
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE
3-Methyladenine potentiates paclitaxel-induced apoptosis and phosphorylation of cyclin-dependent kinase 1 at thr161 in nasopharyngeal carcinoma cell
1 Hengyang Medical School, University of South China, Hengyang, 421001, China
2 Institute for Future Sciences, University of South China, Changsha, 410008, China
3 Medical Research Center, The Affiliated Changsha Central Hospital, Hengyang Medical School, University of South China, Changsha, 410004, China
4 Department of Clinical Laboratory, The Affiliated Changsha Central Hospital, Hengyang Medical School, University of South China, Changsha, 410004, China
* Corresponding Author: BINYUAN JIANG. Email:
# Equal contribution
(This article belongs to the Special Issue: Navigating the Interplay of Cancer, Autophagy, ER Stress, Cell Cycle and Apoptosis: Mechanisms, Therapies, and Future Directions)
BIOCELL 2024, 48(5), 861-872. https://doi.org/10.32604/biocell.2024.048758
Received 17 December 2023; Accepted 27 February 2024; Issue published 06 May 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: Nasopharyngeal carcinoma (NPC) exhibits a significant prevalence in the southern regions of China, and paclitaxel (PTX) is frequently employed as a medication for managing advanced NPC. However, drug resistance is typically accompanied by a poor prognosis. Exploring the synergistic potential of combining multiple chemotherapeutic agents may represent a promising avenue for optimizing treatment efficacy. Methods: This study investigated whether 3-Methyladenine (3-MA) could potentiated the effect of PTX and its potential molecular mechanism. Samples were divided into the following categories: Negative control (NC) with the solvent dimethyl sulfoxide (DMSO, 0.5% v/v), PTX (400 nM), 3-MA (4 mM), and PTX (400 nM) + 3-MA (4 mM). The viability of NPC cells was assessed using both the cell counting kit-8 (CCK-8) assay and the colony formation assay. Microscopic observation was performed to identify morphological cell changes. Flow cytometry was used to assess cell cycle status, mitochondrial membrane potential (MMP), and apoptotic cells. Western blotting was conducted to quantify the protein expression. Results: 3-MA enhanced PTX-specific inhibition of NPC cell proliferation. PTX, either alone or in combination with 3-MA, caused cell cycle halt at the G/M phase in the majority of NPC cells, and the combination treatment of PTX with 3-MA induced a higher rate of NPC cell death compared to PTX alone. Western blotting results revealed the combination of PTX with 3-MA heightened activation of cyclin-dependent kinase 1 (CDK1), a key molecule in shifting cells from mitotic arrest to apoptosis, led to a reduction in Myeloid Cell Leukemia 1 (MCL-1) expression and an increase in Poly (ADP-ribose) polymerase (PARP) cleavage. Conclusion: The concurrent administration of PTX with 3-MA effectively enhances PTX’s inhibitory impact on NPC and activates the apoptosis signal regulated by CDK1.Keywords
Nasopharyngeal carcinoma (NPC) is a prevalent disease in the southern regions of China, with a considerable portion of patients diagnosed at an advanced stage [1,2]. Paclitaxel has been established as the first-line chemotherapy drug for advanced NPC in clinical practice, and drug resistance is typically accompanied by a poor prognosis [3]. Exploring a combination of multiple chemotherapy drugs with diverse pharmacological mechanisms could be a feasible strategy for enhancing treatment outcomes. This approach may offer advantages such as shorter treatment duration, a lower-dose regimen, and reduced resistance risks and side effects [4,5].
3-MA is a well-known Phosphoinositide 3-Kinase (PI3K) inhibitor that targets both class I and class III PI3Ks. It exhibits a long-lasting inhibitory effect on class I PI3K and activates autophagy. In contrast, its inhibition of class III PI3K is transient, inhibiting autophagy [6,7]. Current evidence suggests that the PI3K signaling pathway is among the most commonly dysregulated pathways in cancer and is directly related to cellular quiescence, growth and lifespan [8]. It plays a crucial role in the advancement of the cell cycle, events such as G0/G1 phase transition, S phase entry, and M phase entry all present high PI3K activity [9,10]. Due to 3-MA influencing the activity of class I PI3K and class III PI3K, suggesting complex and comprehensive effects of 3-MA in cell cycle progression.
Paclitaxel, belonging to the taxane class of drugs, is commonly employed in the clinic to treat various cancers, including nasopharyngeal carcinoma [11,12]. It works as an anti-microtubule agent via its binding to tubulin dimers. The permanent attachment of paclitaxel to microtubules disrupts their dynamic regulation, causing mistakes in the spindle-chromosome attachment process. Consequently, cells either undergo mitotic arrest and irregular distribution of genetic material, ultimately leading to mitotic catastrophe [13,14]. Cells undergoing mitotic catastrophe present a rounded cell shape, undergo gradual chromatin condensation, and develop multiple micronuclei. Eventually, this process leads to cell death [15]. Remarkably, mitotic catastrophe serves as a vital mechanism by which cell cycle-targeted chemotherapy suppress tumors [16].
The CDK1 in complex with cyclin B1 (CDK1-cyclin B1) plays a crucial role in regulating the cell cycle advancement in eukaryotes [17]. During mitosis, CDK1 serves as the catalytic subunit, while cyclin B1 acts as a regulatory subunit. The substrates of CDK1 kinase play essential roles in various processes during mitosis, including spindle formation, chromatin condensation, regulation of nuclear envelope integrity, and induction of apoptosis following mitotic errors [18]. During entry into mitosis, CDK1 and cyclin B1 proteins steadily accumulate and form complexes [19]. The activation of CDK1 also involves dynamic regulation of phosphorylation sites, with Y15 and T14 acting as inhibitory sites, while T161 serves as the activation site. CDK1 becomes active only upon phosphorylation of T161 [20]. Additionally, the degradation of cyclin B1 is essential for the rapid inactivation of CDK1 and exit from mitosis. However, when cells experience aberrant mitotic division or permanent cell cycle halt caused by anti-mitotic drugs, the activation of CDK1 and the execution of cell death are needed [21]. The initiation of cell death signaling involves a reduction in anti-apoptotic proteins. MCL-1, a prototypical anti-apoptosis protein, experiences CDK1-mediated phosphorylation at the T92 site, leading to its hydrolysis [22]. Throughout paclitaxel-triggered aberrant mitotic division and subsequent mitotic arrest, the onset of apoptotic signals coincides with MCL-1 hydrolysis and the cleavage of Poly (ADP-ribose) polymerase (PARP) [23,24].
Based on the principle of using non-toxic concentrations for chemosensitizers, we referred to the data from our previous study on the proliferative inhibitory effects of 3-MA on 5–8 F and 6–10 B cells (refer to Suppl. Fig. S1), we selected 3-MA (4 mM), a concentration proven non-cytotoxic to nasopharyngeal carcinoma cell lines, for combination with paclitaxel at an effective inhibitory concentration. This combinatorial approach resulted in a significant augmentation of growth inhibition in NPC cells induced by paclitaxel. Subsequently, we investigated the mechanisms of how 3-MA enhances paclitaxel-mediated cytotoxicity against NPC cells and explored the underlying molecular mechanisms connecting cell cycle and apoptosis.
NPC cell lines 5–8 F and 6–10 B were obtained from Sun Yat-sen University Cancer Center (Guangzhou, Guangdong, China). Prepare complete medium by adding 10% volume of Fetal Bovine Serum (FBS; Biological Industries, 04-121-1A, Kibbutz Beit-Haemek, Kibbutz, Israel) to Dulbecco’s Modified Eagle Medium (DMEM; Biological Industries, 01-052-1A, Kibbutz Beit-Haemek, Kibbutz, Israel), diluted Penicillin-Streptomycin Solution (Biological Industries, 03-031-1B, Kibbutz Beit-Haemek, Kibbutz, Israel) at final concentration of penicillin at 100 U/mL and streptomycin at 100 μg/mL, which was added during cell culture. The cells were cultured in an incubator set at 37°C with a 5% concentration of CO2. 3-MA was purchased from GLPBIO Technology (GC68539, Montclair, CA, USA), and paclitaxel were purchased from MedChemExpress (HY-B0015, Princeton, New Jersey, USA). The cells were treated with 4 mM 3-MA and/or 400 nM paclitaxel for the efficacy analyses, and samples were divided into the following categories: Negative control (NC) with the solvent dimethyl sulfoxide (DMSO, 0.5% v/v), PTX (400 nM), 3-MA (4 mM), and PTX (400 nM) + 3-MA (4 mM).
Cell counting kit-8 (CCK-8) assay
To assess impact on cell growth, in each well of a sterile 96-well cell culture plate containing complete culture medium, 1.5 × 104 cells were plated. Cells were exposed to various drugs after attachment and cultured under conditions specified in the “Cell culture and drugs” section for 24 and 48 h. After 2 h of incubation with 10 µL of CCK-8 reagent (APE-BIO, K1018, Houston, Texas, USA), the experimental results were read using a microplate reader (BioTek Instruments, EPOCH, Winooski, Vermont, USA) set at an absorbance of 450 nm. Three multiple holes were set up in this experiment.
To assess the impact on clone formation ability of cells, in each well of a sterile 6-well cell culture plate containing complete culture medium, 6 × 103 cells were plated. After 8 days, drugs were added at the concentrations specified in the “Cell culture and drugs” section. The cells were cultured for 48 h, followed by PBS washing and fixation with formaldehyde (Beyotime Institute of Biotechnology, P0099, Shanghai, China), and stained with gentian violet (Solarbio, G1075, BeiJing, China). Microscope images were taken of the stained cells (BioTek Instruments, Lionheart, Winooski, Vermont, USA) and the number of clones was calculated by Gen5 software (BioTek Instruments, version 3.12, Lionheart, Winooski, Vermont, USA). Finally, the clone formation rate was calculated and adjusted as follows = Clone formation rate (%)/Clone formation rate in the NC group (%).
To observe the cell morphology changes induced by paclitaxel, in each well of a sterile 24-well cell culture plate containing complete culture medium, 3 × 105 cells were plated. Cells were exposed to various drugs after attachment and cultured under conditions specified in the “Cell culture and drugs” section for 18 h. After the incubation period, observe and photograph under a microscope (Leica Microsystems, DMIL LED, Wetzlar, Hessian, Germany). The proportion of cells displaying a rounded morphology to the total cell count was determined across five randomly chosen microscopic fields (magnification, ×200). To evaluate nuclear condensation induced by paclitaxel in 5-8F and 6-10B cells, we utilized Hoechst 33342 (Beyotime Institute of Biotechnology, C1028, Shanghai, China) at a dilution of 1:100. After removing the cell culture medium and rinse with PBS buffer, 180 µL of diluted Hoechst 33342 solution was added to each well, and incubate in a dark, 37°C incubator for 15 min. Subsequently, the stained nuclei were observed under a microscope. (Lionheart, BioTek Instruments Winooski, Vermont, USA). The images were merged using ImageJ software. (National Institutes of Health, version 1.42q; Bethesda, Maryland, USA).
To measure the related protein expression, in each well of a sterile 12-well cell culture plate containing complete culture medium, 4 × 105 cells were plated. Cells were exposed to various drugs after attachment and cultured under conditions specified in the “Cell culture and drugs” section. Treat the collected cells with pre-cooled lysis buffer (Beyotime Institute of Biotechnology, P0013, Shanghai, China) to obtain protein samples for Western blotting. Protein concentration was determined using the bicinchoninic acid (BCA) method (Beyotime Institute of Biotechnology, P0011, Shanghai, China). The proteins were separated by SDS-PAGE electrophoresis and transferred onto an activated PVDF membrane (MilliporeSigma, ISEQ00010, Burlington, Massachusetts, USA) for subsequent immunoblotting experiments. The PVDF membranes were incubated in blocking buffer (5% skim milk dissolved in PBS with 0.05% Tween-20) at 25°C for 1 h to block nonspecific binding sites. After washing the membrane three times in PBST, the diluted primary antibody was added and incubated overnight at 4°C. Afterwards, the membranes underwent three washes, each lasting 15 min, in PBST, followed by incubation with a diluted secondary antibody at 25°C for 60 min. Following five washes for 3 min each in PBST, the protein bands were detected with electrochemiluminescence (ECL) reagent (Thermo Fisher Scientific, 34075, Waltham, Massachusetts, USA) and imaged. CDK1 antibody (diluted at 1:1000, catalog number: 77055), phosphorylated (p-)CDK1 (Thr161) antibody (diluted at 1:1000, catalog number: 9114), p-CDK1(Tyr15) antibody (diluted at 1:1000, catalog number: 4539), p-CDK1(Thr14) antibody (diluted at 1:1000, catalog number: 2543), MCL-1 antibody (diluted at 1:1000, catalog number: 94296), PARP antibody for detecting both cleaved and full-length PARP (diluted at 1:1000, catalog number: 9532) were produced by Cell Signaling Technology (Danvers, Massachusetts, USA). B-cell lymphoma 2 (Bcl-2) antibody (diluted at 1:2000, catalog number: ET1603-11) and Bcl-2 associated X protein (Bax) (diluted at 1:2000, catalog number: ET1603-34) were purchased from Huaan Biotechnology Company (Hangzhou, Zhejiang, China). The primary antibody for GAPDH (diluted at 1:2000, catalog number: AF0006) was produced by Beyotime Institute of Biotechnology (Shanghai, China) and utilized as a control. Anti-mouse IgG/HRP antibody (diluted at 1:2000, catalog number: 7076) and anti-rabbit IgG/HRP antibody (diluted at 1:2000, catalog number: 7074) were produced by Cell Signaling Technology (Danvers, Massachusetts, USA). The protein gray value was conducted with an image analysis software (National Institutes of Health, ImageJ 1.8.0, Bethesda, Maryland, USA). GAPDH was used as the internal control; protein bands were normalized to GAPDH and subsequently compared with the control group.
To explore the profile of cell cycle phases, in each well of a sterile 6-well cell culture plate containing complete culture medium, 2 × 106 cells were plated. Cells were exposed to various drugs after attachment and cultured under conditions specified in the “Cell culture and drugs” section for 24 h. Subsequently, cells were collected and immobilized in 75% alcohol at 4°C for 24 h. After fixation, cells underwent centrifugation at 500 × g at 4°C for 6 min to eliminate the 75% alcohol, followed by three rinses with cold PBS. Following this, the cells were subjected to staining with propidium iodide (Beyotime Institute of Biotechnology, ST511, Shanghai, China) at a concentration of 50 µg/mL and treated with DNase-free RNase (Beyotime Institute of Biotechnology, ST579, Shanghai, China) at a concentration of 100 µg/mL, samples were incubated in a light-protected 37°C incubator for 20 min. The determination of cell cycle phases involved assessing DNA content using a flow cytometry-based method by BD Accuri C6 (BD Biosciences, San Jose, California, USA), with subsequent data analysis performed using the preloaded software of BD Accuri C6 (BD Biosciences, version 1.0.264.21, San Jose, California, USA). We used the PTX (400 nM) as the control for comparing alterations in the SubG1 phase, whereas the NC group was employed as the control to assess alterations in the G2/M phases.
The JC-1 assay kit (Beyotime Institute of Biotechnology, C2003S, Shanghai, China) was utilized to assess MMP, investigating the initiation of mitochondria-regulated apoptosis. In brief, in each well of a sterile 12-well cell culture plate containing complete culture medium, 3 × 105 cells were plated. Cells were exposed to various drugs after attachment and cultured under conditions specified in the “Cell culture and drugs” section for 48 h. The JC-1 staining procedures were conducted according to the kit instruction. The proportion of MMP changed cells was determined using a flow cytometry-based method by BD Accuri C6 (BD Biosciences, San Jose, California, USA) within 1 h, and data analysis performed using the preloaded software of BD Accuri C6 (BD Biosciences, version 1.0.264.21, San Jose, California, USA). The alterations in the percentage of JC-1 monomer cells were quantified, indicating the dissipation of MMP.
The Annexin V-FITC/PI Detection Kit (Yeasen Biotechnology, 40302ES50, Shanghai, China) was used to perform the cell apoptosis assay, following the manufacturer’s protocol. In brief, in each well of a sterile 12-well cell culture plate containing complete culture medium, 3 × 105 cells were plated. Cells were exposed to various drugs after attachment and cultured under conditions specified in the “Cell culture and drugs” section for 48 h. To label the cells, 150 µL 1X Binding Buffer containing 5 µL of prepared Annexin V-FITC staining solution and 10 µL of prepared PI staining solution were added to each sample tube. The cells were then incubated for at 25°C for 15 min in the absence of light. After adding 400 µL of binding buffer to each sample, they are then analyzed using a flow cytometry-based method by BD Accuri C6 (BD Biosciences, San Jose, USA) within 1 h, and data analysis performed using the preloaded software of BD Accuri C6 (BD Biosciences, version 1.0.264.21, San Jose, USA). Apoptotic cells comprise only early apoptotic cells (FITC-positive, PI-negative) and late apoptotic cells (FITC-positive, PI-positive). The fold change in the percentage of apoptotic cell populations was computed compared with the NC group, and then the fold change between the PTX (400 nM) group and the PTX (400 nM) + 3-MA (4 mM) group was compared.
Each experiment was repeated three times, and findings were expressed as mean ± standard deviation (SD). Statistical assessments were carried out utilizing two-way ANOVA and Tukey’s test. Data statistical analysis and graph generation were conducted using GraphPad Prism (GraphPad Software, GraphPad 8.30, San Diego, California, USA). A p-value less than 0.05 was considered statistically significant.
3-MA potentiates the inhibition of cell proliferation induced by paclitaxel in NPC cells
The findings from the CCK-8 assays indicated ongoing growth in both 5–8 F and 6–10 B cells after a 48-h culture with 3-MA (4 mM), with no significant difference compared to the NC group. Moreover, the suppressive impact of paclitaxel on NPC cell growth was notably amplified by 3-MA (4 mM) after 48 h, as demonstrated by the reduction in absorbance readings at 48 h (Figs. 1A and 1B). Additionally, the clone formation assay also showed that NPC cells simultaneously treated with paclitaxel and 3-MA had the lowest clone formation rate (Figs. 1C–1E).
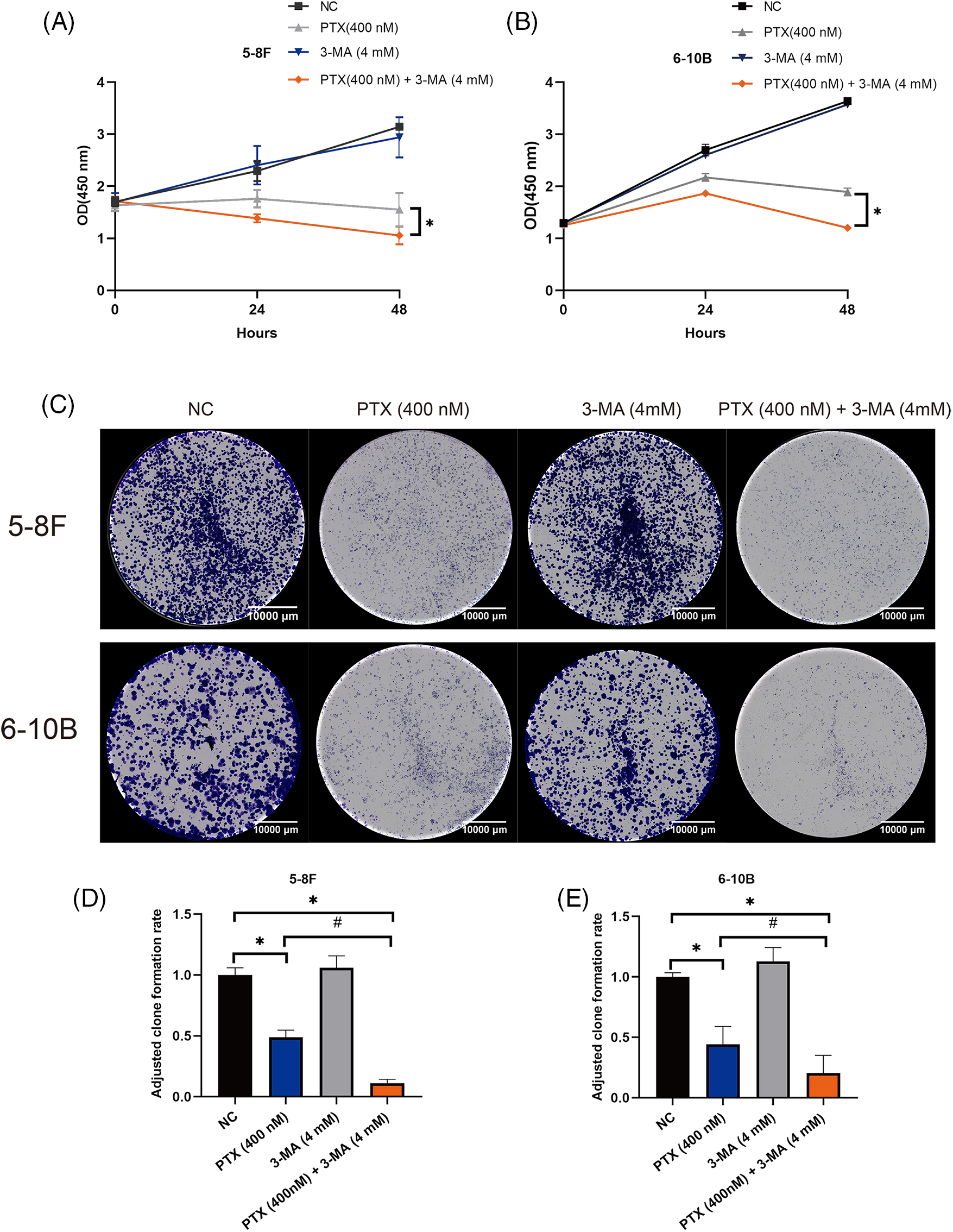
Figure 1: 3-MA potentiates the inhibition of cell growth induced by paclitaxel in NPC cells. The findings from CCK-8 assays demonstrated that 3-MA potentiated the inhibitory effect of paclitaxel on cell growth of 5–8 F and 6–10 B cells (A and B). The data, represented as mean ± SD (n = 3), underwent analysis through two-way ANOVA and Tukey’s test. Compare with PTX (400 nM) group at 48 h, *p < 0.05. Clone formation assays revealed that 3-MA (4 mM) potentiated paclitaxel-induced inhibition of NPC cells clone formation ability (C–E). The data, represented as mean ± SD (n = 3), underwent analysis through two-way ANOVA and Tukey’s test. Compare with NC group, *p < 0.05. Compare with PTX (400 nM) group, #p < 0.05. Scale bar = 10000 μm. OD, optical density.
3-MA augments paclitaxel-specific morphological alterations in NPC cells
The effects of paclitaxel on mitotic catastrophe manifest through alterations in cell morphology and condensation of chromatin [25,26]. Our findings indicated that treatment with 3-MA significantly potentiate paclitaxel-specific morphological changes, representative images (top rows in Figs. 2A and 2B) illustrate that the majority of cells treated with paclitaxel exhibited a rounded morphology. However, combined treatment with paclitaxel and 3-MA, NPC cells exhibit a greater propensity to present a rounded morphology. We assessed the ratio of spherical cells to the overall cell population across five randomly chosen fields of view, which were depicted in the bottom row of Figs. 2A and 2B. Statistical analysis showed that 3-MA significantly increased the number of paclitaxel-induced rounded cells. Additionally, the formation of multiple micronuclei in NPC cells after 20 h, as shown in Figs. 2C and 2D. These data demonstrate that 3-MA enhances NPC cells entering into mitotic catastrophe in the presence of paclitaxel.
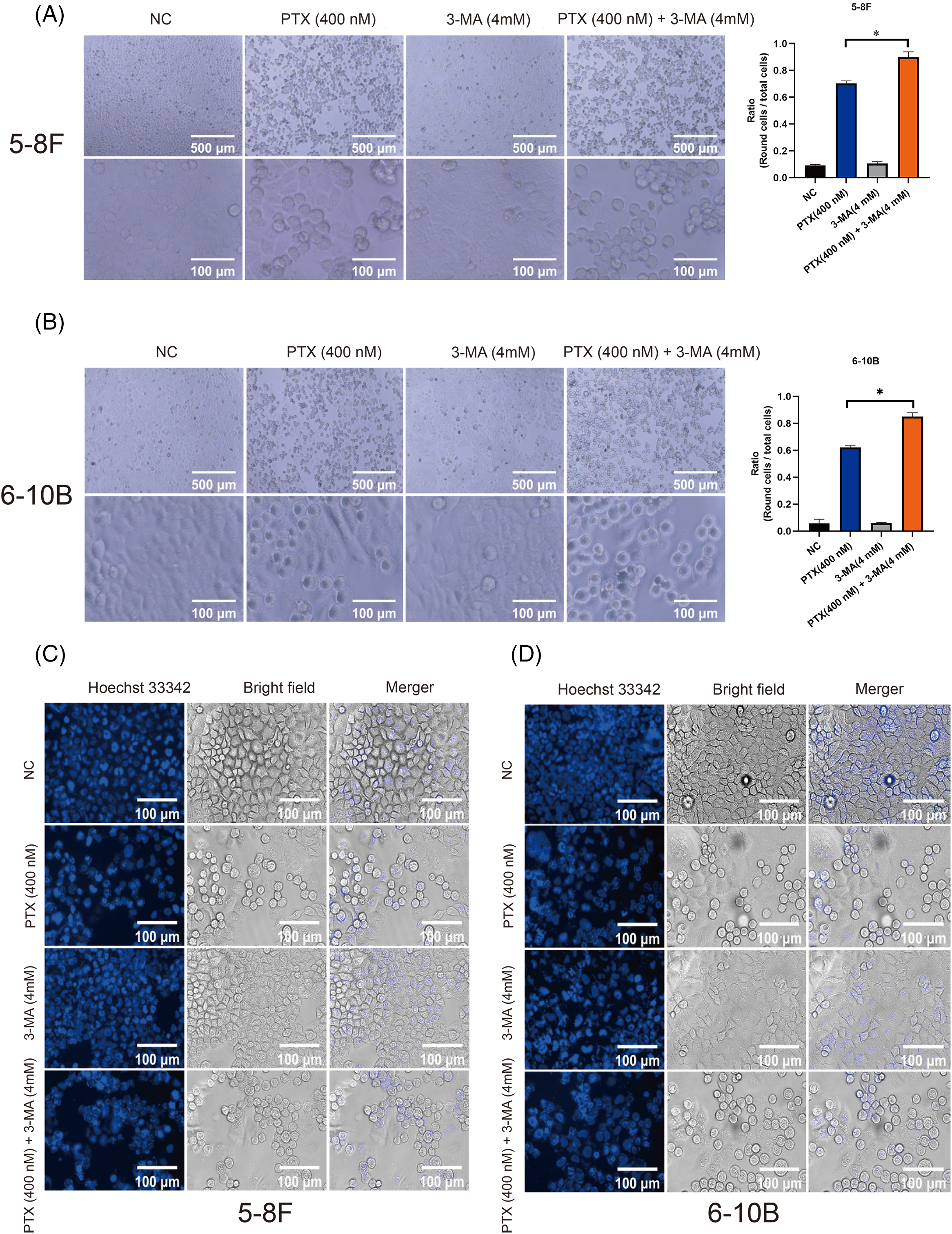
Figure 2: 3-MA amplifies the morphological transformations induced by paclitaxel and promotes nuclear condensation in NPC cells. Treatment with 3-MA significantly augmented the morphological changes induced by paclitaxel in both 5–8 F and 6–10 B cells (A and B). The data, represented as mean ± SD (n = 3), underwent analysis through two-way ANOVA and Tukey’s test. Compare with PTX (400 nM) group, *p < 0.05. Top row scale bar = 500 μm, bottom row scale bar = 100 μm. Additionally, the presence of 3-MA increased nuclear condensation in spherical 5–8 F and 6–10 B cells (C and D). Scale bar = 100 μm.
3-MA does not alter paclitaxel-induced G2/M phase arrest in NPC cells
After 24 h of drug treatment, the profile of cell cycle phases in NPC cells was assessed. Treatment with paclitaxel led to a significant accumulation of NPC cells at the G2/M phase. Similarly, the combination of paclitaxel and 3-MA induced G2/M phase arrest. The PTX-alone and the PTX combined with 3-MA groups did not exhibit any notable difference (Fig. 3A). Furthermore, the inclusion of paclitaxel increased the fraction of cells in the SubG1 phase in both the PTX-alone and the PTX combined with 3-MA groups. Notably, the PTX combined with 3-MA groups exhibited a significantly higher fraction of cells in the SubG1 phase compared to the PTX-alone groups. These cell cycle analyses did not reveal any impact of 3-MA on the distribution of the cell cycle, while it promotes cell death in NPC cells exhibiting G2/M phase arrest, consequently raising the fraction of cells in the SubG1 phase, which may include apoptotic cells.
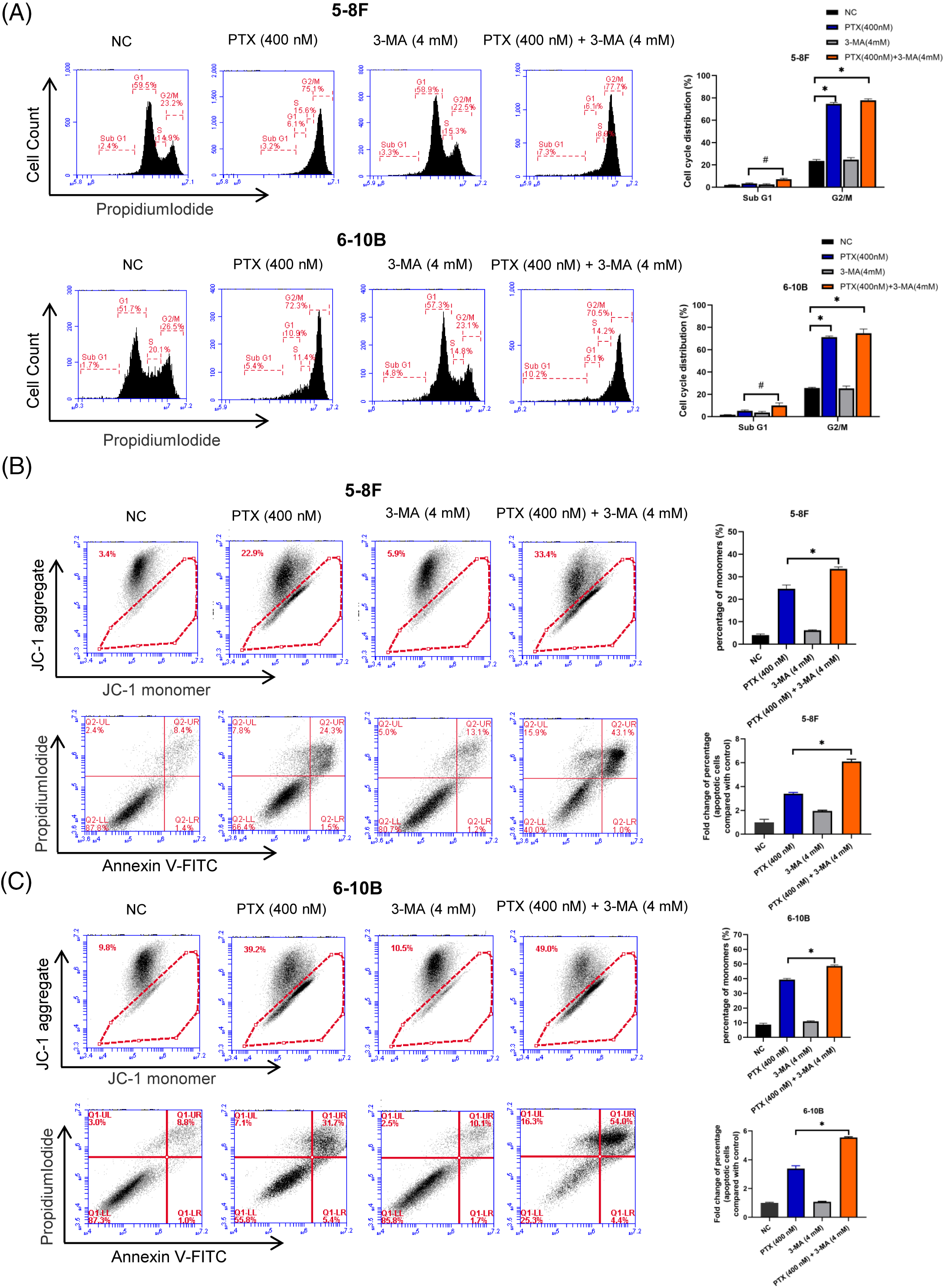
Figure 3: 3-MA potentiates paclitaxel-induced apoptosis of nasopharyngeal carcinoma cells. (A) Paclitaxel treatment resulted in G2/M phase arrest, whereas the addition of 3-MA notably increased the fraction of cells in the SubG1 phase after 24 h. The data, represented as mean ± SD (n = 3), underwent analysis through two-way ANOVA and Tukey’s test. Compare with NC group, *p < 0.05. Compare with PTX (400 nM), #p < 0.05. (B and C) 3-MA significantly potentiate paclitaxel-induced apoptosis. When cells were concurrently treated with paclitaxel and 3-MA, there was a notable increase in the proportion of JC-1 monomers compared to cells treated with paclitaxel alone (Figs. 3B and 3C, top rows), accompanied by a significant rise in the percentage of dead cells compared to the paclitaxel-treated alone group (Figs. 3B and 3C, bottom rows). The data, represented as mean ± SD (n = 3), underwent analysis through two-way ANOVA and Tukey’s test. Compare with PTX (400 nM), *p < 0.05.
3-MA potentiates the apoptosis induced by paclitaxel in NPC cells
Apoptosis has been established as the primary outcome of mitotic catastrophe [27]. Cells undergoing early apoptosis typically exhibit dysregulation of the MMP, leading to the accumulation of monomeric JC1 in degenerating mitochondria and subsequent loss of membrane potential [28]. In both the PTX group (treated with paclitaxel at a concentration of 400 nM) and the PTX group with 3-MA (4 mM), there was an increase in the percentage of JC-1 monomers in NPC cells. Moreover, the PTX combined with 3-MA groups exhibited a higher percentage of JC-1 monomers than with paclitaxel alone (Figs. 3B and 3C, upper parts). Results obtained from the Annexin V-FITC/PI assay demonstrated an elevated proportion of apoptotic cells within the PTX group treated with paclitaxel at a concentration of 400 nM and the PTX group with 3-MA (4 mM). Furthermore, simultaneous treatment of cells with paclitaxel and 3-MA led to a significantly higher percentage of apoptotic cells than with paclitaxel alone. These data demonstrate a significant increase in paclitaxel-induced apoptosis in NPC cells by 3-MA (Figs. 3B and 3C, lower part).
3-MA potentiates cell death induced by paclitaxel and activates CDK1
Taking into account the initiation period of cell death triggered by paclitaxel and the pivotal function of CDK1-cyclin B1 in regulating mitosis, we analyzed the pattern of CDK1-cyclin B1 and CDK1 phosphorylation states at 16 and 24 h. In NPC cells exposed to paclitaxel, a more significant activation of CDK1 was observed (with increased Thr161 phosphorylation and reduced Thr14 and Tyr15 phosphorylation compared to the NC groups). Upon treatment with both paclitaxel and 3-MA, there was a notable increase in Thr161 phosphorylation of CDK1 in cells compared to those treated solely with paclitaxel (Figs. 4A and 4B). Additionally, NPC cells in the PTX-alone and the PTX combined with 3-MA groups showed a significant reduction of the anti-apoptosis protein MCL-1 and increased cleavage of PARP at 24 and 48 h (Figs. 5A and 5B). Bax facilitates apoptosis, whereas Bcl-2 plays an anti-apoptotic role. Hence, we also investigated the protein levels of Bax and Bcl-2. The fold change relative to the negative control (NC) was calculated, and the Bax/Bcl-2 ratio, representing the apoptotic index, reflected cell susceptibility to apoptosis [29]. The Bax/Bcl-2 ratio in the PTX (400 nM) + 3-MA (4 mM) groups exhibited a significant increase compared to the NC groups at 24 and 48 h. In contrast, the Bax/Bcl-2 ratio in the PTX (400 nM) groups showed a significant increase only at 24 h.
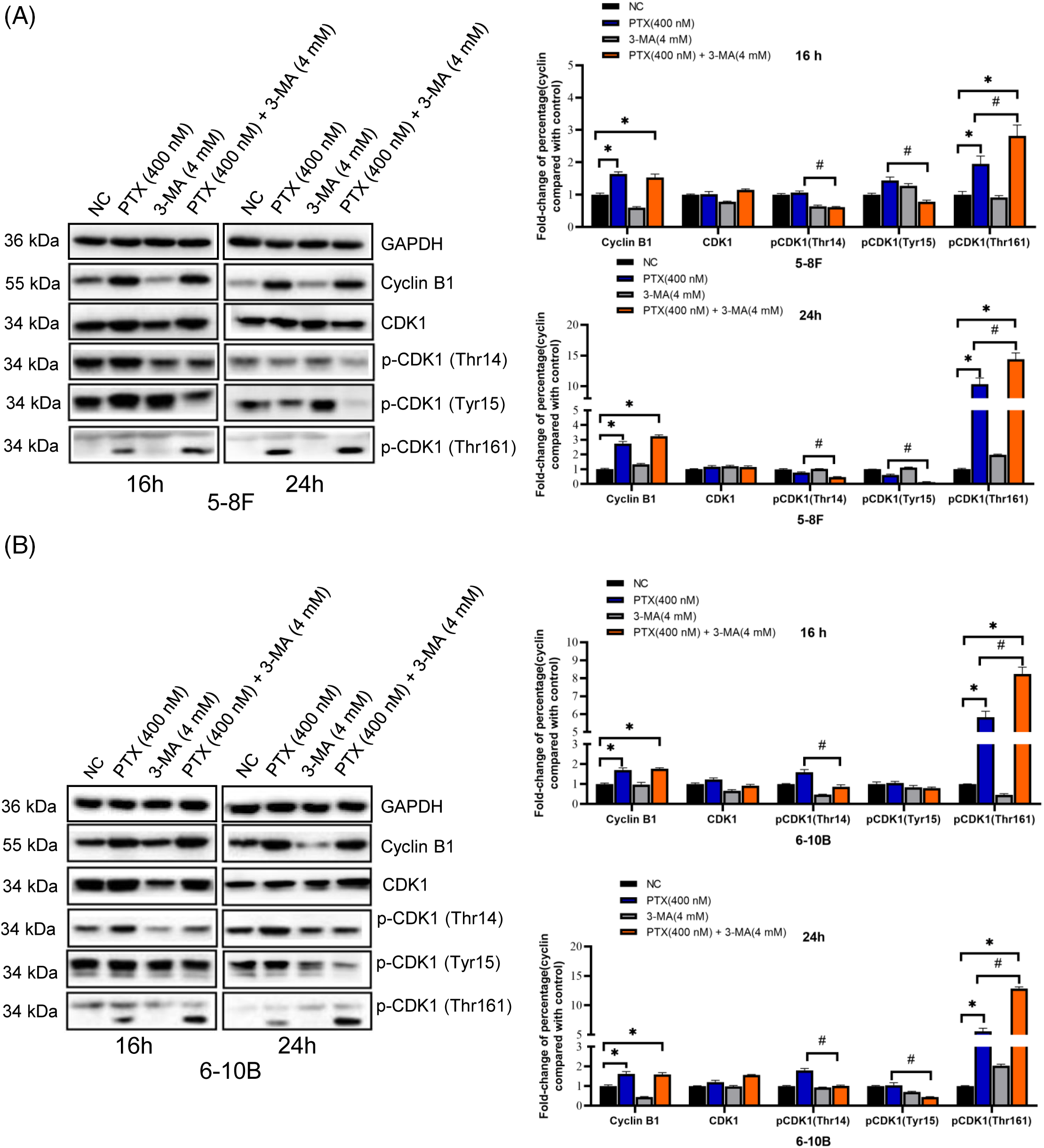
Figure 4: 3-MA promote paclitaxel-induced CDK1 activation at 16 and 24 h. In both the NPC cell lines 5–8 F (A) and 6–10 B (B), the PTX-alone and the PTX combined with 3-MA groups exhibited a significant increase in cyclin B1 protein accumulation compared to the NC groups. Additionally, the phosphorylation pattern of CDK1 shifted from an inhibitory pattern to an activating pattern at 16 and 24 h. The data, represented as mean ± SD (n = 3), underwent analysis through two-way ANOVA and Tukey’s test. Compare with NC group, *p < 0.05. Compare with PTX (400 nM), #p < 0.05. Protein bands were normalized to GAPDH and subsequently compared with the control group. p-, phosphorylated.
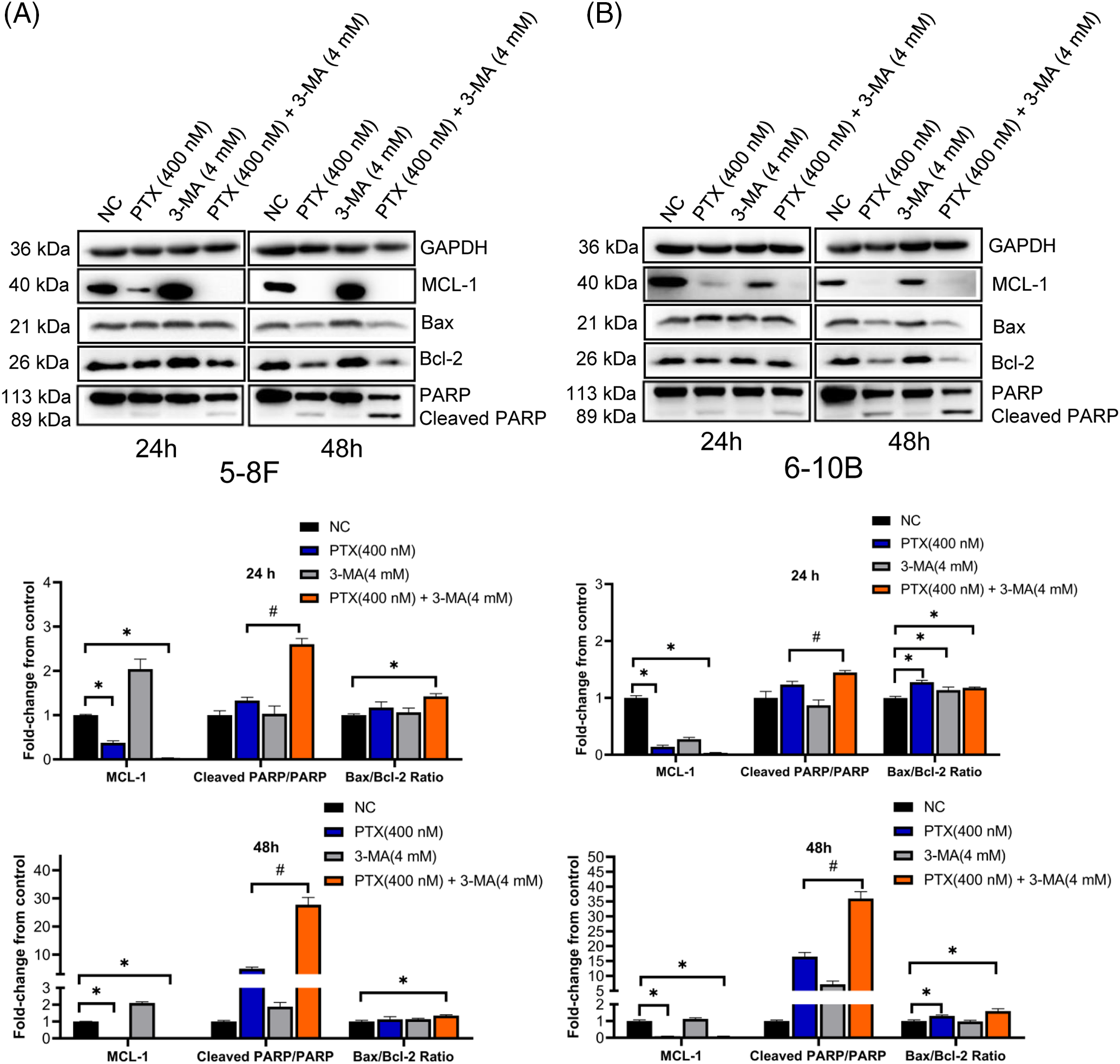
Figure 5: 3-MA enhances paclitaxel-activated apoptosis signal in NPC cells. NPC cell line 5–8 F (A) and 6–10 B (B) exposed to paclitaxel all presented low level MCL-1 expression and high PARP cleavage. The data, represented as mean ± SD (n = 3), underwent analysis through two-way ANOVA and Tukey’s test. Compare with NC group, *p < 0.05. Compare with PTX (400 nM), #p < 0.05. Protein bands were normalized to GAPDH and subsequently compared with the control group.
All these western blot results suggest that the combination of paclitaxel with 3-MA activates the CDK1-related apoptosis cascade and potentiates paclitaxel-induced NPC cell death.
It is now understood that paclitaxel induces abnormalities in the cell division process and triggers the regulatory mechanisms of apoptosis, with CDK1-cyclin B1 playing a key role in determining cell fate between survival and death [30]. Typically, cells undergo prolonged mitotic arrest after exposure to antimitotic drugs, leading to the activation of CDK1 and related cell death signals [31].
In cancer treatment, paclitaxel drug resistance remains a major concern. To enhance cancer therapeutic effects, combining drugs with diverse mechanisms has emerged as a promising and alternative tactic [32]. For instance, a panel of tyrosine kinase inhibitors (lapatinib and poziotinib) potentiates the antitumor properties of paclitaxel treatment in ovarian cancer cells by direct inhibiting the activity of paclitaxel-induced ATP-binding cassette sub-family B member 1 (ABCB1) activity. It has been suggested that combining paclitaxel therapy with the United States Food and Drug Administration (FDA)-approved lapatinib is a promising strategy for patients with recurrent ovarian cancer [33]. Additionally, applying the P-glycoprotein (Pgp) inhibitor NSC23925 in combination with paclitaxel has been shown to improve the long-term clinical outcomes of ovarian cancer patients [34]. In another study, a combination of the neuroleptic agent Penfluridol (PFL) with paclitaxel significantly reduced the survival of paclitaxel-resistant breast cancer cells [35].
In our investigation, we discovered that combining paclitaxel with 3-MA could represent a promising chemotherapy treatment for NPC, given the low toxicity of 3-MA and the effective improvement of paclitaxel lethality. We observed that the addition of 3-MA not only amplified the paclitaxel’s ability to inhibit the replication of nasopharyngeal carcinoma cells but also resulted in a higher number of cells entering mitotic catastrophe within 24 h. Once cells experience mitotic catastrophe, a significant portion of them undergo irreversible apoptosis [36]. The results from flow cytometry analysis of JC-1 and apoptosis further substantiated that 3-MA could induce more apoptosis in nasopharyngeal carcinoma cells. Additionally, by analyzing the phosphorylation of CDK1, we hypothesized that 3-MA could accelerate the transition of nasopharyngeal carcinoma cells, arrested in the G2/M phase by paclitaxel, towards cell cycle regulation, mitotic catastrophe and trigger the apoptotic pathway regulated by CDK1 [37,38]. This shortened the time required for paclitaxel to exert its effects on nasopharyngeal carcinoma cells. However, the potential mechanism behind this phenomenon is still unknown.
3-MA and paclitaxel appear to intersect in their effects on cell cycle progression. We hypothesize that these convergent points could be crucial in understanding how 3-MA enhances paclitaxel-mediated cytotoxicity against NPC cells. It is now understood during the G2 phase, attenuation of PI3K signaling promotes the translocation of Forkhead box O (FoxO) proteins from the cytosol to the nucleus [39]. This, in turn, regulates the expression of Polo-like kinase 1 (PLK1) and cyclin B1 [40]. Increased cyclin B1 may lead to the activation of CDK1, thereby accelerating paclitaxel-induced cell cycle halt and apoptosis. Besides, as cells enter mitosis, CDK1 takes over most of the regulatory functions of mammalian target of rapamycin complex 1 (mTORC1) [41]. This includes phosphorylating Autophagy-related protein 13 (ATG13), Unc-51-like kinase (ULK1), Autophagy-related protein 14 (ATG14), Vacuolar protein sorting 34 (Vps34), and 4E-binding proteins (4E-BPs), resulting in the repression of autophagy to protect nuclear components. Consequently, the dissociation of the autophagy initiation complex Beclin1-Vps34 occurs, releasing a pool of free Beclin1 [42]. The cleavage products of Beclin1 sensitize cells to apoptosis [43]. Importantly, 3-MA inhibits the activity of class III PI3K Vps34 in the Beclin1-Vps34 complex. Last but not least, PI3Ks contribute to metaphase regulation and spindle dynamic [44]. Inhibition of class I PI3K leads to mislocalization of the dynein-associated dynactin and defects in spindle orientation [45]. Importantly, the combination of 3-MA and paclitaxel was found to exacerbate improper spindle-chromosome attachment during metaphase and accelerates the activation of CDK1 and related apoptosis signals. Furthermore, the interplay between the PI3K pathway, autophagy, cell cycle, and apoptosis could be the key to understanding how 3-MA enhances the effect of paclitaxel, particularly the coordination of PI3Ks and CDK1 in cell division. These aspects will be further investigated in our future research.
The current study showed that 5–8 F and 6–10 B cells exposed to paclitaxel and nontoxic doses of 3-MA exhibited increased cell death, potentially through the potentiation and triggering of the CDK1-related apoptosis signal cascade by 3-MA. This suggests that 3-MA could potentially serve as a sensitizer for paclitaxel in NPC chemotherapy.
Acknowledgement: None.
Funding Statement: This study was supported by the Science and Technology Innovation Program of Hunan Province (Grant Numbers: 2021SK1014 and 2022WZ1027), the Colleges and Universities of Hunan Province (Grant Number: HNJG 2020 0440), the Scientific Research Fund of Hunan Provincial Education Department (Grant Number: 21B0411), and the Scientific Research Project of Changsha Central Hospital (Number: YNKY202201).
Author Contributions: XW, YH and YY performed the experiments. LZ, XT analyzed the data. DW, LZ conducted literature search and data interpretation. DW, BJ drafted the initial manuscript. YY and BJ confirmed the authenticity of all the raw data. BJ designed and supervised the study. All authors read and approved the final manuscript.
Availability of Data and Materials: The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics Approval: Not applicable.
Conflicts of Interest: There are no conflicts of interest to report regarding the present study.
References
1. Chang ET, Ye W, Zeng YX, Adami HO. The evolving epidemiology of nasopharyngeal carcinoma. Cancer Epidemiol Biomarkers Prev. 2021;30(6):1035–47. [Google Scholar] [PubMed]
2. Chen YP, Chan ATC, Le QT, Blanchard P, Sun Y, Ma J. Nasopharyngeal carcinoma. Lancet. 2019;394(10192):64–80. [Google Scholar] [PubMed]
3. Guan S, Wei J, Huang L, Wu L. Chemotherapy and chemo-resistance in nasopharyngeal carcinoma. Eur J Med Chem. 2020;207:112758. [Google Scholar] [PubMed]
4. Chang C, Li X, Cao D. Combination of gemcitabine, nab-paclitaxel, and S-1(GAS) as the first-line treatment for patients with locally advanced or advanced pancreatic ductal adenocarcinoma: study protocol for an open-label, single-arm phase I study. BMC Cancer. 2021;21(1):545. [Google Scholar] [PubMed]
5. Nussinov R, Tsai CJ, Jang H. Anticancer drug resistance: an update and perspective. Drug Resist Updat. 2021;59:100796. [Google Scholar] [PubMed]
6. Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem. 2010;285(14):10850–61. [Google Scholar] [PubMed]
7. Zhang Y, Zhou XA, Zhang C, Lai D, Liu D, Wu Y. Vitamin B3 inhibits apoptosis and promotes autophagy of islet β cells under high glucose stress. BIOCELL. 2023;47(4):859–68. doi:10.32604/biocell.2023.026429. [Google Scholar] [CrossRef]
8. Mishra R, Patel H, Alanazi S, Kilroy MK, Garrett JT. PI3K inhibitors in cancer: clinical implications and adverse effects. Int J Mol Sci. 2021;22(7):3464. [Google Scholar] [PubMed]
9. Glaviano A, Foo ASC, Lam HY, Yap KCH, Jacot W, Jones RH, et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol Cancer. 2023;22(1):138. [Google Scholar] [PubMed]
10. Hsin IL, Shen HP, Chang HY, Ko JL, Wang PH. Suppression of PI3K/Akt/mTOR/c-Myc/mtp53 positive feedback loop induces cell cycle arrest by dual PI3K/mTOR inhibitor PQR309 in endometrial cancer cell lines. Cells. 2021;10(11):2916. [Google Scholar] [PubMed]
11. Takeshita N, Enokida T, Okano S, Fujisawa T, Wada A, Sato M, et al. Induction chemotherapy with paclitaxel, carboplatin and cetuximab for locoregionally advanced nasopharyngeal carcinoma: a single-center, retrospective study. Front Oncol. 2022;12:951387. [Google Scholar] [PubMed]
12. Chen Q, Xu S, Liu S, Wang Y, Liu G. Emerging nanomedicines of paclitaxel for cancer treatment. J Control Release. 2022;342:280–94. [Google Scholar] [PubMed]
13. Smith ER, Wang JQ, Yang DH, Xu XX. Paclitaxel resistance related to nuclear envelope structural sturdiness. Drug Resist Updat. 2022;65:100881. [Google Scholar] [PubMed]
14. Zhao B, Gu Z, Zhang Y, Li Z, Cheng L, Li C, et al. Starch-based carriers of paclitaxel: a systematic review of carriers, interactions, and mechanisms. Carbohydr Polym. 2022;291:119628. [Google Scholar] [PubMed]
15. Bai Z, Zhou Y, Peng Y, Ye X, Ma L. Perspectives and mechanisms for targeting mitotic catastrophe in cancer treatment. Biochim Biophys Acta Rev Cancer. 2023;1878(5):188965. [Google Scholar] [PubMed]
16. Chiou LW, Chan CH, Jhuang YL, Yang CY, Jeng YM. DNA replication stress and mitotic catastrophe mediate sotorasib addiction in KRASG12C-mutant cancer. J Biomed Sci. 2023;30(1):50. [Google Scholar] [PubMed]
17. Enserink JM, Chymkowitch P. Cell cycle-dependent transcription: the cyclin dependent kinase cdk1 is a direct regulator of basal transcription machineries. Int J Mol Sci. 2022;23(3):1293. [Google Scholar] [PubMed]
18. Kalous J, Jansova D, Susor A. Role of cyclin-dependent kinase 1 in translational regulation in the M-phase. Cells. 2020;9(7):1568. [Google Scholar] [PubMed]
19. Dumitru AMG, Compton DA. Identifying cyclin A/Cdk1 substrates in mitosis in human cells. Methods Mol Biol. 2022;2415:175–82. [Google Scholar] [PubMed]
20. Haneke K, Schott J, Lindner D, Hollensen AK, Damgaard CK, Mongis C, et al. CDK1 couples proliferation with protein synthesis. J Cell Biol. 2020;219(3):e201906147. [Google Scholar]
21. Ren L, Yang Y, Li W, Zheng X, Liu J, Li S, et al. CDK1 serves as a therapeutic target of adrenocortical carcinoma via regulating epithelial-mesenchymal transition, G2/M phase transition, and PANoptosis. J Transl Med. 2022;20(1):444. [Google Scholar] [PubMed]
22. Hsu JL, Leu WJ, Hsu LC, Ho CH, Liu SP, Guh JH. Phosphodiesterase type 5 inhibitors synergize vincristine in killing castration-resistant prostate cancer through amplifying mitotic arrest signaling. Front Oncol. 2020;10:1274. [Google Scholar] [PubMed]
23. Clarke PR, Allan LA. Destruction’s our delight: controlling apoptosis during mitotic arrest. Cell Cycle. 2010;9(20):4035–6. [Google Scholar] [PubMed]
24. Bhosale PB, Kim HH, Abusaliya A, Jeong SH, Park MY, Kim HW, et al. Inhibition of cell proliferation and cell death by apigetrin through death receptor-mediated pathway in hepatocellular cancer cells. Biomolecules. 2023;13(7):1131. [Google Scholar] [PubMed]
25. Smith ER, Xu XX. Breaking malignant nuclei as a non-mitotic mechanism of taxol/paclitaxel. J Cancer Biol. 2021;2(4):86–93. [Google Scholar] [PubMed]
26. Mascaraque M, Delgado-Wicke P, Damian A, Lucena SR, Carrasco E, Juarranz A. Mitotic catastrophe induced in hela tumor cells by photodynamic therapy with methyl-aminolevulinate. Int J Mol Sci. 2019;20(5):1229. [Google Scholar] [PubMed]
27. Slade D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020;34(5–6):360–94. [Google Scholar] [PubMed]
28. Yu Z, Xiao Z, Guan L, Bao P, Yu Y, Liang Y, et al. Translocation of gasdermin D induced mitochondrial injury and mitophagy mediated quality control in lipopolysaccharide related cardiomyocyte injury. Clin Transl Med. 2022;12(8):e1002. [Google Scholar] [PubMed]
29. Schroer J, Warm D, De Rosa F, Luhmann HJ, Sinning A. Activity-dependent regulation of the BAX/BCL-2 pathway protects cortical neurons from apoptotic death during early development. Cell Mol Life Sci. 2023;80(6):175. [Google Scholar] [PubMed]
30. Maryu G, Yang Q. Nuclear-cytoplasmic compartmentalization of cyclin B1-Cdk1 promotes robust timing of mitotic events. Cell Rep. 2022;41(13):111870. [Google Scholar] [PubMed]
31. Darweesh O, Al-Shehri E, Falquez H, Lauterwasser J, Edlich F, Patel R. Identification of a novel Bax-Cdk1 signalling complex that links activation of the mitotic checkpoint to apoptosis. J Cell Sci. 2021;134(8):jcs244152. [Google Scholar] [PubMed]
32. Pomeroy AE, Schmidt EV, Sorger PK, Palmer AC. Drug independence and the curability of cancer by combination chemotherapy. Trends Cancer. 2022;8(11):915–29. [Google Scholar] [PubMed]
33. McCorkle JR, Gorski JW, Liu J, Riggs MB, McDowell AB, Lin N, et al. Lapatinib and poziotinib overcome ABCB1-mediated paclitaxel resistance in ovarian cancer. PLoS One. 2021;16(8):e0254205. [Google Scholar] [PubMed]
34. Yang X, Shen J, Gao Y, Feng Y, Guan Y, Zhang Z, et al. Nsc23925 prevents the development of paclitaxel resistance by inhibiting the introduction of P-glycoprotein and enhancing apoptosis. Int J Cancer. 2015;137(8):2029–39. [Google Scholar] [PubMed]
35. Gupta N, Gupta P, Srivastava SK. Penfluridol overcomes paclitaxel resistance in metastatic breast cancer. Sci Rep. 2019;9(1):5066. [Google Scholar] [PubMed]
36. Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ. 2008;15(7):1153–62. [Google Scholar] [PubMed]
37. Hou Y, Allan LA, Clarke PR. Phosphorylation of XIAP by CDK1-cyclin-B1 controls mitotic cell death. J Cell Sci. 2017;130(2):502–11. [Google Scholar] [PubMed]
38. Solanki P, Rana N, Jha PC, Manhas A. A comprehensive analysis of the role of molecular docking in the development of anticancer agents against the cell cycle CDK enzyme. BIOCELL. 2023;47(4):707–29. [Google Scholar]
39. Schmitt-Ney M. The FOXO’s advantages of being a family: considerations on function and evolution. Cells. 2020;9(3):787. [Google Scholar] [PubMed]
40. Ke ZB, Cai H, Wu YP, Lin YZ, Li XD, Huang JB, et al. Identification of key genes and pathways in benign prostatic hyperplasia. J Cell Physiol. 2019;234(11):19942–50. [Google Scholar] [PubMed]
41. Odle RI, Florey O, Ktistakis NT, Cook SJ. CDK1, the other ‘master regulator’ of autophagy. Trends Cell Biol. 2021;31(2):95–107. [Google Scholar] [PubMed]
42. Furuya T, Kim M, Lipinski M, Li J, Kim D, Lu T, et al. Negative regulation of Vps34 by Cdk mediated phosphorylation. Mol Cell. 2010;38(4):500–11. [Google Scholar] [PubMed]
43. Li X, Su J, Xia M, Li H, Xu Y, Ma C, et al. Caspase-mediated cleavage of Beclin1 inhibits autophagy and promotes apoptosis induced by S1 in human ovarian cancer SKOV3 cells. Apoptosis. 2016;21(2):225–38. [Google Scholar] [PubMed]
44. Jiang W, Yang X, Shi K, Zhang Y, Shi X, Wang J, et al. MAD2 activates IGF1R/PI3K/AKT pathway and promotes cholangiocarcinoma progression by interfering USP44/LIMA1 complex. Oncogene. 2023;42(45):3344–57. [Google Scholar] [PubMed]
45. Gulluni F, Martini M, de Santis MC, Campa CC, Ghigo A, Margaria JP, et al. Mitotic spindle assembly and genomic stability in breast cancer require PI3K-C2α scaffolding function. Cancer Cell. 2017;32(4):444–59.E7. doi:10.1016/j.ccell.2017.09.002. [Google Scholar] [PubMed] [CrossRef]
Supplementary Materials
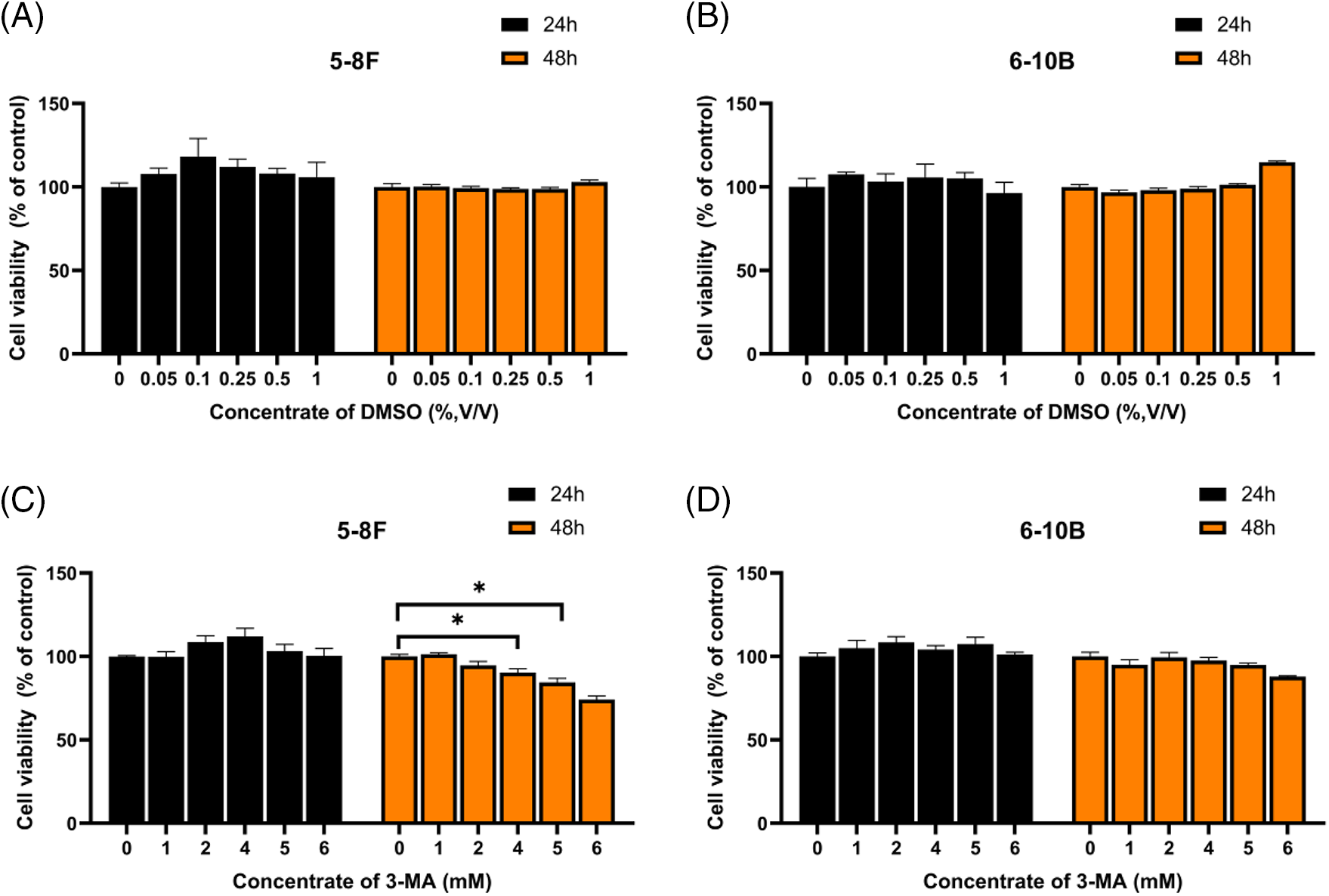
Figure S1: DMSO at concentrations below 1% (v/v) did not exhibit any inhibition of cell proliferation (A and B). At a concentration of 4 mM, 3-MA slightly inhibited the proliferation of 5–8F cells but had no impact on 6–10B cells (C and D). The data, represented as mean ± SD (n = 3), underwent analysis through two-way ANOVA and Tukey’s test. Compare with NC group, *p < 0.05.
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools