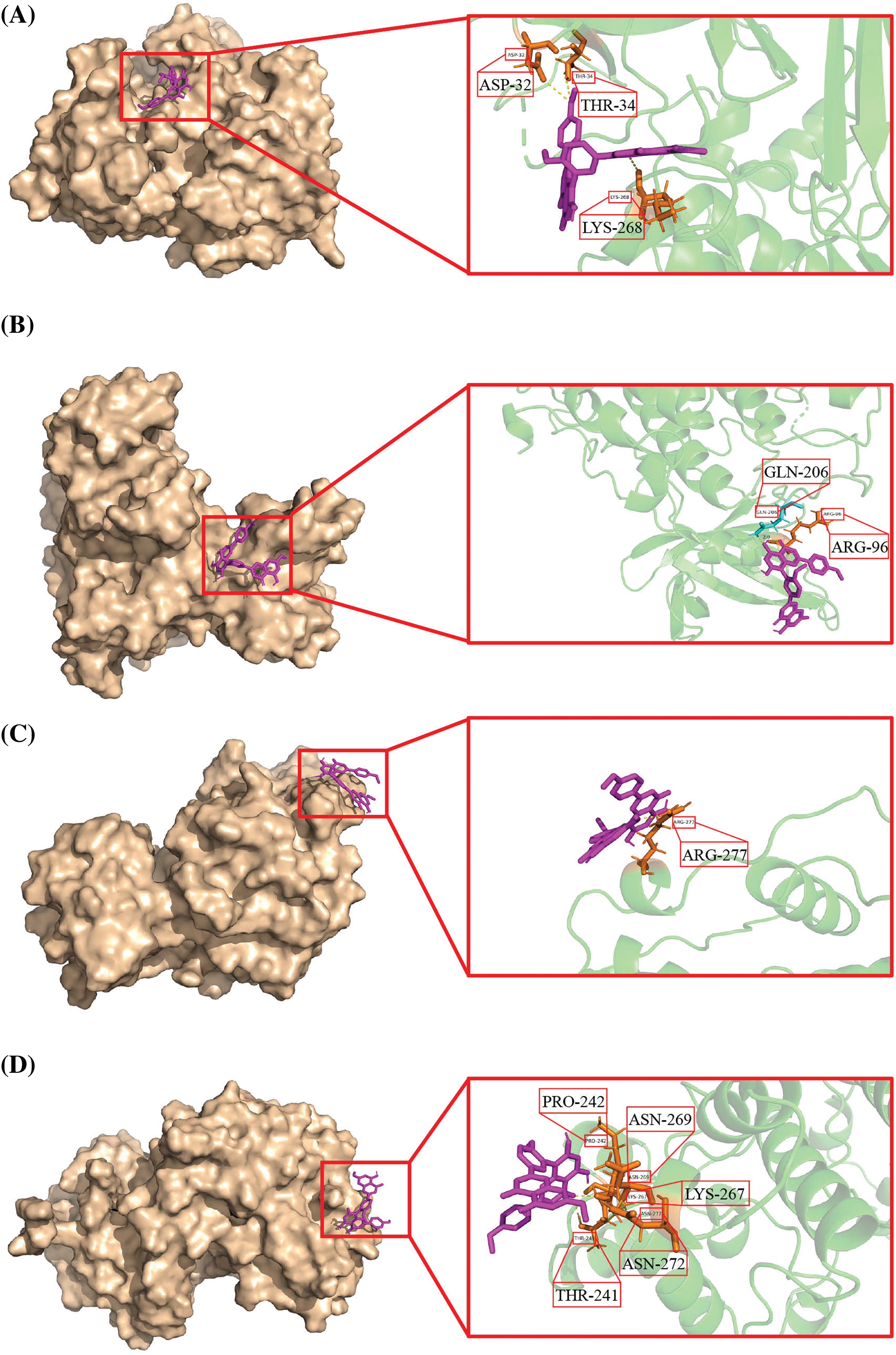
Figure 8: SP docked the target protein by molecular docking. (A) 3D structure of SP binding to AKT. (B) 3D structure of SP binding to GSK-3β. (C) 3D structure of SP binding to ERK. (D) 3D structure of SP binding to p38.
Prior studies have shown that SP treatment could reduce the cellular damage induced by anti-mycetin A in osteoblast MC3T-E1 cells, and by improving the body’s methylglyoxal detoxifying mechanism, SP may also shield it against methylglyoxal-induced glycation [21,24]. However, there is little research on its application as a cancer treatment. We found that SP has cross-targets for HCC in our network pharmacological prediction analysis. Fig. 1B showed that Post-transcriptionalGeneSilencing (PTGS) and GSK-3β are the most critical targets in various proteins involved in the biological process of SP treatment of HCC. In addition, many studies have confirmed that GSK-3β is closely associated with cell migration. This suggests that SP may affect HCC metastasis, which requires further verification. Network pharmacology analysis also showed that SP could induce apoptosis in HCC cells (Figs. 1C, 1D) and that ROS is fundamental to SP against HCC. In addition, we found that the cross-targets of SP and HCC are primarily involved in the PI3K-AKT, MAPK, and NF-κB signaling pathways. According to the network pharmacology prediction results, we hypothesized that SP has a potential therapeutic effect on HCC.
The survival rates of HepG2, Huh-7, Hep3B, THLE-2, 293T, and GES-1 cells were detected in this study, and SP was found to be more effective in killing HCC cells at lower concentrations than 5-FU, with fewer adverse effects on normal cells. The IC50 values of HepG2, Huh-7, and Hep3B were 38.02, 48.89, and 46.69 μM. This indicates that SP has a good killing effect on HCC cells. HepG2 has the lowest IC50 value among these. Thus, HepG2 cells were selected for our further experiments.
Apoptosis participates in the anti-tumor biological process by targeting and destroying cancer cells. Mitochondria, small organelles within cells responsible for energy production, act as gatekeepers, controlling whether cells undergo apoptosis [25]. This study showed that HepG2 cells revealed apoptosis in both the initial and late stages after SP treatment. The duration of SP administration was positively correlated with the degree to which mitochondrial membrane potential was reduced.
The B-cell lymphoma 2 family is widely recognized for regulating apoptosis and maintaining mitochondrial structure and function [26–28]. When apoptosis occurs in cells, the mitochondria release cyto C, cyto CC, and caspase-9 precursors. These proteins are essential for forming apoptotic structures and activating caspase-9, ultimately leading to caspase-3 activation, apoptosis, and a therapeutic effect [29]. In our study, we found that the SP can resulted in the mitochondrion-dependent cell death in HepG2 cells.
The study has demonstrated that SP can activate the NF-κB signaling pathway, and this inhibition is dose-dependent and inhibits osteoclastogenesis induced by receptor activator of Nuclear Factor-κB Ligand (RANKL). Moreover, SP can decrease the upregulation of c-Fos. At the same time, SP can have the potential to suppress osteoclast differentiation and bone resorption activity [20]. Interestingly, our study revealed a fascinating connection between SP-induced apoptosis and the involvement of NF-κB. Furthermore, numerous investigations have validated the interaction between the MAPK and the NF-κB signaling pathway at various temporal stages [30]. In this study, Western Blot analysis results showed that MAPK can regulate STAT3 as an upstream signaling pathway, and induce mitochondrion-dependent apoptosis in HepG2 cells.
ROS accumulation in mitochondria will lead to carcinogenesis. Thus, ROS accumulation in tumor cells is generally high. When ROS accumulates excessively in cancer cells, it promotes the production of oxidative stress, which induces tumor cell death [31]. According to the KEGG analysis suggested that ROS was involved in the anti-HCC mechanism of SP, and the results of this study showed after SP treatment, the ROS level in HepG2 cells was significantly increased. Our previous results showed that the apoptosis could be enhanced through the apoptosis could be enhanced through the modulation of the signaling pathway MAPK/STAT3/NF-κB by SP. Still, it is not clear whether this is related to the changes in intracellular ROS. Therefore, NAC, a ROS scavenger, was used to pretreat HepG2 cells, followed by SP. The results confirmed that SP can upregulate ROS accumulation and regulate the MAPK/STAT3/NF-κB pathway-induced HepG2 cell apoptosis.
Cell cycle dysregulation, which is an established hallmark of cancer, has great significance for cancer treatment [32]. Flow cytometry results showed that many cells were in the G0/G1 after SP treatment. This indicates that these cells are undergoing cell cycle arrest and are not actively dividing. Moreover, the protein expression level was inhibited in the NAC group compared with the SP-treated group. This implies that the ROS-mediated AKT may be related to the SP to arrest the cell cycle in HepG2 cells.
Cell migration is a complex and dynamic process. It affects critical physiological processes, such as individual development and wound healing [33]. Due to cell migration, cancer cells can participate in the circulatory system and migrate from primary tumors to other tissues, causing cancer cell growth, which poses risks to human health and life [34]. AKT is a serine/threonine-protein kinase. After phosphorylation and activation, AKT can regulate the activity of many downstream molecules including GSK-3β, which leads to phosphorylation and nuclear translocation of β-catenin, inhibiting cancer cell migration and invasion [35–37] Through network pharmacology, we previously predicted that GSK-3β, as a critical protein, might have an essential role in the anti-HCC mechanism of SP. Western Blot revealed that SP upregulated the E-cadherin expression level and downregulated p-AKT, p-GSK-3β, N-cadherin, vimentin, SNAI 1, and β-catenin. This result verifies that our prediction was correct. The protein changes in HepG2 cells showed an opposite trend after the addition of NAC pretreatment, and SP inhibited cell migration by accumulating intracellular ROS accumulation.
In this study, we predicted the binding sites of SP to HCC-related proteins by molecular docking. The results showed that SP could successfully dock with AKT, GSK-3β, and some MAPK protein kinases. GSK-3β, as a key target protein screened by PPI, may bind to SP through the key amino acid residue ARG-96 of the GSK-3β protein. Moreover, ERK and p38 can interact with each other, which is consistent with our molecular mechanism results. Although the docking between JNK and SP was not successful in our docking experiment results, considering the limitations of network pharmacological analysis, more experimental verification, and other ways are needed to further explore this issue in the future.
In this study, HepG2, Huh-7, and Hep3B cell lines were selected to detect the anti-HCC effect of SP, and the HepG2 cells were screened by CCK-8 assay as a model system for subsequent studies. However, although HepG2 cells are widely used, they only represent one type of HCC and may not fully reflect all subtypes of HCC. Therefore, SP may have different effects in other HCC cell lines or primary HCC cells. Additional work is needed to precisely assess the efficacy of SP by conducting studies on a broader range of HCC cell models. Clinical trial is the key step to verify the efficacy and safety of drugs. Besides, we found that SP can induce apoptosis and inhibit migration in HepG2 cells, which is consistent with the mechanism of action of commonly used anti-HCC drugs in clinical practice, such as apatinib [38] and lenvatinib [39]. These drugs exert their therapeutic effects by inducing apoptosis and inhibiting migration of HCC cells, and the similar bioactivity of SP suggests that it may have potential for clinical application. However, this hypothesis still needs to be tested, and further in vivo experiments are necessary in the future to examine the expression changes of the relevant target genes at the tissue level.
In the subsequent molecular mechanism analysis, we found that SP exhibited a similar molecular mechanism to apatinib and lenvatinib in the anti-liver cancer process, which was to induce cell apoptosis by regulating AKT, MAPK, STAT3 and NF-κB signaling pathways [38,40,41]. Apoptosis has become a high-profile emerging therapeutic strategy in the field of anti-HCC. This finding not only further confirms the importance of apoptosis in HCC treatment, but also provides theoretical support for the combined application of SP with other drugs.
Moreover, the anti-HCC mechanism of SP is closely related to the actual case situation of HCC patients in clinical practice. It has been demonstrated that the MAPK signaling pathway plays a crucial role in HCC development, which is activated in more than 50% of human HCC cases [42]. Interestingly, this research revealed that SP could regulate this key MAPK signaling pathway and subsequently induce apoptosis in HepG2 cells. The identification of this apoptotic mechanism not only establishes a theoretical foundation for the potential utilization of SP in addressing HCC but also offers significant insights to advance our comprehension of HCC’s pathogenesis and the exploration of novel therapeutic strategies.
This study indicated that SP could induce apoptosis through the ROS-mediated MAPK/STAT3/NF-κB signaling pathway, arrest the cell cycle, and inhibit cell migration through the ROS-mediated AKT signaling pathway in HepG2 cells.
Acknowledgement: None.
Funding Statement: This research was funded by the Heilongjiang Province Key Research and Development Plan Guidance Project (Grant No. GZ20220039), the National Natural Science Foundation of China (Grant No. 82060118), and the Program for Young Talents of Science and Technology in Universities of Inner Mongolia Autonomous Region (Grant No. NJYT24032), the Central Government Supports Local College Reform and Development Fund Talent Training Project (Grant No. 2020GSP16), and Heilongjiang Touyan Innovation Team Program (Grant No. 2019HTY078).
Author Contributions: The authors confirm their contribution to the paper as follows: study conception and design: Yan-Nan Li, Yun-Hong Xiu, and Yan-Jun Tang; data collection: Jing-Long Cao; analysis and interpretation of results: Wen-Shuang Hou and An-Qi Wang; draft manuscript preparation: Yan-Nan Li; revised the final draft: Tian-Zhu Li and Cheng-Hao Jin. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The datasets generated during the analyzed study are available from the corresponding author on reasonable request.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49. doi:10.3322/caac.21660. [Google Scholar] [PubMed] [CrossRef]
2. Numata K. Abdominal ultrasound and treatment of hepatocellular carcinoma. Diagnostics. 2021;11(7):1268. doi:10.3390/diagnostics11071268. [Google Scholar] [PubMed] [CrossRef]
3. Wang Q, Yu X, Zheng Z, Chen F, Yang N, Zhou Y. Centromere protein N may be a novel malignant prognostic biomarker for hepatocellular carcinoma. PeerJ. 2021;9:e11342. doi:10.7717/peerj.11342. [Google Scholar] [PubMed] [CrossRef]
4. Xue XB, Lv TM, Hou JY, Li DQ, Huang XX, Song SJ, et al. Vibsane-type diterpenoids from Viburnum odoratissimum inhibit hepatocellular carcinoma cells via the PI3K/AKT pathway. Phytomedicine. 2023;108:154499. doi:10.1016/j.phymed.2022.154499. [Google Scholar] [PubMed] [CrossRef]
5. Anwanwan D, Singh SK, Singh S, Saikam V, Singh R. Challenges in liver cancer and possible treatment approaches. Biochim Biophys Acta Rev Cancer. 2020;1873(1):188314. doi:10.1016/j.bbcan.2019.188314. [Google Scholar] [PubMed] [CrossRef]
6. Zhan M, Ding Y, Huang S, Liu Y, Xiao J, Yu H, et al. Lysyl oxidase-like 3 restrains mitochondrial ferroptosis to promote liver cancer chemoresistance by stabilizing dihydroorotate dehydrogenase. Nat Commun. 2023;14(1):3123. doi:10.1038/s41467-023-38753-6. [Google Scholar] [PubMed] [CrossRef]
7. Thakur RS, Devaraj E. Lagerstroemia speciosa (L.) Pers. triggers oxidative stress mediated apoptosis via intrinsic mitochondrial pathway in HepG2 cells. Environ Toxicol. 2020;35(11):1225–33. doi:10.1002/tox.22987. [Google Scholar] [PubMed] [CrossRef]
8. Abolfathi H, Arabi M, Sheikhpour M. A literature review of microRNA and gene signaling pathways involved in the apoptosis pathway of lung cancer. Respir Res. 2023;24(1):55. doi:10.1186/s12931-023-02366-w. [Google Scholar] [PubMed] [CrossRef]
9. Żyro D, Śliwińska A, Szymczak-Pajor I, Stręk M, Ochocki J. Pro-apoptotic and genotoxic properties of silver (I) complexes of metronidazole and 4-hydroxymethylpyridine against pancreatic cancer cells in vitro. Cancers. 2020;12(12):3848. doi:10.3390/cancers12123848. [Google Scholar] [PubMed] [CrossRef]
10. Liu WT, Chen DY, Su JY, Zheng RL, Kong R, Zhu B, et al. Quercetin induced HepG2 cells apoptosis through ATM/JNK/STAT3 signaling pathways. BIOCELL. 2023;47(1):187–94. doi:10.32604/biocell.2022.023030. [Google Scholar] [CrossRef]
11. Aggarwal V, Tuli HS, Varol A, Thakral F, Yerer MB, Sak K, et al. Role of reactive oxygen species in cancer progression: molecular mechanisms and recent advancements. Biomolecules. 2019;9(11):735. doi:10.3390/biom9110735. [Google Scholar] [PubMed] [CrossRef]
12. Wang Q, Xu C, Fan Q, Yuan H, Zhang X, Chen B, et al. Positive feedback between ROS and cis-axis of PIASxα/p38α-SUMOylation/MK2 facilitates gastric cancer metastasis. Cell Death Dis. 2021;12(11):986. doi:10.1038/s41419-021-04302-6. [Google Scholar] [PubMed] [CrossRef]
13. Li S, Han X, Lu Z, Qiu W, Yu M, Li H, et al. MAPK cascades and transcriptional factors: regulation of heavy metal tolerance in plants. Int J Mol Sci. 2022;23(8):4463. doi:10.3390/ijms23084463. [Google Scholar] [PubMed] [CrossRef]
14. Cicenas J, Zalyte E, Rimkus A, Dapkus D, Noreika R, Urbonavicius S, et al. JNK, p38, ERK, and SGK1 inhibitors in cancer. Cancers. 2017;10(1):1. doi:10.3390/cancers10010001. [Google Scholar] [PubMed] [CrossRef]
15. Xu X, Lai Y, Hua ZC. Apoptosis and apoptotic body: disease message and therapeutic target potentials. Biosci Rep. 2019;39(1):BSR20180992. doi:10.1042/bsr20180992. [Google Scholar] [PubMed] [CrossRef]
16. D’Arcy MS. Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int. 2019;43(6):582–92. doi:10.1002/cbin.11137. [Google Scholar] [PubMed] [CrossRef]
17. Luo H, Vong CT, Chen H, Gao Y, Lyu P, Qiu L, et al. Naturally occurring anti-cancer compounds: shining from Chinese herbal medicine. Chin Med. 2019;14:48. doi:10.1186/s13020-019-0270-9. [Google Scholar] [PubMed] [CrossRef]
18. Wang K, Chen Q, Shao Y, Yin S, Liu C, Liu Y, et al. Anticancer activities of TCM and their active components against tumor metastasis. Biomed Pharmacother. 2021;133:111044. doi:10.1016/j.biopha.2020.111044. [Google Scholar] [PubMed] [CrossRef]
19. Choi EM, Suh KS, Rhee SY, Kim YS. Sciadopitysin alleviates methylglyoxal-mediated glycation in osteoblastic MC3T3-E1 cells by enhancing glyoxalase system and mitochondrial biogenesis. Free Radic Res. 2014;48(7):729–39. doi:10.3109/10715762.2014.903562. [Google Scholar] [PubMed] [CrossRef]
20. Cao J, Lu Q, Liu N, Zhang YX, Wang J, Zhang M, et al. Sciadopitysin suppresses RANKL-mediated osteoclastogenesis and prevents bone loss in LPS-treated mice. Int Immunopharmacol. 2017;49:109–17. doi:10.1016/j.intimp.2017.05.029. [Google Scholar] [PubMed] [CrossRef]
21. Suh KS, Chon S, Choi EM. The protective effects of sciadopitysin against methylglyoxal-induced cytotoxicity in cultured pancreatic β-cells. J Appl Toxicol. 2018;38(8):1104–11. doi:10.1002/jat.3620. [Google Scholar] [PubMed] [CrossRef]
22. Suh KS, Chon S, Jung WW, Choi EM. Protective effects of sciadopitysin against methylglyoxal-induced degeneration in neuronal SK-N-MC cells. J Appl Toxicol. 2022;42(2):274–84. doi:10.1002/jat.4211. [Google Scholar] [PubMed] [CrossRef]
23. V.H. P, Kuruburu MG, M.K. J, N. AS, Taha Babakr A, Sreenivasan R, et al. Bioactive profiling and evaluation of anti-proliferative and anti-cancerous properties of Shivagutika, an Indian polyherbal formulation synchronizing in vitro and in silico approaches. Front Chem. 2023;11:1195209. doi:10.3389/fchem.2023.1195209. [Google Scholar] [PubMed] [CrossRef]
24. Suh KS, Lee YS, Kim YS, Choi EM. Sciadopitysin protects osteoblast function via its antioxidant activity in MC3T3-E1 cells. Food Chem Toxicol. 2013;58:220–7. doi:10.1016/j.fct.2013.04.028. [Google Scholar] [PubMed] [CrossRef]
25. Zichri SB, Kolusheva S, Shames AI, Schneiderman EA, Poggio JL, Stein DE, et al. Mitochondria membrane transformations in colon and prostate cancer and their biological implications. Biochim Biophys Acta Biomembr. 2021;1863(1):183471. doi:10.1016/j.bbamem.2020.183471. [Google Scholar] [PubMed] [CrossRef]
26. Czabotar PE, Garcia-Saez AJ. Mechanisms of BCL-2 family proteins in mitochondrial apoptosis. Nat Rev Mol Cell Biol. 2023;24(10):732–48. doi:10.1038/s41580-023-00629-4. [Google Scholar] [PubMed] [CrossRef]
27. Bas J, Nguyen T, Gillet G. Involvement of Bcl-xL in neuronal function and development. Int J Mol Sci. 2021;22(6):3202. doi:10.3390/ijms22063202. [Google Scholar] [PubMed] [CrossRef]
28. Means RE, Katz SG. Balancing life and death: BCL-2 family members at diverse ER-mitochondrial contact sites. FEBS J. 2022;289(22):7075–112. doi:10.1111/febs.16241. [Google Scholar] [PubMed] [CrossRef]
29. Zaib S, Hayyat A, Ali N, Gul A, Naveed M, Khan I. Role of mitochondrial membrane potential and lactate dehydrogenase a in apoptosis. Anticancer Agents Med Chem. 2022;22(11):2048–62. doi:10.2174/1871520621666211126090906. [Google Scholar] [PubMed] [CrossRef]
30. Wang Z, Zhao G, Zibrila AI, Li Y, Liu J, Feng W. Acetylcholine ameliorated hypoxia-induced oxidative stress and apoptosis in trophoblast cells via p38 MAPK/NF-κB pathway. Mol Hum Reprod. 2021;27(8):gaab045. doi:10.1093/molehr/gaab045. [Google Scholar] [PubMed] [CrossRef]
31. Moloney JN, Cotter TG. ROS signalling in the biology of cancer. Semin Cell Dev Biol. 2018;80:50–64. doi:10.1016/j.semcdb.2017.05.023. [Google Scholar] [PubMed] [CrossRef]
32. Sava GP, Fan H, Coombes RC, Buluwela L, Ali S. CDK7 inhibitors as anticancer drugs. Cancer Metastasis Rev. 2020;39(3):805–23. doi:10.1007/s10555-020-09885-8. [Google Scholar] [PubMed] [CrossRef]
33. Yamada KM, Sixt M. Mechanisms of 3D cell migration. Nat Rev Mol Cell Biol. 2019;20(12):738–52. doi:10.1038/s41580-019-0172-9. [Google Scholar] [PubMed] [CrossRef]
34. Intlekofer AM, Finley LWS. Metabolic signatures of cancer cells and stem cells. Nat Metab. 2019;1(2):177–88. doi:10.1038/s42255-019-0032-0. [Google Scholar] [PubMed] [CrossRef]
35. Pan J, Fan Z, Wang Z, Dai Q, Xiang Z, Yuan F, et al. CD36 mediates palmitate acid-induced metastasis of gastric cancer via AKT/GSK-3β/β-catenin pathway. J Exp Clin Cancer Res. 2019;38(1):52. doi:10.1186/s13046-019-1049-7. [Google Scholar] [PubMed] [CrossRef]
36. Wan G, Liu Y, Zhu J, Guo L, Li C, Yang Y, et al. SLFN5 suppresses cancer cell migration and invasion by inhibiting MT1-MMP expression via AKT/GSK-3β/β-catenin pathway. Cell Signal. 2019;59:1–12. doi:10.1016/j.cellsig.2019.03.004. [Google Scholar] [PubMed] [CrossRef]
37. Tashiro K, Oikawa M, Miki Y, Takahashi T, Kumamoto H. Immunohistochemical assessment of growth factor signaling molecules: MAPK, Akt, and STAT3 pathways in oral epithelial precursor lesions and squamous cell carcinoma. Odontol. 2020;108(1):91–101. doi:10.1007/s10266-019-00428-4. [Google Scholar] [PubMed] [CrossRef]
38. Zhou C, Yao Q, Zhang H, Guo X, Liu J, Shi Q, et al. Combining transcatheter arterial embolization with iodized oil containing Apatinib inhibits HCC growth and metastasis. Sci Rep. 2020;10(1):2964. doi:10.1038/s41598-020-59746-1. [Google Scholar] [PubMed] [CrossRef]
39. Tan W, Zhang K, Chen X, Yang L, Zhu S, Wei Y, et al. GPX2 is a potential therapeutic target to induce cell apoptosis in lenvatinib against hepatocellular carcinoma. J Adv Res. 2023;44:173–83. doi:10.1016/j.jare.2022.03.012. [Google Scholar] [PubMed] [CrossRef]
40. Wu CH, Hsu FT, Chao TL, Lee YH, Kuo YC. Revealing the suppressive role of protein kinase C delta and p38 mitogen-activated protein kinase (MAPK)/NF-κB axis associates with lenvatinib-inhibited progression in hepatocellular carcinoma in vitro and in vivo. Biomed Pharmacother. 2022;145:112437. doi:10.1016/j.biopha.2021.112437. [Google Scholar] [PubMed] [CrossRef]
41. Weng YS, Chiang IT, Tsai JJ, Liu YC, Hsu FT. Lenvatinib synergistically promotes radiation therapy in hepatocellular carcinoma by inhibiting Src/STAT3/NF-κB-mediated epithelial-mesenchymal transition and metastasis. Int J Radiat Oncol Biol Phys. 2023;115(3):719–32. doi:10.1016/j.ijrobp.2022.09.060. [Google Scholar] [PubMed] [CrossRef]
42. Moon H, Ro SW. MAPK/ERK signaling pathway in hepatocellular carcinoma. Cancers. 2021;13(12):3026. doi:10.3390/cancers13123026. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools