
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Exploring the vital role of microglial membrane receptors in Alzheimer’s disease pathogenesis: a comprehensive review
1 Department of Neurointervention Center, Dalian No. 3 People’s Hospital, Dalian Medical University, Dalian, China
2 Department of Clinical Medicine, China Medical University, Shenyang, China
* Corresponding Authors: YONG-QING JIAO. Email: ; XUN WANG. Email:
(This article belongs to the Special Issue: Exploring the Cellular Mechanisms of Neurodegenerative Diseases)
BIOCELL 2024, 48(7), 1011-1022. https://doi.org/10.32604/biocell.2024.050120
Received 28 January 2024; Accepted 16 April 2024; Issue published 03 July 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Neurodegenerative diseases constitute a broad category of diseases caused by the degeneration of the neurons. They are mainly manifested by the gradual loss of neuron structure and function and eventually can cause death or loss of neurons. As the global population ages rapidly, increased people are being diagnosed with neurodegenerative diseases. It has been established that the onset of Alzheimer’s disease (AD) is closely linked with increasing age and its major pathological features include amyloid-beta plaques (Aβ), Tau hyperphosphorylation, Neurofibrillary tangles (NFTs), neuronal death as well as synaptic loss. The involvement of microglia is crucial in the pathogenesis and progression of AD and exhibits a dual role. For instance, in the early stage of AD, microglia surface membrane proteins or receptors can participate in immunophagocytosis, and anti-inflammatory functions and act as a physical barrier after recognizing various ligands such as Aβ and NFTs. However, in the later stage of the disease, membrane receptors on the surface of microglia can cause its activation to release a substantial quantity of pro-inflammatory factors. Which can amplify the neuroinflammatory response. The rapid decline of normal immune phagocytosis can result in the continuous accumulation of abnormal proteins, leading to neuronal dysfunction and destruction of the formed physical barrier as well as the neurovascular microenvironment. It can also increase the transformation of microglia from anti-inflammatory phenotype M2 to pro-inflammatory phenotype M1, induce severe neuronal injury or apoptosis, and aggravate the progression of AD. Due to few articles have focused on the AD-related membrane protein receptors on microglia, thus in this paper, we have reviewed several representative microglial membrane proteins or receptors about their specific roles and functions implicated in AD, and expect that there will be more in-depth research and scientific research results in the treatment of AD by targeted regulation of microglia membrane protein receptors in the future.Keywords
Abbreviations: Full Name of the Technical Term
| AD | Alzheimer’s disease |
| NFTs | Neurofibrillary tangles |
| Aβ | Amyloid-beta plaques |
| TNF-α | Tumor necrosis factor-α |
| APP | Amyloid precursor protein |
| PSEN1 | Presenilin-1 |
| MG | Microglia |
| HMGB1 | High mobility group box 1 protein |
| ROS | Reactive oxygen species |
| Beclin-1 | Recombinant human beclin 1 protein |
| FcγRIIb | Human Receptor II for the Fc region of immunoglobulin G |
| SCARA-1 | Scavenger receptor A1 |
| CD36 | Cluster of differentiation 36 |
| RAGEs | Receptor for advanced glycation end products |
| PPARγ | Peroxisome proliferator-activated receptor γ |
| DAP-12 | Killer cell activating receptor associated protein 12 |
| PRR | Pattern Recognition Receptor |
| LRR | Leucine-rich repeats |
| TIR | Toll/IL-1R homology |
| ECDs | Extra-cellular domains |
| MyD88 | Myeloid differentiation primary response 88 |
| PS1 | Presenilin 1 |
| MAPK | Mitogen-activated protein kinase |
| MARCKS | Myristoylated alanine-rich C kinase substrates |
| LPS | Lipopolysaccharide |
| p-Tau | Hyperphosphorylated Tau |
| DAP12 | DNAX-activating protein of 12 kDa |
| NF-κB | Nuclear factor-κB |
| NLRP3 | NOD-like receptor family, pyrin domain containing 3 |
| sAPPα | Soluble amyloid precursor protein-α |
| BBG | Brilliant blue-G |
| PD | Parkinson’s disease |
| ALS | Amyotrophic Lateral Sclerosis |
| ERK ½ | Extracellular signal-regulated kinases 1 and 2 |
| DIAPH1 | Diaphanous Homolog 1 |
| TIRAP | Toll-interleukin 1 receptor adaptor protein |
| BBB | Blood-brain barrier |
| SPs | Senile plaques |
| NFTs | Neurofibrillary tangles |
| BMECs | Brain microvascular endothelial cells |
| GPCR | G protein-coupled receptor |
| CX3CL1 | C-X3-C motif ligand 1 |
Alzheimer’s disease is an age-related, insidious, and progressive neurodegenerative disease, which is generally characterized by slow and progressive memory impairment, and then can gradually evolve into irreversible cognitive impairment and executive function loss over time, which has a significant impact on the quality of life of patients [1,2]. It has been established that with the aging of the population, the number of AD cases has increased rapidly, and it is expected that the number of AD patients in the world will exceed 130 million in 30 years [3]. The main pathological features associated with AD are extracellular amyloid-β plaques (Aβ), hyperphosphorylation of tau protein, neurofibrillary tangles (NFTs), neuron death, and synaptic loss in the hippocampus, cortex, and amygdala [4,5]. Aβ plays a critical pathogenic role in AD, all types of AD are characterized by the accumulation of Aβ, which triggers a series of neuroinflammations, culminating in neuronal dysfunction and death [6].
A few prior studies using genetic linkage approaches linked Alzheimer’s disease to familial mutations in proteins associated with beta-amyloid production, amyloid precursor protein (APP), presenilin-1 (PSEN1) and PSEN2, AD symptoms are often accompanied by a significant increase in the level of neuroinflammation in the brain, which is also considered to be an important physiological feature in the development of AD [7].
Microglia (MG) dysfunction is considered an important hallmark and one of the major causes of common neurodegenerative diseases (NDDS) [8–10]. Interestingly, almost all these pathologies are characterized by abnormal aggregation of the pathogenic proteins in the brain. Which can directly activate microglia, trigger microglia-mediated neuroinflammation, and increase oxidative stress. In neuroinflammation, a progressive increase in the circulating proinflammatory cytokines is commonly associated with age-related cognitive decline, enhanced neurodegeneration, and massive release of cytokines by microglia in the brain [11]. The production of different pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) by activated MG can stimulate the expression of β-secretase, thereby significantly increasing APP processing and Aβ deposition [12]. In addition, activated MG and toxin-induced dead neurons can release high mobility group box 1 protein (HMGB1), which can effectively bind to Aβ to form refractory Aβ oligomers and inhibit the clearance of Aβ by MG. Aβ oligomers can act back on MG to stimulate the activated MG to secrete the various pro-inflammatory factors. These include cytokines, chemokines, complement factors, and a variety of free radicals [13,14], a large amount of pro-inflammatory factors or reactive oxygen species (ROS) are released, which can aggravate the process of neuroinflammation [15].
Microglia are central nervous system (CNS) resident immune cells and produce different phenotypes after stress, thus exerting pro-inflammatory, anti-inflammatory, and phagocytic effects [16]. They can also mediate neuroinflammation and thereby play an important role in CNS injury and the progression of neurodegenerative diseases [17]. Neuroinflammation is an important component in the initiation of AD pathology and is tightly regulated by microglia [18]. Besides neuroinflammation, there is also growing evidence to suggest that impaired microglia-mediated phagocytosis can also contribute to AD pathogenesis by exhibiting considerable adverse effects on the brain, including inflammatory responses, clearance defects, Aβ as well as microtubule-associated proteins (Tau) propagation, and synaptic dysfunction [19]. Interestingly, during the early stage of CNS inflammation, microglia activation can inhibit the spread of inflammation and phagocytose various pathogens, aggregated proteins as well and dead neurons, maintain homeostasis in the CNS, and delay AD’s development [20,21], However, insufficient, or excessive phagocytosis can affect neural development, accelerate aging and the pathological process associated AD [22]. For instance, in the early stage of AD, Aβ can be removed from the normal small gelatinous cells through phagocytosis and hydrolysis, but as the disease progresses, Aβ can also affect the phagocytosis of microglia through exerting toxic effects [23]. Moreover, in the post-AD phase, phagocytosis of microglia in the pathological state can further worsen the dendritic dysfunction, by engulfing endangered neurons. Microglia, as one of the immune phagocytic cells in the central nervous system, mostly relies on the various membrane proteins or receptors present on its surface to exert its immune surveillance, pro-inflammatory, and phagocytosis. These membrane proteins or receptors can exhibit diverse effects after recognizing their corresponding ligands. The process of AD is mainly regulated by a variety of microglia membrane proteins. These membrane protein receptors have their unique expression or activation during the different stages of AD. They can modulate the activation or phagocytosis of microglia, thereby exerting an influence on both the occurrence and progression of AD. For example, the expression of recombinant human beclin 1 protein (Beclin-1) is down-regulated in AD, whereas the expression of Human Receptor II for the Fc region of immunoglobulin G (FcγRIIb), scavenger receptor A1 (SCARA-1), cluster of differentiation 36 (CD36), receptor for advanced glycation end products (RAGE), receptor derived from myeloid cells 2 (TREM2) and CD33 is up-regulated in AD [24]. It has been reported that the loss of Beclin-1 decreased Aβ uptake by microglia [25], and FcγRIIb can promote the clearance of Aβ plaque [26]. In addition, SCARA-1 can enhance the ability of microglia to bind and phagocytize Aβ [27], whereas CD36 can promote Aβ phagocytosis through peroxisome proliferator-activated receptor (PPARγ) signaling [28]. Moreover, RAGE can cause an inflammatory response and promote amyloidosis by binding to Aβ [29]. TREM2 acts on the killer cell activating receptor-associated protein 12 (DAP-12) to activate the phagocytosis of microglia without causing an inflammatory response [30], whereas CD33 is associated with a significant decrease in the internalization of Aβ peptides [31]. Based on the reported relationship between microglia membrane protein receptors and the occurrence and development of AD, this paper selected several representative microglia membrane protein receptors related to AD and reviewed their specific structures, functions, and differential effects or roles of microglia induced by them in AD.
Toll-like receptors (TLRs) are evolutionarily conserved receptors and members of the Pattern Recognition Receptor (PRR) family. They belong to type I transmembrane proteins and contain three distinct domains: The N-terminal region which is characterized by a high abundance of leucine-rich repeats (LRR), transmembrane domains, and cytoplasmic Toll/IL-1R homology (TIR) motifs. They can effectively recognize and bind to the corresponding Pathogen Associated Molecular Pattern (PAMP) and Damage Associated Molecular Pattern (DAMP) to induce innate immunity [32]. To date, a total of 11 TLRs types have been identified in diverse human tissues and cells, encompassing dendritic cells (DC), macrophages, neutrophils, B cells, and T cells within the immune system; fibrocytes, epithelial cells, and myocardium within the non-immune system; as well as neurons and glial cells in both the peripheral nervous system and CNS [33]. The TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10 expressed on the cell surface, have been identified to recognize extracellular bacteria-associated ligands and induce disease responses, whereas TLR3, TLR7, TLR8, and TLR9 are located in the endosomes of specific immune system cells [32]. The transmembrane domain of TLRs primarily consists of approximately 20 uncharged hydrophobic residues, while the extracellular domains (ECDs) at the N-terminus of TLRs are glycoproteins comprising 550 to 800 amino acid residues. These ECDs can interact with and recognize molecules produced by invading pathogens. The horseshoe-shaped structure of the N-terminal LRRs enhances the recognition ability towards both exogenous and endogenous pattern molecules, whereas the TIR domain facilitates interactions with a wide range of adaptor molecules for initiating signal transduction. Activation of the innate immune system through TLRs’ recognition of PAMPs subsequently promotes antigen-specific adaptive immunity [34]. TLR binding to the ligands can activate a variety of intracellular downstream signaling cascades to induce the host defense responses. TLR is a transmembrane receptor belonging to class I, which can activate itself through dimerization with a recognized ligand. The nature of the ligand, the specific TLR species involved in activation, and downstream aptamer molecules all have potential influence on TLR signaling. The signaling pathway of TLRs involves at least two distinct pathways: the MyD88-dependent pathway, utilized by all Toll-like receptors (except TLR3), triggers the production of pro-inflammatory cytokines and the other is MyD88-independent pathway, also referred to as the TIR domain-containing adaptor inducible interferon β-dependent pathway, is utilized by TLR3 and TLR4 and is associated with the activation of type 1 interferons [35].
TLRs are extensively expressed in microglia, and their involvement in neuroinflammation has been implicated in a broad spectrum of infectious and non-infectious CNS disorders as well as neurodegenerative diseases [36]. TLRs have also been shown to affect the neurodevelopment and cognitive functions in AD [37]. TLR2 and TLR4 have been studied extensively, but the abundance of TLR2 in the central nervous system is among the highest compared to other Toll-like receptors [38,39]. Interestingly, in animal models of AD, exposure to Aβ can increase TLR2 mRNA levels, whereas upregulation of the TLR2 gene was also found in the temporal cortex of AD patients and the cultured microglia [40,41]. The therapeutic targets of TLR2 can reduce the accumulation of Aβ 1–42 in the hippocampus and thereby alter the progression of memory loss in animal models of AD. TLR2 present on microglia is associated with Aβ phagocytosis. It has been reported that TLR2 deficient APP/Presenilin 1 (PS1) AD mice (A commonly used mouse model of Alzheimer’s disease) can display higher brain Aβ load and rapid cognitive decline accompanied by significantly decreased glial activity in comparison to TLR2 normal mice [42]. Activation of TLR2 by peptidoglycan can induce microglia to phagocytose AD-related Aβ, thereby improving cognitive behavioral performance in mice.
The involvement of TLR4 as a mediator in the neurotoxic activity of DAMPs associated with neuronal damage in AD has been suggested. DAMP. High Mobility Group Box 1 (HMGB1), a Damage-Associated Molecular Pattern (DAMP), is an abundant chromosome-binding protein present in the nucleus of eukaryotic cells. HMGB1 is released by necrotic or hyperactive neurons and exerts its influence on neurite degradation through Toll-like receptor 4 (TLR4). The underlying mechanism involves the binding of HMGB1 to TLR4, leading to the activation of mitogen-activated protein kinase (MAPK). It can affect the phosphorylation of myristoylated alanine-rich C kinase substrates (MARCKS), thus leading to neurite degeneration, which is a fundamental hallmark of AD pathology [32]. The immune memory of microglia can modulate neuropathology and potentially serve as a mechanism for lipopolysaccharide (LPS)-induced immune memory in the brain, with TLR4 playing a pivotal role in this phenomenon [43] both beneficial and detrimental effects of TLR4 in AD have been reported. Notably, TLR4-deficient transgenic AD mice exhibited reduced microglial activation, increased Aβ deposition, and impaired cognitive function. Furthermore, chronic mild stimulation of TLR4 by LPS was observed to attenuate hyperphosphorylated Tau (p-Tau) levels in the brain and improve memory impairment through microglia-dependent autophagy activation in a P301S transgenic mouse model of tau pathology [44]. Significant expression of TLR4 was observed in APP mice, and an increased expression of TLR4 associated with Aβ plaques was also detected in the brains of patients with AD [45]. Moreover, in a mouse model of AD with acute A beta injection, TLR4 was found to be essential for the activation of glial cells that lead to memory impairment [46]. Both TLR2 and TLR4 signaling are involved in the activation of glial cells as well as other inflammatory cytokines and can contribute to inflammation in the injured brain. Collectively, data from multiple TLRs suggest their well-known roles in the activation of microglia, the release of various inflammatory mediators, and potential effects on phagocytosis in AD [47].
Triggering receptor expressed on myeloid cells 2 (TREM2)
TREM2, a member of the immunoglobulin superfamily, is a transmembrane cellular immunomodulatory receptor. The receptor is a unidirectional transmembrane protein that consists of an extracellular type V immunoglobulin (Ig) domain, a transmembrane region composed of lysine residues, and a short cytoplasmic tail region lacking trans-activation signaling function. Interestingly, in the brain tissues, TREM2 was expressed only in microglia [48]. The binding of TREM2 to the DNAX-activating protein of 12 kDa (DAP12) has been observed in several previous studies, which subsequently facilitates downstream signal transduction and modulates microglial function [49]. It also plays a role in microglial proliferation, survival, migration, phagocytosis, and regulation of inflammation. TREM2 has been extensively studied mainly in different neurodegenerative diseases such as AD [50]. TREM2 primarily signals through adaptor protein DAP12 to promote microglial activation, proliferation, and immune response [51]. It also plays an important role in microglial phagocytosis of apoptotic neurons, damaged myelin, and amyloid plaques [50,52]. The TREM2 R47H variant is significantly associated with late-onset AD, thereby conferring a three-fold increased risk of developing AD and being one of the strongest identified risk factors. R47H carriers display increased expression of various genes involved in inflammatory pathways. The localization of the R47H variant in the extracellular domain of TERM2 can adversely affect the interaction with ligands, interfere with the recognition of microglia with damaged neuronal membranes, and affect the phagocytic clearance of microglia [53,54]. The persistent production of abnormal proteins can effectively cause microglial host defense dysfunction, thereby initiating or amplifying inflammatory responses, leading to neurotoxicity and neurodegeneration [55]. The decrease of TREM2 expression can result in the decrease of microglial aggregation around amyloid plaques, which can induce the reduction of plaque phagocytosis as well as clearance, and then lead to diffuse amyloid plaque accumulation. Moreover, several studies have reported increased per-plaque neuroinflammatory dystrophy in TREM2-deficient mice. The microglial processes present in TREM2 can tightly surround early amyloid fibrils and plaques, thus promoting their compaction and insulation. It has been found that in TREM2 or DAP12 haplotype deficient mice and humans with the R47H TREM2 mutation, the ability of microglia to enclose amyloid deposits or regulate amyloid compaction was significantly reduced, and the formation of an insulating neuroprotective microglial barrier was disrupted. Conversely, overexpression of TREM2 could significantly reduce the levels of the soluble and insoluble Aβ, alleviate neuronal damage, and markedly increase the level of synaptophysin, which is related to synapse formation and cognitive functions, to exert a protective effect on the nerves [56]. Knockout of TREM2 has been shown to prevent synaptic loss by directly inhibiting microglial phagocytosis during the early stages of AD (2–6 months), whereas it can inhibit microglial phagocytosis leading to more severe amyloid deposition during the middle and late stages of AD (6–10 months) when amyloid deposition is relatively dominant, accelerated synaptic dysfunction [57]. The initial phase of AD is characterized by an elevation in the activity of phagocytosis-associated and anti-inflammatory genes within microglia, triggered by the signaling pathway involving TREM2/DAP12. Conversely, during the advanced stage, there is a heightened expression of pro-inflammatory genes facilitated through the interaction between TREM2 and nuclear factor-κB (NF-κB) [58]. The findings underscore the significance of early intervention in patients with AD.
The purinergic receptor P2X7, P2X7 receptor
P2X7 receptor (P2X7R), a member of the purinergic ionic P2X receptor family, is the most structurally and functionally distinct P2X subtype, containing a unique cytoplasmic domain. In the central nervous system, P2X7 receptors are mainly expressed in large quantities in microglia, regulate neurotransmitter release, respond to inflammatory signals, and participate in signal transduction. P2X7 receptors typically exist as trimeric, with each subunit containing two hydrophobic domains and a long glycosylated extracellular loop forming an ATP-binding site between each subunit [59]. P2X7 receptor comprises six helical transmembrane domains, with the N and C termini located intracellularly, while 10 conserved cysteines are positioned extracellularly [59]. The structural basis for the unique function of the P2X7 receptor resides in its C-terminal tail, which also plays a crucial role in regulating receptor activity. Specifically, the intracellular C-terminal tail of the P2X7 receptor exhibits interactions with various intracellular molecules including heat shock proteins, cytoskeletal components, kinases, and membranes [60,61]. The P2X7 receptor is a non-selective ATP-gated cation channel receptor, which is activated by extracellular ATP and plays a pivotal role in the regulation, of Ca2+, Na+, and K+ ions. Moreover, it is implicated in apoptosis, inflammation, and other pathological processes [62]. P2X7 exhibits lower sensitivity to Adenosine Triphosphate (ATP) and thus necessitates high extracellular levels of ATP for activation [36,61]. While P2X7 does not play a significant role in the normal functioning of the central nervous system, it becomes relevant under pathological conditions when damaged necrotic cells release substantial amounts of ATP into the extracellular matrix, thereby acting as a damage-related molecular pattern (DAMP). This DAMP accumulates at sites of inflammation and is subsequently released into the extracellular fluid through various pathways such as exocytosis, vesicle transport, and membrane channels. It then interacts with downstream receptors and engages in diverse signal transduction pathways that ultimately result in cellular damage and death. The P2X7 receptor appears to play a pivotal role in pathological processes, particularly when exposed to high levels of ATP, primarily contributing to inflammatory cascades. Notably, it serves as a crucial component in the activation of NLRP3 inflammasome (NOD-like receptor family, pyrin domain containing 3), leading to the subsequent activation of caspase 1 and release of proinflammatory cytokines interleukin-1β (IL-1β) and IL-18 [63,64]. Stimulation of P2X7 also elicits the release of various proinflammatory substances, including TNFα, IL-6, C-C chemokine ligand 2 (CCL2), as well as the generation of excitotoxic levels of glutamate and reactive oxygen species (ROS), these mediators facilitate neuroinflammation, reactive gliosis, and cellular demise.
Either the cortex or hippocampus of AD patients exhibited a significant upregulation of P2X7, both within the core and surrounding amyloid lesions of plaques, as well as near neurofibrillary degeneration [65,66]. Amyloid plaque is a characteristic lesion of AD and consists specifically of Aβ peptide. Aβ peptide is derived from amyloid precursor protein (APP) through amyloid processing by β and γ secretases, while selective cleavage by α secretases produces soluble neuroprotective fragments and soluble amyloid precursor protein α (sAPPα), inhibits the formation of Aβ peptide. Consequently, P2X7 can influence Aβ production through this pathway as well as other pathways. In vitro, short-term stimulation of P2X7 (less than 30 min) has been demonstrated to activate α secretases, which cleaves APP at specific sites within the Aβ peptide sequence [67]. In vivo, treatment with the P2X7 antagonist brilliant blue-G (BBG) for 4 months in an amyloid mouse model (J20 mice) resulted in a significant reduction in Aβ load [68]. Similarly, knockout of P2X7 in APP/PS1 mice, another murine model of AD, also exhibited decreased Aβ plaques and levels of Aβ peptide [65]. The sometimes-inconsistent results observed in vitro and in vivo can be attributed to the diverse roles of P2X7 in the brain, which are contingent upon local ATP concentrations as well as the cofactors and activation status of P2X7-expressing cells [64]. Although there exist certain disparities, the majority of studies have demonstrated that inhibition of P2X7 in a pathological context leads to a reduction in the accumulation of Aβ peptides. In mouse models of AD and tauopathy, the genetic deletion or pharmacological inhibition of P2X7Rs leads to a reduction in the number of amyloid plaques, decreased Tau phosphorylation, and a decline in the abundance of misfolded forms of Tau. Additionally, P2X7R deficiency mitigates various amyloidosis-related alterations in the brains of THYTau22 transgenic mice, encompassing basal synaptic transmission, paired-pulse facilitation, and long-term synaptic plasticity as assessed through hippocampal brain sections [69]. These findings provide compelling evidence supporting the therapeutic potential of P2X7R blockade as a promising treatment strategy for AD.
Receptor for advanced glycation end products, RAGE
RAGEs were initially identified as the sole protein capable of effectively binding to advanced glycation end products (AGEs) and regulating specific signaling pathways. RAGE, a pro-inflammatory pattern recognition receptor, is predominantly expressed in microglia and astrocytes within the CNS. The inflammation, oxidative stress, and cellular dysfunction mediated by it can play an important role in a variety of neurodegenerative diseases such as AD, Parkinson’s disease (PD), and Amyotrophic Lateral Sclerosis (ALS) [70]. RAGE, functioning as a transmembrane receptor, exhibits distinct structural domains including extracellular, transmembrane, and intracellular segments. It exists in the body as both transmembrane and soluble molecules [71]. The RAGE comprises a single V domain and two C domains, which are three distinct immunoglobulin-like extracellular domains that play a pivotal role in the recognition of specific ligands. The intracellular domain possesses a small volume and exhibits high charge density, and it can bind to a variety of intracellular signaling molecules related to RAGE signaling [72]. However, due to its unique structure and form of existence, signal transduction initiated after the interaction of RAGE with its specific ligands can contribute to a variety of physiological processes, such as chemotaxis, angiogenesis, inflammation, apoptosis, and proliferation. RAGE can bind to a variety of different ligands, such as S100, granule calcium, HMGB1, Aβ, etc. [73]. The absence of transmembrane and intracellular fragments in soluble receptors for advanced glycation end products (sRAGE) precludes its signaling functionality [74], the sRAGE, however, exhibits competitive binding to RAGE ligands, thereby effectively antagonizing the pathological effects mediated by RAGE. The intracellular domain of RAGE can bind to the various proteins, such as extracellular signal-regulated kinases 1 and 2 (ERK 1/2), Diaphanous Homolog 1 (DIAPH1), and Toll-interleukin 1 receptor adaptor protein (TIRAP). When RAGE binds simultaneously to the extracellular ligands such as AGEs, it can initiate the downstream signaling events [75,76] and activation of the NF-κB pathway, which can result in the expression of proinflammatory cytokines such as IL-6, IL-9, and TNF-α. Interestingly, these proinflammatory cytokines not only can activate tau phosphorylation and Aβ peptide formation in AD but can also upregulate RAGE and AGEs by exacerbating oxidative stress [77]. AGE can strongly bind to Aβ to form a water-insoluble, non-degradable crosslinker, glycated β-amyloid (Aβ-AGE), and prior studies have demonstrated that Aβ-AGE can further promote the accumulation of Aβ and bind to RAGE to maintain microglia in an activated state [78], which can stimulate them to produce neurotoxic molecules. In addition, compared with Aβ, Aβ-AGE can exhibit stronger neurotoxicity and stronger pathogenicity in the pathogenesis of AD. RAGE can also mediate the transport of Aβ from the periphery to the brain. When RAGE binds Aβ through the blood-brain barrier (BBB), Aβ and RAGE can significantly affect the expression of the various tight adhesion proteins, increase the permeability of the BBB, and further impair the function as well as the integrity of the BBB [79]. There is also a positive feedback regulation reported between RAGE and Aβ, as later can further increase the expression of RAGE, which can promote the entry of Aβ from BBB into the brain tissues, thus forming a vicious cycle (Fig. 1).
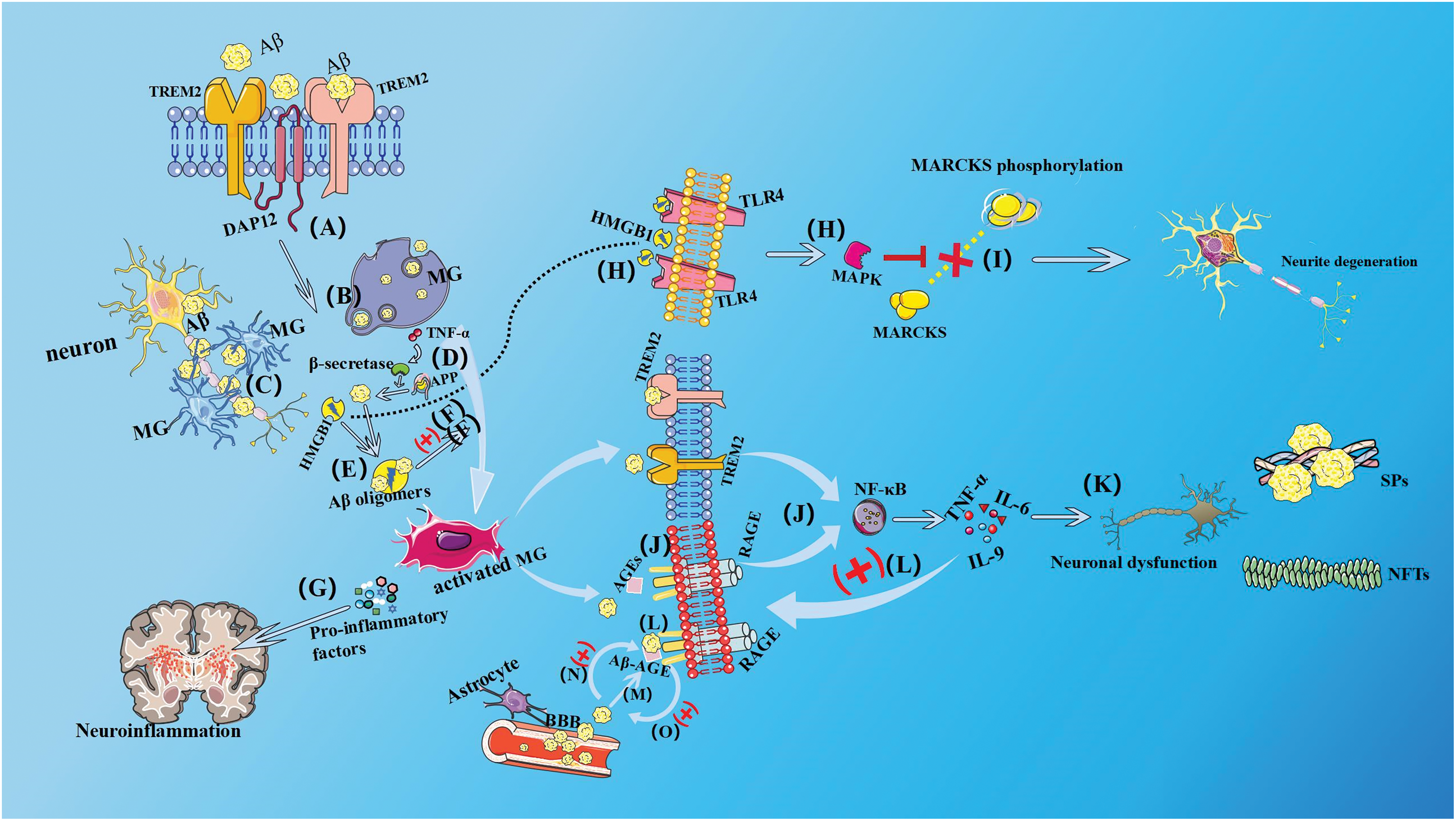
Figure 1: Microglial membrane receptor transduction pathway. (A) Microglia (MG) can mediate downstream signal transduction by binding to the 12-kda DNAx activating protein (DAPl2) after recognizing Aβ via surface TREM2, (B) inhibiting the spread of inflammation, clearing abnormally aggregated proteins such as Aβ through phagocytosis and hydrolysis, and (C) forming cellular bulges tightly surrounding early amyloid fibrils and plaques, promotes its densification and insulation, thereby playing a neuroprotective role and maintaining the homeostasis of the internal environment in the nervous system. (D) The production of pro-inflammatory cytokines such as TNF-α by activated MG stimulates the expression of β-secretase, thereby increasing APP processing and Aβ deposition. (E) Neurons that are induced to die by toxicity release high mobility group egg B1 (HMGB1), HMGB1 combines with Aβ to form a Aβ oligomers that are refractory to degradation, which inhibits the clearance of Aβ by MG, (F), and the Aβ oligomer can feedback on MG, causing activated MG to secrete pro-inflammatory factors. Including cytokines, chemokines, complement factors, and a variety of radicals. (G) A large number of pro-inflammatory factors or reactive oxygen species (ROS) are released, aggravating neuroinflammation. (H) HMGB1 released from necrotic or hyperexcited neurons binds to TLR4, thereby activating mitogen-activated protein kinase (MAPK) and (I) affecting the phosphorylation of myristoylated alanine-rich C kinase substrates (MARCKS), leading to neurite degeneration. (J) When RAGE binds to extracellular ligands such as AGEs simultaneously, RAGE initiates downstream signal transduction and has a synergistic effect with TREM2 to activate nuclear factor κB (NF-κB), which leads to the expression of the pro-inflammatory cytokines IL-6, IL-9, and TNF-α. (K) By exacerbating oxidative stress, these pro-inflammatory cytokines activate tau phosphorylation and Aβ peptide formation in AD, and overactivated microglia immunity disrupts the balance of neuromicroenvironment chemistry, accelerating neuronal dysfunction, and abnormal neurons producing more NFTs, leading to the formation of additional SPs and NFTs. (L) Pro-inflammatory factors can also upregulate RAGE and AGEs, and AGE can bind to Aβ to form A water-insoluble, non-degradable crosslinking, namely, glycated β-amyloid protein (Aβ-AGE), and Aβ-AGE can further promote Aβ accumulation. (M) RAGE can also mediate the transport of Aβ from the peripheral to the brain tissue. When RAGE combines with Aβ, it will affect the expression of tight junction protein, increase the permeability of the blood-brain barrier (BBB), make Aβ more easily penetrate the BBB, and further damage the function and integrity of BBB. (N) There is a positive feedback regulation between RAGE and Aβ. Aβ will further enhance the expression of RAGE, (O) RAGE increases the amount of Aβ entering the brain tissue through the BBB, forming a vicious cycle.
The pathological features of AD include senile plaques (SPs) with neurotoxic Aβ as the primary component and neurofibrillary tangles (NFTs) with abnormally activated tau protein in the nerve cells as the main component [80,81]. Aβ can increase the activation of microglia by binding to RAGE. The over-activated microglia immunity can effectively disrupt the chemical balance of the neural microenvironment and accelerate neuronal dysfunction, which in turn can lead to the formation of additional SPs and NFTs. This can further contribute to neuronal dysfunction, damage, and loss as well as neurovascular dysfunction [82,83]. Neurovascular dysfunction can cause brain microvascular endothelial cells (BMECs) to release different inflammatory mediators such as TNF-α in the early stage of AD, which can also increase cerebral vascular permeability [84]. This facilitates AGEs and other neurotoxic substances to cross the BBB and cause AGE deposition, which in turn can also lead to the significant upregulation of RAGE in BMECs. Moreover, increased ROS production can promote oxidative stress, leading to the secretion of nitric oxide synthase, which can further enhance Aβ deposition in the brain which can stimulate the microglial activation, which in turn can accelerate neurovascular dysfunction. Overall, these appear to be quite complex positive feedback processes, but inhibition of RAGE can prevent neuronal as well as cerebrovascular Aβ damage.
CX3C motif chemokine receptor 1, CX3CR1
In 1975, Tau was first identified as a microtubule-associated protein that is highly expressed in soluble form throughout the neurons of the CNS. It is found mainly in the axons of the neurons. The crucial role of Tau lies in its ability to bind to microtubules, facilitating their assembly and thereby regulating microtubule stability. Consequently, it plays a pivotal role in neurite outgrowth, cellular morphology, and polarity, as well as the intracellular transport of neurotransmitters [85]. Insoluble misfolded tau deposits, consisting of fibrils, are predominantly observed in the cell body and dendrites of neurons affected by AD, and are recognized as a pivotal pathological characteristic of AD. Hyperphosphorylated Tau can induce aberrant aggregation of Tau and compromise its ability to stabilize microtubules, thereby impairing neuronal function [86]. The pathogenic form of Tau can be released from the diseased neurons and subsequently internalized by previously unaffected normal neurons, thereby triggering the production of pathogenic Tau within these normal neurons. This property contributes to disease progression and the development of broader clinical symptoms [87]. Microglial activation is also involved in the advancement of neuropathology associated with Tau. The proportion of morphologically activated microglia in postmortem cortical tissues from patients with AD has shown a strong association with Tau pathology, as microglial activation can contribute to the accumulation of hyperphosphorylated Tau protein and subsequent cognitive decline [88]. The surface receptors of activated microglia which mediated the inflammatory response, which has significant implications for Tau hyperphosphorylation [89].
CX3CR1 is a receptor that is primarily expressed on the microglia and is involved in Tau pathology. CX3CR1 is a G protein-coupled receptor (GPCR). GPCRs, being one of the most prominent protein families, represent a class of receptors characterized by the presence of seven transmembrane domains. GPCRs are capable of detecting extracellular molecules and subsequently transmitting signals to intracellular effector molecules, thereby eliciting cellular responses. Tau can bind to the microglial CX3CR1 to initiate Tau internalization and degradation, whereas chemokine C-X3-C motif ligand 1 (CX3CL1) can compete with Tau for binding CX3CR1, thereby resulting in decreased Tau internalization and increased abnormal aggregation [90]. Accumulating evidence suggests that CX3CR1 and its ligand CX3CL1 can exert opposing effects on Aβ and Tau pathology. Knockdown of CX3CR1 or inhibition of the CX3CL1/CX3CR1 axis can reduce amyloid-beta (Aβ) deposition; however, it may exacerbate Tau pathology, including increased Tau phosphorylation and aggregation. This phenomenon could potentially be associated with the deterioration of behavioral and cognitive impairment [91,92]. CX3CR1 deficiency is linked with reduced Aβ levels, plaque burden, and improved cognitive function [93]. These findings suggest that modulation of the CX3CL1/CX3CR1 axis could play a pivotal role in facilitating microglial phagocytosis and degradation of Tau [94], highlighting its potential as a therapeutic target for preventing tau-related neurodegeneration.
The pathology of AD is characterized by two distinct features, the extracellular neural plaques (SPs) composed of Aβ and the intercellular neurofibrillary tangles (NFTs) which are made up of hyperphosphorylated tau [95]. Neuroinflammation also plays a pivotal role in the pathogenesis of Alzheimer’s disease, and this neuroinflammatory process is primarily orchestrated by diverse innate immune cells residing within the brain, including microglia. Microglia are resident immune cells in the brain and play a crucial role in the immune defense of the central nervous system. They are in a “quiescent state” of inactivity under physiological conditions, surveilling the brain environment and parenchyma. When microglia recognize a specific stimulus in the CNS, they can undergo a transition from a quiescent to an activated state, which is marked by distinct morphological changes and modulation of gene expression, including that of proinflammatory and anti-inflammatory molecules and microglial surface receptors, including TREM2, TLRs, RAGE, and G protein-coupled receptors (GPCRS) such as CX3CR1, which can effectively mediate microglial activation and polarized phenotypes when activated by stimuli [96,97]. The activation of microglia can occur in both the classical M1 state and the alternative M2 state. Existing research predominantly suggests that M1-type microglia exhibit detrimental effects on the nervous system, whereas M2-type microglia confer beneficial outcomes [98]. M1 microglia can significantly promote the release of different proinflammatory cytokines, On the contrary, the M2 microglia can exert potent anti-inflammatory effects by significantly upregulating the expression of diverse anti-inflammatory factors, and M1 and M2 microglia can undergo conversion into each other under certain conditions [99]. When microglia are activated and exhibit the M1 phenotype, they produce a range of enzymes and reactive oxygen species (ROS) that can contribute to persistent tissue inflammation by generating various inflammatory cytokines, thereby resulting in a detrimental neuronal microenvironment [100]. When microglia differentiate into M2 anti-inflammatory phenotype, they gain the ability to secrete various neurotrophins and anti-inflammatory mediators to support the neural microenvironment and hence can attenuate inflammatory response and promote the repair of damaged tissues [101,102].
It has been observed that during the initial stages of AD, activated microglia exhibit phagocytic activity and facilitate the clearance of pathological A beta and Tau, thereby exerting a positive influence on AD pathology [103,104]. A striking feature of microglia in AD is their tendency of general clustering around Aβ deposits, regarded as one of the main pathological features of AD, which can be effectively cleared by activated microglia by phagocytosis. Microglial processes can tightly wrap around the plaque and act as a physical barrier that can prevent the outward extension of amyloid fibrils, a barrier function that promotes the formation of highly dense plaque microzones that have the lowest affinity for soluble Aβ-42. In contrast, regions that are not covered by microglial processes exhibit regions of neurotoxic hot spots with extremely high soluble Aβ-42 affinity [105]. The activation of microglia and subsequent release of a large number of inflammatory factors persist as Alzheimer’s disease (AD) progresses, impairing their ability to phagocytose and degrade neurotoxins. This exacerbates the accumulation of Aβ, proliferation of Tau, and neuronal death, ultimately driving the progression of AD [10]. Interestingly, in the brain of healthy people or the early stage of AD, microglia are M2 type, which can phagocytose Aβ or other abnormal proteins. However, in the aging population or the brain of patients with advanced AD, due to excessive Aβ, microglia can be transformed into M1 type and secrete a large number of inflammatory factors as well as ROS to damage neurons [106], increasing the phagocytosis of microglia has been shown to improve cognitive dysfunction in AD patients [107].
Due to its complex pathogenesis and irreversibility, no cure has been found for AD yet. At present, many drugs are focused on improving the various psychiatric symptoms and cognitive functions, and there are also targeted drugs that can specifically act on the microglia receptors but lack the support of extensive clinical trials. In the early stage of AD, microglia can play the role of immune surveillance, anti-inflammation, immune phagocytosis, and exhibit other functions. Microglia can recognize Aβ and hyperphosphorylated Tau through its surface protein receptors, phagocytose or remove these abnormally accumulated denatured proteins, and form the cell surface protrusions tightly wrapping around the sediment through the cell deformation to form a physical barrier so that the neuroinflammation and lesions are localized. However, it has been found that in the late stage of AD, AD-related ligands can bind to the surface membrane protein receptors of microglia, affect the expression regulation of surface receptors, and thereby cause excessive activation of microglia, resulting in pathological immune phagocytosis as well as amplification of inflammation. At the same time, the transformation from the M2 anti-inflammatory phenotype to the M1 pro-inflammatory phenotype can occur, destroying the neurovascular microenvironment and the physical barrier of microglia. Thus, more neurons are damaged and become apoptotic, which can aggravate the progression of AD.
What we can do is extremely limited, but these findings suggest that we can try to intervene in the early stage of AD by targeting and regulating the surface membrane protein receptors of microglia, thus inhibiting the excessive activation of microglia as well as its phenotypic transformation and delaying the progression of AD to the greatest extent. With the aging of the global population, increased subjects will suffer from this devastating disease. In the future, we can start from the transformation of microglia from M1 pro-inflammatory phenotype to M2 anti-inflammatory phenotype, stabilize the pathological changes of AD, achieve “0” progression of AD course, or even reverse the course of AD, although more studies are needed to achieve the desired course of action. Thus, these strategies can help more AD patients to improve or restore their cognitive function, reduce their family burden, and make their lives better, which is what we expect.
Acknowledgement: None.
Funding Statement: This study was supported by grants from the Science and Technology Innovation Fund Project of Dalian (No. 2021JJ13SN55).
Author Contributions: XW and Y-QJ initiated the work and designed the idea. J-FZ, Y-RJ, and T-LG prepared and collected material and data. J-FZ , Y-RJ, and T-LG wrote the manuscript, J-FZ, Y-RJ contributed further to this work and share first authorship. All authors reviewed the article, read and approved the submitted version.
Availability of Data and Materials: Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Molinuevo J, Ayton S, Batrla R, Bednar M, Bittner T, Cummings J, et al. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol. 2018;136(6):821–53. doi:10.1007/s00401-018-1932-x. [Google Scholar] [PubMed] [CrossRef]
2. Dong K. A review of traditional Chinese medicine in the treatment of Alzheimer’s disease. Aging Commun. 2020;2:1–33. [Google Scholar]
3. Alzheimer’s Association. 2021 Alzheimer’s disease facts and figures. Alzheimer Rep. 2021;17(3):327–406. doi:10.1002/alz.12328. [Google Scholar] [PubMed] [CrossRef]
4. Kumar A, Nemeroff C, Cooper J, Widge A, Rodriguez C, Carpenter L, et al. Amyloid and Tau in Alzheimer’s disease: biomarkers or molecular targets for therapy? Are we shooting the messenger? Am J Psychiatry. 2021;178(11):1014–25. doi:10.1176/appi.ajp.2021.19080873. [Google Scholar] [PubMed] [CrossRef]
5. Wojsiat J, Laskowska-Kaszub K, Mietelska-Porowska A, Wojda U. Search for Alzheimer’s disease biomarkers in blood cells: hypotheses-driven approach. Biomark Med. 2017;11(10):917–31. doi:10.2217/bmm-2017-0041. [Google Scholar] [PubMed] [CrossRef]
6. Zhang R, Li C, Du R, Yuan Y, Zhao B, Zhang Y, et al. Mesenchymal stem cells: as a multi-target cell therapy for clearing β-amyloid deposition in Alzheimer’s disease. BIOCELL. 2022;46(3):583–92. doi:10.32604/biocell.2022.017248. [Google Scholar] [CrossRef]
7. Romagnoli M, Porcellini E, Carbone I, Veerhuis R, Licastro F. Impaired innate immunity mechanisms in the brain of Alzheimer’s disease. Int J Mol Sci. 2020;21(3):11–26. [Google Scholar]
8. Reith W. Neurodegenerative diseases. Radiologe. 2018;58(3):241–58. [Google Scholar] [PubMed]
9. Subhramanyam C, Wang C, Hu Q, Dheen S. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin Cell Dev Biol. 2019;94:112–20. doi:10.1016/j.semcdb.2019.05.004. [Google Scholar] [PubMed] [CrossRef]
10. Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17(3):157–72. doi:10.1038/s41582-020-00435-y. [Google Scholar] [PubMed] [CrossRef]
11. Atienza M, Ziontz J, Cantero J. Low-grade inflammation in the relationship between sleep disruption, dysfunctional adiposity, and cognitive decline in aging. Sleep Med Rev. 2018;42:171–83. doi:10.1016/j.smrv.2018.08.002. [Google Scholar] [PubMed] [CrossRef]
12. Cheng F, Fransson L, Mani K. Proinflammatory cytokines induce accumulation of glypican-1-derived heparan sulfate and the C-terminal fragment of β-cleaved APP in autophagosomes of dividing neuronal cells. Glycobiology. 2020;30(8):539–49. doi:10.1093/glycob/cwaa011. [Google Scholar] [PubMed] [CrossRef]
13. Shi S, Liang D, Chen Y, Xie Y, Wang Y, Wang L, et al. Gx-50 reduces β-amyloid-induced TNF-α, IL-1β, NO, and PGE2 expression and inhibits NF-κB signaling in a mouse model of Alzheimer’s disease. Eur J Immunol. 2016;46(3):665–76. doi:10.1002/eji.v46.3. [Google Scholar] [CrossRef]
14. Cisbani G, Rivest S. Targeting innate immunity to protect and cure Alzheimer’s disease: opportunities and pitfalls. Mol Psychiatr.2021;26(10):5504–15. doi:10.1038/s41380-021-01083-4. [Google Scholar] [PubMed] [CrossRef]
15. He W, Yuan K, Ji B, Han Y, Li J. Protective effects of curcumin against neuroinflammation induced by Aβ25-35 in primary rat microglia: modulation of high-mobility group box 1, toll-like receptor 4 and receptor for advanced glycation end products expression. Ann Transl Med. 2020;8(4):88. doi:10.21037/atm.2019.12.147. [Google Scholar] [PubMed] [CrossRef]
16. Abe N, Nishihara T, Yorozuya T, Tanaka J. Microglia and macrophages in the pathological central and peripheral nervous systems. Cells. 2020;9(9):21–3. [Google Scholar]
17. Zhao J, Ren T, Li X, Guo T, Liu C, Wang X. Research progress on the role of microglia membrane proteins or receptors in neuroinflammation and degeneration. Front Cell Neurosci. 2022;16:831977. doi:10.3389/fncel.2022.831977. [Google Scholar] [PubMed] [CrossRef]
18. Yang G, Tong Y, Wang X, Zhao C, Ba Z, Ahelijiang R, et al. Guizhi Fuling capsule relieves memory deficits by inhibition of microglial neuroinflammation through blocking JAK2/STAT3 pathway in presenilin1/2 conditional double knockout mice. Front Immunol. 2023;14:1185570. doi:10.3389/fimmu.2023.1185570. [Google Scholar] [PubMed] [CrossRef]
19. Ou-Yang P, Cai Z, Zhang Z. Molecular regulation mechanism of microglial autophagy in the pathology of Alzheimer’s disease. Aging Dis. 2023;14(4):1166–77. [Google Scholar] [PubMed]
20. Vilalta A, Brown G. Neurophagy, the phagocytosis of live neurons and synapses by glia, contributes to brain development and disease. FEBS J. 2018;285(19):3566–75. doi:10.1111/febs.14323. [Google Scholar] [PubMed] [CrossRef]
21. Galloway D, Phillips A, Owen D, Moore C. Phagocytosis in the brain: homeostasis and disease. Front Immunol. 2019;10:790. doi:10.3389/fimmu.2019.00790. [Google Scholar] [PubMed] [CrossRef]
22. Butler C, Popescu A, Kitchener E, Allendorf D, Puigdellívol M, Brown G. Microglial phagocytosis of neurons in neurodegeneration, and its regulation. J Neurochem. 2021;158(3):621–39. doi:10.1111/jnc.v158.3. [Google Scholar] [CrossRef]
23. Udeochu J, Shea J, Villeda S. Microglia communication: Parallels between aging and Alzheimer’s disease. Clin Exp Neuroimmunol. 2016;7(2):114–25. doi:10.1111/cen3.2016.7.issue-2. [Google Scholar] [CrossRef]
24. Derecki N, Katzmarski N, Kipnis J, Meyer-Luehmann M. Microglia as a critical player in both developmental and late-life CNS pathologies. Acta Neuropathol. 2014;128(3):333–45. doi:10.1007/s00401-014-1321-z. [Google Scholar] [PubMed] [CrossRef]
25. Jaeger P, Pickford F, Sun C, Lucin K, Masliah E, Wyss-Coray T. Regulation of amyloid precursor protein processing by the Beclin 1 complex. PLoS One. 2010;5(6):e11102. doi:10.1371/journal.pone.0011102. [Google Scholar] [PubMed] [CrossRef]
26. Bacskai B, Kajdasz S, McLellan M, Games D, Seubert P, Schenk D, et al. Non-Fc-mediated mechanisms are involved in clearance of amyloid-beta in vivo by immunotherapy. J Neurosci. 2002;22(18):7873–8. doi:10.1523/JNEUROSCI.22-18-07873.2002. [Google Scholar] [PubMed] [CrossRef]
27. Frenkel D, Wilkinson K, Zhao L, Hickman S, Means T, Puckett L, et al. Scara1 deficiency impairs clearance of soluble amyloid-β by mononuclear phagocytes and accelerates Alzheimer’s-like disease progression. Nat Commun. 2013;4:2030. doi:10.1038/ncomms3030. [Google Scholar] [PubMed] [CrossRef]
28. Yamanaka M, Ishikawa T, Griep A, Axt D, Kummer M, Heneka M. PPARγ/RXRα-induced and CD36-mediated microglial amyloid-β phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J Neurosci. 2012;32(48):17321–31. doi:10.1523/JNEUROSCI.1569-12.2012. [Google Scholar] [PubMed] [CrossRef]
29. Lue L, Yan S, Stern D, Walker D. Preventing activation of receptor for advanced glycation endproducts in Alzheimer’s disease. Curr Drug Targets CNS Neurol Disord. 2005;4(3):249–66. doi:10.2174/1568007054038210. [Google Scholar] [PubMed] [CrossRef]
30. Painter M, Atagi Y, Liu C, Rademakers R, Xu H, Fryer J, et al. TREM2 in CNS homeostasis and neurodegenerative disease. Mol Neurodegener. 2015;10:43. doi:10.1186/s13024-015-0040-9. [Google Scholar] [PubMed] [CrossRef]
31. Ana G, Rudolph ET. The role of innate immune genes in Alzheimer’s disease. Curr Opin Neurol. 2021;34(2):228–36. doi:10.1097/WCO.0000000000000911. [Google Scholar] [PubMed] [CrossRef]
32. Calvo-Rodriguez M, García-Rodríguez C, Villalobos C, Núñez L. Role of toll like receptor 4 in Alzheimer’s disease. Front Immunol. 2020;11:1588. doi:10.3389/fimmu.2020.01588. [Google Scholar] [PubMed] [CrossRef]
33. Caputi V, Giron M. Microbiome-gut-brain axis and toll-like receptors in Parkinson’s disease. Int J Mol Sci. 2018;19(6):16–89. [Google Scholar]
34. Asami J, Shimizu T. Structural and functional understanding of the toll-like receptors. Protein Sci. 2021;30(4):761–72. doi:10.1002/pro.v30.4. [Google Scholar] [CrossRef]
35. Balka K, de Nardo D. Understanding early TLR signaling through the Myddosome. J Leukocyte Biol. 2019;105(2):339–51. doi:10.1002/JLB.MR0318-096R. [Google Scholar] [PubMed] [CrossRef]
36. Miras-Portugal M, Ortega F, Gómez-Villafuertes R, Gualix J, Pérez-Sen R, Delicado E. P2X7 receptors in the central nervous system. Biochem Pharmacol. 2021;187:114472. doi:10.1016/j.bcp.2021.114472. [Google Scholar] [PubMed] [CrossRef]
37. Li G, Hidalgo A. The toll route to structural brain plasticity. Front Physiol. 2021;12:679766. doi:10.3389/fphys.2021.679766. [Google Scholar] [PubMed] [CrossRef]
38. Zhang X, Hei Y, Bai W, Huang T, Kang E, Chen H, et al. Toll-like receptor 2 attenuates traumatic brain injury-induced neural stem cell proliferation in dentate gyrus of rats. Neural Plast. 2020;2020:9814978. [Google Scholar] [PubMed]
39. Kumar V. Toll-like receptors in the pathogenesis of neuroinflammation. J Neuroimmunol. 2019;332:16–30. doi:10.1016/j.jneuroim.2019.03.012. [Google Scholar] [PubMed] [CrossRef]
40. Chakrabarty P, Li A, Ladd T, Strickland M, Koller E, Burgess J, et al. TLR5 decoy receptor as a novel anti-amyloid therapeutic for Alzheimer’s disease. J Exp Med. 2018;215(9):2247–64. doi:10.1084/jem.20180484. [Google Scholar] [PubMed] [CrossRef]
41. Chen S, Tian D, Shen Y, Cheng Y, Fan D, Sun H, et al. Amyloid-beta uptake by blood monocytes is reduced with ageing and Alzheimer’s disease. Transl Psychiat. 2020;10(1):423. doi:10.1038/s41398-020-01113-9. [Google Scholar] [PubMed] [CrossRef]
42. Zhou C, Sun X, Hu Y, Song J, Dong S, Kong D, et al. Genomic deletion of TLR2 induces aggravated white matter damage and deteriorated neurobehavioral functions in mouse models of Alzheimer’s disease. Aging. 2019;11(17):7257–73. doi:10.18632/aging.v11i17. [Google Scholar] [CrossRef]
43. Wendeln A, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. 2018;556(7701):332–8. doi:10.1038/s41586-018-0023-4. [Google Scholar] [PubMed] [CrossRef]
44. Qin Y, Liu Y, Hao W, Decker Y, Tomic I, Menger M, et al. Stimulation of TLR4 attenuates Alzheimer’s disease-related symptoms and pathology in tau-transgenic mice. J Immunol. 2016;197(8):3281–92. doi:10.4049/jimmunol.1600873. [Google Scholar] [PubMed] [CrossRef]
45. Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, et al. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol Biochem. 2007;20(6):947–56. doi:10.1159/000110455. [Google Scholar] [PubMed] [CrossRef]
46. Balducci C, Frasca A, Zotti M, La Vitola P, Mhillaj E, Grigoli E, et al. Toll-like receptor 4-dependent glial cell activation mediates the impairment in memory establishment induced by β-amyloid oligomers in an acute mouse model of Alzheimer’s disease. Brain Behav Immun. 2017;60:188–97. doi:10.1016/j.bbi.2016.10.012. [Google Scholar] [PubMed] [CrossRef]
47. Azam S, Jakaria M, Kim I, Kim J, Haque M, Choi D. Regulation of toll-like receptor (TLR) signaling pathway by polyphenols in the treatment of age-linked neurodegenerative diseases: focus on TLR4 signaling. Front Immunol. 2019;10:1000. doi:10.3389/fimmu.2019.01000. [Google Scholar] [PubMed] [CrossRef]
48. Sheng X, Yao Y, Huang R, Xu Y, Zhu Y, Chen L, et al. Identification of the minimal active soluble TREM2 sequence for modulating microglial phenotypes and amyloid pathology. J Neuroinflamm. 2021;18(1):286. doi:10.1186/s12974-021-02340-7. [Google Scholar] [PubMed] [CrossRef]
49. Konishi H, Kiyama H. Non-pathological roles of microglial TREM2/DAP12: TREM2/DAP12 regulates the physiological functions of microglia from development to aging. Neurochem Intl. 2020;141:104878. doi:10.1016/j.neuint.2020.104878. [Google Scholar] [PubMed] [CrossRef]
50. Jay T, von Saucken V, Landreth G. TREM2 in neurodegenerative diseases. Mol Neurodegener. 2017;12(1):56. doi:10.1186/s13024-017-0197-5. [Google Scholar] [PubMed] [CrossRef]
51. Griciuc A, Patel S, Federico A, Choi S, Innes B, Oram M, et al. TREM2 Acts downstream of CD33 in modulating microglial pathology in Alzheimer’s disease. Neuron. 2019;103(5):820–35. doi:10.1016/j.neuron.2019.06.010. [Google Scholar] [PubMed] [CrossRef]
52. Gratuze M, Leyns C, Holtzman D. New insights into the role of TREM2 in Alzheimer’s disease. Mol Neurodegener. 2018;13(1):66. doi:10.1186/s13024-018-0298-9. [Google Scholar] [PubMed] [CrossRef]
53. Yin J, Liu X, He Q, Zhou L, Yuan Z, Zhao S. Vps35-dependent recycling of Trem2 regulates microglial function. Traffic. 2016;17(12):1286–96. doi:10.1111/tra.2016.17.issue-12. [Google Scholar] [CrossRef]
54. Song W, Joshita S, Zhou Y, Ulland T, Gilfillan S, Colonna M. Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J Exp Med. 2018;215(3):745–60. doi:10.1084/jem.20171529. [Google Scholar] [PubMed] [CrossRef]
55. Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. 2018;21(10):1359–69. doi:10.1038/s41593-018-0242-x. [Google Scholar] [PubMed] [CrossRef]
56. Dardiotis E, Siokas V, Pantazi E, Dardioti M, Rikos D, Xiromerisiou G, et al. A novel mutation in TREM2 gene causing Nasu-Hakola disease and review of the literature. Neurobiol Aging. 2017;53:194. e13–.e22. doi:10.1016/j.neurobiolaging.2017.01.015. [Google Scholar] [PubMed] [CrossRef]
57. Sheng L, Chen M, Cai K, Song Y, Yu D, Zhang H, et al. Microglial Trem2 induces synaptic impairment at early stage and prevents amyloidosis at late stage in APP/PS1 mice. FASEB J. 2019;33(9):10425–42. doi:10.1096/fsb2.v33.9. [Google Scholar] [CrossRef]
58. Ji Z, Liu C, Zhao W, Soto C, Zhou X. Multi-scale modeling for systematically understanding the key roles of microglia in AD development. Comput Biol Med. 2021;133:104374. doi:10.1016/j.compbiomed.2021.104374. [Google Scholar] [PubMed] [CrossRef]
59. Andrejew R, Oliveira-Giacomelli Á, Ribeiro D, Glaser T, Arnaud-Sampaio V, Lameu C, et al. The P2X7 receptor: central hub of brain diseases. Front Mol Neurosci. 2020;13:124. doi:10.3389/fnmol.2020.00124. [Google Scholar] [PubMed] [CrossRef]
60. Illes P. P2X7 receptors amplify CNS damage in neurodegenerative diseases. Int J Mol Sci. 2020;21(17):59–96. [Google Scholar]
61. McCarthy A, Yoshioka C, Mansoor S. Full-length P2X structures reveal how palmitoylation prevents channel desensitization. Cell. 2019;179(3):659–70. doi:10.1016/j.cell.2019.09.017. [Google Scholar] [PubMed] [CrossRef]
62. Wang Z, Ren W, Zhao F, Han Y, Liu C, Jia K. Curcumin amends Ca dysregulation in microglia by suppressing the activation of P2X7 receptor. Mol Cell Biochem. 2020;465:65–73. doi:10.1007/s11010-019-03668-8. [Google Scholar] [PubMed] [CrossRef]
63. Ferrari D, Pizzirani C, Adinolfi E, Lemoli R, Curti A, Idzko M, et al. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol. 2006;176(7):3877–83. doi:10.4049/jimmunol.176.7.3877. [Google Scholar] [PubMed] [CrossRef]
64. Kanellopoulos J, Delarasse C. Pleiotropic Roles of P2X7 in the Central Nervous System. Front Cell Neurosci. 2019;13:401. doi:10.3389/fncel.2019.00401. [Google Scholar] [PubMed] [CrossRef]
65. Martin E, Amar M, Dalle C, Youssef I, Boucher C, Le Duigou C, et al. New role of P2X7 receptor in an Alzheimer’s disease mouse model. Mol Psychiatr. 2019;24(1):108–25. doi:10.1038/s41380-018-0108-3. [Google Scholar] [PubMed] [CrossRef]
66. Di Lauro C, Bianchi C, Sebastián-Serrano Á, Soria-Tobar L, Alvarez-Castelao B, Nicke A, et al. P2X7 receptor blockade reduces tau induced toxicity, therapeutic implications in tauopathies. Prog Neurobiol. 2022;208:102173. doi:10.1016/j.pneurobio.2021.102173. [Google Scholar] [PubMed] [CrossRef]
67. Delarasse C, Auger R, Gonnord P, Fontaine B, Kanellopoulos J. The purinergic receptor P2X7 triggers alpha-secretase-dependent processing of the amyloid precursor protein. J Biol Chem. 2011;286(4):2596–606. doi:10.1074/jbc.M110.200618. [Google Scholar] [PubMed] [CrossRef]
68. Diaz-Hernandez J, Gomez-Villafuertes R, León-Otegui M, Hontecillas-Prieto L, Del Puerto A, Trejo J, et al. In vivo P2X7 inhibition reduces amyloid plaques in Alzheimer’s disease through GSK3β and secretases. Neurobiol Aging. 2012;33(8):1816–28. doi:10.1016/j.neurobiolaging.2011.09.040. [Google Scholar] [PubMed] [CrossRef]
69. Carvalho K, Martin E, Ces A, Sarrazin N, Lagouge-Roussey P, Nous C, et al. P2X7-deficiency improves plasticity and cognitive abilities in a mouse model of Tauopathy. Prog Neurobiol. 2021;206:102139. doi:10.1016/j.pneurobio.2021.102139. [Google Scholar] [PubMed] [CrossRef]
70. Lee J, McDonald T, Fung J, Woodruff T. Absence of receptor for advanced glycation end product (RAGE) reduces inflammation and extends survival in the hSOD1G93A mouse model of amyotrophic lateral sclerosis. Mol Neurobiol. 2020;57(10):4143–55. doi:10.1007/s12035-020-02019-9. [Google Scholar] [PubMed] [CrossRef]
71. Hudson B, Lippman M. Targeting RAGE signaling in inflammatory disease. Annu Rev Med. 2018;69:349–64. doi:10.1146/med.2018.69.issue-1. [Google Scholar] [CrossRef]
72. Syed D, Aljohani A, Waseem D, Mukhtar H. Ousting RAGE in melanoma: a viable therapeutic target? Semin Cancer Biol. 2018;49:20–8. doi:10.1016/j.semcancer.2017.10.008. [Google Scholar] [PubMed] [CrossRef]
73. Jangde N, Ray R, Rai V. RAGE and its ligands: from pathogenesis to therapeutics. Crit Rev Biochem Mol. 2020;55(6):555–75. doi:10.1080/10409238.2020.1819194. [Google Scholar] [PubMed] [CrossRef]
74. Zaki M, Kamal S, Kholousi S, El-Bassyouni H, Yousef W, Reyad H, et al. Serum soluble receptor of advanced glycation end products and risk of metabolic syndrome in Egyptian obese women. Excli J. 2017;16:973–80. [Google Scholar] [PubMed]
75. Bongarzone S, Savickas V, Luzi F, Gee A. Targeting the receptor for advanced glycation endproducts (RAGEa medicinal chemistry perspective. J Med Chem. 2017;60(17):7213–32. doi:10.1021/acs.jmedchem.7b00058. [Google Scholar] [PubMed] [CrossRef]
76. Manigrasso M, Pan J, Rai V, Zhang J, Reverdatto S, Quadri N, et al. Small molecule inhibition of ligand-stimulated RAGE-DIAPH1 signal transduction. Sci Rep. 2016;6:22450. doi:10.1038/srep22450. [Google Scholar] [PubMed] [CrossRef]
77. Reddy V, Aryal P, Soni P. RAGE inhibitors in neurodegenerative diseases. Biomedicines. 2023;11(4):11–31. [Google Scholar]
78. Chellappa R, Palanisamy R, Swaminathan K. RAGE isoforms, its ligands and their role in pathophysiology of Alzheimer’s disease. Curr Alzheimer Res. 2020;17(14):1262–79. [Google Scholar] [PubMed]
79. Cuevas E, Rosas-Hernandez H, Burks S, Ramirez-Lee M, Guzman A, Imam S, et al. Amyloid Beta 25-35 induces blood-brain barrier disruption in vitro. Metab Brain Dis. 2019;34(5):1365–74. doi:10.1007/s11011-019-00447-8. [Google Scholar] [PubMed] [CrossRef]
80. Mizari S, Alyas R, Khan S, Ahmad R, Mehmod R. Targeting microglial neurotransmitter receptors as a therapeutic approach for Alzheimer’s disease. Aging Commun. 2023;5(2):1–8. [Google Scholar]
81. Tallon C, Bell B, Malvankar M, Deme P, Nogueras-Ortiz C, Eren E, et al. Inhibiting tau-induced elevated nSMase2 activity and ceramides is therapeutic in murine Alzheimer’s disease. Res Sq. 2023;12(1):56. [Google Scholar]
82. Wells J, Holmes H, O’Callaghan J, Colgan N, Ismail O, Fisher E, et al. Increased cerebral vascular reactivity in the tau expressing rTg4510 mouse: evidence against the role of tau pathology to impair vascular health in Alzheimer’s disease. J Cereb Blood Flow Metab. 2015;35(3):359–62. doi:10.1038/jcbfm.2014.224. [Google Scholar] [PubMed] [CrossRef]
83. Cai Z, Liu N, Wang C, Qin B, Zhou Y, Xiao M, et al. Role of RAGE in Alzheimer’s disease. Cell Mol Neurobiol. 2016;36(4):483–95. doi:10.1007/s10571-015-0233-3. [Google Scholar] [PubMed] [CrossRef]
84. Qiu L, Ng G, Tan E, Liao P, Kandiah N, Zeng L. Chronic cerebral hypoperfusion enhances Tau hyperphosphorylation and reduces autophagy in Alzheimer’s disease mice. Sci Rep. 2016;6:23964. doi:10.1038/srep23964. [Google Scholar] [PubMed] [CrossRef]
85. Strang K, Golde T, Giasson B. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab Invest. 2019;99(7912–28. doi:10.1038/s41374-019-0197-x. [Google Scholar] [PubMed] [CrossRef]
86. Höglinger G, Respondek G, Kovacs G. New classification of tauopathies. Rev Neurol-France. 2018;174(9):664–8. doi:10.1016/j.neurol.2018.07.001. [Google Scholar] [PubMed] [CrossRef]
87. Mudher A, Colin M, Dujardin S, Medina M, Dewachter I, Alavi Naini S, et al. What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol Com. 2017;5(1):99. doi:10.1186/s40478-017-0488-7. [Google Scholar] [PubMed] [CrossRef]
88. Felsky D, Roostaei T, Nho K, Risacher S, Bradshaw E, Petyuk V, et al. Neuropathological correlates and genetic architecture of microglial activation in elderly human brain. Nat Commun. 2019;10(1):409. doi:10.1038/s41467-018-08279-3. [Google Scholar] [PubMed] [CrossRef]
89. Bolós M, Llorens-Martín M, Jurado-Arjona J, Hernández F, Rábano A, Avila J. Direct evidence of internalization of tau by microglia in vitro and in vivo. J Alzheimers Dis. 2016;50(1):77–87. doi:10.3233/JAD-150704. [Google Scholar] [PubMed] [CrossRef]
90. Castro-Sánchez S, García-Yagüe Á, Kügler S, Lastres-Becker I. CX3CR1-deficient microglia shows impaired signalling of the transcription factor NRF2: implications in tauopathies. Redox Biol. 2019;22:101118. doi:10.1016/j.redox.2019.101118. [Google Scholar] [PubMed] [CrossRef]
91. Puntambekar S, Moutinho M, Lin P, Jadhav V, Tumbleson-Brink D, Balaji A, et al. CX3CR1 deficiency aggravates amyloid driven neuronal pathology and cognitive decline in Alzheimer’s disease. Mol Neurodegener. 2022;17(1):47. doi:10.1186/s13024-022-00545-9. [Google Scholar] [PubMed] [CrossRef]
92. Fan Q, He W, Gayen M, Benoit M, Luo X, Hu X, et al. Activated CX3CL1/Smad2 signals prevent neuronal loss and Alzheimer’s tau pathology-mediated cognitive dysfunction. J Neurosci. 2020;40(5):1133–44. doi:10.1523/JNEUROSCI.1333-19.2019. [Google Scholar] [PubMed] [CrossRef]
93. Hickman S, Allison E, Coleman U, Kingery-Gallagher N, El Khoury J. Heterozygous CX3CR1 deficiency in microglia restores neuronal β-amyloid clearance pathways and slows progression of Alzheimer’s like-disease in PS1-APP mice. Front Immunol. 2019;10:2780. doi:10.3389/fimmu.2019.02780. [Google Scholar] [PubMed] [CrossRef]
94. Stancu I, Cremers N, Vanrusselt H, Couturier J, Vanoosthuyse A, Kessels S, et al. Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019;137(4):599–617. doi:10.1007/s00401-018-01957-y. [Google Scholar] [PubMed] [CrossRef]
95. Iqbal K, Liu F, Gong C. Tau and neurodegenerative disease: the story so far. Nat Rev Neurol. 2016;12(1):15–27. doi:10.1038/nrneurol.2015.225. [Google Scholar] [PubMed] [CrossRef]
96. Salter M, Stevens B. Microglia emerge as central players in brain disease. Nat Med. 2017;23(9):1018–27. doi:10.1038/nm.4397. [Google Scholar] [PubMed] [CrossRef]
97. Yu Y, Ye R. Microglial Aβ receptors in Alzheimer’s disease. Cell Mol Neurobiol. 2015;35(1):71–83. doi:10.1007/s10571-014-0101-6. [Google Scholar] [PubMed] [CrossRef]
98. Guo Q, Wang C, Xue X, Hu B, Bao H. βSOCS1 mediates berberine-induced amelioration of microglial activated states in N9 microglia exposed to β amyloid. Biomed Res Int. 2021;2021:9311855. [Google Scholar] [PubMed]
99. Wang J, Liang J, Deng J, Liang X, Wang K, Wang H, et al. Emerging role of microglia-mediated neuroinflammation in epilepsy after subarachnoid hemorrhage. Mol Neurobiol. 2021;58(6):2780–91. doi:10.1007/s12035-021-02288-y. [Google Scholar] [PubMed] [CrossRef]
100. Liu X, Liu J, Zhao S, Zhang H, Cai W, Cai M, et al. Interleukin-4 is essential for microglia/macrophage M2 polarization and long-term recovery after cerebral ischemia. Stroke. 2016;47(2):498–504. doi:10.1161/STROKEAHA.115.012079. [Google Scholar] [PubMed] [CrossRef]
101. Orihuela R, McPherson C, Harry G. Microglial M1/M2 polarization and metabolic states. Brit J Pharmacol. 2016;173(4):649–65. doi:10.1111/bph.v173.4. [Google Scholar] [CrossRef]
102. Kim S, Son Y. Astrocytes stimulate microglial proliferation and M2 polarization in vitro through crosstalk between astrocytes and microglia. Int J Mol Sci. 2021;22(16):8800. doi:10.3390/ijms22168800. [Google Scholar] [PubMed] [CrossRef]
103. Hansen D, Hanson J, Sheng M. Microglia in Alzheimer’s disease. J Cell Biol. 2018;217(2):459–72. doi:10.1083/jcb.201709069. [Google Scholar] [PubMed] [CrossRef]
104. Feng W, Zhang Y, Wang Z, Xu H, Wu T, Marshall C, et al. Microglia prevent beta-amyloid plaque formation in the early stage of an Alzheimer’s disease mouse model with suppression of glymphatic clearance. Alzheimers Res Ther. 2020;12(1):125. doi:10.1186/s13195-020-00688-1. [Google Scholar] [PubMed] [CrossRef]
105. Condello C, Yuan P, Schain A, Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat Commun. 2015;6:6176. doi:10.1038/ncomms7176. [Google Scholar] [PubMed] [CrossRef]
106. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017;35:441–68. doi:10.1146/immunol.2017.35.issue-1. [Google Scholar] [CrossRef]
107. Pluvinage J, Haney M, Smith B, Sun J, Iram T, Bonanno L, et al. CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature. 2019;568(7751):187–92. doi:10.1038/s41586-019-1088-4. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools