
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW
Targeting cell cycle regulators: A new paradigm in cancer therapeutics
School of Biotechnology, Institute of Science, Banaras Hindu University, Varanasi, UP-221005, India
* Corresponding Author: SAMARENDRA KUMAR SINGH. Email:
# These authors contributed equally
BIOCELL 2024, 48(12), 1639-1666. https://doi.org/10.32604/biocell.2024.056503
Received 24 July 2024; Accepted 30 October 2024; Issue published 30 December 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Dysregulation of the cell cycle is a molecular hallmark of cancer, which leads to uncontrolled proliferation and self-renewal of neoplastic cells. To maintain this phenotype, cells acquire multiple molecular alterations and bypass several cellular checkpoints that are involved in the prevention of genomic instability and uncontrolled cell proliferation. Therefore, targeting cell cycle regulators could prove to be a promising anti-cancer approach. Recent advancements in the understanding of cancer cell susceptibilities have revealed a therapeutic opportunity to selectively target the cell cycle in malignant cells. This review highlights major cell cycle dysregulation in cancerous cells and the latest developments on anti-cancer agents that target cell cycle factors which are either in clinical practice or in clinical trials at present. In light of increasing drug resistance in cancer patients, the article also discusses mechanistic insights for the exploration of probable therapies that could be used to target these cell cycle factors. Additionally, it outlines the future research necessary to develop new therapies and optimize existing ones to achieve maximum efficacy. Thus, this article aims to provide a comprehensive understanding of the advancements in cancer therapies targeting dysregulated cell cycle factors.Graphic Abstract
Keywords
Supplementary Material
Supplementary Material FileAbbreviation
| Ack1 | Activated Cdc42-associated kinase 1 |
| AKT | Ak strain transforming |
| ALL | Acute lymphoblastic leukemia |
| AML | Acute myeloid leukemia |
| APC | Anaphase promoting complex |
| AR | Androgen receptor |
| AS1 | Antisense RNA 1 |
| ATM | Ataxia Telangiectasia Mutated |
| ATR | Ataxia telangiectasia and Rad3-related protein |
| BCL-2 | B-cell lymphoma 2 |
| BCR | BTB-Cul-3-Rbx1 |
| BCR-ABL | Breakpoint cluster-Abelson tyrosine kinase |
| BRCA | Breast cancer gene |
| BUB1B/BUBR1 | Budding inhibited by benzimidazole-related 1 |
| CAMTA1 | Calmodulin binding transcription activator 1 |
| CBX4 | Chromobox protein homolog 4 |
| CCAT1 | Colon cancer-associated transcript 1 |
| CCNA | Cyclin A gene |
| CD133 | Prominin-1 |
| CD44 | Phagocytic-glycoprotein 1 |
| CDC | Cell division cycle |
| CDH1 | Cdc20 homolog 1 |
| CDKi | CDK inhibitors |
| CDKs | Cyclin dependent kinases |
| Cdt1 | Cdc 10 dependent transcript 1 |
| CENP-A | Centromere protein A |
| CeRNAs | Competing endogenous RNAs |
| CHK | Checkpoint kinase |
| CHL-1 | Cell adhesion molecule L1 |
| CIP | CDK interacting protein |
| circRNAs | Circular RNAs |
| CKI | Cyclin-dependent kinase inhibitors |
| c-Myc | cellular-Myelocytomatosis gene |
| COX-2 | Cyclooxygenase-2 |
| CRC | Colorectal cancer |
| CRL4 | Cullin-RING ubiquitin ligase complex 4 |
| CRPC | Castration-resistant prostate cancer |
| CUX1 | Cut like homeobox 1 |
| DBC1 | Deleted in breast cancer 1 |
| DBF4 | Dumbbell former 4 |
| DDR | DNA Damage Repair |
| DNA | Deoxy ribonucleic acid |
| DNMT3A | DNA methyltransferase-3-alpha |
| DSB | Double Strand Break |
| DTL | Denticleless E3 ubiquitin protein ligase homolog |
| DUB | Deubiquitinase |
| DUPA | 2-[3-(1,3-dicarboxy propyl) ureido] pentanedioic acid |
| E2F | Eukaryotic transcription factor |
| EF3 | Elongation factor 3 |
| EMI1 | Early mitotic inhibitor 1 |
| EMS | E2F1 mRNA-stabilizing factor |
| ER | Estrogen receptor |
| ERBB2 | v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2 |
| ERK | Extracellular signal-regulated kinase |
| ETS1 | ETS proto-oncogene 1, transcription factor |
| EZH2 | Enhancer of Zeste Homolog 2 |
| FBW7 | F-box and WD repeat domain-containing 7 |
| FDA | Food and Drug Administration |
| FOXC1 | Forkhead box protein C1 |
| G1 | Gap 1 |
| G2 | Gap 2 |
| GAS5 | Growth arrest-specific 5 |
| GINS | Go, Ichi, Nii, and San; five, one, two, and three in Japanese |
| GSK3β | Glycogen synthase kinase 3β |
| Gαi1 | Guanine nucleotide-binding protein G(i), alpha-1 subunit |
| HDAC | Histone deacetylase complex |
| HER2 | Human epidermal growth factor receptor 2 |
| HGSOC | High grade serous ovarian cancer |
| HL | Hodgkin’s lymphoma |
| HMGA2 | High mobility group AT-hook 2 |
| HMGB1 | High mobility group box 1 |
| Hog1 | High-osmolarity glycerol 1 |
| HPV | Human papilloma virus |
| HR | Hormone receptor |
| HSP90 | Heat shock protein 90 |
| INK4 | Inhibitor of CDK4/6 |
| KIP | Kinase inhibitory protein |
| KRAS | Kirsten rat sarcoma viral oncogene homolog |
| LATS2 | Large tumor suppressor kinase 2 |
| LKB1 | Liver kinase B1 |
| LncRNA | Long non-coding RNA |
| LSCC | Laryngeal squamous cell carcinoma |
| LUCAT1 | Lung cancer-related transcript 1 |
| MAD | Mitotic arrest-deficient |
| MAPK | Mitogen activated protein kinases |
| MCC | Mitotic checkpoint complex |
| MCL-1 | Myeloid cell leukemia-1 |
| MCM | Minichromosome maintenance complex |
| MDM2 | Mouse double minute 2 homolog |
| MET | Mesenchymal Epithelial Transition |
| MGMT | O6-methylguanine-DNA methyltransferase |
| miRNA/miR | MicroRNA |
| MM | Multiple myeloma |
| MMP9 | Matrix metalloproteinase-9 |
| M-phase | Mitotic phase |
| MPS1 | Monopolar spindle 1 |
| MRE | miRNA response element |
| MRN | Mre11, Rad50 and Nbs1 complex |
| mRNA | Messenger RNA |
| MSP1 | Myeloma-derived stromal cell line |
| MTA2 | Nucleosome remodeling and histone deacetylase (NuRD) |
| mTOR | Mammalian target of rapamycin |
| MYT1 | Membrane-associated tyrosine- and threonine-specific cdc2-inhibitory kinase |
| NBS1 | Nijmegen breakage syndrome protein 1, Nibrin |
| ncRNAs | non-coding RNAs |
| NEDD8 | Neural precursor cell expressed, developmentally down regulated 8 |
| NF-kB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NPC | Nasopharyngeal carcinoma |
| NSCLC | Non-small cell lung cancer |
| NTRK2 | Neurotrophic tyrosine receptor kinase 2 |
| ORIs | Origin of replication |
| OTUB1 | OTU deubiquitinase, ubiquitin aldehyde binding 1 |
| OXA | Oxaliplatin |
| PAF | Platelet-activating factor |
| PARP | Poly-ADP ribose polymerase |
| PCAT1 | Prostate cancer-associated transcript 1 |
| PCNA | Proliferating cell nuclear antigen |
| PDK1 | Phosphoinositide-dependent kinase-1 |
| PDX | Patient-derived xenograft |
| PI3K | Phosphatidylinositol 3-kinase |
| PLKs | Polo-Like Kinases |
| Pre-IC | Pre-initiation complex |
| PROTACs | Proteolysis targeting chimeras |
| PTBP1 | Polypyrimidine tract binding protein 1 |
| PTEN | Phosphatase and tensin homolog |
| RAD50 | RAD50 Double Strand Break Repair Protein |
| RAS | Rat sarcoma |
| Rb | Retinoblastoma |
| RNA | Ribo nucleic acid |
| RNATx | RNA therapeutics |
| RPS9 | Ribosomal protein S9 |
| RRM2 | Ribonucleotide reductase subunit M2 |
| RS | Replication stress |
| RSC | Chromatin structure remodelling complex |
| RUNX1 | Runt related transcription factor 1 |
| S. pombe | Schizosaccharomyces pombe |
| SAC | Spindle Assembly checkpoint Spindle assembly checkpoint |
| SCF | Skp-Cullin-F box |
| SCLC | Small cell lung cancer |
| Ser | Serine |
| shRNA | Small hairpin RNA |
| siRNAs | Small interfering RNAs |
| SIRT1 | Sirtuin 1 |
| SKP1 | S phase kinase-associated protein 1 |
| SLC31A1 | Solute carrier family 31 member 1 |
| SMAC | Second mitochondrial activator of caspase |
| SNHG4 | Small Nucleolar RNA Host Gene 4 |
| SOX17 | SRY-box transcription factor 17 |
| S-phase | Synthesis phase |
| SphK2 | Sphingosine kinase 2 |
| STAT3 | Signal transducer and activator of transcription 3 |
| T-ALL | T-cell acute lymphoblastic leukemia |
| TCGA | The Cancer Genome Atlas |
| TEAD | Transcriptional enhanced associate domain |
| TGF- | Transforming growth factor beta |
| Thr | Threonine |
| TME | Tumor microenvironment |
| TNBC | Triple-negative breast cancer |
| TP53 | Tumor protein P53 |
| TTK | Threonine tyrosine kinase |
| Tyr | Tyrosine |
| UBE2N | Ubiquitin conjugating enzyme E2 N |
| USP | Ubiquitin-specific peptidase |
| VEGF | Vascular endothelial growth factor |
| WAF1 | Wildtype p53-activated fragment 1 |
| XRCC5 | X-ray repair cross complementing 5 |
| YAP | Yes-associated protein 1 |
| ZEB1 | Zinc finger E-box binding homeobox factor 1 |
| ZH2 | Zeste homolog 2 |
Cancer is an unregulated condition that is caused by alterations in the genes responsible for the regulation of cell function, leading to continuous and excessive cell division [1,2]. One of the primary factors contributing to this phenomenon is modifications in cell cycle regulators, which compromises their surveillance ability responsible for maintaining the normal functioning of the cell cycle [3]. This continuous division and growth of cancerous cells lead to the invasion of normal tissues and organs. Ultimately, resulting in the spread of cancerous cells (metastasis) (Fig. 1) [4]. The cell cycle is a highly controlled system of functionally linked proteins that carry out accurate and timely duplication of genomic DNA followed by segregation of daughter cells. It is defined by two distinct stages, interphase (consisting of the G1 phase, S-phase, and G2 phase) and M phase [5]. The timely transition of cells from one phase to another phase is guided by the accumulation and degradation of cyclins along with their catalytic partners; cyclin-dependent kinases (CDKs) under the close surveillance of several checkpoints like DNA damage checkpoint, replication stress checkpoint, and mitotic checkpoint [6–8]. Defects in these checkpoints lead to the aforementioned behavior of the cell cycle transforming them into cancerous cells. Abrupt DNA damage checkpoint is one of the most commonly reported defects leading to the accumulation of errors causing mutations [9–11].
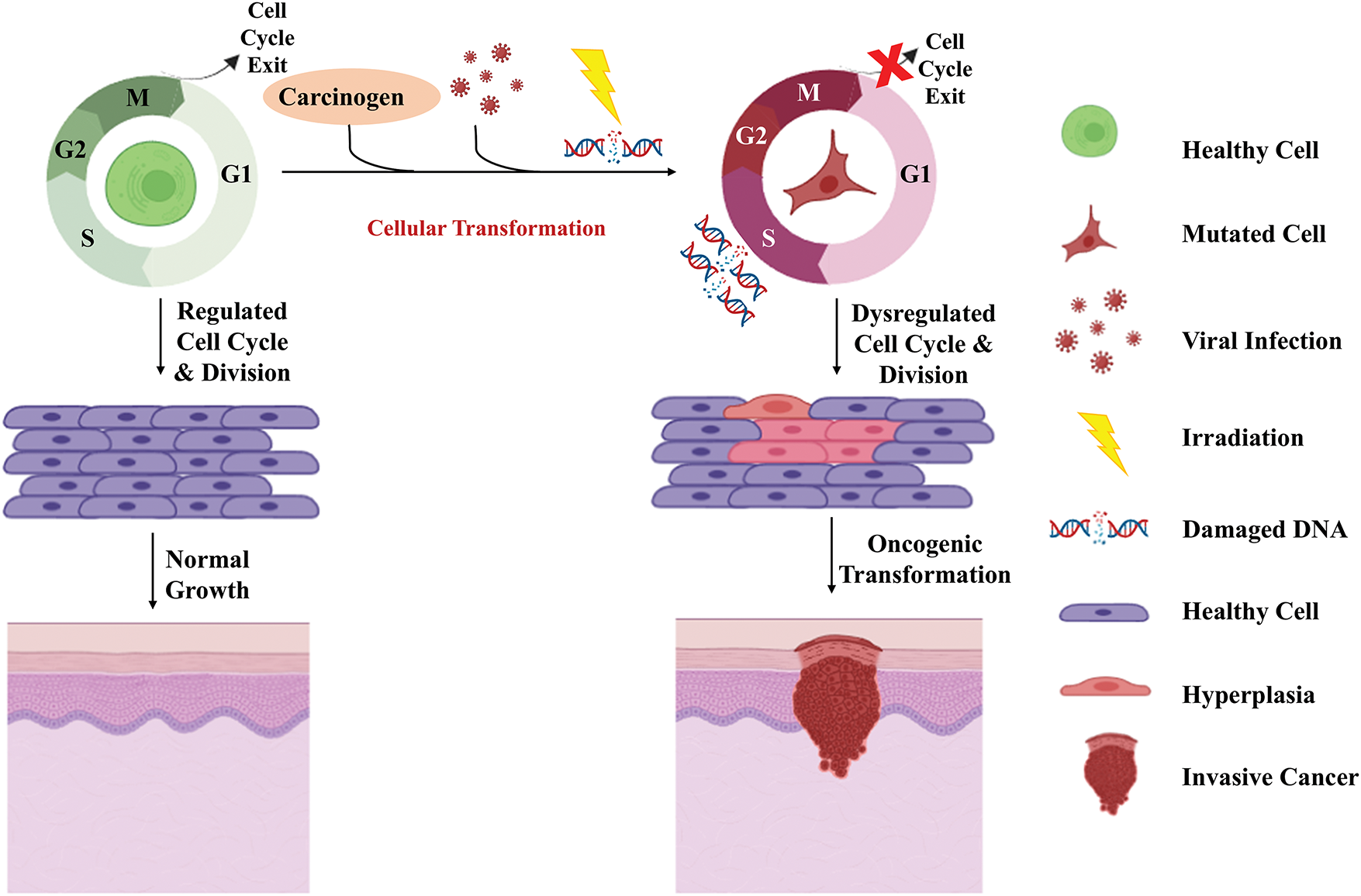
Figure 1: Oncogenic transformation of the normal cells. Exposure to carcinogens, radiations, or onco-viral infections may lead to a dysregulated cell cycle, resulting in the accumulation of mutations, and eventually genomic instability. The dysregulated cell cycle and genomic instability cause the oncogenic transformation of normal cells into neoplastic cells. These neoplastic cells with persistent mutations end up causing hyperplasia followed by an invasive cancerous stage (Created using BioRender graphics).
This review comprehensively discusses the regulators of the cell cycle and their contribution to the oncogenic transformation of normal cells. It also focuses on the cyclins, CDKs, CDK inhibitors (CKIs), major checkpoint regulators, and E3 ubiquitin ligases as therapeutic targets. The probable therapies which could be used to target cell cycle factors, allow the cells to exit the cell cycle and terminate the hyperproliferative nature of the same. Despite the extensive research, there are still some emerging issues like cancer drug resistance, drug non-specificity, immune evasion, and tumor microenvironment (TME) which could be addressed by using novel therapeutics like combination drug therapy, gene therapy, targeting ubiquitin ligase system, etc.
Cell Cycle Factors and Cancer Progression
The well-coordinated regulation of the cell cycle is pivotal to the normal growth and survival of the organism. Furthermore, the research of many diseases, including cancer, revolves around comprehending the mechanistic regulation of the cell cycle. Mutation in genes involved in cell cycle regulation is one of the critical factors responsible for oncogenesis, proliferation, and progression [7,12,13]. The cell cycle regulation involves two important mechanisms. First is the phosphorylation of target proteins by kinases [5] And the second component is checkpoint/s that scrutinize the appropriate progression of each phase and delay the progression, upon encountering any trouble [11,14,15]. The processes are interlinked; phosphorylation-based activation of kinases such as ATM and ATR activates the checkpoint system in the cell cycle which in turn activates the downstream kinases for licensing the cell cycle [13].
Cyclins and cyclin-dependent kinases (CDKs)
Cyclins were discovered by Tim Hunt in Sea Urchins as an essential protein that is degraded after each cycle of the cell division [16,17]. Cyclins A and B were identified through further studies in other organisms such as Schizosacchromyces pombe, Drosophila melanogaster, and Xenopus, that were shown to be required for cell cycle entry [17,18]. Later, Sir Paul Nurse and Lee Hartwell discovered the existence of kinase proteins that in coordination with cyclins phosphorylate the target proteins and aid in cell cycle entry and progression [18,19]. As the cells proceed through the cell cycle, sequentially four major cyclins are involved namely, cyclin D, E, A, and B which regulate the activities of their respective cyclin-dependent kinases (CDKs). For normal functioning of cells, these factors are strictly regulated by various mechanisms including cell cycle-specific transcription and degradation of the gain-of-function/loss-of-function mutations in the viral genes, such as p27, p53, Rb, CCNE1/2, and various others, results in aberrant expression of the cell cycle factors, contributing to tumor development and progression. Consequently, targeting these factors may serve as an effective strategy for developing therapeutics for tumor management and inhibition [20].
The cell cycle is regulated and driven by a set of cyclin proteins and their catalytic partner CDKs [21]. Cyclin D activates and forms a complex with CDK4/6, which phosphorylates proteins like pRb. Upon hyperphosphorylation, pRb gets inactivated leading to the release of transcription factor E2Fs. This process subsequently leads to the activation of E2Fs, which facilitates the transcription of downstream genes including cyclin E, CDKs, and CDCs [22–24]. Cyclin-CDK complexes are inhibited by cyclin-dependent kinase inhibitors (CKIs) [25]. The CKIs involved in the regulation of cyclin-CDKs can be categorized into inhibitors of CDK4/6 (INK4) and kinase inhibitory protein/CDK interacting protein (KIP/CIP). The INK4 inhibitors (p16, p15, p18, p19) inhibit cyclin D-CDK4/6 by inactivating CDK4/6 whereas KIP/CIP inhibitors (p27, p57, p21) promote cyclin D-CDK4/6 complex by inhibiting CDK2, thus promoting the proliferative activity of cells (Fig. 2) [20,26]. During the late G1 phase, cyclin D-CDK4/6 activates cyclin E-CDK2, which phosphorylates pRb and facilitates the G1-S transition (Fig. 2) [20]. The dysregulated activity of CDK4/6 kinases in association with cyclin D [27], results in hyperphosphorylation of pRb that leads to loss of cell cycle regulation thereby, uncontrolled cell proliferation [28,29]. Hyperactivity of cyclin D has been reported in various cancers such as endometrial carcinoma [30], non-small cell lung cancer (NSCLC) [31], head and neck carcinoma [32], pancreatic cancer [33], and colorectal cancer [34]. Overexpression of CDK4 has been reported in uterine cancer [35], colorectal cancer [34], cervical cancer [36], melanoma [37] as well as NSCLC [31]. The CDK4 gene amplification has been found in glioblastomas [38], uterine cervix cancer [39], osteosarcomas [40], rhabdomyosarcomas [41], and liposarcomas [42]. Overexpression and gene amplification of CDK6 have been reported in leukemia [43], gliomas [44], and lymphomas [43]. Targeting CDK4/6 has been a breakthrough in the treatment of breast cancer and can be effective in the treatment of other cancers as well [20].
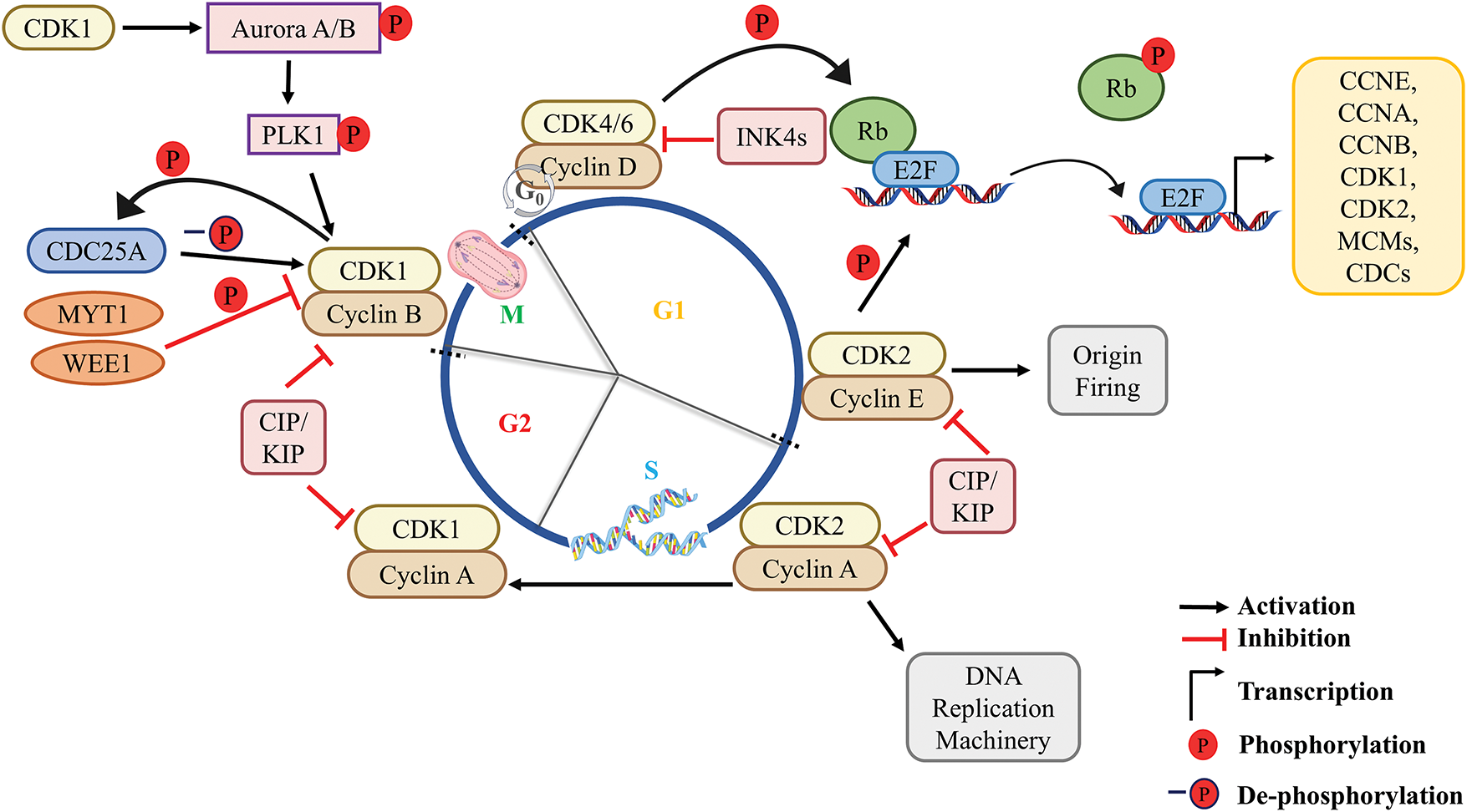
Figure 2: Overview of the molecular mechanism of the cell cycle. In a quiescent cell, the mitogenic signal triggers the activation of cyclin D-CDK4/6 complex resulting in E2F-mediated transcription of downstream genes (CCNE, CCNA, CCNB, CDKs, CDC6, Cdt1, MCMs, etc.). During the late G1 phase, the activated cyclin E-CDK2 complex initiates the loading of replication-related proteins resulting in the onset of origin firing which transits the cell into the S-phase. In the S-phase, cyclin A-CDK2 initiates DNA replication and progresses to the G2 phase. In the G2 phase, accumulated cyclin B in the cytoplasm gets translocated into the nucleus upon being phosphorylated by PLK1. CDC25A removes the inhibitory phosphorylation from CDK1, caused by WEE1 and MYT1. Activated cyclin B-CDK1 complex activates mitotic machinery followed by mitotic exit. P, phosphorylation; -P, dephosphorylation.
CDK2 is involved in G1/S transition [13]. It becomes active when it binds to one of its two regulatory partners, cyclins A or E, to form a functional, heterodimeric complex [2]. Cyclin D-CDK4/6 mediated sequestration of p27/p21, and transcription of cyclin E activates and increases the activity of CDK2 in the late G1 phase [45]. Interestingly, WEE1-mediated phosphorylation of CDK2 at Tyr-15 inhibits its activity. This inhibitory phosphorylation is removed by CDC25A/B phosphatases (Fig. 2). Cyclin E-CDK2 complex phosphorylates and activates various proteins including cyclin A which are needed for DNA replication and cell cycle progression [46]. An E3 ubiquitin ligase system, SCFFBXW7, rapidly breaks down cyclin E during S-phase (Fig. 3) [47]. Active cyclin E-CDK2 complex induces the expression of various factors responsible for DNA replication such as cell division cycle 6 (CDC6), chromatin licensing and DNA replication factor 1 (Cdt1/CDC10 dependent transcript 1), minichromosome maintenance complex (MCM2-7), cyclin A, EMI1 (APC/C inhibitor), etc. which helps in origin recognition, origin firing, DNA unwinding, thereby ensuring single replication per cell cycle [48]. To ensure a single round of replication per cell cycle, two essential steps before DNA synthesis must be carried out: origin licensing and origin firing. Origin licensing involves the assembly of the pre-replication complex (pre-RC) onto the origin of replication (ORIs) and occurs during late mitosis and early G1 phase when cells have low levels of CDK activity [49]. Origin activation occurs during the G1/S transition and generates a pre-initiation complex (pre-IC) [50,51]. Pre-IC formation, in contrast to origin licensing, necessitates high kinase activity; that is achieved through the combined action of two S-phase kinases, CDK2 and CDC7, which bind to the regulatory proteins; cyclin E/A and DBF4, respectively [52–54]. The above kinases work together to phosphorylate several replication factors (CDC45, GINS, replicative MCMs, etc.) and promote replicative protein recruitment to ORIs. DNA polymerases and additional proteins are recruited following helicase activation and subsequent DNA unwinding, ultimately resulting in origin firing [55]. The above events make the entry into the S-phase of the cell cycle.
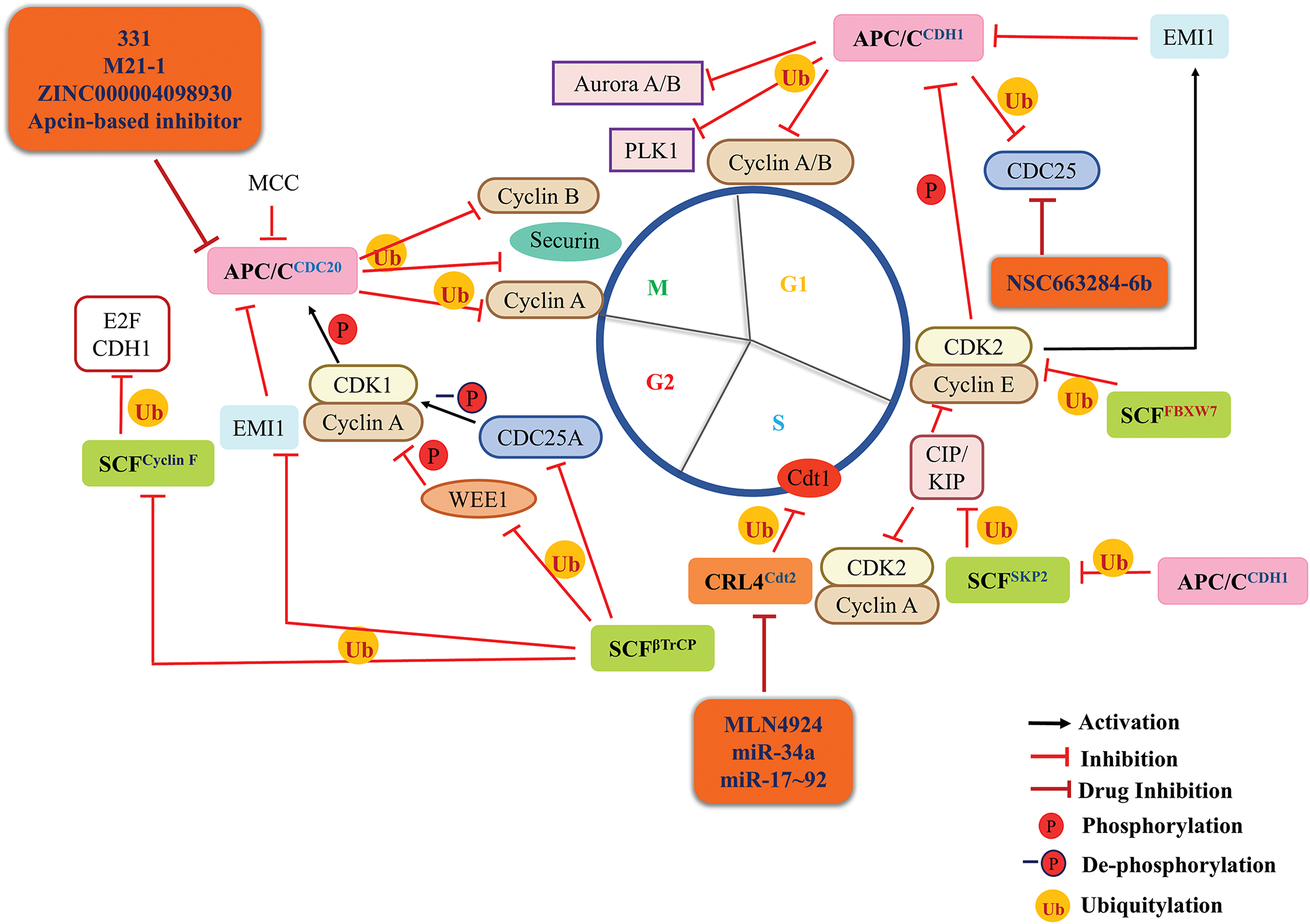
Figure 3: Targeting E3 ubiquitin ligase system as a novel approach for cancer therapeutics. The three major E3 Ubiquitin Ligase complexes that regulate the normal cell cycle progression are Anaphase Promoting Complex or Cyclosome (APC/C, pink round square), Skp-Cullin-F box (SCF, green round square) and Cullin Ring Ubiquitin Ligase 4 (CRL4, peach round square) with their respective adapter proteins. APC/CCDH1 becomes active in anaphase where it modulates the mitotic regulators like Aurora A and PLK1, etc. At the end of the M phase, APC/CCDH1 maintains a low level of mitotic and S-phase cyclins thus stabilizing the G1 phase and preventing premature S-phase occurrence. At the end of the G1 phase, cyclin E-CDK2 complex inactivates APC/CCDH1 and mediates E2F-dependent transcription of EMI1 which further inactivates APC/CCDH1. Degradation of cyclin E by SCFFBXW7 and BTB-Cul-3-Rbx1 (BCR) complex marks the entry of the cell in the S-phase. During S-phase, CRL4Cdt2 degrades Cdt1 and p21 to ensure a single round of DNA replication per cell cycle. During the G2 phase, SCFβTrCP regulates the activity of CDK1 by degrading WEE1 (activation) and CDC25A (inactivation). SCFβTrCP also degrades EMI1 which inhibits the activity of APC/CCDC20. Also, during the G2 phase, SCFcyclin F ensures the degradation of E2F and CDH1. In the early M-phase, APC/CCDC20 mediates the degradation of cyclin A and pushes the cell into metaphase. In metaphase, APC/CCDC20 degrades cyclin B and securin to ensure anaphase onset. During the anaphase, APC/CCDH1 mediates the degradation of cyclin B to exit the cell cycle. In addition, APC/C and SCF E3 ligase control each other to regulate their expression. In cancerous cells, the dysregulated expression of this ubiquitin ligase system leads to prolonged degradation of tumor suppressor proteins like KIP/CIP and constant expression of oncogenic proteins, which could be targeted for cancer therapeutics using several drugs mentioned in orange round boxes (and are under clinical trials). P, Phosphorylation; -P, dephosphorylation; Ub, ubiquitination; black arrow, activation; red hammerhead arrow, inhibition; maroon hammerhead arrow, inhibition by drugs.
At the onset of the S-phase, CDK2 binds to cyclin A2 to form an active cyclin A-CDK2 complex for the progression of DNA replication or S-phase [5,52,56] (Fig. 2). Both E2F-dependent transcription, as well as inactivation of APC/CCDH1 (which degrades cyclin A, Fig. 3), are crucial for cyclin A-CDK2 activity. This inactivation of APC/CCDH1 is a result of cyclin E-CDK2 mediated transcription of EMI1 [57,58]. E2F-mediated transcription results in the accumulation of cyclin A-CDK2 activity, which triggers the cell to initiate DNA replication [13]. Cyclin A-CDK2, later in S-phase also controls the timing of cyclin B-CDK1 activation and mitotic entry of cells by regulating CDH1 levels (Fig. 3) [59].
The Cyclin E1 gene, CCNE1 is highly amplified in many cancers such as ovarian cancers [60], endometrial cancer [61], hepatocellular carcinoma [62], etc. CDK2 is also reported to be overexpressed in prostate cancer [63] and B cell lymphoma [64]. Since FBXW7/FBW7 is directly involved in the degradation of cyclin E [58,65] (Fig. 3), loss of FBXW7 is associated with colon [66,67] and breast cancer [68,69] where cyclin E is not timely degraded resulting in hyperproliferation. Thus, targeting CDK2 could be a better target for cancer therapeutics.
The mitotic cyclins, cyclin A and B (which accumulate in the S-phase) bind to, and activate CDK1 during the G2 phase; cyclin A-CDK1 then propels G2 cells into mitosis [52]. CDK1 accumulates in an inactive form as a result of phosphorylation (inhibitory) at Thr-14 and Tyr-15 by kinases MYT1 and WEE1, respectively. At the end of G2, the accumulated inactive cyclin B-CDK1 complexes are activated by phosphatase CDC25, which removes the inhibitory phosphorylation of CDK1. CDC25 works in a positive feedback loop with active cyclin B-CDK1. As cyclin B-CDK1 activates additional CDC25, which subsequently removes inhibitory phosphorylation from cyclin B-CDK1, resulting in complete activation of the complex (Fig. 2) [20,70]. CDK1 can also be activated by cyclin H-CDK7 mediated phosphorylation at Thr-161 [71]. CDK1 activity is necessary for mitotic entry and multiple mitotic events. Widespread phosphorylation of over a thousand CDK1 substrates, including B-Raf (BRAF, a proto-oncogene), p53, checkpoint kinases (CHK1/2), etc., initiate entry into mitosis once CDK1 activity thresholds are reached [72]. Simultaneously, CDK1 stimulates PLK1, Aurora A/B, the mitotic kinases that phosphorylate other mitotic proteins (Fig. 2). Every cell organelle undergoes structural alterations as a preparatory measure for cytokinesis (completion of cell division). Increased cytoplasmic CDK1 activity during prophase causes centrosome separation and cell rounding. Additionally, the fast nuclear import of activated cyclin B-CDK1 causes chromosomal condensation, APC/C activation, nucleolar disassembly, and nuclear envelope permeabilization [13,20]. The anaphase-promoting complexes (APCCDC20 and APCCDH1) degrade cyclin B during the late mitosis (Fig. 3), resulting in chromosomal separation, completion of mitosis, cytokinesis, and attenuation of CDK1 activity [46].
Amplification of the cyclin B gene (CCNB1) has been closely associated with neuroendocrine prostate cancer [73], lung cancer [74], breast cancer [75], etc. According to The Cancer Genome Atlas (TCGA) database, CDK1 was found to be significantly abundant in 17 out of 24 cancers including cervical cancer, cholangiocarcinoma, urothelial carcinoma, glioblastoma, etc. [76–78]. The deregulation of upstream or downstream mediators of CDK1 is linked with various cancers. Ribosomal protein S9 (RPS9) is an upstream activator of CDK1 and knockdown of RPS9 in colon cancer inhibits the growth of cancerous cells by downregulating CDK1 at the G2/M phase resulting in checkpoint activation which in turn induces apoptosis [79]. Mutations in DNA methyltransferase-3-alpha (DNMT3A), a positive activator of CDK1, increases the expression of CDK1 which induces leukemia [72]. Considering the fact that CDK1 is highly abundant in human cancer, it could act as a potential target for developing novel cancer therapies (Fig. 3).
The cell cycle is a highly orchestrated biological phenomenon that results in DNA replication and cell division. The cell cycle regulatory Checkpoint factors determine whether a cell should continue to the next phase of the cell cycle or abort the process [57,58,80]. They monitor DNA damage, loss of replication fork integrity, and inadequate spindle assembly during different phases of the cycle (Fig. 4) [13].
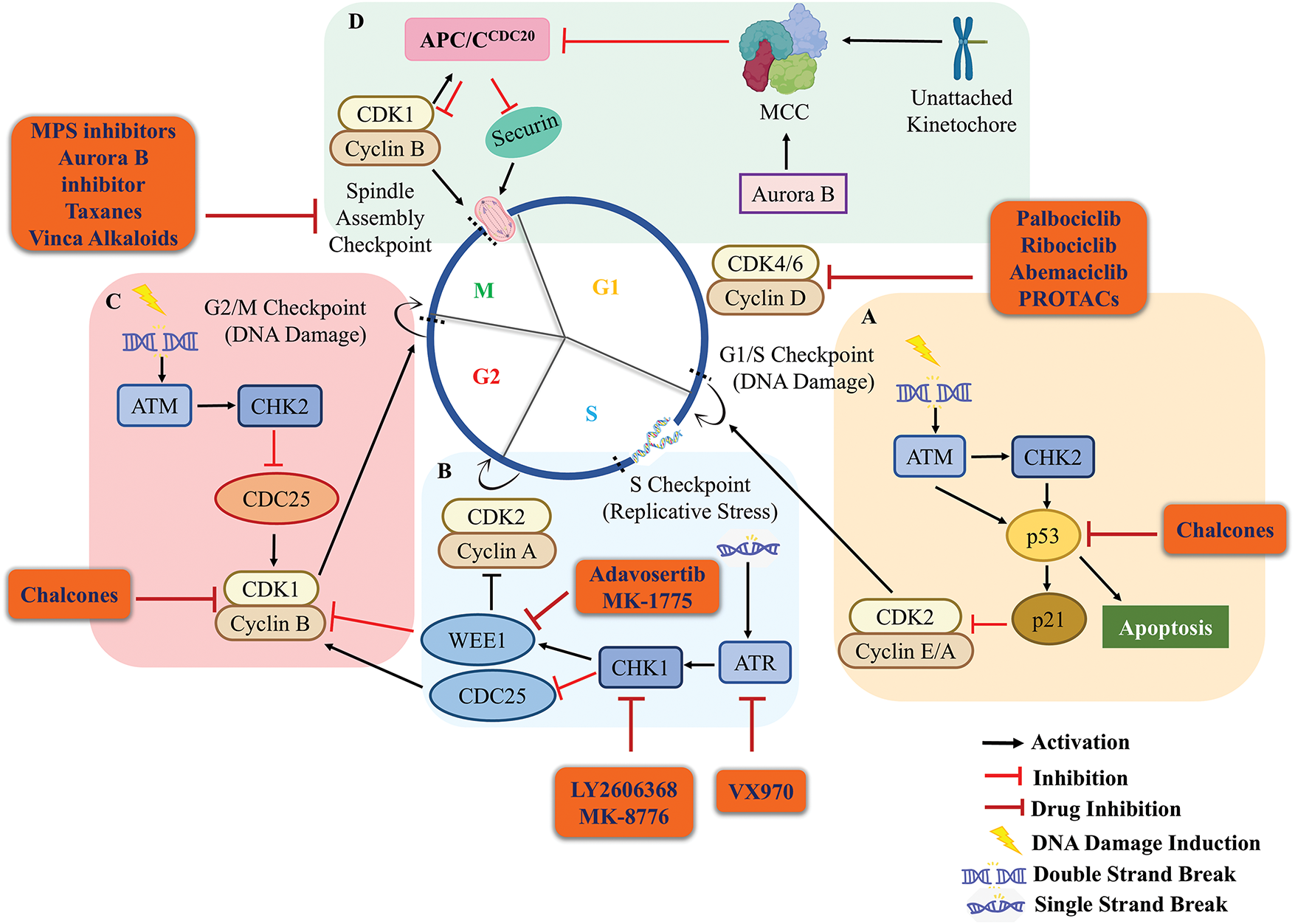
Figure 4: Cell cycle checkpoint regulation and inhibitory effect of drugs on it. (A) During the G1 phase, the DSBs trigger the G1/S checkpoint where ATM is recruited at DSBs by the MRN complex. ATM phosphorylates and activates CHK2 and p53 which in turn activates p21 which inhibits the activity of cyclin E/A-CDK2. This results in the halting of cells in the G1 phase and damage is repaired. If the damage is beyond repair p53 activates the apoptotic machinery of the cells. (B) Stress generated during the replication recruits ATR at the single-strand breaks (SSBs), ATR activates CHK1 which activates WEE1. WEE1 phosphorylates CDK2 thus inactivating cyclin A-CDK2. Thereby, inhibiting the transition of cells from the S to G2 phase to maintain the genomic integrity. (C) In the G2 phase, DNA damage is sensed and ATM-CHK2 signaling gets activated. CHK1/2 in turn inactivates the phosphatase CDC25 which cannot remove the inhibitory phosphorylation of CDK1 caused by WEE1. This aborts the activation of cyclin B-CDK1 and arrests the cells in the G2 phase, thus, limiting the cells from entering into the mitotic phase. (D) During the metaphase of M-phase, chromosomes align at the equator and spindle fibers attach to kinetochores for proper segregation of chromosomes in anaphase. Any unattached kinetochore generates a “wait anaphase” signal which activates the mitotic checkpoint complex (MCC). MCC inhibits the anaphase-promoting complex APCCDC20 inhibiting the degradation of cyclin B-CDK1 and securin. This inhibits the progression of cells from metaphase to anaphase of the cell cycle. During cancer conditions, most of these proteins involved in cell surveillance are aberrantly expressed due to the accumulation of a series of mutations. Most of these mutations upregulate the expression of CDKs and disturb the normal physiology of the cell cycle checkpoints. These aberrations serve as potential targets for cancer therapeutics and small structural molecules/drugs (given in orange round box) have been designed to combat them. The orange round boxes show different drugs that target cell cycle and checkpoint factors to inhibit cancer progression and are under clinical trials. Black arrow, activation; red hammerhead arrow, inhibition; maroon hammerhead arrow, inhibition by drugs.
During interphase, the presence of DNA double-strand breaks (DSBs) activates checkpoint protein kinase Ataxia Telangiectasia Mutated (ATM) signaling cascade.
Upon the initial response, DNA damage sensor complex MRN (MRE1, RAD50, and NBS1) assembles at the double-strand break (DSB) sites, activating ATM which in turn phosphorylates a wide range of substrates. Essential targets in DNA damage response include the protein checkpoint kinase 2 (CHK2; phosphorylation at Thr-68) and the transcription factor p53 (phosphorylation at Ser-15) [81]. Simultaneously, ATM also stabilizes p53 by phosphorylating a p53 degradation E3 ubiquitin ligase machinery, MDM2 [81]. CHK2 also activates p53 by phosphorylating it at Ser-20 [82]. Activated p53 in turn then transactivates p21, a CDK inhibitor, leading to the inactivation of cyclin-CDK complexes particularly in the G1 phase (cyclin E-CDK2), blocking the transition of cells from G1 to S-phase (Fig. 4A) [13]. Mutation or aberration of p53 and p21 thus, results in loss of G1/S checkpoint system [80]. Post DNA replication, ATMs are recruited at DSBs activating CHK2. CHK2 phosphorylates and inactivates CDC25A/C, therefore the latter is not available to dephosphorylate and activate CDK1, ensuring the inactivation of CDK1. This results in the halting of cells at the G2 phase by inhibiting the G2/M transition (Fig. 4C) [13,81].
Cells can exit the cell cycle in two ways: reversibly by undergoing quiescence or irreversibly by apoptosis or senescence [13]. In response to DNA damage, the DNA damage checkpoint can either induce quiescence, senescence, or apoptosis during the interphase of the cell, predominantly through p53-dependent pathways [83]. The most prevalent mutation in cancer is p53 alterations [84]. ATM is a tumor suppressor that induces cell cycle arrest or apoptosis by activating CHK2, p53, DBC1, and other downstream proteins [85]. In contrast, ATM activity and signaling are reported to be elevated in certain cancers such as melanoma, prostate cancer, pancreatic cancer, etc., [13,86]. It is possible that those cancer cells have gained resistance against ATM-induced apoptosis and cell cycle arrest. Further work needs to be done to elucidate this mechanism. In prostate cancer cells, Ack1 kinase phosphorylates androgen receptor (AR) at Thr-267 which mediates the recruitment of phosphorylated AR to the enhancer region of ATM genes resulting in the increased expression of ATM [87]. In pancreatic cancer, overexpression of transcription factor CUX1 serves as the activator for DNA Damage Repair (DDR) genes and upregulates the expression of ATM [88]. Hence, ATM-based anti-cancer therapy should be carefully studied and designed, based on the characteristics of the tumor.
DNA replication stress checkpoint gets activated only during S-phase of the cell cycle. Delay or stalling of DNA replication forks, falling off polymerases from the fork, collision with transcription machinery, DNA damage, etc., eventually exposes single-stranded DNA for too long resulting in replication-induced stress. During the S-phase response to DSBs, DNA resection engages homologous recombination repair leading to the generation of single-stranded DNA. Accumulation of single-stranded DNA activates the replicative stress kinases; ataxia telangiectasia mutated and Rad3-related protein (ATR) and CHK1 [13]. CHK1 phosphorylates the WEE1 and CDC25A/C resulting in their activation and degradation, respectively [20]. WEE1-dependent inhibitory phosphorylation of CDKs, particularly CDK2 and CDK1 results in the inactivation of cyclin-CDK complex, halting the cells in the S-phase. This provides a greater window of time to resolve the replicative stress (RS) and ensures complete duplication of the genome [89]. Simultaneously, the inhibition of CDC25 results in the accumulation of inactivated CDK1 ensuring no premature onset of the mitotic events (Fig. 4B) [13].
Genetic instability, a characteristic of cancer, is a leading cause for intratumor heterogeneity posing a major challenge for therapy development. RS plays a significant role in promoting genomic instability in cancer [90]. Cyclin E is frequently overexpressed in triple-negative breast cancer and ovarian cancer [91,92], amplification of which is linked to induction of replicative stress, as a result of collision between replication and transcription machineries [93]. CCNE1 and CDC25A amplification leads to aberrant origin firing which in turn produces immense replicative stress [94]. The hyperproliferative tendency of cancerous cells is a result of various overexpressing oncogenes which causes replicative stress [9,95]. Due to aforementioned reasons, cancer cell witnesses an increase in RS as they enter the S-phase, thereby increasing their dependency on ATR-mediated S and G2/M checkpoints for the resolution of the same. Thus, blocking ATR has great promise for developing treatment of various cancers [96]. The overexpression of cyclin E1 and CDC25A induces RS, which leads to lagging chromosomes and formation of chromatin bridges [93]. This replication stress is further intensified by inhibiting ATR or WEE1 kinases, ultimately resulting in death of tumor cells [93].
Spindle assembly checkpoint (SAC) which gets activated in M-phase ensures that the duplicated DNA is equally distributed among the two daughter cells [97,98]. The multiprotein SAC machinery is activated if any kinetochore remains unattached to microtubules [13]. SAC allows unattached kinetochores to prevent aneuploidy by limiting premature chromosomal segregation [98]. The CDC20 (also known as CDC20A or p55CDC) stimulates the APC/C during mitosis, and ensures APC/CCDC20 complex ubiquitinates the two anaphase inhibitors; cyclin B and securin to encourage sister chromatid separation and mitotic exit (Fig. 4D) [99]. Unattached kinetochores generate a diffusible signal of ‘wait anaphase’, activating a tetrameric complex; mitotic checkpoint complex (MCC consisting of Mad2, CDC20, BubR1, and Bub3) which turns-off the activity of APC/CCDC20 (Fig. 4D) [97,100]. Once all the kinetochores are attached and bioriented to opposite spindle poles, the MCC disassembles. This frees up the CDC20, which acts as a co-activator, thus, turns on the APC/CCDC20 [101,102]. The re-activated APC/C E3 ubiquitin ligase then marks the cyclin B and securin for degradation, allowing chromosomal segregation and anaphase to occur [13,97].
Cancerous cells are mostly aneuploid, having aberrant chromosomal configurations which are results of mitotic errors. Although, SAC gene mutations have been linked to cancer, they are uncommon [13]. Overexpression of SAC proteins (MPS1, MAD1, MAD2, BUBR1, and CDC20) may result in the delayed mitosis, cohesion fatigue, chromosomal mis-segregation, eventually resulting in chromosomal-instability and aneuploidy. These genetic alterations can contribute to development of various malignancies including colon, pancreatic, oesophageal, oral, tonsillar, gastric, renal, cervical cancers, etc. [8,103,104]. BUB1B which is a downstream protein in SAC signaling pathway, is overexpressed in aggressive breast cancer and knockdown of Bub1b gene suppresses the tumorigenicity in breast cancer cells [105]. MPS1 (an upstream target of SAC) is overexpressed in triple-negative breast cancer, lung cancer, and neuroblastoma [106–108] making it a potential target for cancer therapeutics.
Therapies Available for Targeting Cell Cycle Factors
Till date, indeed there are several therapies available such as gene therapy, stem cell therapy small-molecule inhibitors, etc., that target the growth of cancerous cells. Among them, targeting cell cycle proteins can be an effective strategy for cancer management [20,109,110]. The available drugs that target cell cycle factors like PLK4 inhibitors [111], Threonine Tyrosine Kinase (TTK) inhibitors [112–114], CDK4/6 inhibitors [115], are considered to be promising strategies for the treatment of cancer [116,117], yet very limited success has been achieved (Table 1). For that reason, more mechanistic work needs to be carried out to understand the process in detail which could lead to exploring and developing better and specific targeted therapies to supress and kill cancerous cells.
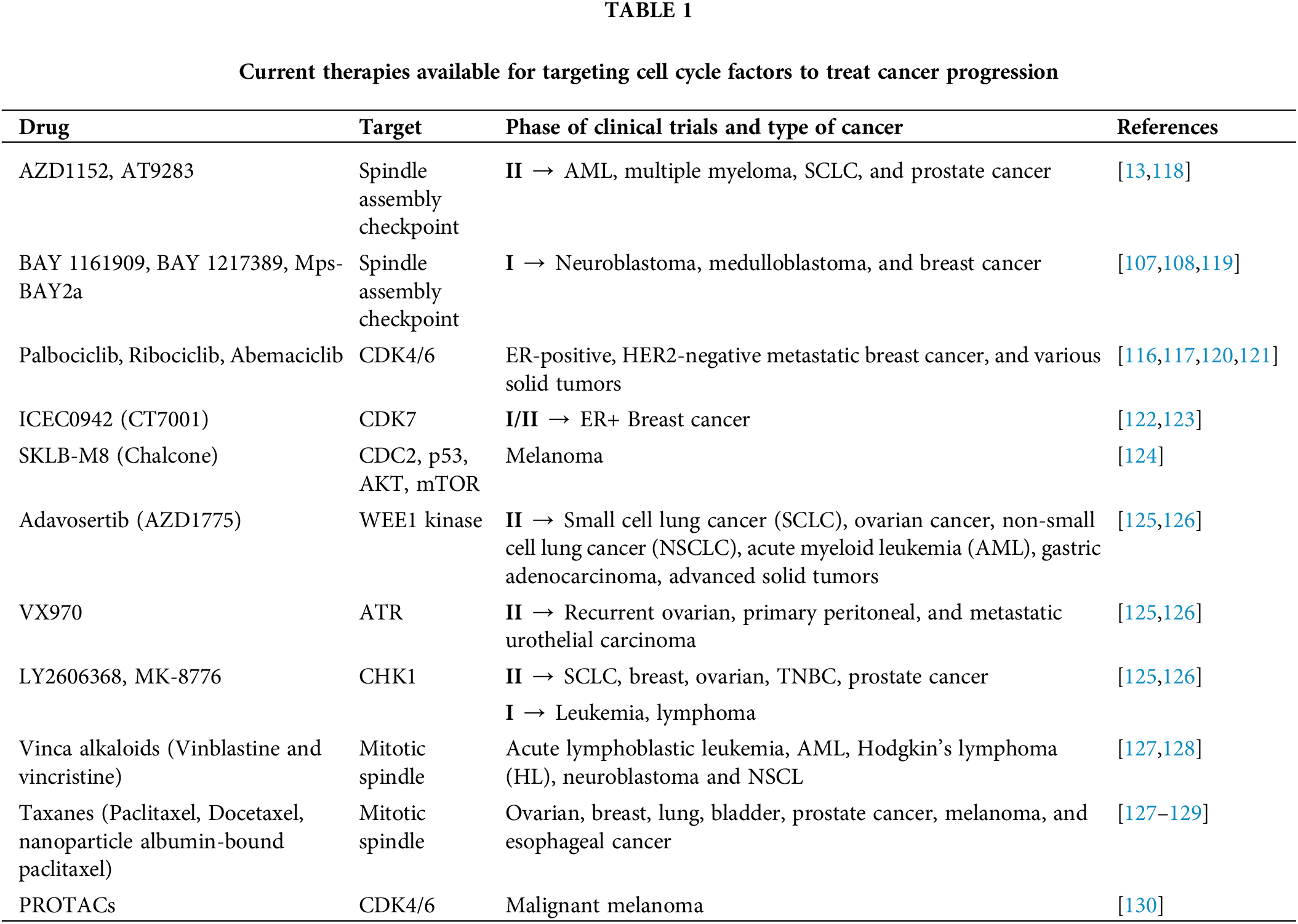
CDK inhibitor class of drugs that have been proven to be effective treatments for cancer. Recently, INX-315; a potent CDK2 inhibitor; that effectively inhibits retinoblastoma protein (Rb) phosphorylation, leading to G1 phase cell cycle arrest [131], has been developed and is under preclinical study in CCNE1-amplified cancers [115]. Various other CDK2 inhibitors are under preclinical or clinical trials such as PE-07104091 (Tagtociclib), BLU-222, etc. [132,133]. To compensate for the overexpression of CDK1 in cancer, some CDK1 inhibitor drugs are already in preclinical testing like RO-3306 and CGP-74514A, which act by inducing G2 arrest, preventing cells with DNA damage from entering mitosis, thereby allowing the required time for DNA repair, and suppressing the tumorigenesis in both cancer cells and patient-derived xenograft (PDX) [134,135]. Another subcategory of CDK4/6 inhibitors; is Palbociclib, Ribociclib, and Abemaciclib, which have demonstrated effectiveness in treating hormone receptor-positive breast cancer. These inhibitors block the cyclin D-CDK4/6 pathway downstream by preventing pRb phosphorylation, causing senescence in cancer cells [117,136]. In patients with advanced-stage hormone receptor-positive, HER2 non-amplified (HR+HER2−) breast cancer, combinatorial therapy with CDK4/6 inhibitors and endocrine therapy is used as a standard of care [137]. ICEC0942 (CT7001) is another drug that targets CDK7 and disrupts CDK7 mediated phosphorylation of RNA polymerase II particularly affecting genes like RUNX1 in T-cell acute lymphoblastic leukemia (T-ALL) facilitating transcription, to force the cell to exit cell cycle. It is in Phase I/II clinical trials for ER+ breast cancer [122] and Acute myeloid leukemia (AML) [138].
Chalcones are precursors of flavonoids, composed of the chemical scaffold 1,3-diphenyl-2-propen-1-one [139,140]. SKLB-M8, a derivative of natural chalcone Millepachine, is a potent growth inhibitor of various melanoma cancer cell lines which downregulates CDC2 expression and upregulates p53 to promote G2/M cell cycle arrest in A2058 and CHL-1 cells, respectively. SKLB-M8 also downregulates AKT and phosphorylates mTOR, resulting in the induction of apoptosis [124].
Cell cycle inhibitors such as CDK and checkpoint kinase inhibitors are often used in combination with chemotherapy to target cell cycle factors for cancer treatment [21,124,140,141]. In addition to CDKs, checkpoint kinases like Aurora A/B and PLKs are other targets for cancer therapy. Promising clinical trial outcomes have been reported, and the first treatment targeting a cell cycle regulator has been approved which has shown quite promising results for treating breast cancers [137]. However, recent research has introduced the possibility of using TTK and PLK4 inhibitors as alternatives to overcome CDK4/6 drug resistance, and further research in this domain could hold great potential for advancement in cancer therapeutics [112,114,142]. BI2536, one of the initial PLK1 inhibitors, inhibits PLK1 and significantly suppresses tumor development in-vivo as well as in-vitro. Although BI2536 demonstrated a satisfactory safety profile in several clinical trials, its monotherapy application resulted in suboptimal response rates. However, when BI2536 was combined with chemotherapy drugs, synergistic effects were observed. For instance, it enhanced the susceptibility of neuroblastoma cells to the anticancer effects of vincristine, which facilitated cell death by blocking B-cell lymphoma 2 (BCL-2) and cleaving caspases 3 and 9. Furthermore, BI2536 exhibited a synergistic impact with eribulin in neuroblastoma and rhabdomyosarcoma, leading to a reduction in tumor development in in-vivo [4]. To modify aberrant ATR expression, four ATR inhibitors M6620 [143], M4344 [144], AZD6738 [145], and BAY1895344 [146] are currently under clinical trials [96,147]. AZD6738 modulates CHK1 phosphorylation, induces ATM-dependent signaling, inhibits homologous recombination repair, and enhances tumor sensitivity to DNA-damaging agents [148]. M6620 significantly reduces the repair of DNA double-strand breaks (DSBs) induced by radiation, leading to increased cell death [149]. M4344 inhibits activation of the ATR signaling pathway, thereby blocking signal transduction from replicative DNA damage. After M4344 treatment, cells undergo replication catastrophe with DNA damage and mitotic defects, leading to cell death [150]. Among all, Adavosertib (AZD1775) which bypasses necessary DNA repair processes, promoting cell death in cancer cells especially those with p53 mutations [151], is in Phase II trials for relapsed small cell lung cancer (SCLC), ovarian cancer, NSCLC, AML, gastric adenocarcinoma, and various advanced solid tumors, that target WEE1 kinase, delaying mitotic entry [125,126]. VX970, which impairs replicative stress tolerance by targeting ATR, is in Phase II trials for recurrent ovarian cancer, primary peritoneal fallopian tube cancer, and metastatic urothelial carcinoma [126,143]. Berzosertib, another potent ATR inhibitor, is currently undergoing Phase I/II clinical trials. When administered in combination with the topoisomerase I inhibitor, topotecan, in small cell lung cancer; cisplatin in neuroendocrine tumors; and benezortib in brain metastases from non-small cell lung cancer, berzosertib significantly enhances the efficacy of chemotherapy. Notably, promising results were observed for high-grade serous ovarian cancer (HGSOC) and chemotherapy-resistant small cell neuroendocrine carcinoma, both of which exhibit elevated levels of replication stress [152]. The drugs LY2606368 and MK-8776, which target CHK1 and regulate replicative stress, are in phase II trials for SCLC, BRCA1/BRCA2-mutated breast, and ovarian cancer [153,154], triple-negative breast cancer (TNBC) [155,156], high-grade serous ovarian cancer (HGSOC), metastatic castration-resistant prostate cancer (CRPC), bladder cancer [157] and advanced solid tumors [158] with homologous recombination repair defects or genetic alterations [159–161]. These drugs have also been tested in phase I—acute leukemia, solid tumors, or lymphoma and phase II—relapsed AML (with or without cytarabine) [126].
Taxanes (paclitaxel, docetaxel, and nanoparticle albumin-bound paclitaxel) are the drugs that target mitotic spindle checkpoint by binding to β-tubulin and prevent microtubule depolymerization resulting in mitotic arrest and subsequent cell death [162]. These are approved for the treatment of various cancers, including ovarian cancer, breast cancer, lung cancer, bladder cancer, prostate cancer, melanoma, and esophageal cancer [128,129,163]. Vinca alkaloids (vinblastine and vincristine) also target mitotic spindle assembly causing metaphase arrest during mitosis and are approved for use in a range of cancers, including ALL (acute lymphoblastic leukemia), AML, Hodgkin’s lymphoma (HL), neuroblastoma, and NSCLC [127,128]. Another class of SAC inhibitors is MPS1 inhibitors, BAY 1161909 and BAY 1217389, [106,164] are under preclinical trial and have recently entered Phase I clinical trials for treating neuroblastoma [108], medulloblastoma [119], and breast cancer in combination with taxanes [107]. Aurora B inhibitors, AZD1152 [165] and AT9283 also target SAC, and are in Phase II of the trial for AML, multiple myeloma (MM), SCLC, prostate cancer, etc. [118,166,167].
Proteolysis targeting chimeras (PROTACs)
PROTACS are bivalent molecules comprising two selective binding moieties (“warheads”) to recruit the protein target and an E3 ligase at its ends, thereby, linking them together to bring the target protein in close proximity to the E3 ligase for its ubiquitination-induced degradation [168]. To combat cancer, PROTACs are used to induce the degradation of tumor-overexpressing oncogenic proteins like CDK2/4/6, WEE1, Aurora A/B as well as E2F1 and CDC20 [169–171]. WEE1-PROTAC (ZNL-20-096) [172], Aurora A-PROTAC (JB170) [173], and CDC20-PROTAC (CP5V) [174] Target upstream regulators of G2/M checkpoint which in turn blocks entry and progression into mitosis. Based on CDK4/6 inhibitors, PROTACs have been developed to target CDK4/6 (BSJ-03-024 [175], BSJ-02-162 [175], and pal-pom [176]), CDK4 only (BSJ-03-132 [175]) and CDK6 only (BSJ-03-123 [177], CP-10 [178], YX-2-107 [179], and Degrader 6 [180]), which have been shown to elicit degradation efficiency and antiproliferative effects in cancerous cells [181].
Developing therapies targeting cell cycle regulators such as CDKs, cyclins, other kinases, and multiple checkpoint proteins could be an effective way to combat cancer with minimal side effects and other diseases caused by re-replication and hyperproliferation leading to abnormal cell growth.
Emerging Trends and Future Directions
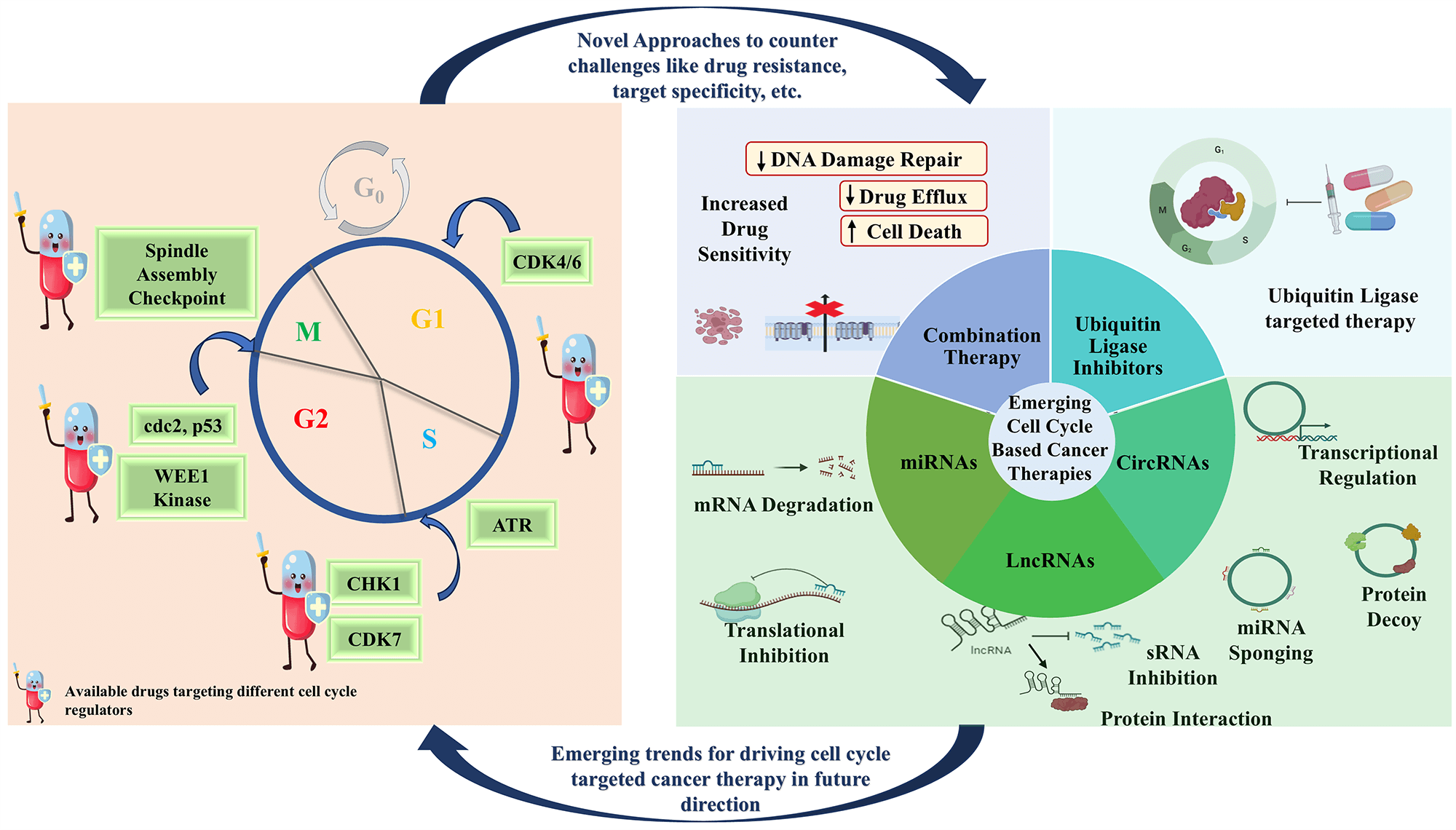
The accomplishment and success story of CDK inhibitors in clinical trials represents the potency of targeting cell cycle proteins for the treatment of cancers. In the ensuing years, compounds that inhibit other cell cycle proteins will be assessed in pre-clinical studies and subsequently on cancer patients (clinical trials). However, despite significant advancements, the efforts of combating solid tumors remain an uphill challenge. One of the roadblocks to this is drug resistance in various cancers that develop over some time. Cancer drug resistance arises due to the intra-tumor heterogeneity resulting from highly diverse genetic, epigenetic, and varying cell cycle dynamics of the cancer cells [182]. As a result of cell cycle heterogeneity, resistant populations of tumor cells persist and proliferate despite the presence of classical chemotherapeutic agents and/or novel targeted drugs. Research has demonstrated that this sub-population of tumor cells adopts a variety of DNA repair mechanisms, reverses or escapes chemical damage to DNA by relying on damaged checkpoints, and avoids apoptosis which eventually results in resistance to a variety of anti-cancerous drugs. In addition to cancer drug resistance, other challenges associated with cancer therapy include a lack of target specificity, an inefficient delivery system, tumor microenvironment (TME), and immune evasion [183], all of which need to be addressed (Fig. 5) to develop an efficient treatment for cancer using methods like Combination therapy and Gene-Based Therapy
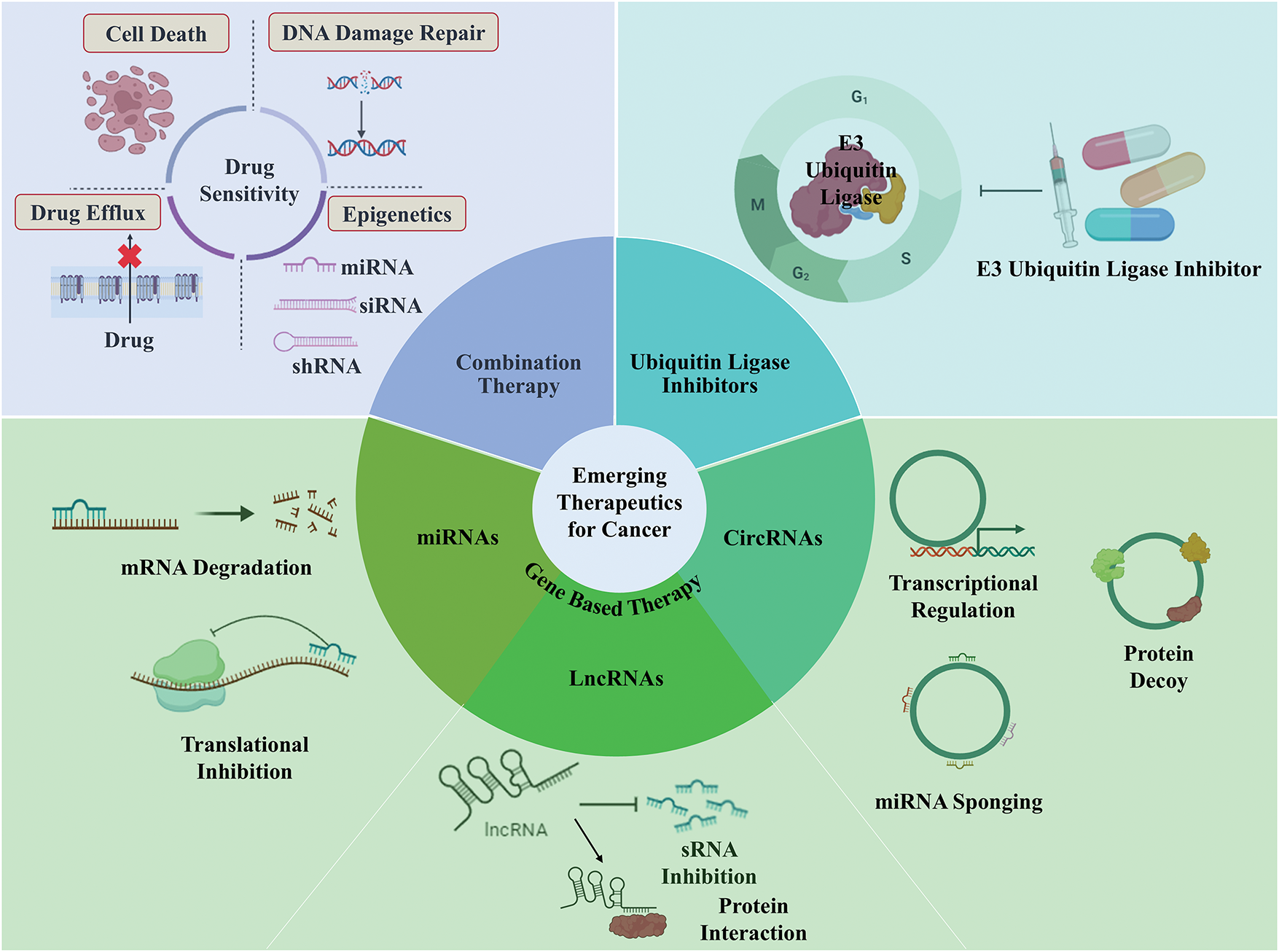
Figure 5: Emerging therapeutics to target cell cycle regulators in cancer. Combination therapy is codelivery of small drugs to overcome cancer drug resistance by inhibiting drug efflux pump hence decreased drug efflux, promoting apoptosis and DNA damage repair. Ubiquitin ligase inhibition, specially E3 inhibition is a more specific interference which ensures the target specificity and also, deubiquitinase inhibitors has also been recognised as a promising therapeutic option. Gene therapy involving inhibition of oncogenic RNA (miRNA, lncRNA, circRNA, and siRNAs) or overexpression of tumor suppressive RNAs to modulate expression of target gene is one of the promising fields in cancer research with minimum side effects. miRNA ensures target gene inhibition either by degrading the target gene or by inhibiting the translation of gene whereas lncRNA and circRNAs can either regulate cell cycle proteins by directly targeting the gene or protein or indirectly via miRNA sponging (Created using BioRender graphics).
Combination therapy for cancer treatment is a promising approach for several reasons. Firstly, it enhances treatment outcomes and achieves superior therapeutic effects, particularly when a synergistic anticancer activity is achieved [184]. Secondly, it overcomes clonal heterogeneity, which is associated with improved response rates [185]. Additionally, combining drug regimens reduces toxicity, as it allows the usage of individual drugs at reduced dosages while maintaining therapeutic efficacy [184]. Another advantage of combination therapies is reducing the emergence of drug resistance [184]. By targeting several molecular pathways, essential for cancer cell survival and abolishing cellular mechanisms associated with adaptive resistance, combination therapy enables concurrent targeting. In one of the studies, the combination of birinapant with the p38 inhibitor (ralimetinib) restored the sensitivity of LKB1- and KRAS-mutated cell lines [186]. Another study by Mortensen et al. in 2020, investigated the impact of the novel heat shock protein 90 (HSP90) inhibitor (onalespib) on enhancing the activity and reversing the resistance of cisplatin in ovarian and head and neck cancer cells. The results showed that the combination of onalespib and cisplatin restored therapeutic activity and enhanced the anti-proliferative, anti-migratory, and apoptotic effects of the chemotherapeutic drug [187]. In 2020, Shi et al. demonstrated sphingosine kinase 2 (SphK2) mediated regorafenib resistance in hepatocellular carcinoma through NF-kB and STAT3 activation. It was also reported that sensitivity to regorafenib was restored with the combination of regorafenib and the SphK2 inhibitor ABC294640 in both in-vitro and xenograft animal models of hepatocellular carcinoma [188]. In 2020, Xu et al. showed that the combination of the HSP90 inhibitor, SNX-2112, with the knockdown of STAT3, is associated with enhanced antiproliferative and apoptotic anticancer activity of cancer stem-like cells in culture and animal models for oesophageal cancer [189].
CDC7 kinase, functioning in collaboration with cyclin-dependent kinase CDK2, is a crucial and rate-limiting factor for initiating DNA replication [54]. However, CDC7 is not essential for the cell division of various cell types, as CDK1 can take over to continue the cell division cycle. CDC7 and CDK1 perform functionally equivalent roles during the G1/S transition, and the presence of at least one of these kinases is necessary for S-phase entry [9,54]. These findings provide insight into the intricate molecular coordination of the cell cycle, and the combination of CDC7 and CDK1 inhibitors could be a potent new strategy for combating cancer [52–54]. A recent combinatorial strategy involving Poly (ADP-ribose) polymerase (PARP) inhibitors and the inhibition of two major cell cycle checkpoint pathways, specifically the DNA damage checkpoints (ATM-CHK2-TP53) and DNA replication stress checkpoints (ATR-CHK1-WEE1), demonstrates synergistic effects in increasing the frequency of DNA errors, compromising the DNA repair machinery, and disrupting the cell cycle. This approach consequently enhances the mortality rate of cancer cells exhibiting DNA repair deficiency or PARP inhibitor resistance [190]. Although cocktail treatments with two or more drugs can often yield additional or synergistic therapeutic effects, they may have their challenges like the possibility of drug-drug interactions and severe side effects. Furthermore, differences in pharmacokinetic profiles among drugs may raise significant concerns and limitations.
Gene-based therapies have proven to be a vital component of precision medicine by utilizing techniques such as gene inhibition, addition, replacement, or editing [191]. This approach has demonstrated considerable success in the treatment of various diseases including neuromuscular diseases and liver metabolic diseases as well [192]. Furthermore, the integration of multi-omics has significantly enhanced our comprehension of the molecular mechanisms associated with tumorigenesis and tumor development, leading to the identification of numerous potential oncological targets. RNA therapeutics, or RNATx, represent a promising approach for treating diseases by targeting or utilizing RNA molecules for therapeutic purposes.
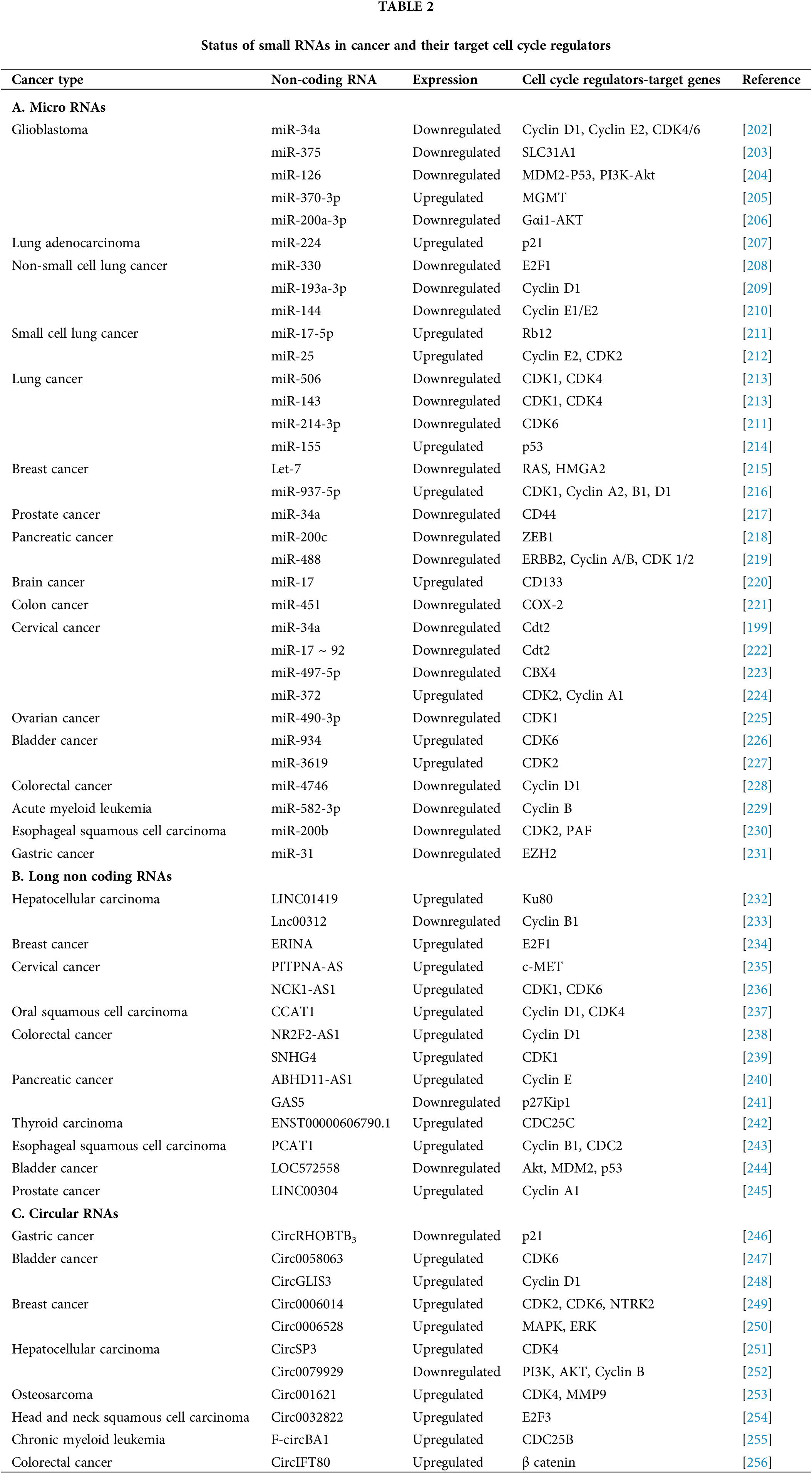
miRNAs are small non-coding RNA molecules that help to maintain homeostatic cell signaling and have been shown to regulate the translation of nearly >60% of total genes including genes involved in cell cycle regulation as well. This is achieved by either repressing translation or destabilizing mRNA of cell cycle proteins. Multiple miRNAs have been found to be aberrantly expressed in cancer due to mutations in genes involved in their biogenesis or dysregulated cell signaling [193,194]. Although miRNAs are dysregulated in nearly every disease, many of them are depleted in cancers. Depleted miRNAs are often classified as tumor-suppressive miRNAs. Restoring these tumor-suppressive miRNAs, such as miR-34a or let-7, can inhibit tumor growth in-vivo and prevent essential oncological processes like invasion, migration, and metastasis. miR-34a, in particular, has been extensively studied for its tumor-suppressive role in cancer. It contributes to p53 function by targeting multiple p53 inhibitors, including histone deacetylases SIRT1 and HDAC1, and MTA2, which deacetylates p53, thereby increasing the transcriptional activity of p53 [195–197]. miR-34a can also induce p53 activation by directly targeting MDM4, a strong p53 transactivation inhibitor [198]. miR-34a also suppresses a S-phase oncogenic factor CDC-10 dependent transcript 2 (Cdt2) and leads to suppression of cervical cancer cells [199]. Due to these anti-cancer properties, miR-34a was the first tumor-suppressive miRNA to be investigated as an anti-cancer agent in clinical trials; however, the study was terminated due to severe immune-related side effects [200]. To address these challenges, a miRNA delivery strategy using an endosomal escape agent DUPA-nigericin was developed which resulted in decreased cell proliferation in-vitro and delayed tumor growth in-vivo. Thus, miR-34a is one such example of a tumor-suppressive miRNA that has shown immense promise towards cancer suppression and treatment [201]. Apart from miR-34a, there are other miRNAs mentioned in Table 2A, which could prove to be quite potent in combating cancers.
In one of the studies performed by Hsu et al., the effective role of a microRNA miR-31-5p in an Oxaliplatin (OXA)-resistant colorectal cancer (CRC) cells both in-vitro and in-vivo has been shown. Oxaliplatin is currently used in first-line chemotherapy to treat Stage III and Stage IV metastatic CRC. They showed that the upregulated expression of FOXC1 and miR-31-5p inhibited LATS2 expression, which led to OXA resistance. They further showed that the knockdown of miR-31-5p results in cancer cell apoptosis, decreased cell proliferation, and enhanced chemosensitivity [257].
Long non-coding RNAs (lncRNAs)
LncRNAs are stake targets due to their capacity to regulate a wide range of oncogenic molecular networks in a cell. These RNAs play a crucial role in controlling gene expression both in the nucleus and cytoplasm by interacting directly or indirectly with DNA, RNA, and proteins [258]. Dysregulated lncRNA expression has been linked with adverse outcomes for cancer patients (Table 2B). When lncRNAs are deregulated during the G1 phase, cells become more susceptible to uncontrolled mitogenic growth and defects in the G1-S transition [258,259].
During the transition from the G1 to S-phase of cell cycle, lncRNAs exert regulatory control over various E2F proteins, influencing cell proliferation and growth in the cancer cells. For instance, lncRNA regulating c-Myc (E2F1 mRNA-stabilizing factor; EMS) is involved in this process. lncRNAs can also directly regulate the E2F mRNA for G1 to S-phase transition. Linc00337, located in the 1p36.31 genomic region, has been reported to coactivate E2F1 mRNA and upregulate its expression leading to cancer progression in pancreatic ductal carcinoma [260]. Additionally, in breast cancer inhibition of linc00337 using small hairpin RNA (shRNA) sensitizes the cancer cells to paclitaxel and hence can be used as a combination therapy to overcome the drug resistance [260]. Apart from E2Fs, lncRNAs can also regulate the cell cycle by targeting cyclin-CDK complexes during G1 progression. They transcriptionally regulate CDK encoding genes as well as directly interact with cyclin and CDKs, affecting the protein phosphorylation and stability. LncRNAs also act as a scaffold that brings together multiple proteins involved in CDK regulation. In stress conditions, the CDC28 lncRNA recruits Hog1 (a stress-activated protein kinase) to CDC28 which in turn helps in recruiting chromatin structure remodelling (RSC) complex, resulting in increased CDC28 transcription and more efficient cell cycle progression upon stress [261]. Another lncRNA, DILA1 has been reported to interact directly with cyclin D1, preventing its phosphorylation at Thr-286. This in turn stabilizes cyclin D1 by suppressing its glycogen synthase kinase 3β (GSK3β)-mediated ubiquitination-dependent degradation. Stabilization of cyclin D1 promotes inactivation of pRb by phosphorylating at Ser-780 which in turn promotes G1/S cell cycle progression. Furthermore, high DILA1 expression is associated with poor prognosis and tamoxifen resistance in breast cancer patients [262]. lncRNA RP11-624L4.1 directly interacts with CDK4 and stabilizes its expression leading to the upregulation of CDK4/6-cyclin D1-Rb-E2F1 pathway, promoting G1 cell cycle progression in nasopharyngeal carcinoma (NPC) cells [263]. Several other lncRNAs have also been reported to be differentially expressed in different cancers (Table 2B) [264–266]. Some of which are hypomethylated and highly expressed in tumors whereas others are hypermethylated and expressed in lower levels in cancers. The S-phase exclusive lncRNAs; LINC00704, LUCAT1, and MIAT, have been reported to be essential for cancer development. Loss-of-function of these lncRNAs accumulates the cells in the G1 phase while decreasing the proportion of cells in the G2/M phase [267–270]. LINC00704 accelerates the cell cycle and contributes to proliferation in nasopharyngeal carcinoma cells by regulating ETS1/CDK6 axis [271] and in papillary thyroid carcinoma by regulating miR-204-5p/HMGB1 axis [268]. LncRNA LUCAT1 also called as SCAL1 has found to be over-expressed [270] in a variety of cancers including choroidal melanoma [265], breast [272], ovarian [273], cervical [274], thyroid [266], and kidney [270] where it contributes to proliferation of ovarian cancer cells by regulating miR-199a-5p expression [275]. LncRNA LUCAT1 contributes to cell proliferation and migration in human pancreatic ductal adenocarcinoma via sponging miR-539 [276]. Hypoxia-induced regulation of PTBP1 by lncRNA LUCATI modulates cancer cell viability and chemotherapy response [277].
Many lncRNAs can also function as competing endogenous RNAs (ceRNAs) by sequestering miRNAs that target cell cycle regulators which could modulate cell proliferation. In thyroid cancer lncRNA MIAT functions as ceRNA to target EZH2 by sponging miR-150-5p [278]. lncRNA H19 (lncH19) regulate E2F1 level by sponging miR-29a in renal cell carcinoma [279].
circRNAs are non-coding, single-stranded, covalently closed circular RNA that differ from traditional linear RNA. Majorly, circRNAs are derived from back splicing of pre-mRNA [280,281]. High-throughput RNA sequencing and circRNA bioinformatics analysis studies have identified thousands of circRNAs in eukaryotes which show tissue and lineage-specific expression [282–284]. Recent studies have shown the emerging roles of circRNA in transcription regulation, miRNA sponging, and protein decoys [285,286]. These circRNA are observed to be dysregulated in many diseases including cancers [287]. Most of the research has been done on the function of circRNAs in solid tumors, where they have been identified as either oncogenes or tumor suppressors [288–291] (Table 2C). Recently, the role of circRNA in tumor progression by regulating cell cycle factors was explored. circRNAs regulate the cell cycle gene expression either indirectly by sponging miRNA or lncRNA involved in cell cycle regulation or by directly interacting with the regulator proteins. CircMYLK has been found to be significantly upregulated in laryngeal squamous cell carcinoma (LSCC). Gain-of-function of circMYLK elevates cyclin D1 levels by sponging miR-195 facilitating LSCC cell proliferation and G1/S cell cycle transition [292]. A CCND1 derived circular RNA, circ-CCND1 is significantly upregulated in LSCC and, is associated with aggressive clinical features with adverse prognosis. circ-CCND1 interacts with HuR protein to stabilize CCND1 mRNA promoting CCND1 translation by sponging miR-646, hence, promotes LSCC tumorigenesis [293]. Another study reported the up-regulated circRNA; circPUM1 which leads to an increase of cyclin D1 and Bcl-2 expression by sponging miR-326 thereby, facilitating cell proliferation, migration, and invasion of lung adenocarcinoma [294]. In 2019, Li et al. reported upregulation of hsa_circ_000984 in NSCLC tissues and cell lines, which exerts oncogenic effects by regulating the expression levels of β-catenin, c-Myc, and cyclin D1, thereby activating Wnt/β-catenin pathway [295]. Also, circ-CMPK1/miR-302e/cyclin D1 signaling pathway has been reported to play a significant role in NSCLC, and can be targeted to develop an effective treatment [291]. In CRC, circ5615 is significantly up-regulated exhibiting an oncogenic role. Silencing of circ5615, leads to sponging of miR-149-5p and upregulation of tankyrase, (a regulator of β-catenin stabilization), thereby inducing β-catenin and cyclin D1 expression [290]. Silencing of hsa_circ_0035483 increases gemcitabine sensitivity and inhibits tumor growth in renal cancer cells via regulation of miR-335/CCNB1 axis [289]. hsa_circ_0136666 upregulates CDK6 expression by sponging miR-1299 to induce G2/M phase transition and promote breast cancer cell proliferation [296]. However, most of the circRNAs have been reported to play oncogenic role, some of them inhibit the cell cycle transition via diverse molecular mechanisms (Table 2C). For example, circNR3C1 has been found to be significantly downregulated in bladder cancer and its upregulation can significantly suppress cell proliferation and cell cycle progression by sponging miR-27a-3p, thereby inhibiting cyclin D1 expression [297].
In recent years, considerable progress has been made in the clinical application of RNA-based therapeutics, primarily through the use of antisense oligonucleotides and small interfering RNAs, with several gaining FDA approval for the treatment of conditions such as Duchenne muscular dystrophy and Spinal muscular atrophy [192]. Nevertheless, there are various limitations associated with RNA therapeutics, including challenges related to RNA delivery, stability, immunogenicity, and off-target effects. These issues need to be addressed before the development of RNA therapeutics for cancers [298]. Currently, several RNA-based potential treatments are in clinical trials and developmental pipelines. Despite all the limitations, non-coding RNA-based therapies hold significant potential in the treatment of cancer. These therapies aim to utilize non-coding RNAs, such as miRNAs, lncRNAs, circRNAs, and small interfering RNAs (siRNAs), to modulate gene expression and control various cellular processes specially cell cycle pathways involved in cancer development and progression. One strategy involves the use of anti-miRNA oligonucleotides and siRNAs to specifically target and inhibit oncogenic miRNAs or key cancer-related genes, thereby reducing tumor growth and promoting cancer cell death [299]. On the other hand, certain tumor-suppressive miRNAs or lncRNAs can be therapeutically delivered to restore their normal function and inhibit cancer. The most challenging task is their effective and accurate delivery mechanism [300]. Moreover, RNA-based therapies can be designed to target specific signaling pathways implicated in cancer, providing a precise and personalized treatment approach. In addition to RNA therapy, alternative gene therapy approaches, such as recombinant protein therapy utilizing appropriate plasmids for genetic correction, have emerged as viable options for the successful targeted delivery of the protein of interest [301].
Targeting ubiquitin ligase system
Ubiquitin ligase system is one of the most implicated systems which involve ubiquitylation of cellular targets leading to control of numerous cellular functions. In cell cycle, ubiquitination mechanism is involved in timely degradation of protein for the transition of cell from one phase to another in most regulated manner (Fig. 3). The deregulation of the components of this elaborated system leads to disease conditions like cancer. Researches over the decades has shown that alteration in the ubiquitin system in cancer can be exploited for the molecular diagnostics and development of novel therapeutics [57,302,303].
APC is one of the major E3 ubiquitin ligases involved in the regulation of timely cell cycle progression by forming two E3 ubiquitin ligase complexes, APCCDC20 and APCCDH1. Recent studies have revealed that CDH1 functions as a tumor suppressor, while CDC20 may act as an oncoprotein [304–307] to promote the development and progression of human cancers, making it a potential target for therapeutics. In 2015, Zhang et al., have identified a compound “331” which inhibits CDC20 expression by upregulating miR-494 to induce cell death in glioma cells without any cytotoxic effect in astrocytes [306]. Another natural ligand-based drug ZINC000004098930 has been shown to target and inhibit CDC20, which has shown promising results for TNBC-targeted treatment [308]. A new apcin-based inhibitor for CDC20, designed by replacing the pyrimidine group with substituted thiazole-containing groups, shows improved antitumor activity [102]. A MAD2 inhibitor, M21-1, also inhibits CDC20 activity by disrupting the CDC20-MAD2 interaction, and consequently, the assembly of the mitotic checkpoint complex increasing the sensitivity of cancer cells to antimitotic drugs in myeloid cell leukemia-1 (MCL-1) [309]. Moreover, a triterpene mixture extracted from the mushroom Poria cocos, polyporenic acid C, can downregulate the expression of CDC20 in some cancer cells and inhibit cancer metastasis [307]. Cdt2 is an essential S-phase regulator protein that forms an E3 ubiquitin ligase system in association with its partner CRL4 to regulate cellular processes like re-replication, DNA damage repair, and proliferation [310–314]. Despite its crucial role, it is amplified in around 37 cancer types. NEDD8 targeting drug MLN4924 inhibits the CRL4Cdt2 complex formation, leading to DNA re-replication by stabilizing Cdt1 in S-phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells [315]. miR-34a and miR-17~92 can also suppress the expression of Cdt2 protein to inhibit CRL4Cdt2 complex formation in cervical cancer cells, thereby suppressing the tumor progression [199,222]. MDM2 is a regulator of p53 and is implicated in the regulation of diverse cellular processes including cell cycle, apoptosis, senescence, and DNA repair [316]. Multiple small molecules have been reported to target MDM2 but none of them showed any promising results in clinical trials [317–319]. Recently another small molecule, AMG-232, a piperidinone-based compound, has been reported to act by binding to the p53 binding pocket in MDM2 [320], which results in the inhibition of p53-MDM2 complex formation. So far in clinical trials, the drug is showing modest safety and tolerability with similar but milder side effects than previous MDM2 inhibitors [321]. Apart from this, other accessory proteins that are involved in maintaining the ubiquitin system have also been observed to be aberrantly expressed during cancer and can be targeted for therapeutic intervention of cancer. MK-1775, a potent small-molecule inhibitor targeting the WEE1 protein, is currently under development for clinical trials in solid tumors [322–324]. A CDC25 inhibitor, NSC663284 derivative 6 has shown anti-proliferative effects in colon and colorectal carcinoma [325,326]. Deubiquitinase molecules (DUBs) have shown significant influence in regulating the specific substrates contributing to cancer progression. Therefore, targeted inhibition of DUBs is a therapeutic strategy for cancer treatment. Knockdown of USP3 leads to suppression of CDC25A, causing a delay in cell cycle progression [327]. Knockdown of USP21, USP39, USP7, and OTUB1 leads to downregulated FOXM1’s transcription activity hence halting the cell cycle in breast cancer [328–331]. Targeting ubiquitin ligase can be a potential therapeutics to deal with drug resistance. However, it may exhibit certain limitations in addressing the same. Firstly, the diversity of the ubiquitin ligase system impedes a comprehensive understanding of their functions and corresponding target genes. Secondly, E3 ubiquitin systems possess the capacity to bind to multiple oncogenes. A single E3 not only interacts with one oncogene or tumor suppressor gene but also has the potential to engage with multiple oncogenes or tumor suppressor genes simultaneously.
The efficacy of cell cycle-targeted therapies in the future will depend on the development of highly specific, least toxic, and potent compounds, targeting particular vulnerabilities of the cancer cells. Screening methodologies utilizing cell cultures, patient-derived xenografts, and genetically modified mouse models will play a critical role in elucidating synthetic lethal interactions between genomic abnormalities and targeted inhibition of specific cell cycle proteins. Novel therapeutic approaches capable of targeting multiple components within the same pathway, such as microRNAs, may also offer therapeutic advantages. Furthermore, the combination of multiple selective inhibitors may significantly enhance clinical efficacy; however, in numerous instances, combination therapies have resulted in a substantial increase in adverse effects, potentially rendering them intolerable in a significant number of patients. Moreover, a very small percentage of patients had exhibited long-term satisfactory responses for currently available targeted therapies. Consequently, genomic technologies will become crucial diagnostic tools for identifying predictive biomarkers in patient sub-groups. Preclinical research and clinical observations (Table S1) have shown the development of drug resistance, characterized by cancer relapse after an initial positive response. Investigating the mechanisms behind drug resistance will offer valuable insights into how existing compounds are countered, potentially leading to improved treatment strategies for patients who have relapsed or become refractory. Furthermore, this research could aid in designing a combination therapy aimed to prevent cancer cells from developing resistance. Another potential therapeutic approach to address drug resistance could be targeting ubiquitin ligases; however, due to its anticipated severe side effects, it requires extensive research before progressing to clinical trials. A critical issue that needs to be addressed for the development of cell cycle-specific novel therapeutic formulations is the immunogenic response. Further research is necessary to identify intersections where immune response could complement cell cycle-based novel therapies. Addressing the aforementioned challenges will create opportunities for managing cancer through therapeutic approaches that induce cancer cells to permanently exit the cell cycle.
The cell cycle machinery, which includes CDKs and cyclin subunits, is tightly regulated by a network of proteins. The past decade has witnessed substantial advancements in their comprehensive role in cancers. For instance, aberrations in CDK4/6, CDK2, and checkpoint proteins such as p53 and Rb, which are crucial for maintaining genomic stability, have been implicated in numerous cancer types. The identification of these aberrations has not only enhanced our mechanistic understanding of cancer biology but also made them highly potent targets for developing cancer therapy. The development of novel compounds through structure-based drug design and high-throughput screening platforms have allowed for the translation of cell cycle studies from bench to bedside. Inhibitors that selectively target CDK4/6, as well as CDK2 and CDK1, are being developed and may offer clinical benefits for certain cancer subtypes specifically. While the first clinical translation of a CDK4/6 selective inhibitor (palbociclib) in breast cancer has been successful, current targeted therapies face the challenge of relatively low response rates and the emergence of drug resistance. To overcome these challenges, novel approaches such as combination therapy, gene therapy, and E3 ubiquitin inhibitors may be employed as valuable therapeutics. Furthermore, a combination of several selective inhibitors may significantly improve clinical efficacy, but it may also increase the risk of adverse effects, limiting their tolerability in many patients. Additionally, developing drugs that target the ubiquitination system represents a promising approach for the treatment of cancer. Although only a limited number of such drugs have been approved by the FDA, a deeper and mechanistic understanding of ubiquitin signalling cascade in regulating cell cycle progression has the potential to expand and enhance current anticancer therapies culminating in improved clinical outcomes. Advances in RNA therapeutics have highlighted the importance of non-coding RNAs (ncRNAs) in cell cycle regulation and their potential as therapeutic targets in cancer. Various strategies have been proposed for harnessing ncRNAs as therapeutic agents, including miRNA mimics that have been tested in clinical trials for cancer treatment. However, other strategies associated with ncRNAs remain in their infancy, therefore, comprehensive investigations are necessary to unlock the potential of ncRNA-based therapeutic agents in targeting cell cycle regulation for advancing future anticancer drug development. Overall, our enhanced understanding of cell cycle control and cancer is guiding current treatments and creating therapeutic opportunities to improve initial treatment with curative intent, either through greater precision or by extending treatment modalities and better-informed treatment decisions that will result in improved outcomes for patients.
Acknowledgement: The authors are thankful to the Director Prof. Sanjay Kumar, Dean Prof. S.M. Singh, Institute of Science, Banaras Hindu University and Co-Ordinator, School of Biotechnology, Institute of Science, Banaras Hindu University for providing space and facilities. Authors are also thankful to Department of Biotechnology, DST-FIST, UGC, and IoE for providing the grant support the School of Biotechnology and the lab.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: The authors confirm their contribution to the paper as follows: Study conception and design: Samarendra Kumari Singh; Draft manuscript preparation: Garima Singh, Sonika Kumari Sharma, Neelu Mishra, Aastha Soni and Manshi Kumari; Review and editing: Samarendra Kumar Singh, Garima Singh, Sonika Kumari Sharma, Neelu Mishra, Aastha Soni and Manshi Kumari; Supervision: Samarendra Kumar Singh. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
Supplementary Materials: The supplementary material is available online at https://doi.org/10.32604/biocell.2024.056503.
References
1. Park M-T, Lee S-J. Cell cycle and cancer. J Biochem Mol Bio. 2003;36(1):60–5. [Google Scholar] [PubMed]
2. Collins K, Jacks T, Pavletich NP. The cell cycle and cancer. Proc Natl Acad Sci U S A. 1997;94(7):2776–8. doi:10.1073/pnas.94.7.2776. [Google Scholar] [PubMed] [CrossRef]
3. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi:10.1016/j.cell.2011.02.013. [Google Scholar] [PubMed] [CrossRef]
4. Cavalu S, Abdelhamid AM, Saber S, Elmorsy EA, Hamad RS, Abdel-Reheim MA, et al. Cell cycle machinery in oncology: a comprehensive review of therapeutic targets. FASEB J. 2024;38(11):e23734. doi:10.1096/fj.202400769R. [Google Scholar] [PubMed] [CrossRef]
5. Stallaert W, Kedziora KM, Taylor CD, Zikry TM, Ranek JS, Sobon HK, et al. The structure of the human cell cycle. Cell Syst. 2022;13(3):230–40. doi:10.1016/j.cels.2021.10.007. [Google Scholar] [PubMed] [CrossRef]
6. Smith HL, Southgate H, Tweddle DA, Curtin NJ. DNA damage checkpoint kinases in cancer. Expert Rev Mol Med. 2020;22(8):e2. doi:10.1017/erm.2020.3. [Google Scholar] [PubMed] [CrossRef]
7. Pedroza-Garcia JA, Xiang Y, De Veylder L. Cell cycle checkpoint control in response to DNA damage by environmental stresses. Plant J. 2022;109(103):490–507. doi:10.1111/tpj.15567. [Google Scholar] [PubMed] [CrossRef]
8. Yuan B, Xu Y, Woo JH, Wang Y, Bae YK, Yoon DS, et al. Increased expression of mitotic checkpoint genes in breast cancer cells with chromosomal instability. Clin Cancer Res. 2006;12(2):405–10. doi:10.1158/1078-0432.CCR-05-0903. [Google Scholar] [PubMed] [CrossRef]
9. González-Garrido C, Prado F. Novel insights into the roles of Cdc7 in response to replication stress. FEBS J. 2023;290(12):3076–88. doi:10.1111/febs.16456. [Google Scholar] [PubMed] [CrossRef]
10. Choi BH, Colon TM, Lee E, Kou Z, Dai W. Abstract 1969: PTEN plays a role in histone modifications during mitosis. Cancer Res. 2021;81(13_Supplement):1969. doi:10.1158/1538-7445.am2021-1969. [Google Scholar] [CrossRef]
11. Roy B, Han SJY, Fontan AN, Jema S, Joglekar AP. Aurora B phosphorylates Bub1 to promote spindle assembly checkpoint signaling. Curr Biol. 2022;32(1):237–47. doi:10.1016/j.cub.2021.10.049. [Google Scholar] [PubMed] [CrossRef]
12. Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol. 2007;19(2):238–45. doi:10.1016/j.ceb.2007.02.009. [Google Scholar] [PubMed] [CrossRef]
13. Matthews HK, Bertoli C, de Bruin RAM. Cell cycle control in cancer. Nat Rev Mol Cell Biol. 2022;23(1):74–88. doi:10.1038/s41580-021-00404-3. [Google Scholar] [PubMed] [CrossRef]
14. Foster DA, Yellen P, Xu L, Saqcena M. Regulation of G1 cell cycle progression: distinguishing the restriction point from a nutrient-sensing cell growth checkpoint(s). Genes Cancer. 2010;1(11):1124–31. doi:10.1177/1947601910392989. [Google Scholar] [PubMed] [CrossRef]
15. Barnum KJ, O’Connell MJ. Cell cycle regulation by checkpoints. Methods Mol Biol. 2014;1170(4):29–40. doi:10.1007/978-1-4939-0888-2_2. [Google Scholar] [PubMed] [CrossRef]
16. Tokunaga Y, Otsuyama KI, Hayashida N. Cell cycle regulation by heat shock transcription factors. Cells. 2022;11(2):203. doi:10.3390/cells11020203. [Google Scholar] [PubMed] [CrossRef]
17. Hunt T. The discovery of cyclin (I). Cell. 2004;116(2 Suppl):S63–4. doi:10.1016/S0092-8674(04)00030-3. [Google Scholar] [PubMed] [CrossRef]
18. Martínez-Alonso D, Malumbres M. Mammalian cell cycle cyclins. Semin Cell Dev Biol. 2020;107(2):28–35. doi:10.1016/j.semcdb.2020.03.009. [Google Scholar] [PubMed] [CrossRef]
19. Uzbekov R, Prigent C. A journey through time on the discovery of cell cycle regulation. Cells. 2022;11(4):704. doi:10.3390/cells11040704. [Google Scholar] [PubMed] [CrossRef]
20. Suski JM, Braun M, Strmiska V, Sicinski P. Targeting cell-cycle machinery in cancer. Cancer Cell. 2021;39(6):759–78. doi:10.1016/j.ccell.2021.03.010. [Google Scholar] [PubMed] [CrossRef]
21. Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov. 2016;6(4):353–67. doi:10.1158/2159-8290.CD-15-0894. [Google Scholar] [PubMed] [CrossRef]
22. Oswald F, Lovec H, Möröy T, Lipp M. E2F-dependent regulation of human MYC: trans-activation by cyclins D1 and A overrides tumour suppressor protein functions. Oncogene. 1994;9(7):2029–36. [Google Scholar] [PubMed]
23. Stevaux O, Dyson NJ. A revised picture of the E2F transcriptional network and RB function. Curr Opin Cell Biol. 2002;14(6):684–91. doi:10.1016/S0955-0674(02)00388-5. [Google Scholar] [PubMed] [CrossRef]
24. Oswald F, Dobner T, Lipp M. The E2F transcription factor activates a replication-dependent human H2A gene in early S phase of the cell cycle. Mol Cell Biol. 1996;16(5):1889–95. doi:10.1128/mcb.16.5.1889. [Google Scholar] [PubMed] [CrossRef]
25. Nataraj SE, Blain SW. A cyclin D-CDK6 dimer helps to reshuffle cyclin-dependent kinase inhibitors (CKI) to overcome TGF-beta-mediated arrest and maintain CDK2 activity. Cell Cycle. 2021;20(8):808–18. doi:10.1080/15384101.2021.1909261. [Google Scholar] [PubMed] [CrossRef]
26. Fassl A, Geng Y, Sicinski P. CDK4 and CDK6 kinases: from basic science to cancer therapy. Science. 2022;375(6577):eabc1495. doi:10.1126/science.abc1495. [Google Scholar] [PubMed] [CrossRef]
27. Gong X, Litchfield LM, Webster Y, Chio LC, Wong SS, Stewart TR, et al. Genomic aberrations that activate D-type cyclins are associated with enhanced sensitivity to the CDK4 and CDK6 inhibitor abemaciclib. Cancer Cell. 2017;32(6):761–76.e6. doi:10.1016/j.ccell.2017.11.006. [Google Scholar] [PubMed] [CrossRef]
28. Susanti NMP, Tjahjono DH. Cyclin-dependent kinase 4 and 6 inhibitors in cell cycle dysregulation for breast cancer treatment. Molecules. 2021;26(15):4462. doi:10.3390/molecules26154462. [Google Scholar] [PubMed] [CrossRef]
29. Wee P, Wang Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers. 2017;9(5):52. doi:10.3390/cancers9050052. [Google Scholar] [PubMed] [CrossRef]
30. Yildirim H, Nergiz D, Sadullahoglu C, Akgunduz Z, Yildirim S, Dogan S, et al. The extent of cyclin D1 expression in endometrial pathologies and relevance of cyclin D1 with the clinicopathological features of endometrioid endometrial carcinoma. Indian J Pathol Microbiol. 2020;63(3):412–7. doi:10.4103/IJPM.IJPM_589_19. [Google Scholar] [PubMed] [CrossRef]
31. Liu Z, Zhao M, Jiang X, Zhang Y, Zhang S, Xu Y, et al. Upregulation of KLHL17 promotes the proliferation and migration of non-small cell lung cancer by activating the Ras/MAPK signaling pathway. Lab Invest. 2022;102(12):1389–99. doi:10.1038/s41374-022-00806-7. [Google Scholar] [PubMed] [CrossRef]
32. Novotný J, Bandúrová V, Strnad H, Chovanec M, Hradilová M, Šáchová J, et al. Analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas and paired normal mucosae reveals cyclin D1 deregulation and compensatory effect of cyclin D2. Cancers. 2020;12(4):792. doi:10.3390/cancers12040792. [Google Scholar] [PubMed] [CrossRef]
33. Tchakarska G, Sola B. The double dealing of cyclin D1. Cell Cycle. 2020;19(2):163–78. doi:10.1080/15384101.2019.1706903. [Google Scholar] [PubMed] [CrossRef]
34. Meiftasari A, Januar Caesar WP, Novarina A, Julika Yovi W, Jenie RI. Ethanolic extract of hedyotis corymbosa and its combination with 5-FU inhibit cyclin D expression on WiDr colorectal cancer cell. Indonesian J Cancer Chemopreventi. 2016;7(1):25–30. doi:10.14499/indonesianjcanchemoprev7iss1pp25-30. [Google Scholar] [CrossRef]
35. Ray-Coquard I, Monk BJ, Lorusso D, Mahdi H, Upadhyay V, Graul R, et al. The promise of combining CDK4/6 inhibition with hormonal therapy in the first-line treatment setting for metastatic or recurrent endometrial adenocarcinoma. Int J Gynecol Cancer. 2023;33(12):1943–9. doi:10.1136/ijgc-2023-004739. [Google Scholar] [PubMed] [CrossRef]
36. Cheung TH, Yu MMY, Lo KWK, Yim SF, Chung TKH, Wong YF. Alteration of cyclin D1 and CDK4 gene in carcinoma of uterine cervix. Cancer Lett. 2001;166(2):199–206. doi:10.1016/S0304-3835(01)00457-8. [Google Scholar] [PubMed] [CrossRef]
37. Garutti M, Targato G, Buriolla S, Palmero L, Minisini AM, Puglisi F. Cdk4/6 inhibitors in melanoma: a comprehensive review. Cells. 2021;10(6):1334. doi:10.3390/cells10061334. [Google Scholar] [PubMed] [CrossRef]
38. Villano JL, Morgan RM, Zhang S, Kolesar J, Xiu J, Seifert H, et al. Association of CDK4 amplification with duration of response to bevacizumab in glioblastoma. J Clin Oncol. 2023;41(16_suppl):2026. doi:10.1200/jco.2023.41.16_suppl.2026. [Google Scholar] [CrossRef]
39. Kim YT, Zhao M. Aberrant cell cycle regulation in cervical carcinoma. Yonsei Med J. 2005;46(5):597–613. doi:10.3349/ymj.2005.46.5.597. [Google Scholar] [PubMed] [CrossRef]
40. Saba KH, Difilippo V, Styring E, Nilsson J, Magnusson L, van den Bos H, et al. CDK4 is co-amplified with either TP53 promoter gene fusions or MDM2 through distinct mechanisms in osteosarcoma. npj Genom Med. 2024;9(1):42. doi:10.1038/s41525-024-00430-y. [Google Scholar] [PubMed] [CrossRef]
41. Park S, Lee J, Do IG, Jang J, Rho K, Ahn S, et al. Aberrant CDK4 amplification in refractory rhabdomyosarcoma as identified by genomic profiling. Sci Rep. 2014;4(1):3623. doi:10.1038/srep03623. [Google Scholar] [PubMed] [CrossRef]
42. Lee S, Park H, Ha SY, Paik KY, Lee SE, Kim JM, et al. CDK4 amplification predicts recurrence of well-differentiated liposarcoma of the abdomen. PLoS One. 2014;9(8):e99452. doi:10.1371/journal.pone.0099452. [Google Scholar] [PubMed] [CrossRef]
43. Liu W, Yi JM, Liu Y, Chen C, Zhang KX, Zhou C, et al. CDK6 is a potential prognostic biomarker in acute myeloid leukemia. Front Genet. 2021;11:600227. doi:10.3389/fgene.2020.600227. [Google Scholar] [PubMed] [CrossRef]
44. Li B, He H, Tao BB, Zhao ZY, Hu GH, Luo C, et al. Knockdown of CDK6 enhances glioma sensitivity to chemotherapy. Oncol Rep. 2012;28(3):909–14. doi:10.3892/or.2012.1884. [Google Scholar] [PubMed] [CrossRef]
45. Zhang J, Gan Y, Li H, Yin J, He X, Lin L, et al. Inhibition of the CDK2 and Cyclin A complex leads to autophagic degradation of CDK2 in cancer cells. Nat Commun. 2022;13(1):2835. doi:10.1038/s41467-022-30264-0. [Google Scholar] [PubMed] [CrossRef]
46. Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer. 2017;17(2):93–115. doi:10.1038/nrc.2016.138. [Google Scholar] [PubMed] [CrossRef]
47. Di Fiore R, Suleiman S, Drago-Ferrante R, Subbannayya Y, Suleiman S, Vasileva-Slaveva M, et al. The role of FBXW7 in gynecologic malignancies. Cells. 2023;12(10):1415. doi:10.3390/cells12101415. [Google Scholar] [PubMed] [CrossRef]
48. Fagundes R, Teixeira LK. Cyclin E/CDK2: DNA replication, replication stress and genomic instability. Front Cell Dev Biol. 2021;9:774845. doi:10.3389/fcell.2021.774845. [Google Scholar] [PubMed] [CrossRef]
49. Fragkos M, Ganier O, Coulombe P, Méchali M. DNA replication origin activation in space and time. Nat Rev Mol Cell Biol. 2015;16(6):360–74. doi:10.1038/nrm4002. [Google Scholar] [PubMed] [CrossRef]
50. Musiałek MW, Rybaczek D. Behavior of replication origins in Eukaryota—Spatio-temporal dynamics of licensing and firing. Cell Cycle. 2015;14(14):2251–64. doi:10.1080/15384101.2015.1056421. [Google Scholar] [PubMed] [CrossRef]
51. Ratnayeke N, Baris Y, Chung M, Yeeles JTP, Meyer T. CDT1 inhibits CMG helicase in early S phase to separate origin licensing from DNA synthesis. Mol Cell. 2023;83(1):26–42.e13. doi:10.1016/j.molcel.2022.12.004. [Google Scholar] [PubMed] [CrossRef]
52. Woo RA, Poon RYC. Cyclin-dependent kinases and S phase control in mammalian cells. Cell Cycle. 2003;2(4):316–24. doi:10.4161/cc.2.4.468. [Google Scholar] [CrossRef]
53. Jones MJK, Gelot C, Munk S, Koren A, Kawasoe Y, George KA, et al. Human DDK rescues stalled forks and counteracts checkpoint inhibition at unfired origins to complete DNA replication. Mol Cell. 2021;81(3):426–41. doi:10.1016/j.molcel.2021.01.004. [Google Scholar] [PubMed] [CrossRef]
54. Suski JM, Ratnayeke N, Braun M, Zhang T, Strmiska V, Michowski W, et al. CDC7-independent G1/S transition revealed by targeted protein degradation. Nature. 2022;605(7909):357–65. doi:10.1038/s41586-022-04698-x. [Google Scholar] [PubMed] [CrossRef]
55. Burgers PMJ, Kunkel TA. Eukaryotic DNA replication fork. Annu Rev Biochem. 2017;86(1):417–38. doi:10.1146/annurev-biochem-061516-044709. [Google Scholar] [PubMed] [CrossRef]
56. Yamazaki H, Takagi M, Kosako H, Hirano T, Yoshimura SH. Cell cycle-specific phase separation regulated by protein charge blockiness. Nat Cell Biol. 2022;24(5):625–32. doi:10.1038/s41556-022-00903-1. [Google Scholar] [PubMed] [CrossRef]
57. Dang F, Nie L, Wei W. Ubiquitin signaling in cell cycle control and tumorigenesis. Cell Death Differ. 2021;28(2):427–38. doi:10.1038/s41418-020-00648-0. [Google Scholar] [PubMed] [CrossRef]
58. Laplante G, Zhang W. Targeting the ubiquitin-proteasome system for cancer therapeutics by small-molecule inhibitors. Cancers. 2021;13(12):3079. doi:10.3390/cancers13123079. [Google Scholar] [PubMed] [CrossRef]
59. Oakes V, Wang W, Harrington B, Lee WJ, Beamish H, Chia KM, et al. Cyclin A/Cdk2 regulates Cdh1 and claspin during late S/G2 phase of the cell cycle. Cell Cycle. 2014;13(20):3302–11. doi:10.4161/15384101.2014.949111. [Google Scholar] [PubMed] [CrossRef]
60. Gorski JW, Ueland FR, Kolesar JM. CCNE1 amplification as a predictive biomarker of chemotherapy resistance in epithelial ovarian cancer. Diagnostics. 2020;10(5):279. doi:10.3390/diagnostics10050279. [Google Scholar] [PubMed] [CrossRef]
61. Zheng X, Chen L, Liu W, Zhao S, Yan Y, Zhao J, et al. CCNE1 is a predictive and immunotherapeutic indicator in various cancers including UCEC: a pan-cancer analysis. Hereditas. 2023;160(1):13. doi:10.1186/s41065-023-00273-0. [Google Scholar] [PubMed] [CrossRef]
62. Aziz K, Limzerwala JF, Sturmlechner I, Hurley E, Zhang C, Jeganathan KB, et al. Ccne1 overexpression causes chromosome instability in liver cells and liver tumor development in mice. Gastroenterology. 2019;157(1):210–26.e12. doi:10.1053/j.gastro.2019.03.016. [Google Scholar] [PubMed] [CrossRef]
63. Yin X, Yu J, Zhou Y, Wang C, Jiao Z, Qian Z, et al. Identification of CDK2 as a novel target in treatment of prostate cancer. Future Oncol. 2018;14(8):709–18. doi:10.2217/fon-2017-0561. [Google Scholar] [PubMed] [CrossRef]
64. Faber AC, Chiles TC. Inhibition of cyclin-dependent kinase-2 induces apoptosis in human diffuse large b-cell lymphomas. Cell Cycle. 2007;6(23):2982–9. doi:10.4161/cc.6.23.4994. [Google Scholar] [PubMed] [CrossRef]
65. Boretto M, Geurts MH, Gandhi S, Ma Z, Staliarova N, Celotti M, et al. Epidermal growth factor receptor (EGFR) is a target of the tumor-suppressor E3 ligase FBXW7. Proc Natl Acad Sci U S A. 2024;121(12):e2309902121. doi:10.1073/pnas.2309902121. [Google Scholar] [PubMed] [CrossRef]
66. Miyamoto H, Kawakami F, Abe S, Sugita H, Matsui H. Comprehensive cancer genomic profiling of liver metastasis led to the unexpected identification of colorectal cancer. Internal Med. 2024;63(1):63–70. doi:10.2169/internalmedicine.1845-23. [Google Scholar] [PubMed] [CrossRef]
67. Takada M, Zhang W, Suzuki A, Kuroda TS, Yu Z, Inuzuka H, et al. FBW7 loss promotes chromosomal instability and tumorigenesis via cyclin E1/CDK2-mediated phosphorylation of CENP-A. Cancer Res. 2017;77(18):4881–93. doi:10.1158/0008-5472.CAN-17-1240. [Google Scholar] [PubMed] [CrossRef]
68. Xiao G, Lu W, Yuan J, Liu Z, Wang P, Fan H. Fbxw7 suppresses carcinogenesis and stemness in triple-negative breast cancer through CHD4 degradation and Wnt/β-catenin pathway inhibition. J Transl Med. 2024;22(1):99. doi:10.1186/s12967-024-04897-2. [Google Scholar] [PubMed] [CrossRef]
69. Jiang X, Wang Y, Guo L, Wang Y, Miao T, Ma L, et al. The FBXW7-binding sites on FAM83D are potential targets for cancer therapy. Breast Cancer Res. 2024;26(1):37. doi:10.1186/s13058-024-01795-9. [Google Scholar] [PubMed] [CrossRef]
70. Ng LY, Ma HT, Poon RYC. Cyclin A-CDK1 suppresses the expression of the CDK1 activator CDC25A to safeguard timely mitotic entry. J Biol Chem. 2023;299(3):102957. doi:10.1016/j.jbc.2023.102957. [Google Scholar] [PubMed] [CrossRef]
71. Massacci G, Perfetto L, Sacco F. The Cyclin-dependent kinase 1: more than a cell cycle regulator. Br J Cancer. 2023;129(11):1707–16. doi:10.1038/s41416-023-02468-8. [Google Scholar] [PubMed] [CrossRef]
72. Wang Q, Bode AM, Zhang T. Targeting CDK1 in cancer: mechanisms and implications. npj Precis Oncol. 2023;7(1):58. doi:10.1038/s41698-023-00407-7. [Google Scholar] [PubMed] [CrossRef]
73. Chen X, Ma J, Wang X, Zi T, Qian D, Li C, et al. CCNB1 and AURKA are critical genes for prostate cancer progression and castration-resistant prostate cancer resistant to vinblastine. Front Endocrinol. 2022;13:1106175. doi:10.3389/fendo.2022.1106175. [Google Scholar] [PubMed] [CrossRef]
74. Żuryń A, Krajewski A, Klimaszewska-Wiśniewska A, Grzanka A, Grzanka D. Expression of cyclin B1, D1 and K in non-small cell lung cancer H1299 cells following treatment with sulforaphane. Oncol Rep. 2019;41(2):1313–23. doi:10.3892/or.2018.6919. [Google Scholar] [PubMed] [CrossRef]
75. Liu NQ, Cao WH, Wang X, Chen J, Nie J. Cyclin genes as potential novel prognostic biomarkers and therapeutic targets in breast cancer. Oncol Lett. 2022;24(4):375. doi:10.3892/ol.2022.13494. [Google Scholar] [PubMed] [CrossRef]
76. Liu F, Zhu C, Ma H, Yang Q. Curcumin targets miR-134-5p to suppress the progression of colorectal cancer through regulating the CDCA3/CDK1 pathway. Naunyn Schmiedebergs Arch Pharmacol. 2024;397(1):109–22. doi:10.1007/s00210-023-02584-5. [Google Scholar] [PubMed] [CrossRef]
77. Wu Y, Wu M, Zheng X, Yu H, Mao X, Jin Y, et al. Discovery of a potent and selective PARP1 degrader promoting cell cycle arrest via intercepting CDC25C-CDK1 axis for treating triple-negative breast cancer. Bioorg Chem. 2024;142:106952. doi:10.1016/j.bioorg.2023.106952. [Google Scholar] [PubMed] [CrossRef]
78. Zhang Y, Sun H, Ji Y, Nie F, Wang R, Han W. Effects of aspirin on colon cancer using quantitative proteomic analysis. Cancer Pathog Ther. 2024;2(2):121–31. doi:10.1016/j.cpt.2023.06.003. [Google Scholar] [PubMed] [CrossRef]
79. Iizumi Y, Oishi M, Taniguchi T, Goi W, Sowa Y, Sakai T. The flavonoid apigenin downregulates CDK1 by directly targeting ribosomal protein S9. PLoS One. 2013;8(8):e73219. doi:10.1371/journal.pone.0073219. [Google Scholar] [PubMed] [CrossRef]
80. Anand SK, Sharma A, Singh N, Kakkar P. Entrenching role of cell cycle checkpoints and autophagy for maintenance of genomic integrity. DNA Repair. 2020;86:102748. doi:10.1016/j.dnarep.2019.102748. [Google Scholar] [PubMed] [CrossRef]
81. Ueno S, Sudo T, Hirasawa A. ATM: functions of ATM kinase and its relevance to hereditary tumors. Int J Mol Sci. 2022;23(1):523. doi:10.3390/ijms23010523. [Google Scholar] [PubMed] [CrossRef]
82. Shi T, van Soest DMK, Polderman PE, Burgering BMT, Dansen TB. DNA damage and oxidant stress activate p53 through differential upstream signaling pathways. Free Radic Biol Med. 2021;172(7):298–311. doi:10.1016/j.freeradbiomed.2021.06.013. [Google Scholar] [PubMed] [CrossRef]
83. Chen J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb Perspect Med. 2016;6(3):a026104. doi:10.1101/cshperspect.a026104. [Google Scholar] [PubMed] [CrossRef]
84. Hafner A, Bulyk ML, Jambhekar A, Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol. 2019;20(4):199–210. doi:10.1038/s41580-019-0110-x. [Google Scholar] [PubMed] [CrossRef]
85. Phan LM, Rezaeian AH. ATM: main features, signaling pathways, and its diverse roles in dna damage response, tumor suppression, and cancer development. Genes. 2021;12(16):845. doi:10.3390/genes12060845. [Google Scholar] [PubMed] [CrossRef]
86. Trenz K, Smith E, Smith S, Costanzo V. ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J. 2006;25(8):1764–74. doi:10.1038/sj.emboj.7601045. [Google Scholar] [PubMed] [CrossRef]
87. Mahajan K, Coppola D, Rawal B, Chen YA, Lawrence HR, Engelman RW, et al. Ack1-mediated androgen receptor phosphorylation modulates radiation resistance in castration-resistant prostate cancer. J Biol Chem. 2012;287(26):22112–22. doi:10.1074/jbc.M112.357384. [Google Scholar] [PubMed] [CrossRef]
88. Vadnais C, Davoudi S, Afshin M, Harada R, Dudley R, Clermont PL, et al. CUX1 transcription factor is required for optimal ATM/ATR-mediated responses to DNA damage. Nucleic Acids Res. 2012;40(10):4483–95. doi:10.1093/nar/gks041. [Google Scholar] [PubMed] [CrossRef]
89. Ngoi NYL, Pham MM, Tan DSP, Yap TA. Targeting the replication stress response through synthetic lethal strategies in cancer medicine. Trends Cancer. 2021;7(10):930–57. doi:10.1016/j.trecan.2021.06.002. [Google Scholar] [PubMed] [CrossRef]
90. Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16(1):2–9. doi:10.1038/ncb2897. [Google Scholar] [PubMed] [CrossRef]
91. Jiang YZ, Ma D, Suo C, Shi J, Xue M, Hu X, et al. Genomic and transcriptomic landscape of triple-negative breast cancers: subtypes and treatment strategies. Cancer Cell. 2019;35(3):428–40.e5. doi:10.1016/j.ccell.2019.02.001. [Google Scholar] [PubMed] [CrossRef]
92. Broggini M, Buraggi G, Brenna A, Riva L, Codegoni AM, Torri V, et al. Cell cycle-related phosphatases CDC25A and B expression correlates with survival in ovarian cancer patients. Anticancer Res. 2000;20(6C):4835–40. [Google Scholar] [PubMed]
93. Kok YP, Guerrero Llobet S, Schoonen PM, Everts M, Bhattacharya A, Fehrmann RSN, et al. Overexpression of Cyclin E1 or Cdc25A leads to replication stress, mitotic aberrancies, and increased sensitivity to replication checkpoint inhibitors. Oncogenesis. 2020;9(10):88. doi:10.1038/s41389-020-00270-2. [Google Scholar] [PubMed] [CrossRef]
94. Jones RM, Mortusewicz O, Afzal I, Lorvellec M, García P, Helleday T, et al. Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene. 2013;32(32):3744–53. doi:10.1038/onc.2012.387. [Google Scholar] [PubMed] [CrossRef]
95. Lecona E, Fernández-Capetillo O. Replication stress and cancer: it takes two to tango. Exp Cell Res. 2014;329(1):26–34. doi:10.1016/j.yexcr.2014.09.019. [Google Scholar] [PubMed] [CrossRef]
96. Bradbury A, Hall S, Curtin N, Drew Y. Targeting ATR as cancer therapy: a new era for synthetic lethality and synergistic combinations? Pharmacol Ther. 2020;207(1):107450. doi:10.1016/j.pharmthera.2019.107450. [Google Scholar] [PubMed] [CrossRef]
97. McAinsh AD, Kops GJPL. Principles and dynamics of spindle assembly checkpoint signalling. Nat Rev Mol Cell Biol. 2023;24(8):543–59. doi:10.1038/s41580-023-00593-z. [Google Scholar] [PubMed] [CrossRef]
98. Lara-Gonzalez P, Pines J, Desai A. Spindle assembly checkpoint activation and silencing at kinetochores. Semin Cell Dev Biol. 2021;117(1):86–98. doi:10.1016/j.semcdb.2021.06.009. [Google Scholar] [PubMed] [CrossRef]
99. Watson ER, Brown NG, Peters JM, Stark H, Schulman BA. Posing the APC/C E3 ubiquitin ligase to orchestrate cell division. Trends Cell Biol. 2019;29(2):117–34. doi:10.1016/j.tcb.2018.09.007. [Google Scholar] [PubMed] [CrossRef]
100. Sitry-Shevah D, Kaisari S, Teichner A, Miniowitz-Shemtov S, Hershko A. Role of ubiquitylation of components of mitotic checkpoint complex in their dissociation from anaphase-promoting complex/cyclosome. Proc Natl Acad Sci U S A. 2018;115(8):1777–82. doi:10.1073/pnas.1720312115. [Google Scholar] [PubMed] [CrossRef]
101. Bassermann F, Eichner R, Pagano M. The ubiquitin proteasome system—implications for cell cycle control and the targeted treatment of cancer. Biochim Biophys Acta. 2014;1843(1):150–62. doi:10.1016/j.bbamcr.2013.02.028. [Google Scholar] [PubMed] [CrossRef]
102. Bhuniya R, Yuan X, Bai L, Howie KL, Wang R, Li W, et al. Design, synthesis, and biological evaluation of apcin-based CDC20 inhibitors. ACS Med Chem Lett. 2022;13(2):188–95. doi:10.1021/acsmedchemlett.1c00544. [Google Scholar] [PubMed] [CrossRef]
103. Bates M, Furlong F, Gallagher MF, Spillane CD, McCann A, O’Toole S, et al. Too MAD or not MAD enough: the duplicitous role of the spindle assembly checkpoint protein MAD2 in cancer. Cancer Lett. 2020;469(4):11–21. doi:10.1016/j.canlet.2019.10.005. [Google Scholar] [PubMed] [CrossRef]
104. Hernando E, Nahlé Z, Juan G, Diaz-Rodriguez E, Alaminos M, Hemann M, et al. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature. 2004;430(7001):797–802. doi:10.1038/nature02820. [Google Scholar] [PubMed] [CrossRef]
105. Koyuncu D, Sharma U, Goka ET, Lippman ME. Spindle assembly checkpoint gene BUB1B is essential in breast cancer cell survival. Breast Cancer Res Treat. 2021;185(2):331–41. doi:10.1007/s10549-020-05962-2. [Google Scholar] [PubMed] [CrossRef]
106. Schulze VK, Klar U, Kosemund D, Wengner AM, Siemeister G, Stöckigt D, et al. Treating cancer by spindle assembly checkpoint abrogation: discovery of two clinical candidates, BAY 1161909 and BAY 1217389, targeting MPS1 kinase. J Med Chem. 2020;63(15):8025–42. doi:10.1021/acs.jmedchem.9b02035. [Google Scholar] [PubMed] [CrossRef]
107. Maia ARR, Linder S, Song JY, Vaarting C, Boon U, Pritchard CEJ, et al. Mps1 inhibitors synergise with low doses of taxanes in promoting tumour cell death by enhancement of errors in cell division. Br J Cancer. 2018;118(12):1586–95. doi:10.1038/s41416-018-0081-2. [Google Scholar] [PubMed] [CrossRef]
108. Simon Serrano S, Sime W, Abassi Y, Daams R, Massoumi R, Jemaà M. Inhibition of mitotic kinase Mps1 promotes cell death in neuroblastoma. Sci Rep. 2020;10(1):1197. doi:10.1038/s41598-020-68829-y. [Google Scholar] [PubMed] [CrossRef]
109. Narayan RS, Molenaar P, Teng J, Cornelissen FMG, Roelofs I, Menezes R, et al. A cancer drug atlas enables synergistic targeting of independent drug vulnerabilities. Nat Commun. 2020;11(1):2935. doi:10.1038/s41467-020-16735-2. [Google Scholar] [PubMed] [CrossRef]
110. Thu KL, Soria-Bretones I, Mak TW, Cescon DW. Targeting the cell cycle in breast cancer: towards the next phase. Cell Cycle. 2018;17(15):1871–85. doi:10.1080/15384101.2018.1502567. [Google Scholar] [PubMed] [CrossRef]
111. Xu B, Li J, Xu D, Ran Q. PLK4 inhibitor plus bortezomib exhibits a synergistic effect on treating multiple myeloma via inactivating PI3K/AKT signaling. Ir J Med Sci. 2023;192(2):561–7. doi:10.1007/s11845-022-03007-9. [Google Scholar] [PubMed] [CrossRef]
112. Ashraf N, Asari A, Yousaf N, Ahmad M, Ahmed M, Faisal A, et al. Combined 3D-QSAR, molecular docking and dynamics simulations studies to model and design TTK inhibitors. Front Chem. 2022;10:1003816. doi:10.3389/fchem.2022.1003816. [Google Scholar] [PubMed] [CrossRef]
113. Thu KL, Silvester J, Elliott MJ, Ba-alawi W, Duncan MH, Elia AC, et al. Disruption of the anaphase-promoting complex confers resistance to TTK inhibitors in triple-negative breast cancer. Proc Natl Acad Sci U S A. 2018;115(7):E1570–7. doi:10.1073/pnas.1719577115. [Google Scholar] [PubMed] [CrossRef]
114. Chan CYK, Chiu DKC, Yuen VWH, Law CT, Wong BPY, Thu KL, et al. CFI-402257, a TTK inhibitor, effectively suppresses hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2022;119(32):e2119514119. doi:10.1073/pnas.2119514119. [Google Scholar] [PubMed] [CrossRef]
115. Dietrich C, Trub A, Ahn A, Taylor M, Ambani K, Chan KT, et al. INX-315, a selective CDK2 inhibitor, induces cell cycle arrest and senescence in solid tumors. Cancer Discov. 2024;14(3):446–67. doi:10.1158/2159-8290.CD-23-0954. [Google Scholar] [PubMed] [CrossRef]
116. Patra D, Bhavya K, Ramprasad P, Kalia M, Pal D. Anti-cancer drug molecules targeting cancer cell cycle and proliferation. Adv Protein Chem Struct Biol. 2023;135(5035):343–95. doi:10.1016/bs.apcsb.2022.11.011. [Google Scholar] [PubMed] [CrossRef]
117. Mir MA, Aisha S, Fatima K, Malik FA. Response of therapy in cell-cycle regulatory genes in breast cancer. Ther. Potential Cell Cycle Kinases Breast Cancer. 2023;335–53. doi:10.1007/978-981-19-8911-7_16. [Google Scholar] [CrossRef]
118. Borah NA, Reddy MM. Aurora kinase B inhibition: a potential therapeutic strategy for cancer. Molecules. 2021;26(7):1981. doi:10.3390/molecules26071981. [Google Scholar] [PubMed] [CrossRef]
119. Alimova I, Ng J, Harris P, Birks D, Donson A, Taylor MD, et al. MPS1 kinase as a potential therapeutic target in medulloblastoma. Oncol Rep. 2016;36(5):2633–40. doi:10.3892/or.2016.5085. [Google Scholar] [PubMed] [CrossRef]
120. Beaver JA, Amiri-Kordestani L, Charlab R, Chen W, Palmby T, Tilley A, et al. FDA approval: palbociclib for the treatment of postmenopausal patients with estrogen receptor-positive, HER2-negative metastatic breast cancer. Clin Cancer Res. 2015;21(21):4760–6. doi:10.1158/1078-0432.CCR-15-1185. [Google Scholar] [PubMed] [CrossRef]
121. Tripathy D, Bardia A, Sellers WR. Ribociclib (LEE011mechanism of action and clinical impact of this selective cyclin-dependent kinase 4/6 inhibitor in various solid tumors. Clin Cancer Res. 2017;23(13):3251–62. doi:10.1158/1078-0432.CCR-16-3157. [Google Scholar] [PubMed] [CrossRef]
122. Patel H, Periyasamy M, Sava GP, Bondke A, Slafer BW, Kroll SHB, et al. ICEC0942, an orally bioavailable selective inhibitor of CDK7 for cancer treatment. Mol Cancer Ther. 2018;17(6):1156–66. doi:10.1158/1535-7163.MCT-16-0847. [Google Scholar] [PubMed] [CrossRef]
123. Whittaker SR, Mallinger A, Workman P, Clarke PA. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol Ther. 2017;173:83–105. doi:10.1016/j.pharmthera.2017.02.008. [Google Scholar] [PubMed] [CrossRef]
124. Wang J, Yang Z, Wen J, Ma F, Wang F, Yu K, et al. SKLB-M8 induces apoptosis through the AKT/mTOR signaling pathway in melanoma models and inhibits angiogenesis with decrease of ERK1/2 phosphorylation. J Pharmacol Sci. 2014;126(3):198–207. doi:10.1254/jphs.14077FP. [Google Scholar] [PubMed] [CrossRef]
125. Ghelli Luserna Di Rorà A, Cerchione C, Martinelli G, Simonetti G. A WEE1 family business: regulation of mitosis, cancer progression, and therapeutic target. J Hematol Oncol. 2020;13(1):126. doi:10.1186/s13045-020-00959-2. [Google Scholar] [PubMed] [CrossRef]
126. Forment JV, O’Connor MJ. Targeting the replication stress response in cancer. Pharmacol Ther. 2018;188(12):155–67. doi:10.1016/j.pharmthera.2018.03.005. [Google Scholar] [PubMed] [CrossRef]
127. Tischer J, Gergely F. Anti-mitotic therapies in cancer. J Cell Biol. 2019;218(1):10–1. doi:10.1083/jcb.201808077. [Google Scholar] [PubMed] [CrossRef]
128. Penna LS, Henriques JAP, Bonatto D. Anti-mitotic agents: are they emerging molecules for cancer treatment? Pharmacol Ther. 2017;173(3):67–82. doi:10.1016/j.pharmthera.2017.02.007. [Google Scholar] [PubMed] [CrossRef]
129. Zasadil LM, Andersen KA, Yeum D, Rocque GB, Wilke LG, Tevaarwerk AJ, et al. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci Transl Med. 2014;6(229):229ra43. doi:10.1126/scitranslmed.3007965. [Google Scholar] [PubMed] [CrossRef]
130. Baur M, Gneist M, Owa T, Dittrich C. Clinical complete long-term remission of a patient with metastatic malignant melanoma under therapy with indisulam (E7070). Melanoma Res. 2007;17(5):329–31. doi:10.1097/CMR.0b013e3282ef4189. [Google Scholar] [PubMed] [CrossRef]
131. Trub AG, Bisi JE, Dietrich C, Taylor M, Strum JC, Goel S, et al. Abstract 5994: iNX-315, a potent and selective CDK2 inhibitor, demonstrates robust antitumor activity in CCNE1-amplified cancers. Cancer Res. 2023;83(7_Supplement):5994. doi:10.1158/1538-7445.am2023-5994. [Google Scholar] [CrossRef]
132. Gerosa R, De Sanctis R, Jacobs F, Benvenuti C, Gaudio M, Saltalamacchia G, et al. Cyclin-dependent kinase 2 (CDK2) inhibitors and others novel CDK inhibitors (CDKi) in breast cancer: clinical trials, current impact, and future directions. Crit Rev Oncol Hematol. 2024;196(5):104324. doi:10.1016/j.critrevonc.2024.104324. [Google Scholar] [PubMed] [CrossRef]
133. Patel MR, Juric D, Henick BS, Duska LR, Wu R, Guo J, et al. BLU-222, an oral, potent, and selective CDK2 inhibitor, in patients with advanced solid tumors: phase 1 monotherapy dose escalation. J Clin Oncol. 2023;41(16):3095. doi:10.1200/jco.2023.41.16_suppl.3095. [Google Scholar] [CrossRef]
134. Wu CX, Wang XQ, Chok SH, Man K, Tsang SHY, Chan ACY, et al. Blocking CDK1/PDK1/β-Catenin signaling by CDK1 inhibitor RO3306 increased the efficacy of sorafenib treatment by targeting cancer stem cells in a preclinical model of hepatocellular carcinoma. Theranostics. 2018;8(14):3737–50. doi:10.7150/thno.25487. [Google Scholar] [PubMed] [CrossRef]
135. Park S, Shim SMI, Nam SH, Andera L, Suh N, Kim I. CGP74514A enhances trail-induced apoptosis in breast cancer cells by reducing X-linked inhibitor of apoptosis protein. Anticancer Res. 2014;34(7):3557–62. [Google Scholar] [PubMed]
136. Fuentes-Antrás J, Bedard PL, Cescon DW. Seize the engine: emerging cell cycle targets in breast cancer. Clin Transl Med. 2024;14(1):e1544. doi:10.1002/ctm2.1544. [Google Scholar] [PubMed] [CrossRef]
137. Morrison L, Loibl S, Turner NC. The CDK4/6 inhibitor revolution—a game-changing era for breast cancer treatment. Nat Rev Clin Oncol. 2024;21(2):89–105. doi:10.1038/s41571-023-00840-4. [Google Scholar] [PubMed] [CrossRef]
138. Clark K, Ainscow E, Peall A, Thomson S, Leishman A, Elaine S, et al. CT7001, a novel orally bio-available CDK7 inhibitor, is highly active in in-vitro and in-vivo models of AML. Blood. 2017;130(Supplement 1):2645. [Google Scholar]
139. Moreira J, Almeida J, Saraiva L, Cidade H, Pinto M. Chalcones as promising antitumor agents by targeting the p53 pathway: an overview and new insights in drug-likeness. Molecules. 2021;26(12):3737. doi:10.3390/molecules26123737. [Google Scholar] [PubMed] [CrossRef]
140. Zhuang C, Zhang W, Sheng C, Zhang W, Xing C, Miao Z. Chalcone: a privileged structure in medicinal chemistry. Chem Rev. 2017;117(12):7762–810. doi:10.1021/acs.chemrev.7b00020. [Google Scholar] [PubMed] [CrossRef]
141. Selvaraj C. Therapeutic targets in cancer treatment: cell cycle proteins. Adv Protein Chem Struct Biol. 2023;135(6):313–42. doi:10.1016/bs.apcsb.2023.02.003. [Google Scholar] [PubMed] [CrossRef]
142. Sultana N, Elford HL, Faridi JS. Abstract 5989: targeting RRM2 & cell cycle for breast cancer treatment. Cancer Res. 2023;83(7_Supplement):5989. doi:10.1158/1538-7445.am2023-5989. [Google Scholar] [CrossRef]
143. Yap TA, O’Carrigan B, Penney MS, Lim JS, Brown JS, De Miguel Luken MJ, et al. Phase I trial of first-in-class ATR inhibitor M6620 (VX-970) as monotherapy or in combination with carboplatin in patients with advanced solid tumors. J Clin Oncol. 2020;38(27):3195–204. doi:10.1200/JCO.19.02404. [Google Scholar] [PubMed] [CrossRef]
144. Burris HA, Berlin J, Arkenau T, Cote GM, Lolkema MP, Ferrer-Playan J, et al. A phase I study of ATR inhibitor gartisertib (M4344) as a single agent and in combination with carboplatin in patients with advanced solid tumours. Br J Cancer. 2024;130(7):1131–40. doi:10.1038/s41416-023-02436-2. [Google Scholar] [PubMed] [CrossRef]
145. Kwon M, Kim G, Kim R, Kim KT, Kim ST, Smith S, et al. Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced gastric cancer. J Immunother Cancer. 2022;10(7):e005041. doi:10.1136/jitc-2022-005041. [Google Scholar] [PubMed] [CrossRef]
146. Harold J, Bellone S, Manavella DD, Mutlu L, McNamara B, Hartwich TMP, et al. Elimusertib (BAY1895344a novel ATR inhibitor, demonstrates in vivo activity in ATRX mutated models of uterine leiomyosarcoma. Gynecol Oncol. 2023;168:157–65. doi:10.1016/j.ygyno.2022.11.014. [Google Scholar] [PubMed] [CrossRef]
147. Huang L, Shao J, Lai W, Gu H, Yang J, Shi S, et al. Discovery of the first ataxia telangiectasia and Rad3-related (ATR) degraders for cancer treatment. Eur J Med Chem. 2024;267:116159. doi:10.1016/j.ejmech.2024.116159. [Google Scholar] [PubMed] [CrossRef]
148. Wilson Z, Odedra R, Wallez Y, Wijnhoven PWG, Hughes AM, Gerrard J, et al. ATR inhibitor AZD6738 (Ceralasertib) exerts antitumor activity as a monotherapy and in combination with chemotherapy and the PARP inhibitor olaparib. Cancer Res. 2022;82(6):1140–52. doi:10.1158/0008-5472.CAN-21-2997. [Google Scholar] [PubMed] [CrossRef]
149. Baschnagel AM, Elnaggar JH, VanBeek HJ, Kromke AC, Skiba JH, Kaushik S, et al. ATR inhibitor M6620 (VX-970) enhances the effect of radiation in non-small cell lung cancer brain metastasis patient-derived xenografts. Mol Cancer Ther. 2021;20(11):2129–39. doi:10.1158/1535-7163.MCT-21-0305. [Google Scholar] [PubMed] [CrossRef]
150. Jo U, Senatorov IS, Zimmermann A, Saha LK, Murai Y, Kim SH, et al. Novel and highly potent ATR inhibitor M4344 kills cancer cells with replication stress, and enhances the chemotherapeutic activity of widely used DNA damaging agents. Mol Cancer Ther. 2021;20(8):1431–41. doi:10.1158/1535-7163.MCT-20-1026. [Google Scholar] [PubMed] [CrossRef]
151. Li S, Juengpanich S, Topatana W, Xie T, Hou L, Zhu Y, et al. Adavosertib-encapsulated metal-organic frameworks for p53-mutated gallbladder cancer treatment via synthetic lethality. Sci Bull. 2024;69(9):1286–301. doi:10.1016/j.scib.2024.02.039. [Google Scholar] [PubMed] [CrossRef]
152. Khamidullina AI, Abramenko YE, Bruter AV, Tatarskiy VV. Key proteins of replication stress response and cell cycle control as cancer therapy targets. Int J Mol Sci. 2024;25(2):1263. doi:10.3390/ijms25021263. [Google Scholar] [PubMed] [CrossRef]
153. Konstantinopoulos PA, Lee LM, Gao B, Miller R, Lee JY, Colombo N, et al. A Phase 2 study of prexasertib (LY2606368) in platinum resistant or refractory recurrent ovarian cancer. Gynecol Oncol. 2022;167(2):213–25. doi:10.1016/j.ygyno.2022.09.019. [Google Scholar] [PubMed] [CrossRef]
154. Burgess BT, Anderson AM, McCorkle JR, Wu J, Ueland FR, Kolesar JM. Olaparib combined with an ATR or Chk1 inhibitor as a treatment strategy for acquired olaparib-resistant BRCA1 mutant ovarian cells. Diagnostics (Basel). 2020;10(2):121. doi:10.3390/diagnostics10020121. [Google Scholar] [PubMed] [CrossRef]
155. Li C, Liao J, Wang X, Chen FX, Guo X, Chen X. Combined aurora kinase A and CHK1 inhibition enhances radiosensitivity of triple-negative breast cancer through induction of apoptosis and mitotic catastrophe associated with excessive DNA damage. Int J Radiat Oncol Biol Phys. 2023;117(5):1241–54. doi:10.1016/j.ijrobp.2023.06.022. [Google Scholar] [PubMed] [CrossRef]
156. Gao F, Wu Y, Wang R, Yao Y, Liu Y, Fan L, et al. Precise nano-system-based drug delivery and synergistic therapy against androgen receptor-positive triple-negative breast cancer. Acta Pharm Sin B. 2024;14(6):2685–97. doi:10.1016/j.apsb.2024.03.012. [Google Scholar] [PubMed] [CrossRef]
157. Isono M, Okubo K, Asano T, Sato A. Inhibition of checkpoint kinase 1 potentiates anticancer activity of gemcitabine in bladder cancer cells. Sci Rep. 2021;11(1):10181. doi:10.1038/s41598-021-89684-5. [Google Scholar] [PubMed] [CrossRef]
158. Gao X, Liao Y, Li Q, Yang L, Zhao C, Teng Y, et al. Abstract 482: discovery of a novel and oral CHK1 inhibitor for the treatment of solid tumors. Cancer Res. 2023;83(7_Supplement):482. doi:10.1158/1538-7445.am2023-482. [Google Scholar] [CrossRef]
159. Ditano JP, Eastman A. Comparative activity and off-target effects in cells of the CHK1 inhibitors MK-8776, SRA737, and LY2606368. ACS Pharmacol Transl Sci. 2021;4(2):730–43. doi:10.1021/acsptsci.0c00201. [Google Scholar] [PubMed] [CrossRef]
160. Franza M, Albanesi J, Mancini B, Pennisi R, Leone S, Acconcia F, et al. The clinically relevant CHK1 inhibitor MK-8776 induces the degradation of the oncogenic protein PML-RARα and overcomes ATRA resistance in acute promyelocytic leukemia cells. Biochem Pharmacol. 2023;214(11):115675. doi:10.1016/j.bcp.2023.115675. [Google Scholar] [PubMed] [CrossRef]
161. Abd El-Hafeez T, Shams MY, Elshaier YAMM, Farghaly HM, Hassanien AE. Harnessing machine learning to find synergistic combinations for FDA-approved cancer drugs. Sci Rep. 2024;14(1):2428. doi:10.1038/s41598-024-52814-w. [Google Scholar] [PubMed] [CrossRef]
162. Xu AP, Xu LB, Smith ER, Fleishman JS, Chen ZS, Xu XX. Cell death in cancer chemotherapy using taxanes. Front Pharmacol. 2024;14:1338633. doi:10.3389/fphar.2023.1338633. [Google Scholar] [PubMed] [CrossRef]
163. Weaver BA. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell. 2014;25(18):2677–81. doi:10.1091/mbc.e14-04-0916. [Google Scholar] [PubMed] [CrossRef]
164. Wengner AM, Siemeister G, Koppitz M, Schulze V, Kosemund D, Klar U, et al. Novel Mps1 kinase inhibitors with potent antitumor activity. Mol Cancer Ther. 2016;15(4):583–92. doi:10.1158/1535-7163.MCT-15-0500. [Google Scholar] [PubMed] [CrossRef]
165. Krex D, Bartmann P, Lachmann D, Hagstotz A, Jugel W, Schneiderman RS, et al. Aurora B kinase inhibition by AZD1152 concomitant with tumor treating fields is effective in the treatment of cultures from primary and recurrent glioblastomas. Int J Mol Sci. 2023;24(5):5016. doi:10.3390/ijms24055016. [Google Scholar] [PubMed] [CrossRef]
166. Shah ET, Molloy C, Gough M, Kryza T, Samuel SG, Tucker A, et al. Inhibition of Aurora B kinase (AURKB) enhances the effectiveness of 5-fluorouracil chemotherapy against colorectal cancer cells. Br J Cancer. 2024;130(7):1196–205. doi:10.1038/s41416-024-02584-z. [Google Scholar] [PubMed] [CrossRef]
167. Xu G, Weng Q, Zhang L, Song Y, Zhan W, Yin Y, et al. Aurora kinase B-instruct release of AZD1152-HQPA from hydrogel to enhance cervical cancer suppression. Adv Funct Mater. 2024;34(7):1333. doi:10.1002/adfm.202309984. [Google Scholar] [CrossRef]
168. Ding Y, Fei Y, Lu B. Emerging new concepts of degrader technologies. Trends Pharmacol Sci. 2020;41(7):464–74. doi:10.1016/j.tips.2020.04.005. [Google Scholar] [PubMed] [CrossRef]
169. Ishida T, Ciulli A. E3 ligase ligands for PROTACs: how they were found and how to discover new ones. SLAS Discov. 2021;26(4):484–502. doi:10.1177/2472555220965528. [Google Scholar] [PubMed] [CrossRef]
170. Nalawansha DA, Crews CM. PROTACs: an emerging therapeutic modality in precision medicine. Cell Chem Biol. 2020;27(8):998–1014. doi:10.1016/j.chembiol.2020.07.020. [Google Scholar] [PubMed] [CrossRef]
171. Liu Z, Hu M, Yang Y, Du C, Zhou H, Liu C, et al. An overview of PROTACs: a promising drug discovery paradigm. Mol Biomed. 2022;3(1):46. doi:10.1186/s43556-022-00112-0. [Google Scholar] [PubMed] [CrossRef]
172. Li Z, Pinch BJ, Olson CM, Donovan KA, Nowak RP, Mills CE, et al. Development and characterization of a wee1 kinase degrader. Cell Chem Biol. 2020;27(1):57–65.e9. doi:10.1016/j.chembiol.2019.10.013. [Google Scholar] [PubMed] [CrossRef]
173. Adhikari B, Bozilovic J, Diebold M, Schwarz JD, Hofstetter J, Schröder M, et al. PROTAC-mediated degradation reveals a non-catalytic function of AURORA-A kinase. Nat Chem Biol. 2020;16(11):1179–88. doi:10.1038/s41589-020-00652-y. [Google Scholar] [PubMed] [CrossRef]
174. Chi J, Li H, Zhou Z, Izquierdo-Ferrer J, Xue Y, Wavelet CM, et al. A novel strategy to block mitotic progression for targeted therapy. EBioMedicine. 2019;49(4):50–4. doi:10.1016/j.ebiom.2019.10.013. [Google Scholar] [PubMed] [CrossRef]
175. Jiang B, Wang ES, Donovan KA, Liang Y, Fischer ES, Zhang T, et al. Development of dual and selective degraders of cyclin-dependent kinases 4 and 6. Angew Chem Int Ed Engl. 2019;58(19):6321–6. doi:10.1002/anie.201901336. [Google Scholar] [PubMed] [CrossRef]
176. Zhao B, Burgess K. PROTACs suppression of CDK4/6, crucial kinases for cell cycle regulation in cancer. Chem Commun. 2019;55(18):2704–7. doi:10.1039/C9CC00163H. [Google Scholar] [PubMed] [CrossRef]
177. Brand M, Jiang B, Bauer S, Donovan KA, Liang Y, Wang ES, et al. Homolog-selective degradation as a strategy to probe the function of CDK6 in AML. Cell Chem Biol. 2019;26(2):300–6.e9. doi:10.1016/j.chembiol.2018.11.006. [Google Scholar] [PubMed] [CrossRef]
178. Su S, Yang Z, Gao H, Yang H, Zhu S, An Z, et al. Potent and preferential degradation of CDK6 via proteolysis targeting chimera degraders. J Med Chem. 2019;62(16):7575–82. doi:10.1021/acs.jmedchem.9b00871. [Google Scholar] [PubMed] [CrossRef]
179. De Dominici M, Porazzi P, Xiao Y, Chao A, Tang HY, Kumar G, et al. Selective inhibition of Ph-positive ALL cell growth through kinase-dependent and -independent effects by CDK6-specific PROTACs. Blood. 2020;135(18):1560–73. doi:10.1182/blood.2019003604. [Google Scholar] [PubMed] [CrossRef]
180. Rana S, Bendjennat M, Kour S, King HM, Kizhake S, Zahid M, et al. Selective degradation of CDK6 by a palbociclib based PROTAC. Bioorg Med Chem Lett. 2019;29(11):1375–9. doi:10.1016/j.bmcl.2019.03.035. [Google Scholar] [PubMed] [CrossRef]
181. Liu J, Peng Y, Wei W. Cell cycle on the crossroad of tumorigenesis and cancer therapy. Trends Cell Biol. 2022;32(1):30–44. doi:10.1016/j.tcb.2021.07.001. [Google Scholar] [PubMed] [CrossRef]
182. Maleki EH, Bahrami AR, Matin MM. Cancer cell cycle heterogeneity as a critical determinant of therapeutic resistance. Genes Dis. 2023;11(1):189–204. doi:10.1016/j.gendis.2022.11.025. [Google Scholar] [PubMed] [CrossRef]
183. Stoletov K, Beatty PH, Lewis JD. Novel therapeutic targets for cancer metastasis. Expert Rev Anticancer Ther. 2020;20(2):97–109. doi:10.1080/14737140.2020.1718496. [Google Scholar] [PubMed] [CrossRef]
184. Bukowska B, Gajek A, Marczak A. Two drugs are better than one. A short history of combined therapy of ovarian cancer. Contemp Oncol. 2015;19(5):350–3. doi:10.5114/wo.2014.43975. [Google Scholar] [PubMed] [CrossRef]
185. Palmer AC, Chidley C, Sorger PK. A curative combination cancer therapy achieves high fractional cell killing through low cross resistance and drug Additivity. eLife. 2019;8:e50036. doi:10.7554/eLife.50036. [Google Scholar] [PubMed] [CrossRef]
186. Colombo M, Marabese M, Vargiu G, Broggini M, Caiola E. Alone or in combination in NSCLCs with different mutations. Front Oncol. 2020;10:532292. doi:10.3389/fonc.2020.532292. [Google Scholar] [PubMed] [CrossRef]
187. Mortensen ACL, Mohajershojai T, Hariri M, Pettersson M, Spiegelberg D. Overcoming limitations of cisplatin therapy by additional treatment with the HSP90 inhibitor onalespib. Front Oncol. 2020;10:532285. doi:10.3389/fonc.2020.532285. [Google Scholar] [PubMed] [CrossRef]
188. Shi W, Zhang S, Ma D, Yan D, Zhang G, Cao Y, et al. Targeting SphK2 reverses acquired resistance of regorafenib in hepatocellular carcinoma. Front Oncol. 2020;10:694. doi:10.3389/fonc.2020.00694. [Google Scholar] [PubMed] [CrossRef]
189. Xu DD, Chen SH, Zhou PJ, Wang Y, Zhao ZD, Wang X, et al. Suppression of esophageal cancer stem-like cells by SNX-2112 is enhanced by STAT3 silencing. Front Pharmacol. 2020;11:532395. doi:10.3389/fphar.2020.532395. [Google Scholar] [PubMed] [CrossRef]
190. Li S, Wang L, Wang Y, Zhang C, Hong Z, Han Z. The synthetic lethality of targeting cell cycle checkpoints and PARPs in cancer treatment. J Hematol Oncol. 2022;15(1):147. doi:10.1186/s13045-022-01360-x. [Google Scholar] [PubMed] [CrossRef]
191. Duan XP, Qin BD, Jiao XD, Liu K, Wang Z, Zang YS. New clinical trial design in precision medicine: discovery, development and direction. Signal Transduct Target Ther. 2024;9(1):57. doi:10.1038/s41392-024-01760-0. [Google Scholar] [PubMed] [CrossRef]
192. Winkle M, El-Daly SM, Fabbri M, Calin GA. Noncoding RNA therapeutics—challenges and potential solutions. Nat Rev Drug Discov. 2021;20(8):629–51. doi:10.1038/s41573-021-00219-z. [Google Scholar] [PubMed] [CrossRef]
193. Melo CA, Melo SA. Biogenesis and physiology of micrornas. In: Fabbri M, editor. Non-coding RNAs and cancer. New York: Springer; 2014. p. 5–24. doi:10.1007/978-1-4614-8444-8_2. [Google Scholar] [CrossRef]
194. Treiber T, Treiber N, Meister G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat Rev Mol Cell Biol. 2019;20(1):5–20. doi:10.1038/s41580-018-0059-1. [Google Scholar] [PubMed] [CrossRef]
195. Vaziri H, Dessain SK, Eaton EN, Imai SI, Frye RA, Pandita TK, et al. hSIR2SIRT1 functions as an NAD-dependent p53 deacetylase. Cell. 2001;107(2):149–59. doi:10.1016/S0092-8674(01)00527-X. [Google Scholar] [PubMed] [CrossRef]
196. Luo J, Nikolaev AY, Imai SI, Chen D, Su F, Shiloh A, et al. Negative control of p53 by Sir2α promotes cell survival under stress. Cell. 2001;107(2):137–48. doi:10.1016/S0092-8674(01)00524-4. [Google Scholar] [PubMed] [CrossRef]
197. Juan LJ, Shia WJ, Chen MH, Yang WM, Seto E, Lin YS, et al. Histone deacetylases specifically down-regulate p53-dependent gene activation. J Biol Chem. 2000;275(27):20436–43. doi:10.1074/jbc.M000202200. [Google Scholar] [PubMed] [CrossRef]
198. Navarro F, Lieberman J. miR-34 and p53: new insights into a complex functional relationship. PLoS One. 2015;10(7):e0132767. doi:10.1371/journal.pone.0132767. [Google Scholar] [PubMed] [CrossRef]
199. Singh G, Sharma SK, Singh SK. miR-34a negatively regulates cell cycle factor Cdt2/DTL in HPV infected cervical cancer cells. BMC Cancer. 2022;22(1):777. doi:10.1186/s12885-022-09879-5. [Google Scholar] [PubMed] [CrossRef]
200. Arenas AM, Andrades A, Patiño-Mercau JR, Sanjuan-Hidalgo J, Cuadros M, García DJ, et al. Opportunities of miRNAs in cancer therapeutics. In: MicroRNA in human malignancies. USA: Academic Press; 2022. p. 153–64. doi:10.1016/B978-0-12-822287-4.00015-3. [Google Scholar] [CrossRef]
201. Abdelaal AM, Sohal IS, Iyer SG, Sudarshan K, Orellana EA, Ozcan KE, et al. Selective targeting of chemically modified miR-34a to prostate cancer using a small molecule ligand and an endosomal escape agent. Mol Ther Nucleic Acids. 2024;35(2):102193. doi:10.1016/j.omtn.2024.102193. [Google Scholar] [PubMed] [CrossRef]
202. Sun F, Fu H, Liu Q, Tie Y, Zhu J, Xing R, et al. Downregulation of CCND1 and CDK6 by miR-34a induces cell cycle arrest. FEBS Lett. 2008;582(10):1564–8. doi:10.1016/j.febslet.2008.03.057. [Google Scholar] [PubMed] [CrossRef]
203. Deng SZ, Lai MF, Li YP, Xu CH, Zhang HR, Kuang JG. Human marrow stromal cells secrete microRNA-375-containing exosomes to regulate glioma progression. Cancer Gene Ther. 2020;27(3–4):203–15. doi:10.1038/s41417-019-0079-9. [Google Scholar] [PubMed] [CrossRef]
204. Chen SR, Cai WP, Dai XJ, Guo AS, Chen HP, Lin GS, et al. Research on miR-126 in glioma targeted regulation of PTEN/PI3K/Akt and MDM2-p53 pathways. Eur Rev Med Pharmacol Sci. 2019;23(8):3461–70. doi:10.26355/eurrev_201904_17711. [Google Scholar] [PubMed] [CrossRef]
205. Ahir BK, Ozer H, Engelhard HH, Lakka SS. Critical reviews in oncology/hematology MicroRNAs in glioblastoma pathogenesis and therapy: a comprehensive review. Crit Rev Oncol Hematol. 2017;120:22–3. [Google Scholar] [PubMed]
206. Liu YY, Chen MB, Cheng L, Zhang ZQ, Yu ZQ, Jiang Q, et al. MicroRNA-200a downregulation in human glioma leads to Gαi1 over-expression, Akt activation, and cell proliferation. Oncogene. 2018;37(21):2890–902. doi:10.1038/s41388-018-0184-5. [Google Scholar] [PubMed] [CrossRef]
207. Wang H, Zhu LJ, Yang YC, Wang ZX, Wang R. MiR-224 promotes the chemoresistance of human lung adenocarcinoma cells to cisplatin via regulating G1/S transition and apoptosis by targeting p21 WAF1/CIP1. Br J Cancer. 2014;111(2):239–54. doi:10.1038/bjc.2014.157. [Google Scholar] [PubMed] [CrossRef]
208. Mohammadi A, Mansoori B, Duijf PHG, Safarzadeh E, Tebbi L, Najafi S, et al. Restoration of miR-330 expression suppresses lung cancer cell viability, proliferation, and migration. J Cell Physiol. 2021;236(1):273–83. doi:10.1002/jcp.29840. [Google Scholar] [PubMed] [CrossRef]
209. Telford BJ, Yahyanejad S, de Gunst T, den Boer HC, Vos RM, Stegink M, et al. Multi-modal effects of 1B3, a novel synthetic miR-193a-3p mimic, support strong potential for therapeutic intervention in oncology. Oncotarget. 2021;12(5):422–39. doi:10.18632/oncotarget.27894. [Google Scholar] [PubMed] [CrossRef]
210. Liang Y, Zhang D, Li L, Xin T, Zhao Y, Ma R, et al. Exosomal microRNA-144 from bone marrow-derived mesenchymal stem cells inhibits the progression of non-small cell lung cancer by targeting CCNE1 and CCNE2. Stem Cell Res Ther. 2020;11(1):87. doi:10.1186/s13287-020-1580-7. [Google Scholar] [PubMed] [CrossRef]
211. Iqbal MA, Arora S, Prakasam G, Calin GA, Syed MA. MicroRNA in lung cancer: role, mechanisms, pathways and therapeutic relevance. Mol Aspects Med. 2019;70(5):3–20. doi:10.1016/j.mam.2018.07.003. [Google Scholar] [PubMed] [CrossRef]
212. Zhao Z, Liu J, Wang C, Wang Y, Jiang Y, Guo M. MicroRNA-25 regulates small cell lung cancer cell development and cell cycle through cyclin E2. Int J Clin Exp Pathol. 2014;7(11):7726–34. [Google Scholar] [PubMed]
213. Hossian AKMN, MacKenzie GG, Mattheolabakis G. Combination of miR-143 and miR-506 reduces lung and pancreatic cancer cell growth through the downregulation of cyclin-dependent kinases. Oncol Rep. 2021;45(4):2. doi:10.3892/or.2021.7953. [Google Scholar] [PubMed] [CrossRef]
214. Van Roosbroeck K, Fanini F, Setoyama T, Ivan C, Rodriguez-Aguayo C, Fuentes-Mattei E, et al. Combining anti-miR-155 with chemotherapy for the treatment of lung cancers. Clinl Cancer Res. 2017;23(11):2891–904. doi:10.1158/1078-0432.CCR-16-1025. [Google Scholar] [PubMed] [CrossRef]
215. Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131(6):1109–23. doi:10.1016/j.cell.2007.10.054. [Google Scholar] [PubMed] [CrossRef]
216. Xiong X, Xu W, Gong J, Wang L, Dai M, Chen G, et al. miR-937-5p targets SOX17 to modulate breast cancer cell cycle and cell proliferation through the Wnt signaling pathway. Cell Signal. 2021;77(1):109818. doi:10.1016/j.cellsig.2020.109818. [Google Scholar] [PubMed] [CrossRef]
217. Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17(2):211–5. doi:10.1038/nm.2284. [Google Scholar] [PubMed] [CrossRef]
218. Ma C, Ding YC, Yu W, Wang Q, Meng B, Huang T. MicroRNA-200c overexpression plays an inhibitory role in human pancreatic cancer stem cells by regulating epithelial-mesenchymal transition. Minerva Med. 2015;106(4):193–202. [Google Scholar] [PubMed]
219. Han D, Zhu S, Li X, Li Z, Huang H, Gao W, et al. The NF-κB/miR-488/ERBB2 axis modulates pancreatic cancer cell malignancy and tumor growth through cell cycle signaling. Cancer Biol Ther. 2022;23(1):294–309. doi:10.1080/15384047.2022.2054257. [Google Scholar] [PubMed] [CrossRef]
220. Schraivogel D, Weinmann L, Beier D, Tabatabai G, Eichner A, Zhu JY, et al. CAMTA1 is a novel tumour suppressor regulated by miR-9/9* in glioblastoma stem cells. EMBO J. 2011;30(20):4309–22. doi:10.1038/emboj.2011.301. [Google Scholar] [PubMed] [CrossRef]
221. Bitarte N, Bandres E, Boni V, Zarate R, Rodriguez J, Gonzalez-Huarriz M, et al. MicroRNA-451 is involved in the self-renewal, tumorigenicity, and chemoresistance of colorectal cancer stem cells. Stem Cells. 2011;29(11):1661–71. doi:10.1002/stem.741. [Google Scholar] [PubMed] [CrossRef]
222. Singh G, Sharma SK, Dorata A, Singh SK. miR-17~92 suppresses proliferation and invasion of cervical cancer cells by inhibiting cell cycle regulator Cdt2. Discov Oncol. 2023;14(1):172. doi:10.1007/s12672-023-00775-3. [Google Scholar] [PubMed] [CrossRef]
223. Chen Y, Du J, Wang Y, Shi H, Jiang Q, Wang Y, et al. Microrna-497-5p induces cell cycle arrest of cervical cancer cells in s phase by targeting cbx4. Onco Targets Ther. 2019;12:10535–45. doi:10.2147/OTT. [Google Scholar] [CrossRef]
224. Tian RQ, Wang XH, Hou LJ, Jia WH, Yang Q, Li YX, et al. MicroRNA-372 is down-regulated and targets cyclin-dependent kinase 2 (CDK2) and cyclin A1 in human cervical cancer, which may contribute to tumorigenesis. J Biol Chem. 2011;286(29):25556–63. doi:10.1074/jbc.M111.221564. [Google Scholar] [PubMed] [CrossRef]
225. Chen S, Chen X, Xiu YL, Sun KX, Zhao Y. MicroRNA-490-3P targets CDK1 and inhibits ovarian epithelial carcinoma tumorigenesis and progression. Cancer Lett. 2015;362(1):122–30. doi:10.1016/j.canlet.2015.03.029. [Google Scholar] [PubMed] [CrossRef]
226. Yan H, Ren S, Lin Q, Yu Y, Chen C, Hua X, et al. Inhibition of UBE2N-dependent CDK6 protein degradation by miR-934 promotes human bladder cancer cell growth. FASEB J. 2019;33(1):12112–23. doi:10.1096/fj.201900499RR. [Google Scholar] [PubMed] [CrossRef]
227. Zhang Q, Miao S, Han X, Li C, Zhang M, Cui K, et al. MicroRNA-3619-5p suppresses bladder carcinoma progression by directly targeting β-catenin and CDK2 and activating p21. Cell Death Dis. 2018;9(10):690. doi:10.1038/s41419-018-0986-y. [Google Scholar] [PubMed] [CrossRef]
228. Ren Y, Li Y, Zhang W, Yang K, Li J, Hu Y, et al. Mir-4746 inhibits the proliferation of colorectal cancer cells in vitro and in vivo by targeting CCND1. Biochem Biophys Res Commun. 2022;594:153–60. doi:10.1016/j.bbrc.2022.01.063. [Google Scholar] [PubMed] [CrossRef]
229. Li H, Tian X, Wang P, Huang M, Xu R, Nie T. MicroRNA-582-3p negatively regulates cell proliferation and cell cycle progression in acute myeloid leukemia by targeting cyclin B2. Cell Mol Biol Lett. 2019;24(1):66. doi:10.1186/s11658-019-0184-7. [Google Scholar] [PubMed] [CrossRef]
230. Zhang HF, Alshareef A, Wu C, Jiao JW, Sorensen PH, Lai R, et al. miR-200b induces cell cycle arrest and represses cell growth in esophageal squamous cell carcinoma. Carcinogenesis. 2016;37(9):858–69. doi:10.1093/carcin/bgw079. [Google Scholar] [PubMed] [CrossRef]
231. Sun K-K, Shen X-J, Yang D, Gan M-Q, Liu G, Zhang Y-F, et al. MicroRNA-31 triggers G 2/M cell cycle arrest, enhances the chemosensitivity and inhibits migration and invasion of human gastric cancer cells by downregulating the expression of zeste homolog 2 (ZH2). Arch Biochem Biophys. 2019;663(5):269–75. doi:10.1016/j.abb.2019.01.023. [Google Scholar] [PubMed] [CrossRef]
232. Liu P, Zhang X, Fu Q, Liu CJ, Luo QK, Yu PF, et al. LINC01419 promotes the proliferation of hepatoma cells by recruiting XRCC5 and regulating its phosphorylation to repair DNA damage. Dis Markers. 2022;9313680. doi:10.1155/2022/9313680. [Google Scholar] [PubMed] [CrossRef]
233. Wu J, Zhou X, Fan Y, Cheng X, Lu B, Chen Z. Long non-coding RNA 00312 downregulates cyclin B1 and inhibits hepatocellular carcinoma cell proliferation in vitro and in vivo. Biochem Biophys Res Commun. 2018;497(1):173–80. doi:10.1016/j.bbrc.2018.02.049. [Google Scholar] [PubMed] [CrossRef]
234. Fang Z, Wang Y, Wang Z, Xu M, Ren S, Yang D, et al. ERINA is an estrogen-responsive LncRNA that drives breast cancer through the E2F1/RB1 pathway. Cancer Res. 2020;80(2):4399–413. doi:10.1158/0008-5472.CAN-20-1031. [Google Scholar] [PubMed] [CrossRef]
235. Guo Q, Li L, Bo Q, Chen L, Sun L, Shi H. Long noncoding RNA PITPNA-AS1 promotes cervical cancer progression through regulating the cell cycle and apoptosis by targeting the miR-876-5p/c-MET axis. Biomed Pharmacother. 2020;128:110072. doi:10.1016/j.biopha.2020.110072. [Google Scholar] [PubMed] [CrossRef]
236. Li H, Jia Y, Cheng J, Liu G, Song F. LncRNA NCK1-AS1 promotes proliferation and induces cell cycle progression by crosstalk NCK1-AS1/MIR-6857/CDK1 pathway. Cell Death Dis. 2018;9(2):198. doi:10.1038/s41419-017-0249-3. [Google Scholar] [PubMed] [CrossRef]
237. Li GH, Ma ZH, Wang X. Long non-coding RNA CCAT1 is a prognostic biomarker for the progression of oral squamous cell carcinoma via miR-181a-mediated Wnt/β-catenin signaling pathway. Cell Cycle. 2019;18(21):2902–13. doi:10.1080/15384101.2019.1662257. [Google Scholar] [PubMed] [CrossRef]
238. Liu J, Qian J, Mo Q, Tang L, Xu Q. LncRNA NR2F2-AS1 silencing induces cell cycle arrest in G0/G1 phase via downregulating Cyclin D1 in colorectal cancer. Cancer Manag Res. 2020;12:1835–43. doi:10.2147/CMAR. [Google Scholar] [CrossRef]
239. Zhou Z, Tan F, Pei Q, Li C, Zhou Y, Li Y, et al. LncRNA SNHG4 modulates colorectal cancer cell cycle and cell proliferation through regulating miR-590-3p/CDK1 axis. Aging. 2021;13(7):9838–58. doi:10.18632/aging.202737. [Google Scholar] [PubMed] [CrossRef]
240. Liu B, Wang W, Sun S, Ding H, Lan L, Li A. Knockdown of lncRNA ABHD11-AS1 suppresses the tumorigenesis of pancreatic cancer via sponging miR-1231. Onco Targets Ther, 13:11347–58. doi:10.2147/OTT.S259598. [Google Scholar] [PubMed] [CrossRef]
241. Luo G, Liu D, Huang C, Wang M, Xiao X, Zeng F, et al. LncRNA GAS5 inhibits cellular proliferation by targeting P27Kip1. Mol Cancer Res. 2017;15(7):789–99. doi:10.1158/1541-7786.MCR-16-0331. [Google Scholar] [PubMed] [CrossRef]
242. Zuo Z, Liu L, Song B, Tan J, Ding D, Lu Y. Silencing of long non-coding RNA ENST00000606790.1 inhibits the malignant behaviors of papillary thyroid carcinoma through the PI3K/AKT pathway. Endocr Res. 2021;46(1):1–9. doi:10.1080/07435800.2020.1804928. [Google Scholar] [PubMed] [CrossRef]
243. Huang L, Wang Y, Chen J, Wang Y, Zhao Y, Wang Y, et al. Long noncoding RNA PCAT1, a novel serum-based biomarker, enhances cell growth by sponging miR-326 in oesophageal squamous cell carcinoma. Cell Death Dis. 2019;10(7):513. doi:10.1038/s41419-019-1745-4. [Google Scholar] [PubMed] [CrossRef]
244. Zhu Y, Dai B, Zhang H, Shi G, Shen Y, Ye D. Long non-coding RNA LOC572558 inhibits bladder cancer cell proliferation and tumor growth by regulating the AKT-MDM2–p53 signaling axis. Cancer Lett. 2016;380(2):269–74. doi:10.1016/j.canlet.2016.04.030. [Google Scholar] [PubMed] [CrossRef]
245. Zhang P, Lu Y, Kong Z, Zhang Y, Fu F, Su X, et al. Androgen-responsive lncRNA LINC00304 promotes cell cycle and proliferation via regulating CCNA1. Prostate. 2019;79(9):994–1006. doi:10.1002/pros.23811. [Google Scholar] [PubMed] [CrossRef]
246. Deng G, Mou T, He J, Chen D, Lv D, Liu H, et al. Circular RNA circRHOBTB3 acts as a sponge for miR-654-3p inhibiting gastric cancer growth. Exp Cancer Res. 2020;39(1):1. doi:10.1186/s13046-019-1487-2. [Google Scholar] [PubMed] [CrossRef]
247. Sun M, Zhao W, Chen Z, Li M, Li S, Wu B, et al. Circ_0058063 regulates CDK6 to promote bladder cancer progression by sponging miR-145-5p. J Cell Physiol. 2019;234(4):4812–24. doi:10.1002/jcp.27280. [Google Scholar] [PubMed] [CrossRef]
248. Wu S, Yang J, Xu H, Wang X, Zhang R, Lu W, et al. Circular RNA circGLIS3 promotes bladder cancer proliferation via the miR-1273f/SKP1/Cyclin D1 axis. Cell Biol Toxicol. 2022;38(1):129–46. doi:10.1007/s10565-021-09591-3. [Google Scholar] [PubMed] [CrossRef]
249. Zhou X, Jian W, Luo Q, Zheng W, Deng X, Wang X, et al. Circular RNA_0006014 promotes breast cancer progression through sponging miR-885-3p to regulate NTRK2 and PIK3/AKT pathway. Aging. 2022;14(7):3105–28. doi:10.18632/aging.203996. [Google Scholar] [PubMed] [CrossRef]
250. Gao D, Qi X, Zhang X, Fang K, Guo Z, Li L. hsa_circRNA_0006528 as a competing endogenous RNA promotes human breast cancer progression by sponging miR-7-5p and activating the MAPK/ERK signaling pathway. Mol Carcinog. 2019;58(4):554–64. doi:10.1002/mc.22950. [Google Scholar] [PubMed] [CrossRef]
251. Li M, Chen H, Xia L, Huang P. Circular RNA circSP3 promotes hepatocellular carcinoma growth by sponging microRNA-198 and upregulating cyclin-dependent kinase 4. Aging. 2021;13(14):18586–605. doi:10.18632/aging.203303. [Google Scholar] [PubMed] [CrossRef]
252. Zheng H, Chen T, Li C, Xu C, Ding C, Chen J, et al. A circular RNA hsa_circ_0079929 inhibits tumor growth in hepatocellular carcinoma. Cancer Manag Res. 2019;11:443–54. doi:10.2147/CMAR. [Google Scholar] [CrossRef]
253. Ji X, Shan L, Shen P, He M. Circular RNA circ_001621 promotes osteosarcoma cells proliferation and migration by sponging miR-578 and regulating VEGF expression. Cell Death Dis. 2020;11(1):18. doi:10.1038/s41419-019-2204-y. [Google Scholar] [PubMed] [CrossRef]
254. Zhang S, Han J, Fu J. The circ_0032822 promotes the proliferation of head and neck squamous cell carcinoma cells through miR-141/EF3 signaling axis. Front Oncol. 2021;11:662496. doi:10.3389/fonc.2021.662496. [Google Scholar] [PubMed] [CrossRef]
255. Tan Y, Huang Z, Wang X, Dai H, Jiang G, Feng W. A novel fusion circular RNA F-circBA1 derived from the BCR-ABL fusion gene displayed an oncogenic role in chronic myeloid leukemia cells. Bioengineered. 2021;12(1):4816–27. doi:10.1080/21655979.2021.1957749. [Google Scholar] [PubMed] [CrossRef]
256. Liu N, Jiang F, Chen Z, Liu X, Fu ZM, Wang BC, et al. circIFT80 Functions as a ceRNA for miR-142, miR-568, and miR-634 and promotes the progression of colorectal cancer by targeting β-catenin. Dis Markers. 2022;8081246. doi:10.1155/2022/8081246. [Google Scholar] [PubMed] [CrossRef]
257. Hsu HH, Kuo WW, Shih HN, Cheng SF, Yang CK, Chen MC, et al. FOXC1 Regulation of miR-31-5p confers oxaliplatin resistance by targeting LATS2 in colorectal cancer. Cancers. 2019;11(10):1576. doi:10.3390/cancers11101576. [Google Scholar] [PubMed] [CrossRef]
258. Pathania AS, Challagundla KB. Exosomal long non-coding RNAs: emerging players in the tumor microenvironment. Mol Ther Nucleic Acids. 2020;23:1371–83. doi:10.1016/j.omtn.2020.09.039. [Google Scholar] [PubMed] [CrossRef]
259. Pathania AS, Chava H, Balusu R, Pasupulati AK, Coulter DW, Challagundla KB. The crosstalk between non-coding RNAs and cell-cycle events: a new frontier in cancer therapy. Mol Ther Oncol. 2024;32(2):200785. doi:10.1016/j.omton.2024.200785. [Google Scholar] [PubMed] [CrossRef]
260. Wang H, Yu S, Peng H, Shu Y, Zhang W, Zhu Q, et al. Long noncoding RNA Linc00337 functions as an E2F1 co-activator and promotes cell proliferation in pancreatic ductal adenocarcinoma. J Exp Cancer Res. 2020;39(1):216. doi:10.1186/s13046-020-01725-5. [Google Scholar] [PubMed] [CrossRef]
261. Nadal-Ribelles M, Solé C, Xu Z, Steinmetz LM, deNadal E, Posas F. Control of Cdc28 CDK1 by a stress-induced lncRNA. Mol Cell. 2014;53(4):549–61. doi:10.1016/j.molcel.2014.01.006. [Google Scholar] [PubMed] [CrossRef]
262. Shi Q, Li Y, Li S, Jin L, Lai H, Wu Y, et al. LncRNA DILA1 inhibits Cyclin D1 degradation and contributes to tamoxifen resistance in breast cancer. Nat Commun. 2020;11(1):5513. doi:10.1038/s41467-020-19349-w. [Google Scholar] [PubMed] [CrossRef]
263. Zhou L, Liu R, Liang X, Zhang S, Bi W, Yang M, et al. lncRNA RP11-624L4.1 is associated with unfavorable prognosis and promotes proliferation via the CDK4/6-Cyclin D1-Rb-E2F1 pathway in NPC. Mol Ther Nucleic Acids. 2020;22:1025–39. doi:10.1016/j.omtn.2020.10.017. [Google Scholar] [PubMed] [CrossRef]
264. Qian Y, Shi L, Luo Z. Long non-coding RNAs in cancer: implications for diagnosis, prognosis, and therapy. Front Med (Lausanne). 2020;7:612393. doi:10.3389/fmed.2020.612393. [Google Scholar] [PubMed] [CrossRef]
265. Wang L, Tang D, Wu T, Sun F. ELF1-mediated LUCAT1 promotes choroidal melanoma by modulating RBX1 expression. Cancer Med. 2020;9(6):2160–70. doi:10.1002/cam4.2859. [Google Scholar] [PubMed] [CrossRef]
266. Luzón-Toro B, Fernández RM, Martos-Martínez JM, Rubio-Manzanares-Dorado M, Antiñolo G, Borrego S. LncRNA LUCAT1 as a novel prognostic biomarker for patients with papillary thyroid cancer. Sci Rep. 2019;9(1):14374. doi:10.1038/s41598-019-50913-7. [Google Scholar] [PubMed] [CrossRef]
267. Yildirim O, Izgu EC, Damle M, Chalei V, Ji F, Sadreyev RI, et al. S-phase enriched non-coding RNAs regulate gene expression and cell cycle progression. Cell Rep. 2020;31(6):107629. doi:10.1016/j.celrep.2020.107629. [Google Scholar] [PubMed] [CrossRef]
268. Lin Y, Jiang J. Long non-coding RNA LINC00704 promotes cell proliferation, migration, and invasion in papillary thyroid carcinoma via miR-204-5p/HMGB1 axis. Open Life Sci. 2020;1591(1):561–71. doi:10.1515/biol-2020-0057. [Google Scholar] [PubMed] [CrossRef]
269. Wang Y, Fu L, Lu T, Zhang G, Zhang J, Zhao Y, et al. Clinicopathological and prognostic significance of long non-coding RNA MIAT in human cancers: a review and meta-analysis. Front Genet. 2021;12:729768. doi:10.3389/fgene.2021.729768. [Google Scholar] [PubMed] [CrossRef]
270. Xing C, Sun SG, Yue ZQ, Bai F. Role of lncRNA LUCAT1 in cancer. Biomed Pharmacother. 2021;134:111158. doi:10.1016/j.biopha.2020.111158. [Google Scholar] [PubMed] [CrossRef]
271. Wang Y, Tan QY, Shen Y, Liu CY, Huang T, Huai D, et al. LINC00704 contributes to the proliferation and accelerates the cell cycle of nasopharyngeal carcinoma cells via regulating ETS1/CDK6 axis. Kashsiung J Med Sci. 2022;38(4):312–20. doi:10.1002/kjm2.12491. [Google Scholar] [PubMed] [CrossRef]
272. Li YL, Wang XM, Qiao GD, Zhang S, Wang J, Cong YZ, et al. Up-regulated lnc-lung cancer associated transcript 1 enhances cell migration and invasion in breast cancer progression. Biochem Biophys Res Commun. 2020;521(2):271–8. doi:10.1016/j.bbrc.2019.08.040. [Google Scholar] [PubMed] [CrossRef]
273. Yu H, Xu Y, Zhang D, Liu G. Long noncoding RNA LUCAT1 promotes malignancy of ovarian cancer through regulation of miR-612/HOXA13 pathway. Biochem Biophys Res Commun. 2018;503(3):2905–100. doi:10.1016/j.bbrc.2018.07.165. [Google Scholar] [PubMed] [CrossRef]
274. Yang T, Xia S. Study of the biological function of LncRNA LUCAT1 on cervical cancer cells by targeting miR-199b-5p. Biosci Rep. 2020;40(4):BSR20200422. doi:10.1042/BSR20200422. [Google Scholar] [PubMed] [CrossRef]
275. Liu HZ, Liu GY, Pang WW, Zhang H, Zeng ZJ, Wang HJ. LncRNA LUCAT1 promotes proliferation of ovarian cancer cells by regulating miR-199a-5p expression. Eur Rev Med Pharmacol Sci. 2020;24(4):1682–7. doi:10.26355/eurrev_202002_20342. [Google Scholar] [PubMed] [CrossRef]
276. Nai Y, Pan C, Hu X, Ma Y. LncRNA LUCAT1 contributes to cell proliferation and migration in human pancreatic ductal adenocarcinoma via sponging miR-539. Cancer Med. 2020;9(2):757–67. doi:10.1002/cam4.2724. [Google Scholar] [PubMed] [CrossRef]
277. Huan L, Guo T, Wu Y, Xu L, Huang S, Xu Y, et al. Hypoxia induced LUCAT1/PTBP1 axis modulates cancer cell viability and chemotherapy response. Mol Cancer. 2020;19(1):11. doi:10.1186/s12943-019-1122-z. [Google Scholar] [PubMed] [CrossRef]
278. Guo K, Qian K, Shi Y, Sun T, Wang Z. LncRNA-MIAT promotes thyroid cancer progression and function as ceRNA to target EZH2 by sponging miR-150-5p. Cell Death Dis. 2021;12(12):1097. doi:10.1038/s41419-021-04386-0. [Google Scholar] [PubMed] [CrossRef]
279. He H, Wang N, Yi X, Tang C, Wang D. Long non-coding RNA H19 regulates E2F1 expression by competitively sponging endogenous miR-29a-3p in clear cell renal cell carcinoma. Cell Biosci. 2017;7(1):65. doi:10.1186/s13578-017-0193-z. [Google Scholar] [PubMed] [CrossRef]
280. Wei W, Mo X, Yan L, Huang M, Yang Y, Jin Q, et al. Circular RNA profiling reveals that circRNA_104433 regulates cell growth by targeting miR-497-5p in gastric cancer. Cancer Manag Res. 2020;12:15–30. doi:10.2147/CMAR. [Google Scholar] [CrossRef]
281. Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20(11):675–91. doi:10.1038/s41576-019-0158-7. [Google Scholar] [PubMed] [CrossRef]
282. Wang PL, Bao Y, Yee MC, Barrett SP, Hogan GJ, Olsen MN, et al. Circular RNA is expressed across the eukaryotic tree of life. PLoS One. 2014;9(6):e90859. doi:10.1371/journal.pone.0090859. [Google Scholar] [PubMed] [CrossRef]
283. Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19(2):141–57. doi:10.1261/rna.035667.112. [Google Scholar] [PubMed] [CrossRef]
284. Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO. Cell-type specific features of circular RNA expression. PLoS Genet. 2013;9(9):e1003777. doi:10.1371/journal.pgen.1003777. [Google Scholar] [PubMed] [CrossRef]
285. Li J, Sun D, Pu W, Wang J, Peng Y. Circular RNAs in Cancer: biogenesis, Function, and Clinical Significance. Trends Cancer. 2020;6(4):319–36. doi:10.1016/j.trecan.2020.01.012. [Google Scholar] [PubMed] [CrossRef]
286. Chen LL. The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol. 2016;17(4):205–11. doi:10.1038/nrm.2015.32. [Google Scholar] [PubMed] [CrossRef]
287. Kristensen LS, Hansen TB, Venø MT, Kjems J. Circular RNAs in cancer: opportunities and challenges in the field. Oncogene. 2018;37(5):555–65. doi:10.1038/onc.2017.361. [Google Scholar] [PubMed] [CrossRef]
288. Bach DH, Lee SK, Sood AK. Circular RNAs in Cancer. Mol Ther Nucleic Acids. 2019;16(Suppl 2):118–29. doi:10.1016/j.omtn.2019.02.005. [Google Scholar] [PubMed] [CrossRef]
289. Yan L, Liu G, Cao H, Zhang H, Shao F. Hsa_circ_0035483 sponges hsa-miR-335 to promote the gemcitabine-resistance of human renal cancer cells by autophagy regulation. Biochem Biophys Res Commun. 2019;519(1):172–8. doi:10.1016/j.bbrc.2019.08.093. [Google Scholar] [PubMed] [CrossRef]
290. Ma Z, Han C, Xia W, wang S, Li X, Fang P, et al. circ5615 functions as a ceRNA to promote colorectal cancer progression by upregulating TNKS. Cell Death Dis. 2020;11(5):356. doi:10.1038/s41419-020-2514-0. [Google Scholar] [PubMed] [CrossRef]
291. Cui D, Qian R, Li Y. Circular RNA circ-CMPK1 contributes to cell proliferation of non-small cell lung cancer by elevating cyclin D1 via sponging miR-302e. Mol Genet Genomic Med. 2020;8(2):e999. doi:10.1002/mgg3.999. [Google Scholar] [PubMed] [CrossRef]
292. Duan X, Shen N, Chen J, Wang J, Zhu Q, Zhai Z. Circular RNA MYLK serves as an oncogene to promote cancer progression via microRNA-195/cyclin D1 axis in laryngeal squamous cell carcinoma. Biosci Rep. 2019;39(9):BSR20190227. doi:10.1042/BSR20190227. [Google Scholar] [PubMed] [CrossRef]
293. Zang Y, Li J, Wan B, Tai Y. circRNA circ-CCND1 promotes the proliferation of laryngeal squamous cell carcinoma through elevating CCND1 expression via interacting with HuR and miR-646. J Cell Mol Med. 2020;24(4):2426–33. doi:10.1111/jcmm.14925. [Google Scholar] [PubMed] [CrossRef]
294. Chen J, Xu S, Chen S, Zong Z, Han X, Zhao Y, et al. CircPUM1 promotes the malignant behavior of lung adenocarcinoma by regulating miR-326. Biochem Biophys Res Commun. 2019;508(3):844–9. doi:10.1016/j.bbrc.2018.11.176. [Google Scholar] [PubMed] [CrossRef]
295. Li XY, Liu YR, Zhou JH, Li W, Guo HH, Ma HP. Enhanced expression of circular RNA hsa_circ_000984 promotes cells proliferation and metastasis in non-small cell lung cancer by modulating Wnt/β-catenin pathway. Eur Rev Med Pharmacol Sci. 2019;23(8):3366–74. doi:10.26355/eurrev_201904_17700. [Google Scholar] [PubMed] [CrossRef]
296. Liu LH, Tian QQ, Liu J, Zhou Y, Yong H. Upregulation of hsa: circ_0136666 contributes to breast cancer progression by sponging miR-1299 and targeting CDK6. J Cell Biochem. 2019;120(8):12684–93. doi:10.1002/jcb.28536. [Google Scholar] [PubMed] [CrossRef]
297. Zheng F, Wang M, Li Y, Huang C, Tao D, Xie F, et al. CircNR3C1 inhibits proliferation of bladder cancer cells by sponging miR-27a-3p and downregulating cyclin D1 expression. Cancer Lett. 2019;460(1):139–51. doi:10.1016/j.canlet.2019.06.018. [Google Scholar] [PubMed] [CrossRef]
298. Damase TR, Sukhovershin R, Boada C, Taraballi F, Pettigrew RI, Cooke JP. The Limitless Future of RNA Therapeutics. Front Bioeng Biotechnol. 2021;9:628137. doi:10.3389/fbioe.2021.628137. [Google Scholar] [PubMed] [CrossRef]
299. Orellana EA, Li C, Lisevick A, Kasinski AL. Identification and validation of microRNAs that synergize with miR-34a-a basis for combinatorial microRNA therapeutics. Cell Cycle. 2019;18(15):1798–811. doi:10.1080/15384101.2019.1634956. [Google Scholar] [PubMed] [CrossRef]
300. Tian H, Cheng L, Liang Y, Lei H, Qin M, Li X, et al. MicroRNA therapeutic delivery strategies: a review. J Drug Deliv Sci Technol. 2024;93(5):105430. doi:10.1016/j.jddst.2024.105430. [Google Scholar] [CrossRef]
301. Su L, Sun Z, Qi F, Su H, Qian L, Li J, et al. GRP75-driven, cell-cycle-dependent macropinocytosis of Tat/pDNA-Ca2+ nanoparticles underlies distinct gene therapy effect in ovarian cancer. J Nanobiotechnol. 2022;20(1):340. doi:10.1186/s12951-022-01530-6. [Google Scholar] [PubMed] [CrossRef]
302. Rizzardi LF, Cook JG. Flipping the switch from g1 to s phase with e3 ubiquitin ligases. Genes Cancer. 2012;3(11–12):634–48. doi:10.1177/1947601912473307. [Google Scholar] [PubMed] [CrossRef]
303. Havens CG, Walter JC. Mechanism of CRL4Cdt2, a PCNA-dependent E3 ubiquitin ligase. Genes Dev. 2011;25(15):1568–82. doi:10.1101/gad.2068611. [Google Scholar] [PubMed] [CrossRef]
304. Wang L, Zhang J, Wan L, Zhou X, Wang Z, Wei W. Targeting Cdc20 as a novel cancer therapeutic strategy. Pharmacol Ther. 2015;151:141–51. doi:10.1016/j.pharmthera.2015.04.002. [Google Scholar] [PubMed] [CrossRef]
305. Xian F, Zhao C, Huang C, Bie J, Xu G. The potential role of CDC20 in tumorigenesis, cancer progression and therapy: a narrative review. Medicine. 2023;102(36):e35038. doi:10.1097/MD.0000000000035038. [Google Scholar] [PubMed] [CrossRef]
306. Zhang L, Niu T, Huang Y, Zhu H, Zhong W, Lin J, et al. Compound 331 selectively induces glioma cell death by upregulating miR-494 and downregulating CDC20. Sci Rep. 2015;5(1):12003. doi:10.1038/srep12003. [Google Scholar] [PubMed] [CrossRef]
307. Cheng S, Castillo V, Sliva D. CDC20 associated with cancer metastasis and novel mushroom-derived CDC20 inhibitors with antimetastatic activity. Int J Oncol. 2019;54(6):2250–6. doi:10.3892/ijo.2019.4791. [Google Scholar] [PubMed] [CrossRef]
308. Liu N, Wang X, Zhu Z, Li D, Lv X, Chen Y, et al. Selected ideal natural ligand against TNBC by inhibiting CDC20, using bioinformatics and molecular biology. Aging. 2021;13(20):23702–25. doi:10.18632/aging.203642. [Google Scholar] [PubMed] [CrossRef]
309. Li J, Dang N, Martinez-Lopez N, Jowsey PA, Huang D, Lightowlers RN, et al. M2I-1 disrupts the in vivo interaction between CDC20 and MAD2 and increases the sensitivities of cancer cell lines to anti-mitotic drugs via MCL-1s. Cell Div. 2019;14(1):5. doi:10.1186/s13008-019-0049-5. [Google Scholar] [PubMed] [CrossRef]
310. Wu X, Yu M, Zhang Z, Leng F, Ma Y, Xie N, et al. DDB2 regulates DNA replication through PCNA-independent degradation of CDT2. Cell Biosci. 2021;11(1):34. doi:10.1186/s13578-021-00540-5. [Google Scholar] [PubMed] [CrossRef]
311. Mazian MA, Yamanishi K, Rahman MZA, Ganasen M, Nishitani H. CRL4Cdt2 ubiquitin ligase, A genome caretaker controlled by Cdt2 binding to PCNA and DNA. Genes. 2022;13(2):266. doi:10.3390/genes13020266. [Google Scholar] [PubMed] [CrossRef]
312. Jin J, Arias EE, Chen J, Harper JW, Walter JC. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell. 2006;23(5):709–21. doi:10.1016/j.molcel.2006.08.010. [Google Scholar] [PubMed] [CrossRef]
313. Kiran S, Wilson B, Saha S, Graff JA, Dutta A. HPVE6-USP46 mediated Cdt2 stabilization reduces set8 mediated h4k20-methylation to induce gene expression changes. Cancers. 2022;14(1):30. doi:10.3390/CANCERS14010030. [Google Scholar] [PubMed] [CrossRef]
314. Dar A, Wu D, Lee N, Shibata E, Dutta A. 14-3-3 proteins play a role in the cell cycle by shielding cdt2 from ubiquitin-mediated degradation. Mol Cell Biol. 2014;34(21):4049–61. doi:10.1128/MCB.00838-14. [Google Scholar] [PubMed] [CrossRef]
315. Lin JJ, Milhollen MA, Smith PG, Narayanan U, Dutta A. NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res. 2010;70(24):10310–20. doi:10.1158/0008-5472.CAN-10-2062. [Google Scholar] [PubMed] [CrossRef]
316. Feroz W, Sheikh AMA. Exploring the multiple roles of guardian of the genome: P53. Egypt J Med Hum Genet. 2020;21(1):831. doi:10.1186/s43042-020-00089-x. [Google Scholar] [CrossRef]
317. Zhu H, Gao H, Ji Y, Zhou Q, Du Z, Tian L, et al. Targeting p53-MDM2 interaction by small-molecule inhibitors: learning from MDM2 inhibitors in clinical trials. J Hematol Oncol. 2022;15(1):91. doi:10.1186/s13045-022-01314-3. [Google Scholar] [PubMed] [CrossRef]
318. Wang S, Chen FE. Small-molecule MDM2 inhibitors in clinical trials for cancer therapy. Eur J Med Chem. 2022;236:114334. doi:10.1016/j.ejmech.2022.114334. [Google Scholar] [PubMed] [CrossRef]
319. Sampson C, Wang Q, Otkur W, Zhao H, Lu Y, Liu X, et al. The roles of E3 ubiquitin ligases in cancer progression and targeted therapy. Clin Transl Med. 2023;13(3):e1204. doi:10.1002/ctm2.1204. [Google Scholar] [PubMed] [CrossRef]
320. Rew Y, Sun D. Discovery of a small molecule MDM2 inhibitor (AMG 232) for treating cancer. J Med Chem. 2014;57(14):6332–41. doi:10.1021/jm500627s. [Google Scholar] [PubMed] [CrossRef]
321. Gluck WL, Gounder MM, Frank R, Eskens F, Blay JY, Cassier PA, et al. Phase 1 study of the MDM2 inhibitor AMG 232 in patients with advanced P53 wild-type solid tumors or multiple myeloma. Invest New Drugs. 2020;38(3):831–43. doi:10.1007/s10637-019-00840-1. [Google Scholar] [PubMed] [CrossRef]
322. Liang L, He Y, Wang H, Zhou H, Xiao L, Ye M, et al. The Wee1 kinase inhibitor MK1775 suppresses cell growth, attenuates stemness and synergises with bortezomib in multiple myeloma. Br J Haematol. 2020;191(1):62–76. doi:10.1111/bjh.16614. [Google Scholar] [PubMed] [CrossRef]
323. Hauge S, Macurek L, Syljuåsen RG. p21 limits S phase DNA damage caused by the Wee1 inhibitor MK1775. Cell Cycle. 2019;18(8):834–47. doi:10.1080/15384101.2019.1593649. [Google Scholar] [PubMed] [CrossRef]
324. Chen CP, Yeh CN, Pan YR, Huang WK, Hsiao YT, Lo CH, et al. Wee1 inhibition by MK1775 potentiates gemcitabine through accumulated replication stress leading to apoptosis in biliary tract cancer. Biomed Pharmacother. 2023;166(1):115389. doi:10.1016/j.biopha.2023.115389. [Google Scholar] [PubMed] [CrossRef]
325. Narwanti I, Yu ZY, Sethy B, Lai MJ, Lee HY, Olena P, et al. 6-Regioisomeric 5,8-quinolinediones as potent CDC25 inhibitors against colorectal cancers. Eur J Med Chem. 2023;258:115505. doi:10.1016/j.ejmech.2023.115505. [Google Scholar] [PubMed] [CrossRef]
326. Tao Y, Hao X, Ding X, Cherukupalli S, Song Y, Liu X, et al. Medicinal chemistry insights into novel CDC25 inhibitors. Eur J Med Chem. 2020;201:112374. doi:10.1016/j.ejmech.2020.112374. [Google Scholar] [PubMed] [CrossRef]
327. Das S, Chandrasekaran AP, Suresh B, Haq S, Kang JH, Lee SJ, et al. Genome-scale screening of deubiquitinase subfamily identifies USP3 as a stabilizer of Cdc25A regulating cell cycle in cancer. Cell Death Differ. 2020;27(11):3004–20. doi:10.1038/s41418-020-0557-5. [Google Scholar] [PubMed] [CrossRef]
328. Zhang Z, Liu W, Bao X, Sun T, Wang J, Li M, et al. USP39 facilitates breast cancer cell proliferation through stabilization of FOXM1. Am J Cancer Res. 2022;12(8):3644–61. [Google Scholar] [PubMed]
329. Arceci A, Bonacci T, Wang X, Stewart K, Damrauer JS, Hoadley KA, et al. FOXM1 deubiquitination by USP21 regulates cell cycle progression and paclitaxel sensitivity in basal-like breast cancer. Cell Rep. 2019;26(11):3076–86. doi:10.1016/j.celrep.2019.02.054. [Google Scholar] [PubMed] [CrossRef]
330. Yi J, Li H, Chu B, Kon N, Hu X, Hu J, et al. Inhibition of USP7 induces p53-independent tumor growth suppression in triple-negative breast cancers by destabilizing FOXM1. Cell Death Differ. 2023;30(7):1799–810. doi:10.1038/s41418-023-01180-7. [Google Scholar] [PubMed] [CrossRef]
331. Karunarathna U, Kongsema M, Zona S, Gong C, Cabrera E, Gomes AR, et al. OTUB1 inhibits the ubiquitination and degradation of FOXM1 in breast cancer and epirubicin resistance. Oncogene. 2016;35(11):1433–44. doi:10.1038/onc.2015.208. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools