
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

MiR-30a-5p alleviates LPS-induced HPMEC injury through regulation of autophagy via Beclin-1
1 Rainbowfish Rehabilitation and Nursing School, Hangzhou Vocational & Technical College, Hangzhou, 310018, China
2 Cancer Institute of Integrated Traditional Chinese and Western Medicine, Key Laboratory of Cancer Prevention and Therapy Combining Traditional Chinese and Western Medicine, Zhejiang Academy of Traditional Chinese Medicine, Hangzhou, 310012, China
3 Geriatric Medicine Center, Department of Endocrinology, Zhejiang Provincial People’s Hospital (Affiliated People’s Hospital), Hangzhou Medical College, Hangzhou, 310015, China
* Corresponding Authors: JUNPING GUO. Email: ; LIJUN WANG. Email:
# Contributed equally
(This article belongs to the Special Issue: MicroRNA as Biomarkers for Disease Diagnosis and Progression)
BIOCELL 2024, 48(3), 431-441. https://doi.org/10.32604/biocell.2023.046484
Received 03 October 2023; Accepted 12 December 2023; Issue published 15 March 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: Sepsis, a type of systemic disease, can impact nearly all organs, tissues and cells. Among them, endothelial cells are amongst the first to be affected and respond to the insult. In this study, we investigated the protective effects of microRNA-30a-5p (miR-30a-5p) on human pulmonary microvascular endothelial cells (HPMECs) treated with lipolysaccharide (LPS). Methods: An in vitro model of sepsis was established in HPMECs with the use of LPS. Transfecting with different tools (mimetic and inhibitor) to modify miR-30a-5p expression. Cell viability, proliferation and apoptosis were detected by the CCK-8 assay, the EdU kit and fluorescence staining, respectively. The autophagy-related protein and mRNA expression, the number of autophagosomes were separately examined through Western blot analysis, qPT-PCR and confocal microscopy. TargetScan and the luciferase reporter assays were used to probe target genes interacting with miR-30a-5p. Results: LPS caused a reduction in the viability and proliferation of HPMECs, as well as an elevation in the number of apoptotic cells. Subsequently, we observed that miR-30a-5p might play a role in preventing LPS-induced inhibition of cell damage and decreasing HPMEC apoptosis, suggesting the potential function of miR-30a-5p in this injury process. Finally, we confirmed that miR-30a-5p exerts its protective effect by regulating cell autophagy, possibly by targeting Beclin-1. Conclusion: Our study provided evidence that autophagy is a crucial aspect in the protective role of miR-30a-5p against LPS-induced HPMEC injury, identifying a promising target for sepsis-related endothelial cell injury.Keywords
Systemic inflammatory response syndrome (SIRS) arises from a disruption in homeostasis following exposure to diverse injuries, including infection, trauma, and surgery, which leads to significantly elevated rates of morbidity and mortality [1]. Sepsis comprises SIRS with a suspected infection source. Hence, the exploration of innovative treatment methods for sepsis holds great significance in the field of medical practice. Endothelial cells (ECs), acting as a distinguishing obstacle separating tissue from blood, potentially exert a crucial influence on the control of inflammatory reactions, immune system function, and overall balance [2]. Inflammatory factors, such as LPS, induce damage and apoptosis in vascular endothelial cells, causing microcirculation disorders and multiple organ dysfunction, as a consequence of sepsis [3]. Therefore, it is thought that the implementation of strategies to protect the vascular endothelium against harm and/or impairment could potentially reduce the incidence of sepsis.
Autophagy is a crucial self-protection mechanism that breaks down intracellular proteins and organelles into smaller molecules for recycling. It helps to sustain cellular metabolism when exposed to various stimuli, such as oxidative stress, toxin exposure, and other factors, thereby promoting cell survival [4]. Under typical physiological circumstances, autophagy is minimal, but it can be triggered by different elements, such as lack of nutrients, low oxygen levels, infections, the development of tumors, tissue harm, and more [5]. The presence of autophagy-related proteins (LC3II/I, Beclin1, and p62) and the quantity of autophagosomes can indicate the initial symptoms of sepsis [6]. Autophagy activation helps to counteract excessive inflammatory responses; therefore, modulation of autophagy is a potential treatment for inflammatory diseases and sepsis.
MicroRNAs (miRNAs) are a group of tiny RNA that control gene expression by attaching to the 3′-untranslated region (UTR) of target genes, leading to either the suppression of translation or the breakdown of the specific transcript [7]. A recent study [8] discovered that septic lung injury can be alleviated through the inhibition of transforming growth factor-β-activated binding protein 2, induced by miR-155 to activate autophagy. During sepsis, renal ischemia/reperfusion (I/R) injury takes place, and research indicates that miR-30a-5p, a type of microRNA, is elevated in renal I/R injury, may mitigate autophagy by controlling the Beclin-1/ATG16 pathway [9]. By performing cecal ligation and puncture (CLP) to create a sepsis model in mice, the presence of adenosine deaminase acting on double stranded RNA 1 (ADAR1) decreases inflammation and organ harm by diminishing the connection between miR-30a and cytokine signaling protein inhibitor 3 (SOCS3) [10]. The liver cells experienced an elevation in miR-30a levels and a reduction in suppressor of cytokine signaling protein 1 (SOCS1) due to sepsis. The study [11] demonstrated that miR-30a inhibits SOCS-1 by targeting it through the JAK/STAT signaling pathway, which impedes the proliferation of liver cells and promotes apoptosis in septic rats. Additional studies have demonstrated that miR-30a, belonging to the miR-30 group, has the ability to inhibit autophagy by targeting Beclin-1 mRNA. This action enhances the responsiveness of gastrointestinal stromal tumor cells to Imatinib [12]. Additionally, it suppressed lung fibrosis by hindering the mitochondrial fission reliant on dynamin 1 Like (DRP-1), thereby impeding type II alveolar epithelial cell (AEC-II) apoptosis [13]. However, the involvement of miR-30a-5p in the regulation of autophagy caused by LPS in HPMECs remains uncertain.
This study investigates the function of miR-30a-5p, originate from the miR-30a precursor, in preventing endothelial cell apoptosis using an in vitro model of LPS-induced HPMECs injury. Additionally, it presents evidence supporting the significant role of autophagy in the protective impact of miR-30a-5p against LPS-induced HPMEC injury.
Shanghai Sixin Biotechnology Co., Ltd. (Shanghai, China) was the source of acquisition for HPMECs. This HPMECs is an immortalized cell line and the cells were cultured in RPMI1640 medium (GIBCO, Grand Island, NY, USA) supplemented with 10% FBS (Procell, Wuhan, China) and 1% streptomycin and penicillin (Gibco, Grand Island, NY, USA). The cells were cultured and kept in the 5% CO2 incubator at a temperature of 37°C.
2 × 105 cells/well HPMECs were used to seed in 6-well plates. RiboBio (Guangzhou, China) provided the miRNA-30a-5p mimic, miRNA-30a-5p inhibitor, and a nonspecific control (NC) that were acquired. The Lipofectamine 2000 Kit (Invitrogen, Carlsbad, CA, USA) was used to transfect all cells as per the instructions provided by the manufacturer.
Assays for the viability and growth of cell
Detect cell viability using the Cell Counting Kite-8 (CCK-8) assay from Beyotime (Jiangsu, China). After 24 h, the cells were placed in 96-well plates with a population of 5,000 cells per well. Based on the findings of prior investigation [14] and our own verification (see Suppl. Fig. S1), the cells underwent pretreatment with 10 μg/mL LPS for a duration of 24 h. Subsequently, 10 μL CCK-8 reagent was introduced to every well, followed by an incubation period of 1–4 h. To determine the optical density, use a microplate reader (Dynex Technologies, Chantilly, VA, USA) to measure the absorbance at 450 nm. The cell viability was normalized in comparison to the control group that was not treated.
The abundance of HPMECs was analyzed using a Click-iT EdU Imaging Kit (Thermo Fisher Scientific, Waltham, MA, USA) in the light of the directions provided by the manufacturer. In short, the cells were exposed to LPS, LPS along with the non-specific control (NC), or LPS along with the miR-30a-5p mimic. Subsequently, they were cultured with 10 μM EdU at 37°C for 2 h. Afterwards, treat with 3.7% formaldehyde for a duration of 15 min and then permeate with 0.5% Triton X-100 (Sigma Aldrich, St. Louis, MO, USA) at ambient temperature for a period of 20 min. Next, the cell nuclei were stained for 15 min using 5 μg/mL DAPI (Invitrogen, Carlsbad, CA, USA). Using an inverted fluorescence microscope (Olympus, Tokyo, Japan), assess the proportion of actively dividing cells in five randomly selected areas on each slide. Compare this to the proportion of actively dividing nuclei in untreated control cells to determine the relative percentage.
Fluorescence staining of living cells
HPMECs were treated with LPS alone, LPS combined with the nonspecific control (NC), or LPS in combination with the miR-30a-5p mimic. Fluorescence staining was utilized for qualitative differentiation, employing either Calcein-AM or propidium iodide (PI). Following three rounds of washing with phosphate buffered saline (PBS), HPMEC should be incubated with 250 μL of diluted solution of Calcein/PI (Dojindo, Kumamoto, Japan) for a duration of 5 min. At last, observe the cells under a fluorescence microscope (Zeiss, Tokyo, Japan).
RIPA lysis reagent, which included proteinase inhibitor and phosphatase inhibitors (Sangon Biotech, Shanghai, China) was used to lyse the entire cells. The quantification was performed using a BCA protein assay reagent kit (Sigma Aldrich, St. Louis, MO, USA). 10% SDS-PAGE was used to separate the protein samples, and PVDF membranes (Millipore, Bedford, MA, USA) were used for transfer. After blocking, immunoblotting was conducted using primary antibodies including anti-Beclin-1, anti-p62, anti-autophagy related 3 (Atg3), anti-β-actin and anti-LC3 antibodies (diluted to 1:1000; Abcam, Cambridge, UK) at a temperature of 4°C overnight. The next day, following three washes with TBST, the membranes were then exposed to the respective secondary antibodies (diluted to 1:5000; Abcam, Cambridge, UK) for a duration of 1 h at ambient temperature. Enhanced chemiluminescence reagents (GE Healthcare Life Sciences, Little Chalfont, UK) were used to measure the immunoreactive bands, which were then analyzed using Image Lab 5.0 (Bio-Rad, Hercules, CA, USA).
RNA preparation and quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR)
TRIzol (Invitrogen, Carlsbad, CA, USA) was used to extract total RNA from cultured cells. The High-Capacity cDNA Reverse Transcription Kit (Vazyme, Nanjing, China) was used to reverse transcribe normalized RNA. The SYBR Premix EX Taq Kit (Takara, Dalian, China) was used for quantitative real-time polymerase chain reaction (qRT-PCR) with the ABI Prism 7900HT Real-Time System (Applied Biosystems Inc., Foster City, CA, USA). The same amount of total RNA (1000 ng) per sample was used to generate cDNA and assume that synthesized cDNA amount will be similar among samples. The relative gene expression levels were calculated by relative quantification (2−ΔΔCt) [15], which was expressed as ratios relative to U6 rRNA. The experiments were conducted three times. Suppl. Table S1 contains the qRT-PCR sequences.
Various therapies were administered to HPMECs and transfected with an adenovirus that expressed green fluorescent protein (GFP)-LC3. Following 48 h of transfection, the cells were immobilized at ambient temperature using 4% paraformaldehyde for a duration of 20 min. Subsequently, they were rinsed three times with PBS, placed on glass slides, and the laser scanning confocal microscope was used for the examination (Zeiss, Tokyo, Japan).
Dual luciferase reporter assay
TargetScan (www.targetscan.org) was used to predict the potential miRNA targeting Beclin-1. The firefly luciferase reporter gene was inserted into the vector pGL3 along with either the amplified Beclin-1 fragment 3′-UTR or the mutated 3′-UTR. HEK293T cells were transfected with either the wild type (WT) 3′-UTR or mutant type (MT) plasmid of Beclin-1, along with the miR-30a-5p mimic. Following a 48-h transfection, the luciferase reporter assay kit (Promega, Madison, WI, USA) was employed to detect luciferase activity.
Statistical analyses were carried out with GraphPad Prism 8.0 (GraphPad Inc., San Diego, CA, USA). The mean ± SD was used to express the experimental data. The datasets were compared with the Student’s t-test and one-way analysis of variance (ANOVA). Statistical significance was indicated by p values less than 0.05.
Cellular damage in HPMECs caused by LPS stimulation
Gram-negative bacteria contain LPS as an essential element in their outer membrane. It activates EC injury and associated syndromes. In the laboratory setting, LPS induces modifications in various functions of ECs, including changes in cell survival and programmed cell death, secretion of malondialdehyde (MDA), and production of tumor necrosis factor-alpha (TNF-α) and interleukin (IL-) 6 [16,17].
To assess the impact of LPS on HPMECs, we employed CCK-8 cell viability and EdU assays to investigate the proliferation of HPMECs cells stimulated by LPS. The viability and proliferation of HPMECs were significantly reduced by treated 10 μg/mL LPS in comparison to the untreated control group, as indicated by the results obtained (Fig. 1A left & middle and Fig. 1B). To visually assess the cytotoxicity on HPMECs, the Calcein-AM staining technique was utilized to distinguish between viable cells (appearing green) and dead cells stained with PI (appearing red). Consistently, the administration of LPS led to increased cell death (Fig. 1C), quantitative statistics on fluorescence intensity are shown in Fig. 1A-right. Therefore, LPS can induced cell damage in HPMECs.
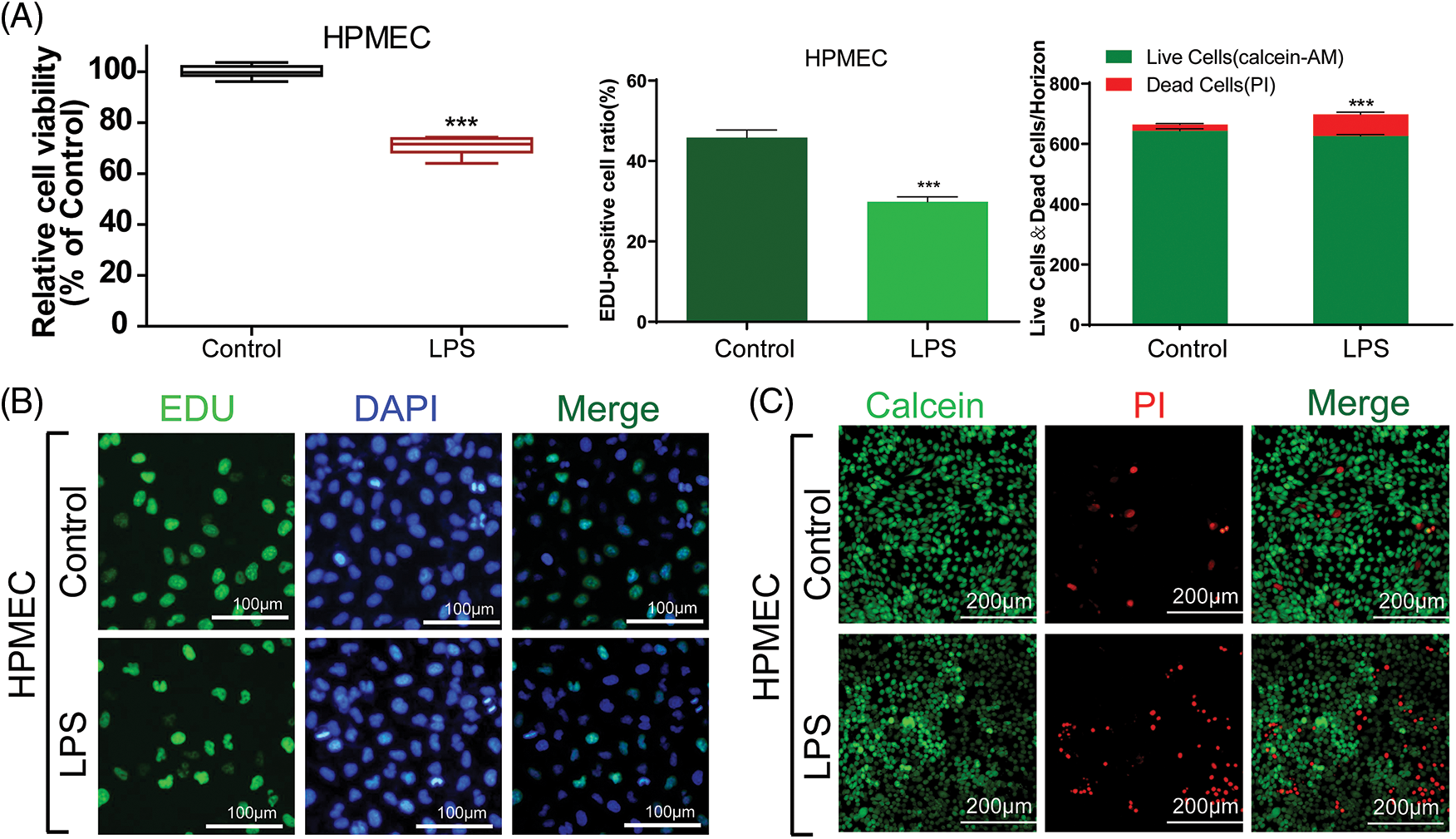
Figure 1: The viability and proliferation of LPS-treated HPMECs. (A) (left) A CCK-8 assay was used to analyze the effect of LPS on the viability of HPMECs, n = 6. (A) (middle) & (B) EdU analysis of cell proliferation following LPS treatment. All scale bars indicate 100 μm, n = 3. (A) (right) & (C) Calcein-AM and PI staining images in HPMECs after LPS treatment. All scale bars indicate 200 μm, n = 3. ***p < 0.001 vs. the control. All data are representative of three independent experiments. LPS, lipopolysaccharide; HPMEC, human pulmonary microvascular endothelial cell; EdU, 5-ethynyl-2′-deoxyuridine; AM, acetoxymethylester; PI, propidium iodide; DAPI, 4′,6-diamidino-2-phenylindole.
MiR-30a-5p offered defense against cellular harm caused by LPS
Several scientists have documented that stimulation with LPS can induce alterations in the cellular expression of miRNAs. Here, we aimed to precisely target the most significantly altered miRNAs. For this purpose, miRNA detecting was performed using HPMECs treated with LPS. This hierarchical cluster analysis conducted through qRT-PCR revealed various differences in the miRNA profiles after LPS treatment. In cells treated with LPS, we noticed a notable growth in miRNAs compared to the control group. Out of the 42 miRNAs analyzed, miR-30a-5p exhibited the most notable decrease in expression in HPMECs after LPS treatment (Fig. 2A). A limited number of reports suggest that certain miRNAs regulate autophagy and apoptosis. Confirmed among them is the regulatory role of miR30a. Consequently, our curiosity was piqued by the investigation of miR-30a-5p.
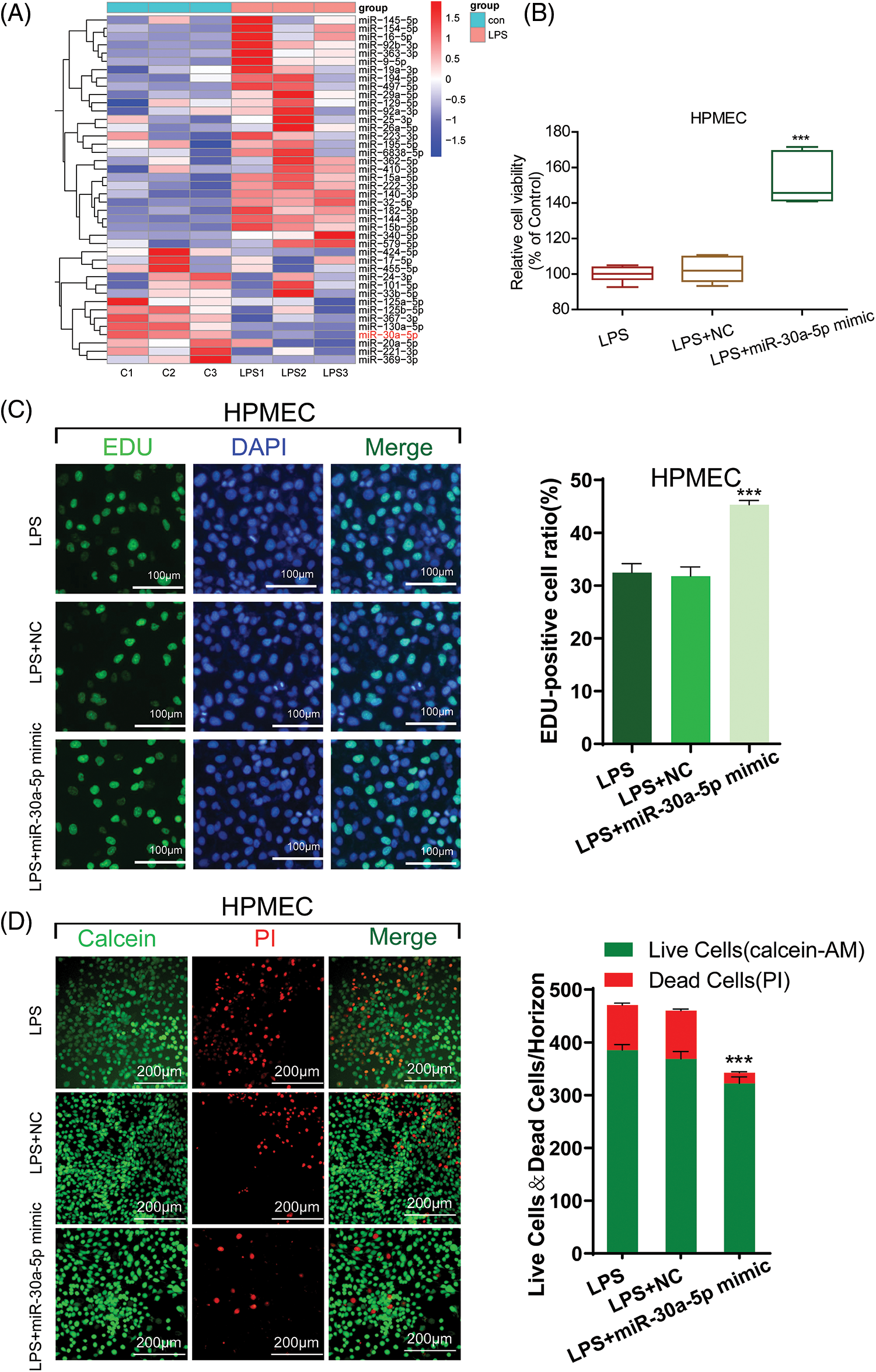
Figure 2: The damage caused by LPS is mitigated by miR-30a-5p. (A) Hierarchical cluster analysis of miRNA expression profiles in HPMECs treated with LPS, n = 3. (B) Cell viability was assessed using the CCK-8 assay, n = 6. (C) Cell proliferation was measured using an EdU assay. All scale bars indicate 100 μm, n = 3. (D) Calcein-AM and PI staining images in HPMECs after different treatments. All scale bars indicate 200 μm, n = 3. ***p < 0.001 vs. LPS+NC. All data are representative of three independent experiments. LPS, lipopolysaccharide; HPMEC, human pulmonary microvascular endothelial cell; EdU, 5-ethynyl-2′-deoxyuridine; AM, acetoxymethylester; PI, propidium iodide; DAPI, 4′,6-diamidino-2-phenylindole; NC, miR-30a-5p nonspecific control.
To assess the impact of miR-30a-5p on LPS-induced damage in HPMECs, we investigated the cellular proliferation of LPS-transfected cells treated with miR-30a-5p mimics or the non-specific control (NC) mimic using CCK-8 and EdU assays. In comparison to the NC group, the transfection of miR-30a-5p mimetics led to heightened activity and proliferation of HPMEC subjected to LPS, whereas the transfection of inhibitor mimetics caused a notable decline in cell viability subsequent to LPS treatment. Nevertheless, there were no noticeable alterations in cell viability or growth, regardless of the presence or absence of the NC control (Fig. 2B, Suppl. Fig. S2 and Fig. 2C). Furthermore, the LPS+miR-30a-5p mimic group exhibited reduced cell mortality as evidenced by Calcein-AM and PI staining (Fig. 2D). Therefore, the expression of miR-30a-5p can offer defense against cell damage induced by LPS.
LPS increases cell damage by activating autophagy in HPMECs
Previous research indicated that LPS has the ability to engage with immune cells, resulting in heightened secretion of inflammatory agents and factors that promote apoptosis, ultimately culminating in autophagy [18], which is the main reason for cell damage. In this way, HPMECs were stimulated by treatment with LPS to create an in vitro model of sepsis. The entire cellular protein was analyzed to quantify autophagy-associated proteins, including p62, Beclin-1, Atg3, and LC3-II buildup. As shown in Fig. 3A, the introduction of LPS led to a notable reduction in the p62 quantity and a rise in the quantities of Beclin-1 and Atg3, along with the buildup of LC3-II in HPMECs. Analysis using confocal microscopy, after LC3 fluorescence staining, demonstrated a rise in the quantity of autophagosomes in HPMECs after being exposed to LPS (Fig. 3B). The findings indicated that autophagy induced by LPS resulted in heightened cellular harm in HPMECs.
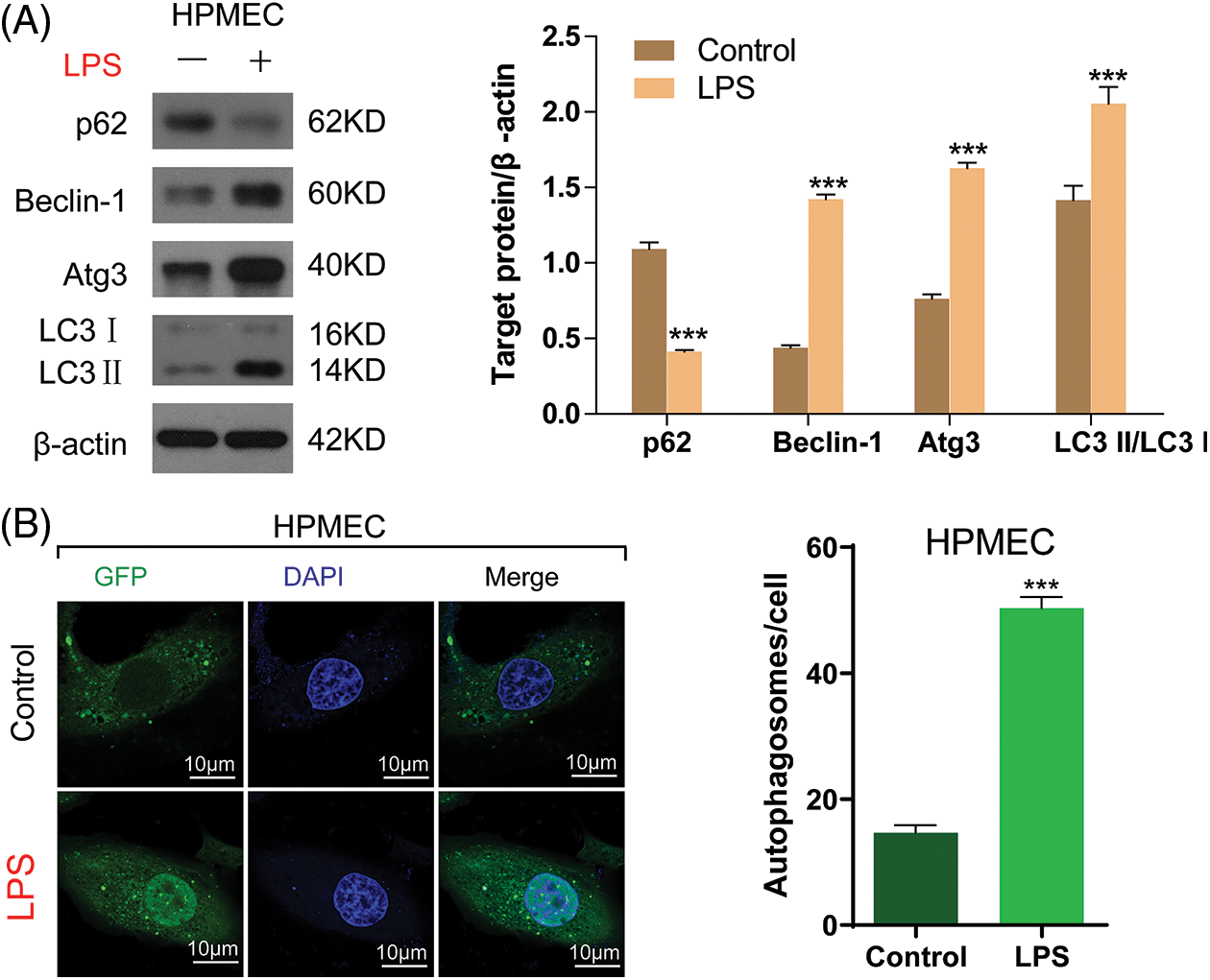
Figure 3: LPS-induced autophagy in HPMECs. (A) p62, Beclin-1, and Atg3 protein levels and LC3-II accumulation after treatment with/without LPS as determined using Western blotting analysis, n = 3. (B) LC3 fluorescence was determined using confocal microscopy analysis. Autophagosomes appear as green dots. All scale bars indicate 10 μm, n = 3. ***p < 0.001 vs. the control. All data are representative of three independent experiments. LPS, lipopolysaccharide; HPMEC, human pulmonary microvascular endothelial cell; p62, sequestosome 1; Atg3, autophagy related 3; LC3-1, cytoplasmic microtubule-associated protein 1A/1B-light chain 3; LC3-II, membrane bound, microtubule-associated protein 1A/1B-light chain 3; DAPI, 4′,6-diamidino-2-phenylindole; GFP, green fluorescent protein.
Autophagy regulation involves the participation of MiR-30a-5p
According to a prior investigation, miR-30a-5p effectively hinders the process of autophagy and is accountable for the decline in imatinib-triggered cell death in chronic myeloid leukemia cells through an autophagy-associated mechanism [19]. To examine if autophagy is induced in HPMECs due to decreased miR-30a-5p expression, HPMECs were transfected with the miR-30a-5p mimic, miR-30a-5p inhibitor, mimic NC, or inhibitor NC. A significant reduction in the levels of miR-30a-5p mRNA expression was noted in the group treated with the miR-30a-5p inhibitor, while an elevation was observed in the miR-30a-5p mimic group, thus confirming the successful establishment of the cellular model (Fig. 4A). In the miR-30a-5p inhibitor group, a significant rise was observed in LC3-II/I, Beclin-1, and Atg3 levels according to Western blotting analysis, in contrast to the inhibitor NC group. Additionally, there was a notable decrease in p62 levels. Furthermore, there was no notable distinction observed between the mimic NC and miR-30a-5p mimic group (Fig. 4B). LC3 fluorescence staining analysis with confocal microscopy also showed an increase in the quantity of autophagosomes in the miR-30a-5p inhibitor group when compared to the inhibitor NC group, but no such increase was observed in the mimic NC and miR-30a-5p mimic group (Fig. 4C). Based on the result, miR-30a-5p is involved in the regulation of autophagy in HPMECs.
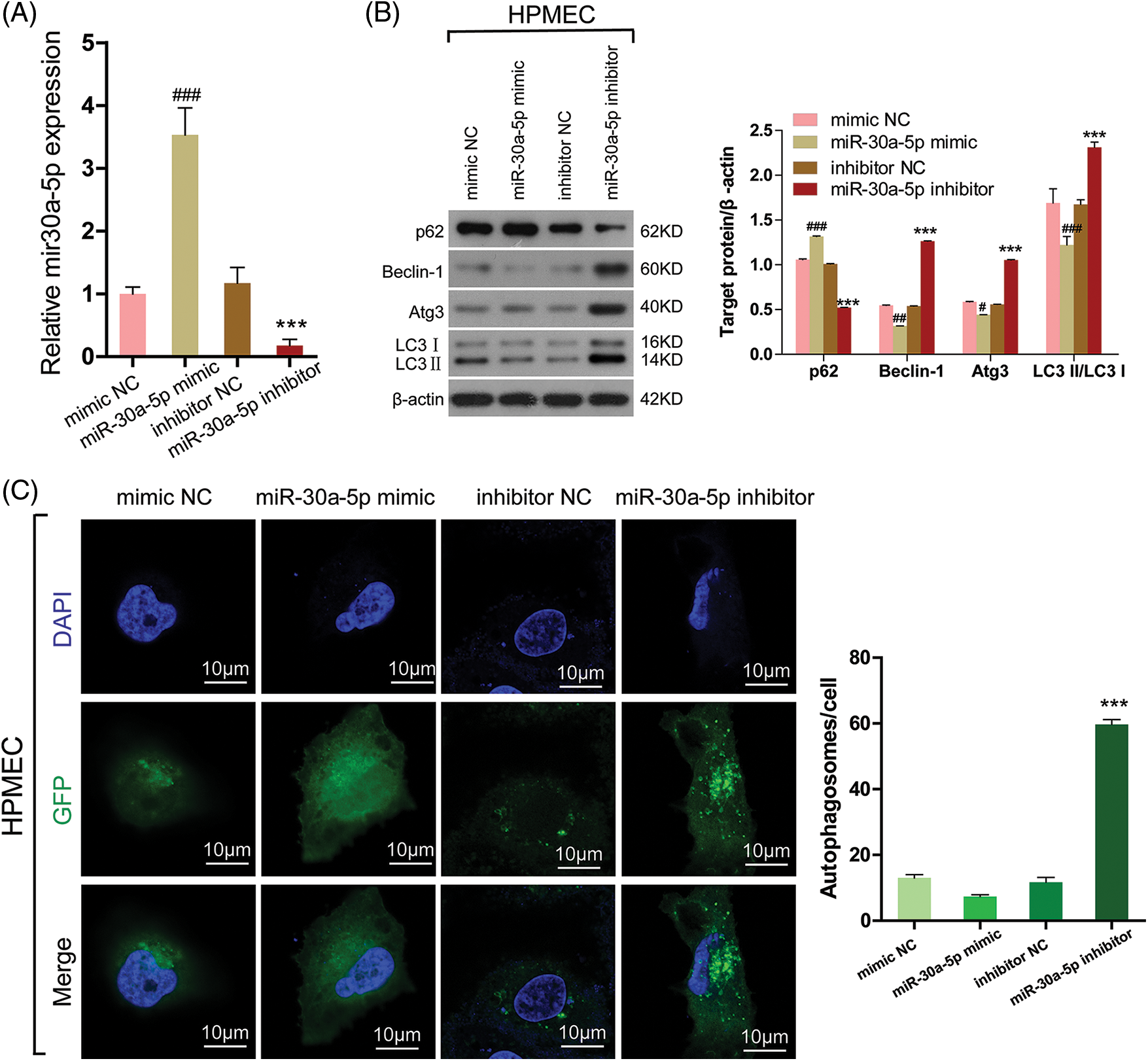
Figure 4: MiR-30a-5p regulates cell autophagy in HPMECs. HPMECs were transfected with miR-30a-5p mimic, miR-30a-5p inhibitor, or nonspecific controls, and then treated with LPS. (A) The efficiency of transfection was validated using qRT-PCR. ###p < 0.001 vs. mimic NC; ***p < 0.001 vs. inhibitor NC, n = 3. (B) Western blotting assessment of the levels of autophagy-related proteins. #p < 0.05, ##p < 0.01, ###p < 0.001 vs. mimic NC; ***p < 0.001 vs. inhibitor NC, n = 3. (C) LC3 fluorescence was determined using confocal microscopy analysis. Autophagosomes appear as green dots. All scale bars indicate 10 μm. ***p < 0.001 vs. inhibitor NC, n = 3. All data are representative of three independent experiments. LPS, lipopolysaccharide; HPMEC, human pulmonary microvascular endothelial cell; p62, sequestosome 1; Atg3, autophagy related 3; LC3-1, cytoplasmic microtubule-associated protein 1A/1B-light chain 3; LC3-II, membrane bound, microtubule-associated protein 1A/1B-light chain 3; DAPI, 4′,6-diamidino-2-phenylindole; GFP, green fluorescent protein; NC, nonspecific control.
During the treatment with LPS, autophagy is suppressed by MiR-30a-5p
In order to learn more about the functional role of miR-30a-5p in autophagy induced by LPS, the expression of autophagy markers was detected using Western blotting. This suggests that the up-regulation of miR-30a-5p in HPMECs treated with LPS can increase the levels of p62, decrease the amount of Beclin-1 and Atg3, and cause a reduction in the LC3-II/LC3I ratio. The illustration depicted in Fig. 5A. Likewise, upregulation of miR-30a-5p hindered the augmentation of autophagosomes in HPMECs treated with LPS (Fig. 5B). Nevertheless, there were no discrepancies observed in the levels of autophagy-associated proteins or the quantity of autophagosomes between the LPS group and the LPS+NC group. Both TargetScan (www.targetscan.org) (Fig. 6A) and prior research have demonstrated that miR-30a-5p has the ability to suppress autophagy by targeting Beclin-1. The luciferase reporter assays (Fig. 6B) further validated this information.
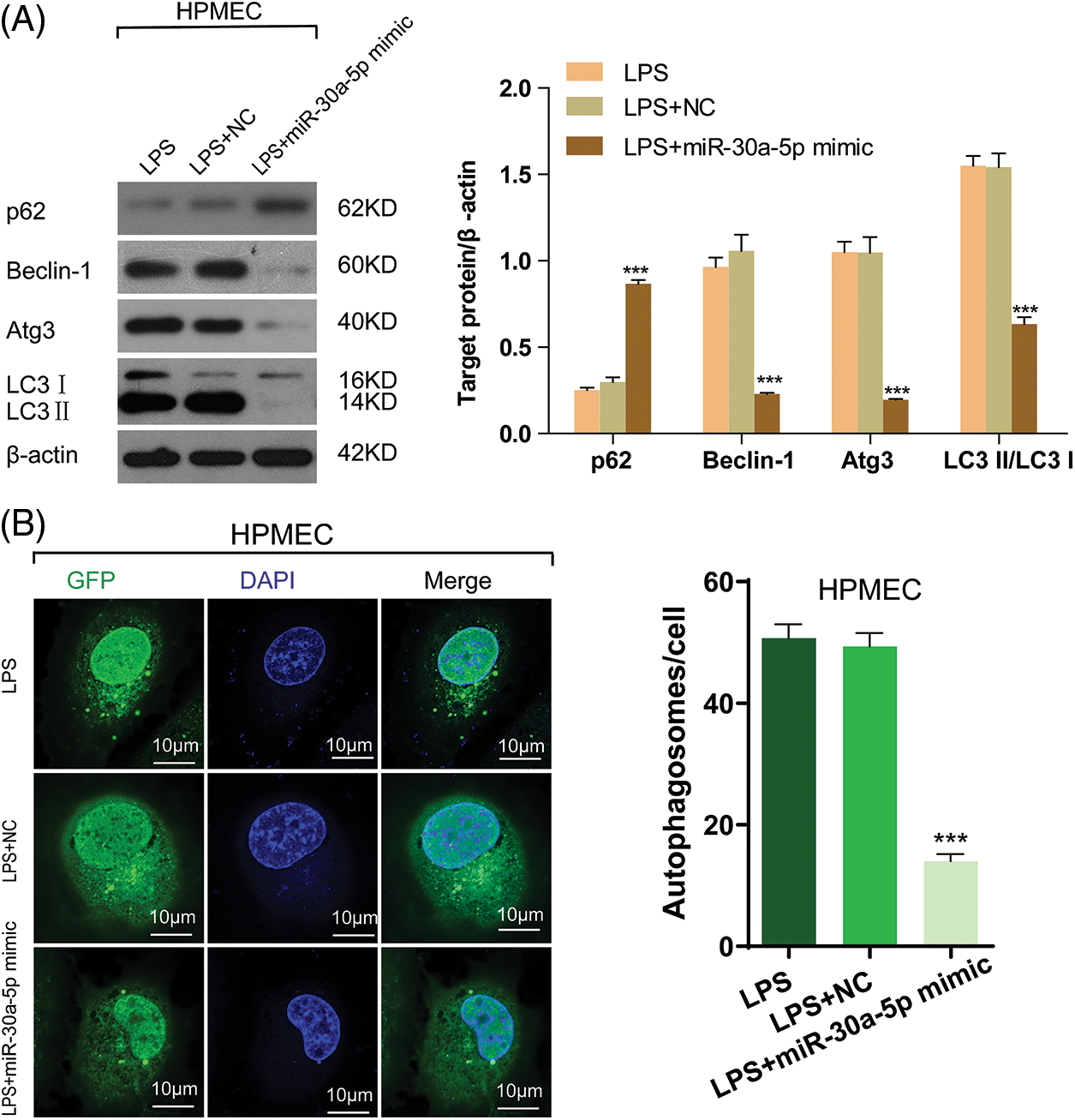
Figure 5: MiR-30a-5p attenuates LPS-induced cell autophagy in HPMECs. HPMECs were transfected with miR-30a-5p mimic or nonspecific controls, and then treated with LPS. (A) Levels of autophagy markers (p62, Beclin-1, and Atg3) and the LC3II/LC3I ratio) in different groups were determined using Western blotting analysis, n = 3. (B) Confocal microscopy analysis of LC3 fluorescence. Autophagosomes appear as green dots. All scale bars indicate 10 μm, n = 3. ***p < 0.001 vs. non-specific control. All data are representative of three independent experiments. LPS, lipopolysaccharide; HPMEC, human pulmonary microvascular endothelial cell; p62, sequestosome 1; Atg3, autophagy related 3; LC3-1, cytoplasmic microtubule-associated protein 1A/1B-light chain 3; LC3-II, membrane bound, microtubule-associated protein 1A/1B-light chain 3; DAPI, 4′,6-diamidino-2-phenylindole; GFP, green fluorescent protein; NC, nonspecific control.
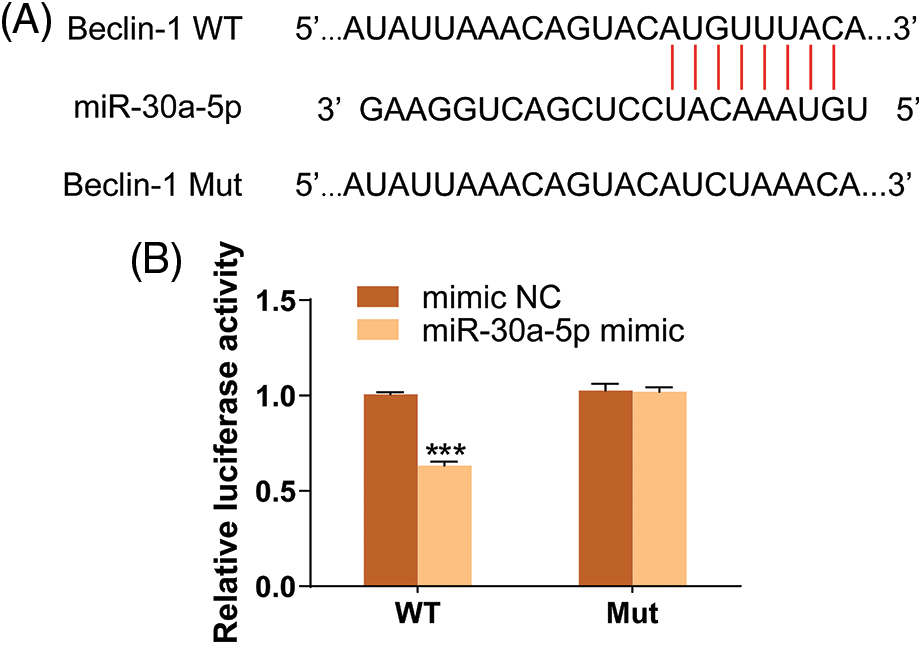
Figure 6: Beclin-1 is a target gene of miR-30a-5p. (A) Result of TargetScan (www.targetscan.org). (B) Result of luciferase reporter assay. N = 3, ***p < 0.001. WT, wild-type; Mut, mutated; NC, nonspecific control.
Organ dysfunction can result from severe sepsis, with injury to ECs being a common cause. When stimulated or injured, ECs will start autophagy in order to preserve cellular metabolism and balance. In the presence of oxidative stress and exposure to toxins, this procedure enhances the viability of cells [4,20]. In general, this stimulated autophagy provides benefits. However, excessive autophagy resulting from a persistent or strong stimulus can lead to cell death and apoptosis, which often proves deleterious. Therefore, regulating autophagy could become a new strategy to treat ECs injury. LPS is a factor that promotes inflammation [21]. It directly induces apoptosis and multiple endothelial responses, and triggers ECs injury [22]. Apoptosis was triggered by LPS in various kinds of ECs, for example HUVEC, HPMEC and normal human microvascular ECs [23]. In our study, we discovered that the introduction of miR-30a-5p through transfection resulted in a notable decrease in the rise of HPMECs apoptosis induced by LPS, as well as a reduction in cell viability and proliferation.
Autophagy is a complex and essential self-protection mechanism for cells. It plays a dual role in the regulation of apoptosis, encompassing both excessive and low levels. Autophagy induced by LPS not only aids in removing pathogens, but also mitigates cellular harm caused by the toxin. It consists of several successive steps, each with a distinct role, such as sequestering, lysosomal transport, degrading and using degraded products. Autophagy in mammalian cells is primarily controlled by proteins known as autophagy-related proteins (Atgs). Atgs, critical proteins responsible for the movement of LC3 towards the isolation membrane [24], are essential in autophagy initiation, autophagosome formation, elongation, maturation, and degradation processes [25]. In the early stages of autophagosome formation, Atg3 acts as an E2-like enzyme. The enzyme facilitates the creation of the Atg8-phosphatidylethanolamine (Atg8-PE) compound, which is essential for the process of autophagy to take place [26]. Autophagy is also tightly regulated by LC3. Activation of autophagic mechanisms may be indicated by alterations in LC3-II or the ratio of LC3-II/LC3-I [27,28]. LC3 is a specific marker of cellular autophagy. Type I (LC3-I, cytoplasmic) and Type II (LC3-II, autophagolysosome membrane) can be distinguished [29]. During the process of autophagy, LC3 undergoes a transformation into soluble LC3-I within the cytoplasm. Subsequently, the autophagy-associated protein Atg7 identifies and triggers the activation of LC3-I, leading to its modification through ubiquitination and resulting in the formation of LC3-II. The quantity of autophagosomes corresponds to the amount of LC3-II. LC3-II is also a frequently-used indicator of autophagy intensity, as it is expressed during the late stages of autophagy [18]. A high proportion of LC3-II/LC3-I suggests a significant amount of autophagosomes can generated through the conversion of LC3-I to LC3-II. P62 serves as a substrate for autophagic degradation and plays a vital role as an indicator, with its expression levels showing an inverse correlation to autophagy levels. To ascertain whether LPS leads to excessive autophagy in HPMECs, we measured autophagy-related protein levels, LC3-II accumulation and autophagosome number. The findings from all these experiments suggested that LPS induced cellular harm through the activation of excessive autophagy, aligning with a prior study [30].
The involvement of miR-30a is essential during autophagy and it controls the expression of different genes associated with autophagy. MiR-30a downregulated Atg5, which suppressed autophagy and reduced fibrosis in airway remodeling in asthma model [31]. MiR-30a-5p reduced A549 injury and lung injury in a model of acute lung inflammation by targeting and controlling the mRNA that encodes human runt-associated transcription factor 2 [32]. SMAD family member 4 (Smad4) is essential to regulate autophagy activation. According to the findings from gene software analysis and the dual luciferase assay, it is indicated that miR-30a has the ability to target and suppress the expression of Smad4 in cell lines of ovarian carcinoma. Additionally, the involvement of the TGF-β/Smad signaling pathway in regulating autophagy inhibited by miR-30a is suggested [33]. Furthermore, Beclin-1 plays a crucial part in vital cancer-related biological processes. Some of these factors comprise of cellular growth, infiltration, spread, and self-degradation [12,34,35]. In a study by Zhang et al. [36], it was found that mice subjected to a high-fat diet exhibited notably reduced levels of autophagy in endothelial cells. The reason for this was the inhibition of Beclin-1 levels by miR-30. In a mouse model of cyclical intermittent hypoxia (CIH), the upregulation of miR-30a’s complementary strand resulted in a notable elevation in Beclin-1 concentrations. Consequently, this led to enhanced autophagy of endothelial cells both in vitro and in vivo, ultimately enhancing ECs viability and resilience against CIH [37]. The administration of LPS in our research led to a decrease in the expression of miR-30a-5p in HPMECs. Inhibition of excessive autophagy by restoring miR-30a-5p levels may protect against LPS-induced cell damage, preventing mortality and promoting viability and proliferation. This indicates that miR-30a-5p has the potential to alleviate injury in HPMECs caused by LPS-induced excessive autophagy. Likewise, restoration of miR-30a-5p levels in HPMECs hindered the LPS-triggered augmentation in autophagy, encompassing the manifestation of diverse autophagic markers and the quantity of autophagosomes. Furthermore, we employed TargetScan and luciferase reporter assays to confirm that miR-30a-5p mitigates LPS-induced HPMEC injury by regulating autophagy through its interaction with Beclin-1.
In conclusion, our study showed that the anti-apoptotic activity of miR-30a-5p safeguards HPMECs from injury induced by LPS. Our findings indicate that miR-30a-5p shows potential as a viable option for addressing sepsis-related harm to the ECs. Nevertheless, the current study is limited by its failure to detect the formation of autophagosomes. Further investigation is needed to confirm the relationship between miR-30a-5p and autophagy in HPMECs exposed to LPS.
Acknowledgement: This work was supported by all the authors.
Funding Statement: This study is supported in part by Zhejiang Provincial Natural Science Foundation of China (Grant No. LTGD23H150001), the Medical and Health Research Project of Zhejiang Province (Grant No. 2023KY212) and the High-Level Talent Research Project of Hangzhou Vocational & Technical College (Grant No. HZYGCC202229).
Author Contributions: The authors confirm their contribution to the paper as follows: study conception and design: LW and JG; data collection: JM and YZ; analysis and interpretation of results: RP, JM and WC; draft manuscript preparation: RP. All authors have read and approved the final manuscript.
Availability of Data and Materials: All data generated or analyzed during this study are included in this published article and its supplementary information files.
Ethics Approval: Not applicable. This study does not include human or animal subjects.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Jaramillo-Bustamante JC, Pineres-Olave BE, Gonzalez-Dambrauskas S. SIRS or not SIRS: is that the infection? A critical review of the sepsis definition criteria. Bol Med Hosp Infant Mex. 2020;77(6):293–302. [Google Scholar] [PubMed]
2. Joffre J, Hellman J, Ince C, Ait-Oufella H. Endothelial responses in sepsis. Am J Respir Crit Care Med. 2020;202(3):361–70. [Google Scholar] [PubMed]
3. Hellenthal KEM, Brabenec L, Wagner NM. Regulation and dysregulation of endothelial permeability during systemic inflammation. Cells. 2022;11(12):1935. [Google Scholar] [PubMed]
4. Galluzzi L, Green DR. Autophagy-independent functions of the autophagy machinery. Cell. 2019;177(7):1682–99. [Google Scholar] [PubMed]
5. Wang L, Cai J, Zhao X, Ma L, Zeng P, Zhou L, et al. Palmitoylation prevents sustained inflammation by limiting NLRP3 inflammasome activation through chaperone-mediated autophagy. Mol Cell. 2023;83(2):281–97.E10. [Google Scholar] [PubMed]
6. Yin X, Xin H, Mao S, Wu G, Guo L. The role of autophagy in sepsis: protection and injury to organs. Front Physiol. 2019;10:1071. [Google Scholar] [PubMed]
7. Kingsley SMK, Bhat BV. Role of microRNAs in sepsis. Inflamm Res. 2017;66(7):553–69. [Google Scholar] [PubMed]
8. Liu F, Nie C, Zhao N, Wang Y, Liu Y, Li Y, et al. MiR-155 alleviates septic lung injury by inducing autophagy via inhibition of transforming growth factor-β-activated binding protein 2. Shock. 2017;48(1):61–8. [Google Scholar] [PubMed]
9. Fang Y, Zou L, He W. miR‐30a‐5p mitigates autophagy by regulating the Beclin‐1/ATG16 pathway in renal ischemia/reperfusion injury. Int J Mol Med. 2021;48(1):144. [Google Scholar] [PubMed]
10. Zhou SX, Li JJ, Zhao W, Wang YT, Jia WY, Liu SS, et al. ADAR1 alleviates inflammation in a murine sepsis model via the ADAR1-miR-30a-SOCS3 axis. Mediators Inflamm. 2020;2020:9607535. [Google Scholar]
11. Yuan FH, Chen YL, Zhao Y, Liu ZM, Nan CC, Zheng BL, et al. microRNA-30a inhibits the liver cell proliferation and promotes cell apoptosis through the JAK/STAT signaling pathway by targeting SOCS-1 in rats with sepsis. J Cell Physiol. 2019;234(10):17839–53. [Google Scholar] [PubMed]
12. Chen W, Li Z, Liu H, Jiang S, Wang G, Sun L, et al. MicroRNA-30a targets BECLIN-1 to inactivate autophagy and sensitizes gastrointestinal stromal tumor cells to imatinib. Cell Death Dis. 2020;11(3):198. [Google Scholar] [PubMed]
13. Liu J, Wang J, Xiong A, Zhang L, Zhang Y, Liu Y, et al. Mitochondrial quality control in lung diseases: current research and future directions. Front Physiol. 2023;14:1236651. [Google Scholar] [PubMed]
14. Durando MM, Meier KE, Cook JA. Endotoxin activation of mitogen-activated protein kinase in THP-1 cells; diminished activation following endotoxin desensitization. J Leukoc Biol. 1998;64(2):259–64. [Google Scholar] [PubMed]
15. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25(4):402–8. [Google Scholar] [PubMed]
16. Shi Q, Wang J, Wang XL, VandeBerg JL. Comparative analysis of vascular endothelial cell activation by TNF-α and LPS in humans and baboons. Cell Biochem Biophys. 2004;40(3):289–303. [Google Scholar] [PubMed]
17. Ahn SK, Choe TB, Kwon TJ. The gene expression profile of human umbilical vein endothelial cells stimulated with lipopolysaccharide using cDNA microarray analysis. Int J Mol Med. 2003;12(2):231–6. [Google Scholar] [PubMed]
18. Abel FL. Myocardial function in sepsis and endotoxin shock. Am J Physiol. 1989;257(6):R1265–81. [Google Scholar] [PubMed]
19. Khalil NA, Desouky MN, Diab IH, Hamed NAM, Mannaa HF. MicroRNA 30a mediated autophagy and imatinib response in egyptian chronic myeloid leukemia patients. Indian J Hematol Blood Transfus. 2020;36(3):491–7. [Google Scholar] [PubMed]
20. Du J, Zhou Y. Propofol reduces lipopolysaccharide‐induced cardiomyocyte injury in sepsis by activating SIRT1‐mediated autophagy. Exp Ther Med. 2023;25(4):187. [Google Scholar] [PubMed]
21. Skrzypczak-Wiercioch A, Sałat K. Lipopolysaccharide-induced model of neuroinflammation: mechanisms of action, research application and future directions for its use. Molecules. 2022;27(17):5481. [Google Scholar] [PubMed]
22. Zhu J, Dong X. Decursin alleviates LPS-induced lung epithelial cell injury by inhibiting NF-κB pathway activation. Allergol Immunopathol. 2023;51(1):37–43. [Google Scholar]
23. Dauphinee SM, Karsan A. Lipopolysaccharide signaling in endothelial cells. Lab Invest. 2006;86(1):9–22. [Google Scholar] [PubMed]
24. Hervas JH, Landajuela A, Anton Z, Shnyrova AV, Goni FM, Alonso A. Human ATG3 binding to lipid bilayers: role of lipid geometry, and electric charge. Sci Rep. 2017;7(1):15614. [Google Scholar] [PubMed]
25. Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy. 2018;14(2):243–51. [Google Scholar] [PubMed]
26. Frudd K, Burgoyne T, Burgoyne JR. Oxidation of Atg3 and Atg7 mediates inhibition of autophagy. Nat Commun. 2018;9(1):95. [Google Scholar] [PubMed]
27. Jamali-Raeufy N, Alizadeh F, Mehrabi Z, Mehrabi S, Goudarzi M. Acetyl-L-carnitine confers neuroprotection against lipopolysaccharide (LPS)-induced neuroinflammation by targeting TLR4/NFκB, autophagy, inflammation and oxidative stress. Metab Brain Dis. 2021;36(6):1391–401. [Google Scholar] [PubMed]
28. Tanida I, Ueno T, Kominami E. LC3 and autophagy. Methods Mol Biol. 2008;445:77–88. [Google Scholar] [PubMed]
29. Sharma P, Kaushal N, Saleth LR, Ghavami S, Dhingra S, Kaur P. Oxidative stress-induced apoptosis and autophagy: balancing the contrary forces in spermatogenesis. Biochim Biophys Acta Mol Basis Dis. 2023;1869(6):166742. [Google Scholar] [PubMed]
30. Guo J, Chen W, Bao B, Zhang D, Pan J, Zhang M. Protective effect of berberine against LPS-induced endothelial cell injury via the JNK signaling pathway and autophagic mechanisms. Bioengineered. 2021;12(1):1324–37. [Google Scholar] [PubMed]
31. Li BB, Chen YL, Pang F. MicroRNA-30a targets ATG5 and attenuates airway fibrosis in asthma by suppressing autophagy. Inflammation. 2020;43(1):44–53. [Google Scholar] [PubMed]
32. Li P, Yao Y, Ma Y, Chen Y. MiR-30a-5p ameliorates LPS-induced inflammatory injury in human A549 cells and mice via targeting RUNX2. Innate Immun. 2021;27(1):41–9. [Google Scholar] [PubMed]
33. Cai Y, An B, Yao D, Zhou H, Zhu J. MicroRNA miR-30a inhibits cisplatin resistance in ovarian cancer cells through autophagy. Bioengineered. 2021;12(2):10713–22. [Google Scholar] [PubMed]
34. Du Q, Ye X, Lu SR, Li H, Liu HY, Zhai Q, et al. Exosomal miR-30a and miR-222 derived from colon cancer mesenchymal stem cells promote the tumorigenicity of colon cancer through targeting MIA3. J Gastrointest Oncol. 2021;12(1):52–68. [Google Scholar] [PubMed]
35. Chen MY, Yadav VK, Chu YC, Ong JR, Huang TY, Lee KF, et al. Hydroxychloroquine (HCQ) modulates autophagy and oxidative DNA damage stress in hepatocellular carcinoma to overcome sorafenib resistance via TLR9/SOD1/hsa-miR-30a-5p/Beclin-1 axis. Cancers. 2021;13(13):3227. [Google Scholar] [PubMed]
36. Zhang T, Tian F, Wang J, Jing J, Zhou SS, Chen YD. Endothelial cell autophagy in atherosclerosis is regulated by miR-30-mediated translational control of ATG6. Cell Physiol Biochem. 2015;37(4):1369–78. [Google Scholar] [PubMed]
37. Bi R, Dai Y, Ma Z, Zhang S, Wang L, Lin Q. Endothelial cell autophagy in chronic intermittent hypoxia is impaired by miRNA-30a-mediated translational control of Beclin-1. J Cell Biochem. 2019;120(3):4214–24. [Google Scholar] [PubMed]
Supplementary Materials

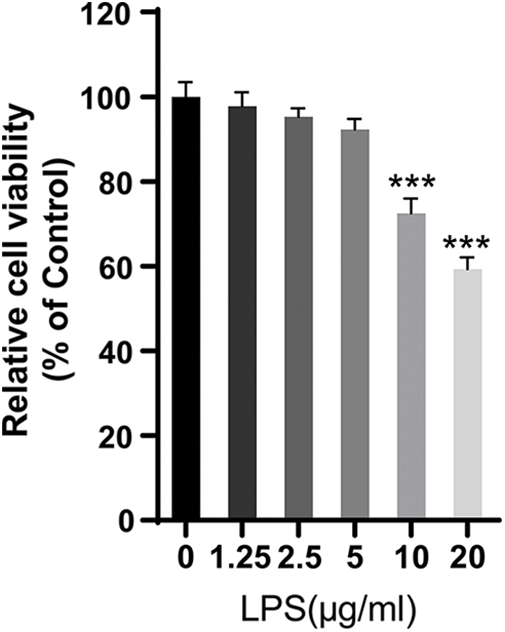
Figure S1: The viability of LPS-treated HPMECs. CCK-8 assay was used to analyze the effect of LPS on the viability of HPMECs. N = 6, ***p < 0.001 vs. the control (0 μg/mL LPS). All data are representative of three independent experiments. LPS, lipopolysaccharide.
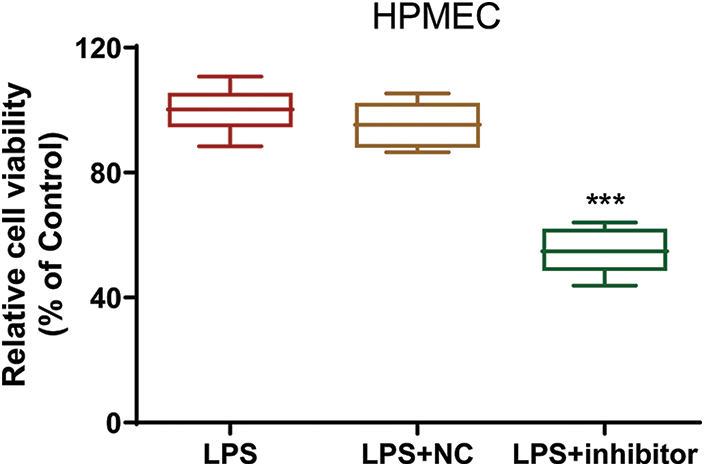
Figure S2: The viability of LPS-treated HPMECs after transfection. Cell viability was assessed with the CCK-8 assay. N = 6, ***p < 0.001 vs. LPS+NC. All data are representative of three independent experiments. LPS, lipopolysaccharide; HPMEC, human pulmonary microvascular endothelial cell; NC, inhibitor nonspecific control; inhibitor, miR-30a-5p inhibitor.
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools