
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Lysine demethylase 5B transcriptionally regulates TREM1 in human cardiac fibroblasts
1 Department of Emergency, Wangjing Hospital of China Academy of Chinese Medical Sciences, Beijing, 100102, China
2 Department of Cardiology, State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100102, China
3 Department of Radiology, Wangjing Hospital of China Academy of Chinese Medical Sciences, Beijing, 100102, China
4 Department of Cardiology, Wangjing Hospital of China Academy of Chinese Medical Sciences, Beijing, 100102, China
* Corresponding Author: JIE YU. Email:
BIOCELL 2024, 48(7), 1105-1113. https://doi.org/10.32604/biocell.2024.050509
Received 08 February 2024; Accepted 10 April 2024; Issue published 03 July 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: A differential gene, triggering receptor expressed on myeloid cells 1 (TREM1), was identified in blood sequencing datasets from myocardial infarction patients and healthy controls. Myocardial fibrosis following myocardial infarction significantly contributes to cardiac dysfunction. Objectives: This study aimed to unveil the intrinsic regulatory mechanism of TREM1 in myocardial fibrosis. Methods: Mimicking pathology by angiotensin II (Ang II) treatment of human cardiac fibroblasts (HCFs), the impacts of TREM1 knockdown on its proliferation, migration, and secretion of the pro-fibrotic matrix were identified. Using the Human Transcription Factor Database (HumanTFDB) website, lysine-specific demethylase 5B (KDM5B) was found to bind to the TREM1 promoter, which was further validated through luciferase reporter and chromatin immunoprecipitation (ChIP). By promoting KDM5B overexpression, its effect on the regulation of TREM1 was examined. Results: TREM1 knockdown suppressed the proliferation, migration, and secretion of the pro-fibrotic matrix in HCFs upon Ang II treatment. KDM5B bound to the TREM1 promoter and upregulated its transcriptional expression. Furthermore, KDM5B overexpression reversed the regulation of the above cellular phenotypes by TREM1 knockdown. Conclusion: This study sheds light on the positive regulation of TREM1 by KDM5B, demonstrating their role in promoting myocardial fibrosis. This finding provides a theoretical foundation for understanding disease pathology and potentially advancing the development of new targeted therapies.Keywords
Myocardial fibrosis refers to a pathological process in which various physical and chemical stimuli, such as ischemic injury, pressure overload, and systemic inflammation, trigger excessive deposition of extracellular matrix (ECM) in the myocardial interstitium [1,2]. Cardiac fibroblasts, constituting 90%–95% of the myocardial interstitium, are indispensable for the normal function of the myocardium under normal conditions [3]. However, under pathological circumstances, such as exposure to angiotensin II (Ang II) [4,5], high glucose [6], and oxidative stress [7], cardiac fibroblasts undergo excessive proliferation and participate in myocardial interstitial remodeling through signal transduction pathways and the regulation of transcription factors [8–10].
Given the tight association between myocardial fibrosis, heart failure, and adverse outcomes [11], gaining a deeper understanding of the pathogenesis of myocardial fibrosis is essential for identifying new therapeutic targets. Nevertheless, there are currently no approved treatments directly targeting the excessive ECM or the cardiac fibroblasts responsible for its production [12].
Myocardial fibrosis following myocardial infarction significantly contributes to cardiac dysfunction in patients [13]. In the healing response after myocardial infarction, damaged tissue is flooded with fibrotic scars created by fibroblasts and myofibroblasts. On the positive side, reparative fibrosis is beneficial in preventing ventricular wall rupture. Nevertheless, fibrosis beyond the damaged site can cause aggravation of cardiac dysfunction [14]. The identification of the differential gene, triggering receptor expressed on myeloid cells 1 (TREM1), was based on bioinformatics analysis of blood sequencing datasets from myocardial infarction patients and healthy controls [15]. Previous reports have shown that TREM1 is upregulated in bleomycin-treated mouse alveolar epithelial cells, exerting pro-fibrotic effects in the lung by inducing cellular senescence [16]. Additionally, reducing TREM1 protein expression attenuates renal inflammation and fibrosis in rats subjected to unilateral ureteral obstruction [17]. The binding of soluble TREM1 to the membrane receptor ring guide receptor 2 activates downstream Smad2/3 and phosphatidylinositol-3-kinase (PI3K)/protein kinase B (Akt) signaling pathways, promoting hepatic stellate cell activation and liver fibrosis [18]. Based on these studies, the hypothesis of TREM1’s role in cardiac fibrosis was proposed.
Investigation via the Human Transcription Factor Database (HumanTFDB) website (http://bioinfo.life.hust.edu.cn/HumanTFDB) reveals that lysine-specific demethylase 5B (KDM5B) binds to the TREM1 promoter, thereby modulating the transcription and expression. KDM5B, a member of the KDM5 enzyme family, acts as a histone demethylase, catalyzing H3K4 demethylation. Interactions between KDM5 family members and transcription factors can localize them to specific genes, and they have also been shown to associate with multiple nuclear receptors and activate or repress target genes [19]. A prior study has demonstrated that KDM5B expression is upregulated in cardiac fibroblasts and myocardial tissue in response to pathological stress. Importantly, KDM5B deficiency significantly improves myocardial fibrosis and prevents adverse cardiac remodeling following myocardial infarction or pressure overload [20]. This study aims to unveil the intrinsic regulatory mechanism of the KDM5B/TREM1 axis in myocardial fibrosis, providing a theoretical foundation for understanding myocardial fibrosis pathology, and potentially advancing the development of new targeted therapies.
Human cardiac fibroblasts (HCFs, Cell Applications, Sigma-Aldrich, Shanghai, China) were maintained in Cardiac Fibroblast Growth Medium (Cell Applications, 316–500, San Diego, CA, USA). Additional 5% fetal bovine serum (Cell Applications, 026–100, San Diego, CA, USA) along with 1% penicillin-streptomycin (Gibco, Thermo Fisher Scientific, 15140122, Shanghai, China) constituted the complete medium. The incubator environment was set at a constant 37°C, 5% CO2. HCFs were exposed to 100 nM Ang II (Abmole, M6240, Shanghai, China) for 48 h to mimic pathological conditions [21].
For mechanistic studies, HCFs were transfected with short hairpin (sh)RNAs or pcDNA 3.1 vectors (Hanbio, Shanghai, China) to receive gene knockdown or overexpression. Scrambled shRNAs or empty vectors served as negative controls (marked as sh-NC and oe-NC). Sequences of shRNAs are listed in Table 1. HCFs (2 × 105/well) were plated one day before transfection. The mixture of Lipofectamine™ 2000 reagent (Thermo Fisher Scientific, 11668027, Shanghai, China) and shRNAs or vectors was supplemented into the wells to facilitate transfection, and the efficiency was identified after 48 h.

HCFs were treated with RIPA buffer (Yeasen, 20101ES60, Shanghai, China), followed by centrifugation at 10,000 × g, 37°C for 5 min. The resulting supernatant was gathered and a spectrophotometer (NanoDrop 2000, Thermo Fisher Scientific, Shanghai, China) was applied for protein quantification. After SDS-PAGE, the separated samples were wet transferred onto the polyvinylidene fluoride (PVDF) membranes (Beyotime, Shanghai, China). After blocking with 5% skimmed milk for 2 h, the above membranes were incubated overnight at 4°C with primary antibodies. TREM1 (11791-1-AP) antibody was purchased from Proteintech (Wuhan, China), while KDM5B (A7772), α-smooth muscle actin (SMA; A1011), connective tissue growth factor (CTGF; A11456), Fibronectin (A16678) and GAPDH (A19056) antibodies were purchased from ABclonal (Wuhan, China). They were applied after a thousand-fold dilution. After the reaction with horseradish peroxidase (HRP)-conjugated anti-rabbit antibody (ABclonal, AS014, 1: 5000; Wuhan, China) for 1.5 h at 37°C, the blots were immersed in enhanced chemiluminescence (ECL) reagent (ABP Biosciences, LLC., FP300; Rockville, MD, USA) at room temperature and the intensity was quantified in ImageJ software (version 1.54).
Quantitative reverse transcription PCR (RT-qPCR)
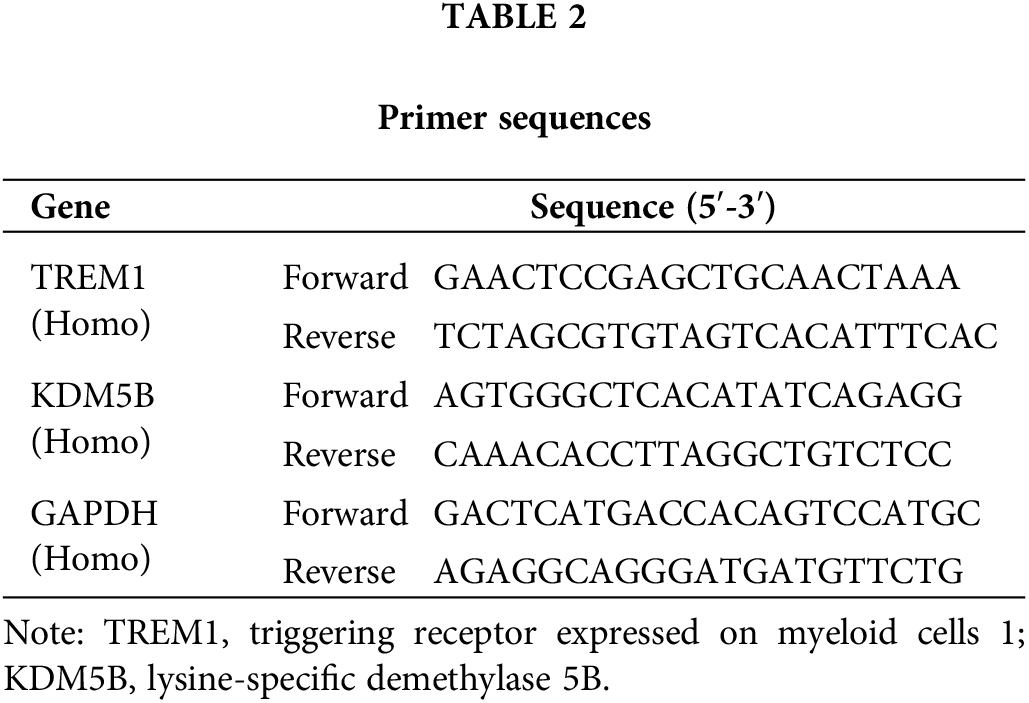
TRIzol (Invitrogen, Thermo Fisher Scientific, 15596018; Shanghai, China) was added in HCFs for lysis and chloroform was added 5 min later. The sample was mixed thoroughly by shaking and centrifuged (12,000 × g, 15 min). The upper phase was gathered and isopropanol precipitated RNA. Following the SYBR Green One-Step qRT-PCR Kit (Beyotime, D7268, Shanghai, China) instructions, the one-step RT-qPCR reaction system, including reaction buffer, enzyme mix, primer mix, template RNA, low ROX, and RNase-free water, was set up on ice. The reaction tube was placed on the Real-Time PCR instrument (7500 Fast, Applied Biosystems, Thermo Fisher Scientific, Shanghai, China). RT was performed at 50°C for 15 min, and the pre-denaturation of the template was set at 95°C for 2 min, followed by a PCR reaction consisting of 40 cycles of 95°C for 15 s and 60°C for 30 s. Analysis proceeded using the Delta Delta cycle threshold (∆∆Ct) method [22]. See Table 2 for primer sequences.
Cell counting kit-8 (CCK-8) assay
HCFs (5 × 103 per well) in a 96-well plate were stimulated with Ang II as afore-described. CCK-8 solution (10 μL; Yeasen, 40203ES76, Shanghai, China) was supplemented. A microplate reader (UH5700, Hitachi, Shanghai, China) was utilized for data acquisition at 450 nm.
5-ethynyl-2′-deoxyuridine (EdU) assay
HCFs (5 × 103 per well) in a 96-well plate were treated with Ang II for 48 h. They were incubated with a medium containing 10 μM EdU (Sangon Biotech, E607204, Shanghai, China) for 2 h, and paraformaldehyde (4%; Life-ilab, AC28L112, Shanghai, China) was added for fixation at room temperature. HCFs were permeabilized with Triton X-100 (0.5%; Invitrogen, Thermo Fisher Scientific, HFH10, Shanghai, China) for 10 min. The staining reaction mixture was prepared according to the instruction manual and it was used to treat HCFs for 30 min in the dark. Visualization of cell nuclei applied 4′,6-diamidino-2-phenylindole (DAPI, Biosharp, BL105A, Hefei, China) for 5 min. Photographs were obtained under a microscope (ECLIPSE 80i, Nikon, Tokyo, Japan) and positive cells were computed using ImageJ software (version 1.54).
After HCFs were starved overnight and reached full confluency, scratches were created using a cell scraper. The remaining HCFs in the wound were removed by rinsing with PBS. HCFs were maintained at 37°C with medium containing 1% FBS and Ang II for 48 h and images were photographed initially and after 48 h under a microscope (ECLIPSE E100, Nikon, Tokyo, Japan).
The TREM1 promoter fragment (−674~−689) was mutated, and the mutant along with wild fragments was constructed into the pGL3-basic reporter vector (Promega, E1751, Beijing, China). Co-transfection of KDM5B overexpression plasmids/oe-NC and the prepared reporter vector into HCFs proceeded. The values of Firefly and Renilla luciferase were acquired from the Dual-Luciferase Reporter Assay System (Promega, E1910, Beijing, China). The ratio of these two luciferase activity reflects the activity of transcription.
Chromatin immunoprecipitation (ChIP)
Formaldehyde was supplemented to the wells, mixed gently, and kept for 10 min to cross-link the target protein and DNA. DNA was fragmented by incubating at 37°C with micrococcal nuclease (MNase; Beyotime, D7201M, Shanghai, China) for 20 min and sonicated 3 times for 15 s each time. A small portion of the supernatant from the sonicated samples (centrifugation at 4°C, 12,000 × g, 5 min) was taken as Input, and KDM5B/lgG antibodies (CST, 15327 and 2729, 1:50, Shanghai, China) were incubated with the rest at 4°C overnight. Next, Protein A/G beads (Beyotime, P2083S, Shanghai, China) were supplemented to precipitate the protein-DNA complex in a slow oscillation for 1 h at 4°C. The beads were gathered via magnetic force and samples were eluted using buffer (1% SDS, 0.1 M NaHCO3) thrice at room temperature. The supernatants were combined and 5 M NaCl was applied to remove cross-links at 65°C for 2 h, including the previously retained Input, and then qPCR proceeded as described above.
Following treatment with Ang II for 48 h, fixation of HCFs proceeded at room temperature with 4% paraformaldehyde (Life-ilab, AC28L112, Shanghai, China) for 15 min. Permeability with Triton X-100 (Invitrogen, Thermo Fisher Scientific Inc., HFH10, Shanghai, China) for 5 min and block with 1% BSA (Yeasen, 36102ES60, Shanghai, China) for 1 h were followed at room temperature. HCFs were incubated with Collagen I antibody (ABclonal, A5786, 1:200; Wuhan, China) overnight at 4°C, and then replaced with FITC-conjugated antibody (Abcam, ab6717, 1:3000, Shanghai, China) in the dark. Visualization of cell nuclei applied DAPI for 5 min. Photographs were obtained under a microscope (ECLIPSE 80i, Nikon, Tokyo, Japan), and the level of Collagen I was semi-quantified using ImageJ software (version 1.54).
The obtained data were analyzed for significance in the form of means ± standard deviations (n ≥ 3) in GraphPad Prism (version 9.0). The significance test for the two groups used the unpaired t-test. The rest used analysis of variance together with Tukey’s test. p < 0.05 represents significant.
TREM1 level in response to Ang II
HCFs were subjected to Ang II to simulate pathological conditions. Ang II treatment induced significant increases in TREM1 levels, both mRNA and protein, in HCFs (Figs. 1A, 1B). By knocking down TREM1 expression in HCFs, its regulatory function was specifically studied in the subsequent assays. The sh-TREM1-1 group was chosen because its knockdown efficacy was better than that in the sh-TREM1-2 (Figs. 1C, 1D). Despite Ang II treatment, the sh-TREM1-1 group exhibited a significant reduction in TREM1 levels compared to the Ang II + sh-NC group (Figs. 1E, 1F).
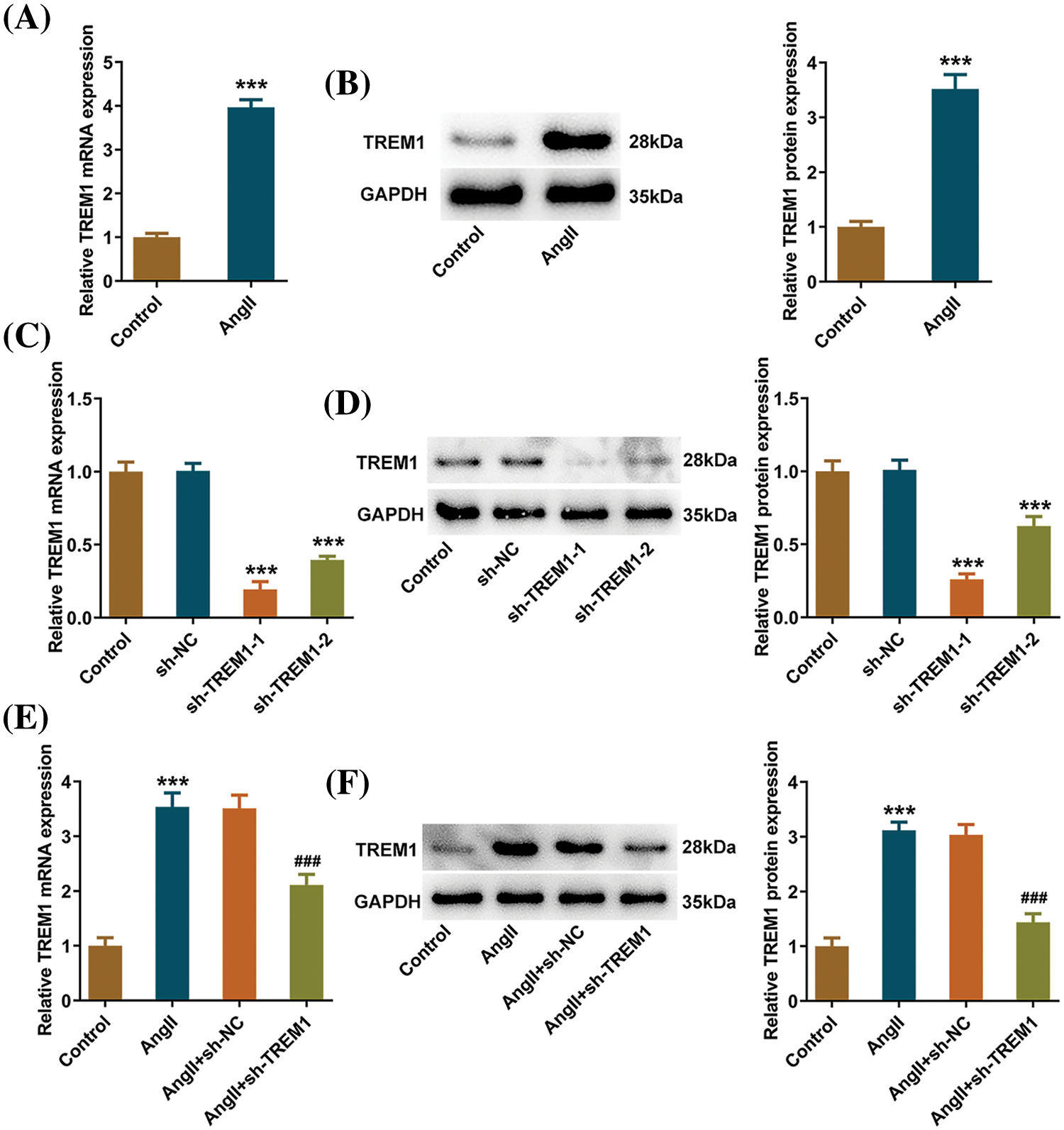
Figure 1: Triggering receptor expressed on myeloid cells 1 (TREM1) level in response to angiotensin II (Ang II). (A) Quantitative reverse transcription PCR (RT-qPCR) and (B) western blotting reveal the impact of Ang II treatment on TREM1 levels in human cardiac fibroblasts (HCFs). (C) The mRNA and (D) protein levels of TREM1 in HCFs following transfection. (E) TREM1 mRNA and (F) protein levels in HCFs with TREM1 knockdown resulted from Ang II treatment. ***p < 0.001 vs. control; ###p < 0.001 vs. Ang II + short hairpin RNA (sh)-negative control (NC).
Impacts of TREM1 on the profibrotic phenotypes
Afterward, the impact of TREM1 knockdown on HCF viability and proliferation phenotypes was evaluated. CCK8 assay was used to determine the viability of each group of HCFs. Ang II treatment led to a marked increase in HCF viability, reflecting enhanced cell proliferation. TREM1 knockdown significantly reduced the level of cell proliferation in contrast with the Ang II + sh-NC group (Fig. 2A). EdU staining exhibited that the Ang II group had significantly more HCFs during the proliferation period than the control group, and the knockdown of TREM1 was beneficial in alleviating the increase in proliferation (Fig. 2B). Furthermore, HCF migration upon TREM1 knockdown was evaluated using scratch analysis. Ang II treatment enhanced the migration potential of HCFs, and the migration of HCFs with TREM1 knockdown was suppressed in contrast with the Ang II + sh-NC (Fig. 2C).
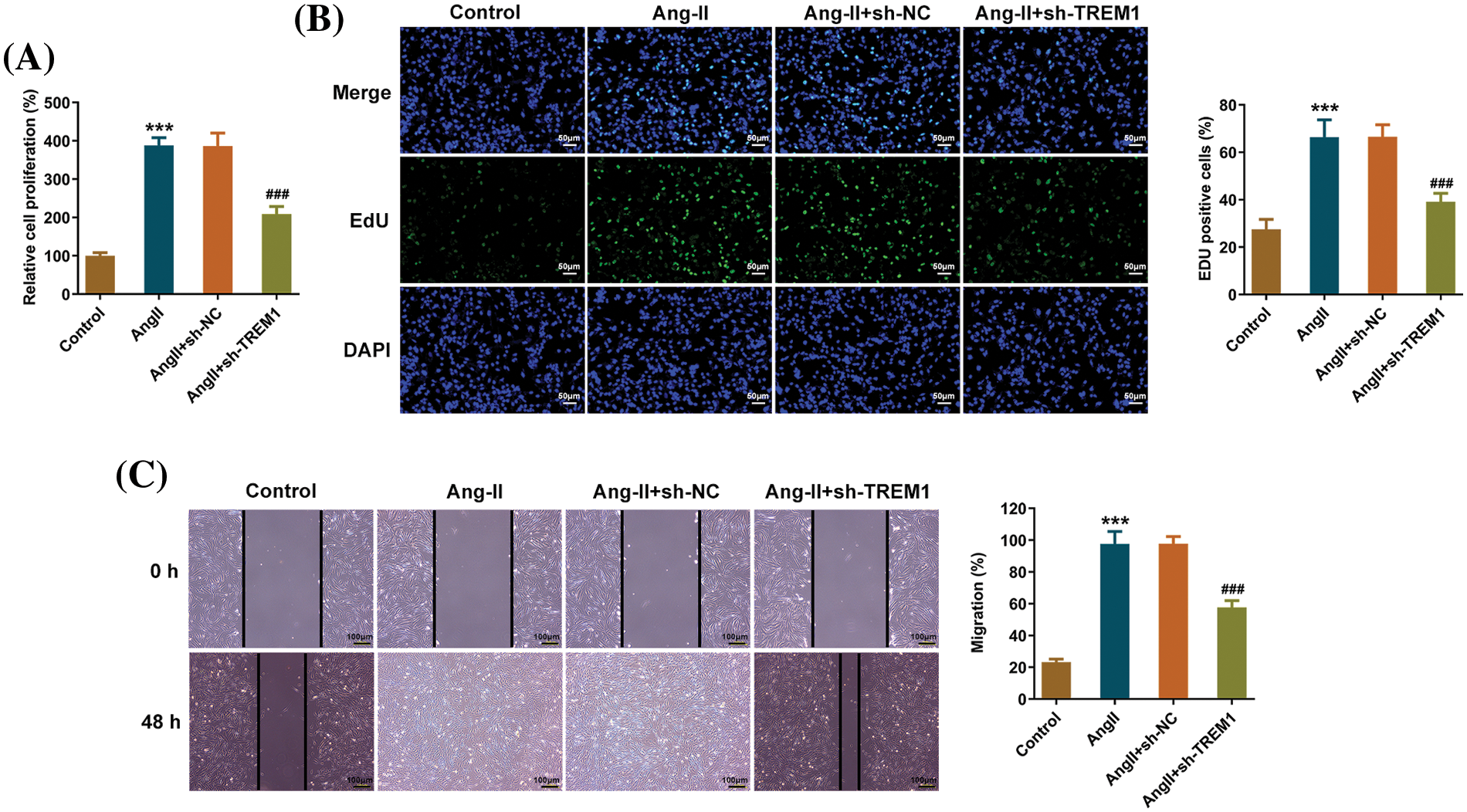
Figure 2: Impacts of triggering receptor expressed on myeloid cells 1 on the proliferation and migration. (A) Cell counting kit-8 assay determines the viability of each group of human cardiac fibroblasts (HCFs), reflecting the level of cell proliferation. (B) 5-ethynyl-2′-deoxyuridine (EdU) staining shows the number of HCFs during the proliferation period, scale bar: 50 μm. (C) Scratch analysis reveals the migration potential of HCFs, scale bar: 100 μm. ***p < 0.001 vs. control; ###p < 0.001 vs. angiotensin II (Ang II) + short hairpin RNA (sh)-negative control (NC).
Extracellular matrices such as Collagen I contribute to the fibrotic process, and the level of Collagen I in HCFs was used to assess fibrotic propensity. IF results demonstrated that the Collagen I level was increased in the Ang II group, whereas TREM1 knockdown inhibited the increase of Collagen I level (Fig. 3A). Additionally, levels of several other proteins involved in fibrosis were measured to assist in assessing the degree of pro-fibrosis. α-SMA, CTGF, and fibronectin proteins increased sharply upon Ang II treatment, and TREM1 knockdown hindered the increase in the levels of these fibrosis-related proteins (Fig. 3B). These revealed that TREM1 knockdown weakened the pro-fibrotic phenotypes of HCFs, suggesting that TREM1 knockdown was beneficial to avoid myocardial fibrosis tendencies.
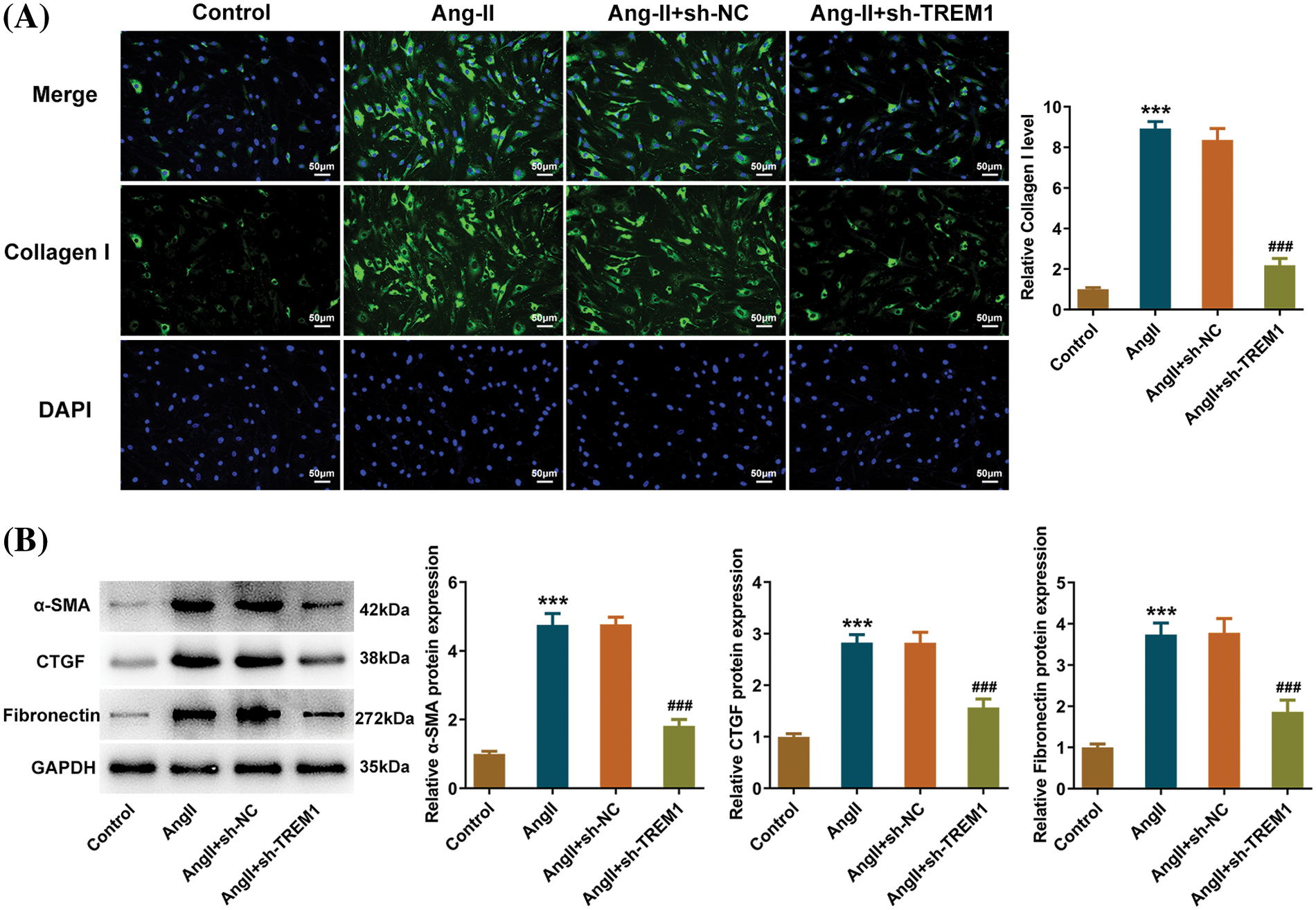
Figure 3: Impacts of triggering receptor expressed on myeloid cells 1 on the pro-fibrotic matrix. (A) Immunofluorescence assay demonstrates the level of Collagen I in human cardiac fibroblasts, scale bar: 50 μm. (B) Fibrosis-related protein levels were assessed using western blotting. ***p < 0.001 vs. control; ###p < 0.001 vs. angiotensin II (Ang II) + short hairpin RNA (sh)-negative control (NC).
KDM5B positively promotes the transcriptional expression of TREM1
Furthermore, to explore the function of KDM5B, its level upon Ang II treatment was examined. Ang II could elevate KDM5B level in HCFs (Figs. 4A, 4B), and the binding site of KDM5B and TREM1 promoter was analyzed via the JASPAR (jaspar.genereg.net) (Fig. 4C). Through transfection, KDM5B was overexpressed or knocked down in HCFs (Figs. 4D, 4E), and sh-KDM5B-1 was selected for the subsequent assays. It was found that KDM5B overexpression increased TREM1 levels, while KDM5B knockdown decreased TREM1 levels (Figs. 4F, 4G). To confirm the relationship between TREM1 and KDM5B, luciferase reporter gene and ChIP assays were performed. Luciferase reporter gene results revealed that TREM1 promoter activity was low in both groups with mutant sites. In the wild-type groups, TREM1 promoter activity was higher in the TREM1-WT + oe-KDM5B group (Fig. 4H). In the ChIP assay, the KDM5B antibody was used to catch TREM1 DNA bound to the KDM5B protein. The result revealed that TREM1 was more enriched in the KDM5B antibody group compared to the lgG, indicating the binding of KDM5B to the TREM1 promoter (Fig. 4I).
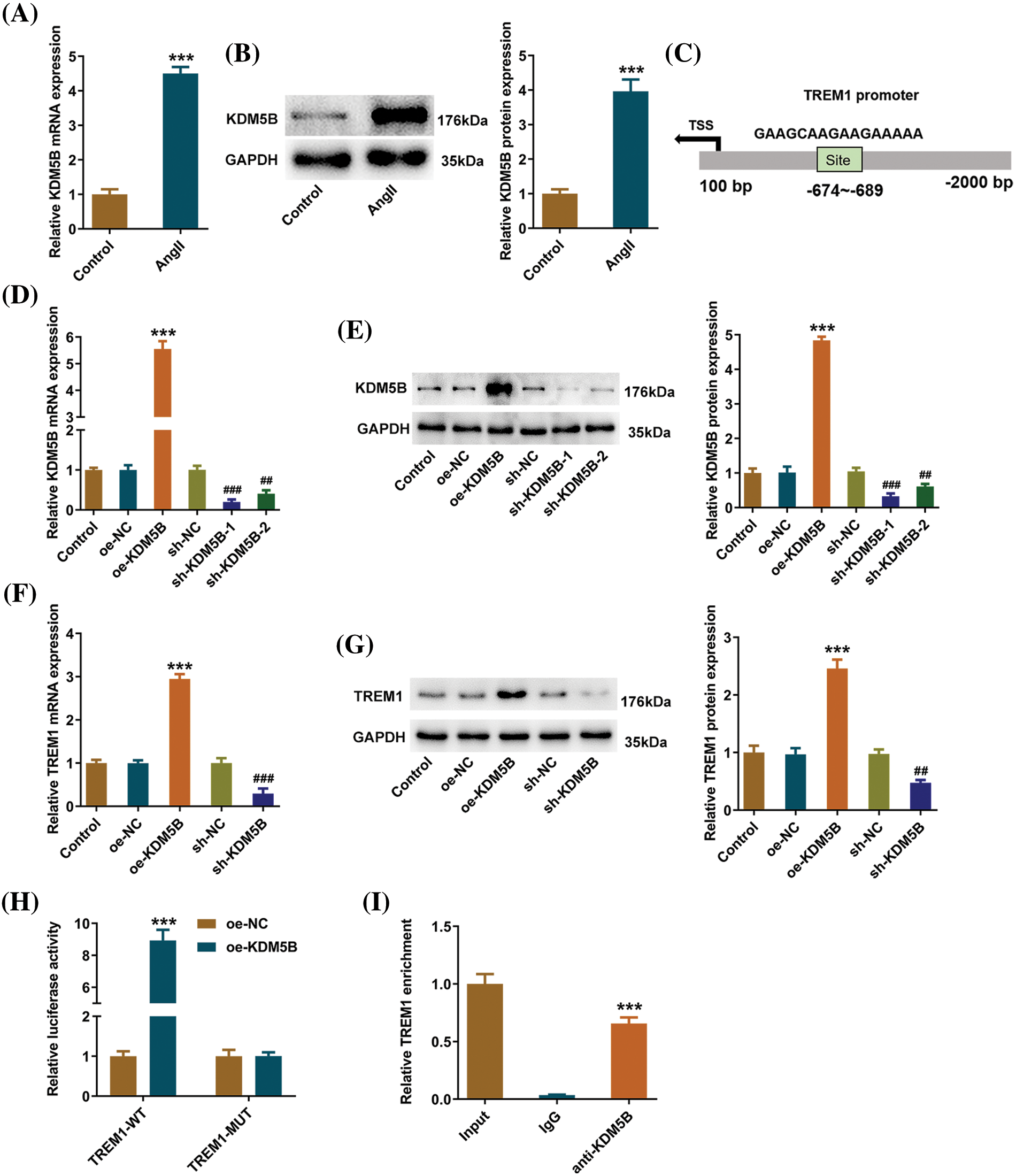
Figure 4: Lysine-specific demethylase 5B (KDM5B) positively promotes the transcriptional expression of triggering receptor expressed on myeloid cells 1 (TREM1). (A) The impact of angiotensin II treatment on KDM5B mRNA and (B) protein levels in human cardiac fibroblasts. (C) The binding site of KDM5B and TREM1 promoter. (D) KDM5B mRNA and (E) protein levels following transfection. (F) TREM1 mRNA and (G) protein levels following transfection. (H) Luciferase reporter assay reveals the regulation of TREM1 on KDM5B. (I) Chromatin immunoprecipitation reveals the binding between KDM5B and TREM1. ***p < 0.001 vs. control, overexpression (oe)-negative control (NC) or lgG; ##p < 0.01, ###p < 0.001 vs. sh-NC.
KDM5B reverses the effects of TREM1 knockdown
Afterward, to assess the regulation of KDM5B on TREM1, HCFs were co-transfected with sh-TREM1 and oe-KDM5B. Additional KDM5B overexpression promoted cell proliferation, as obtained from CCK8 (Fig. 5A) and EdU results (Fig. 5B). Scratch assay reflected that KDM5B overexpression also promoted cell migration (Fig. 5C).
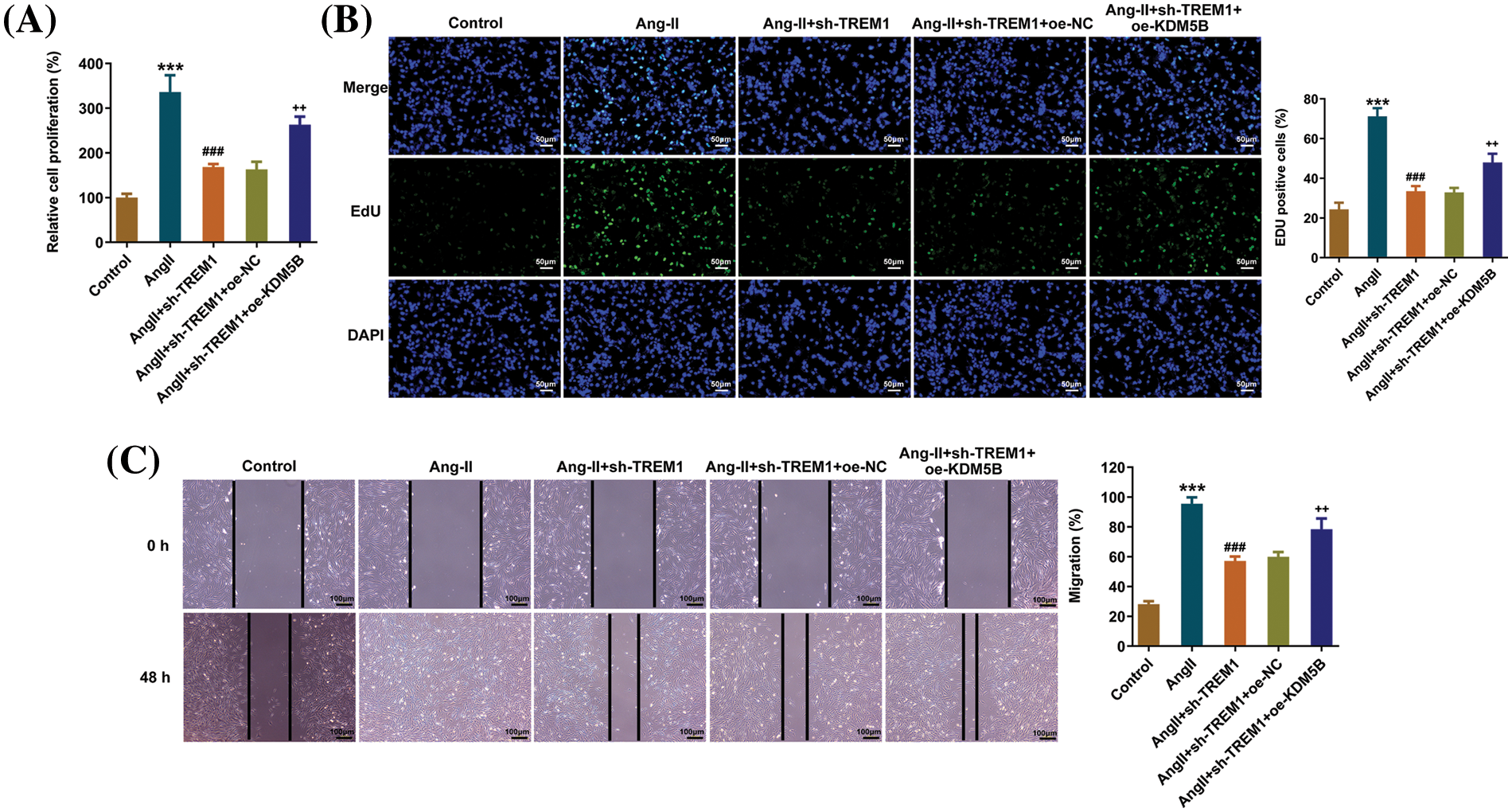
Figure 5: Lysine-specific demethylase 5B (KDM5B) reverses the impacts of triggering receptor expressed on myeloid cells 1 (TREM1) knockdown on human cardiac fibroblast (HCF) proliferation and migration. HCFs were co-transfected with short hairpin RNA (sh)-TREM1 and overexpression (oe)-KDM5B. Additional KDM5B overexpression promoted cell proliferation, as reflected by the (A) cell counting kit-8 and (B) 5-ethynyl-2′-deoxyuridine (EdU) assays, scale bar: 50 μm. (C) Scratch assay reflects the impact of KDM5B overexpression on cell migration, scale bar: 100 μm. ***p < 0.001 vs. control; ###p < 0.001 vs. Angiotensin II (Ang II); ++p < 0.01 vs. Ang II + sh-TREM1 + oe-negative control (NC).
IF and western blot displayed that additional KDM5B overexpression elevated Collagen I level (Fig. 6A), as well as α-SMA, CTGF, and Fibronectin protein levels (Fig. 6B). KDM5B overexpression partially reversed the alteration in the fibrotic phenotype of HCFs resulting from TREM1 knockdown.
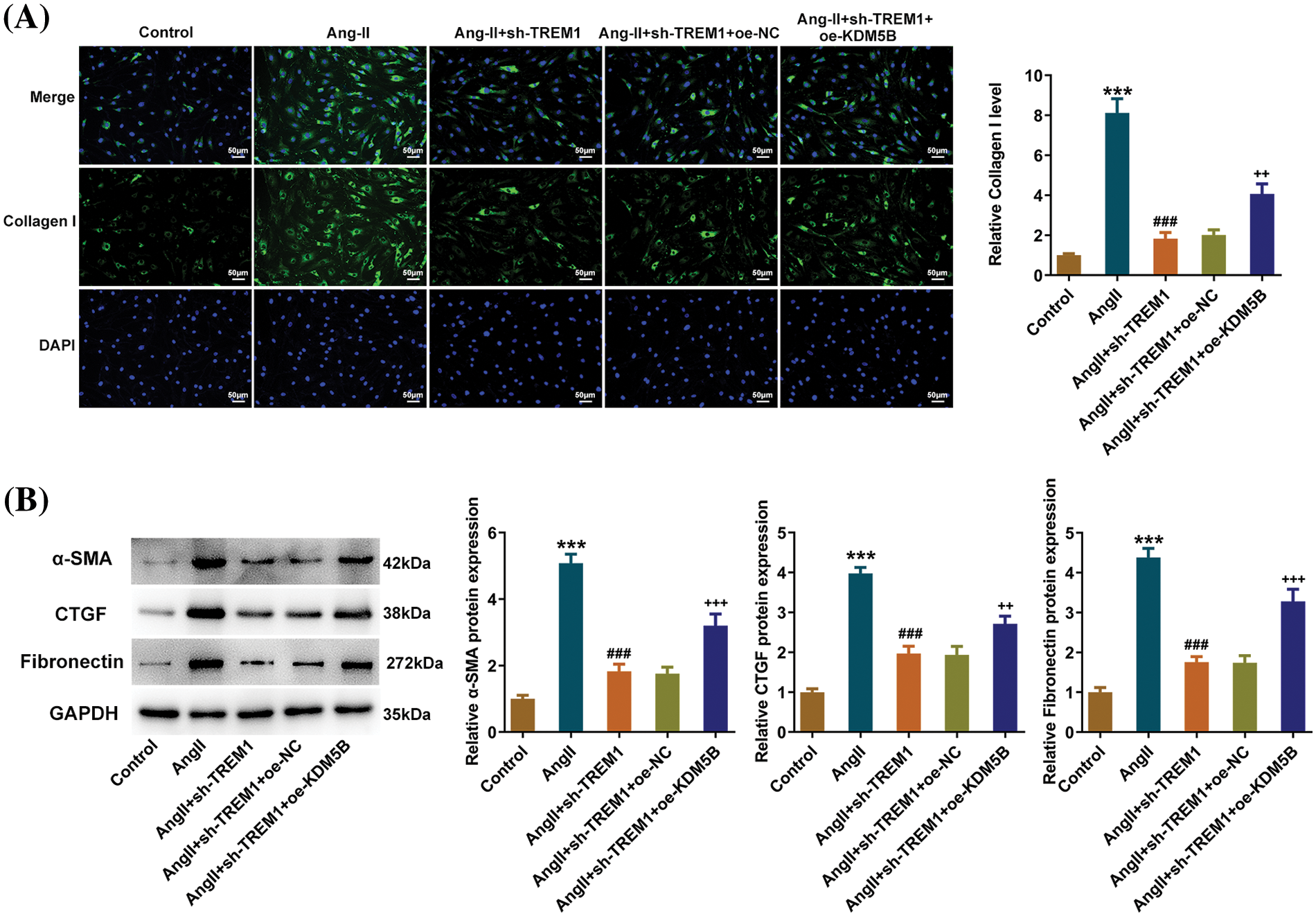
Figure 6: Lysine-specific demethylase 5B (KDM5B) reverses the effects of triggering receptor expressed on myeloid cells 1 (TREM1) knockdown on the secretion of the pro-fibrotic matrix. (A) Immunofluorescence assay demonstrates the impact of additional KDM5B overexpression on the level of Collagen I, scale bar: 50 μm. (B) Western blotting reveals the impact of additional KDM5B overexpression on fibrosis-related protein levels. ***p < 0.001 vs. control; ###p < 0.001 vs. Angiotensin II (Ang II); ++p < 0.01, +++p < 0.001 vs. Ang II + short hairpin RNA (sh)-TREM1 + overexpression (oe)-negative control (NC).
Cardiac fibrosis represents a common feature of nearly all end-stage heart diseases [23], characterized by the accumulation of ECM in the myocardium. This pathological process increases the risk of arrhythmias and disrupts cardiac function to progression to heart failure [1]. Given the critical need to halt cardiac fibrosis progression to preserve cardiac function, there is a lack of approved therapies directly targeting the ECM. Ang II, as a locally regulated growth hormone, can promote the conversion of HCFs into myofibroblasts and promote the secretion of collagens and ECM [24]. Given studies showing that Ang II-mediated activation is a pivotal factor in myocardial fibrosis, Ang II was utilized to treat HCFs to construct the research model. In our study, we discovered that KDM5B regulates TREM1 transcription, leading to the promotion of fibroblast proliferation, migration, and secretion of the pro-fibrotic matrix. Under pathological stimulation, cardiac fibroblasts proliferate and migrate to the injured site, differentiate into myofibroblasts, and form scar tissue to replace the necrotic area. In the absence of cardiomyocyte death, continued activation of myofibroblasts will lead to excessive accumulation of ECM proteins in the interstitial space and form myocardial fibrosis [25]. Consequently, reducing TREM1 expression or its inactivation emerges as a potential strategy to inhibit fibrosis and target the ECM.
This study innovatively discovered the role of TREM1 in cardiac fibrosis. This finding can be supported by previous studies that found its involvement in cardiac dysfunction. For example, research indicates that TREM1 gene knockout reduced cardiomyocyte apoptosis and alleviated myocardial ischemia-reperfusion injury [26], while downregulation of TREM1 expression reversed doxorubicin-induced cardiotoxicity in rats [27]. Moreover, Boufenzer et al. [28] reported the potential of the synthetic peptide LR12, which suppresses TREM1 expression, to improve cardiac function after myocardial infarction. The mechanism is to hinder neutrophil aggregation and chemokine production. This suggests that immune activation following TREM1 inhibition may reduce post-myocardial infarction inflammatory responses, leading to improved prognosis. Additionally, serum TREM1 levels combined with interleukin-1β proved to be a valuable predictor of inflammatory cascade response in a study of patients undergoing cardiac surgery [29], highlighting TREM1’s potential as a predictor of heart disease.
It is noted that the regulatory axis of KDM5B and TREM1 was revealed in this article. From the perspective of a single gene, research on KDM58 is mainly concentrated in the field of oncology [19,30,31]. Fortunately, there is growing recognition of the function of KDM5B in cardiovascular diseases. For instance, elevated KDM5B levels have been observed in mouse hearts following aortic coarctation and in activated cardiac fibroblasts. The KDM5B inhibitor TK-129 has shown efficacy in inhibiting isoproterenol-triggered myocardial fibrosis in mouse models [32]. These findings indicate a detrimental role for high levels of KDM5B and TREM1 in the development of cardiac disease, and inhibiting their expression may benefit disease control.
In conclusion, our study sheds light on the positive regulation of TREM1 by KDM5B, demonstrating its role in promoting fibroblast proliferation, migration, and pro-fibrotic matrix secretion in myocardial fibrosis. However, the clinical significance of this regulatory axis in myocardial fibrosis, including its role in disease diagnosis and prognosis, warrants further investigation and exploration.
Acknowledgement: None.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: CL and JC contributed to the study concept, methods, and writing. XC, WY, and JY contributed to methods and analysis. All authors approve the final version.
Availability of Data and Materials: The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Liu M, López de Juan Abad B, Cheng K. Cardiac fibrosis: myofibroblast-mediated pathological regulation and drug delivery strategies. Adv Drug Deliv Rev. 2021;173:504–19. doi:10.1016/j.addr.2021.03.021. [Google Scholar] [PubMed] [CrossRef]
2. Frangogiannis NG. Cardiac fibrosis. Cardiovasc Res. 2021;117(6):1450–88. doi:10.1093/cvr/cvaa324. [Google Scholar] [PubMed] [CrossRef]
3. Ricketts SN, Qian L. The heart of cardiac reprogramming: the cardiac fibroblasts. J Mol Cell Cardiol. 2022;172:90–9. doi:10.1016/j.yjmcc.2022.08.004. [Google Scholar] [PubMed] [CrossRef]
4. Lu W, Zhu H, Wu J, Liao S, Cheng G, Li X. Rhein attenuates angiotensin II-induced cardiac remodeling by modulating AMPK-FGF23 signaling. J Transl Med. 2022;20(1):305. doi:10.1186/s12967-022-03482-9. [Google Scholar] [PubMed] [CrossRef]
5. Ock S, Ham W, Kang CW, Kang H, Lee WS, Kim J. IGF-1 protects against angiotensin II-induced cardiac fibrosis by targeting αSMA. Cell Death Dis. 2021;12(7):688. doi:10.1038/s41419-021-03965-5. [Google Scholar] [PubMed] [CrossRef]
6. Lu Y, Wang Q, Zhang C. Hyperglycemia-induced myocardial fibrosis may be associated with pyroptosis and apoptosis of cardiomyoctes in diabetic mice. BIOCELL. 2023;47(2):393–400. doi:10.32604/biocell.2023.024944. [Google Scholar] [CrossRef]
7. Yao H, He Q, Huang C, Wei S, Gong Y, Li X, et al. Panaxatriol saponin ameliorates myocardial infarction-induced cardiac fibrosis by targeting Keap1/Nrf2 to regulate oxidative stress and inhibit cardiac-fibroblast activation and proliferation. Free Radic Biol Med. 2022;190:264–75. doi:10.1016/j.freeradbiomed.2022.08.016. [Google Scholar] [PubMed] [CrossRef]
8. Liu J, Zhuang T, Pi J, Chen X, Zhang Q, Li Y, et al. Endothelial forkhead box transcription factor p1 regulates pathological cardiac remodeling through transforming growth factor-β1-endothelin-1 signal pathway. Circulation. 2019;140(8):665–80. doi:10.1161/CIRCULATIONAHA.119.039767. [Google Scholar] [PubMed] [CrossRef]
9. Kumar S, Wang G, Zheng N, Cheng W, Ouyang K, Lin H, et al. HIMF (Hypoxia-induced mitogenic factor)-IL (interleukin)-6 signaling mediates cardiomyocyte-fibroblast crosstalk to promote cardiac hypertrophy and fibrosis. Hypertension. 2019;73(5):1058–70. doi:10.1161/HYPERTENSIONAHA.118.12267. [Google Scholar] [PubMed] [CrossRef]
10. Janbandhu V, Tallapragada V, Patrick R, Li Y, Abeygunawardena D, Humphreys DT, et al. Hif-1a suppresses ROS-induced proliferation of cardiac fibroblasts following myocardial infarction. Cell Stem Cell. 2022;29(2):281–97. doi:10.1016/j.stem.2021.10.009. [Google Scholar] [PubMed] [CrossRef]
11. Ko T, Nomura S, Yamada S, Fujita K, Fujita T, Satoh M, et al. Cardiac fibroblasts regulate the development of heart failure via Htra3-TGF-β-IGFBP7 axis. Nat Commun. 2022;13(1):3275. doi:10.1038/s41467-022-30630-y. [Google Scholar] [PubMed] [CrossRef]
12. Tallquist MD. Cardiac fibroblast diversity. Annu Rev Physiol. 2020;82:63–78. doi:10.1146/annurev-physiol-021119-034527. [Google Scholar] [PubMed] [CrossRef]
13. Cho N, Razipour SE, McCain ML. Featured article: tGF-β1 dominates extracellular matrix rigidity for inducing differentiation of human cardiac fibroblasts to myofibroblasts. Exp Biol Med. 2018;243(7):601–12. doi:10.1177/1535370218761628. [Google Scholar] [PubMed] [CrossRef]
14. Talman V, Ruskoaho H. Cardiac fibrosis in myocardial infarction—from repair and remodeling to regeneration. Cell Tissue Res. 2016;365(3):563–81. doi:10.1007/s00441-016-2431-9. [Google Scholar] [PubMed] [CrossRef]
15. Ji Z, Zhang R, Yang M, Zuo W, Yao Y, Qu Y, et al. Accuracy of triggering receptor expressed on myeloid cells 1 in diagnosis and prognosis of acute myocardial infarction: a prospective cohort study. PeerJ. 2021;9:e11655. doi:10.7717/peerj.11655. [Google Scholar] [PubMed] [CrossRef]
16. Xiong JB, Duan JX, Jiang N, Zhang CY, Zhong WJ, Yang JT, et al. TREM-1 exacerbates bleomycin-induced pulmonary fibrosis by aggravating alveolar epithelial cell senescence in mice. Int Immunopharmacol. 2022;113:109339. doi:10.1016/j.intimp.2022.109339. [Google Scholar] [PubMed] [CrossRef]
17. Mao Y, Yu J, Da J, Yu F, Zha Y. Acteoside alleviates UUO-induced inflammation and fibrosis by regulating the HMGN1/TLR4/TREM1 signaling pathway. PeerJ. 2023;11:e14765. doi:10.7717/peerj.14765. [Google Scholar] [PubMed] [CrossRef]
18. Liu T, Chen S, Xie X, Liu H, Wang Y, Qi S, et al. Soluble TREM-1, as a new ligand for the membrane receptor Robo2, promotes hepatic stellate cells activation and liver fibrosis. J Cell Mol Med. 2021;25(24):11113–27. doi:10.1111/jcmm.17033. [Google Scholar] [PubMed] [CrossRef]
19. Xhabija B, Kidder BL. KDM5B is a master regulator of the H3K4-methylome in stem cells, development and cancer. Semin Cancer Biol. 2019;57(12):79–85. [Google Scholar] [PubMed]
20. Wang B, Tan Y, Zhang Y, Zhang S, Duan X, Jiang Y, et al. Loss of KDM5B ameliorates pathological cardiac fibrosis and dysfunction by epigenetically enhancing ATF3 expression. Exp Mol Med. 2022;54(12):2175–87. doi:10.1038/s12276-022-00904-y. [Google Scholar] [PubMed] [CrossRef]
21. Chen Y, Huang M, Yan Y, He D. Tranilast inhibits angiotensin II-induced myocardial fibrosis through S100A11/transforming growth factor-β (TGF-β1)/smad axis. Bioengineered. 2021;12(1):8447–56. doi:10.1080/21655979.2021.1982322. [Google Scholar] [PubMed] [CrossRef]
22. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25(4):402–8. doi:10.1006/meth.2001.1262. [Google Scholar] [PubMed] [CrossRef]
23. Czubryt MP, Hale TM. Cardiac fibrosis: pathobiology and therapeutic targets. Cell Signal. 2021;85:110066. doi:10.1016/j.cellsig.2021.110066. [Google Scholar] [PubMed] [CrossRef]
24. Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res. 2016;119(1):91–112. doi:10.1161/CIRCRESAHA.116.303577. [Google Scholar] [PubMed] [CrossRef]
25. Schlittler M, Pramstaller PP, Rossini A, De Bortoli M. Myocardial fibrosis in hypertrophic cardiomyopathy: a perspective from fibroblasts. Int J Mol Sci. 2023;24(19):14845. doi:10.3390/ijms241914845. [Google Scholar] [PubMed] [CrossRef]
26. Zhang H, Ye J, Wang X, Liu Z, Chen T, Gao J. Muscone inhibits the excessive inflammatory response in myocardial infarction by targeting TREM-1. Evid Based Complement Alternat Med. 2022;2022(12):9112479. [Google Scholar] [PubMed]
27. Yao L, Gui M, Li J, Lu B, Wang J, Zhou X, et al. Shengxian decoction decreases doxorubicin‐induced cardiac apoptosis by regulating the TREM1/NF‐κB signaling pathway. Mol Med Rep. 2021;23(3):219. doi:10.3892/mmr.2024.13159. [Google Scholar] [PubMed] [CrossRef]
28. Boufenzer A, Lemarié J, Simon T, Derive M, Bouazza Y, Tran N, et al. TREM-1 mediates inflammatory injury and cardiac remodeling following myocardial infarction. Circ Res. 2015;116(11):1772–82. doi:10.1161/CIRCRESAHA.116.305628. [Google Scholar] [PubMed] [CrossRef]
29. Stoppelkamp S, Veseli K, Stang K, Schlensak C, Wendel HP, Walker T. Identification of predictive early biomarkers for sterile-SIRS after cardiovascular surgery. PLoS One. 2015;10(8):e0135527. doi:10.1371/journal.pone.0135527. [Google Scholar] [PubMed] [CrossRef]
30. Jose A, Shenoy GG, Sunil Rodrigues G, Kumar NAN, Munisamy M, Thomas L, et al. Histone demethylase KDM5B as a therapeutic target for cancer therapy. Cancers. 2020;12(8):2121. doi:10.3390/cancers12082121. [Google Scholar] [PubMed] [CrossRef]
31. Zhang SM, Cai WL, Liu X, Thakral D, Luo J, Chan LH, et al. KDM5B promotes immune evasion by recruiting SETDB1 to silence retroelements. Nature. 2021;598(7882):682–7. doi:10.1038/s41586-021-03994-2. [Google Scholar] [PubMed] [CrossRef]
32. Tang K, Jiao LM, Qi YR, Wang TC, Li YL, Xu JL, et al. Discovery of novel pyrazole-based KDM5B inhibitor TK-129 and its protective effects on myocardial remodeling and fibrosis. J Med Chem. 2022;65(19):12979–3000. doi:10.1021/acs.jmedchem.2c00797. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools