
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

α7nAChR inhibition by methyllycaconitine citrate promotes cell pyroptosis by triggering the polyol pathway in cervical cancer cells
1 Department of Gynecology, Zhuhai Women’s and Children’s Hospital, Zhuhai, 519000, China
2 Institute of Genetic Research, Zhuhai Women’s and Children’s Hospital, Zhuhai, 519000, China
3 Department of Pediatric Haematology and Rheumatalogy, Zhuhai Center For Maternal and Child Health Care, Zhuhai, 519000, China
* Corresponding Authors: DAQIANG YE. Email: ; XIUFU TANG. Email:
# These authors contributed equally to this work
BIOCELL 2024, 48(2), 283-291. https://doi.org/10.32604/biocell.2023.045429
Received 26 August 2023; Accepted 30 November 2023; Issue published 23 February 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: α7 nicotinic acetylcholine receptor (α7nAChR) has been demonstrated to be involved in numerous of inflammatory diseases. Cell pyroptosis is a kind of cell death accompanied by inflammation. Objectives: The objective of this work is to explore the function of α7nAChR on cell pyroptosis in cervical cancer cells. Methods: Immunoblotting, quantitative polymerase chain reaction, and enzyme-linked immunosorbent assay were employed to examine the function of α7nAChR on cell pyroptosis and metabolic changes. Results: Herein, we found that α7nAChR inhibition led to cell pyroptosis in HeLa and SiHa of cervical cancer cells, which was attributed to the upregulation of the polyol pathway. Inhibition of α7nAChR in HeLa and SiHa cells induced the activation of NACHT, LRR and PYD domains-containing protein 3 inflammasomes, leading to enhanced cleaved caspase-1 expression to activate gasdermin D and interleukin-18/1β mature and secretion. Moreover, α7nAChR activation in cervical cancer cells decreased the aldo-keto reductase family 1 member B1 (AKR1B1)-mediated polyol pathway, resulting in reduced sorbitol production. Suppression of AKR1B1 could overcome cell pyroptosis caused by α7nAChR inhibition. Mechanically, α7nAChR inhibition could increase the level of phosphorylation of nuclear factor-kappa B (NF-κB) in HeLa cells, while α7nAChR activation caused inhibition of NF-κB phosphorylation, which further reduced NF-κB binding to the AKR1B1 promoter and consequently abolished AKR1B1 transactivation. In addition, inhibition of NF-κB activity could reverse the promotion of α7nAChR inhibition on cell pyroptosis. Conclusion: This work enriches the metabolic function of α7nAChR in cancer cells and reveals the novel mechanism of α7nAChR-mediated cell pyroptosis.Keywords
Nicotinic acetylcholine receptors (nAChRs) play a critical function in serials of human cancer development, confirmed by a large number of experimental studies, including initiation, tumor growth, invasion, proliferation, and angiogenesis [1,2]. Currently, α7nAChR was found to be of particular significance for its dual role in inflammation [3,4], while its anti-inflammatory function has also emerged as a novel therapeutic strategy for inflammation-associated diseases [5]. For instance, activation of α7 nicotinic acetylcholine receptor could alleviate the dextran sulfate sodium-induced colitis model by improving intestinal mucosal barrier function and decreasing pro-inflammatory factors, such as IL6 and tumor necrosis factor-α expression in the intestine [6,7], in addition, α7nAChR was also reported to regulate inflammatory reaction through cell pyroptosis, even in cancer [8,9], however, the mechanism through which α7nAChR regulated cell pyroptosis remained to be addressed.
Recently, alteration of metabolic reprogramming was able to initiate the development of tumorigenesis and was demonstrated to regulate cell pyroptosis. For instance, in addition to mitochondrial hexokinase 2 (HK2) activated the BCL-xL/BCL-2-associated death promoter/BCL2-associated X (BAD/BAX)-caspase 3 cascade to cleavage of GSDME to induce pyroptosis in cancer cells [10], lactate dehydrogenase A (LDHA), a key limited enzyme in tumor glycolysis pathway that converted pyruvate into lactate, promoted pyroptosis through H3K18lac-mediated high mobility group box 1 (HMGB1) upregulation [11]. In addition, under tumor glycolysis-remodeled acid microenvironment, malate dehydrogenase 1 (MDH1) promiscuously catalyzes α-ketoglutarate (α-KG) into L-2-hydroxyglutarate (L-2HG), accumulating ROS level, which induced the oxidation of death receptor DR6, leading to endocytosis to recruit pro-caspase-8 and GSDMC, subsequently inducing pyroptosis [12]; moreover, a major endogenous product of lipid peroxidation 4-hydroxynonenal (HNE) was also shown to suppress NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome activation [13]. These findings suggested metabolism is necessary for cell pyroptosis. However, the other metabolic changes underlying this pyroptosis remain to be further explored.
Polyol pathway, like glycolysis, converts glucose into sorbitol [14], which is mediated by key limited enzymes aldo-keto reductase (AR), such as aldo-keto reductase family 1 member B1 (AKR1B1) and AKR1B10 [15]. In tumors, loss of AKR1B1 expression could inhibit epithelial-mesenchymal transition and cell growth, respectively [16,17], while upregulation of AKR1B1 expression increased tumor migration and proliferation and enhanced chemosensitivity in malignant glioblastoma [18]. Inhibition of AKR1B1 by the clinically approved antidiabetic drug, including epalrestat or sorbinil, could improve the resistance of epidermal growth factor receptor-tyrosine kinase inhibitor, retarding tumor development by reducing glutathione de novo synthesis [19]; moreover, depletion of AKR1B1 expression or epalrestat treatment in HeLa cell largely blocked cell proliferation, migration, and invasion [20]. Recently, a study revealed that linarin could protect against liver injury in type 2 diabetes mellitus by the inhibition of AKR1B1-mediated alleviating oxidative stress and inflammation [21]. In this work, we revealed that inhibition of α7nAChR was found to promote cell pyroptosis to aggregate inflammation by triggering the polyol pathway. α7nAChR inhibition by methyllycaconitine citrate (MC) in cervical cancer cells induced cell proptosis and AR-mediated polyol pathway, respectively. Mechanically, α7nAChR inhibition activated phosphorylation of nuclear factor-kappa B (NF-κB), initiating aldose reductase (AR) transactivation verified by luciferase and ChIP assay. In addition, AR suppression could overcome the promotion of α7nAChR inhibition on cell pyroptosis. Taken together, this work suggested α7nAChR is a critical modulator linking metabolism reprogramming and cell pyroptosis.
Chemical reagents and antibodies
Reagents: Dulbecco’s modified eagle medium (DMEM; C11965500BT), fetal bovine serum (FBS, cat no. 10099141C), and trypsin (25200056) were from GIBCO (NY, USA), MC (HY-N2332A), nelonicline (HY-16748) and ponalrestat (HY-106697) were purchased from MedChemExpress (NJ, USA). LDH (ab102526) was from Abcam (Cambridge, UK). Trizol reagent (15596-018) was purchased from Invitrogen (Carlsbad, CA, USA). mRNA was detected using the All-in-oneTM cDNA synthesis kit (AORT-0060) and All-in-oneTM quantitative polymerase chain reaction (qPCR) mix (QP002) (GenecopoeiaTM, Rockville, MD, USA), respectively. Human interleukin (IL)-1β enzyme-linked immunosorbent assay (ELISA) Kit (KE00021) and Human Total IL-18 ELISA Kit (KE00193) were purchased from Proteintech (Chicago, IL, USA). D-Sorbitol Assay (Colorimetric) (ab118968) was purchased from Abcam (Cambridge, UK).
Antibodies: NLRP3(68102-1-Ig, 1:1500), apoptosis-associated speck-like protein containing a CARD (ASC; 67494-1-Ig, 1:2000), caspase-1(22915-1-AP, 1:2000), gasdermin D (GSDMD; 66387-1-Ig, 1:2000), GSDME (67731-1-Ig, 1:2000), interleukin (IL)-1β (16806-1-AP, 1:2000), IL-18 (10663-1-AP, 1:2000), AKR1B1 (67498-1-Ig, 1:2000), NF-κB (66535-1-Ig, 1:2000) and phosphor-NF-κB (39675, 1:2000) were from Proteintech (Chicago, IL). HRP-conjugated goat anti-rabbit (111-035-003, 1:3000) and goat anti-mouse (115-035-003, 1:3000) were purchased from Jackson Immunoresearch and α-tubulin (AC007, 1:4000) were purchased from Ray (Beijing, China).
HeLa and SiHa cells of cervical cancer cells were purchased from the China Center for Type Culture Collection (Beijing, China) and cultured in DMEM supplemented with 10% FBS. The α7nAChR antagonist MC was used to treat cells at final concertation of 20 µM for 48 h, while NF-κB inhibitor BAY 11-7082 (HY-13453, MCE) and AR inhibitor ponalrestat were used at 10 and 5 µM, respectively, for the treatment.
After digestion, cells were collected to re-seeded into a 96-well plate overnight and treated further. The LDH level was tested after treatment with or without MC for 48 h.
Enzyme-linked immunosorbent assay (ELISA)
The level of IL-1β, IL-18, and sorbitol in the medium content from HeLa and SiHa cells received with or without stimulation were quantified by corresponding commercial kit.
Quantitative polymerase chain reaction analysis
After treatment, the whole cell was lysed to isolate total RNA to conduct a qPCR assay to detect the expression of indicated genes. The primer sequences used in this study are listed in Suppl. Table S1.
The whole cell protein was extracted, and western blotting was performed. Briefly, the whole cell lysate was separated and transferred onto polyvinylidene fluoride membranes, and blocked with phosphate-buffered saline with Tween 20 (PBST) and 5% milk for 1 h at room temperature. Indicated antibodies were diluted and added to the membrane to incubate overnight at 4°C. Next, another 1 h incubation was required for horseradish peroxidase-conjugated secondary antibodies at room temperature. The bands were visualized using a chemiluminescent imaging system after washing with PBST three times.
SiRNA targeting NF-κB (si-NF-κB) or control siRNA (si-CTL), purchased from genepharm (Shanghai, China), combined with luciferase plasmid containing AKR1B1 promoter and Renilla plasmid were transfected into cells for 24 h, and stimulated with or without MC/nelonicline for further 24 h. Relative luciferase unit (RLU) were examined using Duo-Lite Luciferase Assay System (Vazyme, cat no. DD1205-01).
Chromatin immunoprecipitation (ChIP) assay
After serum-starvation for 18 h, cells were incubated with or without MC/nelonicline for 1 h. After isolation of chromatin, chromatin was immunoprecipitated with anti- NF-κB using SimpleChIP® Enzymatic Chromatin IP Kit (Magnetic Beads; CST, cat no. 9003) After purification of the chromatin DNA fragments, qPCR was used to analyze the effect of α7nAChR inhibition/activation on NF-κB binding to AKR1B1 promoter with primer synthesized from.
All statistical differences were examined using GraphPad Prism 9 software. A p-value < 0.05 was significant.
Inhibition of α7nAChR-promoted increased cell pyroptosis
α7nAChR has been demonstrated to act as an important anti-inflammatory agent in various disease, such as preeclampsia [22] and cancer [23]. The Gene Expression Profiling Interactive Analysis (GEPIA) dataset showed that no significant difference was observed in α7nAChR expression in cervical squamous cell carcinoma and endocervical adenocarcinoma in comparison with control, and the Kaplan–Meier survival analysis indicated there is no difference in overall survival for patients with high α7nAChR expression in comparison with those with low α7nAChR expression (Figs. 1A and 1B). To further uncover the possible role of α7nAChR in cervical cancer cells, HeLa and SiHa cells were employed to determine the possible function of α7nAChR in cell pyroptosis. Morphologically, an increased number of dead cells were obtained in response to MC stimulation compared with the DMSO group, and inhibition of α7nAChR in HeLa and SiHa cells could significantly increase relative cell death as confirmed by LDH assay (Figs. 1C and 1D). Moreover, IL-1β/IL-18 were drastically enhanced at mRNA levels in response to α7nAChR inhibition (Fig. 1E). Moreover, the quantified band from WB and ELISA analysis also demonstrated that a drastically enhanced IL-1β/IL-18 levels were obtained in HeLa and SiHa cells in response to α7nAChR inhibition (Fig. 1F, Suppl. Fig. S1A). These results suggested α7nAChR inhibition could induce cell pyroptosis in cervical cancer cells to aggravate inflammation.
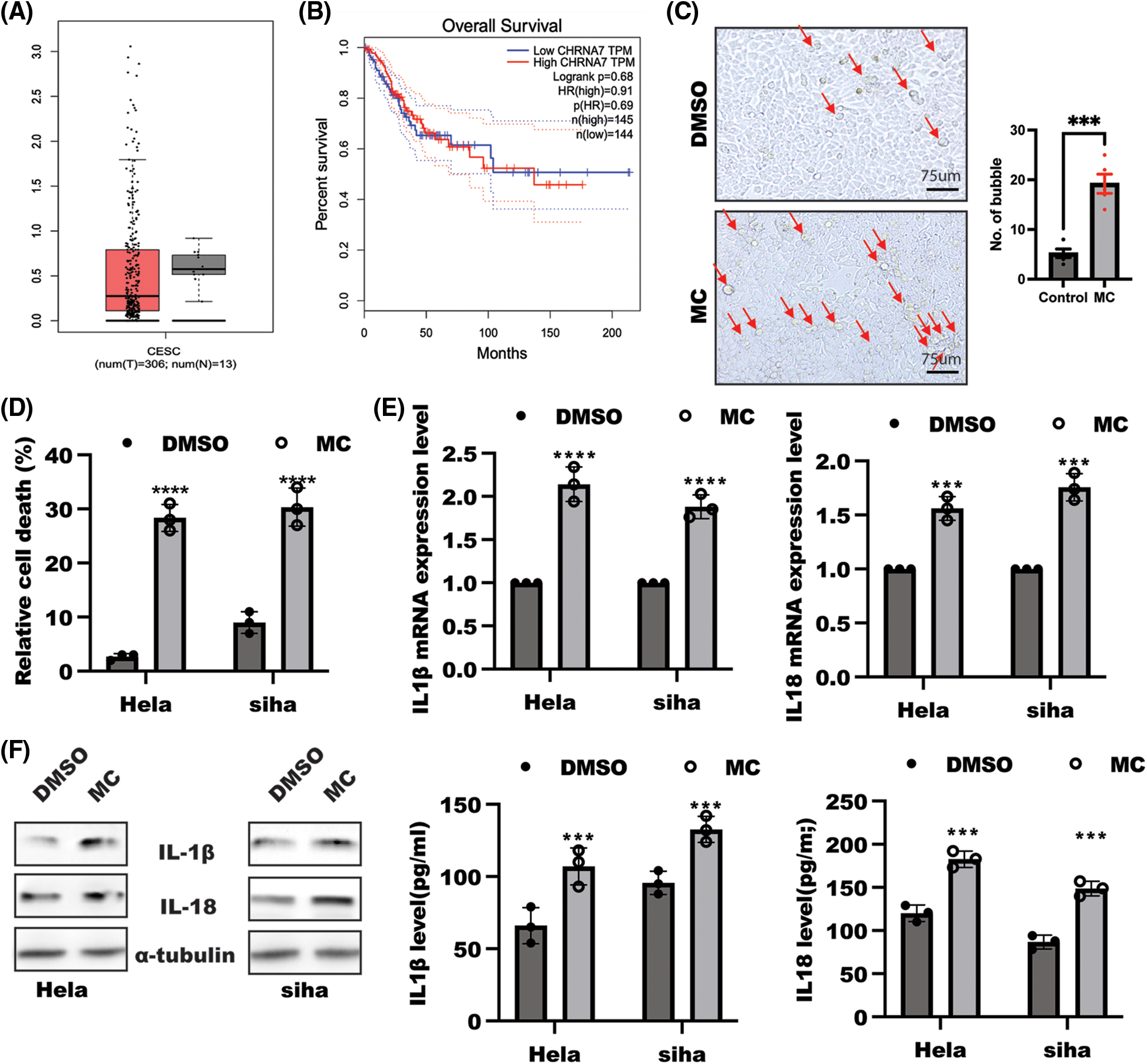
Figure 1: Inhibition of α7 nicotinic acetylcholine receptor (α7nAChR) induced cell pyroptosis. (A) α7nAChR gene expression in cervical cancer (n = 306) and healthy control (n = 13) (GEPIA database). (B) Kaplan–Meier curves analysis of overall survival in patients with high α7nAChR expression (n = 145) in comparison with low α7nAChR (n = 144) group. (C) Bright field of HeLa cells treated with or without methyllycaconitine citrate (MC) (20 µM) for 48 h and the number of bubbles was estimated using t-test, scale bar: 250 µm, ***p < 0.001. (D) HeLa and SiHa cells were seeded at a density of 105/mL overnight, and then stimulated with or without α7nAChR inhibitor, MC (20 µM) for 48 h after a serum starved for 12 h; the content of LDH was used to examine relative cell death. Data was displayed as means ± SD and determined by t-test, n = 3, ****p < 0.0001 vs. the DMSO group. (E) qPCR was used to test the Interleukin (IL)-1β and IL-18 mRNA levels in HeLa and SiHa cells treated as indicated. Data was displayed as mean ± SD and examined by one sample t-test, n = 3, ****p < 0.0001, ***p < 0.001 vs. the DMSO group. (F) WB (left panel) and enzyme-linked immunosorbent assay (right panel) were used to examine IL-1β and IL-18 expression. Data was displayed as mean ± SD and analyzed by t-test, n = 3, ***p < 0.001 vs. the DMSO group.
α7nAChR inhibition contributed to NLRP3 inflammasome activation
Next, we sought to discuss the possible change of NLRP3 inflammasome activation in response to α7nAChR inhibition in cervical cancer cells. Real-time PCR analysis suggested a significant enhancement in NLRP3, ASC, caspase1, and GSDMD mRNA levels in HeLa cells subjected to the inhibition of α7nAChR (Fig. 2A), while α7nAChR activation caused by nelonicline could decrease NLRP3, ASC, and caspase 1 mRNA levels (Fig. 2B). Consistently, the further work displayed that the level of NLRP3and GSDMD proteins, were markedly ablated in HeLa cells after nelonicline treatment, while inhibition of α7nAChR by MC triggered NLRP3 inflammasome activation (Fig. 2C, >Suppl. Fig. S1B). Collectively, these results suggest that α7nAChR inhibition in cervical cancer cells led to cell pyroptosis through activation of NLRP3 inflammasome.
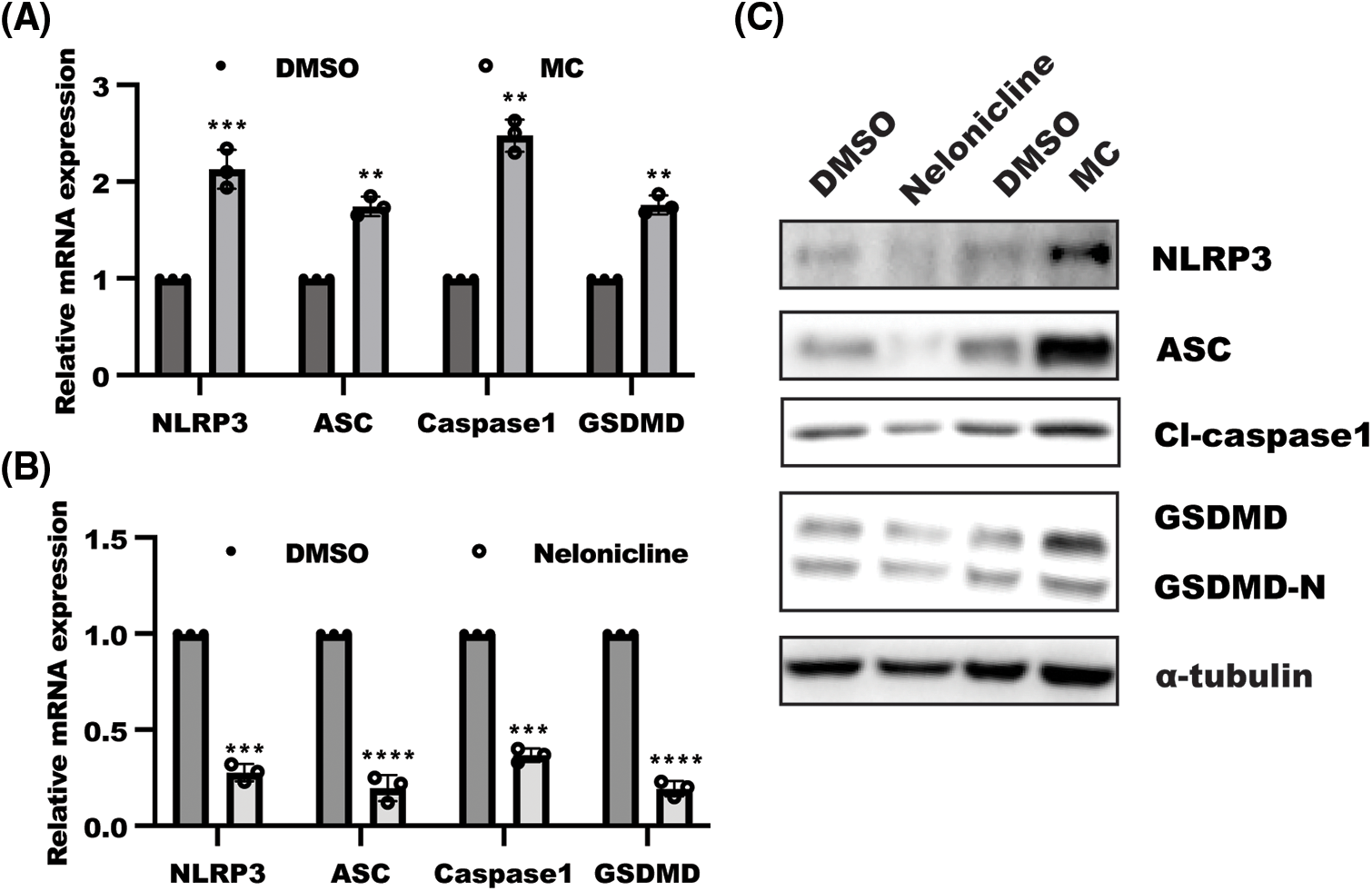
Figure 2: α7 nicotinic acetylcholine receptor (α7nAChR) suppressed NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasomes activation. HeLa cells were treated with (A) methyllycaconitine citrate (MC) and (B) nelonicline for 48 h, and RNA was isolated to examine the level of pyroptosis-related genes. Data was displayed as mean ± SD and analyzed by one sample t-test, n = 3, ****p < 0.0001, ***p < 0.001, **p < 0.01 vs. the DMSO group. (C) Western blotting was performed in HeLa cells treated as indicated for 48 h. NLRP3 inflammasomes, including NLRP3, apoptosis-associated speck-like protein containing a CARD (ASC), and caspase1 and gasdermin D, were detected in response to α7nAChR activation and inhibition.
α7nAChR inhibition promoted cell pyroptosis through AKR1B1
Metabolic reprogramming is a critical factor in tumor development. Accumulating evidences demonstrated the obvious role of AKR1B1, a limited enzyme of the polyol pathway that catalyzes glucose into sorbitol, in the promotion of inflammation [24,25]. Therefore, we focused our attention to exploring whether the polyol pathway is involved in α7nAChR modulating cell pyroptosis. As shown in Fig. 3A, α7nAChR inhibition in HeLa and SiHa cells led to an upregulation of sorbitol production, which was attributed to enhanced expression of AKR1B1 mRNA and protein caused by α7nAChR inhibition (Figs. 3B–3C, Suppl. Fig. S1C). Most importantly, the addition of AR inhibitor ponalrestat (ARI, 5 µM) could reverse the effect of α7nAChR inhibitor MC on cell pyroptosis, resulting in decreasing IL-1β and IL-18 levels (Figs. 3D–3E, Suppl. Fig. S1D). Taken together, these results suggest that α7nAChR suppressed NLRP3 inflammasome activation by reducing AR-mediated sorbitol production.
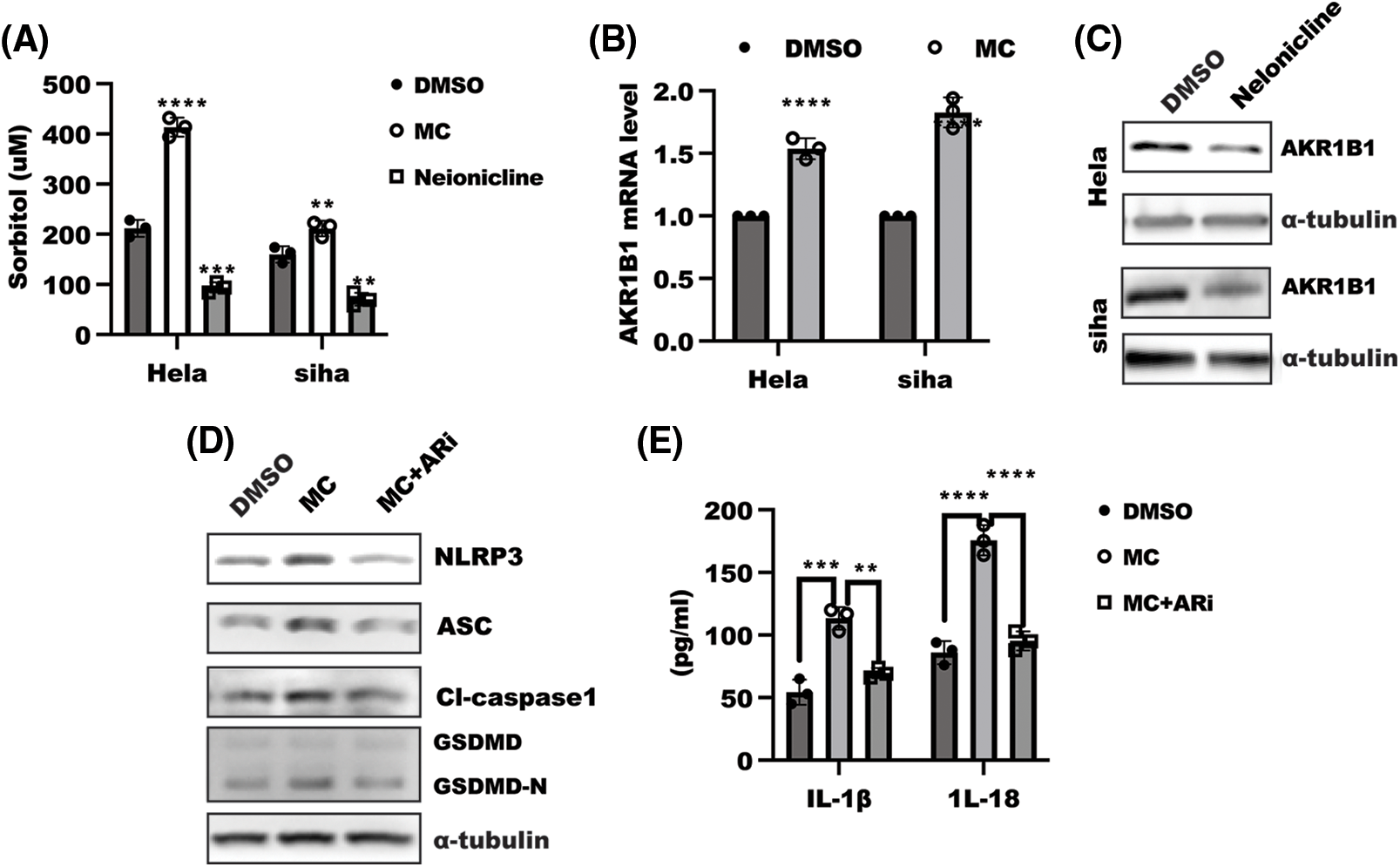
Figure 3: α7 nicotinic acetylcholine receptor (α7nAChR) regulated cell pyroptosis through aldo-keto reductase family 1 member B1 (AKR1B1). (A) enzyme-linked immunosorbent assay (ELISA) was used to detect the level of sorbitol in HeLa and SiHa cells treated as indicated for 48 h. Data was displayed as mean ± SD and tested using one-way ANOVA with multiple comparisons, followed by Dunnett post-hoc test for significance vs. dimethylsulfoxide (DMSO). n = 3, ****p < 0.0001, ***p < 0.001, **p < 0.01. (B) after stimulation for 48 h, as indicated, total RNA from HeLa and SiHa cells were harvested to detect the level of pyroptosis-related genes. Data was shown as mean ± SD. and was determined with one-sample t-test, ****p < 0.0001 vs. DMSO group. (C–D) Western blotting was performed in HeLa cells treated as indicated for 48 h. NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasomes, including NLRP3, apoptosis-associated speck-like protein containing a CARD (ASC), and caspase1 and gasdermin D, were detected in response to α7nAChR combined with aldose reductase (AR) inhibition. (E) ELISA assays were conducted to examine interleukin (IL)-1β and IL-18 secretion levels in cells. Data was shown as mean ± SD and determined using one-way ANOVA with multiple comparisons, followed by the Dunnett post hoc test for significance. n = 3, ****p < 0.0001, ***p < 0.001, **p < 0.01.
The importance of NF-κB for α7nAChR-mediated AKR1B1 expression
Next, to unravel the mechanism through which α7nAChR modulated AKR1B1 expression. AR was confirmed to be a direct target of NF-κB [26], which led us to analyze the influence of NF-κB in α7nAChR-mediated AR transactivity. Luciferase reporter assay showed that α7nAChR inhibition led to a significant increase, while α7nAChR activation drastically decreased AKR1B1 transactivity. Loss of NF-κB expression could reverse the enhanced AKR1B1 luminescence caused by α7nAChR inhibition (Fig. 4A). Further work showed that α7nAChR inhibition increased, while activation of α7nAChR largely blocked the binding of NF-κB to AKR1B1 promoter (Fig. 4B).
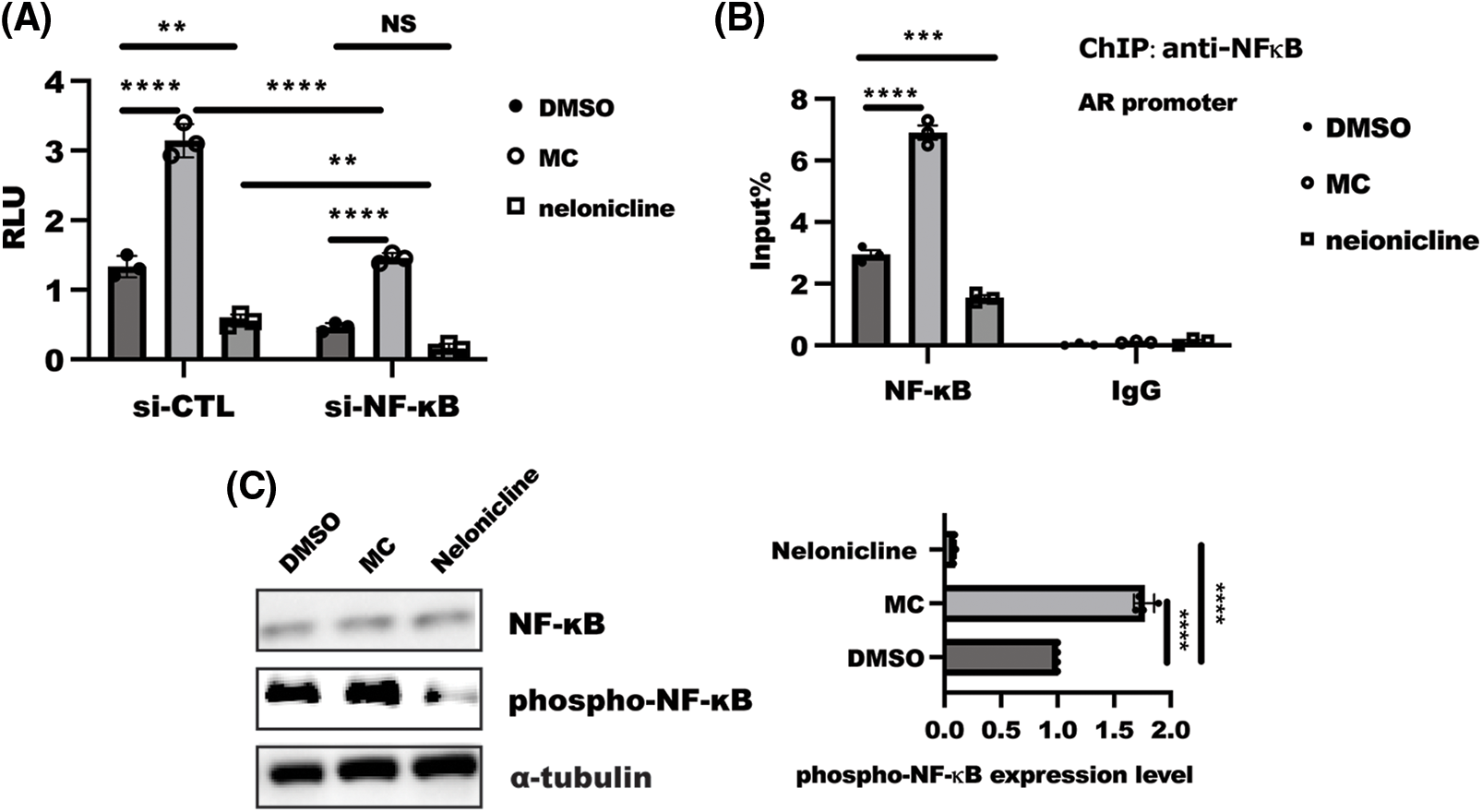
Figure 4: NF-κB was required for α7 nicotinic acetylcholine receptor (α7nAChR)-mediated aldo-keto reductase family 1 member B1(AKR1B1). (A) after co-transfection with indicated siRNA combined with AKR1B1-Luc and Renilla plasmid, a further 24-h treatment with MC or nelonicline treatment was required before detection. Data are shown as the mean ± SD and test using two-ANOVA with multiple comparisons, followed by the Bonferroni post-hoc test for significance. ****p < 0.0001, **p < 0.01, n = 3. (B) HeLa cells were serum starved for 24 h and then treated with MC/nelonicline for an additional 1 h. ChIP analysis of NF-κB binding to AKR1B1 gene promoter. Data presented the mean ± SD two-ANOVA combined with Dunnett’s multiple comparison were employed to determine significance, ****p < 0.001, ***p < 0.01; (C) HeLa cells were serum starved overnight, then subsequently following treatment by MC/nelonicline for 1 h, respectively. SDS-PAGE was used to analyze proteins expression. the data of quantified band was displayed as means ± SD and determined with one-way ANOVA test, n = 3, ****p < 0.0001, ***p < 0.001.
Phosphorylation of NF-κB is a critical event for NLRP3 inflammasome activation [14]. To understand the driving forces controlling α7nAChR-mediated AKR1B1 transcription better, we focused our attention on analyzing the influence of α7nAChR on the phosphorylation of NF-κB. The inhibition of α7nAChR could increase the phosphorylation of NF-κB, while activation of α7nAChR in HeLa cells drastically reduced the phosphorylation of NF-κB (Fig. 4C). Taken together, these findings suggested α7nAChR regulated AR expression through NF-κB.
Inhibition of NF-κB could rescue activation of cell pyroptosis by α7nAChR inhibition
To test the hypothesis that α7nAChR inhibition contributed to NF-κB-mediated AR-dependent cell pyroptosis, we treated HeLa cells with NF-κB inhibitor BAY 11-7082 (10 µM) and analyzed cellular pyroptosis and polyol pathway. As expected, inhibition of NF-κB alone significantly decreased baseline cell pyroptosis and AR expression, and this NF-κB inhibition-mediated effect could reverse the promotion of α7nAChR inhibition on cell pyroptosis, including IL-1β and IL-18 levels, leading to reduced relative cell death (Figs. 5A–5C, Suppl. Fig. S1E), in addition, specific depletion of NF-κB expression could overcome the effect of MC on IL-1β and IL-18 level (Fig. 5D).
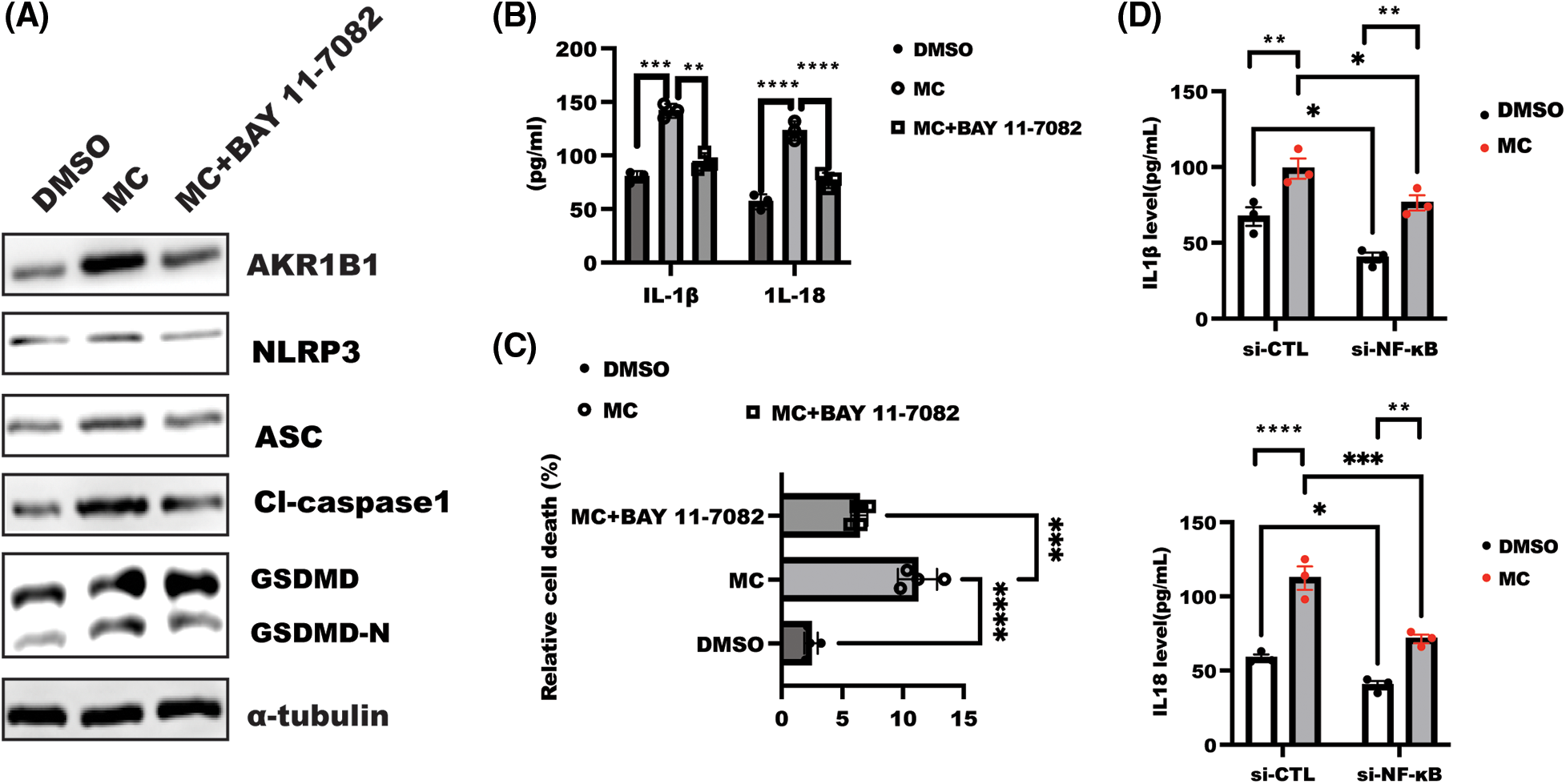
Figure 5: Nuclear factor-kappa B (NF-κB) inhibition rescued α7 nicotinic acetylcholine receptor (α7nAChR) inhibition-mediated cell pyroptosis. (A) After overnight serum-starvation, HeLa cells were stimulated with BAY 11-7082 (10 µM) for 1 h, followed by treatment with MC for 48 h as indicated. The lysate was collected to detect indicated proteins. (B) Interleukin (IL)-1β and IL-18 were examined in the supernatant from the indicated group treated as described in A. Data was displayed as the mean ± SD. and examined by one-way ANOVA with Dunnett post hoc test for significance. n = 3, ****p < 0.0001, ***p < 0.001, **p < 0.01. (C) HeLa cells were stimulated as described in A, and LDH was tested to analyze relative cell death in the indicated group. Data was shown as mean ± SD and tested using one-way ANOVA with Dunnett post hoc test for significance. n = 3, ****p < 0.0001, ***p < 0.001. (D) After transfection with siRNA for 18 h, HeLa cells were incubated with or without MC for a further 24 h, and the level of IL-1β and IL-18 was tested using enzyme-linked immunosorbent assay assay. Data are displayed as mean ± SD and examined by two ANOVA with multiple comparisons, followed by the Dunnett post hoc test for significance vs. DMSO. n = 3, ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05.
In this study, we addressed a novel insight that α7nAChR inhibition promoted cell pyroptosis through the NF-κB-mediated polyol pathway in cervical cancer cells, thus aggravating inflammation. α7nAChR inhibitor treatment in HeLa and SiHa cells induced a drastic enhancement of cell pyroptosis and also increased AR expression. This inhibitor-mediated suppression of AR could rescue the cell pyroptosis caused by α7nAChR inhibition. Mechanically, α7nAChR inhibition in cervical cancer cells enhanced NF-κB phosphorylation, which further induced NF-κB binding to the AR promoter, leading to the initiation of AR transactivation. AR inhibition could overcome the influence of α7nAChR inhibition on cell pyroptosis, alleviating inflammation. This work revealed the metabolic function of α7nAChR and enriched the mechanism of α7nAChR in cell pyroptosis in cervical cancer cells.
NLRP3 inflammasome complex derived the activation of caspase-1, which further induced pro-IL-1β, pro-IL-18, and GSDMD to cleave into maturation, aggravating inflammation. In this work, we found increased cleaved-caspase-1 expression caused by enhanced NLRP3 inflammasome activation in response to α7nAChR inhibition. However, more in-depth work is required to explore the possible role of α7nAChR in other NLRPs inflammasomes. In addition to caspase-1-mediated canonical pyroptosis, whether caspase 4/5-mediated noncanonical pyroptosis is involved in α7nAChR-mediated pyroptosis remains unknown.
Aldo-keto reductase has been reported to participate in serial aspects of cancer. For instance, AKR1B10 facilitates the Warburg effect by regulating LDHA expression, which promotes acquired resistance to pemetrexed in lung cancer-derived brain metastasis [27], and AKR1B1 could suppress cell proliferation through p38 MAPK activation to control the Bcl-2/BAX/caspase-3 apoptosis signaling in glioma cell [28]. Moreover, AKR1B1 promoted tumor aggressiveness through a positive feedback loop that activates the EMT program in basal-like breast cancer progression [29]. In this work, we found that AR inhibition and reduced sorbitol production, caused by α7nAChR activation, could reduce cell pyroptosis, leading to alleviate inflammation, suggesting that α7nAChR regulated cell pyroptosis in dependent of AR-mediated polyol pathway. Interestingly, in line with this, another preliminary work from our lab has implied that sorbitol, a product of the polyol pathway, could activate NF-κB to trigger NLRP3 inflammasome activation (data not shown). However, the mechanism through which AKR1B1 regulated NLRP3 inflammasome activation remains unclear. In addition, the investigation is needed to reveal whether other metabolic changes involved in α7nAChR regulated cell pyroptosis, including glycolysis, glutaminolysis, and de novo lipogenesis.
The study showed that Nuclear factor erythroid 2-related factor 2 (NRF2) plays an essential role in the AKR1B10 gene regulation [30–32], and NF-κB could direct transactive AKR1B1 [33]. Interestingly, α7nAChR inhibition in cervical cancer cells led to increase NF-κB phosphorylation, which further enhance the binding of NF-κB to the AKR1B1 promoter to initiate transactivation. However, how α7nAChR regulates NF-κB phosphorylation through classical or non-classical pathways, remains to be addressed. In addition to NF-κB, whether α7nAChR inhibition led to the disassociation of Kelch-like ECH-associated protein1-NRF2 complex to release and induce the binding of NRF2 to ARE to initiate AKR1B1 transactivation also needs to be addressed. Moreover, our present findings might not exclude the hypothesis that α7nAChR directly regulates NRF2 and/or AR; this could be uncovered in the future. In summary, this work suggested that alteration of α7nAChR activity is a critical event for cell pyroptosis in dependent of NF-κB-mediated polyol pathway, and inhibition of polyol pathway might be a sufficient strategy to reduce cell pyroptosis to alleviate inflammation.
Acknowledgement: None.
Funding Statement: This work was supported by the Zhuhai Science and Technology Plan (ZH22036201210174PWC).
Author Contributions: The authors confirm their contribution to the paper as follows: QFT and DQY conceived and designed the experiments; JYX, GW, and PL performed experiments and analyzed data; QFT and DQY wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: All data generated or analyzed during this study are included in this published article (and its supplementary information files).
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Bele T, Turk T, Krizaj I. Nicotinic acetylcholine receptors in cancer: limitations and prospects. Biochim Biophys Acta Mol Basis Dis. 2024;1870(1):166875. [Google Scholar] [PubMed]
2. Sansone L, Milani F, Fabrizi R, Belli M, Cristina M, Zaga V, et al. Nicotine: from discovery to biological effects. Int J Mol Sci. 2023;24(19):14570. [Google Scholar] [PubMed]
3. Wu YJ, Wang L, Ji CF, Gu SF, Yin Q, Zuo J. The role of α7nAChR-mediated cholinergic anti-inflammatory pathway in immune cells. Inflammation. 2021;44(3):821–34. [Google Scholar] [PubMed]
4. Hajiasgharzadeh K, Somi MH, Sadigh-Eteghad S, Mokhtarzadeh A, Shanehbandi D, Mansoori B, et al. The dual role of alpha7 nicotinic acetylcholine receptor in inflammation-associated gastrointestinal cancers. Heliyon. 2020;6(3):e03611. [Google Scholar] [PubMed]
5. Hone AJ, McIntosh JM. Nicotinic acetylcholine receptors: therapeutic targets for novel ligands to treat pain and inflammation. Pharmacol Res. 2023;190(1):106715. [Google Scholar] [PubMed]
6. Pu W, Su Z, Wazir J, Zhao C, Wei L, Wang R, et al. Protective effect of α7 nicotinic acetylcholine receptor activation on experimental colitis and its mechanism. Mol Med. 2022;28(1):104. [Google Scholar] [PubMed]
7. Ye Z, Zhu Y, Tang N, Zhao X, Jiang J, Ma J, et al. α7 nicotinic acetylcholine receptor agonist GTS-21 attenuates DSS-induced intestinal colitis by improving intestinal mucosal barrier function. Mol Med. 2022;28(1):59. [Google Scholar] [PubMed]
8. Fu H, Shen QR, Zhao Y, Ni M, Zhou CC, Chen JK, et al. Activating α7nAChR ameliorates abdominal aortic aneurysm through inhibiting pyroptosis mediated by NLRP3 inflammasome. Acta Pharmacol Sin. 2022;43(10):2585–95. [Google Scholar] [PubMed]
9. Tang H, Li J, Zhou Q, Li S, Xie C, Niu L, et al. Vagus nerve stimulation alleviated cerebral ischemia and reperfusion injury in rats by inhibiting pyroptosis via α7 nicotinic acetylcholine receptor. Cell Death Discov. 2022;8(1):54. [Google Scholar] [PubMed]
10. Cai J, Yi M, Tan Y, Li X, Li G, Zeng Z, et al. Natural product triptolide induces GSDME-mediated pyroptosis in head and neck cancer through suppressing mitochondrial hexokinase-II. J Exp Clin Cancer Res. 2021;40(1):190. [Google Scholar] [PubMed]
11. Yao X, Li C. Lactate dehydrogenase A mediated histone lactylation induced the pyroptosis through targeting HMGB1. Metab Brain Dis. 2023;38(5):1543–53. [Google Scholar] [PubMed]
12. Zhang JY, Zhou B, Sun RY, Ai YL, Cheng K, Li FN, et al. The metabolite α-KG induces GSDMC-dependent pyroptosis through death receptor 6-activated caspase-8. Cell Res. 2021;31(9):980–97. [Google Scholar] [PubMed]
13. Hsu CG, Chavez CL, Zhang C, Sowden M, Yan C, Berk BC. The lipid peroxidation product 4-hydroxynonenal inhibits NLRP3 inflammasome activation and macrophage pyroptosis. Cell Death Differ. 2022;29(9):1790–803. [Google Scholar] [PubMed]
14. Huang L, Tang X, Yang F, Pan W, Liang X, Xu Z, et al. Shikonin contributes to intestinal epithelial cell differentiation through PKM2/NRF2-mediated Polyol pathway. Pharmacol Res—Modern Chin Med. 2021;1(12):100004. [Google Scholar]
15. Zhou Y, Lin Y, Li W, Liu Q, Gong H, Li Y, et al. Expression of AKRs superfamily and prognostic in human gastric cancer. Medicine. 2023;102(8):e33041. [Google Scholar] [PubMed]
16. Schwab A, Siddiqui A, Vazakidou ME, Napoli F, Bottcher M, Menchicchi B, et al. Polyol pathway links glucose metabolism to the aggressiveness of cancer cells. Cancer Res. 2018;78(7):1604–18. [Google Scholar] [PubMed]
17. Liu L, Zhu L, Cheng Z, Sun Y, Zhou Y, Cao J. Aberrant expression of AKR1B1 indicates poor prognosis and promotes gastric cancer progression by regulating the AKT-mTOR pathway. Aging. 2023;15(18):9661–75. [Google Scholar] [PubMed]
18. Meng M, Yang L, Zhou H, Cheng Q, Peng R, Wang Z, et al. LINC00978 regulates metabolic rewiring to promote the malignancy of glioblastoma through AKR1B1. Cancer Lett. 2023;567:216277. [Google Scholar] [PubMed]
19. Zhang KR, Zhang YF, Lei HM, Tang YB, Ma CS, Lv QM, et al. Targeting AKR1B1 inhibits glutathione de novo synthesis to overcome acquired resistance to EGFR-targeted therapy in lung cancer. Sci Transl Med. 2021;13(614):eabg6428. [Google Scholar] [PubMed]
20. Ji J, Xu MX, Qian TY, Zhu SZ, Jiang F, Liu ZX, et al. The AKR1B1 inhibitor epalrestat suppresses the progression of cervical cancer. Mol Biol Rep. 2020;47(8):6091–103. [Google Scholar] [PubMed]
21. Wang T, Shan MY, Tang CY, Cheng MY, Chen B, Yan J, et al. Linarin ameliorates diabetic liver injury by alleviating oxidative stress and inflammation through the inhibition of AKR1B1. Comb Chem High Throughput Screen. 2023. doi:10.2174/1386207326666230412084201 [Google Scholar] [PubMed] [CrossRef]
22. Han X, Li W, Li P, Zheng Z, Lin B, Zhou B, et al. Stimulation of alpha7 nicotinic acetylcholine receptor by nicotine suppresses decidual M1 macrophage polarization against inflammation in lipopolysaccharide-induced preeclampsia-like mouse model. Front Immunol. 2021;12:642071. [Google Scholar] [PubMed]
23. Hsu CC, Tsai KY, Su YF, Chien CY, Chen YC, Wu YC, et al. alpha7-Nicotine acetylcholine receptor mediated nicotine induced cell survival and cisplatin resistance in oral cancer. Arch Oral Biol. 2020;111(5):104653. [Google Scholar] [PubMed]
24. Yadav UC, Ramana KV, Srivastava SK. Aldose reductase inhibition suppresses airway inflammation. Chem Biol Interact. 2011;191(1–3):339–45. [Google Scholar] [PubMed]
25. Np S, Rajdev B, Jain S, Gangasani JK, Vaidya JR, Naidu V. Molecular dissection of anti-colon cancer activity of NARI-29: special focus on H2O2 modulated NF-κB and death receptor signaling. Free Radic Res. 2023;57(4):1–17. [Google Scholar]
26. Jung KA, Choi BH, Nam CW, Song M, Kim ST, Lee JY, et al. Identification of aldo-keto reductases as NRF2-target marker genes in human cells. Toxicol Lett. 2013;218(1):39–49. [Google Scholar] [PubMed]
27. Duan W, Liu W, Xia S, Zhou Y, Tang M, Xu M, et al. Warburg effect enhanced by AKR1B10 promotes acquired resistance to pemetrexed in lung cancer-derived brain metastasis. J Transl Med. 2023;21(1):547. [Google Scholar] [PubMed]
28. Huang YK, Chang KC, Li CY, Lieu AS, Lin CL. AKR1B1 represses glioma cell proliferation through p38 MAPK-mediated Bcl-2/BAX/Caspase-3 apoptotic signaling pathways. Curr Issues Mol Biol. 2023;45(4):3391–405. [Google Scholar] [PubMed]
29. Wu X, Li X, Fu Q, Cao Q, Chen X, Wang M, et al. AKR1B1 promotes basal-like breast cancer progression by a positive feedback loop that activates the EMT program. J Exp Med. 2017;214(4):1065–79. [Google Scholar] [PubMed]
30. Liu Z, Zhong L, Krishack PA, Robbins S, Cao JX, Zhao Y, et al. Structure and promoter characterization of aldo-keto reductase family 1 B10 gene. Gene. 2009;437(1–2):39–44. [Google Scholar] [PubMed]
31. Nishinaka T, Miura T, Shimizu K, Terada T. Identification and characterization of functional antioxidant response elements in the promoter of the aldo-keto reductase AKR1B10 gene. Chem Biol Interact. 2017;276:160–6. [Google Scholar] [PubMed]
32. Nishinaka T, Miura T, Okumura M, Nakao F, Nakamura H, Terada T. Regulation of aldo-keto reductase AKR1B10 gene expression: involvement of transcription factor Nrf2. Chem Biol Interact. 2011;191(1–3):185–91. [Google Scholar] [PubMed]
33. Yang B, Hodgkinson A, Oates PJ, Millward BA, Demaine AG. High glucose induction of DNA-binding activity of the transcription factor NFκB in patients with diabetic nephropathy. Biochim Biophys Acta. 2008;1782(5):295–302. [Google Scholar] [PubMed]
Supplementary Materials
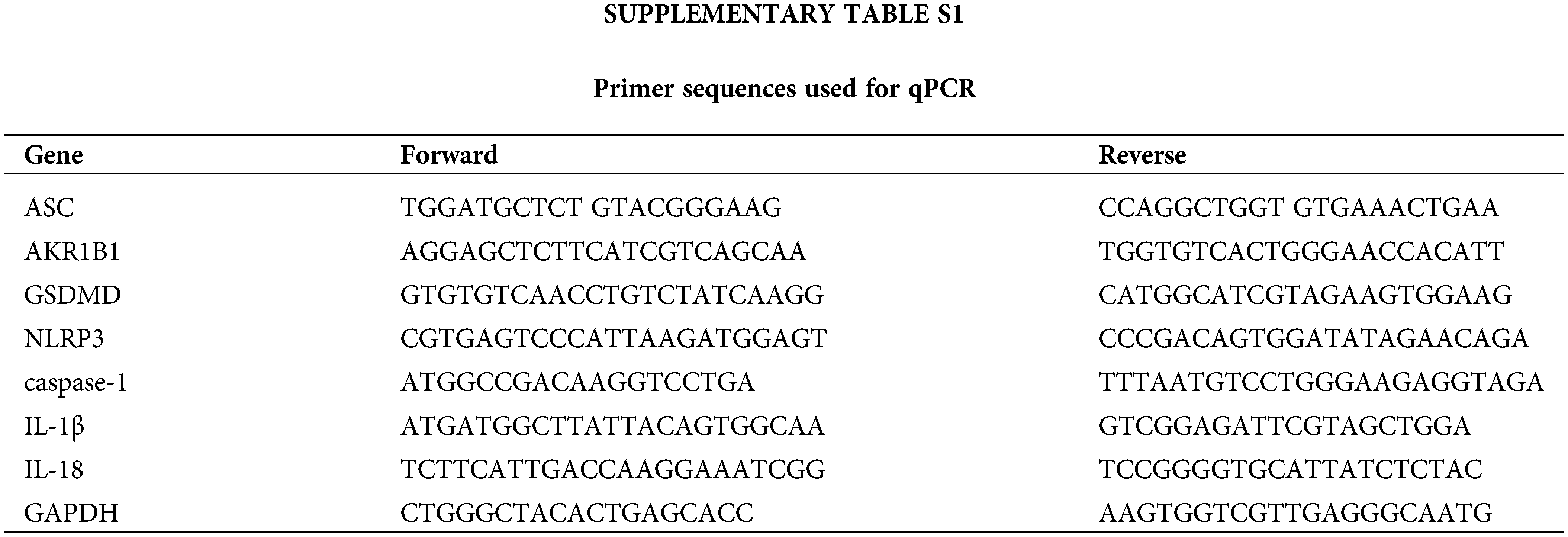
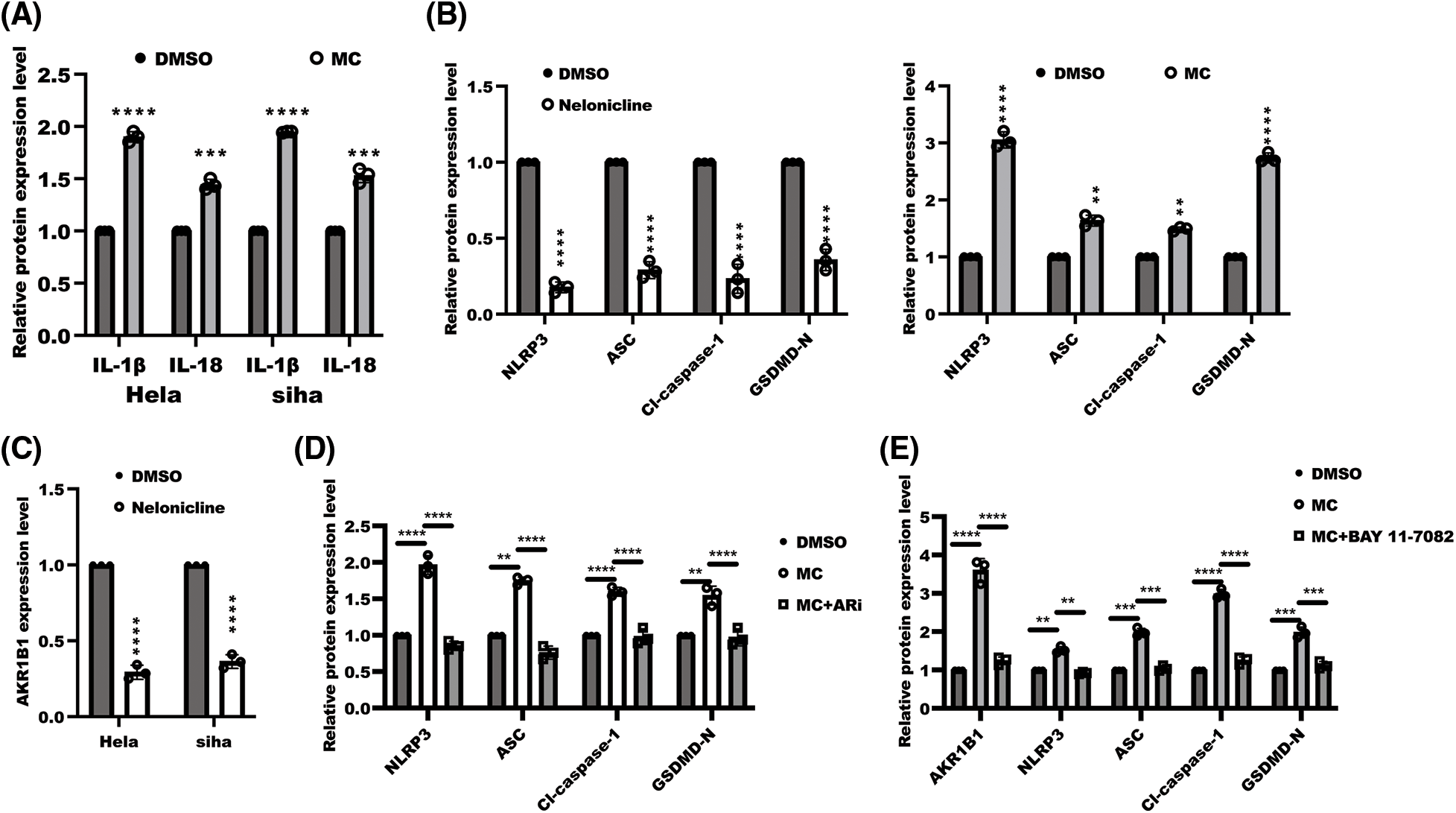
Figure S1: The quantitation of WB band. The band intensity was measured and tested by statistical analysis as followed: (A) the data of quantified band from Fig. 1F was displayed as means±s.d. and determined by one sample t-test, n = 3, ****p < 0.0001, ***p < 0.001; (B) the data of quantified band from Fig. 2C was displayed as means ± s.d. and examined with one sample t-test, n = 3, ****p < 0.0001, ** p <0.01; (C) the data of quantified AKR1B1 band intensity from Fig. 3C was displayed as means ± s.d. and determined by one sample t-test, ****p < 0.0001, n = 3; (D) the data of quantified band from Fig. 3D was displayed as means ± s.d. and determined with one-ANOVA test, n = 3, ****p < 0.0001, **p < 0.01; (E) the data of quantified band from Fig. 5A was displayed as means ± s.d. and examined with one sample t-test, n = 3, ****p < 0.0001, **p < 0.01.
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools