
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Gp78 regulates PMP22 and causes ER stress and autophagy in EV71-VP1-overexpressing mouse Schwann cells
1 Pediatric Emergency Department, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangdong Provincial Clinical Research Center for Child Health, Guangzhou, China
2 Department of Child Health Care, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangdong Provincial Clinical Research Center for Child Health, Guangzhou, China
3 Pediatric Neurology Department, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangdong Provincial Clinical Research Center for Child Health, Guangzhou, China
* Corresponding Authors: SIDA YANG. Email: ; PEIQING LI. Email:
# These authors are co-first authors and contributed equally to this work
BIOCELL 2024, 48(4), 653-664. https://doi.org/10.32604/biocell.2024.044856
Received 10 August 2023; Accepted 23 January 2024; Issue published 09 April 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: During Enterovirus type 71 (EV71) infection, the structural viral protein 1 (VP1) activates endoplasmic reticulum (ER) stress associated with peripheral myelin protein 22 (PMP22) accumulation and induces autophagy. However, the specific mechanism behind this process remains elusive. Methods: In this research, we used the VP1-overexpressing mouse Schwann cells (SCs) models co-transfected with a PMP22 silencing or Autocrine motility factor receptor (AMFR/gp78) overexpressing vector to explore the regulation of gp78 on PMP22 and its relationship with autophagy and apoptosis. Results: The activity of gp78 could be influenced by EV71-VP1, leading to a decrease in the ubiquitination and degradation of PMP22, resulting in PMP22 accumulation in ER. In VP1-overexpressing mouse SCs, all three ER stress sensors, including pancreatic endoplasmic reticulum kinase (PERK), activating transcription factor 6 (ATF6) and inositol-requiring enzyme 1 (IRE1) and the related downstream signals (C/EBP-homologous protein (CHOP) and Caspase 12) were activated, as well as the ER-resident chaperone Glucose-regulated protein 78 (GRP78). In addition, VP1 upregulated the autophagy marker Microtubule-associated protein 1 light chain 3 beta (LC3B), while PMP22 silencing or gp78 overexpression reversed the phenomenon. Meanwhile, PMP22 silencing or gp78 overexpression increased proliferation of EV71-VP1-transfected mouse SCs. Conclusion: Gp78 could regulate PMP22 accumulation through ubiquitination degradation and cause ER stress and autophagy in EV71-VP1-overexpressing mouse SCs. Therefore, the gp78/PMP22/ER stress axis might emerge as a promising therapeutic target for myelin and neuronal damage induced by EV71 infection.Keywords
Supplementary Material
Supplementary Material FileEnterovirus 71 (EV71) stands out as a significant contributor to hand-foot-and-mouth disease (HFMD) among the pediatric population [1,2]. Since the early 1980s, EV71 has triggered numerous extensive global outbreaks among children, particularly in the Asia-Pacific region, evolving into a significant public health concern within the impacted nations [3,4]. Although most infected patients exhibit a mild and self-limiting illness, EV71 can sometimes cause fatal neurological diseases, including meningitis, brainstem encephalitis, encephalomyelitis, and acute flaccid paralysis [5,6]. Even more importantly, there is still no effective antiviral therapy against EV71. Therefore, it is crucial to explore the mechanisms of EV71 infection-induced neurologic injury.
Belonging to the Picornaviridae family, EV71 is a single-stranded, positive-sense RNA virus. The capsids of EV71 are comprised of four structural proteins (VP1 to VP4), with VP1 assuming a pivotal role in both virion formation and intracellular entry [7]. VP1 can also promote viral replication by inducing cell autophagy [8]. Previous studies demonstrated that EV71 possesses strong neurotropism, and retrograde axonal transport is the major transmission route [9–11]. Clinical research by the authors’ group revealed the manifestations and the neuroimaging changes of EV71-infected patients and indicated that the neural invasion of EV71 infection was from peripheral to central nerves [12,13].
The neuron’s axons are wrapped by the Schwann cells (SCs) membrane (myelin), and SCs play central roles in the propagation of the nervous signals in the peripheral nerves [14]. Myelination increases the speed of action potential propagation and plays an important role in the development and regeneration of neuron axons [15]. Peripheral myelin protein 22 (PMP22) is a tetraspan membrane protein that constitutes 2%–5% of total peripheral myelin protein [16]. It plays crucial roles in developing and maintaining compact myelin sheaths in the peripheral nervous system [17]. Accumulated mutated PMP22 in the endoplasmic reticulum (ER) leads to inherited neuropathies like Charcot-Marie-Tooth (CMT) disease [17,18]. A previous study by the authors’ group showed that EV71 VP1 could upregulate the expression of ER stress-mediated PMP22, induce the accumulation of PMP22 in the ER, and ultimately promote mouse SC autophagy [8]. Methyltransferase-like protein 3 (METTL3) is required for SC autophagy during EV71 infection [19]. Nevertheless, the mechanism through which PMP22 accumulates in the ER is unknown.
In normal SCs, approximately 80% of the recently synthesized PMP22 undergoes degradation predominantly within a 30-min timeframe, facilitated by the ubiquitin–proteasome system (UPS)-endoplasmic reticulum-associated degradation (ERAD) system [20,21]. Recently, it has been demonstrated that the UPS plays a role in supervising crucial quality control stages in the formation of protein complexes [22]. Autocrine motility factor receptor (AMFR/gp78), as an ER membrane-anchored ubiquitin ligase (E3), functions as a crucial element in ERAD. Its role involves averting the harmful accumulation of various misfolded proteins through a multifactorial mechanism [23]. As demonstrated in an earlier study using a mouse model of CMT disease type 1A, diminished proteasome function in neuropathological nerves is linked to the buildup of ubiquitinated substrates. Additionally, there is a recruitment of components from the proteasomal pathway to the aggregates [24]. Therefore, a decreased proteasome activity could lead to insufficient ubiquitylation and degradation of PMP22 and promote PMP22 aggregate formation, negatively impact cellular proteostasis, and result in SCs dysfunction. Our team recently discovered positive regulation of myelination and ubiquitin-specific protease binding enrichment in EV infected patients with neurological involvement, as well as gp78. Pleckstrin homology containing family A, number 4 (PLEKHA4), which is a regulator of the E3 ubiquitin ligase and controlling disheveled ubiquitination, was also found to be significantly enriched [25]. Those results indicated that abnormal PMP22 ubiquitination and degradation may be involved in EV induced neuronal damage.
The ER is a vital subcellular component in eukaryotic cells, which plays an important role in the synthesis, post-translational modifications, and proper folding of proteins, which are essential for cell survival. When the cells are subjected to various pathogenic stress signals, several misfolded and unfolded proteins gather within the ER, resulting in ER dysfunction, which is called endoplasmic reticulum stress (ERs) [26]. During this process, ER has two ways to deal with accumulated proteins: the unfolded protein response (UPR) and the ERAD, which promotes the refolding degradation of the misfolded and unfolded proteins, achieving ER homeostasis. Three transmembrane proteins of the ER—activating transcription factor 6 (ATF6), pancreatic endoplasmic reticulum kinase (PERK), and inositol-requiring enzyme 1α (IRE1)—initiate the UPR by activating their associated downstream signals [27]. If the ERs persists and the accumulated proteins surpass the processing potential of the UPR, the cellular functions deteriorate, ultimately leading to cell death [28]. Evidence shows that the ERs is a major contributor to many diseases, including diabetes, depression, neurodegeneration, cancer, and infectious diseases [27,29,30]. Previous studies demonstrated that EV71 could induce ERs and modify the outcome to assist viral replication [26,31]. In addition, through ER stress-induced upregulation of PMP22, EV71 VP1 stimulates autophagy in mouse SCs [8]. Still, the underlying mechanism and the specific signal pathways remain ambiguous.
In this study, VP1-overexpressing mouse SCs were used to characterize the potential role of PMP22 on ERs and autophagy caused by EV71 infection and explore the effect of gp78-associated ERAD on PMP22 accumulation in the ER lumen. This study will help understand the molecular mechanism related to the neurotoxicity of EV71 and could provide some indication for future research on possible drug targets against EV71 infection.
Study subject and sampling in clinical analysis
An observational and forward-looking cohort investigation took place from January 2018 to December 2019 at the Guangzhou Women and Children’s Medical Center. The clinical attributes, along with the demographic and neurological symptoms of the enrolled patients, were previously detailed in other studies [25]. Pediatric patients confirmed to have an EV infection based on positive EV-Rt-PCR results were included in our study. The samples for testing included cerebrospinal fluid (CSF), anal swabs, or throat swabs. Individuals co-infected with other pathogens were not considered for the study. Patients were divided into control and NS groups according to whether they had neurological symptoms. The controls were matched by age with the patients of the NS group. For RNA-seq and MeRIP-seq analysis, lumbar puncture was performed to collect CSF samples from all individuals.
MeRIP-seq and RNA sequencing combination analysis
The CSF samples underwent total RNA extraction and purification using the Zymo RNA clean and concentrator-5 kit (Zymo Research, cat. No. R1013, Irvine, CA, USA). Subsequently, the purified RNA was utilized to generate an RNA-seq library. To conduct MeRIP-seq analysis, we subjected purified RNA to hyperthermia, cutting it into 200 nt lengths. The resulting fragments were then introduced into the precipitation buffer alongside the anti-m6A Antibody (Sigma-Aldrich, ABE572, St. Louis, Missouri, USA). Following magnetic separation and the removal of the supernatant, an RNA enzyme inhibitor was introduced and allowed to incubate for 1–3 h at 4°C. Following rinsing with high and low salt precipitation buffers, the RNA present in the eluate underwent purification and was employed for the generation of a library. Subsequently, RNA Sequencing was performed using NovaSeq (Illumina NovaSeq 6000) PE150 following the guidelines provided by the manufacturer. Subsequently, the combined analysis of MeRIP-seq and RNA-seq data (MeRIP-seq input library data) was conducted following the previously outlined methodology [25]. The level of gene expression was computed based on fragment length, and the read counts were assigned to these fragments, subsequently normalized using a variation of the FPKM method. In this context, we screened and organized differentially expressed genes based on criteria such as log2FC (log2FC greater than 0 for upregulated genes; log2FC less than 0 for downregulated genes), p-value, and False Discovery Rates (FDR) derived from RNA-seq differences. Differential RNA methylation analysis utilized the tool called “exome Peak”.
Ethics approval and EV71 isolation
Approval for this study was granted by the ethics committee at Guangzhou Women and Children’s Medical Center under the reference numbers Nos. 2017122501, VIVA07201904, and 2021019A01. All actions conducted with human participants adhered to ethical standards set by the institution, national research guidelines, and the 1964 Helsinki Declaration, along with subsequent amendments or equivalent ethical standards. Informed written consent was acquired from the legal guardians of the patients who were enrolled in the study. EV71, sourced from the Center for Disease Control and Prevention of Guangdong Province in Guangzhou, China, underwent transfection with the VP1 plasmid in mouse SCs, following a previously outlined procedure [8].
Mouse SCs (#M1700) were cultured in SCs medium (#1701), both products purchased from Sciencell Research Laboratories, Inc. (Carlsbad, CA, USA), following the manufacturer’s provided guidelines. SCs stably expressed SC marker nerve growth factor receptor (NGFR) in confocal imaging (Suppl. Fig. S1). Cell viability was 90% according to Trypan blue (Sigma, cat. no. T8154, St. Louis, Missouri, USA) staining. For the experiments, the cell concentration was standardized to 1 × 105/ml.
EV71-VP1 and gp78 overexpression vectors
RNA from EV71 was isolated employing TRIzol (Invitrogen Inc., #15596-026, Carlsbad, CA, USA), and cDNA was synthesized through reverse transcription using the ImProm-IITM Reverse Transcription System (Promega, A3800, Madison, WI, USA). Separate amplification of VP1 and gp78 cDNAs was conducted through PCR (TOYOBO Co., Ltd., Tokyo, Japan) employing the primers specified in Suppl. Table S1. After the separation and purification of PCR amplicons, they were subsequently subcloned into the pEGFP-c3 expression vector (Takara Bio, Otsu, Japan). To verify the successful cloning, sequencing was carried out by Sangon Biotech. Comparison of the results was conducted with the VP1 and gp78 cDNA sequences documented in the GenBank database (GenBank accession numbers: U55763 and Query-12825).
Small interfering RNA (siRNA) against PMP22
SiRNA targeting PMP22 (siPMP22, sc-42037) was utilized at a concentration of 100 nmol and introduced into the cells using the siRNA transfection reagent (sc-37007), both products obtained from Santa Cruz Biotechnology, Inc. (CA, USA). The transfection process took place 24 h before the commencement of the experiments.
Experimental grouping and cell transfection
The mouse SCs were divided into five groups. The PEGFP-C3+EV71-VP1 overexpression group (VP1 group) was the experimental group. In order to investigate the function of PMP22 and gp78 on EV71 infection, the VP1 group cells were also transfected with siPMP22 (VP1+siPMP22 group) or gp78 overexpression vector (VP1+OE-gp78 group). Blank control (Ctrl group) consisted of normal cells, while negative control (NC group) included cells transfected with PEGFP-C3. The cell lines were passaged the day prior to transfection to achieve a confluence level of 70%–80%. During transfection, Lipofectamine 2000 (#11668019) and Opti-MEM medium (#31985070) were employed, following the provided manufacturer’s instructions. Both products were purchased from Invitrogen Inc., Carlsbad, CA, USA.
Cell counting Kit‑8 (CCK‑8) assay
Cell harvesting occurred 48 h post-transfection, spanning the period from 0 to 72 h. The cell concentration was standardized to 1 × 105/m, and approximately 1 × 104 cells were collected. Following the combination with 10 µl of CellTiter96AQ Single solution cell proliferation detection reagent (1:10, Promega, #G3582, Madison, WI, USA), the cells underwent a 4-h incubation period. Subsequently, a Multiscan MK3 microplate reader (Thermo Fisher Scientific, Waltham, MA, USA) was utilized to scan and quantify the optical density at 490 nm.
PBS was used to rinse cultured cells, eliminating the culture medium. Following this, the cells were subjected to fixation in 4% paraformaldehyde for 25 min at 4°C, followed by two washes with PBS. Afterward, the cells underwent labeling for DNA fragmentation through terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL; Promega, #G3250, Madison, WI, USA), adhering to the guidelines provided by the manufacturer. Cellular images were acquired employing an inverted light microscope CKX41 U-CTR30-2 (Olympus, Tokyo, Japan) and a fluorescence microscope DMI6000B (Leica Microsystems, Wetzlar, Germany).
The cells were subjected to RNA extraction using Trizol reagent (Invitrogen Inc., #15596-026, Carlsbad, CA, USA), following the provided instructions. The obtained RNA was then reverse transcribed into cDNA utilizing PrimeScript II 1st Strand cDNA Synthesis Kit (Takara Bio, #D6210A, Otsu, Japan). Utilizing SYBR® Premix Ex Taq™ II (Perfect Real Time from Takara Bio), qPCR was conducted in accordance with the provided manufacturer’s guidelines. The primer sequences can be found in Suppl. Table S2. β-actin was utilized as the internal reference.
Lysis of mouse SCs was carried out by employing a RIPA lysis and extraction buffer (#89900). The protein concentration within the lysates was assessed using the Micro BCA Protein Assay Kit (#23235). All products were purchased from Thermo Fisher Scientific, Waltham, MA, USA. Subsequently, the proteins underwent separation via SDS‑PAGE and transfer onto polyvinylidene fluoride membranes. These membranes underwent a 1-h blocking step at room temperature (RT), followed by incubation with a primary antibody as detailed in Suppl. Table S3. After three cold TBST washes (a mixture of Tween 20 and Tris-Buffered Saline), the membranes underwent incubation with a peroxidase-conjugated secondary antibody (rabbit anti-mouse IgG (H+L), 1:4000; SouthernBiotech, #6170-05, Birmingham, AL, USA or goat anti-rabbit IgG (H+L chain-specific), 1:5000; SouthernBiotech, #4050-05, Birmingham, AL, USA) at room temperature for 1 h. Following three TBST washes, protein expression was identified using an enhanced chemiluminescent development reagent (BeyoECL Plus, Beyotime, Beijing, China). Densitometry analysis was performed utilizing Quantity One software (Bio-Rad, Hercules, CA, USA), and the intensities of the bands were standardized against those of GADPH.
Following transfection, cells were cultured on sterile coverslips and left to incubate overnight at 37°C for immunofluorescence assays. For immunofluorescence staining, the cells underwent fixation with 4% paraformaldehyde for 30 min, exposure to 0.2% Triton X-100 for 5 min at 4°C, and then underwent three washes with ice-cold PBS. After blocking with 10% normal goat serum from Jackson ImmunoResearch (West Grove, PA, USA) for half an hour, the cells were exposed overnight at 4°C to primary antibodies (anti-EV71 polyprotein VP1 (1:1000, Bioss Antibodies, Inc., bs-2297R, Woburn, MA, USA), anti-LC3B (1:1000, Sigma, St., L7543, Louis, MO, USA), recombinant anti-SQSTM1/p62 (phospho S349) antibody (1:1000, Abcam, ab211324, Cambridge, United Kingdom), or anti-SQSTM1/p62 (1:1000, Abcam, ab56416, Cambridge, United Kingdom). The cells were incubated with goat anti-mouse IgG (H+L) secondary antibody DyLight 594 (1:1000; Thermo Fisher Scientific, #35510, Waltham, MA, USA) at room temperature for 1 h following removal of the primary antibody and three PBS rinses. DAPI staining was performed before visualization employing a Leica confocal microscope (Leica Microsystems, Wetzlar, Germany).
Co-immunoprecipitation (Co-IP) experiment
Co-IP assays were conducted with the gp78 antibody (bs-1219R), PMP22 antibody (bs-0235R), both acquired from Bioss Antibodies, Inc., Woburn, MA, USA, and the ubiquitin antibody (Abcam, ab134953, Cambridge, UK). Subsequently, co-precipitated proteins were analyzed through western blotting. Lysis of the cells was achieved by employing ice-cold IP lysis buffer for 5 min, followed by centrifugation at 13,000 ×g for 10 min to precipitate cellular debris. Transferring the supernatant to a fresh tube, the determination of protein concentration was conducted using the Micro BCA Protein Assay Kit (Thermo Fisher Scientific, #23235, Waltham, MA, USA). The protein lysate from the mouse SCs that had been transfected with the EV71-VP1 overexpression vector was regarded as the experimental group, and the protein lysate from empty vector-transfected cells was used as control. The Co-IP procedure was executed utilizing a Thermo Scientific Pierce Co-IP kit (Thermo Fisher Scientific, #26149, Waltham, MA, USA) in accordance with the provided manufacturer’s guidelines. The protein samples were separated by SDS-PAGE, with non-specific mouse IgG utilized as a negative control.
The experiments were replicated thrice, and the results were expressed as means ± standard error (SE). Analysis of statistics was carried out employing SPSS 22.0 for Windows from IBM (Armonk, NY, USA). Comparison of data across multiple groups was conducted through one-way ANOVA and Dunnett’s post hoc test or adjusted using the Bonferroni method. Statistical significance was determined for two-sided p-values less than 0.05.
AMFR/gp78 of ubiquitin-specific protease binding enrichment and positive regulation of myelination in EV-infected children with neurological involvement
In our cohort study in children with EV infection [25], the gene ontology (GO) molecular functional and biological process enrichment results of ubiquitin-related input RNA genes both showed the significant enrichment of AMFR/gp78 in children with neurological involvement (NS group) (p = 0.003 and p = 0.01, respectively) (Table 1), whose ubiquitination substrate proteins include PMP22 [32]. Furthermore, in contrast to individuals with EV infection lacking neurological symptoms, we detected an enhancement in positive myelination regulation and ubiquitin-specific protease binding within the NS cohort. This was achieved through the combined examination of MeRIP-seq and RNA-seq data (Suppl. Fig. S2). In addition, PLEKHA4 enrichment was recognized as a gene specific to nerve tissue in the NS group (Suppl. Table S4), which was a regulator of the E3 ubiquitin ligase and controlled disheveled ubiquitination [33,34]. These findings indicated that active myelination and abnormal protein ubiquitination occur when EV infection involves nerves.
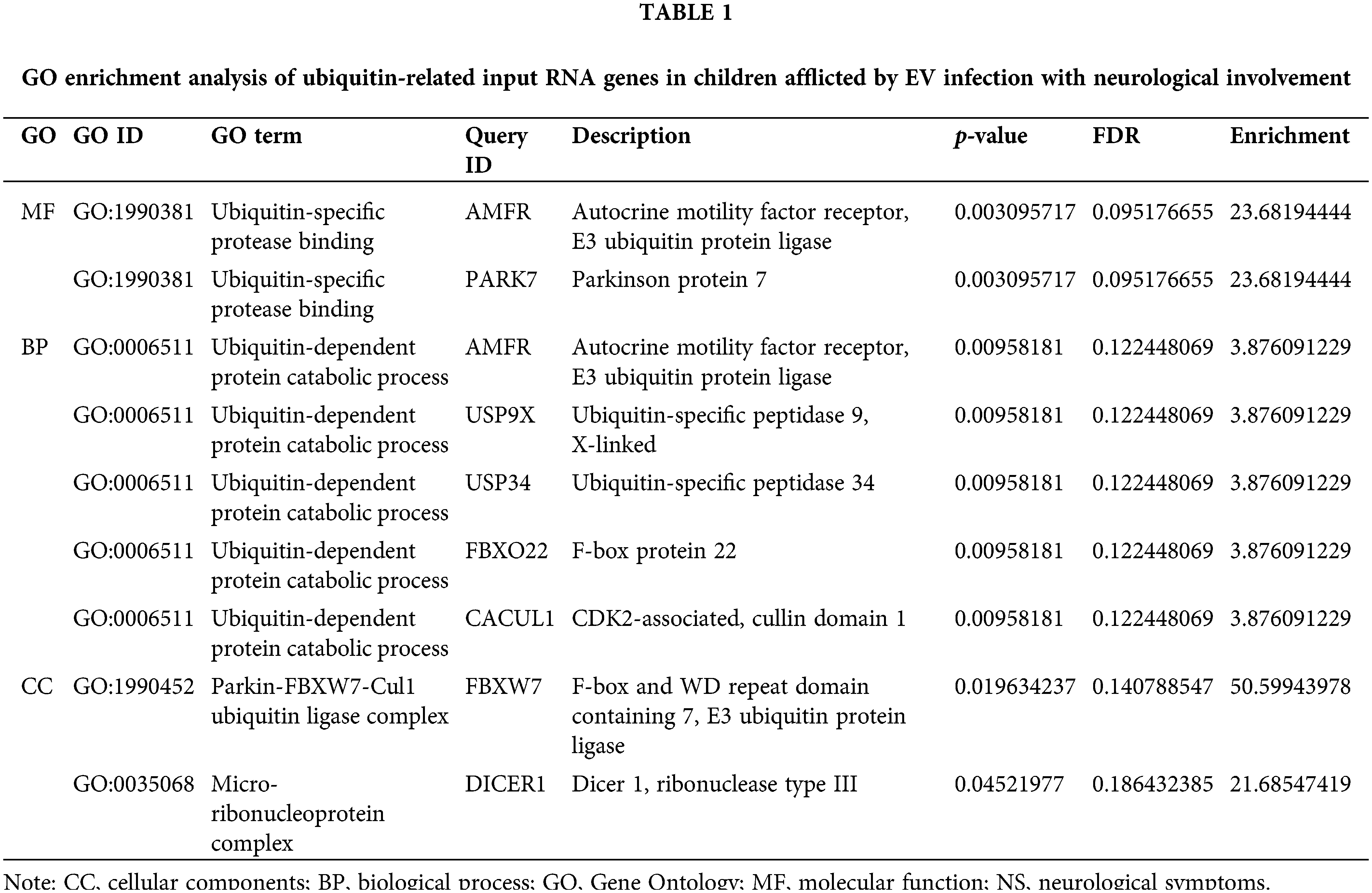
Gp78 participates in PMP22 regulation in VP1-overexpressing mouse Schwann cells
As an important myelin protein in childhood, PMP22 is essential for myelin growth and repair, and 80% of it is degraded by the UPS-ER PQC (protein quality control) system. When overexpressed PMP22 exceeds the degradation capacity of this system, it will induce PMP22 aggregation and cause disease [20]. In order to test our hypothesis that gp78 participates in VP1-induced PMP22 accumulation, we knocked down PMP22 and overexpressed gp78 in VP1-overexpressing mouse SCs. Immunofluorescence staining revealed prominent fluorescent signals for VP1 in all experimental groups, with no significant differences in average fluorescence intensity observed among the VP1, VP1+PMP22si, and VP1+OE-gp78 groups (Fig. 1). This result confirmed the successful establishment of VP1 overexpressing mouse SC models. As shown in Figs. 2A and 2B, the expression of PMP22 significantly increased in VP1-overexpressing cells at both the mRNA and protein concentrations when compared to the Ctrl group. The protein level exhibited an elevation in gp78 expression; however, there was no concurrent increase observed at the mRNA quantity (Figs. 2A and 2B). Compared with VP1-overexpressing cells, the expression level of PMP22 was significantly decreased in gp78-overexpressing cells (Figs. 2A and 2B, p < 0.001). On the other hand, PMP22 silencing resulted in a downregulated tendency of gp78 expression in VP1-overexpressing cells (Fig. 2B). These data indicate that gp78 participates in PMP22 regulation in VP1-overexpressing mouse SCs.
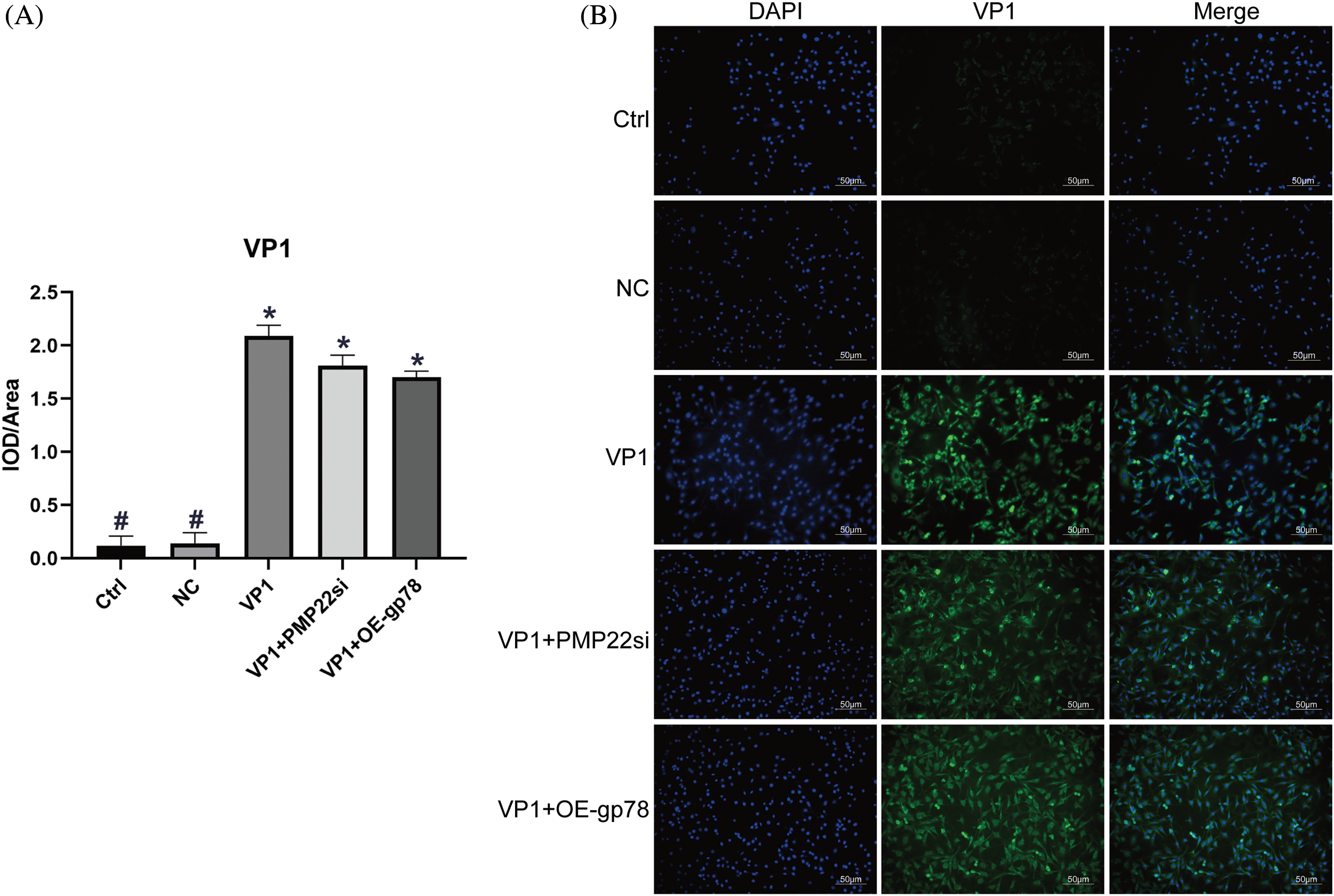
Figure 1: Immunofluorescence staining of VP1 in mouse Schwann cells (200). VP1 overexpressing Mouse SCs were transfected with siPMP22 or gp78 overexpression vector, respectively. Mouse SCs were transfected with pEGFP-c3-VP1 for 36 h. Cells transfected with pEGFP-C3 served as negative control (NC). (A) Quantification of relative VP1 expression level by immunofluorescence staining analysis. The data are presented as means ± SE. *p less than 0.05 compared to the Ctrl group. #p less than 0.05 compared to the VP1 group. (B) Representative images of VP1 stain by immunofluorescence assay. Mouse SCs were subjected to laser scanning confocal microscopy after staining with VP1 stain (emitting green fluorescence) and a nuclear dye (DAPI, emitting blue fluorescence). The scale bars represent a length of 50 μm.
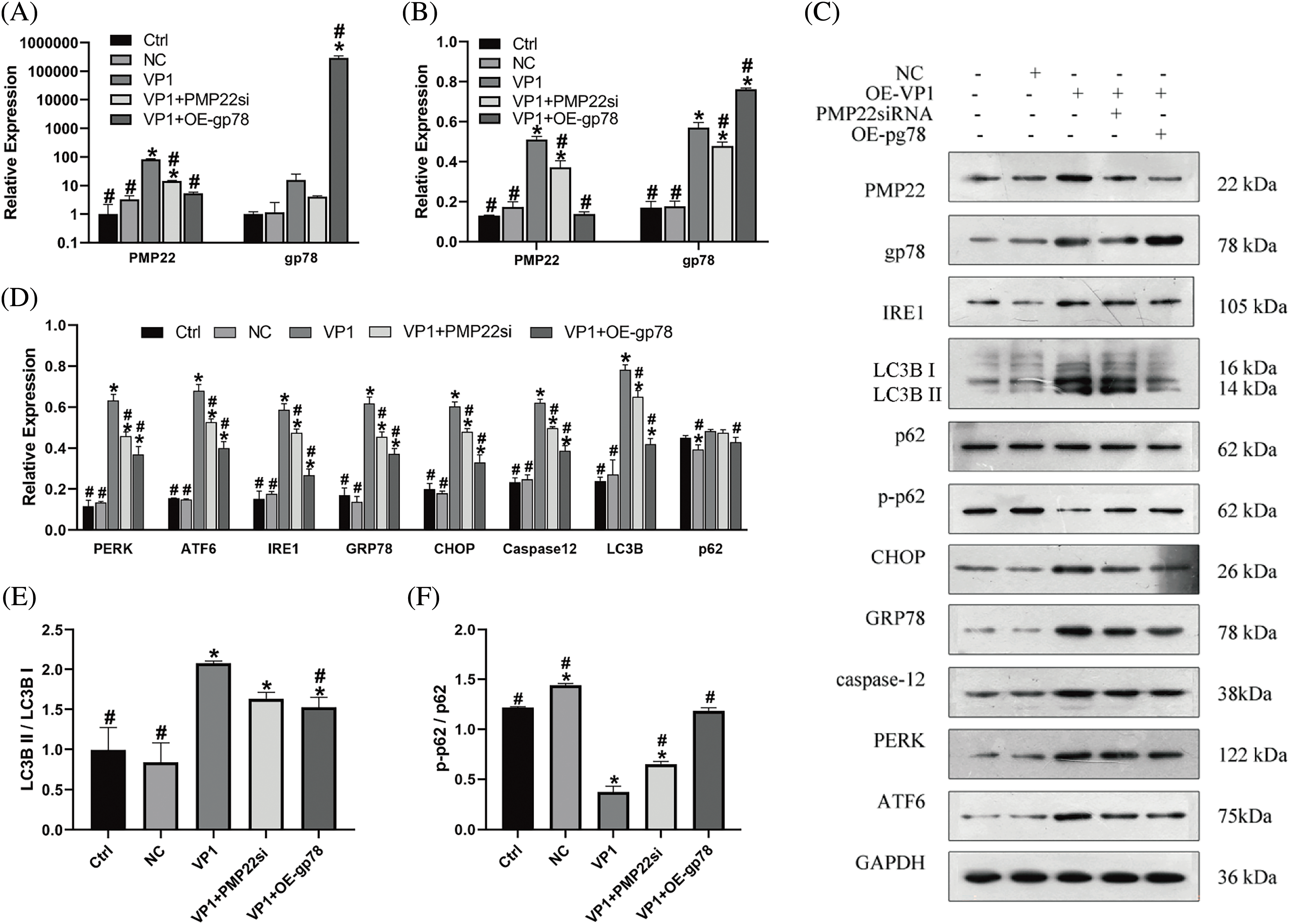
Figure 2: Effect of PMP22 Silencing and gp78 overexpression in VP1 overexpressing mouse Schwann cells. VP1 overexpressing Mouse SCs were transfected with siPMP22 or gp78 overexpression vector, respectively. Mouse SCs underwent transfection with pEGFP-c3-VP1 over a duration of 36 h, utilizing PEGFP-C3-transfected cells as the negative control (NC). (A) mRNA expression levels of PMP22 and gp78. (B) Quantification of relative PMP22 and gp78 expression levels by western blot analysis. (C) Representative images of PMP22, gp78, ER stress-associated signal molecules, and autophagy markers expression by western blot analysis. (D) Quantification of relative ER stress-associated signal molecules and autophagy markers expression levels by western blot analysis. (E) Ratio of LC3B II to LC3B I. (F) Ratio of p-p 62 to p62. The results are presented as means ± SE from three distinct repetitions. *p < 0.05 compared to the Ctrl group. #p < 0.05 compared to the VP1 group.
PMP22 silencing and gp78 overexpression alleviates VP1-induced ERs
The authors’ group previously indicated that PMP22 was involved in VP1-mediated ERs and mouse SCs autophagy [8]. In this study, we detected all three ER transmembrane receptors and related downstream signal molecules. From the western blot analysis (Fig. 2 and Suppl. Table S5), the protein expression levels of PERK, ATF6, and IRE1 were significantly increased in VP1-overexpressing mouse SCs compared with the Ctrl and NC groups (Figs. 2C and 2D) (p < 0.001), the same with two downstream signals of ERs, caspase12 and CHOP. The ER-resident chaperone GRP78 also had the same tendency (Figs. 2C and 2D). Furthermore, PMP22 silencing resulted in significantly reduced expression of PERK, ATF6, IRE1, and GRP78 at the protein level compared with VP1-overexpressing cells (Figs. 2C and 2D). the downstream signals, CHOP, and caspase 12, also had a significantly reduced expression level in PMP22 silencing combined with VP1-overexpressing cells. The same phenomenon was observed in gp78-overexpressing cells compared with VP1-overexpressing mouse SCs (Figs. 2C and 2D, p < 0.001). In comparison with the PMP22 silencing group, the declined tendency of PERK, ATF6, IRE1, and GRP78 expression was more significant in gp78-overexpressing cells, the same with CHOP and caspase12 (Figs. 2C and 2D). Therefore, PMP22 silencing and gp78 overexpression alleviate VP1-induced ERs.
PMP22 silencing and gp78 overexpression reduce VP1-induced autophagy
In order to describe the effect of VP1 on mouse SCs autophagy and investigate whether PMP22 and gp78 are involved in this process, the autophagy markers LC3B and p62 were measured in this study. Compared with the Ctrl and NC groups, the western blot showed that VP1 overexpression upregulated the expression of LC3B (Fig. 2D) (p < 0.001). When co-transfected with PMP22 silencing or gp78 overexpression, the LC3B expression level was reduced compared with VP1-overexpressing cells (Fig. 2D) (p = 0.02; p < 0.001). Notably, VP1 overexpression did not change the expression of p62; PMP22 silencing also showed the same result. While gp78 overexpression reduced the expression level of p62 in VP1-overexpressing cells (Fig. 2D) (p = 0.03). Furthermore, the LC3B II to LC3B I ratio in gp78-overexpressing cells was also significantly lower compared with VP1-overexpressing cells (Fig. 2E) (p = 0.03). Nevertheless, the ratio of p-p62 to p62 reduced significantly in VP1-overexpressing cells, and PMP22 silencing or gp78 overexpression reversed the phenomenon (Fig. 2F) (p < 0.001). Immunofluorescence analysis demonstrated similar results (Suppl. Figs. S3–S5).
VP1 reduces the ubiquitination degradation of PMP22 through gp78
In order to explore the regulation relationship between gp78 and PMP22, Co-IP assays were performed in VP1-transfected and empty vector-transfected mouse SCs (NC group). As shown in Figs. 3A and 3B, PMP22 could be precipitated by the gp78 and ubiquitin antibody in VP1-transfected and empty vector-transfected mouse SCs. In addition, gp78 and ubiquitin could also be precipitated by the PMP22 antibody (Fig. 3C). Those results indicated that the expression of PMP22 might be regulated by gp78 through ubiquitination. As revealed by quantification analysis, the interaction between gp78 and PMP22 was significantly reduced in VP1-overexpressing cells compared with the negative control (Fig. 3D) (p < 0.001). A similar trend was observed between PMP22 and ubiquitin (Fig. 3E). Fig. 3F also demonstrated the same result. From those data, we speculated that VP1 could reduce the ubiquitination degradation of PMP22 through gp78, resulting in PMP22 accumulation.
Figure 3: Co-immunoprecipitation analysis of the relationship between PMP22 and gp78. SCs underwent transfection with pEGFP-c3-VP1 over a duration of 36 h, utilizing PEGFP-C3-transfected cells as the negative control (NC). Positive and negative references were employed as input and non-specific mouse IgG, respectively. (A) Representative western blot image of PMP22 expression precipitated by gp78 antibody. (B) Representative western blot image of PMP22 expression precipitated by ubiquitin protein antibody. (C) Representative western blot image of gp78 and ubiquitin protein expression precipitated by PMP22 antibody. (D) Quantification of relative PMP22 expression. (E) Quantification of relative ubiquitinated PMP22 expression. (F) Quantification of relative gp78 and ubiquitin expression. Results are presented as means ± SE. Statistical significance is denoted by *p < 0.05 compared to the NC group.
PMP22 silencing or gp78 overexpression improves the proliferation rate of cells overexpressing EV71-VP1
In order to explore the influence of EV71-VP1 on cell growth and further elucidate the impact of PMP22 silencing or gp78 overexpression on this process, EV71-VP1 was transfected into mouse SCs, combined with PMP22 silencing or gp78 overexpression. All cells were cultured and collected at 24, 48, and 72 h. Cell proliferation was evaluated through the utilization of the CCK-8 test. The findings demonstrated a notable decline in the growth rate of cells transfected with VP1 over time when compared to both the Ctrl and NC groups (Fig. 4A). We also applied the TUNEL assay to evaluate cell apoptosis (Figs. 4B and 4C). The apoptotic cell proportion of VP1-overexpressing cells was significantly increased compared to the Ctrl and NC groups (Fig. 4B) (38% vs. 4%, p = 0.003; 38% vs. 3%, p = 0.003). Furthermore, PMP22 silencing did not decrease apoptotic cell proportion in EV71-VP1-overexpressing cells (25% vs. 38%, p = 0.16). The results were similar in gp78 overexpression cells (20% vs. 38%, p = 0.05). In addition, PMP22 silencing and gp78 overexpression improved the proliferation rate of EV71-VP1-overexpressing cells at 48 and 72 h (Fig. 4D).
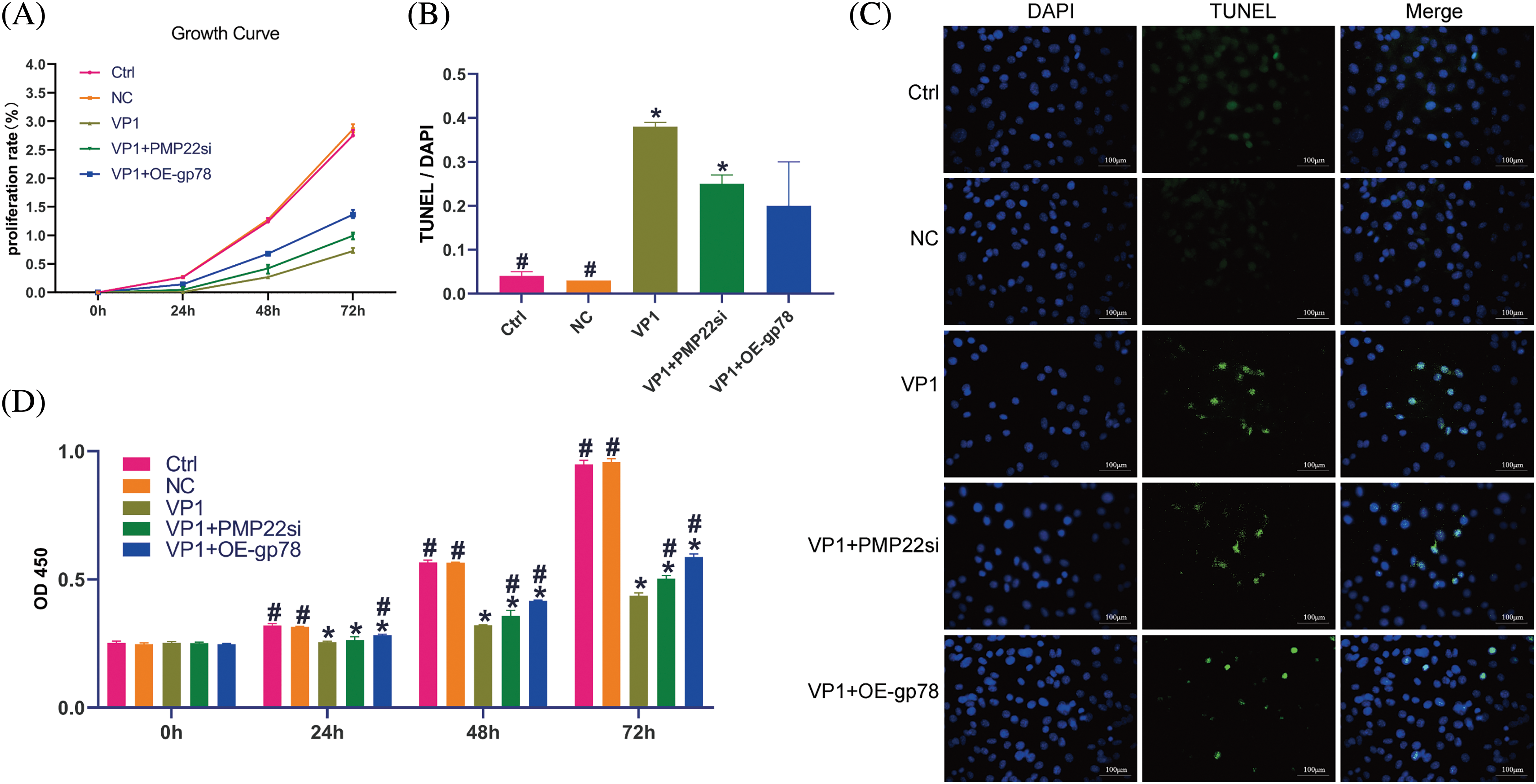
Figure 4: Effect of PMP22 silencing and gp78 overexpression on proliferation and apoptosis of mouse SCs overexpressing VP1. VP1-overexpressing mouse SCs underwent transfection with either siPMP22 or a gp78 overexpression vector. Mouse SCs underwent transfection with pEGFP-c3-VP1 over a duration of 36 h, utilizing PEGFP-C3-transfected cells as the negative control (NC). For the CCK-8 test, all cells were collected after transfection for 48 h, at a series of times from 0~72 h. (A) Growth curve of different experiment cells, the cell proliferation rate was evaluated by the CCK-8 method. (B) The apoptotic cell proportion of different experiment cells. The proportion was represented by the percentage of DNA fragmentation. (C) Fluorescence microscope observation of different experiment group mouse SCs. DNA fragmentation was assessed by TUNEL assay; green-staining cells have DNA fragmentation. Nuclear (DAPI) was stained by blue fluorescence. Scale bars are 100 μm. (D) Cell proliferation status in different incubation times from 0 to 72 h. The results are expressed as means ± SE. Statistical significance is denoted by *p < 0.05 compared to the Ctrl group and #p < 0.05 compared to the VP1 group.
This research demonstrated that EV71-VP1 has the potential to impact gp78 activity, leading to a decrease in the ubiquitination and degradation of PMP22. This results in the buildup of PMP22 within the ER, triggering ER stress and autophagy in mouse SCs overexpressing EV71-VP1 (more details in Fig. 5). VP1-overexpressing cells exhibited activation of all three ER stress sensors (ATF6, PERK, and IRE1) along with their corresponding downstream signals (Caspase 12 and CHOP). LC3B was also upregulated, while PMP22 silencing or gp78 overexpression reversed the phenomenon. PMP22 silencing or gp78 overexpression increased proliferation of EV71-VP1-transfected cells. Those findings help understand how EV71 interacts with host cells, suggesting possible therapeutic targets against EV71 infection-related neurologic injury.
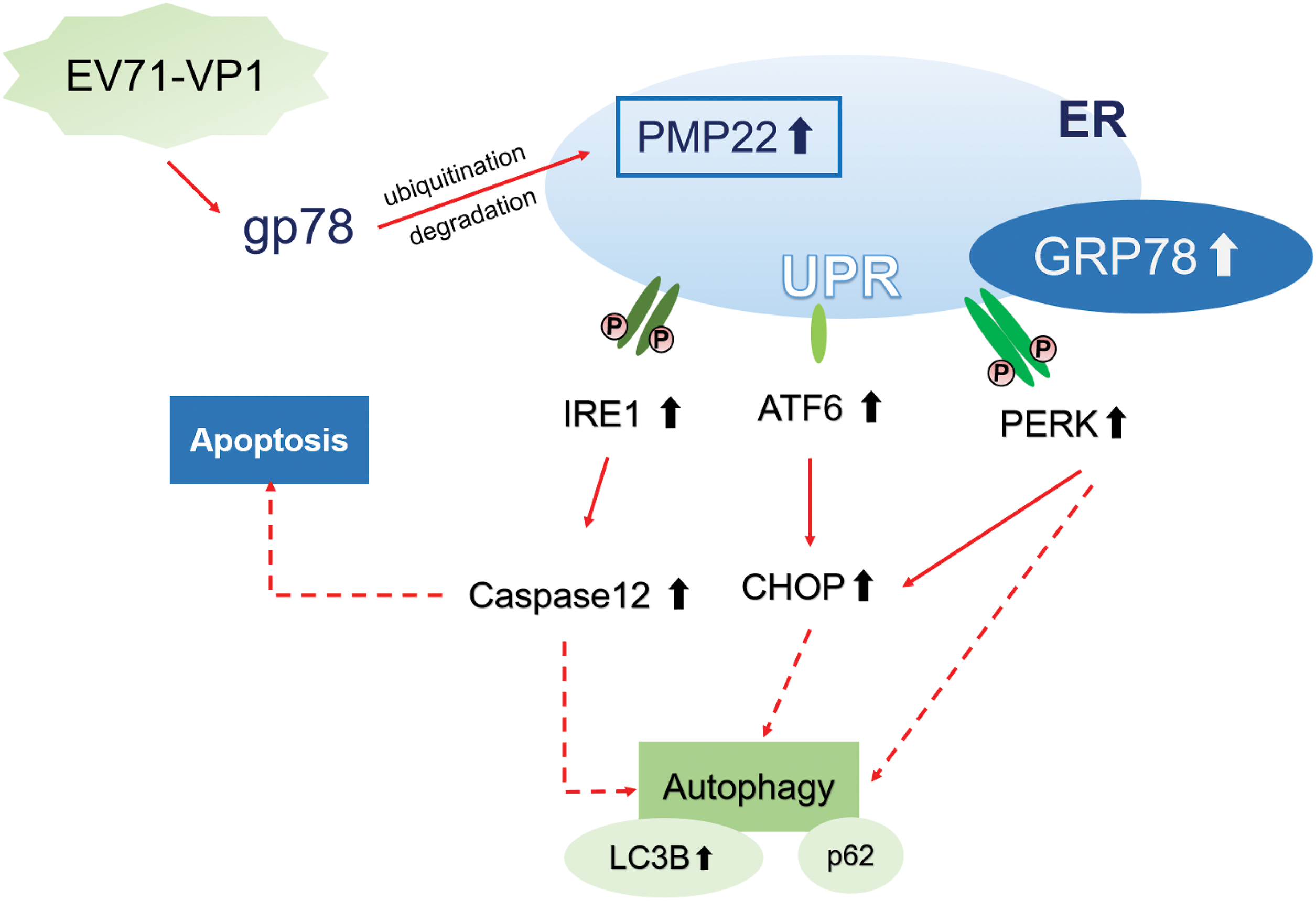
Figure 5: EV71-VP1 induces the PMP22 accumulation through gp78 and causes ER stress and autophagy in mouse Schwann cells. EV71-VP1 could affect the activity of gp78 to decrease the ubiquitination and degradation of PMP22, resulting in PMP22 accumulation in ER. Accumulated PMP22 triggered the UPR, induced the up-regulation of three ER stress sensors (ATF6, PERK, and IRE1) and the related downstream signals (CHOP and Caspase 12), as well as the ER-resident chaperone GRP78. In addition, VP1 upregulated the autophagy marker LC3B but did not change the expression of p62.
Previous studies showed that several viruses could induce ERs and activate the UPR signaling pathways, such as the West Nile virus and dengue virus [35,36]. The authors’ group also previously reported ERs activation in autophagy of EV71-VP1-overexpressing mouse SCs and PMP22 were involved in this process, but without exploring the exact pathways involved. In this study, it was found that EV71-VP1 could affect the activity of gp78 to induce decreased ubiquitination and degradation of PMP22, resulting in the accumulation of PMP22 in the ER, causing a series of ERs responses in the mouse SCs. Accumulating evidence shows that gp78 is crucially involved in regulating cell division, ERAD pathway, maintenance of cellular proteostasis, and mitophagy [32]. Furthermore, gp78 is involved in neuroprotection and several neurodegenerative diseases [32]. In the classical Parkinson’s disease model, gp78 was significantly decreased, and its overexpression could protect neurons against MPP+-induced cell death [37]. In the case of CMT disease, a previous study also demonstrated that gp78 employs the proteasomal pathway to break down mutated PMP22, which is the causative factor for the disease [17]. Moreover, gp78 is involved in promoting innate immune responses to microbial infections. Depleting gp78 leads to a significant reduction in vesicular stomatitis virus infection and a simultaneous amplification of type I interferon signaling [38]. In another study, it was found that gp78 has the ability to mediate the ubiquitination of NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3), thereby inhibiting the activation of the inflammasome [39]. In the present study, the overexpression of gp78 could help reduce the ERs induced by EV71-VP1, alleviate autophagy, and reduce the apoptosis of SCs. These results indicated the protective role of gp78 during EV71 infection, and its depletion could aggravate nervous system damage. Whether medications targeting gp78 would be helpful to alleviate EV71-induced myelin cells and neuronal damage needs further study.
Our results revealed that the increase in ubiquitination was not sufficient to cause the degradation of the significantly elevated PMP22 protein, although EV71 infection can simultaneously upregulate PMP22 and induce ubiquitination to some extent. Excessive accumulation of PMP22 induces autophagy and causes myelin damage [40,41]. This was further evidenced by a significant reduction in the damage of the infected cells after overexpression of gp78. PMP22 silencing improves the proliferation rate of EV71-VP1-overexpressing cells in our study. Previous research found that inhibiting PMP22 boosts etoposide-induced cell apoptosis, and the negative regulation of PMP22 by MiR-139-5p hinders cell proliferation in Gastric Cancer [42,43]. During peripheral nerve regeneration, the dysregulated interaction of miR‑29a‑3p and PMP22 within Schwann cells (SCs) could impact cellular proliferation and migration [44]. All those results indicate that PMP22 can be a potential treatment target. The present study showed that the accumulation of PMP22 triggered the UPR and all three ERs sensors (ATF6, PERK, and IRE1), and their related signaling pathways were activated. In addition, the ER-resident chaperone GRP78 was also activated. A different investigation uncovered that EV71 infection could potentially lead to the elevation and abnormal redistribution of GRP78 into the cytosol, thereby aiding in virus replication [31]. Some other viruses can activate a subset or only one protein of the UPR pathways. For instance, the IRE1 pathway is induced in severe acute respiratory syndrome virus infection [45], and rotavirus activates the IRE1 and ATF6 pathways [46]. In addition, raised PERK-P and phosphorylated eukaryotic initiation factor 2 alpha (eIF2α-P) are detected in mice affected by prion disease [47]. Furthermore, mice subjected to GSK2606414, a highly selective inhibitor of PERK, showed no clinical symptoms associated with prion disease. Neuroprotection was evident throughout the brain [48], indicating a promising application of PERK inhibition in the prevention of neurologic diseases and neurodegeneration. In this study, activated UPR was observed in VP1-overexpressing SCs and related to autophagy and cell apoptosis. It could partly explain the mechanisms of central nervous system damage during EV71 infection. The potential therapeutic impact of inhibiting UPR, as well as the associated sensors and signals, in neuroprotection during EV71 infection requires further exploration.
An earlier investigation demonstrated that EV71 infection has the ability to trigger ERs but alters the outcome to support viral replication [26]. In this research, the heightened expression of the autophagy marker LC3B confirmed that autophagy was induced in VP1-overexpressing cells. Furthermore, through the CCK‑8 and TUNEL assays, delayed cell proliferation and increased apoptosis were observed in mouse SCs and were probably caused by VP1-induced ERs. Those data showed that VP1-induced ERs accelerated cell death, contributing to the central nervous system lesion in some EV71 cases. Therefore, it can be speculated that EV71 infection could induce excessive ERs, leading to cell death and facilitating virus invasion.
There are some limitations to this study. In GO analysis of clinical samples from patients, myelination was upregulated, and our clinical samples were from children. Previous studies have shown that when the nervous system is damaged in adults, myelination is upregulated [14,49,50], but whether the same mechanism is observed in children is unknown. When a child’s nervous system is damaged, the body usually initiates a series of repair and regeneration mechanisms to rebuild the myelin sheath of the damaged nerve fibers, resulting in myelination upregulation [51]. This upregulated myelination facilitates the protection and repair of nerve fibers. However, upregulated myelination does not imply a re-establishment of normal function since it appears that remyelination after demyelination never restores the myelin sheath to its original characteristics [51]. In conclusion, our study showed that the overexpression of VP1 in cells actually leads to apoptosis of myelin cells. We intend to conduct further future studies using clinical samples from adults.
In conclusion, the current investigation strongly indicates that gp78 controls the build-up of PMP22 in the ER and induces ERs in EV71-VP1-overexpressing cells. This process is associated with autophagy activation and cell apoptosis. Unlike apoptosis, autophagy is a protective mechanism for cells, but both were activated in VP1-overexpressing cells. Based on those findings, it can be speculated that in the early stage of EV71 infection, EV71-VP1 activates autophagy to eliminate the ERs, but the constant ERs ultimately lead to cell apoptosis. Increased gp78 or reduced aggregation of PMP22 could help protect mouse SCs from VP1-induced cell damage. Exploring the therapeutic potential of the gp78/PMP22/ER stress axis appears to be a promising strategy for addressing EV71 infection and warrants further investigation before advancing to clinical trials.
Acknowledgement: None.
Funding Statement: The study was supported by Guangdong Natural Science Foundation (Grant Numbers 2020A1515010014, 2022A1515012411); Science and Technology Key Project for People’s Livelihood of Guangzhou, China (Grant Number 202206010060); Guangzhou Science and Technology Bureau Basic Research Project (SL2024A03J01288); and Innovative Project of Children’s Research Institute, Guangzhou Women and Children’s Medical Center, China (Grant Numbers Pre-NSFC-2019-002, NKE PRE-2019-015).
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: P. Li; data collection: S. Yang; analysis and interpretation of results: G. Liu, K. Feng, S. Li, D. Hu; draft manuscript preparation: D. Zhu., P. Li. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Ethics Approval: This study was approved by the ethics committee of Guangzhou Women and Children’s Medical Center (Nos. 2017122501, VIVA07201904 and 2021019A01). Written informed consent was obtained from the legal guardians of the enrolled patients.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Supplementary Materials: The supplementary material is available online at https://doi.org/10.32604/biocell.2024.044856.
References
1. Huang J, Liao Q, Ooi MH, Cowling BJ, Chang Z, Wu P, et al. Epidemiology of recurrent hand, foot and mouth disease, China, 2008–2015. Emerg Infect Dis. 2018;24(3):432–42. [Google Scholar] [PubMed]
2. Wang J, Hu T, Sun D, Ding S, Carr MJ, Xing W, et al. Epidemiological characteristics of hand, foot, and mouth disease in Shandong, China, 2009–2016. Sci Rep. 2017;7(1):8900. [Google Scholar] [PubMed]
3. Gonzalez G, Carr MJ, Kobayashi M, Hanaoka N, Fujimoto T. Enterovirus-associated hand-foot and mouth disease and neurological complications in japan and the rest of the world. Int J Mol Sci. 2019;20(20):5201. [Google Scholar] [PubMed]
4. Li J, Sun Y, Du Y, Yan Y, Huo D, Liu Y, et al. Characterization of coxsackievirus A6- and enterovirus 71-associated hand foot and mouth disease in Beijing, China, from 2013 to 2015. Front Microbiol. 2016;7:391. [Google Scholar] [PubMed]
5. Wu W, Li B, Xie T. Children with severe enterovirus A71 infection. BMC Pediatr. 2023;23(1):172. [Google Scholar] [PubMed]
6. Ai J, Xie Z, Liu G, Chen Z, Yang Y, Li Y, et al. Etiology and prognosis of acute viral encephalitis and meningitis in Chinese children: a multicentre prospective study. BMC Infect Dis. 2017;17(1):494. [Google Scholar] [PubMed]
7. Yuan S, Li G, Wang Y, Gao Q, Wang Y, Cui R, et al. Identification of positively charged residues in enterovirus 71 capsid protein VP1 essential for production of infectious particles. J Virol. 2016;90(2):741–52. [Google Scholar] [PubMed]
8. Li P, Yang S, Hu D, Wei D, Lu J, Zheng H, et al. Enterovirus 71 VP1 promotes mouse Schwann cell autophagy via ER stress‐mediated PMP22 upregulation. Int J Mol Med. 2019;44(2):759–67. [Google Scholar] [PubMed]
9. Cordey S, Petty TJ, Schibler M, Martinez Y, Gerlach D, van Belle S, et al. Identification of site-specific adaptations conferring increased neural cell tropism during human enterovirus 71 infection. PLoS Pathog. 2012;8(7):e1002826. [Google Scholar] [PubMed]
10. Chen CS, Yao YC, Lin SC, Lee YP, Wang YF, Wang JR, et al. Retrograde axonal transport: a major transmission route of enterovirus 71 in mice. J Virol. 2007;81(17):8996–9003. [Google Scholar] [PubMed]
11. Jin Y, Li D, Sun T, Du Y, Gao Y, Ding R, et al. Pathological features of enterovirus 71-associated brain and lung damage in mice based on quantitative proteomic analysis. Front Microbiol. 2021;12:663019. [Google Scholar] [PubMed]
12. Yang SD, Li PQ, Li YM, Li W, Lai WY, Zhu CP, et al. Clinical manifestations of severe enterovirus 71 infection and early assessment in a Southern China population. BMC Infect Dis. 2017;17(1):153. [Google Scholar] [PubMed]
13. Yang SD, Li PQ, Huang YG, Li W, Ma LZ, Wu L, et al. Factors associated with fatal outcome of children with enterovirus A71 infection: a case series. Epidemiol Infect. 2018;146(6):788–98. [Google Scholar] [PubMed]
14. Wang Q, Chen FY, Ling ZM, Su WF, Zhao YY, Chen G, et al. The effect of schwann cells/schwann cell-like cells on cell therapy for peripheral neuropathy. Front Cell Neurosci. 2022;16:836931. [Google Scholar] [PubMed]
15. Johnson JH, Al Khalili Y. Histology, myelin. Treasure Island (FLStatPearls; 2022. [Google Scholar]
16. Vanoye CG, Sakakura M, Follis RM, Trevisan AJ, Narayan M, Li J, et al. Peripheral myelin protein 22 modulates store-operated calcium channel activity, providing insights into Charcot-Marie-Tooth disease etiology. J Biol Chem. 2019;294(32):12054–65. [Google Scholar] [PubMed]
17. Hara T, Hashimoto Y, Akuzawa T, Hirai R, Kobayashi H, Sato K. Rer1 and calnexin regulate endoplasmic reticulum retention of a peripheral myelin protein 22 mutant that causes type 1A Charcot-Marie-Tooth disease. Sci Rep. 2014;4:6992. [Google Scholar] [PubMed]
18. Pipis M, Won S, Poh R, Efthymiou S, Polke JM, Skorupinska M, et al. Post-transcriptional microRNA repression of PMP22 dose in severe Charcot-Marie-Tooth disease type 1. Brain. 2023;146(10):4025–32. [Google Scholar] [PubMed]
19. Xiao Y, Yang Y, Hu D. Knockdown of METTL3 inhibits enterovirus 71-induced apoptosis of mouse Schwann cell through regulation of autophagy. Pathog Dis. 2021;79(6):ftab036. [Google Scholar] [PubMed]
20. Okamoto Y, Takashima H. The current state of charcot-marie-tooth disease treatment. Genes. 2023;14(7):1391. [Google Scholar] [PubMed]
21. Marinko JT, Carter BD, Sanders CR. Direct relationship between increased expression and mistrafficking of the Charcot-Marie-Tooth-associated protein PMP22. J Biol Chem. 2020;295(34):11963–70. [Google Scholar] [PubMed]
22. Pla-Prats C, Thoma NH. Quality control of protein complex assembly by the ubiquitin-proteasome system. Trends Cell Biol. 2022;32(8):696–706. [Google Scholar] [PubMed]
23. Chen Z, Du S, Fang S. gp78: a multifaceted ubiquitin ligase that integrates a unique protein degradation pathway from the endoplasmic reticulum. Curr Protein Pept Sci. 2012;13(5):414–24. [Google Scholar] [PubMed]
24. Fortun J, Li J, Go J, Fenstermaker A, Fletcher BS, Notterpek L. Impaired proteasome activity and accumulation of ubiquitinated substrates in a hereditary neuropathy model. J Neurochem. 2005;92(6):1531–41. [Google Scholar] [PubMed]
25. Zhu D, Song Y, Hu D, Li S, Liu G, Li P, et al. Characterization of enterovirus associated m6A RNA methylation in children with neurological symptoms: aa prospective cohort study. Front Neurosci. 2021;15:791544. [Google Scholar] [PubMed]
26. Su R, Yin J, Ruan X, Chen Y, Wan P, Luo Z. Featured interactome of homocysteine-inducible endoplasmic reticulum protein uncovers novel binding partners in response to ER stress. Comput Struct Biotechnol J. 2023;21:4478–87. [Google Scholar] [PubMed]
27. Chen X, Shi C, He M, Xiong S, Xia X. Endoplasmic reticulum stress: molecular mechanism and therapeutic targets. Signal Transduct Target Ther. 2023;8(1):352. [Google Scholar] [PubMed]
28. Hagenlocher C, Siebert R, Taschke B, Wieske S, Hausser A, Rehm M. ER stress-induced cell death proceeds independently of the TRAIL-R2 signaling axis in pancreatic beta cells. Cell Death Discov. 2022;8(1):34. [Google Scholar] [PubMed]
29. Li S, Kong L, Yu X. The expanding roles of endoplasmic reticulum stress in virus replication and pathogenesis. Crit Rev Microbiol. 2015;41(2):150–64. [Google Scholar] [PubMed]
30. Mao J, Hu Y, Ruan L, Ji Y, Lou Z. Role of endoplasmic reticulum stress in depression (review). Mol Med Rep. 2019;20(6):4774–80. [Google Scholar] [PubMed]
31. Jheng JR, Wang SC, Jheng CR, Horng JT. Enterovirus 71 induces dsRNA/PKR-dependent cytoplasmic redistribution of GRP78/BiP to promote viral replication. Emerg Microbes Infect. 2016;5(3):e23. [Google Scholar] [PubMed]
32. Joshi V, Upadhyay A, Kumar A, Mishra A. Gp78 E3 ubiquitin ligase: essential functions and contributions in proteostasis. Front Cell Neurosci. 2017;11:259. [Google Scholar] [PubMed]
33. Shami Shah A, Batrouni AG, Kim D, Punyala A, Cao W, Han C, et al. PLEKHA4/kramer attenuates dishevelled ubiquitination to modulate wnt and planar cell polarity signaling. Cell Rep. 2019;27(7):2157–70.e8. [Google Scholar] [PubMed]
34. Shami Shah A, Cao X, White AC, Baskin JM. PLEKHA4 promotes Wnt/β-catenin signaling-mediated G1-S transition and proliferation in melanoma. Cancer Res. 2021;81(8):2029–43. [Google Scholar] [PubMed]
35. Ambrose RL, Mackenzie JM. West Nile virus differentially modulates the unfolded protein response to facilitate replication and immune evasion. J Virol. 2011;85(6):2723–32. [Google Scholar] [PubMed]
36. Umareddy I, Pluquet O, Wang QY, Vasudevan SG, Chevet E, Gu F. Dengue virus serotype infection specifies the activation of the unfolded protein response. Virol J. 2007;4:91. [Google Scholar] [PubMed]
37. Wang Q, Jiao F, Zhang P, Yan J, Zhang Z, He F, et al. CDK5-mediated phosphorylation-dependent ubiquitination and degradation of E3 ubiquitin ligases GP78 accelerates neuronal death in Parkinson’s disease. Mol Neurobiol. 2018;55(5):3709–17. [Google Scholar] [PubMed]
38. Jacobs JL, Zhu J, Sarkar SN, Coyne CB. Regulation of mitochondrial antiviral signaling (MAVS) expression and signaling by the mitochondria-associated endoplasmic reticulum membrane (MAM) protein Gp78. J Biol Chem. 2014;289(3):1604–16. [Google Scholar] [PubMed]
39. Xu T, Yu W, Fang H, Wang Z, Chi Z, Guo X, et al. Ubiquitination of NLRP3 by gp78/Insig-1 restrains NLRP3 inflammasome activation. Cell Death Differentiation. 2022;29(8):1582–95. [Google Scholar] [PubMed]
40. Lin DS, Huang YW, Lee TH, Chang L, Huang ZD, Wu TY, et al. Rapamycin alleviates protein aggregates, reduces neuroinflammation, and rescues demyelination in globoid cell leukodystrophy. Cells. 2023;12(7):993. [Google Scholar] [PubMed]
41. Watila MM, Balarabe SA. Molecular and clinical features of inherited neuropathies due to PMP22 duplication. J Neurol Sci. 2015;355(1–2):18–24. [Google Scholar] [PubMed]
42. Hou J, Wang L, Zhao J, Zhuo H, Cheng J, Chen X, et al. Inhibition of protein PMP22 enhances etoposide-induced cell apoptosis by p53 signaling pathway in Gastric Cancer. Int J Biol Sci. 2021;17(12):3145–57. [Google Scholar] [PubMed]
43. Hou J, Zhuo H, Chen X, Cheng J, Zheng W, Zhong M, et al. MiR-139-5p negatively regulates PMP22 to repress cell proliferation by targeting the NF-κB signaling pathway in gastric cancer. Int J Biol Sci. 2020;16(7):1218–29. [Google Scholar] [PubMed]
44. Shen Y, Cheng Z, Chen S, Zhang Y, Chen Q, Yi S. Dysregulated miR-29a-3p/PMP22 modulates schwann cell proliferation and migration during peripheral nerve regeneration. Mol Neurobiol. 2021;59(2):1058–72. [Google Scholar] [PubMed]
45. Xia B, Shen X, He Y, Pan X, Liu FL, Wang Y, et al. SARS-CoV-2 envelope protein causes acute respiratory distress syndrome (ARDS)-like pathological damages and constitutes an antiviral target. Cell Res. 2021;31(8):847–60. [Google Scholar] [PubMed]
46. Trujillo-Alonso V, Maruri-Avidal L, Arias CF, Lopez S. Rotavirus infection induces the unfolded protein response of the cell and controls it through the nonstructural protein NSP3. J Virol. 2011;85(23):12594–604. [Google Scholar] [PubMed]
47. Halliday M, Radford H, Sekine Y, Moreno J, Verity N, le Quesne J, et al. Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 2015;6(3):e1672. [Google Scholar] [PubMed]
48. Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med. 2013;5(206):206ra138. [Google Scholar] [PubMed]
49. Chapman TW, Hill RA. Myelin plasticity in adulthood and aging. Neurosci Lett. 2020;715:134645. [Google Scholar] [PubMed]
50. McNamara NB, Munro DAD, Bestard-Cuche N, Uyeda A, Bogie JFJ, Hoffmann A, et al. Microglia regulate central nervous system myelin growth and integrity. Nature. 2023;613(7942):120–9. [Google Scholar] [PubMed]
51. Franklin RJ, Goldman SA. Glia disease and repair-remyelination. Cold Spring Harb Perspect Biol. 2015;7(7):a020594. [Google Scholar] [PubMed]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools