
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

OPA3 overexpression modulates lipid droplet production and sensitizes colorectal cancer cells to bevacizumab treatment
Department of Colorectal and Anal Surgery, Ningbo First Hospital, Ningbo, 315010, China
* Corresponding Author: HONGBIAO WU. Email:
(This article belongs to the Special Issue: Intestinal Epithelial Cells in Health and Disease)
BIOCELL 2024, 48(6), 971-980. https://doi.org/10.32604/biocell.2024.049466
Received 08 January 2024; Accepted 07 March 2024; Issue published 10 June 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: Colorectal cancer (CRC) represents a substantial risk to public health. Bevacizumab, the first US FDA-approved antiangiogenic drug (AAD) for human CRC treatment, faces resistance in patients. The role of lipid metabolism, particularly through OPA3-regulated lipid droplet production, in overcoming this resistance is under investigation. Methods: The protein expression pattern of OPA3 in CRC primary/normal tissues was evaluated by bioinformatics analysis. OPA3-overexpressed SW-480 and HCT-116 cell lines were established, and bevacizumab resistance and OPA3 effects on cell malignancy were examined. OPA3 protein/mRNA expression and lipid droplet-related genes were measured with Western blot and qRT-PCR. OPA3 subcellular localization was detected using immunofluorescence. Proliferation and apoptosis were assessed via colony formation and flow cytometry. Tube formation assays were conducted to assess the angiogenic potential of human umbilical vein endothelial cells (HUVECs). Lipid analysis was used to measure the phosphatidylcholine (PC) and lysophosphatidylcholine (LPC) levels in CRC cells. Results: Bioinformatics analysis revealed that OPA3 was downregulated in CRC. Overexpression of OPA3 inhibited CRC cell proliferation, stimulated apoptosis, and suppressed the angiogenic ability of HUVECs. OPA3 effectively reversed the resistance of CRC cells to bevacizumab and decreased lipid droplet production in CRC cells. Additionally, OPA3 reversed the bevacizumab-induced lipid droplet production in CRC cells, thereby increasing CRC cell sensitivity to bevacizumab treatment. Conclusion: This study suggests that OPA3 modulates lipid metabolism in CRC cells and reduces resistance to bevacizumab in CRC cells. Therefore, OPA3 may be a potential therapeutic target against the AAD resistance in CRC.Graphic Abstract
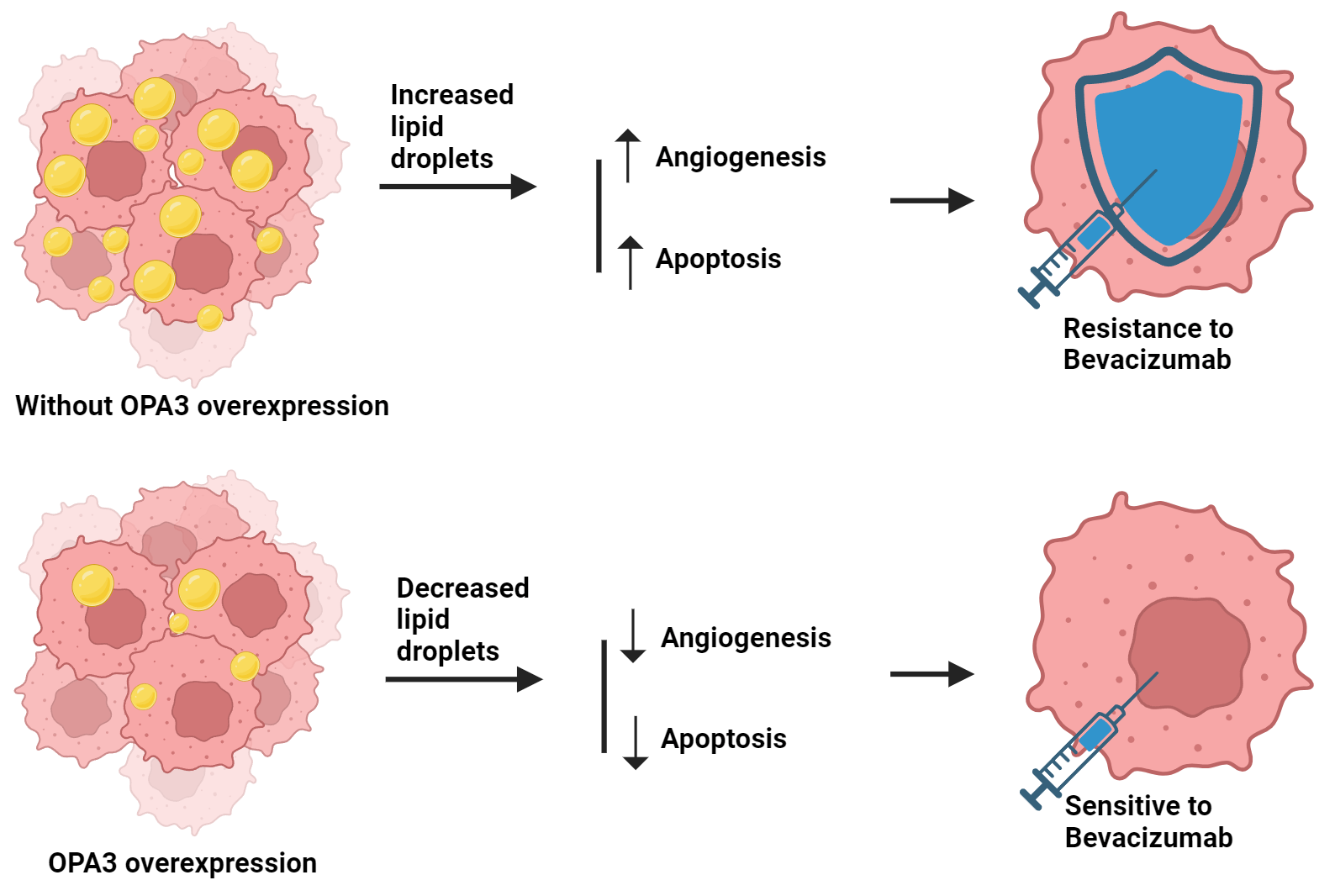
Keywords
Based on the most recent data published by GLOBOCAN (https://gco.iarc.fr/), China experienced an approximate total of 555,477 newly diagnosed cases of colorectal cancer (CRC) and 286,162 fatalities associated with CRC in 2020 [1–3]. Effective strategies to reduce the burden of CRC are urgently needed. Angiogenesis is crucial to tumor growth, since it results in the formation of new blood vessels. Factors that regulate angiogenesis include vascular endothelial growth factor (VEGF), platelet-derived endothelial cell growth factor (PD-ECGF), angiopoietin, thrombin sensitivity element (TSP), and erythropoietin-producing hepatocellular receptor (Eph) [4,5]. Blocking the pathologic angiogenesis is indicated to contribute to CRC therapy with improved survival outcomes [6].
Antiangiogenic drugs (AADs), such as bevacizumab, inhibit VEGF signaling to prevent new blood vessel formation in the tumor microenvironment, ultimately limiting the supply of nutrients and oxygen in tumor growth [7]. As one of the representative AADs and the first anti-angiogenic monoclonal antibodies approved in the world, bevacizumab has been extensively utilized in the treatment of various malignancies [8]. However, high concentrations of bevacizumab have been shown to induce drug resistance in cancer cells, resulting in the activation of VEGF signaling and tolerance to hypoxia in CRC [9,10].
In recent years, cancer has been increasing recognized as metabolic disorders with dysregulated lipid metabolism [11]. Increased lipid uptake, storage, and lipogenesis are seen in different types of cancers, contributing to the advancement of tumors. A lipid droplet (LD) is an organelle that affects the cell physiology in diverse aspects such as energy storage and response to cellular stress [12], and LD accumulation is one of the metabolic characteristics associated with cancer [13]. Lipid metabolism is crucial in cancer resistance, as higher lipid levels can lead to the formation of cancer stem cells [14]. Additionally, AADs are revealed to cause tumor hypoxia, leading to the uptake of free fatty acids and fatty acid oxidation, and targeting the lipid metabolic pathways can potentially reduce AAD resistance and improve the treatment efficacy [15,16]. The outer mitochondrial membrane lipid metabolism regulator OPA3 (OPA3) is implicated in the regulation of lipid metabolism, linking lipid uptake with lipid storage in the liver [17,18]. However, whether OPA3 affects the bevacizumab resistance in CRC remains unknown.
Based on these findings, we hypothesize that OPA3 may reverse the resistance of CRC cell lines to bevacizumab by modulating LD production. Our study results may offer new perspectives on developing therapeutic strategies to combat AAD resistance in CRC.
OPA3 protein expression was analyzed in 97 CRC primary tumor samples and 100 normal tissues using UALCAN (http://ualcan.path.uab.edu/), which utilized data from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) dataset for colorectal cancer.
Human normal colon mucosal epithelial cell line (NCM460) was purchased from Gibco Biological (CM-H203, Shanghai, China). Human CRC cell line SW480 was purchased from ABIOWELL (AW-CCH107, Changsha, China). Human CRC cell lines (HCT-8, Caco-2 and HCT-116) were purchased from MED-BIO-CHEM (CLH0095-RT; CLH0041-RT; CLH0090-RT, Shanghai, China). The American Type Culture Collection (ATCC, Manassas, Virginia, USA) provided the human umbilical vein endothelial cells (HUVECs, PCS-100-010) that we used. At 37°C, with 5% CO2 and 95% air, all cells were grown in Roswell Park Memorial Institute (RPMI)-1640 media (#11875093, Gibco, Carlsbad, CA, USA) + 10% Fetal Bovine Serum (FBS, #A3161001C, Gibco, Carlsbad, CA, USA) +1% penicillin–streptomycin (#15140163, Gibco, Carlsbad, CA, USA). To generate a resistance model, 250 μg/mL of bevacizumab (A0420B, purity, 95%, Meilunbio, Dalian, China) was employed.
SW480 and HCT-116 cells (2.5 × 105) were grown in 6-well plates overnight. For OPA3 overexpression, lentiviral OPA3 overexpression vectors (Over-OPA3) were designed and synthesized by Abace Biotechnology (Beijing, China) based on the pLenti-CMV-puro backbone. The Over-OPA3 and lentiviral blank control (Over-NC) were transfected into SW480 and HCT-116 cells using Lipofectamine 2000 transfection reagent (#11668019, Invitrogen, Carlsbad, CA, USA) following manufacturer’s instructions. Cells were infected with lentivirus with 8 µg/ml polybrene (#ST1380, Beyotime, Shanghai, China). After 36 h, 0.7 μg/mL puromycin (#ST551, Beyotime) was added to select the infected cells, and the overexpression efficacy was examined using quantitative real-time polymerase chain reaction (qRT‒PCR).
TRIzol® reagent (15596018, ThermoFisher, USA) was employed to isolate RNA, which was subsequently reverse transcribed into cDNA utilizing an iScript™ cDNA Synthesis Kit (1708890, Bio-Rad Laboratories, USA). Following this, PCR was conducted using SYBR™ Green HiScript® III RT SuperMix (R323, Vazyme, China), with three cycles consisting of repeats. PCR results were calculated using the 2−ΔΔCT method with GAPDH as the internal control. Sequences of primers were Human OPA3: F: 5′-AGTGTATCACTGGGTGGAG-3′, R: 5′-GATGAAGATGGTGGCTTCG-3′; Human lysophosphatidylcholine acyltransferase 2 (LPCAT2): F: 5′-TCTACGCCCGGCTCAACTAT-3′, R: 5′-GACAATCTGGACCCGCCTC-3′; Human perilipin 2 (PLIN2): F: 5′-GTTGATCCACAACCGAGTG-3′, R: 5′-TAGGCTGAGGACATGAGGT-3′; Human vascular endothelial growth factor A (VEGF): F: 5′-TCACCAAGGCCAGCACATAG-3′, R: 5′-GGCTCCAGGGCATTAGACAG-3′; Human GAPDH: F: 5′-TCAAGATCATCAGCAATGCC-3′, R: 5′-CGATACCAAAGTTGTCATGGA-3′.
Proteins were prepared according to standard protocols. They were denatured and covered by SDS‒PAGE and loaded on polyvinylidene fluoride membranes (IEVH85R, Millipore, USA). Quantitative analysis was performed using a BCA Protein Assay Kit (23225, ThermoFisher, USA). Primary antibodies were added and cultured with the membranes at 4°C overnight and then mixed with secondary antibody (ab7090, 1/1000, Abcam, Cambridge, UK) for 1 h at room temperature. The bands were visualized using BeyoECL Plus (P0018M, Beyotime, China). The primary antibodies (Abcam, UK) used included anti-OPA3 (ab69163, 1/1000), anti-lysophosphatidylcholine acyltransferase 2 (LPCAT2, ab224244, 1/1000), anti-perilipin 2 (PLIN2) (ab108323, 1/1000) and GAPDH (ab9484, 1/1000).
The cell lines NCM460, SW480, HCT-8, Caco-2, and HCT-116 were prepared, fixed for 15 min with 4% paraformaldehyde (#P0099, Beyotime Institute of Biotechnology, Shanghai, China), and then treated for 20 min with 0.5% Triton X-100 solution (#ST797, Beyotime Institute of Biotechnology, Shanghai, China). Following that, the cells were blocked for 30 min at room temperature using regular goat serum (#16210064, Gibco, USA). Anti-OPA3 antibody (ab230205, 1/200, Abcam, UK) was cultivated in cells for an overnight period at 4°C. Subsequently, the cells were incubated with secondary antibodies (ab150075, Abcam, UK) and the nuclei were stained with DAPI solution (62248, ThermoFisher, USA) following three rounds of washing in phosphate-buffered saline (PBS) containing 0.05% Tween 20 (PBST). Following a one-hour incubation period at room temperature, the samples were washed three times with PBST and then once more with distilled water. The findings were examined under a fluorescent microscope (Olympus IX73, Japan) upon sealing.
HCT-116 and SW-480 cells transfected with OPA3/NC were placed in 6-well plates. The cells were cultivated for a period of 14 days, with the media being replaced every 3 days. Following colony formation, cells were treated with 4% paraformaldehyde (#P0099, Beyotime Institute of Biotechnology, Shanghai, China) for fixation and stained with crystal violet dye solution (C0121, Beyotime Institute of Biotechnology, Shanghai, China). Cells were imaged and the number of colonies was counted manually.
An Annexin V-FITC/PI Apoptosis Detection Kit (#A211-01, Vazyme, Nanjing, Jiangsu, China) was used to measure the apoptosis of CRC cells. After using pancreatic enzymes without EDTA (#C0205, Beyotime Institute of Biotechnology, Shanghai, China) to digest the cells, the cells were centrifuged (SW41 rotor; Beckman Coulter, Inc., CA, USA) twice at 4°C for 5 min at 1000 rpm. After removing the supernatant, the cells were collected at a certain concentration (106/mL) and thrice washed with cold PBS. Subsequently, the cells were reconstituted in 100 μL 1 × Binding Buffer, along with 5 μL FITC-conjugated Annexin V and 5 μL propidium iodide (PI) and allowed to incubate for 10 min at room temperature under dark conditions. The cells were then put on ice and extra PBS was added. Within an hour, the findings were seen with BD FACS Aria II flow cytometry (BD Biosciences, San Jose, CA, USA).
A 96-well plate was coated with Matrigel (#354149, Corning, Inc., NY, USA) for one hour at 37°C. In a 5:1 coculture, human umbilical vein endothelial cells (HUVECs) and SW-480/HCT-116 cells were used. The cells were then put to the solidified Matrigel gel (#354149, Corning, Inc., NY, USA) after being resuspended in a single-cell suspension at 1.5 × 105/mL in mixed media. After a 24-h incubation period at 37°C, the plate was examined under a light microscope (Olympus BX53, Tokyo, Japan) to detect any alterations related to angiogenesis.
High-performance liquid chromatography-tandem mass spectrometry (HPLC‒MS/MS)
To analyze the lipids in the cells, the Folch method was used to extract the lipids after 72 h of transfection [19]. Internal standards were set for lysophosphatidylcholine (LPC) and phosphatidylcholine (PC). The lipids were solubilized in a CHCl3/MeOH/H2O (60/40/4.5) mixture and analyzed using a Zorbax RX-C18 1200 HPLC (Agilent Technologies), along with 1200 HPLC (Agilent Technologies) using a binary gradient as previously described [20]. A QQQ 6460 mass spectrometer (Agilent Technologies) was used for positive electrospray ionization mass spectrometry, and Agilent MassHunter Workstation software was used to analyze the chromatograms. The concentrations of PC and LPC were expressed as pmoles/µg, and calculated as the peak ratio of PC/LPC to the internal standard.
To quantify the triglyceride level, cells were washed with precooled PBS and incubated with isopropanol to isolate the lipids. The extracts were dissolved in a mixture of 1% Triton X-100 in chloroform (PHR1552, Sigma-Aldrich, USA) and dried before being dissolved in water for 1 h at 55°C. A triglyceride assay kit (BC0625, Solarbio, Beijing, China) was used to calculate the triglyceride level, which was expressed as μg/mg of protein.
To isolate the LDs, the confluent monolayers were homogenized in Tris-NaCl-EDTA (TNE) buffer (#351-302-101, Quality Biological, MD, USA) and centrifuged. The postnuclear supernatant was moved to an ultracentrifuge tube and a stepwise gradient sample was prepared. The sample was centrifuged at 4°C and 28,000 rpm using the Beckman SW41 rotor (Beckman Coulter, Inc., CA, USA) for 2 h, and the LD fraction was collected using a Pasteur pipette. The LD fraction was washed with TNE (pH 11) and precipitated by MeOH/CHCl3. Then the fraction was mixed with Radio Immunoprecipitation Assay (RIPA) buffer (#P0013E, Beyotime Institute of Biotechnology, Shanghai, China) for quantification.
Data processing was performed with SPSS 22.0. At least three experimental replicates were conducted and each biological sample was run in triplicate. Variable data were expressed as the mean ± standard deviation (SD). After normalization, Student’s t test and one-way Analysis of Variance (ANOVA) were used for comparisons between groups. p < 0.05 indicated statistical significance.
The expression of OPA3 was reduced in CRC, as observed from the analysis of Clinical Proteomic Tumor Analysis Consortium (CPTAC) samples in the UALCAN database. The expression of OPA3 was markedly minimal in primary tumors of CRC compared to the normal samples (Fig. 1A). The significant reduction of OPA3 expression in CRC cells was also demonstrated by both qRT‒PCR and Western blot tests (Figs. 1B and 1C). Immunofluorescence assays reaffirmed that the OPA3 is mainly located in the cell nuclei, and its expression showed a substantial decrease in CRC cells, particularly in SW-480 and HCT-116 cell lines, which displayed the greatest reduction (Fig. 1D). For further experiments, these two cell lines were chosen as in vitro models. When treated with bevacizumab, the relative expression of OPA3 increased in both SW-480 and HCT-116 cells, reaching a peak at 24 h, and then reduced (Fig. 1E). Collectively, these data show that OPA3 is downregulated in CRC tissues and cells, and is upregulated in response to bevacizumab treatment.
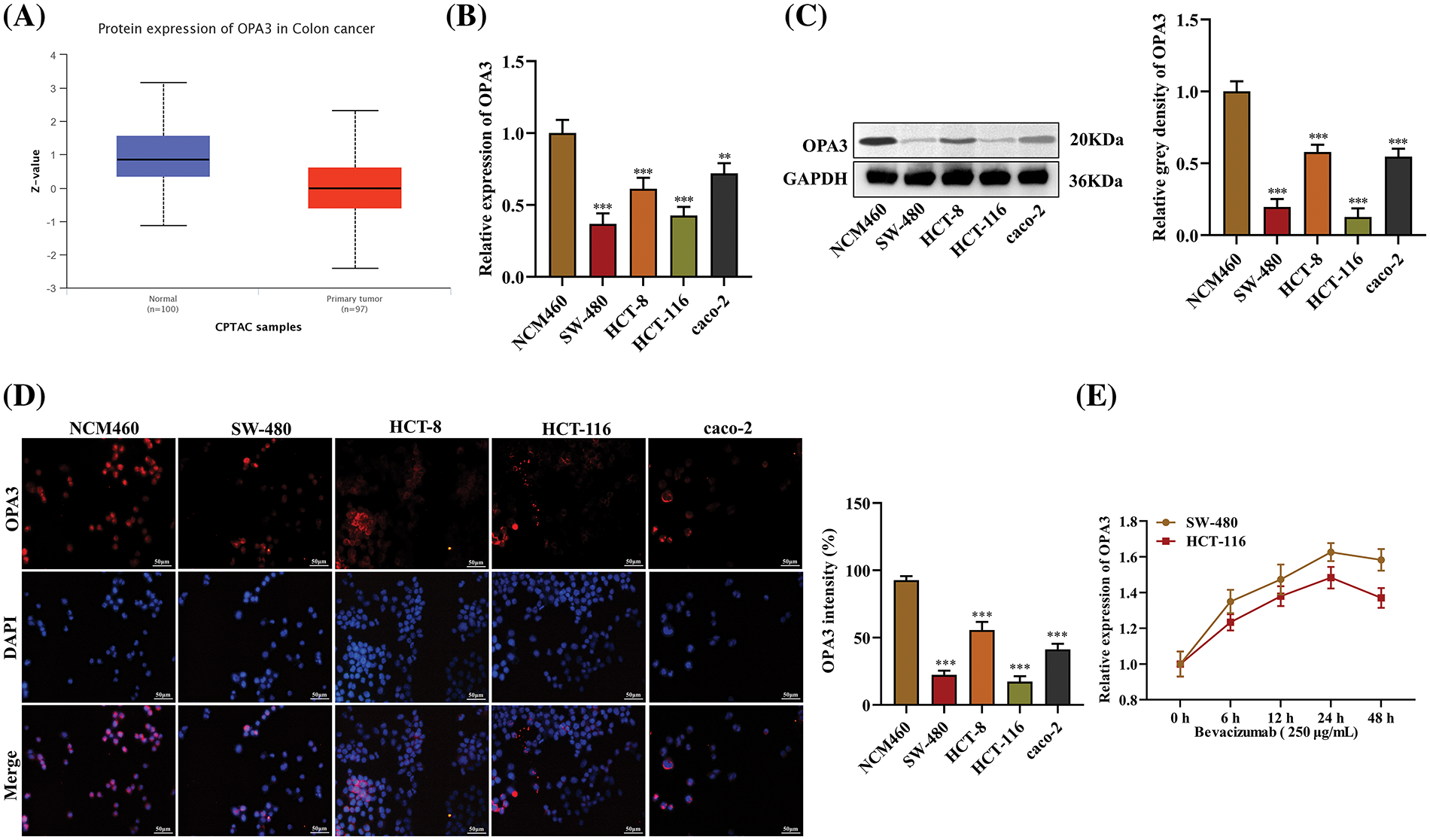
Figure 1: Decreased expression of outer mitochondrial membrane lipid metabolism regulator OPA3 (OPA3) in colorectal cancer (CRC). (A) In the UALCAN database, OPA3 expression patterns were observed in colon cancer tissues and normal tissues. (B) Comparison of OPA3 levels in human normal colon mucosal epithelial cell line (NCM460) and CRC cell lines (SW-480, HCT-8, HCT-116, cao-2) using qRT‒PCR analysis. (C) OPA3 expression was detected by Western blot in NCM460, SW-480, HCT-8, HCT-116, and cao-2 cell lines. (D) Assessment of OPA3 protein expression in NCM460, SW-480, HCT-8, HCT-116, and cao-2 cell lines via immunofluorescence (Bar: 50 μm). (E) qRT‒PCR analysis was carried out to monitor the relative expression of OPA3 in SW-480 and HCT-116 cells after bevacizumab treatment for 0, 6, 12, 24, and 48 h. The data are presented as the mean ± standard deviation (n = 3). **p < 0.01, ***p < 0.001.
OPA3 suppresses CRC cell proliferation, enhances apoptosis, and represses the angiogenesis of HUVECs
To examine the functional significance of OPA3 in CRC, we transfected SW-480 and HCT-116 cell lines with Over-OPA3/NC (overexpression efficiency shown in Fig. 2A) and confirmed significant upregulation of OPA3 protein expression in the Over-OPA3 group using Western blot (Fig. 2B). The number of CRC cell colonies declined in the Over-OPA3 group, indicating suppressed cell proliferation after OPA3 overexpression (Fig. 2C). Flow cytometry analysis showed a boosted apoptotic rate in the Over-OPA3 group (Fig. 2D). The tube formation assay showed that overexpressing OPA3 markedly inhibited the tube-forming ability of HUVECs when cocultured with CRC cells, leading to a reduction in the number of loops (Fig. 2E). Our findings show that OPA3 suppresses CRC cell proliferation, enhances apoptosis, and represses angiogenesis, suggesting it as a promising therapeutic target for CRC.
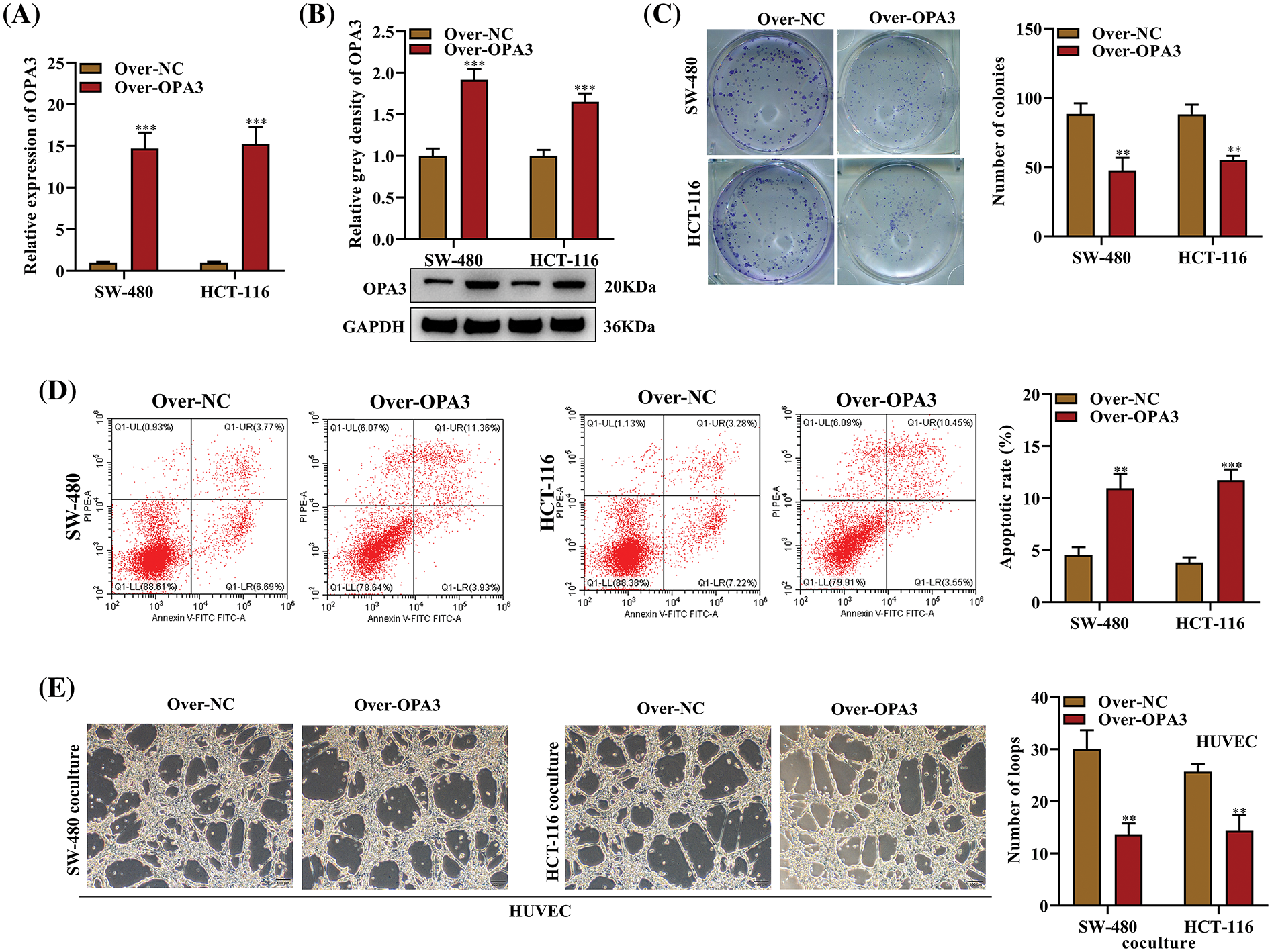
Figure 2: Impact of OPA3 on CRC cell growth, apoptosis, and the angiogenic capacity of human umbilical vein endothelial cells (HUVECs). The role of OPA3 in CRC development in vitro was studied using SW-480 and HCT-116 cell lines transfected with Over-OPA3/NC. (A) Quantitative assessment of OPA3 mRNA expression levels in SW-480 and HCT-116 cell lines with Over-OPA3/NC transfection using qRT-PCR. (B) Protein levels of OPA3 post-Over-OPA3/NC transfection analyzed by Western blot. (C) Number of colonies of cells with Over-OPA3/NC transfection was counted in colony formation assays. (D) Rate of apoptosis post-Over-OPA3/NC transfection analyzed by flow cytometry. (E) Tube formation ability of SW-480 and HCT-116 cell lines cocultured with CRC cells following Over-OPA3/NC transfection was evaluated using a tube formation assay. The data are presented as the mean ± standard deviation (n = 3). **p < 0.01, ***p < 0.001.
OPA3 reverses the resistance of CRC cells to bevacizumab
Over-OPA3/NC was transfected into bevacizumab-resistant SW-480 and HCT-116 cell lines in order to examine the role of OPA3 in CRC resistance to bevacizumab. Our findings demonstrated that overexpression of OPA3 reversed the resistance of CRC cells to bevacizumab, thereby enhancing the medication’s efficacy. We observed a reduction in the quantity of colonies in the bevacizumab group, as determined by colony formation assays; this reduction was further accentuated by transfection with Over-OPA3 (Fig. 3A). The flow cytometry analysis demonstrated a substantial increase in the apoptotic rate induced by bevacizumab treatment; this was further augmented through the overexpression of OPA3 (Fig. 3B). As displayed in Fig. 3C, the overexpression of OPA3 substantially reduced the number of loops in the bevacizumab group in comparison to the bevacizumab treatment group that received only bevacizumab. This finding suggests that bevacizumab has an anti-angiogenic effect. Based on these results, OPA3 may represent a viable target for ameliorating bevacizumab resistance and enhancing the therapeutic efficacy of the drug in colorectal cancer cells.
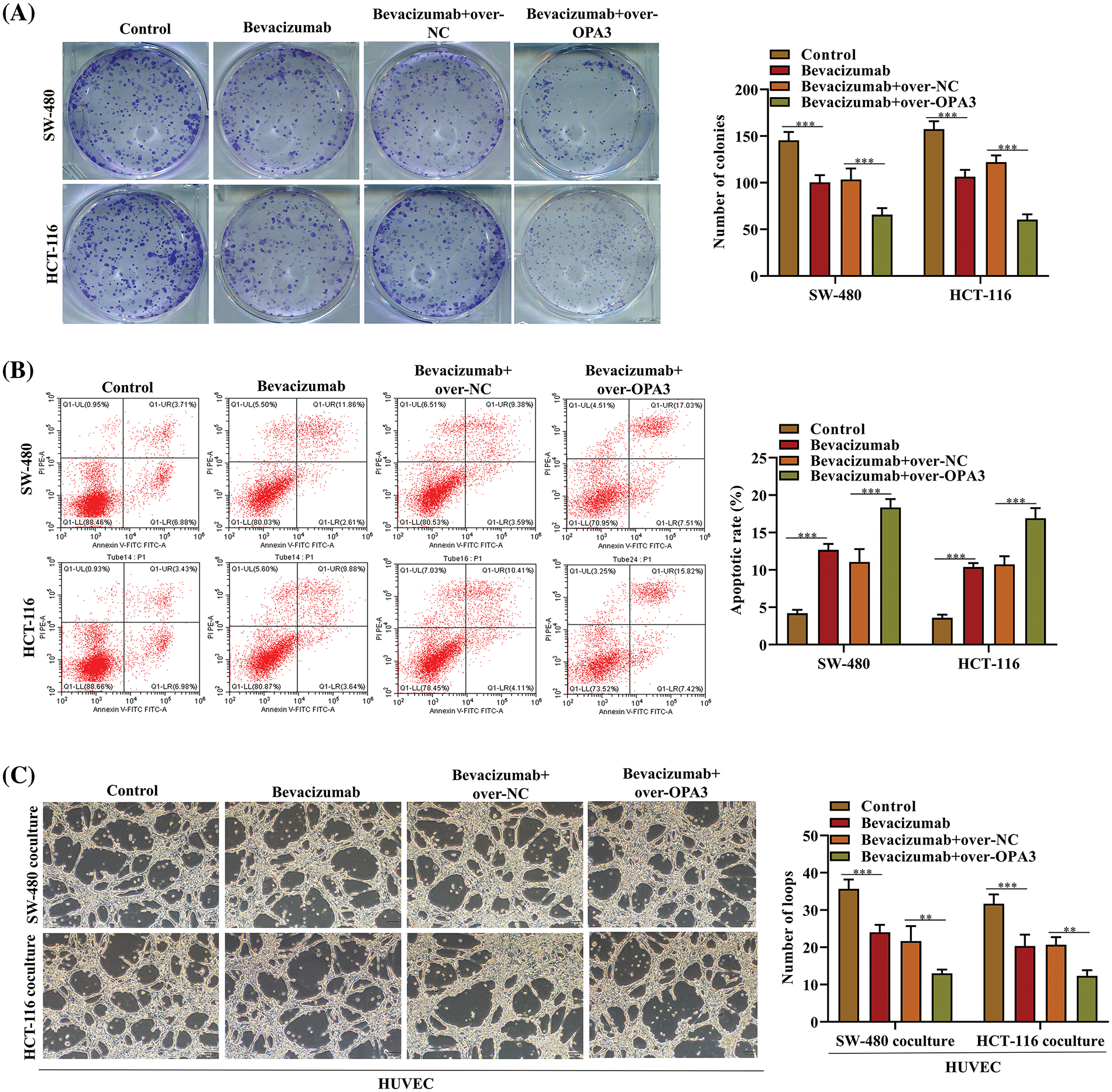
Figure 3: Effects of OPA3 on bevacizumab resistance in CRC cells. (A) Colony formation assays were carried out to evaluate the proliferation post-bevacizumab/NC treatment and Over-OPA3/NC transfection. (B) SW-480 and HCT-116 cell lines were treated with bevacizumab/NC and transfected with Over-OPA3/NC and their apoptosis rate was determined using flow cytometry. (C) Tube-formation ability was assessed by tube formation assay in cell lines treated with bevacizumab/NC and transfected with Over-OPA3/NC. The data are presented as the mean ± standard deviation (n = 3). **p < 0.01, ***p < 0.001.
OPA3 reduces LD production in CRC cells
Cellular lipids are stored and metabolized by LDs, which are critical for cancer development [21]. To investigate the impact of OPA3 on LD production, we examined the levels of PC and LPC, which are key components of the phospholipid monolayer of LDs [22,23]. We found that overexpression of OPA3 considerably diminished the levels of PC in both cell lines SW-480 and HCT-116, while LPC levels were not affected (Figs. 4A and 4B). Furthermore, although triglyceride levels did not show a significant difference in the Over-OPA3 group (Fig. 4C), the number of LDs was markedly lowered after OPA3 overexpression compared to the control group (Fig. 4D). These data provide credence to the possibility that OPA3 contributes to the regulation of LD in CRC cells.
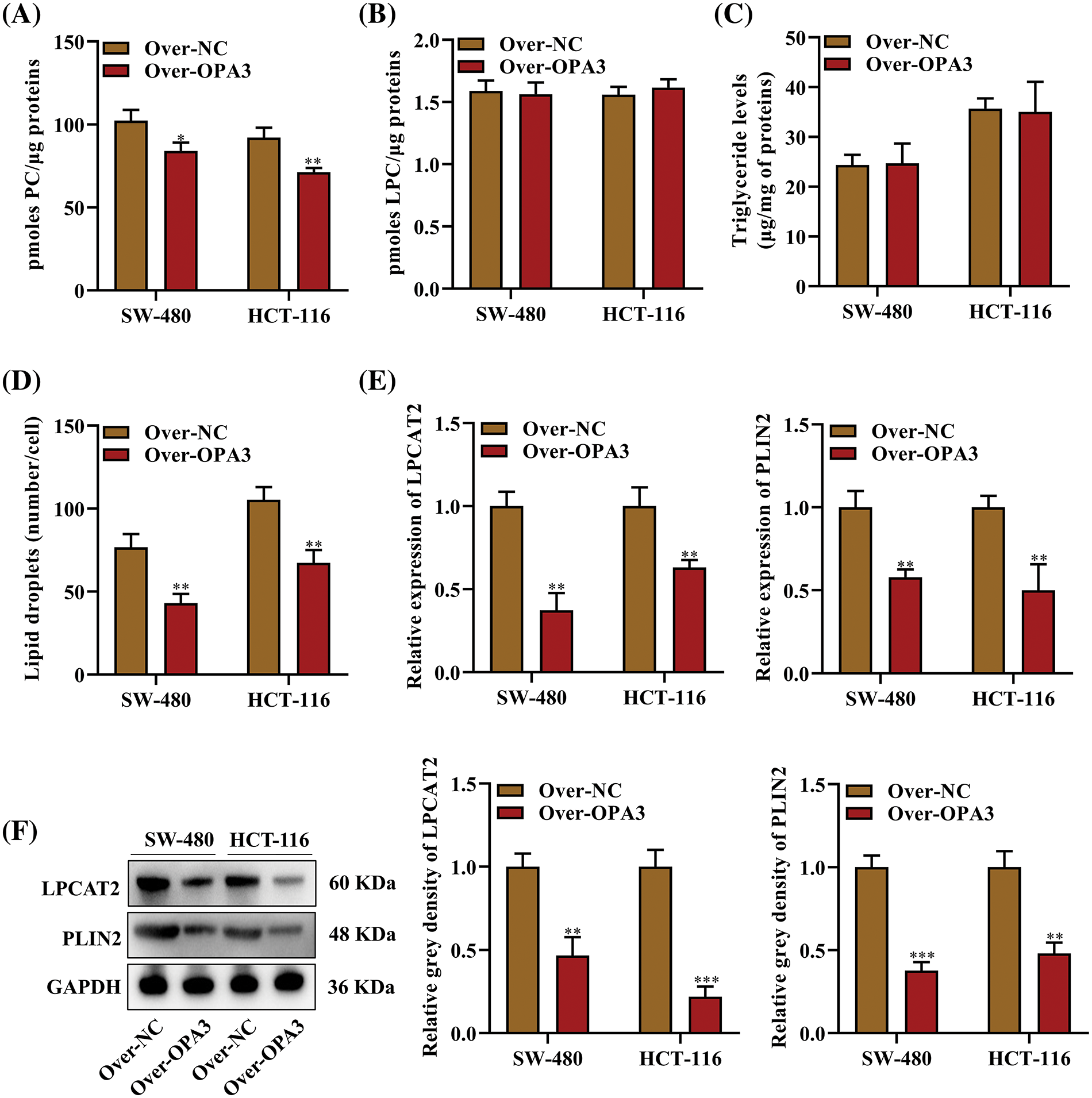
Figure 4: The role of OPA3 in lipid droplet (LD) formation in CRC cells. (A) Phosphatidylcholine (PC) levels in SW-480 and HCT-116 cell lines post-Over-OPA3/NC transfection, denoted as protein content per sample. (B) Lysophosphatidylcholine (LPC) level per sample after Over-OPA3/NC transfection. (C) Triglyceride content per sample post-Over-OPA3/NC transfection. (D) Number of LDs per cell following Over-OPA3/NC transfection. (E) qRT‒PCR measurement of lysophosphatidylcholine acyltransferase 2 (LPCAT2) and perilipin 2 (PLIN2) mRNA post-Over-OPA3/NC transfection. (F) After Over-OPA3/NC transfection, SW-480 and HCT-116 cells were evaluated by Western blotting for the expression of LPCAT2 and PLIN2. The data are presented as the mean ± standard deviation (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001.
Further analysis of LD metabolism was performed by determining the expression of two primary regulators, LPCAT2 and PLIN2. LPCAT2 is involved in controlling the growth and stability of LDs [20], and PLIN2 is regarded as a metabolic activity indicator in cells and an essential element for LD formation [24]. qRT-PCR results showed a notable reduction in the expression of LPCAT2 and PLIN2 in SW-480 and HCT-116 cells following transfection with Over-OPA3 (Fig. 4E). Additionally, Western blot analysis confirmed a decline in the protein levels of LPCAT2 and PLIN2 upon overexpression of OPA3 (Fig. 4F). From these observations, it can be inferred that OPA3 might contribute to the reduction of LD generation in CRC cells via the downregulation of LPCAT2 and PLIN2 expression.
OPA3 reverses bevacizumab-induced LD production in CRC cells
CRC resistance to AAD is linked to lipid metabolism. AADs are said to boost the reduction of oxygen and nutrients in tumor growth by aiding the transition from relying on glucose to relying on lipids for metabolism [25]. To explore the association between the inhibitory impact of OPA3 on LDs and the resistance of SW-480 and HCT-116 cell lines to bevacizumab, we evaluated the quantities of PC, LDs, LPCAT2, and PLIN2 in cells exposed to bevacizumab/NC treatment. The treatment with bevacizumab resulted in elevated PC levels in comparison to the control group. Conversely, this rise was markedly counteracted by the OPA3 overexpression (Fig. 5A). In addition, LDs were enhanced in the over-NC + bevacizumab group compared to the over-NC + control group but were reduced in the Over-OPA3 + bevacizumab group compared with bevacizumab treatment alone (Fig. 5B). Both LPCAT2 and PLIN2 mRNA and protein expression levels in SW-480 and HCT-116 cell lines was elevated due to bevacizumab treatment, and OPA3 overexpression was demonstrated to reverse the bevacizumab-induced up-regulation of LPCAT2 and PLIN2 mRNA and protein in SW-480 and HCT-116 cell lines (Figs. 5C and 5D). In addition, we measured VEGF levels in HUVECs cocultured with SW-480 and HCT-116 cells, and it was found that overexpression of OPA3 substantially diminished VEGF levels (Fig. 5E). Moreover, we also demonstrated that OPA3 overexpression enhanced the bevacizumab-induced downregulation of VEGF in HUVECs (Fig. 5F). These results indicate that OPA3 reverses bevacizumab-induced LD production in CRC cells.
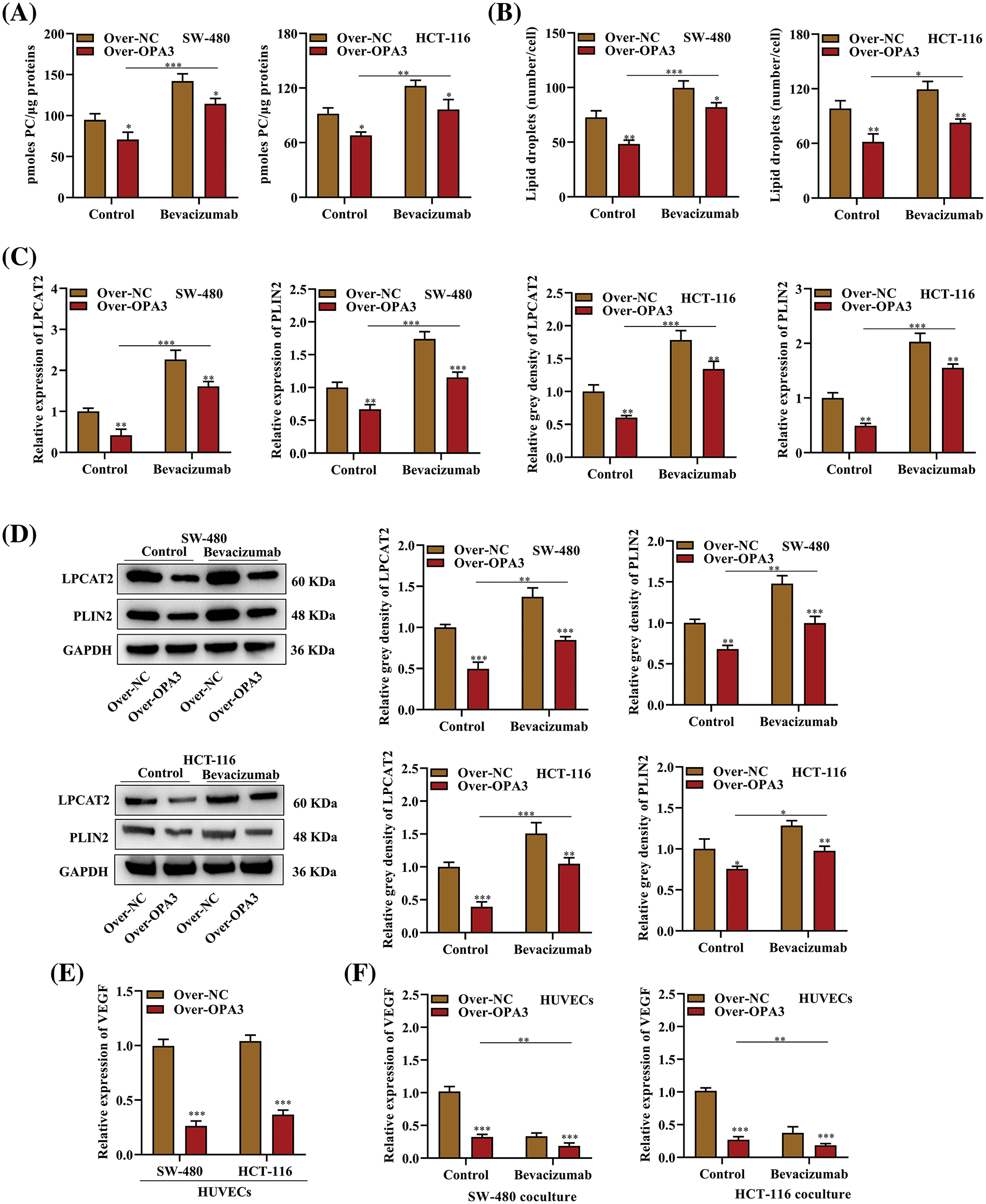
Figure 5: The effects of OPA3t on bevacizumab-induced LD production in CRC cells. (A) PC content per sample in SW-480 and HCT-116 cell lines transfected with Over-OPA3/NC and treated with bevacizumab/NC. (B) After transfection with Over-OPA3/NC and treatment with bevacizumab/NC, SW-480 and HCT-116 cells were evaluated for number of LDs per cell. (C) The expression of LPCAT2 and PLIN2 mRNA in SW-480 and HCT-116 cell lines transfected with Over-OPA3/NC and treated with bevacizumab/NC was investigated by qRT-PCR. (D) After Over-OPA3/NC transfection and bevacizumab/NC treatment, in SW-480 and HCT-116 cell lines were analyzed by Western blotting for LPCAT2 and PLIN2 protein expression. (E) HUVECs cocultured with SW-480 and HCT-116 cell lines transfected with Over-OPA3/NC were analyzed using qRT-PCR to detect VEGF expression. (F) The expression of VEGF in HUVECs was detected through qRT-PCR analysis after Over-OPA3/NC transfection and bevacizumab/NC treatment in combination with SW-480 and HCT-116 cell lines. The data are presented as the mean ± standard deviation (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001.
CRC typically occurs in a lipid-rich environment, and recent research suggests that lipid metabolism is specifically altered in CRC. The amount and make-up of oxylipins, fatty acids, triacylglycerols, and polar lipids are possible biomarkers for the detection of CRC [26]. Studies have shown that signatures of triglyceride species differentiate tumor tissue from normal tissue, with increased levels of lipogenic enzymes [27–30]. OPA3 is reported as a relevant regulator of lipid metabolism [18]. We further investigated the regulation of LDs via OPA3 and its role in CRC development. Our results indicate that overexpression of OPA3 suppressed cell proliferation and increased the apoptotic rate in CRC. Additionally, OPA3 overexpression also reduced the production of LDs in CRC cells and increased the sensitivity of CRC cells to bevacizumab.
Overexpression of OPA3 significantly reduced the ability of HUVECs cocultured with SW-480 and HCT-116 cells to form tubes, as measured by the number of loops. This reduction was comparable to the original anti-angiogenic efficacy of bevacizumab, suggesting that OPA3 has the potential to overcome bevacizumab resistance. Evidence suggests that bevacizumab resistance is linked to kinase signaling [31], and several molecules have been identified to promote bevacizumab resistance in non-small cell lung cancer, such as estrogen and hypoxia [32,33]. This is the first study to demonstrate a connection between OPA3 and resistance to bevacizumab in CRC. In the tube formation assays, the number of loops observed in the bevacizumab group was further decreased by overexpression of OPA3, suggesting that OPA3 can reverse the resistance of CRC cells to bevacizumab and enhance its anti-angiogenic efficacy.
In addition to inhibiting insulin-like growth factor1, OPA3 is also an inhibitor of postnatal growth retardation and hepatic steatosis [18]. Therefore, we conducted further assays to investigate the regulatory mechanism behind OPA3-mediated bevacizumab resistance, particularly with respect to LD-related markers. A higher level of PC was detected in bevacizumab-treated cells compared with controls. However, the overexpression of OPA3 significantly mitigated this increase. The increase in LDs induced by bevacizumab treatment was significantly reversed by overexpressing OPA3. The levels of LPCAT2 and PLIN2 mRNA and protein increased in the bevacizumab group were also counteracted after OPA3 overexpression. The collected data indicate that OPA3 reduces bevacizumab resistance by restraining the irregular formation of LDs in CRC cells.
Fundamentally, bevacizumab antagonizes the VEGF released by endothelial cells [34,35]. A study on brown fat in neonatal mice suggested that LD accumulation was a concurrent phenomenon of decreased VEGF levels in interscapular brown adipose tissue injected with an anti-VEGF antibody [36]. The AADs promotes the switch from glucose-dependent to lipid-dependent metabolism, and thereby affecting the genes in the lipid metabolism pathway. In our study, we found that OPA3 overexpression markedly impaired VEGF in HUVECs and enhanced the bevacizumab-induced reduction in VEGF expression in HUVECs. The anti-angiogenic therapy targeting VEGF can induce the alteration in lipid metabolism in tumor, and the lipid metabolic reprograming is associated with the treatment resistance in cancer [37–39]. This may explain why OPA3 reverses resistance to bevacizumab by reducing LD production. However, alterations in LD number and triglyceride level did not completely match, raising questions for future studies on whether OPA3 reduces LD production by interfering with the lipid storage ability of CRC cells [40,41] or accelerating lipolysis [42].
We also acknowledge that this study had some limitations. First, the role of OPA3 was only explored using two CRC cell lines due to the relatively lower expression pattern, and its effects on other CRC cell lines were unclear. Second, involvement of OPA3 in CRC tumor growth as well as bevacizumab resistance in vivo remains unclear. Third, the altered lipid metabolism reflects one aspect of the mechanism related to bevacizumab resistance, and the potential mechanisms of OPA3 to regulate VEGF expression against bevacizumab resistance in CRC are not fully understood.
In conclusion, OPA3 is a differentially expressed gene in CRC with anti-tumor effects on CRC cell proliferation and apoptosis. OPA3 also reverses the resistance of CRC cell lines to bevacizumab by regulating LD production, which might provide a new therapeutic target against bevacizumab resistance in CRC.
Acknowledgement: None.
Funding Statement: The study did not receive any financial support.
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Hongbiao Wu, Dongfang Liu; analysis and interpretation of results: Dongfang Liu, Hongbiao Wu; draft manuscript preparation: Dongfang Liu, Hongbiao Wu. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics Approval: Not applicable.
Conflicts of Interest: No conflicts of interest have been disclosed.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J Clin. 2021;71(3):209–49 [Google Scholar] [PubMed]
2. Chen H, Lu B, Dai M. Colorectal cancer screening in China: status, challenges, and prospects—China, 2022. China CDC Weekly. 2022;4(15):322–8 [Google Scholar] [PubMed]
3. Jiang L, Ji W, Gong Y, Li J, Liu J. MiR-520f-3p inhibits epithelial-mesenchymal transition of colorectal cancer cells by targeting yes-associated protein 1. BIOCELL. 2023;47(8):1803–10. doi:10.32604/biocell.2023.029516. [Google Scholar] [CrossRef]
4. Liu Y, Hong K, Weng W, Huang S, Zhou T. Association of vascular endothelial growth factor (VEGF) protein levels and gene polymorphism with the risk of chronic kidney disease. Libyan J Med. 2023;18(1):2156675 [Google Scholar] [PubMed]
5. Xu WL, Li SL, Niu AG, Shi BJ, Zhong ZY, Li YC. The role of PD-ECGF and VEGF in proliferative and involuted mechanism of the infantile capillary hemangiomas. Zhonghua Zheng Xing Wai Ke Za Zhi. 2011;27(3):182–6 [Google Scholar] [PubMed]
6. Saclarides TJ. Angiogenesis in colorectal cancer. Surg Clin North Am. 1997;77(1):253–60. doi:10.1016/S0039-6109(05)70543-0 [Google Scholar] [PubMed] [CrossRef]
7. Sun Q, Wang Y, Ji H, Sun X, Xie S, Chen L, et al. Lenvatinib for effectively treating antiangiogenic drug-resistant nasopharyngeal carcinoma. Cell Death Dis. 2022;13(8):724. doi:10.1038/s41419-022-05171-3 [Google Scholar] [PubMed] [CrossRef]
8. Fukunaga A, Takata K, Itoh S, Yamauchi R, Tanaka T, Yokoyama K, et al. Complete tumor necrosis confirmed by conversion hepatectomy after atezolizumab-bevacizumab treatment for advanced-stage hepatocellular carcinoma with lung metastasis. Clin J Gastroenterol. 2023;16(2):224–8. doi:10.1007/s12328-022-01744-z [Google Scholar] [PubMed] [CrossRef]
9. Huang H, Song J, Liu Z, Pan L, Xu G. Autophagy activation promotes bevacizumab resistance in glioblastoma by suppressing Akt/mTOR signaling pathway. Oncol Lett. 2018;15(2):1487–94 [Google Scholar] [PubMed]
10. Mesange P, Poindessous V, Sabbah M, Escargueil AE, de Gramont A, Larsen AK. Intrinsic bevacizumab resistance is associated with prolonged activation of autocrine VEGF signaling and hypoxia tolerance in colorectal cancer cells and can be overcome by nintedanib, a small molecule angiokinase inhibitor. Oncotarget. 2014;5(13):4709–21. doi:10.18632/oncotarget.1671 [Google Scholar] [PubMed] [CrossRef]
11. Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018;38(1):27. [Google Scholar]
12. Zadoorian A, Du X, Yang H. Lipid droplet biogenesis and functions in health and disease. Nat Rev Endocrinol. 2023;19(8):443–59 [Google Scholar] [PubMed]
13. Luo W, Wang H, Ren L, Lu Z, Zheng Q, Ding L, et al. Adding fuel to the fire: the lipid droplet and its associated proteins in cancer progression. Int J Biol Sci. 2022;18(16):6020–34 [Google Scholar] [PubMed]
14. Germain N, Dhayer M, Boileau M, Fovez Q, Kluza J, Marchetti P. Lipid metabolism and resistance to anticancer treatment. Biology. 2020;9(12):474. doi:10.3390/biology9120474 [Google Scholar] [PubMed] [CrossRef]
15. Iwamoto H, Abe M, Yang Y, Cui D, Seki T, Nakamura M, et al. Cancer lipid metabolism confers antiangiogenic drug resistance. Cell Metab. 2018;28(1):104–17 [Google Scholar] [PubMed]
16. Zheng Y, Zhou R, Cai J, Yang N, Wen Z, Zhang Z, et al. Matrix stiffness triggers lipid metabolic cross-talk between tumor and stromal cells to mediate bevacizumab resistance in colorectal cancer liver metastases. Cancer Res. 2023;83(21):3577–92. doi:10.1158/0008-5472.CAN-23-0025 [Google Scholar] [PubMed] [CrossRef]
17. Gaudet P, Livstone MS, Lewis SE, Thomas PD. Phylogenetic-based propagation of functional annotations within the gene ontology consortium. Brief Bioinform. 2011;12(5):449–62. doi:10.1093/bib/bbr042 [Google Scholar] [PubMed] [CrossRef]
18. Wells T, Davies JR, Guschina IA, Ball DJ, Davies JS, Davies VJ, et al. Opa3, a novel regulator of mitochondrial function, controls thermogenesis and abdominal fat mass in a mouse model for Costeff syndrome. Hum Mol Genet. 2012;21(22):4836–44. doi:10.1093/hmg/dds315 [Google Scholar] [PubMed] [CrossRef]
19. Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226(1):497–509. doi:10.1016/S0021-9258(18)64849-5. [Google Scholar] [CrossRef]
20. Cotte AK, Aires V, Fredon M, Limagne E, Derangere V, Thibaudin M, et al. Lysophosphatidylcholine acyltransferase 2-mediated lipid droplet production supports colorectal cancer chemoresistance. Nat Commun. 2018;9(1):322. doi:10.1038/s41467-017-02732-5 [Google Scholar] [PubMed] [CrossRef]
21. Cruz ALS, Barreto EA, Fazolini NPB, Viola JPB, Bozza PT. Lipid droplets: platforms with multiple functions in cancer hallmarks. Cell Death Dis. 2020;11(2):105. doi:10.1038/s41419-020-2297-3 [Google Scholar] [PubMed] [CrossRef]
22. van der Veen JN, Kennelly JP, Wan S, Vance JE, Vance DE, Jacobs RL. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim Biophys Acta Biomembr. 2017;1859(9):1558–72. doi:10.1016/j.bbamem.2017.04.006 [Google Scholar] [PubMed] [CrossRef]
23. Penno A, Hackenbroich G, Thiele C. Phospholipids and lipid droplets. Biochim Biophys Acta. 2013;1831(3):589–94. doi:10.1016/j.bbalip.2012.12.001 [Google Scholar] [PubMed] [CrossRef]
24. Tsai TH, Chen E, Li L, Saha P, Lee HJ, Huang LS, et al. The constitutive lipid droplet protein PLIN2 regulates autophagy in liver. Autophagy. 2017;13(7):1130–44. doi:10.1080/15548627.2017.1319544 [Google Scholar] [PubMed] [CrossRef]
25. Wang J, Millstein J, Yang Y, Stintzing S, Arai H, Battaglin F, et al. Impact of genetic variants involved in the lipid metabolism pathway on progression free survival in patients receiving bevacizumab-based chemotherapy in metastatic colorectal cancer: a retrospective analysis of FIRE-3 and MAVERICC trials. EClinicalMedicine. 2023;57:101827. doi:10.1016/j.eclinm.2023.101827 [Google Scholar] [PubMed] [CrossRef]
26. Pakiet A, Kobiela J, Stepnowski P, Sledzinski T, Mika A. Changes in lipids composition and metabolism in colorectal cancer: a review. Lipids Health Dis. 2019;18(1):29 [Google Scholar] [PubMed]
27. Ecker J, Benedetti E, Kindt ASD, Horing M, Perl M, Machmuller AC, et al. The colorectal cancer lipidome: identification of a robust tumor-specific lipid species signature. Gastroenterol. 2021;161(3):910–23.e19. [Google Scholar]
28. Li C, Wang Y, Liu D, Wong CC, Coker OO, Zhang X, et al. Squalene epoxidase drives cancer cell proliferation and promotes gut dysbiosis to accelerate colorectal carcinogenesis. Gut. 2022;71(11):2253–65 [Google Scholar] [PubMed]
29. Piccinin E, Cariello M, Moschetta A. Lipid metabolism in colon cancer: role of liver X receptor (LXR) and Stearoyl-CoA Desaturase 1 (SCD1). Mol Aspects Med. 2021;78:100933 [Google Scholar] [PubMed]
30. Coleman O, Ecker M, Haller D. Dysregulated lipid metabolism in colorectal cancer. Curr Opin Gastroenterol. 2022;38(2):162–7 [Google Scholar] [PubMed]
31. Ramezani S, Vousooghi N, Joghataei MT, Chabok SY. The role of kinase signaling in resistance to bevacizumab therapy for glioblastoma multiforme. Cancer Biother Radiopharm. 2019;34(6):345–54. doi:10.1089/cbr.2018.2651 [Google Scholar] [PubMed] [CrossRef]
32. Patel SA, Herynk MH, Cascone T, Saigal B, Nilsson MB, Tran H, et al. Estrogen promotes resistance to bevacizumab in murine models of NSCLC. J Thorac Oncol. 2021;16(12):2051–64. doi:10.1016/j.jtho.2021.07.007 [Google Scholar] [PubMed] [CrossRef]
33. Mao XG, Wang C, Liu DY, Zhang X, Wang L, Yan M, et al. Hypoxia upregulates HIG2 expression and contributes to bevacizumab resistance in glioblastoma. Oncotarget. 2016;7(30):47808–20. doi:10.18632/oncotarget.10029 [Google Scholar] [PubMed] [CrossRef]
34. Gao F, Yang C. Anti-VEGF/VEGFR2 monoclonal antibodies and their combinations with PD-1/PD-L1 inhibitors in clinic. Curr Cancer Drug Targets. 2020;20(1):3–18. doi:10.2174/1568009619666191114110359 [Google Scholar] [PubMed] [CrossRef]
35. Raghav K, Liu S, Overman MJ, Willett AF, Knafl M, Fu SC, et al. Efficacy, safety, and biomarker analysis of combined PD-L1 (Atezolizumab) and VEGF (Bevacizumab) blockade in advanced malignant Peritoneal Mesothelioma. Cancer Discov. 2021;11(11):2738–47. doi:10.1158/2159-8290.CD-21-0331 [Google Scholar] [PubMed] [CrossRef]
36. Jo DH, Park SW, Cho CS, Powner MB, Kim JH, Fruttiger M, et al. Intravitreally injected anti-vegf antibody reduces brown fat in neonatal mice. PLoS One. 2015;10(7):e0134308 [Google Scholar] [PubMed]
37. Morandi A, Indraccolo S. Linking metabolic reprogramming to therapy resistance in cancer. Biochim Biophys Acta Rev Cancer. 2017;1868(1):1–6 [Google Scholar] [PubMed]
38. Curtarello M, Tognon M, Venturoli C, Silic-Benussi M, Grassi A, Verza M, et al. Rewiring of lipid metabolism and storage in ovarian cancer cells after anti-VEGF therapy. Cells. 2019;8(12):1601. doi:10.3390/cells8121601 [Google Scholar] [PubMed] [CrossRef]
39. Bacci M, Lorito N, Smiriglia A, Morandi A. Fat and furious: lipid metabolism in antitumoral therapy response and resistance. Trends Cancer. 2021;7(3):198–213 [Google Scholar] [PubMed]
40. Bostrom P, Rutberg M, Ericsson J, Holmdahl P, Andersson L, Frohman MA, et al. Cytosolic lipid droplets increase in size by microtubule-dependent complex formation. Arterioscler Thromb Vasc Biol. 2005;25(9):1945–51 [Google Scholar] [PubMed]
41. Schott MB, Weller SG, Schulze RJ, Krueger EW, Drizyte-Miller K, Casey CA, et al. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J Cell Biol. 2019;218(10):3320–35. doi:10.1083/jcb.201803153 [Google Scholar] [PubMed] [CrossRef]
42. Yin H, Li W, Mo L, Deng S, Lin W, Ma C, et al. Adipose triglyceride lipase promotes the proliferation of colorectal cancer cells via enhancing the lipolytic pathway. J Cell Mol Med. 2021;25(8):3963–75. doi:10.1111/jcmm.16349 [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools