
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE
MKT-077 normalizes mitochondrial function and mitigates cardiac pathology in mdx mice
1 Department of Biochemistry, Cell Biology, and Microbiology, Mari State University, Yoshkar-Ola, 424001, Russia
2 Laboratory of Mitochondrial Transport, Institute of Theoretical and Experimental Biophysics, Russian Academy of Sciences, Pushchino, 142290, Russia
* Corresponding Author: MIKHAIL V. DUBININ. Email:
(This article belongs to the Special Issue: Mitochondrial Dysfunction in Metabolic and Neuromuscular Diseases: Mechanisms and Therapeutic Strategies)
BIOCELL 2024, 48(12), 1815-1825. https://doi.org/10.32604/biocell.2024.058068
Received 03 September 2024; Accepted 06 November 2024; Issue published 30 December 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Objectives: Duchenne muscular dystrophy (DMD) is characterized by the development of cardiac pathology secondarily expressed in mitochondrial dysfunction. DMD treatment includes support for mitochondrial function. The purpose of this work was to evaluate the effects of the lipophilic cation MKT-077, capable of modulating mitochondrial activity, on the structure and function of cardiac mitochondria in mdx and wild-type mice, as well as the state of this organ. Methods: Animals were divided into 4 groups: wild type (WT), WT + 5 mg/kg MKT-077, mdx, mdx + 5 mg/kg MKT-077. MKT-077 was administered intraperitoneally daily for 28 days. Finally, we assessed the parameters of the functioning of the cardiac mitochondria of mice, the expression of genes encoding proteins involved in mitochondrial communication, as well as the histology and ultrastructure of the myocardium and heart rate. Results: MKT-077 was shown to reverse mitochondrial hyperfunctionalization in mdx mice, reducing respiratory parameters to WT levels. MKT-077 also inhibited mitochondrial respiration in the hearts of WT mice. MKT-077 administration was accompanied by a tendency to normalize calcium retention capacity in the cardiac mitochondria of mdx mice. Myocardial ultrastructure and gene signature of mdx + MKT077 animals demonstrated increased mitophagy. We noted changes in the sarcoplasmic reticulum (SR)/mitochondria contacts, accompanied by differential changes in the level of genes encoding proteins involved in the communication of these organelles. This effect of MKT-077 was accompanied by normalization of the relative heart weight of mdx mice, a decrease in the level of fibrosis, and a tendency toward normalization of heart rate. Conclusion: MKT-077-induced reversal of cardiac mitochondrial hyperfunctionalization in mdx mice may promote a healthy-like state of the myocardium in these animals.Keywords
Duchenne muscular dystrophy (DMD) is an inherited disorder caused by the loss of the dystrophin protein in striated muscles [1]. DMD is caused secondarily by dysfunction of the cell’s bioenergetic apparatus including mitochondria, which produce metabolic energy in the form of ATP, regulate ionic homeostasis, and perform a variety of other functions [2,3]. Mitochondrial dysfunction in skeletal muscle fibers is expressed in the early stages of DMD in the suppression of oxidative phosphorylation (OXPHOS), dysregulation of calcium homeostasis, and disruption of the ultrastructure of organelles [2,3]. Cardiac mitochondria also show similar pathological changes [4,5]. However, they are more typical for the late stages of the pathology, while the early DMD stages are characterized by signs of adaptation of cardiac mitochondria. In particular, mitochondria from cardiomyocytes of dystrophin-deficient mdx mice demonstrate an increase in the OXPHOS efficiency, improved calcium recycling, and an increase in the resistance of organelles to the pathological phenomenon of the mitochondrial permeability transition (MPT) pore [2,6]. All these changes are also accompanied by restructuring in protein systems that regulate the calcium transport in mitochondria [2]. It can be assumed that these phenomena in the early stages of pathology are capable of compensating for the dysfunction of the sarcoplasmic reticulum (SR) characteristic of DMD [7], as well as masking metabolic dysfunction, which has been suggested in several studies [8,9]. However, along with this, cardiac mitochondria of mdx mice exhibit changes in ultrastructure, as well as an increase in reactive oxygen species (ROS) production, leading to oxidative stress conditions [2,6]. Moreover, skeletal muscles of mdx mice show suppression of mitochondrial K+ homeostasis, which is due to the dysfunction of mitochondrial potassium channels, contributing to ROS overproduction and stress conditions [10]. All these events contribute to the development of fibrosis, cardiac hypertrophy, and electrophysiological disorders [6,10].
Improved mitochondrial function results in the amelioration of dystrophin-deficient pathology in both skeletal muscles and the heart, as demonstrated in mdx mice. This can be achieved using modulators of mitochondrial membrane channels and MPT pore [6,10–12], mitochondrial biogenesis [13] mitophagy [14], and antioxidants [15,16].
We recently showed that one of the pathways that contributes to the amelioration of mitochondrial dysfunction in skeletal muscles of mdx mice is the modulation of mitochondrial calcium levels by administration of the rhodacyanine dye MKT-077 [17]. MKT-077 is thought to act by inhibiting the activity of glucose-regulated protein 75 (GRP75, also known as HSP70 or mortalin) which mediates calcium transport into the mitochondria and, according to recent data, is involved in the pathogenesis of DMD [18,19]. In addition, we have identified an inhibitory effect of MKT-077 on mitochondrial OXPHOS, which could be attributed to the agent’s known ability to accumulate in mitochondria and impair function [20–22]. Nevertheless, in our experiment, this approach improved skeletal muscle structure and function in model mdx mice [17]. However, nothing is known about the effect of MKT-077 on cardiac pathology in mdx mice, which would make it possible to assess the complex therapeutic potential of this agent. Therefore, the aim of this work is to study the effect of MKT-077 administration on the functioning of cardiac mitochondria in mdx mice, their ultrastructure, and the state of the heart of these animals. Similar experiments were conducted on wild-type control mice to assess possible side effects of MKT-077.
Male dystrophin-deficient C57BL/10ScSn-mdx mice (mdx) and wild-type C57BL10 mice (WT) were obtained from the Russian Academy of Sciences Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry («Bio-model», Pushchino, Russia). Animals were housed under a controlled temperature (22 ± 1°C) with a 12:12 h light-dark cycle (light on 07.00–19.00 h local time). Water and food were available ad libitum. The study with mice was in line with the European Convention for the Protection of Vertebrates used for experimental and other purposes (Strasbourg, 1986). All mouse experimentation was approved by the Mari State University Ethics Committee (protocol No. 01/2023 of 30.11.2023). The mice (WT = 22.7 ± 0.4 g and mdx = 23.7 ± 0.4 g) were randomly divided into the following groups: (1) vehicle-treated WT mice (n = 10); (2) MKT-077-treated WT mice (WT + MKT077, n = 8); (3) vehicle-treated mdx mice (n = 10); (4) MKT-077-treated mdx mice (mdx + MKT077, n = 10). The treatment of animals began at 8 weeks of age. Fig. 1 shows the experimental design. MKT-077 (1-Ethyl-2-[[3-ethyl-5-(3-methyl-2(3H)-benzothiazolylidene)-4-oxo-2-thiazolidinylidene]methyl]-pyridinium chloride, also known as FJ-776, Fig. 2) was from Chem Scene (CS-0003758, Monmouth Junction, South Brunswick Township, NJ, USA). A solution of MKT-077 in sterile saline (or sterile saline alone in the case of control animals) was administered daily for 4 weeks at a dose of 5 mg/kg body weight, which corresponded to 150–200 μL of solution.

Figure 1: The experimental protocol for the study on WT and mdx mice (with signs of cardiomyopathy). IP: intraperitoneal; ECG: electrocardiography; TEM: transmission electron microscopy.
Figure 2: Structure of MKT-077.
Combined anesthesia was used to immobilize mice [23]. The animal was kept at rest until it lost the righting reflex. ECG recording was performed in standard lead II for 10 min using a pulse monitor based on an ECG sensor (BiTronics LAB, Moscow, Russia). ECG recording was initiated after confirmation of the depth of surgical anesthesia by a negative tail pinch reflex test [24].
Transmission electron microscopy
Left ventricular pieces (three samples per group from different mice) were taken for electron microscopic examination and processed as described in detail previously [6,10]. 60–70 nm thick ultrathin sections were prepared by use of a Leica EM UC6 ultramicrotome (Leica, Wetzlar, Germany). Samples stained with uranyl acetate and lead citrate were examined using a JEM-1400 electron microscope (JEOL, Tokyo, Japan). The obtained images were analyzed using ImageJ 1.53c (Wayne Rasband, National Institutes of Health, Bethesda, Rockville, MD, USA). Morphometric analysis of mitochondria-associated membranes (MAM contacts) consisted of manual contouring of mitochondria along their outer membrane (cross-sections) and SR membranes in contact with mitochondria within 30 nm [25]. The results are presented as total MAM contact length/mitochondrial perimeter (in %). For morphometry, 25 images per animal were analyzed. The average values obtained from each animal were used for statistical analysis.
Heart tissue samples (four samples per group from different mice) were taken for histological analysis and processed as described in detail previously [6]. 5 μm thick sections (at least 15 samples per heart) were prepared using a Minux S710 rotary microtome (RWD, Shenzhen, China). The preparations were stained with Sirius red (GC307014, Servicebio, Wuhan, China) to assess the level of fibrosis. EVOS M5000 imaging system (Thermo Fisher Scientific, Waltham, MA, USA) and ImageJ version 1.53 (National Institutes of Health, Bethesda, MD, USA) were used for visualization and analysis of the preparations. The connective tissue area (fibrosis) was expressed as a percentage of the Sirius red-stained area to the total myocardial area.
Quantitative real-time PCR (qRT-PCR)
Deep frozen heart samples (about 100 mg, six samples per group from different mice) were used to obtain total RNA. RNA was extracted using the ExtractRNA kit (#BC032, Evrogen, Moscow, Russia). The qPCRmix-HS SYBR mixture (PK147L, Eurogen, Moscow, Russia) and the DTLite5 amplifier (DNA-Technology LLC, Moscow, Russia) were used for qRT-PCR. Gene-specific primer selection and analysis were performed using Primer-BLAST [26]. Assessment of gene expression levels (normalized to the Rplp2 mRNA level), was carried out using comparative CT method [27]. The following primers were utilized: Hspa9 forward, 5′-GACAAGGATGCCCAAGGTTC-3′; Hspa9 reverse, 5′-GTAAAGACGCCTCCCAGAGT-3′; Itpr1 forward, 5′-GCAACTGCTGGAGGAGAATG-3′; Itpr1 reverse, 5′-GTTCAAGCTCCTGCTCTGTG-3′; Itpr2 forward, 5′-CAGTCGATTTGGACAGCCAG-3′; Itpr2 reverse, 5′-GGCCACGACATCCTGTAACT-3′; Itpr3 forward, 5′-CCCTCCAAAGATGAGTCGCT-3′; Itpr3 reverse, 5′-CAGACCACAGATGAAGCACG-3′; Parkin forward, 5′-AGCCAGAGGTCCAGCAGTTA-3′; Parkin reverse, 5′-GAGGGTTGCTTGTTTGCAGG-3′; Pink1 forward, 5′-TTGCCCCACACCCTAACATC-3′; Pink1 reverse, 5′-GCAGGGTACAGGGGTAGTTCT-3′; Vdac1 forward, 5′-CTCGCCGAACACTGGGAAA-3′; Vdac1 reverse, 5′-CCCAGCGATGTCAAAGTCCA-3′; Rplp2 forward, 5′-CGGCTCAACAAGGTCATCAGTGA-3′; Rplp2 reverse, 5′-AGCAGAAACAGCCACAGCCCCAC-3′.
Isolation of cardiac mitochondria and functional assessment of organelles
Mitochondria were isolated from the total mouse heart using differential centrifugation [10]. The Bradford assay revealed on average 15–20 mg mitochondrial protein/mL in the resulting suspension. Mitochondrial O2 consumption was recorded using an Oxygraph Plus system (Hansatech Instruments, King’s Lynn, UK) in a buffer consisting of 120 mM KCl, 2.5 mM K-malate + 2.5 mM K-glutamate as substrates, 5 mM NaH2PO4 and 10 mM HEPES-KOH (pH 7.4). Each experiment consisted of a sequential addition of 0.1 mg mitochondrial protein/mL, 100 μM ADP, and 50 μM 2,4-dinitrophenol (2,4-DNP). Mitochondrial O2 consumption was expressed as nmol O2/min/mg protein. The respiratory control ratio (RCR = State 3/State 4) was used as a quantitative indicator of mitochondrial quality. The ability of mitochondria to absorb and retain Ca2+ was assessed using the arsenazo III indicator and a Multiscan Go spectrometer (Thermo Fisher Scientific) in a buffer consisting of 210 mM mannitol, 70 mM sucrose, 10 μM EGTA, 50 μM arsenazo III, 1 mM KH2PO4, 2.5 mM K-malate + 2.5 mM K-glutamate as substrates and 10 mM HEPES-KOH (pH 7.4) [10]. Each experiment consisted of a sequential addition of 5 μM calcium chloride (Са2+) pulses to buffer supplemented with mitochondria (0.25 mg protein/mL). Са2+ was added until spontaneous release from the mitochondria, reflecting MPT pore induction.
Statistical analyses were performed in GraphPad Prism 8.0.1 (GraphPad Software Inc., San Diego, CA, USA). Tests for significance among groups were performed using a 1-way analysis of variance with post hoc Tukey correction. Significance was set at p < 0.05.
Effect of MKT-077 on mitochondrial function and ultrastructure
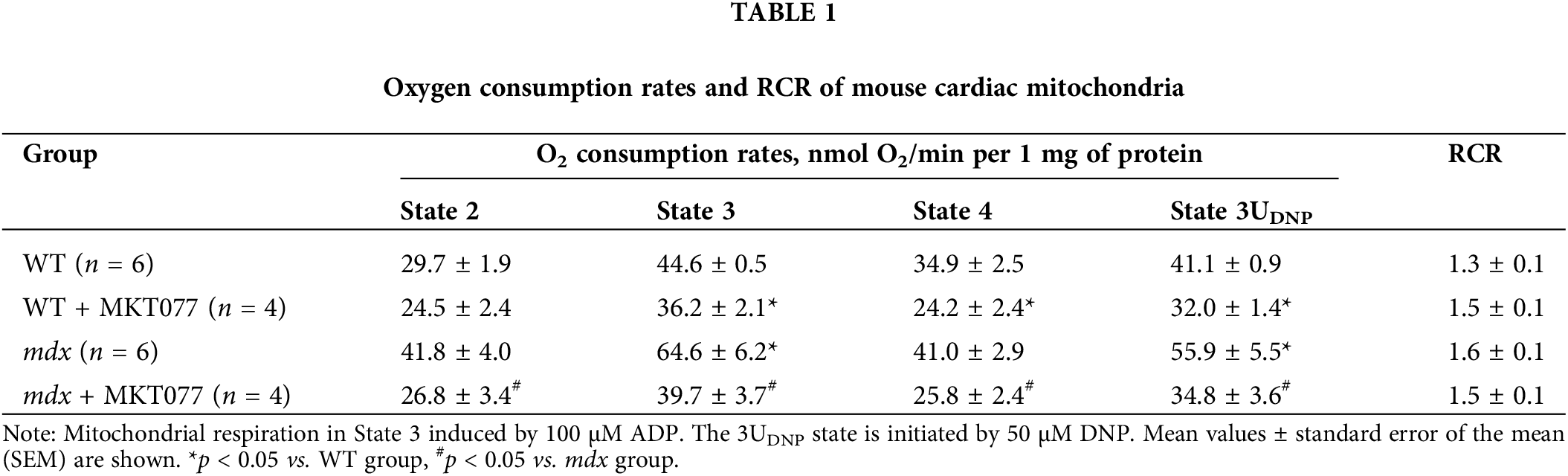
MKT-077 is known to have direct or indirect (via GRP75 modulation) effects on mitochondrial function [20,28,29]. In this work we assessed the effects of daily intraperitoneal administration of 5 mg/kg MKT-077 for 4 weeks on cardiac mitochondrial function in mdx mice and healthy WT animals (Fig. 1). We have previously shown that cardiac mitochondria from mdx mice up to 12 weeks of age are characterized by an increase in the OXPHOS intensity [6,10]. Indeed, Table 1 shows that cardiac mitochondria of mdx mice exhibit an increase in State 3 respiration rate (phosphorylating state), as well as in the State 3UDNP (with 2,4-DNP uncoupler), which allows one to estimate the maximum rate of electron transport along the mitochondrial electron transport chain. These events are also accompanied by a tendency to increase the RCR, reflecting OXPHOS coupling efficiency and the quality of organelles. MKT-077 significantly reduces the mitochondrial respiration rates in mdx mice in all functional states. Moreover, MKT-077-treated WT animals show a similar pattern. All this indicates that MKT-077 suppresses oxygen consumption by cardiac mitochondria. At the same time, it can be seen that despite the decrease in the rate of ADP-stimulated respiration in MKT-077-treated animals, the RCR does not change significantly, which is due to the corresponding reduction in State 4 respiration rate.
It is known that MKT-077 is able to modulate mitochondrial calcium homeostasis [28]. This has been demonstrated by our group in the skeletal muscle mitochondria of mdx mice and may be due to MKT-077-induced blockade of GRP75 activity [17]. Therefore, in the next part, we assessed the effect of the administration of this agent on the ability of the cardiac mitochondria of experimental animals to transport and accumulate Ca2+ in the matrix. Fig. 3 shows that mdx mitochondria can accumulate a greater amount of externally added Ca2+ compared to WT mitochondria, which is accompanied by a 1.5-fold increase in Ca2+ retention capacity (CRC) compared to WT mitochondria. This agrees with previous data [6,10] and indicates an increase in mdx cardiac mitochondria resistance to MPT pore opening, whose induction is involved in cell death [30]. MKT-077 does not affect the CRC of WT cardiac mitochondria, but in the case of MKT-077-treated mdx mice, we found a tendency toward a decrease in the ability to accumulate and retain Ca2+ in the matrix, which is also expressed in a tendency toward a decrease in the CRC parameter to the level of WT mice.
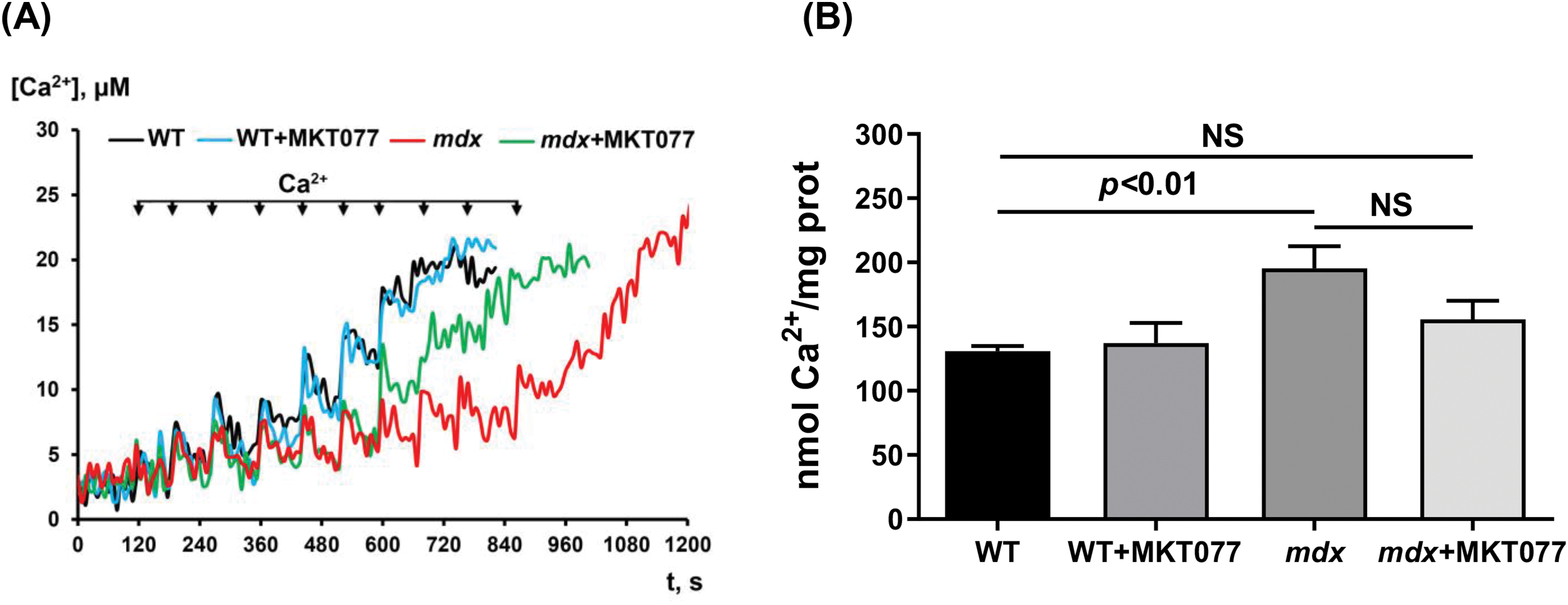
Figure 3: Effect of MKT-077 on mitochondrial calcium homeostasis. (А) Changes in [Ca2+] in the medium upon the pulse addition of 5 μM Ca2+ to cardiac mitochondria. (B) CRC of the cardiac mitochondria. Mean values ± SEM are shown (n = 4). NS is not significant.
We also assessed the effect of MKT-077 administration on the ultrastructure of cardiomyocytes, with a particular focus on the mitochondrial apparatus. Fig. 4 shows typical electron micrographs of cardiomyocytes from experimental animals. In the WT group, the contractile apparatus of cardiomyocytes is represented by parallel-oriented bundles of myofibrils. All components are clearly visible in the structure of sarcomeres. Myofibrils are usually located along the long axis of the cell. Mitochondria, located in regular rows along the myofibrils, have a round or ellipsoid shape in cross-section. The bulk of mitochondria have a clearly defined outer membrane and numerous parallel-oriented cristae. In certain areas of the cardiomyocyte, mitochondria are in a state of moderate swelling, their matrix becomes clearer and slightly increases in volume. Numerous lipid inclusions are found in the sarcoplasm of cardiomyocytes. The SR forms flattened cisterns. The WT + MKT077 group exhibits features in the morphological organization of cardiomyocytes that distinguish them from the control WT group. Myofibril bundles generally retain a parallel orientation and transverse striation. The cisterns of the SR expand and are filled with an electron-transparent substance. Small lipid inclusions are few in number. Mitochondrial changes are polymorphic. Some of them contain an unevenly contrasted matrix, others are electron-dense with numerous cristae. Mitochondria with numerous electron-dense granules with a diameter of 20–50 nm are found in some areas of cardiomyocytes. Altered mitochondria, with focal lysis of cristae, are quite frequently detected in the WT + MKT077 group. The perimeter of mitochondria in the heart of WT + MKT077 mice does not change, but their number is increased (Fig. 5).
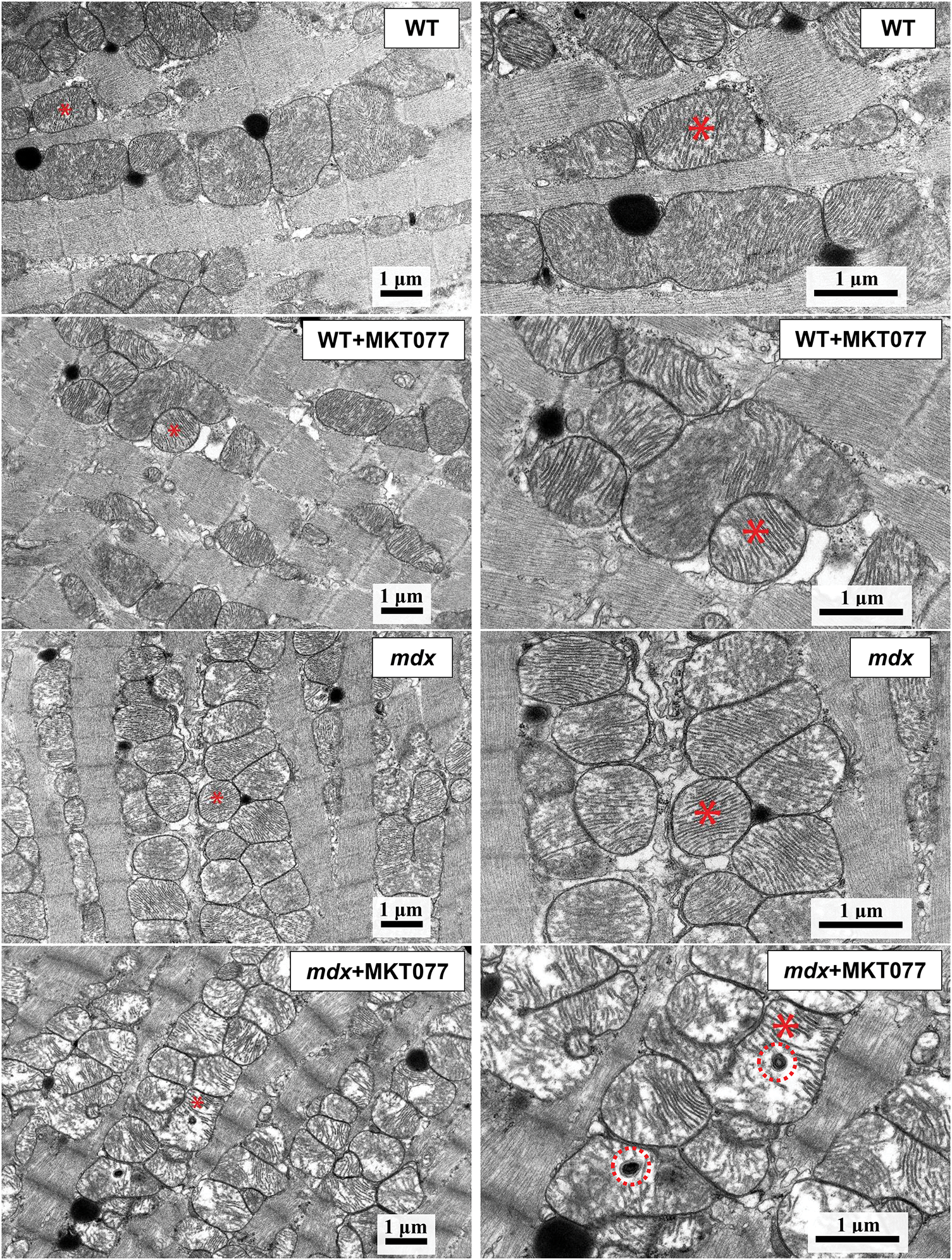
Figure 4: Images of mouse cardiomyocytes obtained by electron microscopy. The same mitochondria at low (left side) and high (right side) magnification are marked with an asterisk. Red circles in the lower right panel indicate micromitophagosomes.
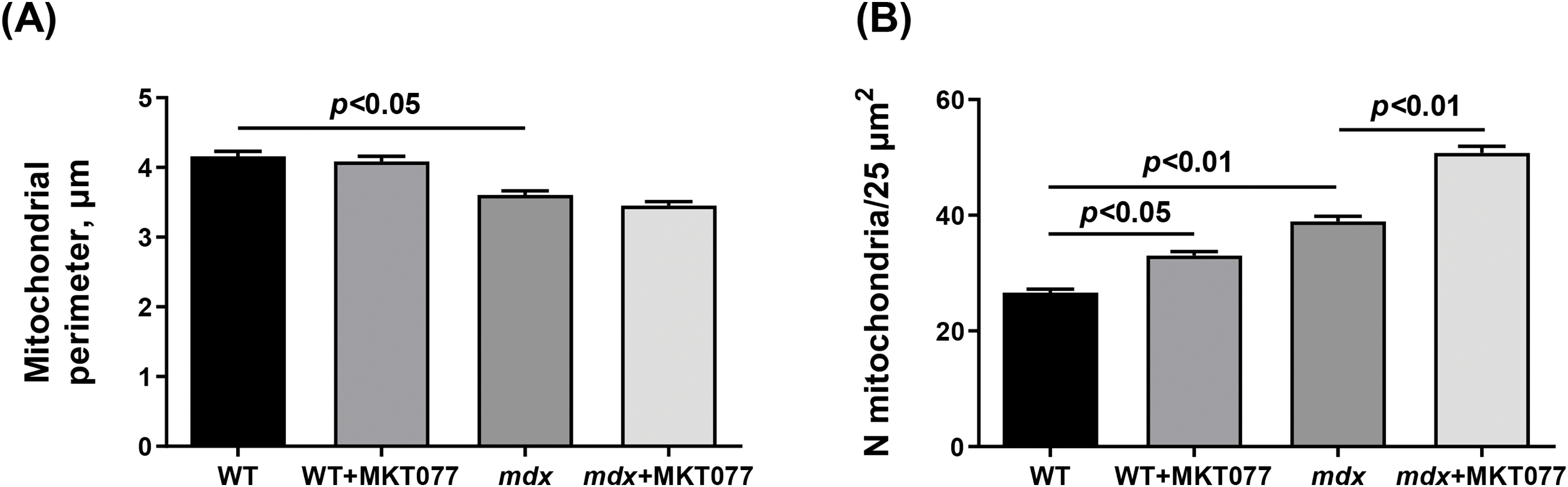
Figure 5: Electron micrograph (Fig. 4) profiles: mitochondrial perimeter (A) and number of mitochondria per plate (B). Mean values ± SEM are shown (n = 3).
Electron microscopic analysis revealed similar destructive changes in the mdx and mdx + MKT077 groups. They are usually local in nature and occur in individual areas of the cardiomyocyte, adjacent to unchanged areas (Fig. 4). The arrangement of myofibrils and mitochondria is less ordered. Myofibrils have a looser structure. Mitochondria are not arranged correctly along the myofibrils but are redistributed into clusters of varying sizes. The number of these organelles in the heart of mdx mice is increased compared to the WT group, with MKT-077 administration leading to a further significant increase in their number (Fig. 5B). The mitochondrial perimeter in the heart of mdx mice is reduced and MKT-077 has no effect on this parameter (Fig. 5A). There is a significant expansion of sarcoplasm between the myofibrils. The sarcoplasm shows proliferation and dilation of the SR cisterns and an increase in glycogen content. Mitochondria are at different stages of destructive changes from swelling, compaction of their internal space, deformation, and fragmentation of cristae to ruptures of the outer mitochondrial membrane. There are specific morphological features that distinguish the mdx + MKT077 mice from the mdx group. Micromitophagosomes containing fragments of cristae surrounded by multilayer membranes and at different stages of formation are observed inside and in close proximity to mitochondria. This may indicate increased mitophagy in the hearts of mdx mice treated with MKT-077. A similar pattern was previously observed in skeletal muscle mitochondria from MKT-077-treated mice and was accompanied by an increase in the Pink1 expression [17], which may indirectly reflect increased mitophagy in the skeletal muscles, and this phenomenon was most pronounced in WT mice. The same picture is observed in the heart. In particular, we note an increase in the Pink1 mRNA level, which is statistically significant in WT mice, whereas the mdx + MKT077 animals show only a tendency to increase in the Pink1 expression (Fig. 6A). It is interesting to note that the level of Parkin, encoding the E3 ubiquitin ligase recruited by Pink1 in the first steps of mitophagy, on the contrary, significantly decreases in both groups of animals (Fig. 6B). It should be noted here that multiple cellular systems are involved in the initiation and development of mitophagy [31], as well as autophagy in general [32], and they are significantly altered in DMD conditions [14], so further studies are required to deeply understand the effect of MKT-077 on this process in healthy and dystrophin-deficient cells.
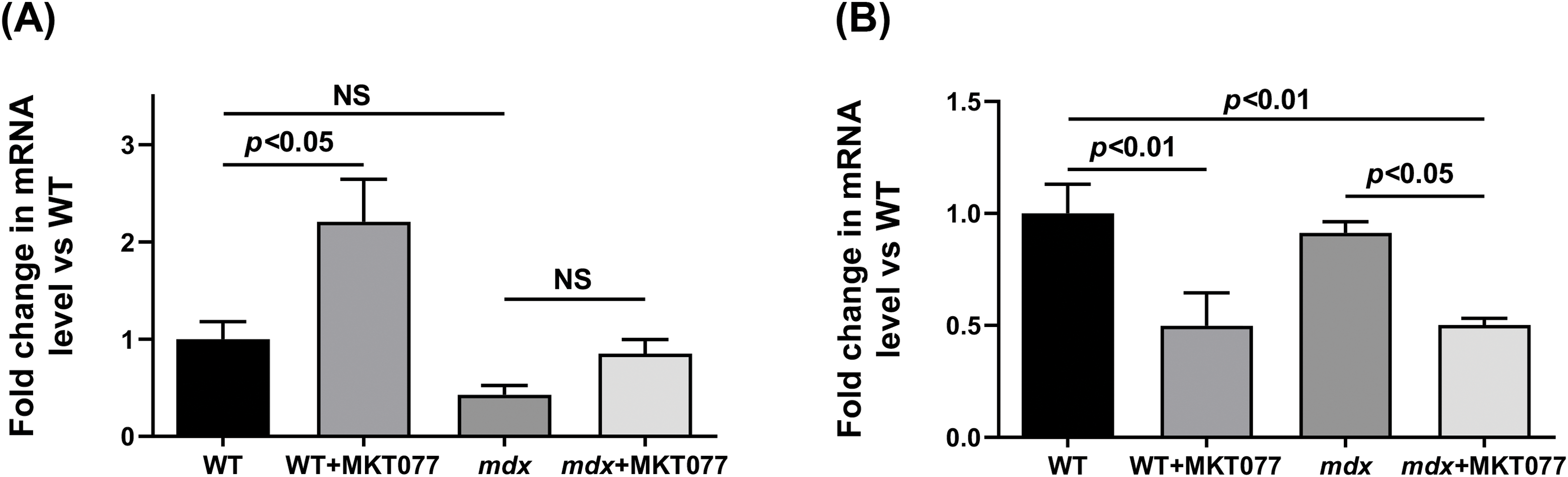
Figure 6: The relative expressions of Pink1 (A) and Parkin (B) in mouse hearts. Mean values ± SEM are shown (n = 6). NS is not significant.
Effect of MKT-077 on the area of MAM contacts and the expression level of genes encoding proteins associated with their formation
Regulation of the functional activity of mitochondria is ensured by the transport of calcium from the SR to the mitochondria through mitochondria-associated membranes (or MAM contacts) [33]. The main proteins that form MAM contacts are the voltage-dependent anion channel (VDAC) of the outer mitochondrial membrane, the inositol-1,4,5-triphosphate receptor (IP3R) of the sarcoplasmic reticulum, and the glucose-regulated protein 75 (GRP75) that binds them [33]. One should note that GRP75 directly regulating Ca2+ transfer from SR to mitochondria is one of the targets of MKT-077 in mammalian cells [28]. Therefore, in this work, we also assessed the possible effects of MKT-077 administration on changes in the area of MAM contacts and the expression level of genes encoding proteins involved in the formation of MAM contacts in the cardiomyocytes of experimental mice. Cardiomyocytes of mdx mice show significant proliferation and expansion of the SR cisterns that surround mitochondria (Fig. 4), which is accompanied by an increase in the MAM contact area (Fig. 7) and is consistent with known data [34]. MKT-077 treatment is accompanied by an increase in the area of MAM contacts in cardiomyocytes of WT mice, which corresponds to data obtained on the skeletal muscles of these animals [17]. At the same time, we note a tendency toward a decrease in the area of MAM contacts in the case of MKT077-treated mdx mice (p = 0.09 vs. mdx group). We assessed how morphological changes in MAM contacts relate to the expression of genes encoding proteins associated with their formation. One can see that the hearts of mdx mice show a reduced level of the HSPA9 gene expression encoding GRP75 (Fig. 8A). A similar pattern is observed for the Itpr2 gene encoding IP3R2 of SR (Fig. 8C), the predominant isoform found in the heart [35]. The expression of the Itpr1 and Itpr3 genes, encoding the IP3R1 and IP3R3 isoforms, respectively, does not change in the myocardium of mdx mice (Fig. 8B,D). On the other hand, we note an increase in the VDAC1 expression in mdx mice (Fig. 8E). One could assume that these changes may contribute to structural rearrangements in MAM contacts in cardiomyocytes of mdx mice, which were recently described in detail in the work of Angebault et al. [34].
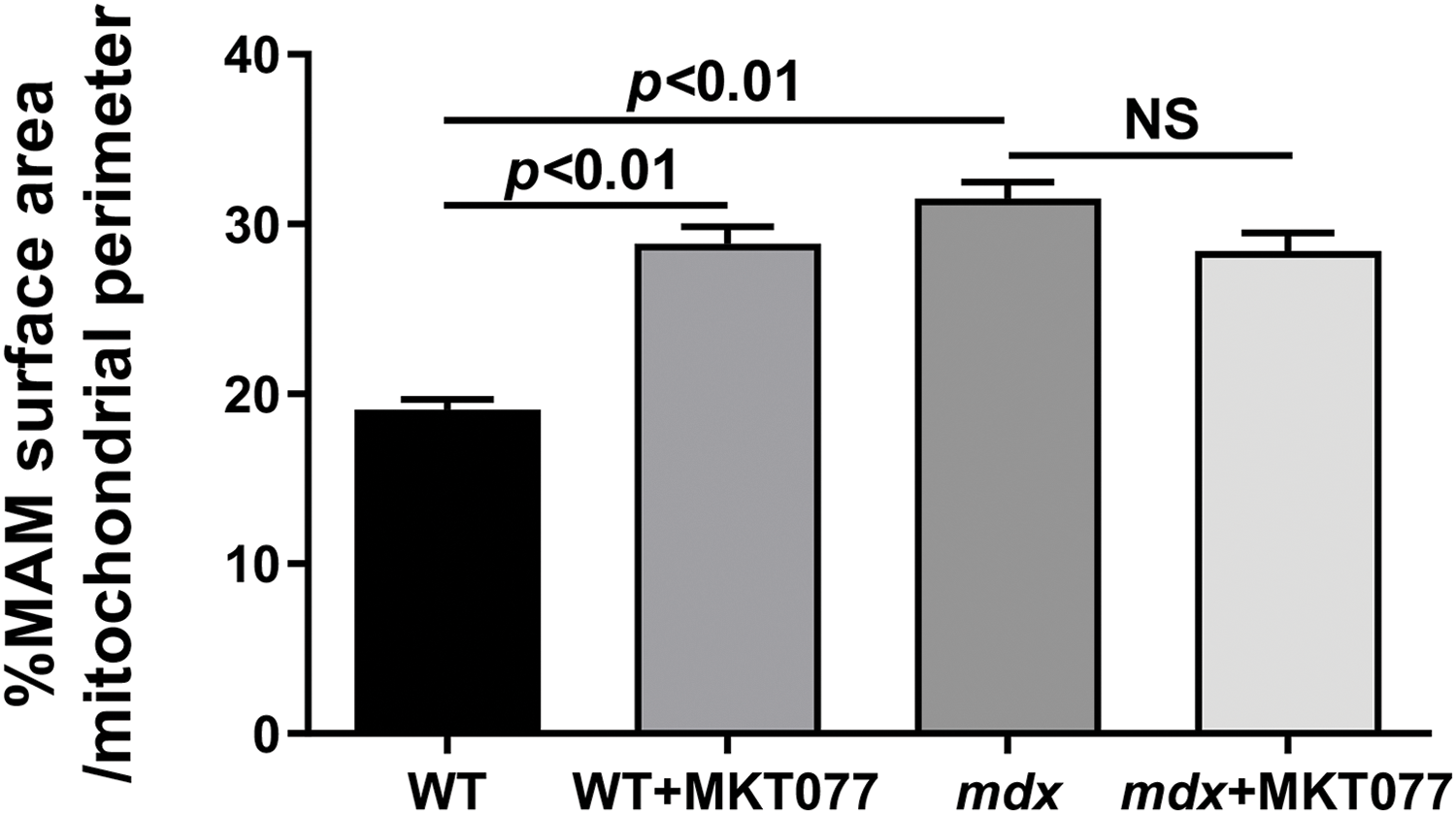
Figure 7: % of MAM surface area per mitochondrion perimeter in each microscopic field calculated from electron micrograph profiles (Fig. 4). Mean values ± SEM are shown (n = 3). NS is not significant.
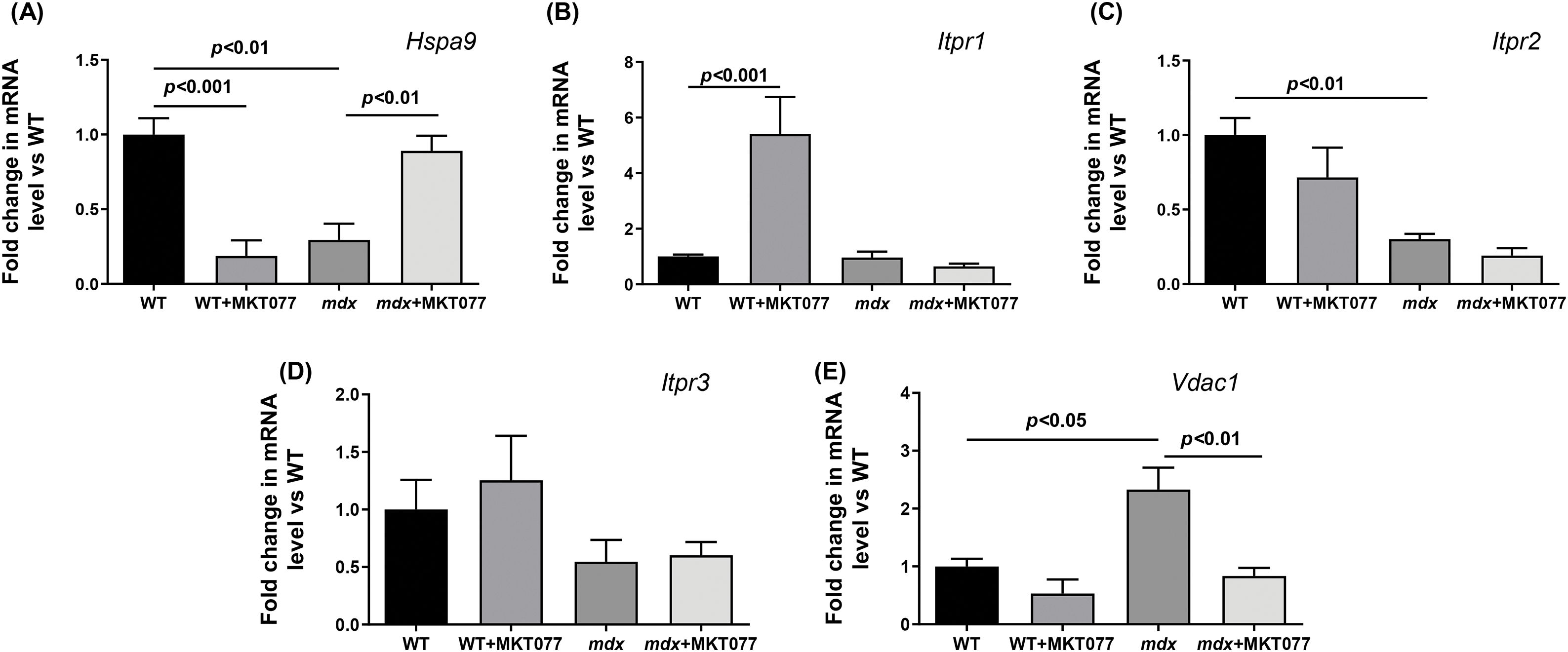
Figure 8: The relative expressions of Hspa9 (A), Itpr1 (B), Itpr2 (C), Itpr3 (D), and VDAC1 (E) in mouse hearts. Mean values ± SEM are shown (n = 6).
MKT-077 treatment does not affect the level of all three IP3R isoforms, but causes an increase in HSPA9 gene expression in the mdx hearts, and, on the contrary, decreases VDAC1 expression. In wild-type mice, MKT-077 does not affect the expression of IP3R2 and IP3R3, as well as VDAC1. At the same time, we note an increase in the expression of IP3R1 and, conversely, a decrease in the expression of the HSPA9 gene in the heart of WT + MKT077 mice. These results may indicate a differential effect of MKT-077 on the IP3R-GRP75-VDAC1 axis in the heart of experimental mice, contributing to morphological changes in MAM contacts. However, these data require confirmation at the protein level using Western blotting and more advanced techniques such as proximity ligation assay, which allows to assessment of the interaction of these and many other proteins in this region of the cell (according to the proteomic data, the MAM contact region contains up to 1000 proteins [36].
Effect of MKT-077 on histopathology and cardiac function in mdx mice
Finally, we assessed the effect of MKT-077 on cardiac health and function in mice. It is known that DMD (including mdx mice) is characterized by cardiac hypertrophy, as well as fibrosis leading to changes in the functioning of this organ [37]. One can see that mdx mice exhibit an increase in both absolute and relative heart weight (Fig. 9A,B). Histological images show that this is also accompanied by a slight increase in fibrous tissue deposition in the myocardium of mdx mice (Fig. 10). We note an increase in heart rate in mdx mice compared to the WT group, which agrees with known data (Fig. 11) [38]. MKT-077 reduces the relative heart weight of mdx mice, which is accompanied by alleviation of fibrosis and a tendency (p = 0.08 vs. mdx group) to normalize the heart rate of these animals.
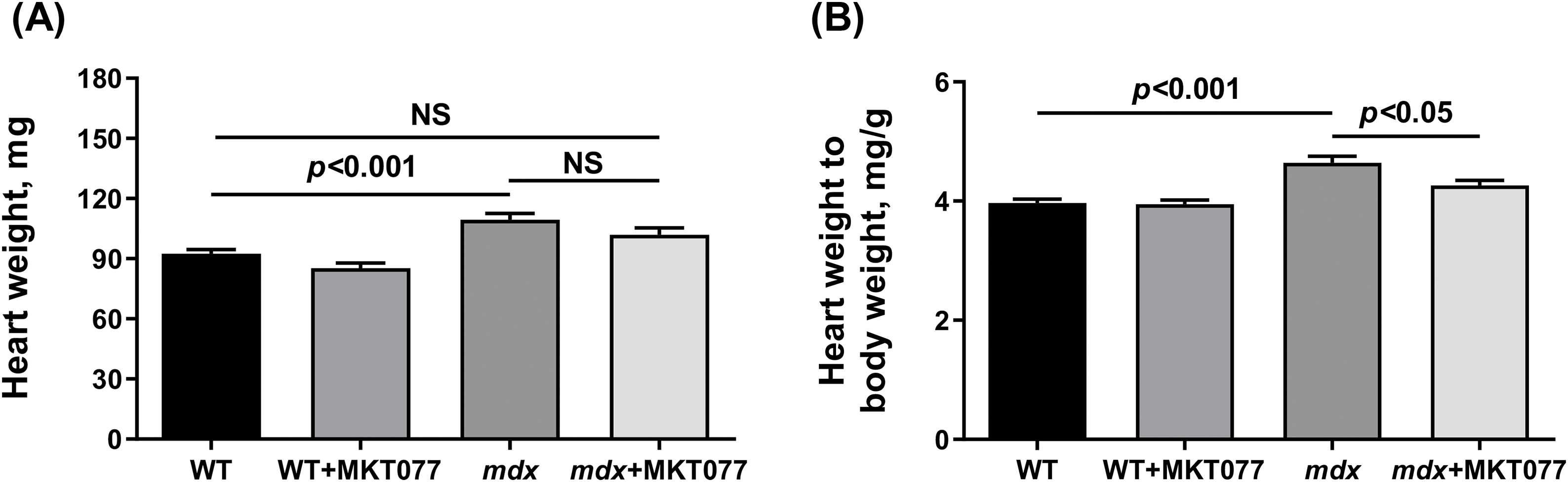
Figure 9: Absolute (A) and relative (B) weight of mouse hearts. Mean values ± SEM are shown (n = 8–10). NS is not significant.
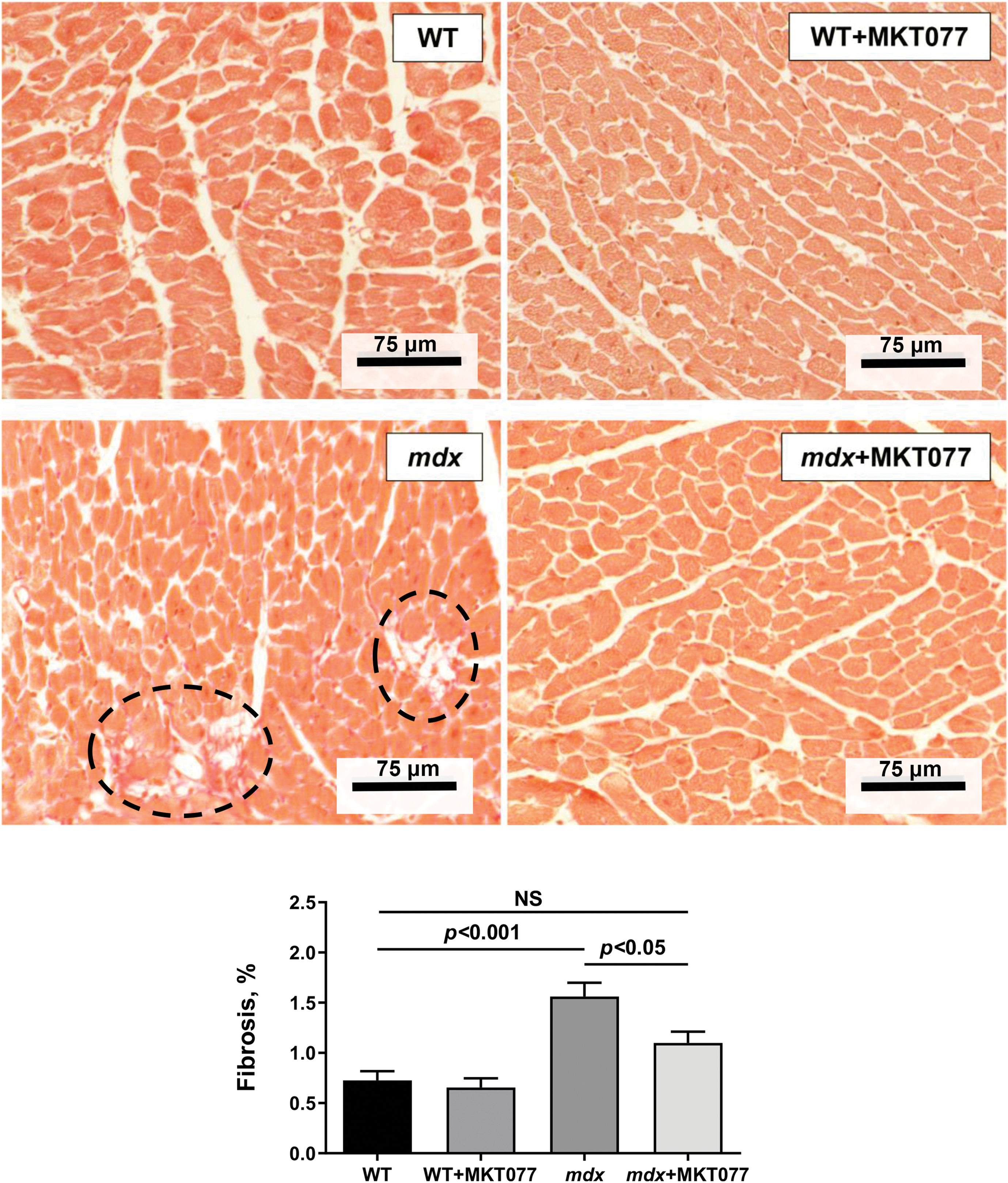
Figure 10: Typical histological images of heart tissue (sections of the left ventricle are shown) in experimental groups of mice. Areas of connective tissue are marked with black dotted lines. Mean values ± SEM are shown (n = 4). NS is not significant.
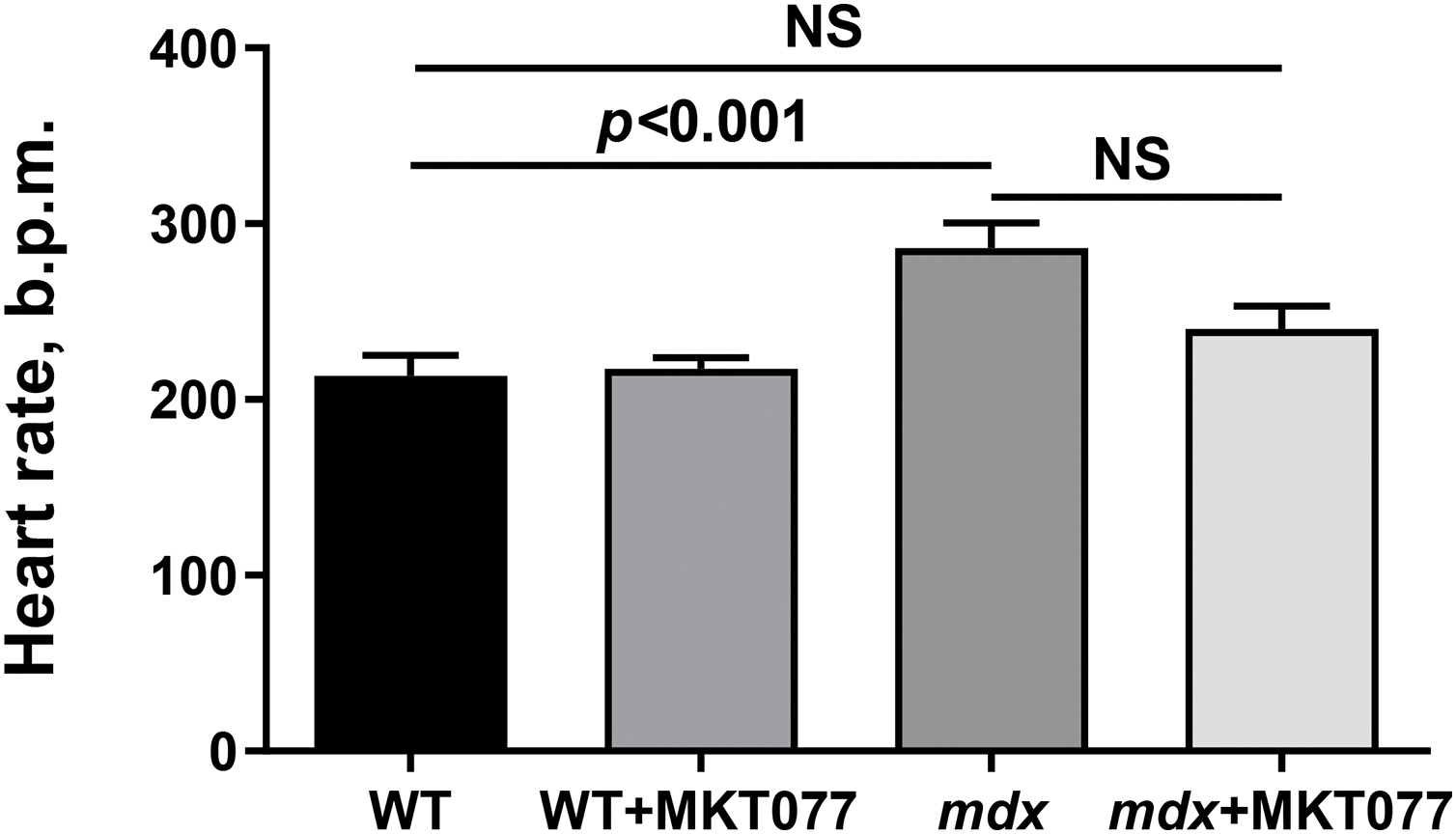
Figure 11: Effect of MKT-077 on the heart rate of mice. Mean values ± SEM are shown (n = 4). NS is not significant.
DMD is characterized by significant dysfunction of the bioenergetic apparatus of striated muscles, represented by mitochondria. This also applies to the cardiac muscle. Currently, supporting mitochondrial function is considered one of the strategies for the treatment of DMD, which has been confirmed both in animal models and in humans [2–5]. In particular, we have recently shown that rhodacyanine dye MKT-077 at a dose of 5 mg/kg body weight is able to alleviate mitochondrial dysfunction and skeletal muscle pathology in mdx mice by blocking the activity of GRP75 preventing mitochondrial calcium overload [17]. In this work, we assessed the effect of MKT-077 on mitochondrial function and cardiac pathology in mdx mice, as well as the state of the heart and cardiac mitochondria in healthy WT mice. It was found that the same dose and administration regimen of MKT-077 causes depression of cardiac mitochondrial respiration in virtually all functional states and this effect is detected in both WT and mdx mice (Table 1). This is largely consistent with previous data showing suppression of OXPHOS in mouse skeletal muscle mitochondria induced by MKT-077 administration [17]. One should note that the cardiac mitochondria of mdx mice, in contrast to skeletal muscle mitochondria, exhibit hyperfunctionalization, which is expressed in an increase in the OXPHOS intensity, and in this case MKT-077 reduces the respiratory parameters of cardiac mitochondria to the level of WT animals. Moreover, hyperfunctionalization of cardiac mitochondria in mdx mice is characterized by an increased ability to accumulate Ca2+, and MKT-077 treatment is accompanied by a tendency to normalize this parameter to the level of WT mice (Fig. 3). All this may indicate that the hearts of MKT-077-treated mdx mice demonstrate restoration of mitochondrial function to the level of healthy group. Here, it is important to note early data demonstrating suppression of mitochondrial respiration in the liver of rats treated with 15 mg MKT-077/kg body weight for 5 days [22]. However, this effect was observed only in rat liver mitochondria but not in kidney and cardiac mitochondria. Experiments on isolated liver mitochondria revealed a possible MKT-077-induced inhibition of the activity of mitochondrial respiratory chain complexes, which may result in suppression of OXPHOS [20]. One could assume that the decrease in OXPHOS efficiency in the mitochondria of the mouse heart that we identified is due to similar mechanisms and, in contrast to the previous work [22], is associated with a longer administration of MKT-077 to mice for 28 days (the summary dose of MKT-077 in our work is almost twice as high as in the work [22].
MKT-077 was initially introduced into biology and experimental medicine as a promising anti-cancer drug. In particular, in vitro experiments have shown that MKT-077 treatment of cancer cells leads to their death through suppression of mitochondrial respiration and disruption of mitochondrial ultrastructure [20,21]. It was later shown that MKT-077 administration also resulted in a decrease in mitochondrial DNA levels in rat hearts [22], which may indicate mitochondrial degradation and possibly mitophagy. Indeed, in this work, we also note changes in the ultrastructure of mitochondria in cardiomyocytes of both WT and mdx mice. In particular, the myocardium of MKT-077-treated mice shows the appearance of micromitophagosomes, which may reflect an increase in mitophagy (Fig. 4). In this case, we also note an increase in Pink1 mRNA levels in the hearts of MKT077-treated mice, which is statistically significant in WT animals and may indirectly indicate an increase in mitophagy (Fig. 6). This phenomenon, apparently also observed in the skeletal muscles of MKT-077-treated mice [17], may lead to the elimination of damaged organelles from the heart of mice, which are known to be efficient ROS producers [39] contributing to the development of oxidative stress, which is especially important in the case of mdx mice.
One of the specific targets of MKT-077 in mammalian cells is the GRP75 protein mediating communication between the SR and mitochondria via MAM contacts formed with the participation of this protein [28]. We have found that MKT-077 administration is accompanied by changes in the expression of genes encoding GRP75, as well as the IP3R isoforms of SR and mitochondrial VDAC1 that contact this protein in both the hearts of WT and mdx mice (Fig. 8). These events are accompanied by ultrastructural rearrangements in the myocardium, leading to changes in the area of MAM contacts (Fig. 7), which are consistent with our recent data obtained on skeletal muscles of MKT-077-treated mice [17]. In this case, we also presumably observe extensive adaptation of cardiomyocytes in WT mice, expressed in an increase in the area of MAM contacts, which can be considered as a response to MKT-077-induced disruption of SR-to-mitochondria Ca2+ transfer, and a similar picture is found in control mdx mice (Fig. 7) with chronic dysregulation of Ca2+ homeostasis. Moreover, considering the known involvement of MAM contacts in the regulation of mitochondrial function [33], one could assume that this action of MKT-077 contributes to the above-described changes in mitochondrial functioning and suppression of respiratory function.
The obtained results indicate that MKT-077 administration suppresses mitochondrial function in mice hearts. In the case of mdx mice, whose cardiac mitochondria exhibit hyperfunctionalization, we observe normalization of respiratory parameters to the level of WT animals and a tendency toward normalization of calcium homeostasis. This effect of MKT-077 is also accompanied by the elimination of cardiac hypertrophy in mdx mice, as evidenced by the normalization of the relative heart weight of the animals (Fig. 9B), as well as the level of fibrosis (Fig. 10). Moreover, the positive effect of MKT-077 is accompanied by an improvement in cardiac function, in particular, we note a tendency towards normalization of heart rate in treated mdx mice (Fig. 11). Thus, one could assume that MKT-077-induced elimination of mitochondrial hyperfunctionalization in the mdx heart may contribute to a healthy-like state of the myocardium of these animals. On the other hand, the duration of this positive effect is still unclear. Firstly, mitochondrial hyperfunctionalization in mdx mice hearts can be considered as an adaptive response of the organ, contributing to the maintenance of cardiac function and its elimination can potentially exacerbate the development of pathology and this requires further verification in a longer experiment, possibly on larger cohorts. Secondly, it has previously been shown that discontinuation of MKT-077 administration results in the elimination of the mitochondrial effects of this agent in rats [22], which is likely due to its susceptibility to rapid metabolism [40]. This suggests a continuous regimen of MKT-077 administration or the use of its more stable analogues.
Other important limitations of our study include the lack of detailed mechanistic explanations for how MKT-077 exerts its effects. The mitochondria-protective effect of MKT-077, as well as the possible targets of this agent, were first described earlier in vitro on nerve cell cultures [28,29], but to date, nothing is known about the effects of this agent on cultured cardiomyocytes and about the differences in the degree of MKT-077 accumulation in WT and mdx cardiomyocytes. Such experiments will allow us to describe in more detail the mechanism of action of MKT-077. Moreover, our studies focused on striated muscle and did not address the potential impact of MKT-077 on non-target tissue, which should also be assessed to identify undesirable effects in other tissues and organs.
The obtained data demonstrate changes in mitochondrial function and the nature of their interaction with SR, as well as the contribution of these secondary changes to cardiac pathology in mdx mice. The rhodacyanine derivative MKT-077 promotes a healthy-like state of the myocardium in mdx mice, apparently through modulation of mitochondrial function. These results, along with our recent data demonstrating mitochondrial dysfunction amelioration and slowing of skeletal muscle pathology development in MKT-077-treated mdx mice, provide evidence for the therapeutic effect of this compound and overall support the efficacy of a mitochondria-targeted approach for the treatment of secondary manifestations of DMD.
Acknowledgement: The authors acknowledge the Moscow State University Development Program (PNR 5.13) and the “Superresolution microscopy and spectroscopy” core facility at Belozersky Institute of Physico-Chemical Biology for granting access to their equipment.
Funding Statement: This work was supported by a grant from the Russian Science Foundation (23-75-10006).
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Mikhail V. Dubinin; data collection: Mikhail V. Dubinin, Irina B. Mikheeva, Anastasia E. Stepanova, Natalia V. Mikina, Daniil V. Sushentsov, Vyacheslav A. Sharapov, Alena A. Cherepanova, Valentin V. Loskutov; analysis and interpretation of results: Mikhail V. Dubinin, Konstantin N. Belosludtsev; draft manuscript preparation: Mikhail V. Dubinin. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Ethics Approval: The study with mice was in line with the European Convention for the Protection of Vertebrates used for experimental and other purposes (Strasbourg, 1986). The research protocol was approved by the Mari State University Ethics Committee (Protocol No. 01/2023 of 30.11.2023).
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Duan D, Goemans N, Takeda S, Mercuri E, Aartsma-Rus A. Duchenne muscular dystrophy. Nat Rev Dis Primers. 2021;7(1):13. doi:10.1038/s41572-021-00248-3. [Google Scholar] [PubMed] [CrossRef]
2. Dubinin MV, Belosludtsev KN. Ion channels of the sarcolemma and intracellular organelles in Duchenne muscular dystrophy: a role in the dysregulation of ion homeostasis and a possible target for therapy. Int J Mol Sci. 2023;24(3):2229. doi:10.3390/ijms24032229. [Google Scholar] [PubMed] [CrossRef]
3. Budzinska M, Zimna A, Kurpisz M. The role of mitochondria in Duchenne muscular dystrophy. J Physiol Pharmacol. 2021;72(2):157–66. [Google Scholar]
4. Gandhi S, Sweeney HL, Hart CC, Han R, Perry CGR. Cardiomyopathy in Duchenne muscular dystrophy and the potential for mitochondrial therapeutics to improve treatment response. Cells. 2024;13(14):1168. doi:10.3390/cells13141168. [Google Scholar] [PubMed] [CrossRef]
5. Ignatieva E, Smolina N, Kostareva A, Dmitrieva R. Skeletal muscle mitochondria dysfunction in genetic neuromuscular disorders with cardiac phenotype. Int J Mol Sci. 2021;22(14):7349. doi:10.3390/ijms22147349. [Google Scholar] [PubMed] [CrossRef]
6. Dubinin MV, Starinets VS, Talanov EY, Mikheeva IB, Belosludtseva NV, Serov DA, et al. Effect of the non-immunosuppressive MPT pore inhibitor alisporivir on the functioning of heart mitochondria in dystrophin-deficient mdx mice. Biomedicines. 2021;9(9):1232. doi:10.3390/biomedicines9091232. [Google Scholar] [PubMed] [CrossRef]
7. Valladares D, Utreras-Mendoza Y, Campos C, Morales C, Diaz-Vegas A, Contreras-Ferrat A, et al. IP3 receptor blockade restores autophagy and mitochondrial function in skeletal muscle fibers of dystrophic mice. Biochim Biophys Acta Mol Basis Dis. 2018;1864(11):3685–95. doi:10.1016/j.bbadis.2018.08.042. [Google Scholar] [PubMed] [CrossRef]
8. Mourkioti F, Kustan J, Kraft P, Day JW, Zhao MM, Kost-Alimova M, et al. Role of telomere dysfunction in cardiac failure in Duchenne muscular dystrophy. Nat Cell Biol. 2013;15(8):895–904. doi:10.1038/ncb2790. [Google Scholar] [PubMed] [CrossRef]
9. Chang AC, Ong SG, LaGory EL, Kraft PE, Giaccia AJ, Wu JC, et al. Telomere shortening and metabolic compromise underlie dystrophic cardiomyopathy. Proc Natl Acad Sci U S A. 2016;113(46):13120–5. doi:10.1073/pnas.1615340113. [Google Scholar] [PubMed] [CrossRef]
10. Dubinin MV, Starinets VS, Chelyadnikova YA, Belosludtseva NV, Mikheeva IB, Penkina DК, et al. Effect of large-conductance calcium-dependent K+ channel activator NS1619 on function of mitochondria in the heart of dystrophin-deficient mice. Biochemistry. 2023;88(2):189–201. doi:10.1134/S0006297923020037. [Google Scholar] [PubMed] [CrossRef]
11. Bellissimo CA, Delfinis LJ, Hughes MC, Turnbull PC, Gandhi S, DiBenedetto SN, et al. Mitochondrial creatine sensitivity is lost in the D2.mdx model of Duchenne muscular dystrophy and rescued by the mitochondrial-enhancing compound olesoxime. Am J Physiol Cell Physiol. 2023;324(5):1141–57. doi:10.1152/ajpcell.00377.2022. [Google Scholar] [PubMed] [CrossRef]
12. Stocco A, Smolina N, Sabatelli P, Šileikytė J, Artusi E, Mouly V, et al. Treatment with a triazole inhibitor of the mitochondrial permeability transition pore fully corrects the pathology of sapjezebrafish lacking dystrophin. Pharmacol Res. 2021;165:105421. doi:10.1016/j.phrs.2021.105421. [Google Scholar] [PubMed] [CrossRef]
13. Giovarelli M, Zecchini S, Catarinella G, Moscheni C, Sartori P, Barbieri C, et al. Givinostat as metabolic enhancer reverting mitochondrial biogenesis deficit in Duchenne muscular dystrophy. Pharmacol Res. 2021;170:105751. doi:10.1016/j.phrs.2021.105751. [Google Scholar] [PubMed] [CrossRef]
14. Reid AL, Alexander MS. The interplay of mitophagy and inflammation in Duchenne muscular dystrophy. Life. 2021;11(7):648. doi:10.3390/life11070648. [Google Scholar] [PubMed] [CrossRef]
15. Ren S, Yao C, Liu Y, Feng G, Dong X, Gao B, et al. Antioxidants for treatment of Duchenne muscular dystrophy: a systematic review and meta-analysis. Eur Neurol. 2022;85(5):377–88. doi:10.1159/000525045. [Google Scholar] [PubMed] [CrossRef]
16. Buyse GM, Van der Mieren G, Erb M, D’hooge J, Herijgers P, Verbeken E, et al. Long-term blinded placebo-controlled study of SNT-MC17/idebenone in the dystrophin deficient mdx mouse: cardiac protection and improved exercise performance. Eur Heart J. 2009;30(1):116–24. doi:10.1093/eurheartj/ehn406. [Google Scholar] [PubMed] [CrossRef]
17. Dubinin MV, Stepanova AE, Mikheeva IB, Igoshkina AD, Cherepanova AA, Talanov EY, et al. Reduction of mitochondrial calcium overload via MKT077-induced inhibition of glucose-regulated protein 75 alleviates skeletal muscle pathology in dystrophin-deficient mdx mice. Int J Mol Sci. 2024;25(18):9892. doi:10.3390/ijms25189892. [Google Scholar] [PubMed] [CrossRef]
18. Cosemans G, Merckx C, De Bleecker JL, De Paepe B. Inducible heat shock protein 70 levels in patients and the mdx mouse affirm regulation during skeletal muscle regeneration in muscular dystrophy. Front Biosci (Schol Ed). 2022;4(3):19. doi:10.31083/j.fbs1403019. [Google Scholar] [PubMed] [CrossRef]
19. Lim JH, Bang MS. The effect of steroid on heat shock protein 70 expression in mdx mice. J Korean Acad Rehabil Med. 2009;33(1):1–4. [Google Scholar]
20. Modica-Napolitano JS, Koya K, Weisberg E, Brunelli BT, Li Y, Chen LB. Selective damage to carcinoma mitochondria by the rhodacyanine MKT-077. Cancer Res. 1996;56(3):544–50. [Google Scholar] [PubMed]
21. Koya K, Li Y, Wang H, Ukai T, Tatsuta N, Kawakami M, et al. MKT-077, a novel rhodacyanine dye in clinical trials, exhibits anticarcinoma activity in preclinical studies based on selective mitochondrial accumulation. Cancer Res. 1996;56(3):538–43. [Google Scholar] [PubMed]
22. Weisberg EL, Koya K, Modica-Napolitano J, Li Y, Chen LB. In vivo administration of MKT-077 causes partial yet reversible impairment of mitochondrial function. Cancer Res. 1996;56(3):551–5. [Google Scholar] [PubMed]
23. Astashev ME, Serov DA, Tankanag AV. Anesthesia effects on the low frequency blood flow oscillations in mouse skin. Skin Res Technol. 2019;25(1):40–6. doi:10.1111/srt.2019.25.issue-1. [Google Scholar] [CrossRef]
24. Tsukamoto A, Serizawa K, Sato R, Yamazaki J, Inomata T. Vital signs monitoring during injectable and inhalant anesthesia in mice. Exp Anim. 2015;64(1):57–64. doi:10.1538/expanim.14-0050. [Google Scholar] [PubMed] [CrossRef]
25. Thoudam T, Ha CM, Leem J, Chanda D, Park JS, Kim HJ, et al. Pdk4 augments ER-mitochondria contact to dampen skeletal muscle insulin signaling during obesity. Diabetes. 2019;68:571–86. doi:10.2337/db18-0363. [Google Scholar] [PubMed] [CrossRef]
26. Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden T. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012;13:134. doi:10.1186/1471-2105-13-134. [Google Scholar] [PubMed] [CrossRef]
27. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protoc. 2008;3:1101–8. doi:10.1038/nprot.2008.73. [Google Scholar] [PubMed] [CrossRef]
28. Wen B, Xu K, Huang R, Jiang T, Wang J, Chen J, et al. Preserving mitochondrial function by inhibiting GRP75 ameliorates neuron injury under ischemic stroke. Mol Med Rep. 2022;25(5):165. doi:10.3892/mmr. [Google Scholar] [CrossRef]
29. Liang T, Hang W, Chen J, Wu Y, Wen B, Xu K, et al. ApoE4 (Δ272–299) induces mitochondrial-associated membrane formation and mitochondrial impairment by enhancing GRP75-modulated mitochondrial calcium overload in neuron. Cell Biosci. 2021;11(1):50. doi:10.1186/s13578-021-00563-y. [Google Scholar] [PubMed] [CrossRef]
30. Baev AY, Vinokurov AY, Potapova EV, Dunaev AV, Angelova PR, Abramov AY. Mitochondrial permeability transition, cell death and neurodegeneration. Cells. 2024;13(7):648. doi:10.3390/cells13070648. [Google Scholar] [PubMed] [CrossRef]
31. Mao W, Zong G, Gao Y, Qu S, Cheng X. Integrative analyses of mitophagy-related genes and mechanisms associated with type 2 diabetes in muscle tissue. Curr Issues Mol Biol. 2024;46(9):10411–29. doi:10.3390/cimb46090619. [Google Scholar] [PubMed] [CrossRef]
32. Yamamoto H, Zhang S, Mizushima N. Autophagy genes in biology and disease. Nat Rev Genet. 2023;24(6):382–400. doi:10.1038/s41576-022-00562-w. [Google Scholar] [PubMed] [CrossRef]
33. Barazzuol L, Giamogante F, Calì T. Mitochondria associated membranes (MAMsarchitecture and physiopathological role. Cell Calcium. 2021;94:102343. doi:10.1016/j.ceca.2020.102343. [Google Scholar] [PubMed] [CrossRef]
34. Angebault C, Panel M, Lacôte M, Rieusset J, Lacampagne A, Fauconnier J. Metformin reverses the enhanced myocardial SR/ER-mitochondria interaction and impaired complex I-driven respiration in dystrophin-deficient mice. Front Cell Dev Biol. 2021;8:609493. doi:10.3389/fcell.2020.609493. [Google Scholar] [PubMed] [CrossRef]
35. Garcia MI, Boehning D. Cardiac inositol 1,4,5-trisphosphate receptors. Biochim Biophys Acta Mol Cell Res. 2017;1864(6):907–14. doi:10.1016/j.bbamcr.2016.11.017. [Google Scholar] [PubMed] [CrossRef]
36. Poston CN, Krishnan SC, Bazemore-Walker CR. In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J Proteom. 2013;79:219–30. doi:10.1016/j.jprot.2012.12.018. [Google Scholar] [PubMed] [CrossRef]
37. Statile CJ, Taylor MD, Mazur W, Cripe LH, King E, Pratt J, et al. Left ventricular noncompaction in Duchenne muscular dystrophy. J Cardiovasc Magn Reson. 2013;15(1):67. doi:10.1186/1532-429X-15-67. [Google Scholar] [PubMed] [CrossRef]
38. Chu V, Otero JM, Lopez O, Sullivan MF, Morgan JP, Amende I, et al. Electrocardiographic findings in mdx mice: a cardiac phenotype of Duchenne muscular dystrophy. Muscle Nerve. 2002;26(4):513–9. doi:10.1002/mus.v26:4. [Google Scholar] [CrossRef]
39. Casati SR, Cervia D, Roux-Biejat P, Moscheni C, Perrotta C, De Palma C. Mitochondria and reactive oxygen species: the therapeutic balance of powers for Duchenne muscular dystrophy. Cells. 2024;13(7):574. doi:10.3390/cells13070574. [Google Scholar] [PubMed] [CrossRef]
40. Tatsuta N, Suzuki N, Mochizuki T, Koya K, Kawakami M, Shishido T, et al. Pharmacokinetic analysis and antitumor efficacy of MKT-077, a novel antitumor agent. Cancer Chemother Pharmacol. 1999;43(4):295–301. doi:10.1007/s002800050898. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools