
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

The heterogeneity of tumor-associated macrophages and strategies to target it
1 Institute of Cancer, Xinqiao Hospital, Third Military Medical University, Chongqing, 400037, China
2 Chongqing Key Laboratory of Immunotherapy, Xinqiao Hospital, Third Military Medical University, Chongqing, 400037, China
* Corresponding Author: DEGAO CHEN. Email:
BIOCELL 2024, 48(3), 363-378. https://doi.org/10.32604/biocell.2023.046367
Received 27 September 2023; Accepted 30 November 2023; Issue published 15 March 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Tumor-associated macrophages (TAMs) are emerging as targets for tumor therapy because of their primary role in promoting tumor progression. Several studies have been conducted to target TAMs by reducing their infiltration, depleting their numbers, and reversing their phenotypes to suppress tumor progression, leading to the development of drugs in preclinical and clinical trials. However, the heterogeneous characteristics of TAMs, including their ontogenetic and functional heterogeneity, limit their targeting. Therefore, in-depth exploration of the heterogeneity of TAMs, combined with immune checkpoint therapy or other therapeutic modalities could improve the efficiency of tumor treatment. This review focuses on the heterogeneous ontogeny and function of TAMs, as well as the current development of tumor therapies targeting TAMs and combination strategies.Keywords
Macrophages are fundamental components of the innate immune system and are responsible for pathogen clearance, tissue homeostasis, and inflammation. Tumor-associated macrophages (TAMs), a major component of the tumor immune microenvironment (comprising over 50% of solid tumors), are involved in tumor initiation, progression, metastasis and angiogenesis [1–3]. Moreover, TAMs predominantly shape tumor microenvironment, anti-tumor immunity and immunotherapy efficacy [4–6]. Therefore, targeting TAMs has emerged as a potential strategy for revitalizing the anti-tumor immune response in tumor therapy.
Based on technological advances in single-cell RNA sequencing and lineage tracing, TAMs have been suggested to be a heterogeneous population with heterogeneous origins and functions [7]. This means that the ontogeny of TAMs includes tissue-resident macrophages (TRMs) originating from yolk-sac-derived erythro-myeloid progenitors, alongside differentiation from peripheral monocyte infiltration. The differentiation from peripheral monocyte infiltration implies that TAMs can affect tumorigenesis and progression through various mechanisms, including fostering tumor cell proliferation, distant metastasis, angiogenesis, and suppression of the antitumor T cell immunity. Understanding the heterogeneity of TAMs will help decipher the phenotypic and functional heterogeneity, enabling the development of antitumor strategies by targeting TAMs.
To date, there have been many developments in antitumor treatment strategies targeting TAMs. These include endeavors to reduce the infiltration of TAMs [8–10], depletion of TAMs [11–13], reversal of the phenotype of TAMs [14–16], and the combine effort of immune checkpoint inhibitors or other antitumor strategies [17,18]. Nevertheless, breakthroughs in drugs targeting TAMs are still pending and efforts to clarify the heterogeneity of TAMs are required. In this review, we summarized the heterogeneous ontogeny and function of TAMs, tumor therapies targeting TAMs and strategies for combination therapy.
The heterogeneous ontogeny of TAMs
The ontogeny of TAMs is mainly derived from infiltrating peripheral monocytes in response to the tumor microenvironment (TME). However, the discovery of TRMs within tumors, originating from embryonic sources, has been questioned [19] (Fig. 1). TRM originates from yolk sac progenitor cells and, during organogenesis, enters specific tissues to differentiate into macrophages, establishing stable spatial and functional relationships with specific cells within the tissues [20].
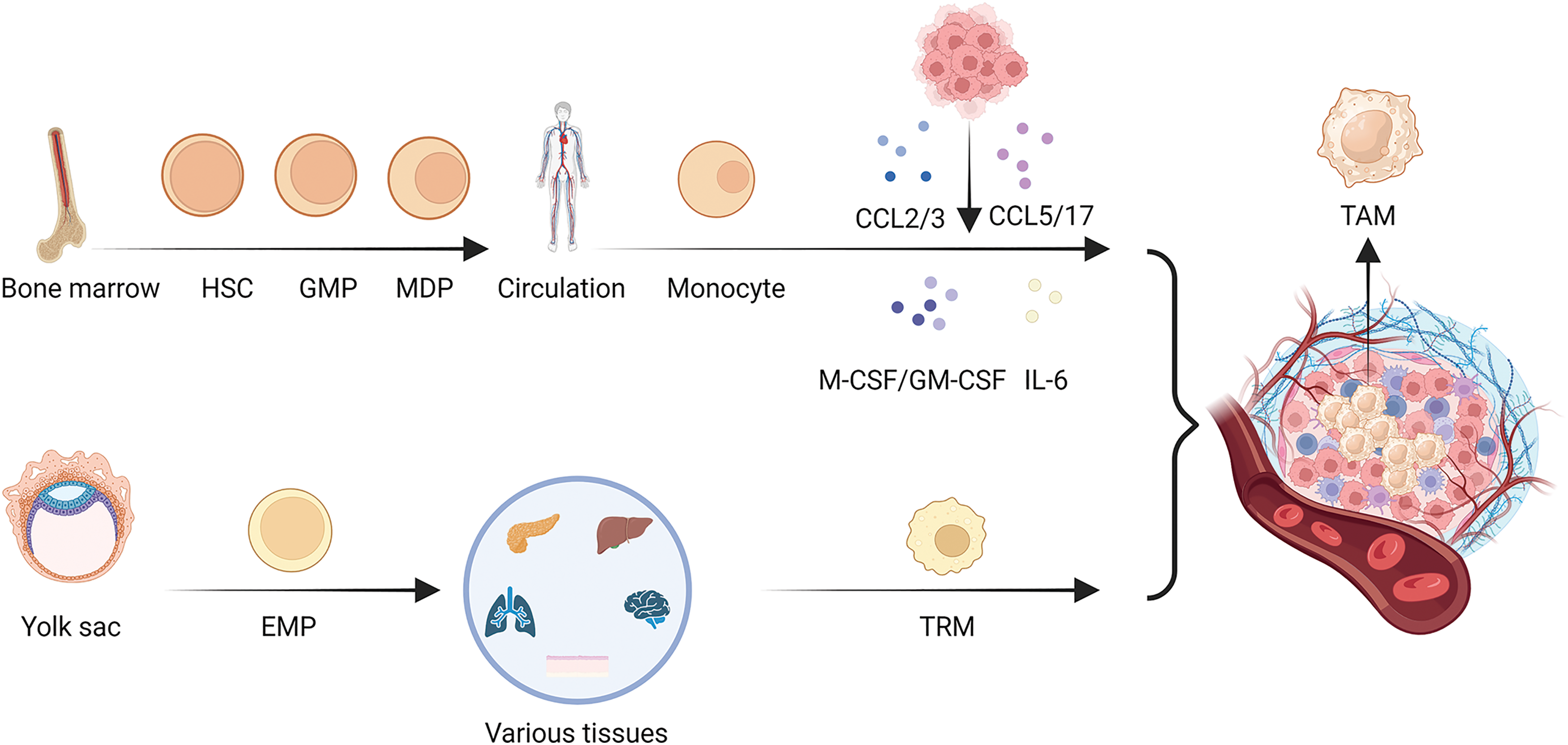
Figure 1: Heterogeneity in the origin of TAMs. There are two main origins of TAMs, in addition to the well-known bone marrow origin, there is yolk sac origin of TAMs. As the main source of TAMs, monocytes first develop from hematopoietic stem cells (HSCs), then differentiate into granulocyte-macrophage progenitors (GMPs), further develop into monocyte-dendritic cell progenitors (MDPs) and leave the bone marrow for the circulation, developing into monocytes. In the process of tumor development, monocytes are influenced by various chemokines and cytokines secreted by tumor cells and can form TAMs in the tumor microenvironment, while TRMs are mainly developed from the yolk sac-derived erythroid-myeloid progenitor cells (EMPs), and are resided in various tissues in the body, including lung, liver, pancreas, brain, skin, etc. When tumor occurs in an organ, TRMs will form TAMs and play a role in regulating the growth of the tumor.
Myeloid lineage formation involves the development of HSCs and the process of differentiation of GMPs and MDPs in the bone marrow [21–23]. Once monocytes derived from the bone marrow enter circulation, chemotactic signals produced by cancer cells or stromal cells cause circulating monocytes to be drawn into the TME. Various chemokines have been elucidated, such as CCL2 [24], CCL3 [25], CCL5 [26], and CCL17 [27].
Monocytes and macrophages express CCR2, a CCL2 receptor. Upregulated levels of CCL2 and CCR2 promote macrophages accumulation [24]. CCL3 functions as a chemokine that induces macrophages migration. Additionally, CCL3 may bind to CCR1 and CCR5 on macrophages, which polarize them to take on the traits and functionality of M2-type macrophages [28,29]. CCL5 attracts M2-type macrophages and encourages them to accumulate at sites of inflammation or tumor. In addition, CCL5 can lead to the polarization and activation of M2-type macrophages, inducing them to exhibit anti-inflammatory properties through the CCL5/CCR5 axis [30]. The binding of CCL17 to CCR4 promotes M2 macrophages migration. Further studies have shown that CCL17 can induce the transformation of M2 macrophages into a pro-inflammatory phenotype [27]. The CCL family plays an integral role in TAMs developmental heterogeneity.
In addition, various cytokines, including Macrophage Colony-Stimulating Factor (M-CSF), Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF), and Interleukin-6 (IL-6) [31,32], are involved in the differentiation of monocytes into macrophages [33,34].
M-CSF, which binds to CSF1R mainly expressed on macrophage, promotes the differentiation and survival of monocytes into macrophages, whereas GM-CSF not only promotes differentiation, but also enhances antigen presentation by macrophages [34,35]. IL-6 is one of the important factors for the differentiation of monocytes into macrophages, contributing to the activation of macrophages into a pro-inflammatory type and release of pro-inflammatory cytokines, such as tumor necrosis factor α (TNF-α) and IL-12 [33]. Furthermore, in tumor-bearing mice, angiotensin 2 (Ang2) overproduction increases the number of macrophage progenitors and hematopoietic stem cells, providing a fresh supply of TAMs [36]. Thus, it is evident that numerous cytokines such as M-CSF, GM-CSF, and IL-6 synergize with the CCL family and play an important role in the origin of TAMs.
TAMs originate from the bone marrow. In mice, the development of EMPs present in the fetal liver primarily arises from yolk sac hematopoietic endothelial cells. EMPs are a special type of hematopoietic stem cells that exist in the hematopoietic system during the fetal stage. These cells can differentiate into various types of blood cell progenitor cells, including red blood cells, macrophages, and other types of white blood cells [37]. Unlike monocyte-derived macrophages, EMP-derived macrophage precursors initially colonize the embryonic yolk sac, accompany organogenesis and eventually matures into TRMs [38].
TRMs and inflammatory monocytes are sources of TAMs in pancreatic ductal adenocarcinomas (PDAC). A sizeable fraction of macrophages in the pancreas settle during embryonic development and expand through in situ proliferation as PDAC develops. In PDAC, TAMs generated from monocytes are more effective at presenting antigens; however, embryo-derived TAMs display a pro-fibrotic transcriptional profile, suggesting their function in generating and modifying extracellular matrix components [39]. TRMs play an indispensable role in the development of PDAC.
Additionally, alveolar macrophages increase early in neoplasia and inhibit cytotoxic T cell responses in an oncogenic Kras-driven lung cancer mouse model, suggesting that by changing the TME, a subgroup of TRMs could assist in neoplastic development [40]. Similarly, early in the development of non-small cell lung carcinoma (NSCLC), TRMs gather near tumor cells to encourage the transition of the epithelium to the mesenchyme and the invasiveness of the tumor cells, causing a strong regulatory T cell response that shields tumor cells from the immune system [41]. Furthermore, in both healthy mammary glands and primary sites of breast cancer, there was a population of FOLR2+ TRMs. In cancer stroma, FOLR2+ macrophages congregate in the perivascular regions where they interact with CD8+ T cells [42]. This suggests that TRMs play an important role in TME change in NSCLC and breast cancer.
TRMs reside in tissues for long periods of time and are mainly derived from macrophage precursor cells during embryonic development. Monocyte derived TAMs mainly differentiate from the circulation and migrate from peripheral blood monocytes into the TME [43]. Thus, the heterogeneous categorization of TAMs according to their origin and function, to be elaborated next, has important implications for basic research and clinical applications.
The functional heterogeneity of TAMs
Macrophages are thought to phagocytose tumors and activate antitumor immunity [44]. However, it has been shown that macrophages encourage tumor development by boosting cancer cell invasion and metastasis [45], stimulating angiogenesis [46], and supplying growth factors conducive to cancer [47]. Moreover, they have been found to spread to malignant tumors [48]. However, different macrophage populations with opposing pro- and anti-tumorigenic activities may coexist in the same cancer type [49]. Although some studies have classified macrophages into M1 antitumor and M2 tumor-promoting types, such classifications are simplistic and difficult to perform within the TME. It is more advantageous to consider macrophages as existing along a spectrum of phenotypes, wherein molecular and genetic features coexist [50] (Fig. 2).
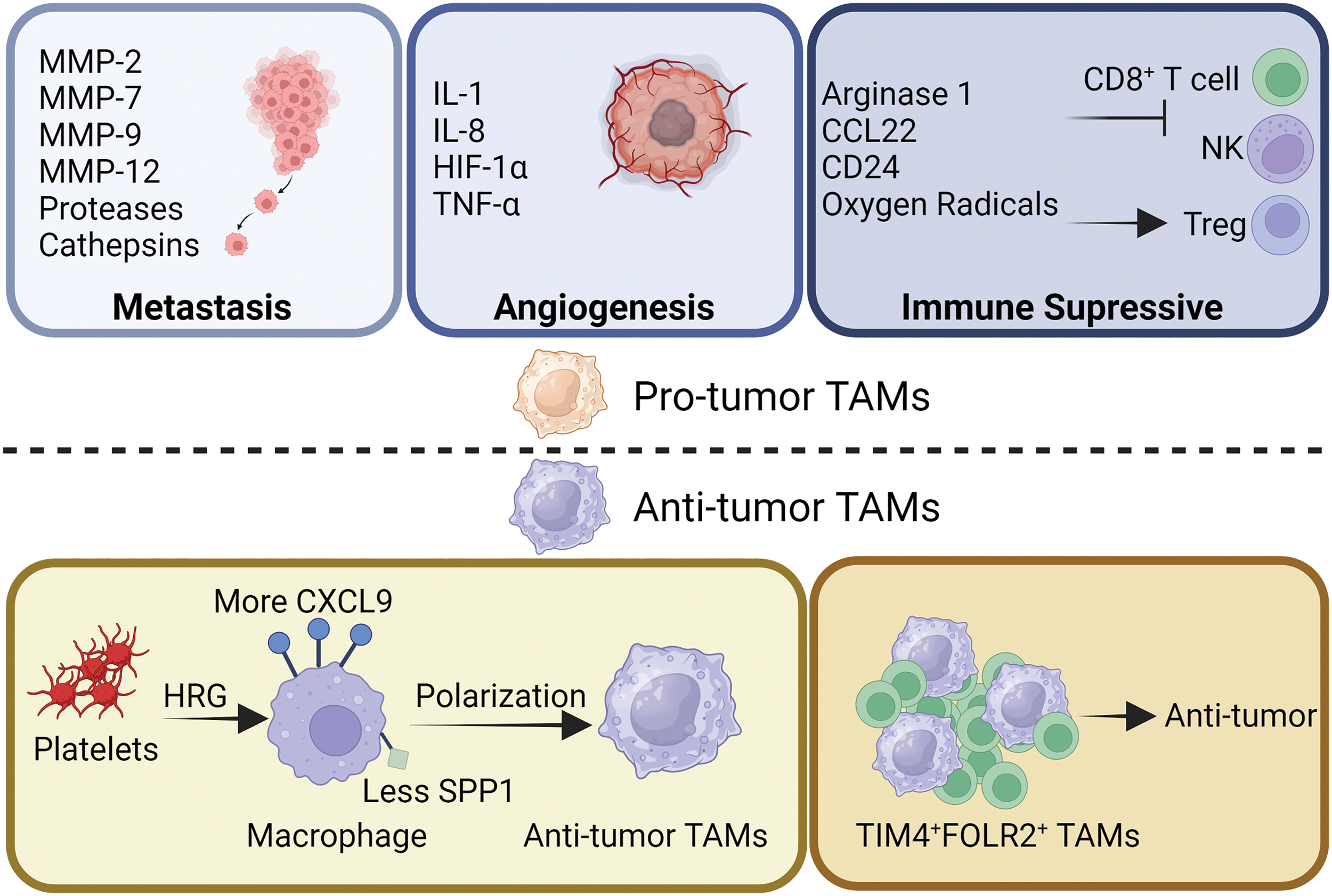
Figure 2: Functional heterogeneity of TAMs. TAMs have long been known to promote tumor development. During tumor metastasis, TAMs promote tumor cell metastasis and invasion by secreting various types of matrix metalloproteinases, proteases and cathepsins to degrade the matrix. TAMs inhibit the tumor suppressor function of CD8+ T cells and NK cells by secreting arginase1, CCL22, oxygen radicals, and expressing CD24, and increase the tumor-promoting effect of Tregs. In addition, TAMs are also able to secrete IL-1/8, Hypoxia-Inducible Factor 1 (HIF-1), and TNF-α to promote tumor angiogenesis. However, TAMs have also been reported to have tumor suppressor function. Platelets, by secreting HRG, are able to increase the expression of CXCL9 on the surface of macrophages and decrease the expression of secreted phosphoprotein-1 (SPP1), thus contributing to the development of macrophages toward TAMs with anti-tumor properties. In addition, a subpopulation of TAMs identified as TIM4+FOLR2+ has been reported to have antitumor activity.
TAMs’ pro-tumoral behaviors, such as tumor initiation, angiogenesis, metastasis, drug resistance, and antitumor immunosuppression, have been well investigated [51]. By fostering genetic instability, feeding cancer stem cells, facilitating metastasis, impeding the TME through metabolites, and suppressing protective adaptive immunity, TAMs contributed to tumor progression at multiple levels [4,52].
TAMs are involved in all stages of tumor progression, are strongly correlated with poor prognosis, and play a crucial role in the interaction between cancer cells and the TME [4,53]. There is a strong correlation between TAMs infiltration and tumor proliferation. TAMs contribute to tumor invasion and metastasis by releasing extracellular matrix (ECM) degrading enzymes. Several metalloproteinases, such as matrix metalloproteinases-2 (MMP-2), MMP-9, serine proteases and cathepsins, have been demonstrated to break down the ECM, allowing tumor cells to escape and spread, and facilitating the migration of tumor cells [54,55]. Thus, tumor metastasis and TAM infiltration are inextricably linked.
To exert their proangiogenic effects, TAMs coordinate the expression of several cytokines, such as IL-1, IL-8 and tumor necrosis factor α (TNF-α). Endothelial cell proliferation, matrix remodeling, and spatial or temporal vascularization all aided by the coordinated expression of these molecules [56,57]. Under hypoxic conditions, together with the upregulation of hypoxia-inducible transcription factors, TAMs appear to reshape TME into proangiogenic, prometastatic, and immunosuppressive situation [58]. Consecutively, TAMs further strengthen tumor viability by promoting angiogenesis.
In addition, immune killing mediated by tumor-specific T cell and natural killer (NK) cells can be modulated by TAMs. TAMs can limit CD8+ T cell proliferation directly through arginase 1 and oxygen radicals. To further suppress the antitumor immune response of T cells, TAMs recruit Tregs via CCL22 [59]. TAMs are also engaged in tumor immune escape, for instance, CD24 interacts with sialic acid binding Ig like lectin 10 (Siglec-10) on TAMs to facilitate the immune escape of tumor cell [60]. Overall, TAMs exhibit immunosuppressive effects that promote tumor proliferation.
Although a large number of studies have reported that TAMs promote cancer growth and invasion, there is a growing number of articles that focus on the key role played by different TAMs subpopulations in supporting cancer suppressive immune responses [42,61–63].
Prior to the initiation of tumor development, healthy mammary glands were populated with FOLR2+ macrophages. FOLR2+ macrophages gather with CD8+ T cell aggregates in the cancer stroma close to the vasculature, indicating better survival in patients with breast cancer [42]. Similarly, a particular group of TIM4+FOLR2+ macrophages that localize in the T-cell zone of tumor-associated tertiary lymphoid structures (TLS) is topographically different from typical TAMs, which is positively related to a better prognosis in several tumor types [64]. These results suggest that FOLR2+ macrophages are associated with better survival prognosis.
In the TME, the level of histidine-rich glycoprotein (HRG), which is mainly derived from the plasma or platelets, is low [65]. Reversing TAM polarization by HRG depends heavily on the downregulation of placental growth factors. In order to reduce cancer development and metastasis, HRG is crucial in inducing antitumor immune responses and vascular normalization by regulating macrophage polarization [63]. HRG plays a key role in macrophages and induces antitumor effects.
Furthermore, the expression of two genes, CXCL9 and SPP1, which determine macrophage polarity, represents a fundamental characteristic of the TME. The quantity of antitumor immune cells in cancer and treatment efficiency may be determined by the CXCL9:SPP1 ratio [61].
There is significant heterogeneity in TAMs across different cancer types. Recognizing and differentiating various TAM subtypes is crucial for providing personalized treatment strategies for patients with cancer.
Tumor therapy by targeting TAMs
Because of their pro-tumoral functions, TAMs have been considered potential targets for tumor suppression, and multiple strategies targeting TAMs, such as decreasing TAMs infiltration, inducing TAMs depletion, and TAMs reprograming (Fig. 3) (Table 1), are currently undergoing preclinical research and clinical trials.
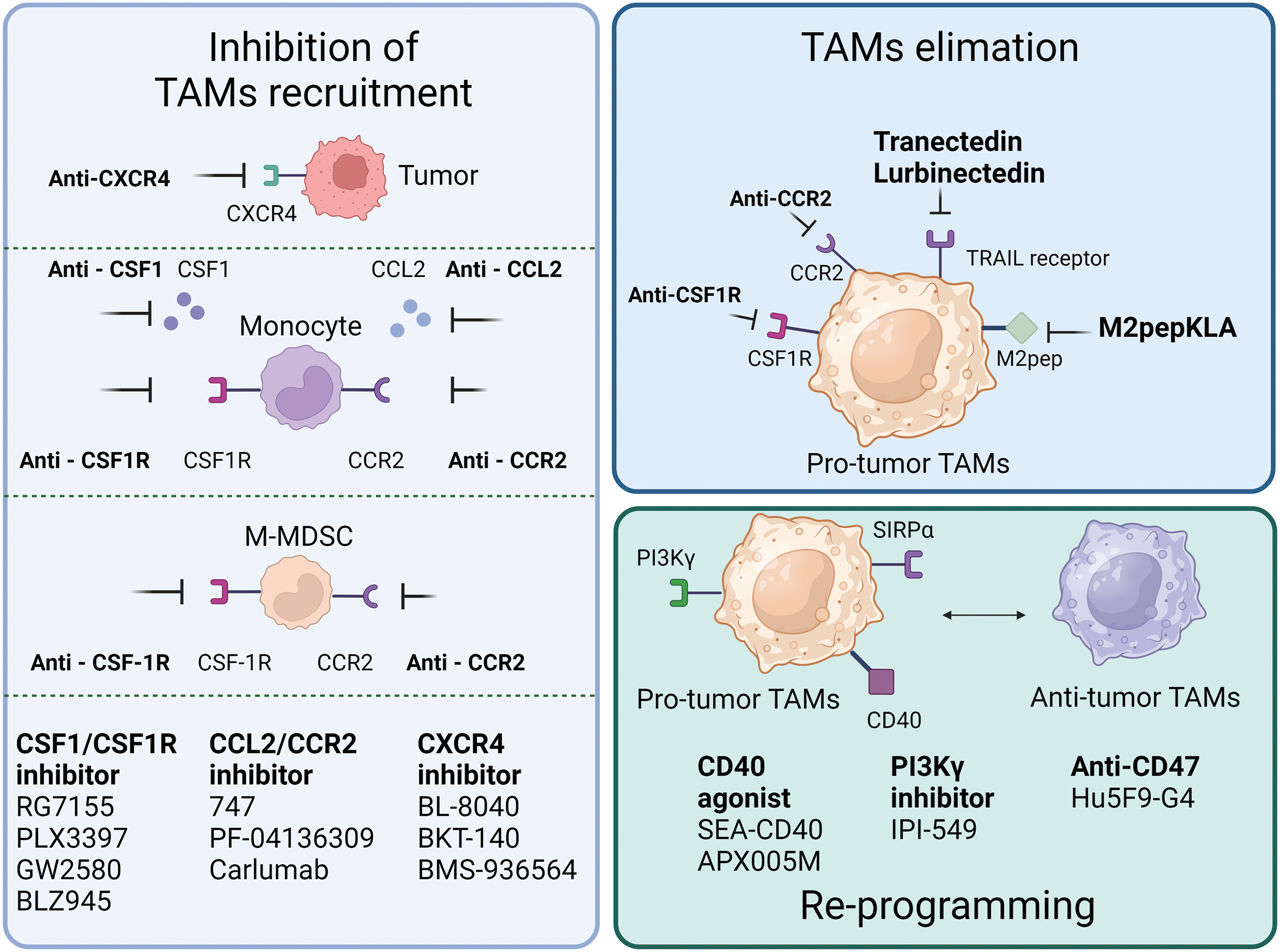
Figure 3: Single-agent tumor therapy strategies targeting TAMs. The current mainstream therapeutic strategies for targeting TAMs are mainly in the following three categories: one is to inhibit the recruitment of TAMs, and common targets mainly include inhibition of CSF-1R and CCR2 expression on monocytes and M-MDSC, inhibition of CXCR4 expression on tumor cells, as well as the release of CSF1, CCL2 and other factors. Secondly, targeting CSF1R, CCR2, TRAIL, M2pep expressed on pro-tumor-associated TAMs to remove TAMs. Lastly, reprogramming of pro-tumorigenic TAMs to tumor-suppressive TAMs through activation of CD40, inhibition of PI3Kγ, CD47, etc.
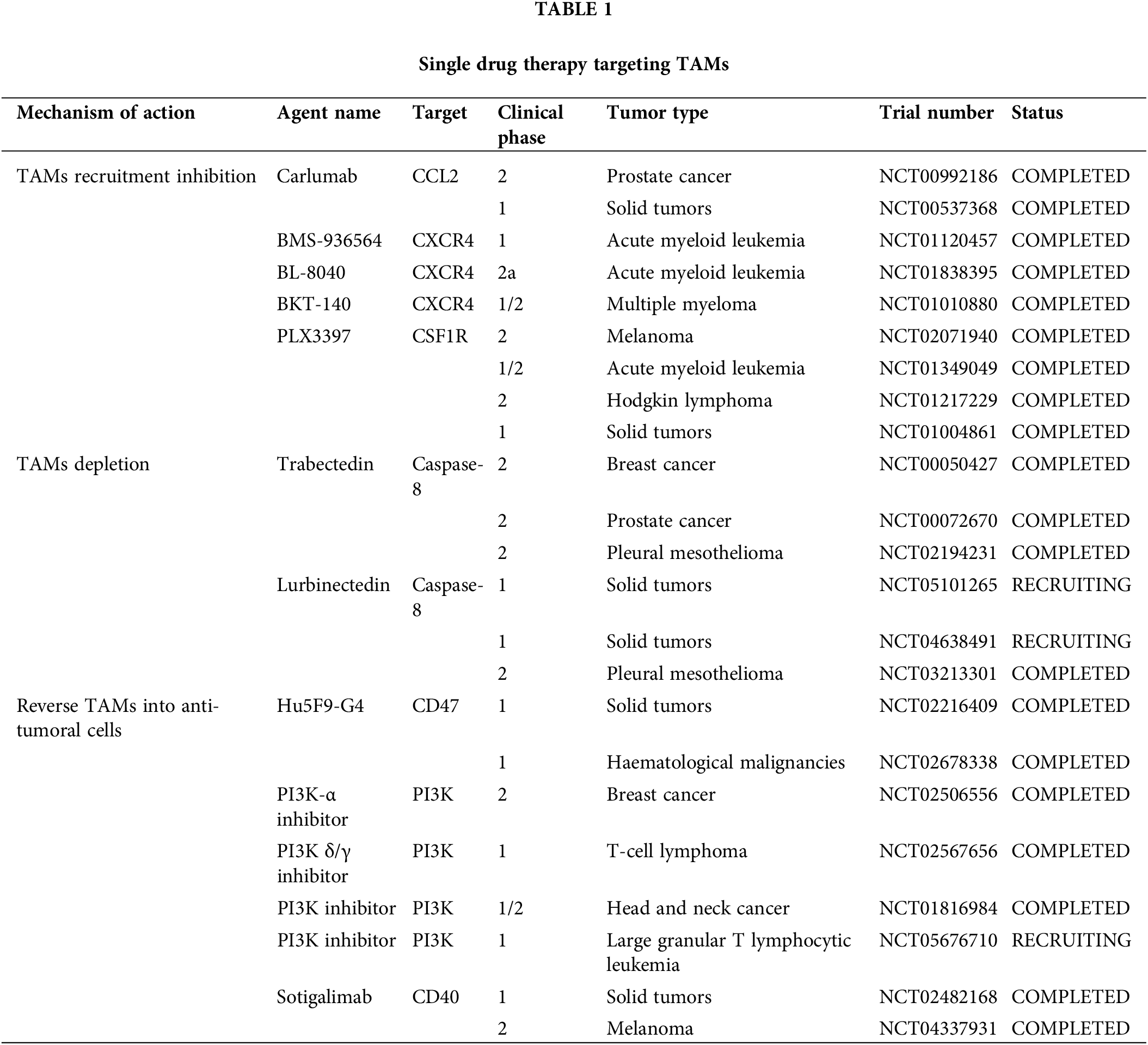
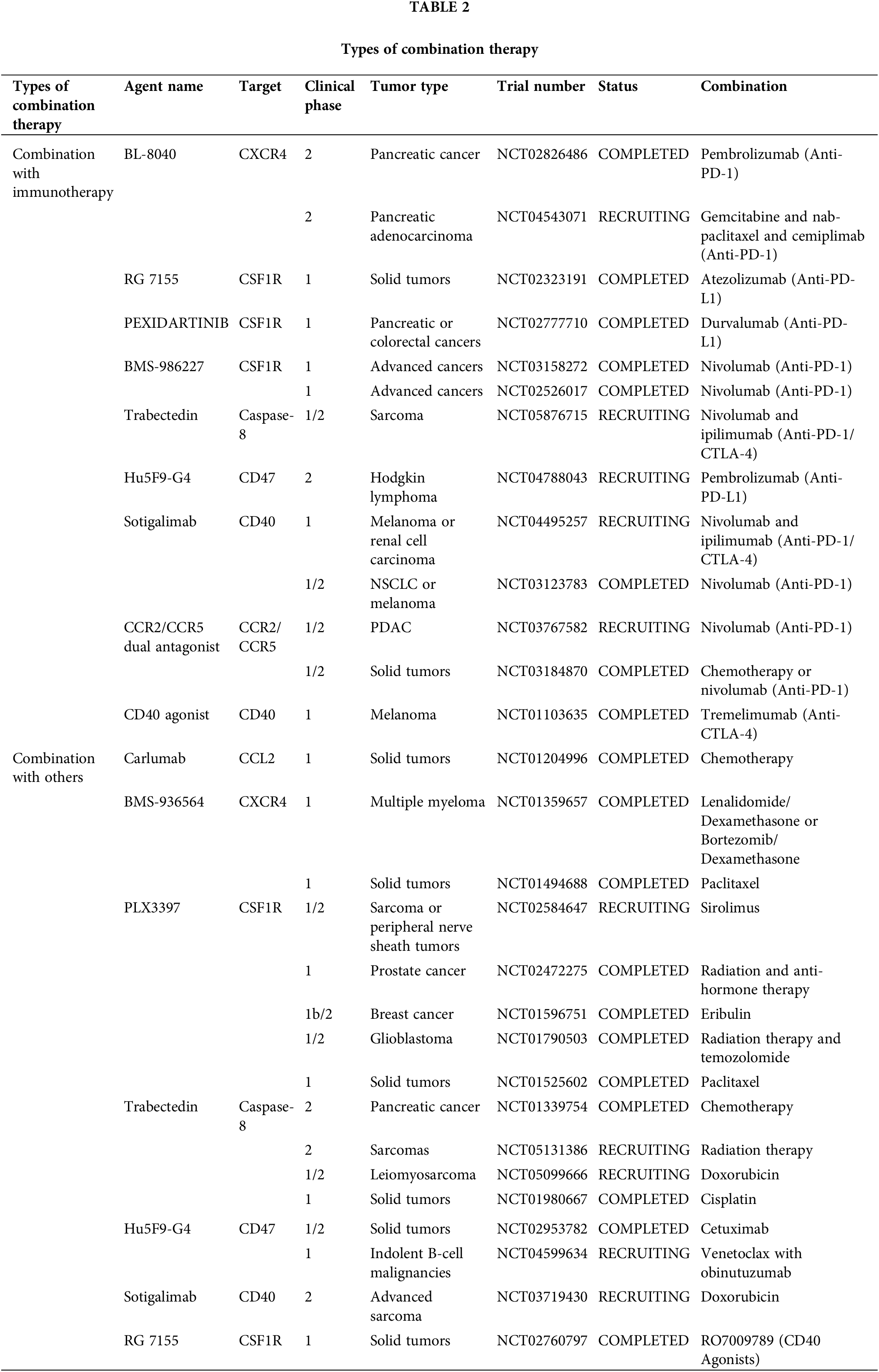
In animal models of esophageal squamous cell carcinoma, blocking the CCL2-CCR2 axis significantly reduces carcinogenesis, which is partly caused by the suppression of M2 polarization and TAMs recruitment [66]. The human anti-CCL2 antibody, carlumab, formerly known as CNTO888, prevents CCL2 from interacting with CCR2 [67]. In patients with prostate cancer, the administration of CNTO888 dramatically reduces microvascular density, decreases CD68+ macrophage infiltration, and considerably slows tumor growth [68]. However, in a phase 2 (NTC00992186) clinical trial, carlumab did not exhibit anticancer efficacy when used alone in metastatic castration-resistant prostate cancer, despite its good tolerability [69]. Inhibition of TAMs recruitment by CCL2 is an attractive target; however, more clinical trials are needed to validate this hypothesis.
The recruitment of the TAMs is further aided by the increase of stromal cell-derived factor 1α (SDF-1α/CXCR4) that is brought on by hypoxia, inflammation and tumor progression [70]. In a hepatic cell carcinoma mice model, AMD3100’s inhibition of CXCR4 alleviated local immunosuppression and increased the efficiency of anti-programmed cell death-1 (PD-1) treatment [56,71]. Furthermore, because CXCR4 is a unique vascular marker of angiogenesis and is expressed by TAMs via the ERK pathway, CXCR4 inhibition may work in conjunction with anti-angiogenic medications [72,73]. Targeting CXCR4 in various cancers involves the use of several antagonists, including BL-8040, BKT-140, and ulocuplumab (BMS-936564) [74–76]. In patients with metastatic pancreatic cancer, BL-8040’s safety and effectiveness are being studied in one ongoing phase 2 clinical research (NCT04543071). Another phase 2 clinical trial, NCT02826486, demonstrated the effectiveness and safety of BL-8040 in patients with PDAC [77]. A single-arm trial (NCT01010880) found that administering BKT-140 to patients with multiple myeloma improved their prognosis, while maintaining safety. Additionally, ulocuplumab, a human monoclonal anti-CXCR4 antibody, prevents CXCR4 from attaching to CXCL12. Ulocuplumab treatment is safe, has manageable side effects, and is associated with a high response rate in patients with myeloma [78]. It has also been studied in hematological cancers (NCT01120457 and NCT01359657). These results suggested that CXCR4 is a promising target for cancer treatment.
In addition to the aforementioned signals, there is a negative correlation between CSF1R+ macrophages within tumors and patient survival, across a wide range of tumor types. Blocking CSF1R signaling is an appealing method to remove TAMs [12,79]. To counteract the effects of colony-stimulating factor 1 (CSF1), a primary differentiation factor of TAMs, the monoclonal humanized antibody RG7155 was engineered [80]. CD163+ macrophages can be eliminated by RG7155 therapy and this effect is reproduced in differentiated macrophages in vitro. However, there are substantial interpatient variations in the extent to which TAMs are depleted, spanning from 40% to 90% [81]. Macrophage polarization by IL-4 results in an immunoregulatory CD206+ macrophage phenotype, whereas macrophage polarization by IL-10 results in an immunosuppressive CD163+ macrophage phenotype with tissue-remodeling abilities [82,83]. CD206+ macrophages developed in vitro require less CSF1 for survival than CD163+ macrophages. Furthermore, CD206+ macrophages are resistant to CSF-1R signaling suppression and are less dependent on STAT6 signaling, whereas STAT1 activation is critical for their survival. It is possible that the IL-4-regulated component maintains STAT1 signaling, which is necessary for CD206+ macrophage survival because RG7155 effectively blocks active CSF-1R signaling via STAT1 [81]. Clinical objective responses were observed in individuals with diffuse-type giant cell tumor (Dt-GCT) after treatment with RG7155, which was associated with dramatic decreases in CSF-1R+CD163+ macrophages [80]. Patients with Dt-GCT had been further studied to determine the efficacy, safety, and tolerability of RG7155. Twenty-four (86%) of the twenty-eight patients reached an objective improvement, with two (7%) showing a complete improvement [84]. RG7155, a CSF1-targeting drug, is a promising candidate for the treatment of patients with clinical tumors.
Most patients with Dt-GCT treated with another specific inhibitor of CSF1R, PLX3397, experienced durable reductions [85]. It can also cross the blood-brain barrier in mouse models. Although PLX3397 is safe and easily passes the barrier, it has been proven ineffective against Glioblastoma (GBM). Further combined studies are required [86]. However, it is crucial to understand that this medication has a boxed warning regarding its potential for significant and even fatal liver damage [87]. Several PLX3397 trials, either alone or in combination with other treatments, have been terminated or abandoned due to various reasons, including subpar business decisions or insufficient clinical outcomes (NCT01826448, NCT01499043, NCT01349036, NCT02452424).
It is possible to directly counteract the detrimental effects of TAMs on the immune system’s response against tumors by eliminating TAMs from the body. M2pep specifically binds to murine M2-like TAMs [88]. Although the binding affinity between M2pep and M2 macrophages is limited, M2pep, combined with the proapoptotic peptide KLA (M2pepKLA), was used to decrease the TAM population; however, frequent administration and high doses are required [89]. Intravenous injection of M2pep specifically targets TAMs in mice with colon or breast carcinoma. This strategy shows promise for selective targeting of TAMs [90].
Trabectedin kills cancer cells directly by obstructing several transcription factors, such as DNA repair pathways and DNA-binding proteins. Moreover, myelomonocytes are the only cell type in which trabectedin triggers caspase-8-dependent apoptosis via expression of Fas and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptors [91]. In 2015, the Food and Drug Administration (FDA) authorized trabectedin for the treatment of leiomyosarcoma or liposarcoma [92]. The selective depletion of mononuclear phagocytes has been suggested to be a crucial element in its anticancer effect. In a phase 2 study, patients with advanced breast cancer treated with trabectedin showed better objective response rates and longer progression-free survival (NCT00050427). According to current clinical trials, trabectedin is a promising candidate for the treatment of tumors by targeting macrophages.
Lurbinectedin, a trabectedin analog, demonstrates strong apoptotic potential in macrophages. As a result, it significantly reduces the number of macrophages in the TME and circulation in mouse models [93]. These trials recruited patients with advanced solid tumors to assess their safety and efficacy (NCT05101265, NCT05072106, and NCT04638491). However, because macrophages play an important role in host defense and the regulation of homeostasis, neither trabectedin nor lurbinectedin can completely prevent the adverse effects associated with unselective macrophage consumption.
Reverse TAMs into antitumoral cells
Although tumor suppression can be achieved by reducing the number of TAMs in the TME, reversing tumor-promoting TAMs into antitumor immune cells seems to be a smarter strategy, as shown in experimental investigations [6,94]. TAMs can be phagocytosed and can inhibit tumor growth by triggering antitumor immune responses. This provides evidence that the therapeutic manipulation of macrophage plasticity is a viable option for reviving the anticancer effects of TAMs. Thus, reprogramming TAMs into an “immune-beneficial” phenotype offers an opportunity to change the immunosuppressive TME, providing a more potent strategy for optimizing treatment options [95].
Signal regulatory proteins (SIRP) is linked to poor patient survival and can recognize CD47, which has been found to be overexpressed in cancer cells, as a “don’t eat me” signal. Following therapy with CD47 antibodies, cancer cell phagocytosis by macrophages was restored [96]. Notably, cholangiocarcinoma exhibits excessive CD47 expression. Disruption of the CD47-SIRP connection prevents cholangiocarcinoma progression by encouraging the phagocytosis of TAMs [97]. Hu5F9-G4, a CD47-targeting antibody, has been tested in several phase 1/2 clinical trials, where it demonstrated its efficiency and safety in both hematological and solid malignancies (NCT02216409, NCT02678338, NCT02953782).
Apart from the CD47-SIRPα signal, phosphoinositide 3 kinase γ (PI3Kγ) is involved in different forms of macrophage. During the process of inflammation and tumor formation, PI3Kγ, mainly expressed in myeloid cells, regulates a key switch between immunological activation and repression. To enhance immune suppression during tumor progression, PI3Kγ signaling suppressed nuclear factor kappa B (NF-κB) activation via Akt and mTor while boosting C/EBP activity [98]. Without directly targeting cancer cells, a phase 1 clinical trial (NCT02637531) preliminary proved that inhibiting PI3Kγ by a selective inhibitor can enhance T cell cytotoxicity [99]. Therefore, PI3Kγ also plays an important role in macrophage polarization.
Generally, CD40 is expressed on antigen-presenting cells. IL-12, which is essential for T-cell priming, is released as a result of the binding of CD40 and CD40L, which also boosts major histocompatibility complex (MHC) expression [100]. CD40 agonist treatment of macrophages resulted in considerable TAM reprogramming, which creates an inflammatory environment that prompts effective T cell responses. In a phase 1 clinical trial, the CD40 agonist SEA-CD40 showed anticancer efficacy, causing a complete response in a patient with follicular lymphoma [101]. In addition, phase 1/2 trials (NCT02482168 and NCT04337931) have evaluated the safety and tolerability of the CD40 agonistic APX005M in solid tumors.
Lysosomal acidity is a characteristic of macrophages, and M2-like macrophages have more acidic lysosomes than M1 macrophages [102]. Using chloroquine, a proven anti-malarial drug, in combination with alkalinity, can increase the pH value within lysosome in M2-like macrophage and switch TAMs toward M1 phenotype through lysosomal calcium induced p38 and NF-κB activation [102]. Moreover, in preclinical tumor models, chloroquine mediates macrophage-dependent antitumor T cell immunity. These results suggest that lysosomal acidity could be a target for reprogramming the phenotype of TAMs.
Combination strategies based on TAMs
Broadly speaking, targeting TAMs is also a form of immunotherapy because macrophages are an important component of innate immune cells. Various therapeutic modalities have been reported in combination with targeted TAMs, including a combination of immune checkpoint inhibitors, chemotherapy, radiotherapy, and targeted therapy (Fig. 4) (Table 2).
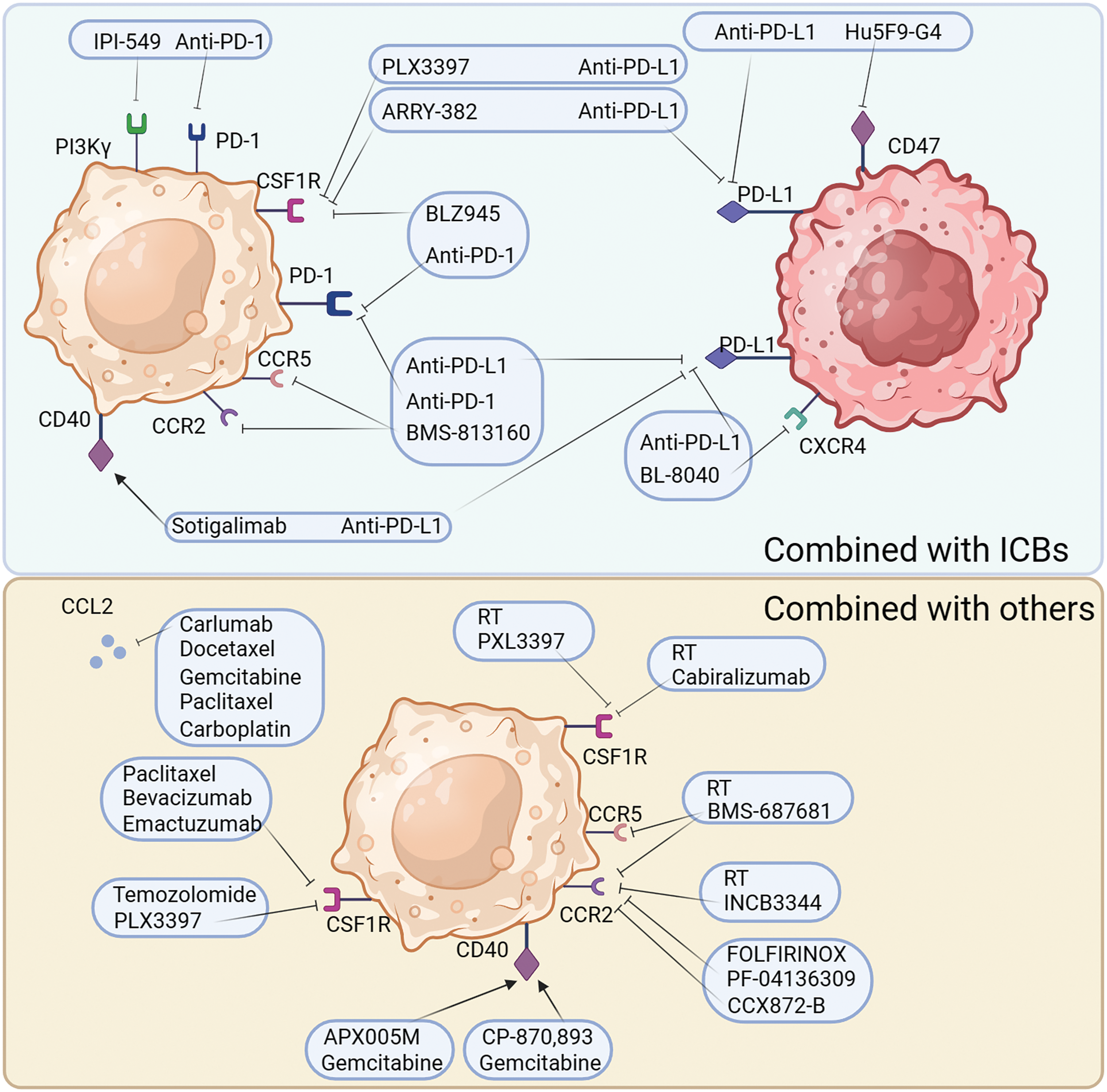
Figure 4: Combination tumor therapy strategies for targeted TAMs. The treatment of targeted TAMs is mainly synergistic immunotherapy or other common clinical treatment. Common modalities of combination immunotherapy are: PD-1 in combination with PI3Kγ antagonists, PD-L1 in combination with CD40/CD47/CXCR4 inhibitors, PD-L1/PD-1 in combination with CSF1R receptor inhibitors, and PD-L1/PD-1 in combination with CCR2/CCR5 receptor inhibitors. Other common combinations include, radiotherapy combined with CSF1R antagonists, radiotherapy combined with CCR2/5 antagonists, chemotherapeutic agents combined with CCL2 antagonists and CSF1R antagonists, and chemotherapy combined with CD40 agonists.
Targeting TAMs combined with immune checkpoint therapy
Currently, high levels of TAM are found in the TME of immunotherapy-resistant patients. During cancer progression, massive and rapid shifts occur in macrophage subpopulations, which are associated with the effectiveness of immunotherapy. These findings imply that, to improve immunotherapies, an improved comprehension of diverse TAMs and their functions in immunotherapy is essential [103,104].
PD-1, PD-1 ligand 1 (PD-L1), and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) antagonists are ineffective as single treatments in halting the progression of PDAC. However, the combination of immune checkpoint blockers (ICBs) with CSF1R inhibition strongly evokes tumor regression, even in advanced malignant tumors. In pancreatic tumors, inhibition of CSF1/CSF1R signaling reduces the number of CD206+ TAMs and reprograms macrophages to aid in antitumor immunity [105].
Osteopontin (OPN), which is produced by cancer cells, activates the CSF1-CSF1R pathway in macrophages, promoting PD-L1 expression in HCC and facilitating the chemotactic migration of macrophages. OPNhigh tumor-bearing mice live significantly longer when treated with a combination of anti-PD-L1 and the CSF1R inhibitor PLX3397 [106]. Additionally, treatment with the CSF1R inhibitor PLX3397 increases CD8+ T cell infiltration in lung squamous cell carcinoma mouse models, which improves the efficiency of PD-1 monoclonal antibodies and slows tumor growth [107]. Although there was little therapeutic effect, pembrolizumab combined with ARRY-382 was well tolerated in a phase 1b/2 study (NCT02880371). Another CSF1R inhibitor, BLZ945, in combination with PD-1 monoclonal antibodies has been shown to increase the efficiency of immunotherapy. In several advanced solid tumors, a CSF1R inhibitor combined with a PD-1/PD-L1 antagonist showed improved efficacy and fewer side effects in several completed clinical trials (NCT02777710, NCT02323191, NCT03158272, and NCT02526017). However, some combination therapies were terminated because of poor results (NCT02452424, NCT02880371, NCT02829723, and NCT03599362). The use of CSF1 inhibitors can improve the TME; however, the use of PD-1/PD-L1 inhibitors in combination with PD-1 inhibitors in clinical practice has shown no effect, indicating that there are more mechanisms that need to be explored.
Considering the limited efficiency of immunotherapies and the crucial immunosuppressive role of TAMs in the TME, a CCR2/CCR5 antagonist, in combination with anti-PD-1/PD-L1, may be a promising treatment. BMS-813160, a dual CCR2/CCR5 antagonist, is currently being tested in a number of clinical trials, in combination with immunotherapies, to treat advanced cancers, such as PDAC, HCC, and NSCLC (NCT03496662, NCT04123379, NCT03767582). From this perspective, CCR2/CCR5 antagonist are expected to improve the efficacy of immunotherapy.
Recent data suggests that the TME is modified by CXCR4. Immunotherapy sensitivity and T cell infiltration are improved by inhibiting CXCR4 and its ligand CXCL12 [108]. The CXCR4 antagonist BL-8040 (motixafortide), in combination with pembrolizumab, was shown to be safe and effective and to have immunobiological effects in a phase 2a clinical study of patients with metastatic PDAC (NCT02826486). However, some trials involving a combination of CXCR4 and immunotherapy involving metastatic pancreatic cancer or acute myeloid leukemia have been terminated or suspended due to poor results (NCT02907099, NCT03154827). Therefore, the efficacy of CXCR4 antagonists in combination with immunotherapeutic drugs needs to be verified in clinical trials.
In tumor cell evasion of antibody-dependent clearance by phagocytes, the upregulation of CD47 appears to play a significant and perhaps ubiquitous role. Blocking CD47 alone does not improve antitumor efficiency in a mouse model of melanoma. However, the combination of CD47 antibody and ICBs strongly enhanced antitumor effects [109]. Similarly, in MC38-bearing mice, tumor targeting and treatment efficacy were dramatically improved using bispecific anti-PD-L1-SIRP, by inhibiting CD47 and PD-L1, compared with that of monotherapy [110]. Magrolimab (Hu5F9-G4), a CD47 blocker, and anti-PDL1 together resulted in a stable disease in 56% of the patients in an ovarian cancer phase 1b trial (NCT03558139). A study on atezolizumab in combination with Hu5F9-G4 in patients with acute myeloid leukemia was terminated because of sponsor-related issues (NCT03922477). In another phase 2 clinical trial, the study of Hodgkin’s lymphoma treated with magrolimab and pembrolizumab is still ongoing (NCT04788043). These results suggest that CD47 blockers can effectively improve the efficacy of immunotherapy.
PI3Kγ, which is overexpressed in myeloid cells, can be selectively targeted pharmacologically to restore sensitivity to ICBs. The aim of reshaping the TME and promoting cytotoxic T cell-mediated tumor regression may be achieved without directly targeting cancer cells. This goal is pursued through the targeting of PI3Kγ using a selective inhibitor, currently under examination in a phase 1 clinical study (NCT02637531). To overcome resistance to ICBs in patients with high levels of suppressive myeloid cell infiltration in tumors, new combination methods have been being developed using a selective small molecule PI3Kγ inhibitor, such as IPI-549 [111].
Preclinical results suggest that the combination of CD40 agonists with anti-PD1 might weaken anti-PD-1 resistance. In a phase 2 clinical trial, a fraction of patients with melanoma who received nivolumab plus APX005M (sotigalimab), a CD40 agonist, experienced persistent and prolonged responses owing to the favorable safety profile of the combination [112]. In a phase 1 trial, individuals with metastatic melanoma were treated with tremelimumab in addition to the monoclonal antibody CP-870,893, a CD40 agonist (NCT01103635). In patients with melanoma, the combination of APX005M and nivolumab demonstrated therapeutic efficacy, with generally favorable tolerability and safety (NCT03123783). Phase I/2 trials are being conducted to test this combination in patients with melanoma, NSCLC, or renal cancer (NCT02706353 and NCT03502330). Therefore, CD40 is a promising target for improving immunotherapy efficacy.
Some of the aforementioned trials have suggested that immune checkpoint inhibitors in combination with TAM-related inhibitors are effective in improving the survival prognosis of patients, both in terms of reducing the side effects of the drugs and increasing the efficiency of the therapy. However, efforts to improve efficiency and overcome drug resistance in combination therapies are needed in the future.
Targeting TAMs combined with others
Apart from the combination of targeting TAMs with immunotherapy, TAM-targeted therapy is also used in traditional therapies, such as radiation therapies and chemotherapies.
Reducing macrophage infiltration into the TME is a key strategy to overcome radioresistance and maximize the therapeutic benefits of radio therapy (RT). Radiation results in elevated serum CSF1 levels in patients with prostate cancer. CSF1/CSF1R signaling is significant in the recruitment of TAMs, which can reduce the efficiency of radiotherapy. CSF1 inhibitors in combination with radiation in clinical trials may result in better outcomes. In another clinical trial, CCL2 levels were found to be reduced in patients treated with GW2580 or PLX3397 [113]. This confirms the results of a recent study that showed that blocking CSF1 resulted in a dramatic decrease in CCL2 [114]. The number of M2 macrophages recruited to bladder tumors and metastases after RT can be reduced by suppressing the CCL2-CCR2 interaction with INCB3344 [115]. It has been established that decreasing macrophage infiltration can improve the efficacy of RT when PXL3397, a CSF1 inhibitor, is administered [116]. When paired with RT and PD-1 therapy in a PDAC mouse model, the dual inhibitor BMS-687681, which targets both CCR2 and CCR5, significantly improved the survival rate [117]. Therefore, drugs targeting TAMs may play an important role in improving the efficacy of RT and reducing its side effects.
Radiotherapy dose also has a significant impact on the efficacy of TAM-targeted drugs. High-dose radiotherapy can induce angiogenesis by stimulating M2-polarized TAMs and creating an environment conducive to tumor growth. Medium-dose radiotherapy can effectively stimulate phagocytosis by macrophages, whereas site-specific low-dose radiotherapy is more likely to improve the efficacy of combination therapy [118].
During chemotherapy, drug cytotoxicity causes tumor cells to create monocyte recruitment factors that increase macrophage infiltration into tumor lesions. The TME can be reprogrammed to enhance antitumor immunity and respond to cytotoxic therapy by blocking macrophage recruitment in combination with chemotherapy [100]. Recruitment of CCR2+ monocytes may be affected by the myelosuppressive effects of conventional chemotherapy. Following chemotherapy in a mouse model of breast cancer, intravital imaging revealed an increase in the number of TAMs. This increase is mediated by an increase in stromal CCL2 production [119]. Chemotherapy alone is insufficient to prevent the recruitment of inflammatory monocytes to the bone marrow. Systemic administration of the CCL2 antagonist, carlumab, in mice significantly attenuated prostate tumor growth and decreased CD68+ macrophage infiltration and the density of the tumor microvasculature in preclinical studies [68]. The human IgG1 anti-CCL2 monoclonal antibody carlumab has been demonstrated anticancer efficacy in both animal models and human beings together with the application of classical chemotherapy [120]. When used in combination with docetaxel, carlumab also induced prostate cancer regression and a significant decrease in tumor burden [67]. In conclusion, targeting CCL2 can reduce the recruitment, thereby effectively improving the efficacy of chemotherapy.
FOLFIRINOX is the traditional treatment for pancreatic cancer. PF-04136309, a CCR2 inhibitor, blocks CCR2-positive monocytes from leaving the bone marrow and migrating to peripheral circulation, resulting in a better control of the primary tumor [121]. Inhibiting CCR2 reduced the number of TAMs and Treg cells in pancreatic tumors and increased the number of CD8+ T cells. BMS-813160 is a CCR2/CCR5 dual antagonist. In a phase 1/2 research, patients with colorectal and pancreatic cancer responded well to the combination therapy of BMS-813160 with chemotherapy (NCT03184870). However, when patients with PDAC were treated with a CCR2 inhibitor, the number of CXCR2+ neutrophils increased as a compensatory response. The prognosis of patients with PDAC worsens with an increase in CXCR2+ neutrophils. Therefore, the individualized use of CCR2 inhibitors in different scenarios to effectively avoid side effects is extremely important in combination with chemotherapy.
Tumor regression is seen in certain patients after combining a CD40 agonist with gemcitabine treatment, which is promising because CD40 activation by CP-870,893 can reverse the immunosuppressive TME and generate anticancer T cell responses [122]. Combination therapy with an anti-CD40 monoclonal antibody and CSF-1R inhibitor significantly reduced tumor growth in a mouse model of melanoma. Various TAMs subpopulations that secrete MMP9 or CCL17/22, indicative of an M2-like state, are present in primary tumors. The frequency of these subsets was lowered by combination therapy, and a new inflammatory TAM population was induced, which secreted TNF-α and IL-6 [123]. The combined stimulation of CSF-1R and CD40 on macrophages is sufficient to generate a proinflammatory TME that re-energizes an efficient T-cell response in transplanted tumors that are either sensitive or insensitive to immune checkpoint blockade [124]. A phase 2 clinical trial is ongoing to assess the effectiveness and safety of combining doxorubicin with APX005M (another CD40 agonist) for the treatment of sarcoma (NCT03719430). As CD40 can convert TAMs to do antitumor activity, CD40-targeting drugs combined with chemotherapy can effectively improve the efficacy of chemotherapy.
Both CSF1 and IL-34 are produced by tumors in response to conventional chemotherapy, leading to an increase in the number of immunosuppressive TAMs in a CSF1R-dependent manner [125]. A phase 1b/2 study of the application of the CSF1R inhibitor PLX3397 with Temozolomide and RT in patients with glioblastoma has been completed (NCT01790503). Another randomized phase 2 trial has finished examining the negative effects of emactuzumab, an anti-CSF-1R monoclonal antibody, in combination with paclitaxel and bevacizumab. Emactuzumab can also reduce the number of TAMs in individuals with solid tumors, whether alone or in combination with paclitaxel (NCT01494688). In summary, TAM-targeting drugs, with either conventional therapy or immunotherapy, can play a role as a therapeutic adjuvant by modulating the TME.
As the most abundant immune cell population in the TME, TAMs are attracting increasing attention from researchers, especially in the fields of tumor immunity and immunotherapy. With the advancement of research techniques, our understanding of TAMs has deepened. Unlike traditional M1/M2 typing, TAMs allow for greater population delineation by single-cell RNA sequencing in the TME, leading us to recognize that there is extreme heterogeneity in TAMs. Embryonic stem cell derived TRMs are important components of the heterogeneous ontogeny of TAMs. In addition, the tumor microenvironmental clues induced transcriptional and epigenetic differences in different TAM subpopulations, which determined the functional heterogeneity of pro-tumoral and antitumoral TAMs. Based on these findings, drugs against different TAM targets have been developed and tested in clinical trials, including single agents and combinations of immune checkpoint inhibitors. In the future, the heterogeneity of TAMs in different tumor types will be further revealed, and precision immunotherapy targeting TAMs will be developed.
Acknowledgement: The authors acknowledge the use of Biorender that is used to create schematic Figs. 1–4.
Funding Statement: This work was supported by the National Natural Science Foundation of China (82003018).
Author Contributions: The authors confirm contribution to the paper as follows: study conception and manuscript revision: Degao Chen, Bo Zhu; draft manuscript preparation: Hao Lv. All authors reviewed the manuscript and approved the final version of the manuscript.
Availability of Data and Materials: Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell. 2019;35(4):588–602.E10. doi:10.1016/j.ccell.2019.02.009. [Google Scholar] [PubMed] [CrossRef]
2. Chevrier S, Levine JH, Zanotelli VRT, Silina K, Schulz D, Bacac M, et al. An immune atlas of clear cell renal cell carcinoma. Cell. 2017;169(4):736–49.E18. doi:10.1016/j.cell.2017.04.016. [Google Scholar] [PubMed] [CrossRef]
3. Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21(8):938–45. doi:10.1038/nm.3909. [Google Scholar] [PubMed] [CrossRef]
4. Chen D, Zhang X, Li Z, Zhu B. Metabolic regulatory crosstalk between tumor microenvironment and tumor-associated macrophages. Theranostics. 2021;11(3):1016–30. doi:10.7150/thno.51777. [Google Scholar] [PubMed] [CrossRef]
5. Gunassekaran GR, Poongkavithai Vadevoo SM, Baek MC, Lee B. M1 macrophage exosomes engineered to foster M1 polarization and target the IL-4 receptor inhibit tumor growth by reprogramming tumor-associated macrophages into M1-like macrophages. Biomater. 2021;278:121137. doi:10.1016/j.biomaterials.2021.121137. [Google Scholar] [PubMed] [CrossRef]
6. Hao X, Zheng Z, Liu H, Zhang Y, Kang J, Kong X, et al. Inhibition of APOC1 promotes the transformation of M2 into M1 macrophages via the ferroptosis pathway and enhances anti-PD1 immunotherapy in hepatocellular carcinoma based on single-cell RNA sequencing. Redox Biol. 2022;56:102463. doi:10.1016/j.redox.2022.102463. [Google Scholar] [PubMed] [CrossRef]
7. Mulder K, Patel AA, Kong WT, Piot C, Halitzki E, Dunsmore G, et al. Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity. 2021;54(8):1883–900.E5. doi:10.1016/j.immuni.2021.07.007. [Google Scholar] [PubMed] [CrossRef]
8. Iwamoto H, Izumi K, Mizokami A. Is the C-C motif ligand 2-C-C chemokine receptor 2 axis a promising target for cancer therapy and diagnosis? Int J Mol Sci. 2020;21(23):9328. doi:10.3390/ijms21239328. [Google Scholar] [PubMed] [CrossRef]
9. Jin L, Guo Y, Mao W, Wang J, Jin L, Liu X, et al. Total glucosides of paeony inhibit breast cancer growth by inhibiting TAMs infiltration through NF-κB/CCL2 signaling. Phytomedicine. 2022;104(1):154307. doi:10.1016/j.phymed.2022.154307. [Google Scholar] [PubMed] [CrossRef]
10. Wang Y, Wang Z, Jia F, Xu Q, Shu Z, Deng J, et al. CXCR4-guided liposomes regulating hypoxic and immunosuppressive microenvironment for sorafenib-resistant tumor treatment. Bioact Mater. 2022;17:147–61. doi:10.1016/j.bioactmat.2022.01.003. [Google Scholar] [PubMed] [CrossRef]
11. Kawaguchi Y, Ohshio Y, Watanabe A, Shiratori T, Okamoto K, Ueda K, et al. Depletion of tumor-associated macrophages inhibits lung cancer growth and enhances the antitumor effect of cisplatin. Cancer Sci. 2023;114(3):750–63. doi:10.1111/cas.15671. [Google Scholar] [PubMed] [CrossRef]
12. Li Z, Ding Y, Liu J, Wang J, Mo F, Wang Y, et al. Depletion of tumor associated macrophages enhances local and systemic platelet-mediated anti-PD-1 delivery for post-surgery tumor recurrence treatment. Nat Commun. 2022;13(1):1845. doi:10.1038/s41467-022-29388-0. [Google Scholar] [PubMed] [CrossRef]
13. Lu X, Meng T. Depletion of tumor-associated macrophages enhances the anti-tumor effect of docetaxel in a murine epithelial ovarian cancer. Immunobiol. 2019;224(3):355–61. doi:10.1016/j.imbio.2019.03.002. [Google Scholar] [PubMed] [CrossRef]
14. Khalaji A, Yancheshmeh FB, Farham F, Khorram A, Sheshbolouki S, Zokaei M, et al. Don’t eat me/eat me signals as a novel strategy in cancer immunotherapy. Heliyon. 2023;9(10):e20507. doi:10.1016/j.heliyon.2023.e20507. [Google Scholar] [PubMed] [CrossRef]
15. Kuo TC, Chen A, Harrabi O, Sockolosky JT, Zhang A, Sangalang E, et al. Targeting the myeloid checkpoint receptor SIRPalpha potentiates innate and adaptive immune responses to promote anti-tumor activity. J Hematol Oncol. 2020;13(1):160. doi:10.1186/s13045-020-00989-w. [Google Scholar] [PubMed] [CrossRef]
16. Schurch CM, Roelli MA, Forster S, Wasmer MH, Bruhl F, Maire RS, et al. Targeting CD47 in anaplastic thyroid carcinoma enhances tumor phagocytosis by macrophages and is a promising therapeutic strategy. Thyroid. 2019;29(7):979–92. doi:10.1089/thy.2018.0555. [Google Scholar] [PubMed] [CrossRef]
17. Park DJ, Sung PS, Lee GW, Cho S, Kim SM, Kang BY, et al. Preferential expression of programmed death ligand 1 protein in tumor-associated macrophages and its potential role in immunotherapy for hepatocellular carcinoma. Int J Mol Sci. 2021;22(9):4710. doi:10.3390/ijms22094710. [Google Scholar] [PubMed] [CrossRef]
18. Wu Q, Zhou W, Yin S, Zhou Y, Chen T, Qian J, et al. Blocking triggering receptor expressed on myeloid cells-1-positive tumor-associated macrophages induced by hypoxia reverses immunosuppression and anti-programmed cell death ligand 1 resistance in liver cancer. Hepatol. 2019;70(1):198–214. doi:10.1002/hep.30593. [Google Scholar] [CrossRef]
19. Zhang XM, Chen DG, Li SC, Zhu B, Li ZJ. Embryonic origin and subclonal evolution of tumor-associated macrophages imply preventive care for cancer. Cells. 2021;10(4):903. doi:10.3390/cells10040903. [Google Scholar] [CrossRef]
20. Lazarov T, Juarez-Carreno S, Cox N, Geissmann F. Physiology and diseases of tissue-resident macrophages. Nature. 2023;618(7966):698–707. doi:10.1038/s41586-023-06002-x. [Google Scholar] [PubMed] [CrossRef]
21. Evren E, Ringqvist E, Tripathi KP, Sleiers N, Rives IC, Alisjahbana A, et al. Distinct developmental pathways from blood monocytes generate human lung macrophage diversity. Immunity. 2021;54(2):259–75 e257. doi:10.1016/j.immuni.2020.12.003. [Google Scholar] [PubMed] [CrossRef]
22. Liu Y, Cao X. The origin and function of tumor-associated macrophages. Cell Mol Immunol. 2015;12(1):1–4. doi:10.1038/cmi.2014.83. [Google Scholar] [PubMed] [CrossRef]
23. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344(6186):921–5. doi:10.1126/science.1252510. [Google Scholar] [PubMed] [CrossRef]
24. Zhou C, Weng J, Liu C, Liu S, Hu Z, Xie X, et al. Disruption of SLFN11 deficiency-induced CCL2 signaling and macrophage M2 polarization potentiates anti-PD-1 therapy efficacy in hepatocellular carcinoma. Gastroenterol. 2023;164(7):1261–78. doi:10.1053/j.gastro.2023.02.005. [Google Scholar] [PubMed] [CrossRef]
25. Xu L, Chen Y, Nagashimada M, Ni Y, Zhuge F, Chen G, et al. CC chemokine ligand 3 deficiency ameliorates diet-induced steatohepatitis by regulating liver macrophage recruitment and M1/M2 status in mice. Metabolis. 2021;125:154914. doi:10.1016/j.metabol.2021.154914. [Google Scholar] [PubMed] [CrossRef]
26. Nie Y, Huang H, Guo M, Chen J, Wu W, Li W, et al. Breast phyllodes tumors recruit and repolarize tumor-associated macrophages via secreting CCL5 to promote malignant progression, which can be inhibited by CCR5 inhibition therapy. Clin Cancer Res. 2019;25(13):3873–86. doi:10.1158/1078-0432.CCR-18-3421. [Google Scholar] [PubMed] [CrossRef]
27. Miller JE, Ahn SH, Marks RM, Monsanto SP, Fazleabas AT, Koti M, et al. IL-17A modulates peritoneal macrophage recruitment and M2 polarization in endometriosis. Front Immunol. 2020;11:108. doi:10.3389/fimmu.2020.00108. [Google Scholar] [PubMed] [CrossRef]
28. Yang X, Walton W, Cook DN, Hua X, Tilley S, Haskell CA, et al. The chemokine, CCL3, and its receptor, CCR1, mediate thoracic radiation-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2011;45(1):127–35. doi:10.1165/rcmb.2010-0265OC. [Google Scholar] [PubMed] [CrossRef]
29. Vidyarthi A, Agnihotri T, Khan N, Singh S, Tewari MK, Radotra BD, et al. Predominance of M2 macrophages in gliomas leads to the suppression of local and systemic immunity. Cancer Immunol Immunother. 2019;68(12):1995–2004. doi:10.1007/s00262-019-02423-8. [Google Scholar] [PubMed] [CrossRef]
30. Zeng Z, Lan T, Wei Y, Wei X. CCL5/CCR5 axis in human diseases and related treatments. Genes Dis. 2022;9(1):12–27. doi:10.1016/j.gendis.2021.08.004. [Google Scholar] [PubMed] [CrossRef]
31. Fuentelsaz-Romero S, Cuervo A, Estrada-Capetillo L, Celis R, Garcia-Campos R, Ramirez J, et al. GM-CSF expression and macrophage polarization in joints of undifferentiated arthritis patients evolving to rheumatoid arthritis or psoriatic arthritis. Front Immunol. 2020;11:613975. doi:10.3389/fimmu.2020.613975. [Google Scholar] [PubMed] [CrossRef]
32. Li Z, Xiao J, Xu X, Li W, Zhong R, Qi L, et al. M-CSF, IL-6, and TGF-β promote generation of a new subset of tissue repair macrophage for traumatic brain injury recovery. Sci Adv. 2021;7(11). doi:10.1126/sciadv.abb6260. [Google Scholar] [PubMed] [CrossRef]
33. Weng YS, Tseng HY, Chen YA, Shen PC, Al Haq AT, Chen LM, et al. MCT-1/miR-34a/IL-6/IL-6R signaling axis promotes EMT progression, cancer stemness and M2 macrophage polarization in triple-negative breast cancer. Mol Cancer. 2019;18(1):42. doi:10.1186/s12943-019-0988-0. [Google Scholar] [PubMed] [CrossRef]
34. Ushach I, Zlotnik A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. J Leukoc Biol. 2016;100(3):481–9. doi:10.1189/jlb.3RU0316-144R. [Google Scholar] [PubMed] [CrossRef]
35. Amorim A, De Feo D, Friebel E, Ingelfinger F, Anderfuhren CD, Krishnarajah S, et al. IFNγ and GM-CSF control complementary differentiation programs in the monocyte-to-phagocyte transition during neuroinflammation. Nat Immunol. 2022;23(2):217–28. doi:10.1038/s41590-021-01117-7. [Google Scholar] [PubMed] [CrossRef]
36. Cortez-Retamozo V, Etzrodt M, Newton A, Ryan R, Pucci F, Sio SW, et al. Angiotensin II drives the production of tumor-promoting macrophages. Immunity. 2013;38(2):296–308. doi:10.1016/j.immuni.2012.10.015. [Google Scholar] [PubMed] [CrossRef]
37. Atkins MH, Scarfo R, McGrath KE, Yang D, Palis J, Ditadi A, et al. Modeling human yolk sac hematopoiesis with pluripotent stem cells. J Exp Med. 2022;219(3):e20211924. doi:10.1084/jem.20211924. [Google Scholar] [PubMed] [CrossRef]
38. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518(7540):547–51. doi:10.1016/j.exphem.2015.06.130. [Google Scholar] [CrossRef]
39. Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity. 2017;47(2):323–38.E6. doi:10.1016/j.immuni.2017.07.014. [Google Scholar] [PubMed] [CrossRef]
40. Prieto LI, Sturmlechner I, Graves SI, Zhang C, Goplen NP, Yi ES, et al. Senescent alveolar macrophages promote early-stage lung tumorigenesis. Cancer Cell. 2023;41(7):1261–75.E6. doi:10.1016/j.ccell.2023.05.006. [Google Scholar] [PubMed] [CrossRef]
41. Casanova-Acebes M, Dalla E, Leader AM, LeBerichel J, Nikolic J, Morales BM, et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature. 2021;595(7868):578–84. doi:10.1038/s41586-021-03651-8. [Google Scholar] [PubMed] [CrossRef]
42. Nalio Ramos R, Missolo-Koussou Y, Gerber-Ferder Y, Bromley CP, Bugatti M, Nunez NG, et al. Tissue-resident FOLR2+ macrophages associate with CD8+ T cell infiltration in human breast cancer. Cell. 2022;185(7):1189–207.E25. doi:10.1016/j.cell.2022.02.021. [Google Scholar] [PubMed] [CrossRef]
43. Aegerter H, Lambrecht BN, Jakubzick CV. Biology of lung macrophages in health and disease. Immunity. 2022;55(9):1564–80. doi:10.1016/j.immuni.2022.08.010. [Google Scholar] [PubMed] [CrossRef]
44. Khan F, Pang L, Dunterman M, Lesniak MS, Heimberger AB, Chen P. Macrophages and microglia in glioblastoma: heterogeneity, plasticity, and therapy. J Clin Invest. 2023;133(1):e163446. doi:10.1172/JCI163446. [Google Scholar] [PubMed] [CrossRef]
45. Yin H, Zhang X, Yang P, Zhang X, Peng Y, Li D, et al. RNA m6A methylation orchestrates cancer growth and metastasis via macrophage reprogramming. Nat Commun. 2021;12(1):1394. doi:10.1038/s41467-021-21514-8. [Google Scholar] [PubMed] [CrossRef]
46. Qiu S, Xie L, Lu C, Gu C, Xia Y, Lv J, et al. Gastric cancer-derived exosomal miR-519a-3p promotes liver metastasis by inducing intrahepatic M2-like macrophage-mediated angiogenesis. J Exp Clin Cancer Res. 2022;41(1):296. doi:10.1186/s13046-022-02499-8. [Google Scholar] [PubMed] [CrossRef]
47. Mortezaee K, Majidpoor J. Roles for macrophage-polarizing interleukins in cancer immunity and immunotherapy. Cell Oncol. 2022;45(3):333–53. doi:10.1007/s13402-022-00667-8. [Google Scholar] [PubMed] [CrossRef]
48. Guillot J, Dominici C, Lucchesi A, Nguyen HTT, Puget A, Hocine M, et al. Sympathetic axonal sprouting induces changes in macrophage populations and protects against pancreatic cancer. Nat Commun. 2022;13(1):1985. doi:10.1038/s41467-022-29659-w. [Google Scholar] [PubMed] [CrossRef]
49. Ali HR, Chlon L, Pharoah PD, Markowetz F, Caldas C. Patterns of immune infiltration in breast cancer and their clinical implications: a gene-expression-based retrospective study. PLoS Med. 2016;13(12):e1002194. doi:10.1371/journal.pmed.1002194. [Google Scholar] [PubMed] [CrossRef]
50. Varol C, Mildner A, Jung S. Macrophages: development and tissue specialization. Annu Rev Immunol. 2015;33(1):643–75. doi:10.1146/annurev-immunol-032414-112220. [Google Scholar] [PubMed] [CrossRef]
51. Pittet MJ, Michielin O, Migliorini D. Clinical relevance of tumour-associated macrophages. Nat Rev Clin Oncol. 2022;19(6):402–21. doi:10.1038/s41571-022-00620-6. [Google Scholar] [PubMed] [CrossRef]
52. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14(7):399–416. doi:10.1038/nrclinonc.2016.217. [Google Scholar] [PubMed] [CrossRef]
53. Franklin RA, Li MO. Ontogeny of tumor-associated macrophages and its implication in cancer regulation. Trends Cancer. 2016;2(1):20–34. doi:10.1016/j.trecan.2015.11.004. [Google Scholar] [PubMed] [CrossRef]
54. Liu Y, Wang J, Zhang J, Marbach S, Xu W, Zhu L. Targeting tumor-associated macrophages by MMP2-sensitive apoptotic body-mimicking nanoparticles. ACS Appl Mater Interfaces. 2020;12(47):52402–14. doi:10.1021/acsami.0c15983. [Google Scholar] [PubMed] [CrossRef]
55. Zhang N, Liu C, Jin L, Zhang R, Wang T, Wang Q, et al. Ketogenic diet elicits antitumor properties through inducing oxidative stress, inhibiting MMP-9 expression, and rebalancing M1/M2 tumor-associated macrophage phenotype in a mouse model of colon cancer. J Agric Food Chem. 2020;68(40):11182–96. doi:10.1021/acs.jafc.0c04041. [Google Scholar] [PubMed] [CrossRef]
56. Biasci D, Smoragiewicz M, Connell CM, Wang Z, Gao Y, Thaventhiran JED, et al. CXCR4 inhibition in human pancreatic and colorectal cancers induces an integrated immune response. Proc Natl Acad Sci USA. 2020;117(46):28960–70. doi:10.1073/pnas.2013644117. [Google Scholar] [PubMed] [CrossRef]
57. Salmaninejad A, Valilou SF, Soltani A, Ahmadi S, Abarghan YJ, Rosengren RJ, et al. Tumor-associated macrophages: role in cancer development and therapeutic implications. Cell Oncol. 2019;42(5):591–608. doi:10.1007/s13402-019-00453-z. [Google Scholar] [PubMed] [CrossRef]
58. Winning S, Fandrey J. Oxygen sensing in innate immune cells: how inflammation broadens classical hypoxia-inducible factor regulation in myeloid cells. Antioxid Redox Signal. 2022;37(13–15):956–71. doi:10.1089/ars.2022.0004. [Google Scholar] [PubMed] [CrossRef]
59. Zhang W, Jiang X, Zou Y, Yuan L, Wang X. Pexidartinib synergize PD-1 antibody through inhibiting treg infiltration by reducing TAM-derived CCL22 in lung adenocarcinoma. Front Pharmacol. 2023;14:1092767. doi:10.3389/fphar.2023.1092767. [Google Scholar] [PubMed] [CrossRef]
60. Chen C, Zhang L, Ruan Z. GATA3 encapsulated by tumor-associated macrophage-derived extracellular vesicles promotes immune escape and chemotherapy resistance of ovarian cancer cells by upregulating the CD24/Siglec-10 axis. Mol Pharm. 2023;20(2):971–86. doi:10.1021/acs.molpharmaceut.2c00557. [Google Scholar] [PubMed] [CrossRef]
61. Bill R, Wirapati P, Messemaker M, Roh W, Zitti B, Duval F, et al. CXCL9: SPP1 macrophage polarity identifies a network of cellular programs that control human cancers. Science. 2023;381(6657):515–24. doi:10.1126/science.ade2292. [Google Scholar] [PubMed] [CrossRef]
62. Palmieri EM, Menga A, Martin-Perez R, Quinto A, Riera-Domingo C, De Tullio G, et al. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an M1-like phenotype and inhibits tumor metastasis. Cell Rep. 2017;20(7):1654–66. doi:10.1016/j.celrep.2017.07.054. [Google Scholar] [PubMed] [CrossRef]
63. Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C, et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell. 2011;19(1):31–44. doi:10.1016/j.ccr.2010.11.009. [Google Scholar] [PubMed] [CrossRef]
64. Bugatti M, Bergamini M, Missale F, Monti M, Ardighieri L, Pezzali I, et al. A population of TIM4+FOLR2+ macrophages localized in tertiary lymphoid structures correlates to an active immune infiltrate across several cancer types. Cancer Immunol Res. 2022;10(11):1340–53. doi:10.1158/2326-6066.CIR-22-0271. [Google Scholar] [PubMed] [CrossRef]
65. Yin Y, Dai H, Sun X, Xi Z, Zhang J, Pan Y, et al. HRG inhibits liver cancer lung metastasis by suppressing neutrophil extracellular trap formation. Clin Transl Med. 2023;13(6):e1283. doi:10.1002/ctm2.1283. [Google Scholar] [PubMed] [CrossRef]
66. Yang H, Zhang Q, Xu M, Wang L, Chen X, Feng Y, et al. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol Cancer. 2020;19(1):41. doi:10.1186/s12943-020-01165-x. [Google Scholar] [PubMed] [CrossRef]
67. Loberg RD, Ying C, Craig M, Day LL, Sargent E, Neeley C, et al. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res. 2007;67(19):9417–24. doi:10.1158/0008-5472.CAN-07-1286. [Google Scholar] [PubMed] [CrossRef]
68. Loberg RD, Ying C, Craig M, Yan L, Snyder LA, Pienta KJ. CCL2 as an important mediator of prostate cancer growth in vivo through the regulation of macrophage infiltration. Neoplasia. 2007;9(7):556–62. doi:10.1593/neo.07307. [Google Scholar] [PubMed] [CrossRef]
69. Pienta KJ, Machiels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, et al. Phase 2 study of carlumab (CNTO 888a human monoclonal antibody against CC-chemokine ligand 2 (CCL2in metastatic castration-resistant prostate cancer. Invest New Drugs. 2013;31(3):760–8. doi:10.1007/s10637-012-9869-8. [Google Scholar] [PubMed] [CrossRef]
70. Chen Y, Huang Y, Reiberger T, Duyverman AM, Huang P, Samuel R, et al. Differential effects of sorafenib on liver versus tumor fibrosis mediated by stromal-derived factor 1 alpha/C-X-C receptor type 4 axis and myeloid differentiation antigen-positive myeloid cell infiltration in mice. Hepatology. 2014;59(4):1435–47. doi:10.1002/hep.26790. [Google Scholar] [PubMed] [CrossRef]
71. Chen Y, Ramjiawan RR, Reiberger T, Ng MR, Hato T, Huang Y, et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology. 2015;61(5):1591–602. doi:10.1002/hep.27665. [Google Scholar] [PubMed] [CrossRef]
72. Heidegger I, Fotakis G, Offermann A, Goveia J, Daum S, Salcher S, et al. Comprehensive characterization of the prostate tumor microenvironment identifies CXCR4/CXCL12 crosstalk as a novel antiangiogenic therapeutic target in prostate cancer. Mol Cancer. 2022;21(1):132. doi:10.1186/s12943-022-01597-7. [Google Scholar] [PubMed] [CrossRef]
73. Meng YM, Liang J, Wu C, Xu J, Zeng DN, Yu XJ, et al. Monocytes/Macrophages promote vascular CXCR4 expression via the ERK pathway in hepatocellular carcinoma. Oncoimmunology. 2018;7(3):e1408745. doi:10.1080/2162402X.2017.1408745. [Google Scholar] [PubMed] [CrossRef]
74. Huynh C, Dingemanse J, Meyer Zu Schwabedissen HE, Sidharta PN. Relevance of the CXCR4/CXCR7-CXCL12 axis and its effect in pathophysiological conditions. Pharmacol Res. 2020;161:105092. doi:10.1016/j.phrs.2020.105092. [Google Scholar] [PubMed] [CrossRef]
75. Rioja-Blanco E, Arroyo-Solera I, Alamo P, Casanova I, Gallardo A, Unzueta U, et al. CXCR4-targeted nanotoxins induce GSDME-dependent pyroptosis in head and neck squamous cell carcinoma. J Exp Clin Cancer Res. 2022;41(1):49. doi:10.1186/s13046-022-02267-8. [Google Scholar] [PubMed] [CrossRef]
76. Bockorny B, Semenisty V, Macarulla T, Borazanci E, Wolpin BM, Stemmer SM, et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nat Med. 2020;26(6):878–85. doi:10.1038/s41591-020-0880-x. [Google Scholar] [PubMed] [CrossRef]
77. Bockorny B, Macarulla T, Semenisty V, Borazanci E, Feliu J, Ponz-Sarvise M, et al. Motixafortide and pembrolizumab combined to nanoliposomal irinotecan, fluorouracil, and folinic acid in metastatic pancreatic cancer: the COMBAT/KEYNOTE-202 trial. Clin Cancer Res. 2021;27(18):5020–7. doi:10.1158/1078-0432.CCR-21-0929. [Google Scholar] [PubMed] [CrossRef]
78. Ghobrial IM, Liu CJ, Redd RA, Perez RP, Baz R, Zavidij O, et al. A phase Ib/II trial of the first-in-class anti-CXCR4 antibody ulocuplumab in combination with lenalidomide or bortezomib plus dexamethasone in relapsed multiple myeloma. Clin Cancer Res. 2020;26(2):344–53. doi:10.1158/1078-0432.CCR-19-0647. [Google Scholar] [PubMed] [CrossRef]
79. Rao R, Han R, Ogurek S, Xue C, Wu LM, Zhang L, et al. Glioblastoma genetic drivers dictate the function of tumor-associated macrophages/microglia and responses to CSF1R inhibition. Neuro Oncol. 2022;24(4):584–97. doi:10.1093/neuonc/noab228. [Google Scholar] [PubMed] [CrossRef]
80. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25(6):846–59. doi:10.1016/j.ccr.2014.05.016. [Google Scholar] [PubMed] [CrossRef]
81. Pradel LP, Ooi CH, Romagnoli S, Cannarile MA, Sade H, Ruttinger D, et al. Macrophage susceptibility to emactuzumab (RG7155) treatment. Mol Cancer Ther. 2016;15(12):3077–86. doi:10.1158/1535-7163.MCT-16-0157. [Google Scholar] [PubMed] [CrossRef]
82. Aendekerk JP, Jiemy WF, Raveling-Eelsing E, Bijnens N, Abdul-Hamid MA, Strating IM, et al. CD163 and CD206 expression define distinct macrophage subsets involved in active ANCA-associated glomerulonephritis. J Autoimmun. 2022;133:102914. doi:10.1016/j.jaut.2022.102914. [Google Scholar] [PubMed] [CrossRef]
83. Kvorning SL, Nielsen MC, Andersen NF, Hokland M, Andersen MN, Moller HJ. Circulating extracellular vesicle-associated CD163 and CD206 in multiple myeloma. Eur J Haematol. 2020;104(5):409–19. doi:10.1111/ejh.13371. [Google Scholar] [PubMed] [CrossRef]
84. Cassier PA, Italiano A, Gomez-Roca CA, Le Tourneau C, Toulmonde M, Cannarile MA, et al. CSF1R inhibition with emactuzumab in locally advanced diffuse-type tenosynovial giant cell tumours of the soft tissue: a dose-escalation and dose-expansion phase 1 study. Lancet Oncol. 2015;16(8):949–56. doi:10.1016/S1470-2045(15)00132-1. [Google Scholar] [PubMed] [CrossRef]
85. Tap WD, Wainberg ZA, Anthony SP, Ibrahim PN, Zhang C, Healey JH, et al. Structure-guided blockade of CSF1R kinase in tenosynovial giant-cell tumor. N Engl J Med. 2015;373(5):428–37. doi:10.1056/NEJMoa1411366. [Google Scholar] [PubMed] [CrossRef]
86. Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy foundation early phase clinical trials consortium phase II study. Neuro Oncol. 2016;18(4):557–64. doi:10.1093/neuonc/nov245. [Google Scholar] [PubMed] [CrossRef]
87. Lamb YN. Pexidartinib: first approval. Drugs. 2019;79(16):1805–12. doi:10.1007/s40265-019-01210-0. [Google Scholar] [PubMed] [CrossRef]
88. Sun Y, Cronin MF, Mendonca MCP, Guo J, O’Driscoll CM. M2pep-modified cyclodextrin-siRNA nanoparticles modulate the immunosuppressive tumor microenvironment for prostate cancer therapy. Mol Pharm. 2023;20(11):5921–36. doi:10.1021/acs.molpharmaceut.3c00769. [Google Scholar] [PubMed] [CrossRef]
89. Ngambenjawong C, Cieslewicz M, Schellinger JG, Pun SH. Synthesis and evaluation of multivalent M2pep peptides for targeting alternatively activated M2 macrophages. J Control Release. 2016;224:103–11. doi:10.1016/j.jconrel.2015.12.057. [Google Scholar] [PubMed] [CrossRef]
90. Cieslewicz M, Tang J, Yu JL, Cao H, Zavaljevski M, Motoyama K, et al. Targeted delivery of proapoptotic peptides to tumor-associated macrophages improves survival. Proc Natl Acad Sci USA. 2013;110(40):15919–24. doi:10.1073/pnas.1312197110. [Google Scholar] [PubMed] [CrossRef]
91. Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23(2):249–62. doi:10.1016/j.ccr.2013.01.008. [Google Scholar] [PubMed] [CrossRef]
92. Barone A, Chi DC, Theoret MR, Chen H, He K, Kufrin D, et al. FDA approval summary: trabectedin for unresectable or metastatic liposarcoma or leiomyosarcoma following an anthracycline-containing regimen. Clin Cancer Res. 2017;23(24):7448–53. doi:10.1158/1078-0432.CCR-17-0898. [Google Scholar] [PubMed] [CrossRef]
93. Belgiovine C, Bello E, Liguori M, Craparotta I, Mannarino L, Paracchini L, et al. Lurbinectedin reduces tumour-associated macrophages and the inflammatory tumour microenvironment in preclinical models. Br J Cancer. 2017;117(5):628–38. doi:10.1038/bjc.2017.205. [Google Scholar] [PubMed] [CrossRef]
94. Dai X, Lu L, Deng S, Meng J, Wan C, Huang J, et al. USP7 targeting modulates anti-tumor immune response by reprogramming tumor-associated macrophages in lung cancer. Theranostics. 2020;10(20):9332–47. doi:10.7150/thno.47137. [Google Scholar] [PubMed] [CrossRef]
95. van der Heide D, Weiskirchen R, Bansal R. Therapeutic targeting of hepatic macrophages for the treatment of liver diseases. Front Immunol. 2019;10:2852. doi:10.3389/fimmu.2019.02852. [Google Scholar] [PubMed] [CrossRef]
96. Chen SH, Dominik PK, Stanfield J, Ding S, Yang W, Kurd N, et al. Dual checkpoint blockade of CD47 and PD-L1 using an affinity-tuned bispecific antibody maximizes antitumor immunity. J Immunother Cancer. 2021;9(10). doi:10.1136/jitc-2021-003464. [Google Scholar] [PubMed] [CrossRef]
97. Vaeteewoottacharn K, Kariya R, Pothipan P, Fujikawa S, Pairojkul C, Waraasawapati S, et al. Attenuation of CD47-SIRPalpha signal in cholangiocarcinoma potentiates tumor-associated macrophage-mediated phagocytosis and suppresses intrahepatic metastasis. Transl Oncol. 2019;12(2):217–25. doi:10.1016/j.tranon.2018.10.007. [Google Scholar] [PubMed] [CrossRef]
98. He S, Wang S, Liu S, Li Z, Liu X, Wu J. Baicalein potentiated M1 macrophage polarization in cancer through targeting PI3Kγ/NF-κB signaling. Front Pharmacol. 2021;12:743837. doi:10.3389/fphar.2021.743837. [Google Scholar] [PubMed] [CrossRef]
99. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature. 2016;539(7629):443–7. doi:10.1038/nature20554. [Google Scholar] [PubMed] [CrossRef]
100. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. 2018;17(12):887–904. doi:10.1038/nrd.2018.169. [Google Scholar] [PubMed] [CrossRef]
101. Coveler AL, Smith DC, Phillips T, Curti BD, Goel S, Mehta AN, et al. Phase 1 dose-escalation study of SEA-CD40: a non-fucosylated CD40 agonist, in advanced solid tumors and lymphomas. J Immunother Cancer. 2023;11(6). doi:10.1136/jitc-2022-005584. [Google Scholar] [PubMed] [CrossRef]
102. Chen D, Xie J, Fiskesund R, Dong W, Liang X, Lv J, et al. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat Commun. 2018;9(1):873. doi:10.1038/s41467-018-03225-9. [Google Scholar] [CrossRef]
103. Binnewies M, Pollack JL, Rudolph J, Dash S, Abushawish M, Lee T, et al. Targeting TREM2 on tumor-associated macrophages enhances immunotherapy. Cell Rep. 2021;37(3):109844. doi:10.1016/j.celrep.2021.109844. [Google Scholar] [PubMed] [CrossRef]
104. Gubin MM, Esaulova E, Ward JP, Malkova ON, Runci D, Wong P, et al. High-dimensional analysis delineates myeloid and lymphoid compartment remodeling during successful immune-checkpoint cancer therapy. Cell. 2018;175(4):1014–30.E19. doi:10.1016/j.cell.2018.09.030. [Google Scholar] [PubMed] [CrossRef]
105. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74(18):5057–69. doi:10.1158/0008-5472.CAN-13-3723. [Google Scholar] [PubMed] [CrossRef]
106. Zhu Y, Yang J, Xu D, Gao XM, Zhang Z, Hsu JL, et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut. 2019;68(9):1653–66. doi:10.1136/gutjnl-2019-318419. [Google Scholar] [PubMed] [CrossRef]
107. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci U S A. 2018;115(17):E4041–50. doi:10.1073/pnas.1720948115. [Google Scholar] [PubMed] [CrossRef]
108. Seo YD, Jiang X, Sullivan KM, Jalikis FG, Smythe KS, Abbasi A, et al. Mobilization of CD8+ T cells via CXCR4 blockade facilitates PD-1 checkpoint therapy in human pancreatic cancer. Clin Cancer Res. 2019;25(13):3934–45. doi:10.1158/1078-0432.CCR-19-0081. [Google Scholar] [PubMed] [CrossRef]
109. Sockolosky JT, Dougan M, Ingram JR, Ho CC, Kauke MJ, Almo SC, et al. Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc Natl Acad Sci USA. 2016;113(19):E2646–54. doi:10.1073/pnas.1604268113. [Google Scholar] [PubMed] [CrossRef]
110. Liu X, Liu L, Ren Z, Yang K, Xu H, Luan Y, et al. Dual targeting of innate and adaptive checkpoints on tumor cells limits immune evasion. Cell Rep. 2018;24(8):2101–11. doi:10.1016/j.celrep.2018.07.062. [Google Scholar] [PubMed] [CrossRef]
111. Song Y, Bugada L, Li R, Hu H, Zhang L, Li C, et al. Albumin nanoparticle containing a PI3Kγ inhibitor and paclitaxel in combination with α-PD1 induces tumor remission of breast cancer in mice. Sci Transl Med. 2022;14(643):eabl3649. doi:10.1126/scitranslmed.abl3649. [Google Scholar] [PubMed] [CrossRef]
112. Weiss SA, Sznol M, Shaheen M, Berciano-Guerrero MA, Munoz-Couselo E, Rodriguez-Abreu D, et al. A phase II trial of the CD40 agonist sotigalimab (APX005M) in combination with nivolumab in subjects with metastatic melanoma with disease progression on anti-PD-1. Clin Cancer Res. 2023. doi:10.1158/1078-0432.CCR-23-0475. [Google Scholar] [PubMed] [CrossRef]
113. Xu J, Escamilla J, Mok S, David J, Priceman S, West B, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013;73(9):2782–94. doi:10.1158/0008-5472.CAN-12-3981. [Google Scholar] [PubMed] [CrossRef]
114. El Chartouni C, Benner C, Eigner M, Lichtinger M, Rehli M. Transcriptional effects of colony-stimulating factor-1 in mouse macrophages. Immunobiol. 2010;215(6):466–74. doi:10.1016/j.imbio.2009.08.002. [Google Scholar] [PubMed] [CrossRef]
115. Chiang Y, Tsai YC, Wang CC, Hsueh FJ, Huang CY, Chung SD, et al. Tumor-derived C-C motif ligand 2 induces the recruitment and polarization of tumor-associated macrophages and increases the metastatic potential of bladder cancer cells in the postirradiated microenvironment. Int J Radiat Oncol Biol Phys. 2022;114(2):321–33. doi:10.1016/j.ijrobp.2022.06.054. [Google Scholar] [PubMed] [CrossRef]
116. Shiao SL, Ruffell B, DeNardo DG, Faddegon BA, Park CC, Coussens LM. TH2-polarized CD4+ T cells and macrophages limit efficacy of radiotherapy. Cancer Immunol Res. 2015;3(5):518–25. doi:10.1158/2326-6066.CIR-14-0232. [Google Scholar] [PubMed] [CrossRef]
117. Wang J, Saung MT, Li K, Fu J, Fujiwara K, Niu N, et al. CCR2/CCR5 inhibitor permits the radiation-induced effector T cell infiltration in pancreatic adenocarcinoma. J Exp Med. 2022;219(5):e20211631. doi:10.1084/jem.20211631. [Google Scholar] [PubMed] [CrossRef]
118. Becherini C, Lancia A, Detti B, Lucidi S, Scartoni D, Ingrosso G, et al. Modulation of tumor-associated macrophage activity with radiation therapy: a systematic review. Strahlenther Onkol. 2023. doi:10.1007/s00066-023-02097-3. [Google Scholar] [PubMed] [CrossRef]
119. Nakasone ES, Askautrud HA, Kees T, Park JH, Plaks V, Ewald AJ, et al. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell. 2012;21(4):488–503. doi:10.1016/j.ccr.2012.02.017. [Google Scholar] [PubMed] [CrossRef]
120. Brana I, Calles A, LoRusso PM, Yee LK, Puchalski TA, Seetharam S, et al. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: an open-label, multicenter phase 1b study. Target Oncol. 2015;10(1):111–23. doi:10.1007/s11523-014-0320-2. [Google Scholar] [PubMed] [CrossRef]
121. Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17(5):651–62. doi:10.1016/S1470-2045(16)00078-4. [Google Scholar] [PubMed] [CrossRef]
122. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–6. doi:10.1126/science.1198443. [Google Scholar] [PubMed] [CrossRef]
123. Perry CJ, Munoz-Rojas AR, Meeth KM, Kellman LN, Amezquita RA, Thakral D, et al. Myeloid-targeted immunotherapies act in synergy to induce inflammation and antitumor immunity. J Exp Med. 2018;215(3):877–93. doi:10.1084/jem.20171435. [Google Scholar] [PubMed] [CrossRef]
124. Hoves S, Ooi CH, Wolter C, Sade H, Bissinger S, Schmittnaegel M, et al. Rapid activation of tumor-associated macrophages boosts preexisting tumor immunity. J Exp Med. 2018;215(3):859–76. doi:10.1084/jem.20171440. [Google Scholar] [PubMed] [CrossRef]
125. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1(1):54–67. doi:10.1158/2159-8274.CD-10-0028. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools