
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Cholic acid mitigates osteoarthritis by inhibiting the NF-κB/PERK/SIRT1 signaling pathway
1 The Minda Hospital of Hubei Minzu University, Hubei Provincial Key Laboratory of Occurrence and Intervention of Rheumatic Disease, Enshi, 445000, China
2 The Obstetrics and Gynecology of Jianshi County Hospital of Traditional Chinese Medicine, Enshi, 445300, China
* Corresponding Authors: YUFANG DAI. Email: ; LINCHONG SU. Email:
(This article belongs to the Special Issue: Bioinformatics Study of Diseases)
BIOCELL 2024, 48(7), 1095-1104. https://doi.org/10.32604/biocell.2024.028421
Received 16 December 2022; Accepted 06 July 2023; Issue published 03 July 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Introduction: Cholic acid (CA) is a natural steroid useful in treating chronic bronchitis and cholecystitis. On the other hand, its potential impact on osteoarthritis (OA) is unknown. Objective: Using an in vitro and in vivo osteoarthritis model, we sought to assess the chondroprotective properties of CA. Methods: We employed the Cell Counting Kit-8 to measure the impact of CA on chondrocyte activity to assess the toxicity of the cells. Multiple molecular biology experimental techniques were used to investigate potential signaling pathways that CA may use to prevent inflammation and give chondrocytes protection. Furthermore, how CA affects the OA model in Sprague-Dawley (SD) rats was evaluated. Results: CA significantly suppressed the up-regulation of the interleukin-1 β (IL-1β), cyclooxygenase-2 (COX-2), and matrix metalloproteinase 13 (MMP-13) and the downregulation of aggrecan and type II collagen A1 (COL2A) in chondrocytes treated tumor necrosis factor-alpha (TNF-α). Differentially expressed genes (DEG) enrichment revealed IL-17, TNF, chemokine, cytokine-cytokine receptor, toll-like receptor, and nucleotide oligomerization domain-like receptor were the primary signaling pathways. The enriched DEGs included CXCL6, CCL20, MMP3, CXCL3, CXCL11, CCL5, CXCL10, MMP9, MMP13, and CXCL2; these DEGs are involved in inflammatory responses and their expression induced by TNF-α was reversed by CA treatment. CA inhibits p65 nuclear translocation and inhibitory subunit kappa B alpha (IκBα) phosphorylation induced by TNF-α. Furthermore, CA attenuated protein expression of protein kinase RNA-like endoplasmic reticulum kinase (PERK), inositol-requiring transmembrane kinase/endoribonuclease 1α (IRE1α), glucose regulatory protein 78 (GRP78), and sirtuin 1 (SIRT1), and down-regulation of phosphorylation of AMP-activated protein kinase-α (p-AMPKα) in TNF-α-treated chondrocytes. Conclusions: CA significantly ameliorated cartilage degradation in the OA rat model. CA alleviated the inflammatory response through the nuclear factor kappa B/PERK/SIRT1 axis and ameliorated cartilage degradation.Keywords
The incidence and prevalence of osteoarthritis (OA), a prevalent joint disease, have been found to rise with age [1]. The main characteristic of OA is cartilage breakdown, which can cause patients to experience joint pain, stiffness, and limits in their range of motion. These symptoms can drastically lower the patient’s quality of life and raise their risk of impairment [2]. An enormous rise in inflammatory mediators has been shown in the knee joint cavities of OA patients, according to mounting data [3,4]. These cytokines play a role in the pathophysiology of OA by inducing the synthesis of matrix-degrading enzymes (matrix metalloproteinase, MMPs) that lead to cartilage disintegration and the loss of proteoglycans [5]. Since inflammation is the core pathological factor of OA, treating OA may benefit by addressing the inflammatory process. While glucocorticoid drugs are now the first-line therapy treatments for osteoarthritis (OA), they only provide symptom relief; therefore, it is necessary to search for new potential small-molecule drugs for treating OA.
Natural bile acid called cholic acid (CA) is utilized to address genetic abnormalities linked to synthetic bile acid. Due to their numerous biological functions, which include anti-inflammatory and antioxidative qualities, bile acids have drawn a lot of interest. The main active ingredient in animal bile acid, chenodeoxycholic acid, strongly reduces the activity of MMPs. In an OA model, it also considerably reduces bone loss and joint erosion in vivo by blocking catabolic enzymes [6]. Natural dihydroxy hydrophilic bile acid ursodeoxycholic acid reduces oxidative stress and prevents the synthesis of MMPs, which in turn prevents cartilage degradation and relieves pain [7]. Comparable in its therapeutic effects on cartilage are the actions of CA and other bile acid members. For instance, by controlling the changes in genes and proteins linked to cartilage, CA-modified glucosamine encourages the production of cartilage [8]. On the other hand, CA increased the frequency and intensity of inflammatory erosive arthritis in different research [9].
Our work thus illustrated the chondroprotective and inflammatory antagonism properties of CA in in vitro, and in vivo. In addition, RNA-seq was used to analyze mRNA levels to find other molecular pathways. Our findings suggested that the antagonizing effects of CA on chondrocyte degradation are related to the nuclear factor kappa B/protein kinase RNA-like endoplasmic reticulum kinase/sirtuin 1 (NF-κB/PERK/SIRT1) pathway.
Sprague Dawley (SD) rats’ femoral heads were used to harvest cartilage tissue (purchased from the Experimental Animal Center of Three Gorges University, China). The rats were raised in cages with five rats each, fed water daily, and had their bedding changed every three days. From these samples, the primary chondrocytes were separated [10,11]. In brief, the tissue was chopped into tiny pieces and placed in a centrifuge tube. After adding 2% collagenase II (Gibco, 17101015, Grand Island, USA) to the tube, the samples were broken down and incubated. Following digestion, cells were centrifuged and then triple-washed in phosphate-buffered saline (PBS). The cells were then again resuspended in DMEM/F12 medium (Gibco, C11330500BT, Grand Island, USA) containing 10% fetal bovine serum (FBS) (Gibco, CB14808348, Grand Island, USA) and streptomycin and penicillin (North China Pharmaceutical, Shijiazhuang, China), and cultured in 37°C and 5% CO2 incubator. For subsequent experiments, second-to fourth-generation cells are used The animal experiments in the present study were approved by the Ethics Committee of the Minda Hospital of Hubei Minzu University (Hubei University for Nationalities Biomedical Research Ethics Approval Document Number: (2022) 32).
Trypsin (Sigma T3924, Missouri, USA) was used to treat the cells, and the resulting suspension was cultured in the plate. After obtaining CA from Selleck Chemicals LLC (Selleck, catalog no. s3742, Houston, USA), it was diluted in 0.1% dimethyl sulfoxide (DMSO) to produce a stock solution with a concentration of 20 millimolar. The growth medium was then supplemented with 0.1% DMSO to serve as the control, and this stock solution of CA was diluted in it to reach different concentrations. For 48 h, the cytotoxicity of 0–100 μM CA was synthesized and assessed. Three PBS washes were performed on the cells (Solarbio, P1020, Beijing, China). After being treated with CA for 48 h, each well of the 96-well plates was filled with a serum-free medium supplemented with 10% CCK-8 (Beyotime, C0037, Shanghai, China) as the culture media. After three hours of dark incubation, the absorbance of the plates was surveyed at 490 nm using a Bio-Rad microplate reader (Bio-Rad, Hercules, CA, USA).
The chondrocytes were cultivated in DMEM/F12 media and conducted experimental processing. After a 16-h cell starvation phase, the cells were treated with 10 ng/mL TNF-α with or without 10 μM CA. The cells were sequentially fixed and penetrated. Subsequently, the cells were treated with the primary antibody NF-κB p65 (RELA) (Cell Signaling Technology, #8242, MA, USA) for an hour at 37°C using a blocking solution, and then a secondary antibody (Thermo Fisher Scientific, A-11001, Waltham, MA, USA) tagged with fluorescence.
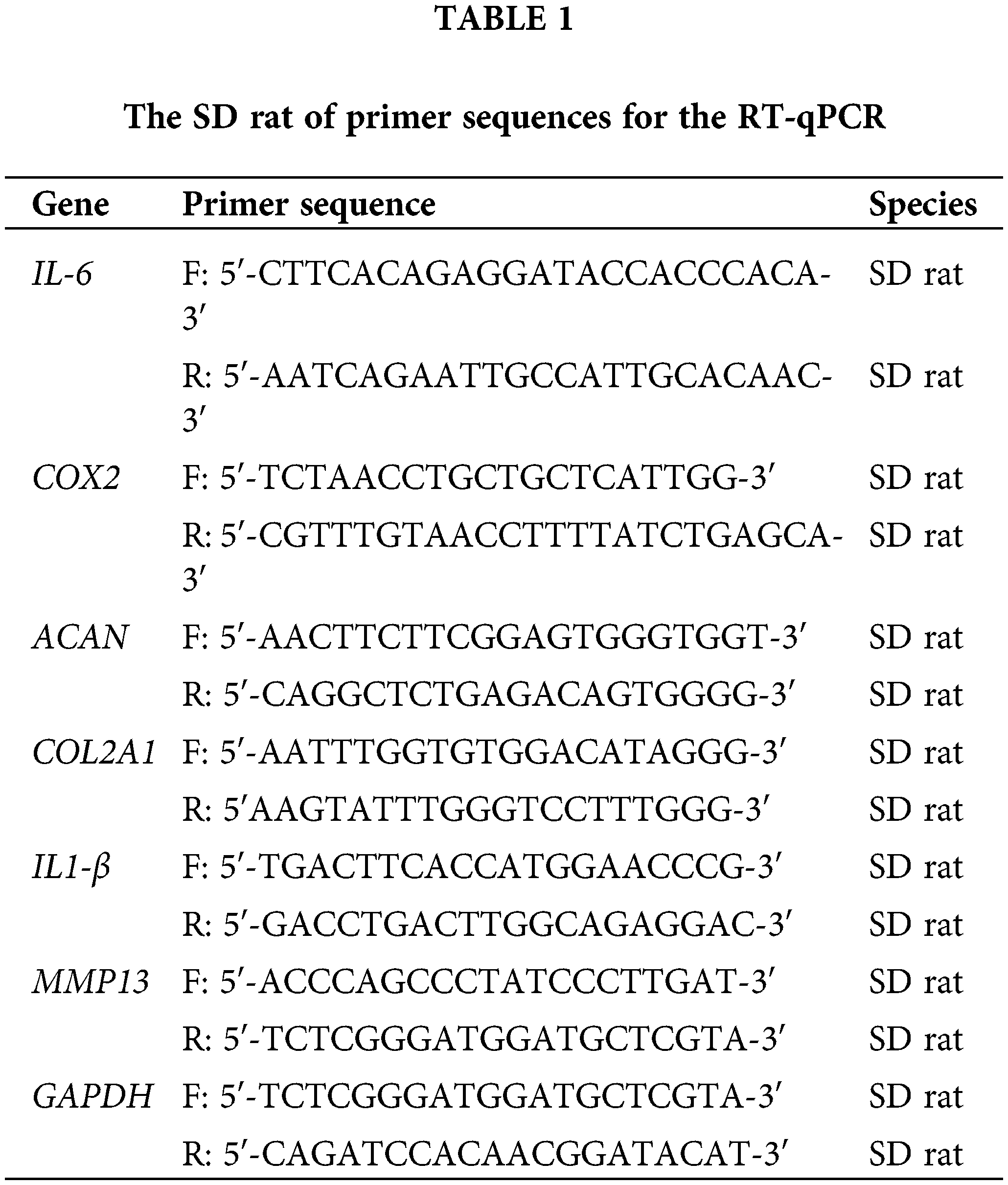
The chondrocytes were cultivated in DMEM/F12 supplemented with 10% FBS in 6-well plates. From the cells in the different treatment groups, total RNA was obtained by RNA extraction kit (Thermo Fisher Scientific, 12183555, Waltham, MA, USA). Following room temperature cell lysis, cDNA synthesis was performed by the RT-First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, K1622, Waltham, MA, USA). The gene expression type II collagen A1 (COL2), interleukin-1β (IL-1β), and cyclooxygenase were detected by qPCR. The internal control was Glyceraldehyde 3-phosphate dehydrogenase (GAPDH). By this, the primer sequences for the RT-qPCR experiment were created (Table 1). Invitrogen Shanghai generated the primers, using the 2−ΔΔCt technique.
The chondrocytes were sown in a 10-cm dish. The cells underwent two 4°C PBS washes following treatment. To extract the whole protein, standard cell lysate and 1% phenylmethylsulfonyl fluoride (Beyotime, ST507, Shanghai, China) were used. The proteins were separated using a 10% gel before being transferred onto a polyvinylidene difluoride membrane. The membrane was then blocked, primary antibodies were added, and the mixture was incubated for an additional night at 4°C: collagen II (Santa Cruz, sc-7763, TEXAS, USA), ACAN (Zen Bio, 381869, Sichuan, China), MMP-13 (Santa Cruz, sc-30073, Shanghai, China), t-IkBα (Zen Bio, 380682, Sichuan, China), p-IkBα (Zen Bio, 530614, Sichuan, China), p-AMPKα (Zen Bio, 310044, Sichuan, China), AMPKα (Zen Bio, 380431, Sichuan, China), p-IRE1α (Zen Bio, 381161, Sichuan, China), IRE1α (Zen Bio, 220399, Sichuan, China), PERK (Zen Bio, 340751, Sichuan, China), p-PERK (Zen Bio, 340846, Sichuan, China), GRP78 (Zen Bio, 380795, Sichuan, China), Sirt1 (Zen Bio, 510656, Sichuan, China), and GAPDH (Zen Bio, 384404, Sichuan, China). After that, the membrane was treated for one hour at 37°C with a secondary antibody (Proteintech, RGAM001, Wuhan, China). Pierce™ ECL Western Blotting Substrate (Thermo Fisher Scientific, Waltham, MA, USA) was exposed to a developer to produce the images. Using Quantum One Software (Bio-Rad, Hercules, CA, USA), the images were examined.
For RNA sequencing analysis, total RNA was isolated from samples belonging to different experimental groups and then enriched with mRNA [12,13]. Using RNA-sequencing (RNA-seq), the changes in the mRNA expression profiles of cells from various treatment groups were evaluated. The BGISEQ-500 platform from BGI Co., Ltd. (Shenzhen, China) was used for sequencing, and triple testing was performed on each sample. Differentially expressed genes (DEGs) in cartilage were found through the analysis of the sequencing data using the R programming language. Genes displaying notable differential expression were identified by DEG analysis.
To create an OA model, an anterior cruciate ligament (ACL) transection was carried out in SD rats, as previously mentioned [14]. The SD rat’s thigh was shaved, and the ACL was then severed following a little incision made in the medial patellar ligament to reveal it. Rapid suture closure of the knee joint cavity was then carried out. To prevent infection and lower animal mortality, penicillin was administered every day for the first three days following surgery. The rats in this study were divided into four groups at random, each with six rats: a group for osteoarthritis (OA), a group for Dexamethasone (DEX), a group for therapy with CA, and a control group (healthy). After four weeks, a daily intra-articular injection containing 1 mM CA was given to the knee for one week, and then injections were given every three days for the next week. The knee joint of the OA group was injected with saline. Cartilage samples were collected for microscopic examination four weeks after the start of CA treatment to evaluate the treatment’s effectiveness. Samples of articular cartilage were gathered for histological examination.
Specimens of articular cartilage were removed from each group and formaldehyde was fixed and decalcified to prepare paraffin slices. The cartilage specimens were all stained with Safranin-O/green (Solarbio, G1371, Beijing, China) and hematoxylin-eosin (Solarbio, G1120, Beijing, China). According to the previously mentioned histological ratings, pathological alterations in the cartilage from each group were assessed [10,15]. The statistical analysis findings are expressed according to each group’s average score. Using an optical microscope (Olympus, IX73, Japan), the microscopic characteristics of the cartilage surface from each group of specimens were examined.
Software called SPSS 22.0 (Statistical Package for the Social Sciences) was used to examine the data. The findings, derived from a representative experiment carried out in triplicate, were displayed as the mean ± standard deviation. A one-way analysis of variance was used for group comparisons, with a significance level of p < 0.05.
Cytotoxicity of cholic acid to chondrocytes
To activate the isolated chondrocytes in this investigation, several concentrations of CA ranging from 1–100 μM were used. Using the CCK-8 assay, the chondrocytes viability effect of CA treatment was evaluated. Based on optical density readings, the results showed that chondrocytes exposed over an extended period to a medium containing 0–10 μM CA did not exhibit any discernible toxicity. In contrast, the addition of 10 μM CA to the control group (DMSO, *p < 0.05) slightly increased cell viability and proliferation (Fig. 1a). Moreover, a dose-dependent decrease of cell viability was seen at CA doses of 20, 50, and 100 μM (*p < 0.05; Fig. 1a). 20.49 µM was the 50% inhibitory concentration (IC50) of CA (Fig. 1b). Based on the findings, 10 μM CA was selected for additional research.
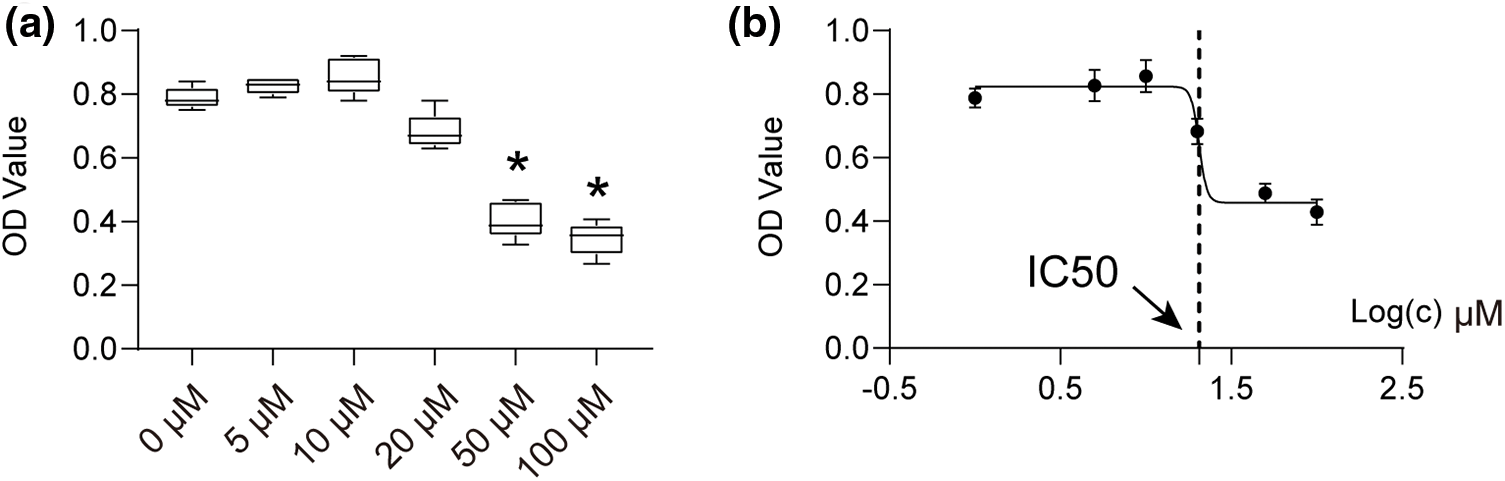
Figure 1: The cell vitality of chondrocytes treated with CA. (a) Effects of various concentrations of CA on the proliferative activity of chondrocytes, n = 3, *p < 0.05. (b) The IC50 converts numerical values to log display, n = 5.
Cholic acid restrained TNF-α-induced chondrocytes inflammatory phenotype
Compared with the control group, the results of the TNF-α treatment indicated a minor rise in the mRNA level of IL-1β, IL-6, COX2, and MMP-13 after 24 h. However, there was no significant change in the gene expression of ACAN and COL2A1 after 24 h (*p < 0.05, Fig. 2a). At 48 h, TNF-α, however, markedly increased the mRNA levels of COX2, MMP-13, IL-1β, IL-6, and ACAN while considerably decreasing the mRNA levels of COL2A1 and ACAN (*p < 0.05, Fig. 2b). On the other hand, 48-h CA therapy reversed the effects of TNF-α. As illustrated in Fig. 2b, the combined treatment of TNF-α and CA led to a decrease in the mRNA levels of IL-1β, IL-6, COX2, and MMP-13, and an increase in the expression levels of COL2A1 and ACAN relative to the TNF-α group (#p < 0.05). After 48 h, the results of the gene expression analysis and the western blotting analysis were comparable. The downregulation of COL2A1 and ACAN, as well as the overexpression of MMP-13, caused by TNF-α, were successfully stopped by treatment with CA, returning gene expression levels to those seen in the control group (Figs. 2c, 2d). Moreover, the DEX reversed the TNF-α-induced elevation of IL-1β, IL-6, and MMP-13 gene and protein production, but it was unable to reverse the TNF-α-induced downregulation of COL2A and ACAN (Figs. 2e, 2f). On the other hand, 10 μM CA treatment effectively reversed the TNF-α-induced production of IL-1β and MMP-13 proteins and restored the downregulation of COL2A and ACAN expression (Figs. 2e, 2f). To protect chondrocytes, CA has anti-inflammatory qualities and encourages the formation of cartilage extracellular matrix (ECM).
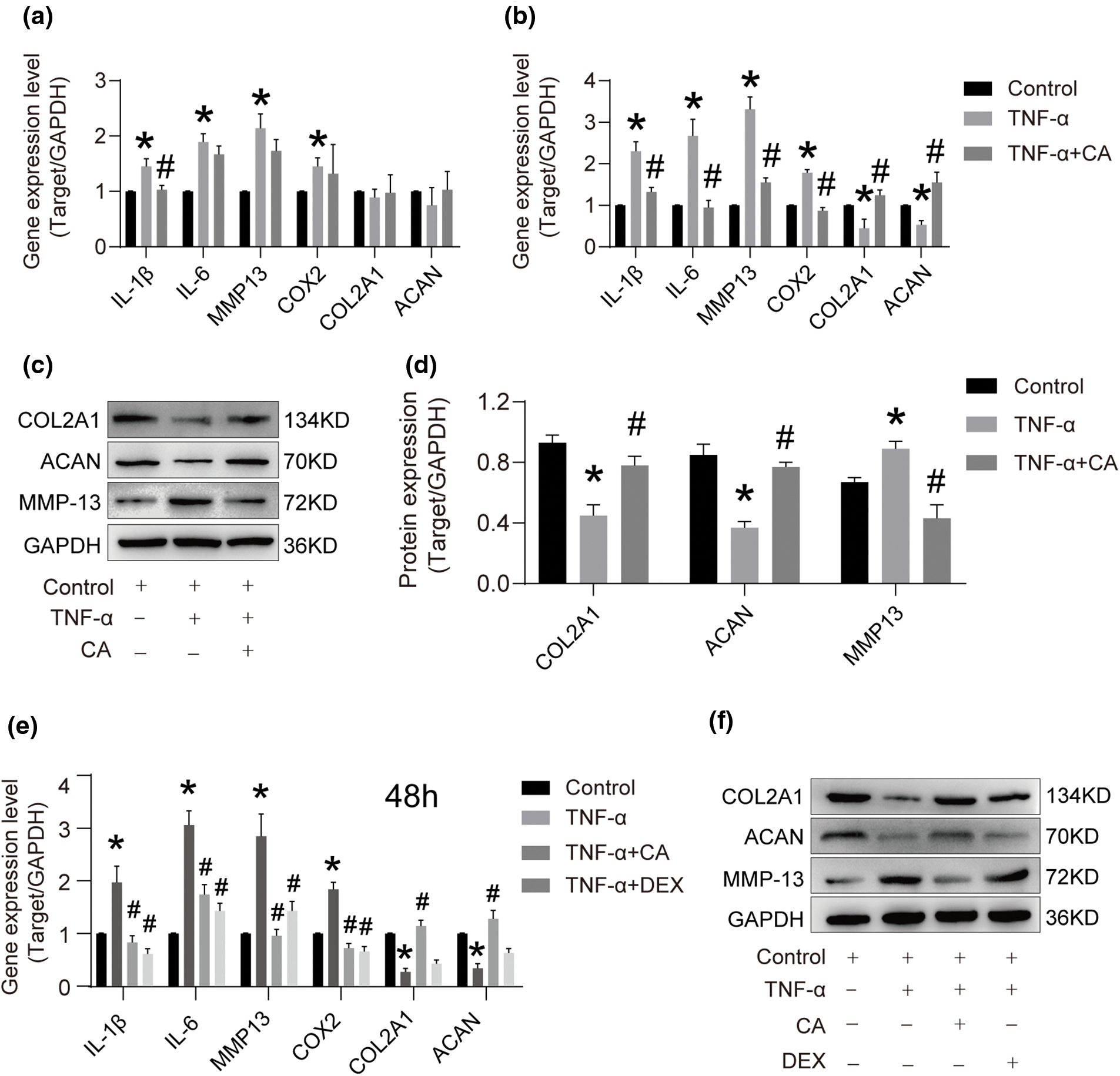
Figure 2: The effect of CA on the gene and protein level. (a) After being stimulated with TNF-α, chondrocytes were treated with or without 10 μM CA for 24 h (n = 3). RT-qPCR was used to determine the mRNA expression levels of IL-1β, IL-6, MMP13, COX2, COL2A1, and ACAN (n = 3). (b) TNF-α activated chondrocytes, which were subsequently subjected to either 10 μM CA or no treatment for 48 h (n = 3). RT-qPCR was used to determine the mRNA expression levels of IL-1β, IL-6, MMP13, COX2, COL2A1, and ACAN (n = 3). (c) Western blotting for cartilage matrix markers, COL2A1, ACAN, and MMP13 throughout a 48-h period, n = 3. (d) Semi-quantitative employing Image J program and cartilage matrix content (IOD) intensity. (e) Chondrocytes were stimulated with TNF-α and then treated with or without 10 μM CA and DEX for 48 h. RT-qPCR was used to determine the mRNA expression levels of IL-1β, IL-6, MMP13, COX2, COL2A1, and ACAN (n = 3). (f) Western blotting for markers of cartilage matrix, COL2A1, ACAN, and MMP13 with different treatments for 48 h, n = 3. *p < 0.05 is considered a significant difference from the control group; #p < 0.05 is considered a significant difference from TNF-α group. ACAN, aggrecan; CA, cholic acid; COX2, cyclooxygenase 2; COL2A1, type II collagen A1; DEX, dexamethasone as positive control; IL-1β, interleukin 1β; MMP1, matrix metalloprotein 13; RT-qPCR, real time-quantitative polymerase chain reaction; TNF-α, tumor necrosis factor α.
The mRNA-sequence analysis results
Comparing the control and TNF-α groups (upregulated expression: 191, downregulated expression: 91) and the TNF-α and TNF-α plus CA groups (upregulated expression: 79, downregulated expression: 123), differentially expressed genes (DEGs) were found (Fig. 3a). p-value (−log10) > 3 and fold change (FC: log2) > 1 were used to establish significance. 112 frequent DEGs were found for additional research after Venn diagram analysis (Fig. 3b). Functional annotations from Gene Ontology were applied to the shared 112 DEGs (Fig. 3c) [16]. The majority of the genes that were found to be involved in biological processes showed enrichment in binding and catalytic activity, molecular function regulation, cell-cell part and organelle, membrane of the component, cellular process, and biological regulation for biological process (cellular component). The 112 differentially expressed genes (DEGs) were also subjected to KEGG signaling pathway enrichment analysis, which showed a notable enrichment in functions linked to IL-17, TNF, chemokine, cytokine-cytokine receptor, toll-like receptor, nucleotide oligomerization domain (NOD)-like pathways, and other similar pathways (Fig. 3d).
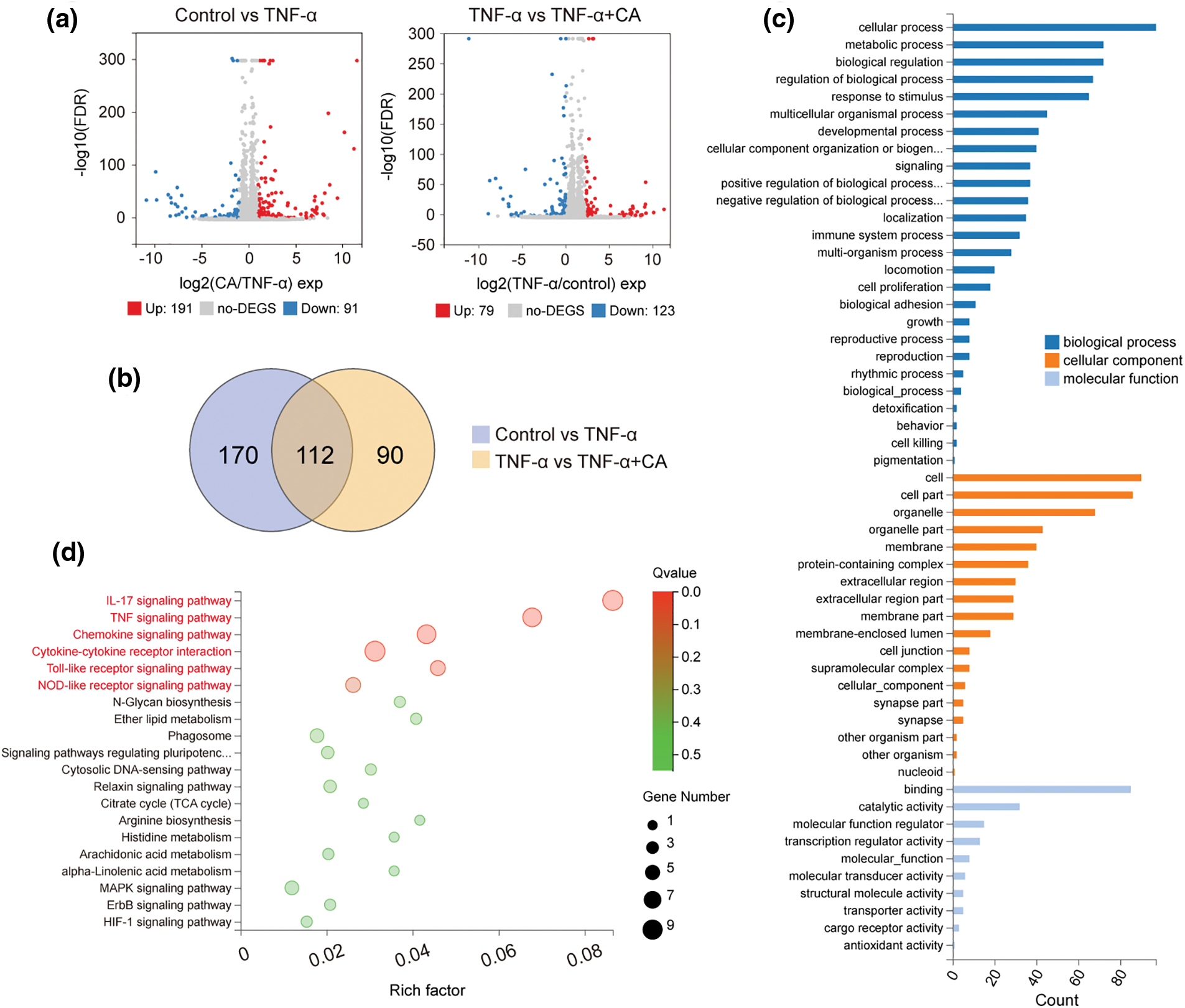
Figure 3: Gene expression profiles in TNF-conditioned cultures, either with or without CA. (a) The values of FC (fold change: log2) > 1 and p (−log10) > 3 were deemed significant in the volcano map of DEGs in control vs. TNF-α (up-regulation: 191 and down-regulation: 91); and TNF-α vs. TNF-α plus CA (up-regulation: 79 and down-regulation: 123). (b) The above DEGs’ Venn interaction reveals 112 common DEGs. (c) GO enrichment of these often chosen DEGs according to three criteria: molecular function, cellular component, and biological process. (d) A comprehensive evaluation of the KEGG pathway enrichment analysis; a higher degree of enrichment is indicated by a bigger q value (−log10). The six most abundant paths were indicated in red: IL-17, TNF, Chemokine, cytokine-cytokine receptor, Toll-like receptor, and NOD-like receptor signaling pathways. Tumor necrosis factor-alpha, or TNF-α; CA, cholic acid; DEG, differentially expressed genes; GO, Gene Ontology; IL-17, interleukin 17; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Analysis and expression of DEG in the top six enriched pathways
Moreover, extra analyses of differential gene expression (DEG) were carried out using the previously determined top six enriched pathways as a basis. The enriched DEGs in the TNF-α vs. control group and the TNF-α + CA vs. TNF-α group were among the top six pathways whose fold changes in gene expression were computed (see Fig. 4a). The enhanced DEGs were linked to inflammatory reactions; these included CXCL6, CCL20, MMP3, CXCL3, CXCL11, CCL5, CXCL10, MMP9, MMP13, and CXCL2. It was discovered that CA therapy reversed their gene expression during TNF-α stimulation (see Fig. 4b). According to this research, CA may have an anti-inflammatory effect on chondrocytes. Furthermore, additional DEG analyses were conducted using the top six enriched pathways indicated earlier. The TNF-α vs. control group and the TNF-α + CA vs. TNF-α group’s fold changes in the gene expression of the top six pathways involving the enriched DEGs were computed. The DEGs that were enriched, which included CXCL6, CCL20, MMP3, CXCL3, CXCL11, CCL5, CXCL10, MMP9, MMP13, and CXCL2, were all associated with inflammatory reactions. The administration with CA reversed their gene expression when stimulated by TNF-α (Fig. 4b). This result implies that CA has anti-inflammatory characteristics in chondrocytes.
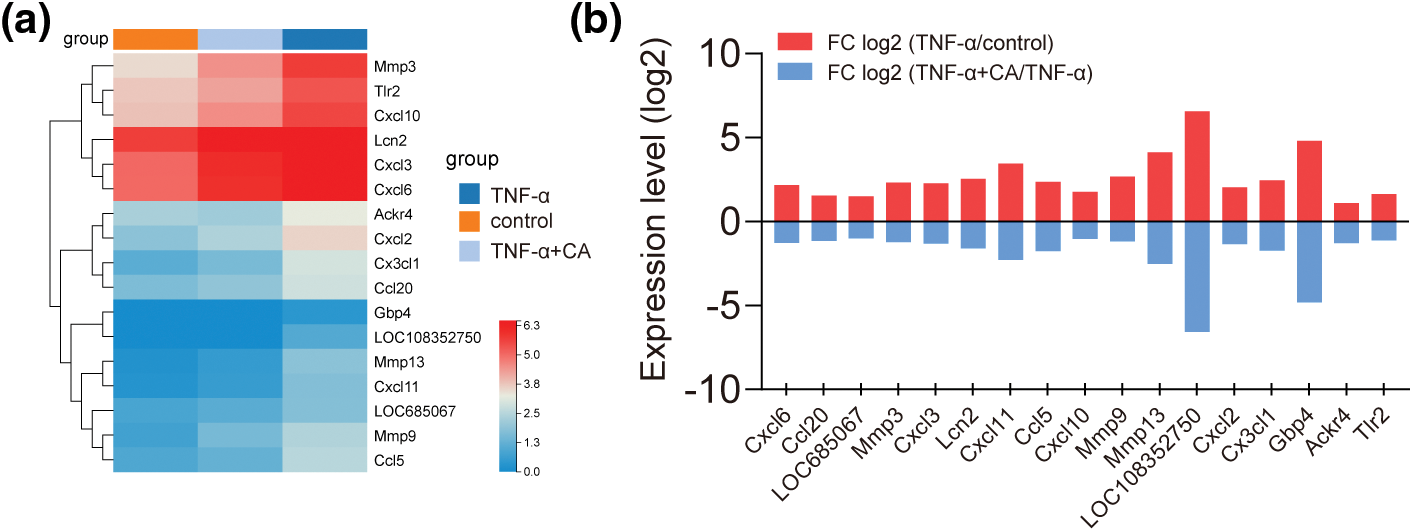
Figure 4: Analysis of top six enriched pathways. (a) Heatmap (b) Expression fold changes (log2) of differentially expressed genes in selected top six pathways indicate inflammation suppression with cholic acid treatment.
Cholic acid restores TNF-α-induced nuclear factor-κB, ER, NFκB, and AMPK signal pathway
According to immunostaining analysis, p65 was mostly found in the cytoplasm in the control group, but after TNF-α treatment, p65 moved to the nucleus (Fig. 5a). The application of CA considerably reduced the nuclear translocation of p65 caused by TNF-α. Furthermore, there was an increase in the ratio of phosphorylated-IκBα/total-IκBα (p/t-IκBα; *p < 0.05; Figs. 5b, 5c) after TNF-α treatment. Supplementing with CA restored the rise in p/t-IκBα caused by TNF-α (*p < 0.05; Figs. 5b, 5c). Endoplasmic reticulum (ER) stress modulators (PERK, IRE1α, and GRP78) were used to study the effect of CA on cell viability. The phosphorylation of IRE1α (p/t-IRE1α) and PERK (p/t-PERK) as well as the protein production of GRP78 in chondrocytes stimulated by TNF-α showed a substantial increase when compared to the control group. On the other hand, the effects of TNF-α treatment were lessened when CA was also administered (*p < 0.05; Figs. 5d, 5f, 5h, 5i). In addition, the TNF-α group showed a significant decrease in SIRT1 protein expression and AMPK-α phosphorylation (p/t-AMPKα) when compared to the control group. Treatment with CA reduced the increase in AMPKα phosphorylation and SIRT1 protein expression brought on by TNF-α stimulation (*p < 0.05; Figs. 5e, 5g, 5j).
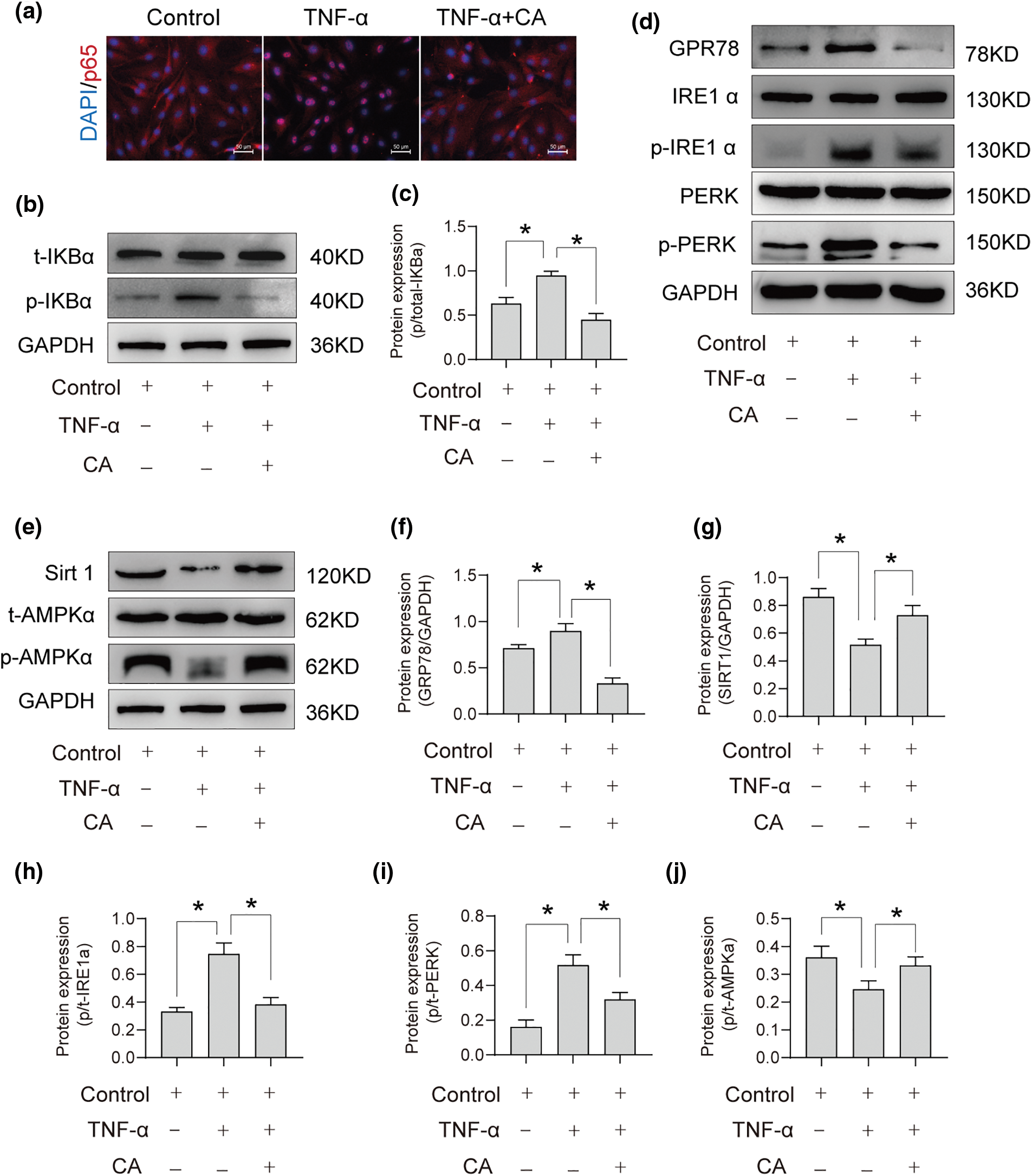
Figure 5: The participation of the CA signaling pathway in anti-inflammation. CA treatment under TNF-α conditions involves different pathways. (a) Immunofluorescence staining was used to verify that CA inhibits p65 nuclear translocation in cells induced by TNF-α, scale bar = 200 μm, n = 3. (b and c) Protein expression profile of phosphorylated IκBα cultured under conditions of TNF-α with or without CA, n = 3. (d) Protein expression of CPR78, IREα, p-IREα, PERK, p-PERK response cultured under conditions of TNF-α with or without CA, n = 3. (e) Phosphorylated AMPKα protein expression profile and SIRT1 expression in CA, cultured under TNF-α conditions, n = 3. (f–j) Statistical analysis of GRP78/GAPDH, SIRT1/GAPDH, p/t-IRE1α, p/t-PERK, p/t-AMPKα, and obtained from gray semi-quantification. *p < 0.05 represents significant differences.
Cholic acid prevents articular cartilage degradation in osteoarthritis model of rats
Knee joint specimens from SD rats in the model and therapy groups were examined morphologically and pathologically. The osteoarthritis (OA) group’s articular cartilage showed irregularities, whereas the group receiving CA treatment showed a considerably higher cartilage structure score than the OA group. Microscopic examination revealed that the OA group had uneven shape and less articular cartilage thickness than the control group of healthy rats (Fig. 6). In contrast to the OA group, the CA group’s articular cartilage thickness increased significantly and their shape became more regular as a result of the additional CA therapy. In the CA group, smooth, clear tissue filled the cartilage defect. Analyzing the pathological alterations in rat knee joints showed that there were notable differences between the OA and healthy groups, with the OA group showing lower cartilage matrix content (IOD) and notable pathological abnormalities (*p < 0.05). Additionally, compared to the OA group, there was a significant rise in both measures with additional CA therapy (#p < 0.05). The results of this investigation show that the degeneration of articular cartilage in its native environment can be successfully suppressed by treatment with CA.
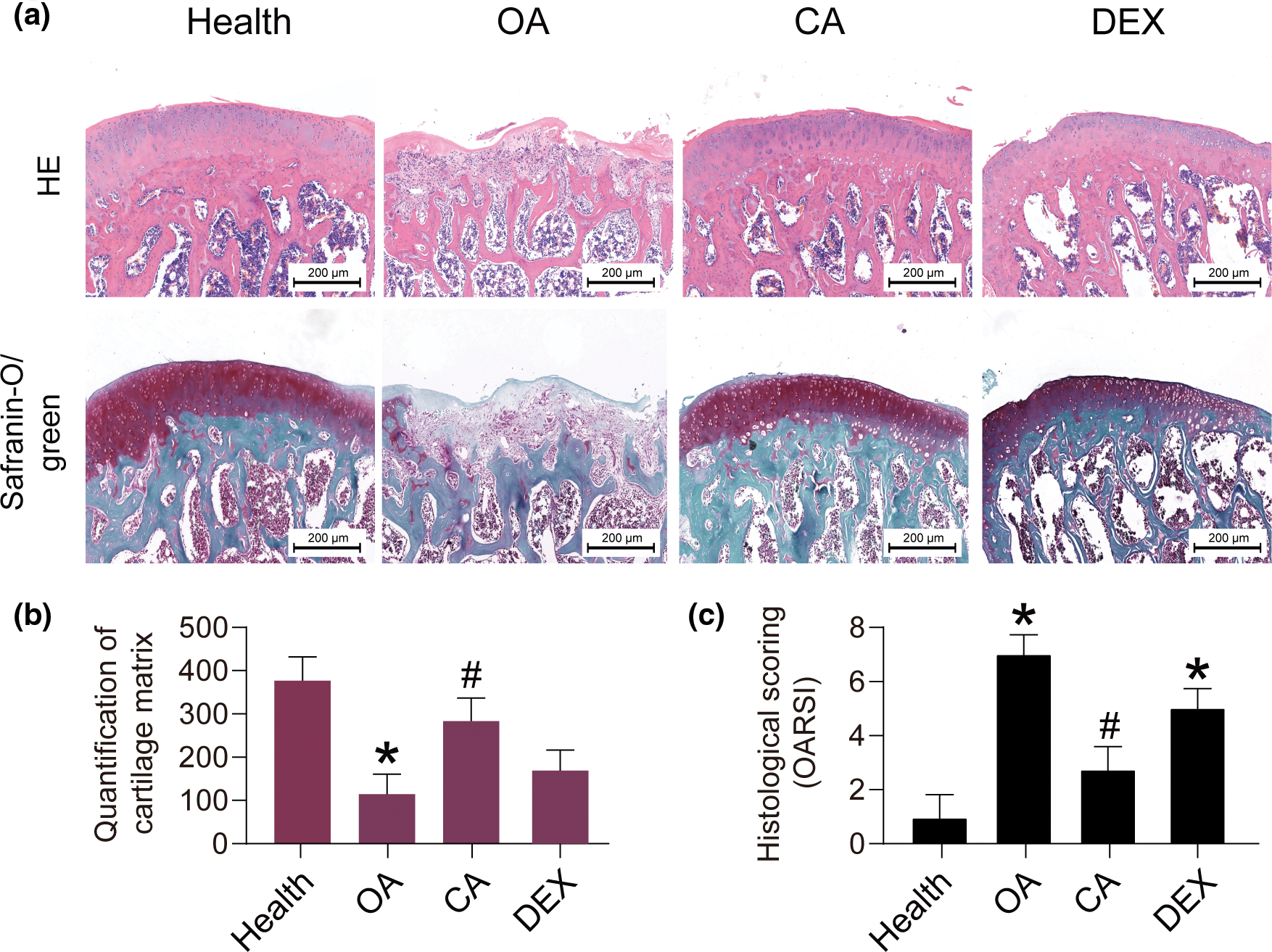
Figure 6: The organizational evaluation of CA treatment in SD rat model. (a) Staining analysis of extracellular matrix components of tissues (H&E and Safranin-O/green), scale bar = 200 μm, n = 3. (b and c) The semi-quantification of Safranin-O/green staining and histological scoring by osteoarthritis research society international (OARSI), *p < 0.05 represents significant differences compare to health, and #p < 0.05 represents significant differences compared to OA. DEX, dexamethasone as positive control. p/t-AMPKα, phosphorylated/total AMP-activated protein kinase; CA, cholic acid; H&E, Hematoxylin and Eosin; OA, osteoarthritis.
OA is a chronic inflammatory disease mainly characterized by joint pain and cartilage dysfunction [17]. Excessive production and accumulation of inflammatory cytokines in the knee joint contribute to OA pathogenesis owing to the induction of the production of matrix-degrading enzymes to degrade the ECM [18–20]. Thus, this study explored the anti-inflammatory effect of CA in vitro and in vivo.
The production of TNF-α increased excessively in patients with OA, which causes apoptosis of chondrocytes, and the release of catabolic enzymes. Moreover, TNF-α causes apoptosis and aging of chondrocytes, which blocks cartilage regeneration and repair in OA. The results showed that CA inhibits the inflammation response in rat chondrocytes. CA significantly suppressed the upregulated expression of IL-1β, IL-6, COX-2, and MMP-13, and downregulated expression of COL2A1 and ACAN in chondrocytes when treated with TNF-α. These results suggest that CA could ameliorate TNF-α-induced OA in vitro, which was confirmed by high-throughput sequencing. The enriched DEGs in TNF-α vs. control and TNF-α plus CA vs. TNF-α obtained from RNA-seq, namely, CXCL6, CCL20, MMP3, CXCL3, CXCL11, CCL5, CXCL10, MMP9, MMP13, and CXCL2 were all involved in inflammatory responses. Expression of pro-inflammatory genes induced by TNF-α was reversed by CA. The unique combination of matrix molecules, including ACAN and COL2A1 provides cartilage from damage due to load [21–23]. The integrity of ECM is the basis for protecting chondrocytes from stimuli. Studies have found that other members of bile acids protect the matrix of chondrocytes from MMP degradation and suppress bone destruction in an OA rabbit model [6]. MMP-13 is the main enzyme for cartilage ECM degradation, which has been identified as a key mechanism of OA pathogenesis [24–26]. Studies have confirmed that MMP-13 drug inhibitors inhibit articular cartilage degradation and increase in the synthesis of COL2 and ACAN.
The NF-κB signaling pathway is a classic signaling pathway related to cellular inflammation [27]. Activation of that pathway occurs through shifting of the p65 and p50 dimethyl components to the nucleus and their binding to specific target sequences, thus inducing the expression of inflammatory genes [28,29]. NF-κB signaling is activated by OA stimulation, including inflammatory and mechanical load [30]. It has been validated that the importance of NF-κB signaling in OA disease. Inhibiting NF-κB disturbed the expression of TNF-α-induced catabolic genes in chondrocytes [31]. Knockout of p65 with small interference RNA results in a decrease in inflammatory factor concentrations in synovial fluid, leading to reduced cartilage damage caused by injury in an animal model of OA [32]. This study reveals the potential molecular mechanism of CA treatment for OA.
Both PERK and IRE1 are the primary signaling molecules produced in the response to ER stress [33]. A reduction in the PERK activity was observed in the chondrocytes, accompanied by a decrease in COL2A1 expression, leading to the degradation of the ECM [34]. Therefore, PERK can be regarded as a therapeutic target for OA treatment [34]. Chronic PERK activity caused by severe or persistent ER stress activates downstream transcription factor C/EBP homologous protein (CHOP) and promotes apoptosis signaling [35]. The molecular chaperone GRP78 plays a vital role in the PERK cavity domain that maintains kinase activity in an inactive state [33].
Sirtuin 1 (SIRT1) and AMP-activated protein kinase (AMPK) are proven therapeutic targets for OA [36]. SIRT1 expression was downregulated in articular chondrocytes of patients with OA compared with that in healthy people [16]. Various studies have shown that SIRT1 activation reduced inflammatory cytokines and degradation enzymes in chondrocytes through the NF-κB signaling pathway. SIRT1 up-regulated expression inhibited TNF-α-induced activation of COX-2, and MMP-13 through the NF-κB pathway [37]. Moreover, SIRT1 overexpression reduced IL-1β-induced upregulated expression of MMP-13 and p65 [38]. SIRT1 inhibited NF-κB degradation by activating AMPK. The SIRT1/AMPK signaling pathway in chondrocytes was abnormally down-regulated in OA [38]. Studies have shown that the inhibition of SIRT1/AMPK promoted the pro-apoptotic response and oxidative stress [36]. In addition, abnormalities in SIRT1/AMPK signaling promoted NF-κB to upregulate the expression of MMP-13, IL-8, and COX-2 in chondrocytes. Moreover, SIRT1 enhanced the anti-stress ability of chondrocytes to promote cell survival, indicating that SIRT1 prevents OA by inhibiting the apoptosis of chondrocytes [36].
This study has several limitations. First, we did not validate the pharmacological effects of CA on human primary cells or human-derived cell line C28. Second, the signaling pathway of CA has not been validated at the animal level. Third, we did not take into account the bioavailability of orally administered CA.
In summary, CA has a therapeutic effect in the OA rat model. CA regulates ER stress and exhibits anti-inflammatory effects on rat chondrocytes by regulating NF-κB/PERK/SIRT1 signaling, which may be used to guide the OA treatment.
Acknowledgement: None.
Funding Statement: This work was financially supported by the Open Fund for Hubei Provincial Key Laboratory of Occurrence and Intervention of Rheumatic Diseases (PT022311).
Author Contributions: The authors confirm their contribution to the paper as follows: study conception and design: Jiaoe Sheng, Yufang Dai; data collection: Zumin Yi, Qguoqing Yan; analysis and interpretation of results: Sanshan He, Qingchao Wu, Xia Huang; draft manuscript preparation: Jiaoe Sheng, Linchong Su. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The datasets used or analyzed during the current study are available from the first author upon reasonable request.
Ethics Approval: Animal experiments were approved by the Ethics Committee of the Minda Hospital of Hubei Minzu University (Hubei University for Nationalities Biomedical Research Ethics Approval Document Number: (2022) 32).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Tong L, Yu H, Huang X, Shen J, Xiao G, Chen L, et al. Current understanding of osteoarthritis pathogenesis and relevant new approaches. Bone Res. 2022;10(1):60. doi:10.1038/s41413-022-00226-9. [Google Scholar] [PubMed] [CrossRef]
2. He Y, Li Z, Alexander PG, Ocasio-Nieves BD, Yocum L, Lin H, et al. Pathogenesis of osteoarthritis: risk factors, regulatory pathways in chondrocytes, and experimental models. Biology. 2020;9(8):194. doi:10.3390/biology9080194. [Google Scholar] [PubMed] [CrossRef]
3. Knights AJ, Redding SJ, Maerz T. Inflammation in osteoarthritis: the latest progress and ongoing challenges. Curr Opin Rheumatol. 2023;35(2):128–34. doi:10.1097/BOR.0000000000000923. [Google Scholar] [PubMed] [CrossRef]
4. Wang T, He C. Pro-inflammatory cytokines: the link between obesity and osteoarthritis. Cytokine Growth Factor Rev. 2018;44:38–50. doi:10.1016/j.cytogfr.2018.10.002. [Google Scholar] [PubMed] [CrossRef]
5. Matta C, Fellows CR, Quasnichka H, Williams A, Jeremiasse B, Allaway D, et al. Clusterin secretion is attenuated by the proinflammatory cytokines interleukin-1β and tumor necrosis factor-α in models of cartilage degradation. J Orthop Res. 2021;39(5):1017–29. doi:10.1002/jor.v39.5. [Google Scholar] [CrossRef]
6. Yan ZW, Dong J, Qin CH, Zhao CY, Miao LY, He CY. Therapeutic effect of chenodeoxycholic acid in an experimental rabbit model of osteoarthritis. Mediat Inflamm. 2015;2015(4):780149. [Google Scholar]
7. Moon SJ, Jeong JH, Jhun JY, Yang EJ, Min JK, Choi JY, et al. Ursodeoxycholic Acid ameliorates pain severity and cartilage degeneration in monosodium iodoacetate-induced osteoarthritis in rats. Immune Netw. 2014;14(1):45–53. doi:10.4110/in.2014.14.1.45. [Google Scholar] [PubMed] [CrossRef]
8. Xue J, Song W, Yao H, Hou S, Liu S, Wang Y, et al. Effects of cholic acid modified glucosamine on chondrogenic differentiation; 2016. doi:10.1039/c6ra09547j. [Google Scholar] [CrossRef]
9. Hayer S, Halilbasic E, Niederreiter B, Willburger M, Blüml S, Mandl P, et al. Effects of cholic acid and its derivatives in experimental arthritis. Ann Rheum Dis. 2013;72(Suppl 3):A818. [Google Scholar]
10. Xu K, Sha Y, Wang S, Chi Q, Liu Y, Wang C, et al. Effects of Bakuchiol on chondrocyte proliferation via the PI3K-Akt and ERK1/2 pathways mediated by the estrogen receptor for promotion of the regeneration of knee articular cartilage defects. Cell Prolif. 2019;52(5):e12666. doi:10.1111/cpr.v52.5. [Google Scholar] [CrossRef]
11. Xu K, Pan X, Sun Y, Xu W, Njunge L, Yang L. Psoralen activates cartilaginous cellular functions of rat chondrocytes in vitro. Pharm Biol. 2015;53(7):1010–5. doi:10.3109/13880209.2014.952835. [Google Scholar] [PubMed] [CrossRef]
12. Pan L, Feng F, Wu J, Fan S, Han J, Wang S, et al. Demethylzeylasteral targets lactate by inhibiting histone lactylation to suppress the tumorigenicity of liver cancer stem cells. Pharmacol Res. 2022;181(10127):106270. [Google Scholar] [PubMed]
13. Xu H, Li L, Qu L, Tu J, Sun X, Liu X, et al. Atractylenolide-1 affects glycolysis/gluconeogenesis by downregulating the expression of TPI1 and GPI to inhibit the proliferation and invasion of human triple-negative breast cancer cells. Phytother Res. 2023;37(3):820–33. doi:10.1002/ptr.v37.3. [Google Scholar] [CrossRef]
14. Wang C, Gao Y, Zhang Z, Chen C, Chi Q, Xu K, et al. Ursolic acid protects chondrocytes, exhibits anti-inflammatory properties via regulation of the NF-κB/NLRP3 inflammasome pathway and ameliorates osteoarthritis. Biomed Pharmacother. 2020;130(1):110568. [Google Scholar] [PubMed]
15. Wang C, Al-Ani MK, Sha Y, Chi Q, Dong N, Yang L, et al. Psoralen protects chondrocytes, exhibits anti-inflammatory effects on synoviocytes, and attenuates monosodium iodoacetate-induced osteoarthritis. Int J Biol Sci. 2019;15(1):229–38. doi:10.7150/ijbs.28830. [Google Scholar] [PubMed] [CrossRef]
16. Oh H, Kwak JS, Yang S, Gong MK, Kim JH, Rhee J, et al. Reciprocal regulation by hypoxia-inducible factor-2α and the NAMPT-NAD+-SIRT axis in articular chondrocytes is involved in osteoarthritis. Osteoarthr Cartilage. 2015;23(12):2288–96. doi:10.1016/j.joca.2015.07.009. [Google Scholar] [PubMed] [CrossRef]
17. Zheng L, Zhang Z, Sheng P, Mobasheri A. The role of metabolism in chondrocyte dysfunction and the progression of osteoarthritis. Ageing Res Rev. 2021;66:101249. doi:10.1016/j.arr.2020.101249. [Google Scholar] [PubMed] [CrossRef]
18. Floramo JS, Molchanov V, Liu H, Liu Y, Craig SEL, Yang T. An integrated view of stressors as causative agents in OA pathogenesis. Biomolecules. 2023;13(5):721. doi:10.3390/biom13050721. [Google Scholar] [PubMed] [CrossRef]
19. Motta F, Barone E, Sica A, Selmi C. Inflammaging and osteoarthritis. Clin Rev Allergy Immunol. 2023;64(2):222–38. [Google Scholar] [PubMed]
20. Gilbert SJ, Bonnet CS, Blain EJ. Mechanical cues: bidirectional reciprocity in the extracellular matrix drives mechano-signalling in articular cartilage. Int J Mol Sci. 2021;22(4):13595. [Google Scholar] [PubMed]
21. He L, He T, Xing J, Zhou Q, Fan L, Liu C, et al. Bone marrow mesenchymal stem cell-derived exosomes protect cartilage damage and relieve knee osteoarthritis pain in a rat model of osteoarthritis. Stem Cell Res Ther. 2020;11(1):276. doi:10.1186/s13287-020-01781-w. [Google Scholar] [PubMed] [CrossRef]
22. Liu Z, Wang K, Jiang C, Chen Y, Liu F, Xie M, et al. Morusin alleviates aortic valve calcification by inhibiting valve interstitial cell senescence through Ccnd1/Trim25/Nrf2 axis. Adv Sci. 2024. doi:10.1002/advs.202307319:e2307319. [Google Scholar] [PubMed] [CrossRef]
23. Wang C, Wang S, Wang Z, Han J, Jiang N, Qu L, et al. Andrographolide regulates H3 histone lactylation by interfering with p300 to alleviate aortic valve calcification. Br J Pharmacol. 2024. doi:10.1111/bph.16332. [Google Scholar] [PubMed] [CrossRef]
24. Grillet B, Pereira RVS, van Damme J, Abu El-Asrar A, Proost P, Opdenakker G. Matrix metalloproteinases in arthritis: towards precision medicine. Nat Rev Rheumatol. 2023;19(6):363–77. doi:10.1038/s41584-023-00966-w. [Google Scholar] [PubMed] [CrossRef]
25. Hu Q, Ecker M. Overview of MMP-13 as a promising target for the treatment of osteoarthritis. Int J Mol Sci. 2021;22(4):1742. doi:10.3390/ijms22041742. [Google Scholar] [PubMed] [CrossRef]
26. Wilkinson DJ, Falconer AMD, Wright HL, Lin H, Yamamoto K, Cheung K, et al. Matrix metalloproteinase-13 is fully activated by neutrophil elastase and inactivates its serpin inhibitor, alpha-1 antitrypsin: implications for osteoarthritis. Febs J. 2022;289(1):121–39. doi:10.1111/febs.v289.1. [Google Scholar] [CrossRef]
27. Kunnumakkara AB, Shabnam B, Girisa S, Harsha C, Banik K, Devi TB, et al. Inflammation, NF-κB, and chronic diseases: how are they linked? Crit Rev Immunol. 2020;40(1):1–39. doi:10.1615/CritRevImmunol.v40.i1. [Google Scholar] [CrossRef]
28. Ilchovska DD, Barrow DM. An overview of the NF-kB mechanism of pathophysiology in rheumatoid arthritis, investigation of the NF-kB ligand RANKL and related nutritional interventions. Autoimmun Rev. 2021;20(2):102741. doi:10.1016/j.autrev.2020.102741. [Google Scholar] [PubMed] [CrossRef]
29. Jimi E, Fei H, Nakatomi C. NF-κB signaling regulates physiological and pathological chondrogenesis. Int J Mol Sci. 2019;20(24):6275. doi:10.3390/ijms20246275. [Google Scholar] [PubMed] [CrossRef]
30. Choi MC, Jo J, Park J, Kang HK, Park Y. NF-κB signaling pathways in osteoarthritic cartilage destruction. Cells. 2019;8(7):734. doi:10.3390/cells8070734. [Google Scholar] [PubMed] [CrossRef]
31. Lianxu C, Hongti J, Changlong Y. NF-κBp65-specific siRNA inhibits expression of genes of COX-2, NOS-2 and MMP-9 in rat IL-1β-induced and TNF-α-induced chondrocytes. Osteoarthr Cartilage. 2006;14(4):367–76. doi:10.1016/j.joca.2005.10.009. [Google Scholar] [PubMed] [CrossRef]
32. Yan H, Duan X, Pan H, Holguin N, Rai MF, Akk A, et al. Suppression of NF-κB activity via nanoparticle-based siRNA delivery alters early cartilage responses to injury. Proc Natl Acad Sci U S A. 2016;113(41):E6199–208. [Google Scholar] [PubMed]
33. Kopp MC, Larburu N, Durairaj V, Adams CJ, Ali MMU. UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat Struct Mol Biol. 2019;26(11):1053–62. doi:10.1038/s41594-019-0324-9. [Google Scholar] [PubMed] [CrossRef]
34. Li YH, Tardif G, Hum D, Kapoor M, Fahmi H, Pelletier JP, et al. The unfolded protein response genes in human osteoarthritic chondrocytes: PERK emerges as a potential therapeutic target. Arthritis Res Ther. 2016;18:172. doi:10.1186/s13075-016-1070-6. [Google Scholar] [PubMed] [CrossRef]
35. Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell. 2018;69(2):169–81. doi:10.1016/j.molcel.2017.06.017. [Google Scholar] [PubMed] [CrossRef]
36. Ma CH, Chiua YC, Wu CH, Jou IM, Tu YK, Hung CH, et al. Homocysteine causes dysfunction of chondrocytes and oxidative stress through repression of SIRT1/AMPK pathway: a possible link between hyperhomocysteinemia and osteoarthritis. Redox Biol. 2018;15:504–12. doi:10.1016/j.redox.2018.01.010. [Google Scholar] [PubMed] [CrossRef]
37. Moon MH, Jeong JK, Lee YJ, Seol JW, Jackson CJ, Park SY. SIRT1, a class III histone deacetylase, regulates TNF-α-induced inflammation in human chondrocytes. Osteoarthr Cartilage. 2013;21(3):470–80. doi:10.1016/j.joca.2012.11.017. [Google Scholar] [PubMed] [CrossRef]
38. Matsushita T, Sasaki H, Takayama K, Ishida K, Matsumoto T, Kubo S, et al. The overexpression of SIRT1 inhibited osteoarthritic gene expression changes induced by interleukin-1beta in human chondrocytes. J Orthop Res. 2013;31(4):531–7. doi:10.1002/jor.v31.4. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools