
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE
Propofol suppressed cell proliferation through inhibition of SREBP1c-mediated De novo lipogenesis in colorectal cancer cells
1 Department of Anesthesiology, Shenzhen Hospital, Southern Medical University, Shenzhen, 518000, China
2 The Third School of Clinical Medicine, Southern Medical University, Guangzhou, 510630, China
3 Department of Anesthesia, Zhuhai Center for Maternal and Child Health Care, Zhuhai, 519000, China
4 Department of Anesthesia, Zhuhai Doumen Maternal and Child Health Care Hospital, Zhuhai, 519000, China
* Corresponding Authors: JIEXIAN ZHOU. Email: ; YOUTAN LIU. Email:
# These authors contributed equally to this work
BIOCELL 2024, 48(12), 1773-1780. https://doi.org/10.32604/biocell.2024.056374
Received 21 July 2024; Accepted 12 October 2024; Issue published 30 December 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: De novo lipogenesis (DNL) is a critical event for the development of tumors, in the present work, we revealed the role of propofol in colorectal cancer (CRC) cell proliferation. Methods: Western blotting (WB), Real-time PCR, and luciferase combined with chromatin immunoprecipitation (ChIP) were used to identify the mechanism underlying propofol-modulated cell proliferation in CRC cells. Results: Herein, we showed that propofol suppressed cell proliferation, which was attributed to the inhibition of DNL characterized by reduced fatty acid synthase (FASN), acetyl-coA carboxylase alpha (ACCA), and stearoyl-coA desaturase-1 (SCD1) expression. Mechanically, propofol stimulation decreased sterol regulatory element-binding proteins-1c (SREBP-1c) mature and nuclear translocation, which further decreased SCD1 transactivation confirmed by luciferase and ChIP analysis, while no significant difference in total SREBP1c was observed. What’s more, supplementation of Monounsaturated fatty acid (MuFA) could reverse the inhibitory effect of propofol on cell proliferation. Conclusion: Taken together, these results suggested propofol modulated cell proliferation is dependent on SREBP1c-mediated DNL.Keywords
Abbreviation
| ACAT1 | Acetyl-CoA acetyltransferase 1 |
| AMPK | Adenosine 5′-monophosphate (AMP)-activated protein kinase |
| AKT | Protein kinase B |
| ACCA | Acetyl-coA carboxylase alpha |
| CRC | Colorectal cancer |
| ChIP | Chromatin immunoprecipitation |
| CCK-8 | Cell counting kit-8 |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| DNL | De novo lipogenesis |
| ER | Endoplasmic reticulum |
| FASN | Fatty acid synthase |
| HIF1α | Hypoxia-inducible factor 1-alpha |
| KLF2 | Kruppel-like factor 2 |
| MuFA | Monounsaturated fatty acid |
| mTOR | Mammalian target of rapamycin |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRP3 | NOD-, LRR- and pyrin domain-containing protein 3 |
| OSCC | Oral squamous cell carcinoma |
| OA | Oleic acid |
| PPARs | Peroxisome proliferator-activated receptors |
| PI3K | Phosphoinositide 3-kinase |
| qPCR | Quantitative PCR |
| SREBP-1c | Sterol regulatory element-binding proteins -1c |
| SCAP | SREBP cleavage-activating protein |
| SCD1 | Stearoyl-coA desaturase-1 |
| SIRT1 | Silent mating type information regulation 2 homolog-1 |
| SFA | Saturated fatty acids |
| TM2D1 | TM2 domain containing 1 |
| WB | Western blotting |
More and more evidence found that anesthetic techniques are critical in treating postoperative recurrence and can improve survival rates for patients with cancers [1–3]. Propofol, a widely used intravenous anesthetic, has been shown to suppress tumor progression, including proliferation and metastasis in colorectal cancer (CRC) [4], colon cancer [5], and oral squamous cell carcinoma (OSCC) [6]. The critical molecules, including silent mating type information regulation 2 homolog-1 (SIRT1), hypoxia-inducible factor 1-alpha (HIF1α) [7] and Phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) signaling pathways [5] or circular RNA [6], were reported to involved in propofol-modulated proliferation. However, the mechanism of propofol-modulated cell proliferation in tumors has not been completely identified yet.
Metabolic reprogramming of De novo lipogenesis (DNL) plays a vital role in tumor progression, which has been listed as a hallmark of cancer [8,9]. DNL is a multi-step fatty acid metabolism mediated by critical rate-limiting enzymes, such as FASN, ACCA, and SCD1 [10]. These key lipogenic enzyme genes were directly transactivated by SREBP-1c [11,12]. In particular, SCD1 was demonstrated to play a critical role in tumors by modulating the balance between monounsaturated fatty acids (MuFA)/saturated fatty acids (SFA) in cellular lipids to influence cell membrane structure, energy metabolism, and signaling [13]. For instance, inhibition of SCD1 could induce the anti-tumor activity of CD8+ T cells through enhancement of interferon-γ (IFN-γ) production dependent on acetyl-CoA acetyltransferase 1 (ACAT1) [14] or β-catenin signaling in cancer cells and endoplasmic reticulum (ER) stress in T cells [15]. What’s more, the depletion of SCD1 expression in cervical cancer cells led to a decline in cell proliferation, cell migration, and invasion [16]. Interestingly, propofol has been demonstrated to inhibit glycolysis in ovarian tumors and CRC cells [17,18]. However, no available studies about the function of DNL in propofol-modulated proliferation were reported at present, which remains to be addressed.
Recently, the work has revealed that propofol-induced apoptosis and ferroptosis inhibit cell proliferation by targeting the TM2 domain containing 1 (TM2D1) [19], NF-κB/HIF-1α [20]. The metabolic reprogramming of DNL was also found to be involved in tumor progress, such as tumor growth [21,22]. In this work, we would explore the possible crosstalk between DNL and propofol in cell proliferation, which could extend the novel role of propofol in metabolic reprogramming and supply a novel unreported approach to propofol in cell proliferation.
Reagents: Dulbecco’s Modified Eagle’s Medium (DMEM) (GIBCO, C11885500BT, Shanghai, China) and Fetal bovine serum (FBS) (GIBCO, 10099141C) were purchased from GIBCO (Shanghai, China). Propofol (MedChemExpress, HY-B0649, Wuhan, China) and Oleic acid (MedChemExpress, HY-N1446) were purchased from MedChemExpress (Wuhan, China). Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, P0028, Shanghai, China), BeyoRT™ II First Strand cDNA Synthesis Kit (Beyotime, D7168M), BeyoFast™ SYBR Green qPCR Mix (Beyotime, D7265), BCA Protein Assay Kit (Beyotime, P0011), Lipo8000™ transfection reagents (Beyotime, C0533) and BeyoECL Moon (Beyotime, P0018FS) were from Beyotime (Shanghai, China). The dual Luciferase Assay System (Vazyme, DD1205-01, Nanjing, China) kit was from Vazyme (Nanjing, China). ChIP Kit (Abcam, ab500, Cambridge, UK) and Human monounsaturated fatty acid (MFA) elisa kit (Abcam, ab242305) were purchased from Abcam (Cambridge, UK). Plasmid was purchased from Youbio (Changsha, China), and RNAiso Plus (Takara, 9108Q, Dalian, China) was from Takara (Dalian, China). All ultrapure reagents were from Sigma (St Louis, MO, USA).
Antibodies: FASN monoclonal antibody (Proteintech, 66591-1-Ig, 1:2000, Wuhan, China), ACCA monoclonal antibody (Proteintech, 67373-1-Ig, 1:2000), SCD polyclonal antibody (Proteintech, 28678-1-AP, 1:2000), SREBP-1c (Proteintech, 66875-1-Ig, 1:2000), Lamin A/C polyclonal antibody (Proteintech, 10298-1-AP, 1:4000) and PCNA polyclonal antibody (Proteintech, 10205-2-AP, 1:2000) were from Proteintech Company (Wuhan, China). Histone H3.1 (MG4) mouse monoclonal antibody (Ray, RM2005 1:4000, Beijing, China) and β-actin (MG3) Mouse Monoclonal Antibody (Ray, RM2001, 1:4000) were from Beijing Ray Antibody Biotech (Beijing, China). Peroxidase-AffiniPure Goat Anti-Mouse IgG (H+L) (Jackson immunoresearch, 115-035-003 1:2000, PA, USA) and Peroxidase-AffiniPure Goat Anti-Rabbit IgG (H+L) (Jackson immunoresearch, 111-035-003, 1:2000) were from Jackson immunoresearch Laboratories.
Cells, treatment, and transfection
HT29 and CaCO2 cells were cultured in DMEM at 37°C in a 5% CO2 incubator supplied with 10% FBS, 100 units/mL penicillin, and streptomycin (Beyotime, C0222), myco-Zero™ mycoplasma removal agent (MRA) (Beyotime, C0280S) to clear mycoplasma, for treatment, cells were treated with propofol at a concentration of 10 μg/mL according to the Dai et al.’s work [23]. For transfection, plasmids were constructed from Youbio (Changsha, China) and transfected into cells using Lipo8000™ transfection reagents.
After treatment with or without propofol for 48 h, the total RNA was extracted using RNAiso plus combined with recombinant DNase I (RNase-free) (Takara, No. 2270A, Dalian, China), reverse transcription and quantitative PCR (qPCR) was performed to test indicated gene expression with BeyoRT™ II First Strand cDNA Synthesis Ki and BeyoFast™ SYBR Green qPCR Mix, respectively, according to the manufacturer’s instruction. The primers sequence used in this study were from Dong et al.’s work [24] and Zou et. al [25] were synergized from GenePharma and listed as follows: human-FASN-F: TACGACTACGGCCCTCATTT; human-FASN-R: CCATGAAGCTCACCCAGTTATC; human-ACCA-F: GAGGTGGATCGGAGATTTCATAG; human-ACCA-R: AGGCTCCAGATGACGATAGA; human-SCD1-F: CCTGCAGAATGGAGGAGATAAG; human-SCD1-R: GCCTTCCTTATCCTTGTAGGTG; human-UBC-F (internal reference.): 5′-ATTTGGGTCGCGGTTCTTG-3′; human-UBC-R (internal reference.): 5′-TGCCTTGACATTCTCGATGGT-3′.
Cell counting kit (CCK)-8 assay
Cells were digested and reseeded in a 96-well plate at a density of 105 cells/mL overnight, cells were treated with or without propofol for the indicated time. Each group was repeated three times, medium was replaced with fresh medium containing 10% CCK8 (Biosharp, BS350B) to incubate for about 30 min to 1 h. The absorbance of the indicated group was tested with a microplate reader (Thermo Scientific™ Varioskan™ LUX, VLBL0TD2, Waltham, MA USA) at 450 nm.
Nuclear and cytosolic fraction extraction
After cell confluence reached 80%, the cells were treated with or without propofol for 1 h, and cell fraction was isolated using a Nuclear and Cytoplasmic Protein Extraction Kit and diluted in a loading buffer (Biosharp, BL502B, Hefei, China). The immunoblotting analysis was performed to detect indicated protein. Histone H 3.1/Lamin A/C and β-actin were taken as an internal control for nuclear and cytosolic fractions, respectively.
Monounsaturated fatty acid (MuFA) detection
As described in the Qi et al.’s work [26], after treatment, the supernatant from the indicated group was collected to centrifuge to remove cell debris, 15 uL sample was added into a 96-well plate to incubate at 90°C for 30 min and placed into 4°C for 5 min. Sulfuric acid was added to incubate for 10 min at 90°C, and subsequent subjected to 4°C for 5 min. 100 uL of vanillin was added and mixed to incubated for 37°C for 15 min to determine the level of MuFA according to the manufacturer’s instruction.
As described in Qi et al.’s study [27], briefly, after treatment, the cells were fixed and stained with 0.5% Oil-Red-O (Biosharp, BL987A) in isopropanol diluted with 40% water, which was infiltered for 1 h at room temperature. Subsequently, the cells were washed with 75% ethanol and captured under a microscope. Extraction of Oil-Red-O-stained triglyceride drops with isopropanol was quantified and measured with a microplate reader at 570 nm.
Subcellular fractionation was isolated with a Nuclear and Cytoplasmic Protein Extraction Kit according to the manufacturer’s instructions. Protein was determined using a BCA Protein Assay Kit and subjected to SDS-PAGE using 2x loading buffer (Beyotime, P0015B). Briefly, isolated nuclear and cytoplasmic protein, BCA assay was performed to determine to load equal protein in SDS-PAGE. After separation, the protein was transferred into the nitrocellulose membrane (Beyotimes, FFN08), and incubation was performed with 5% milk for 1 h at room temperature. The primary antibodies were diluted and added to incubate indicated bands overnight at 4°C. The secondary antibodies were added to incubate for another 1 h after washing for 15 min with PBST (Biosharp, BL345A). The protein bands were detected and imaged using the BeyoECL Moon kit.
Cells were transfected with SCD1 reporter plasmid combined with pGL4.74 renilla plasmid, purchased from Youbio (Changsha, China), using Lipo8000™ transfection reagents for 24 h, a further 24-h incubation with or without propofol was performed. The luciferase relative luciferase unit (RLU) was measured using the Duo-Lite Luciferase Assay System kit.
Cells were stimulated with or without propofol for another 1 h after 24-h starvation, subsequently followed by fixation and isolation nuclear fraction, immunoprecipitation was performed with an anti-SREBP1c antibody to precipitate chromatin DNA fragments using a ChIP Kit according to the manufacturer’s protocol. qPCR was used to amplify the SCD1 promoter sequence with specific primer listed as follows: forward: 5′-AAGGCTCCTACAGACACGGA-3′, reverse: 5′-CAGGACCATATTGCCCTCGG-3′.
All statistical analyses were conducted using GraphPad Prism 10 (GraphPad Prism Software Inc, Boston, MA, USA). One sample t test and two sample t test were used to determine the difference in analysis of mRNA and cell viability/MuFA level, respectively. Two-way ANOVA with multiple comparisons, followed by Bonferroni post hoc test for significance for luciferase and ChIP experiment. A p-value less than 0.05 was considered statistically significant.
Propofol induced cell proliferation inhibition
We analyzed the effects of propofol on tumor cell proliferation, and a time course assessment of the influence of propofol on cell viability was performed in HT29 and CaCO2 cells of CRC cells. As demonstrated in Fig. 1A, no significant difference was obtained in HT29 cells at 24 h, while a large reduction of cell proliferation was observed in HT29 and CaCO2 cells at 48 and 72 h, respectively (Fig. 1A,B). What’s more, immunoblotting analysis showed that cell proliferation index PCNA was decreased in HT29, and CaCO2 cells received propofol treatment for 72 h (Fig. 1C). Together, propofol displayed a suppressive effect on cell proliferation in CRC cells.
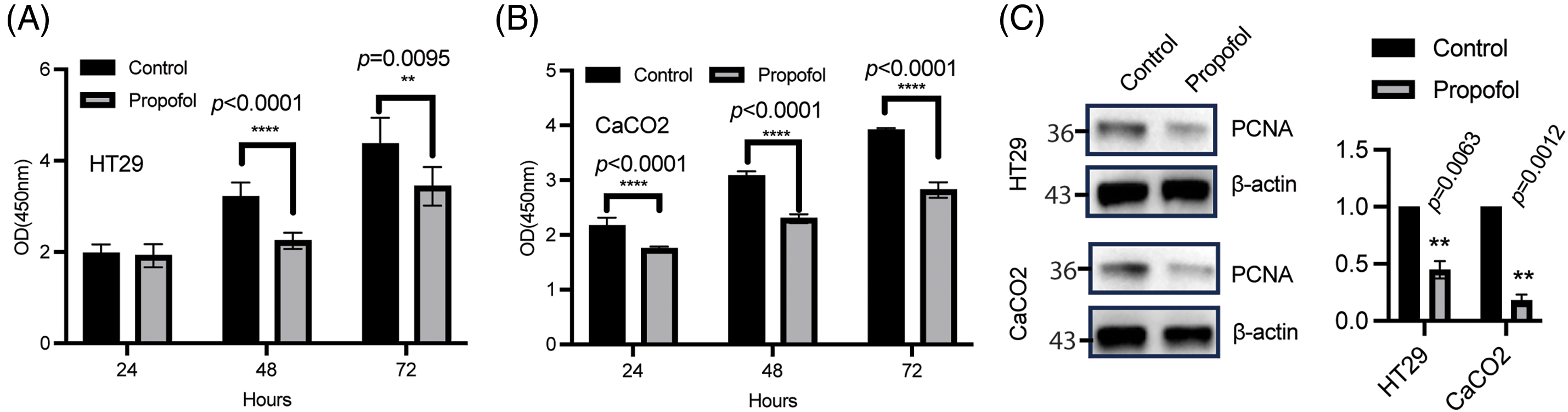
Figure 1: Propofol reduced cell proliferation. HT29 (A) and CaCO2 (B) cells were plated overnight, followed by treatment with or without propofol (10 μg/mL) for the indicated time. Fresh medium containing 10% CCK8 was used to incubate cells for about 1 h and the 96-well plate was tested with a microplate reader. Data was listed as mean ± SD, n = 6, and determined using a two-sample t test for each timepoint, **p < 0.01, ****p < 0.0001. (C) the whole protein was harvested from HT29 (upper) and CaCO2 (bottom) treated with or without propofol for 72 h, PCNA expression was analyzed by immunoblotting and the band intensity was quantified to test using one sample t test, n = 3, **p < 0.01. The control group was normalized as 1. β-actin was taken as loading control.
Propofol regulated cell proliferation through DNL
Metabolic reprogramming is critical for cell proliferation to achieve enough energy. The above work demonstrated propofol has an inhibitory effect on cell proliferation. DNL has been shown to sustain rapid proliferation and resistance to cellular stress [28], which focused us to ask whether oleic acid (OA), a kind of MuFA, addition could overcome the influence of propofol on cell proliferation using CCK8 assay. As expected, we found that cell proliferation was significantly reversed in HT29 and CaCO2 cells treated with propofol treatment after the addition of monounsaturated fatty acid (MuFA) (Fig. 2A), indicating propofol-regulated cell proliferation independent of MuFA. Moreover, propofol treatment could induce a significant reduction of total free fatty acid concentrations (Fig. 2B). The further results showed that several key enzymes involved in DNL, including FASN, ACCA, and SCD1, were drastically downregulated in HT29 and CaCO2 cells in response to propofol treatment at the mRNA level (Fig. 2C). In line with this, the immunoblotting analysis demonstrated that propofol treatment in CRC cells led to a strong inhibition of FASN, ACCA, and SCD1 expression (Fig. 2D). Moreover, oil-red staining and quantitation analysis showed that propofol could inhibit the accumulation of lipid droplets (Fig. 2E) These results implied that propofol modulated cell proliferation through DNL.
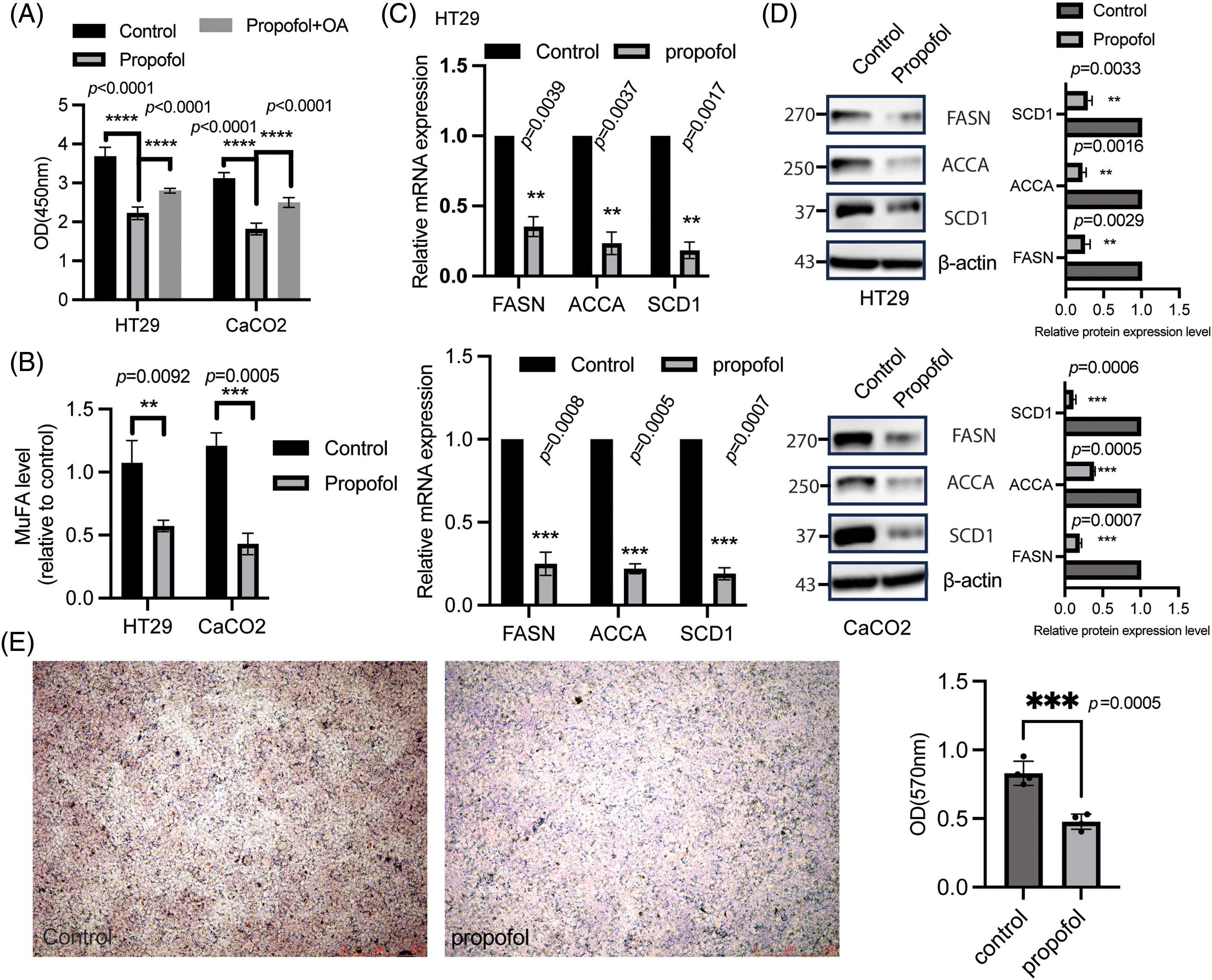
Figure 2: Propofol downregulated DNL. (A) HT29 and CaCO2 cells were seeded in a 96-well plate overnight, followed by treatment with or without propofol (10 μg/mL) for 1 h, and subsequent incubation with or without OA for another 72 h. CCK8 assay was employed to test cell proliferation. Data was displayed as mean ± SD, n = 3, and one-way ANOVA was used to determine the difference. ****p < 0.0001. (B) the level of MuFA was measured in supernatant from HT29 and CaCO2 cells received with or without propofol treatment for 72 h. Data was listed as mean ± SD, n = 3, statical difference was determined using a two-sample t test. **p < 0.01, ***p < 0.001. (C) the total RNA was isolated in HT29 and CaCO2 cells treated with or without propofol using RNAiso Plus according to the manufacturer’s protocol. Realtime PCR was employed to detect indicated gene expression. Data was displayed as mean ± SD, n = 3, and the difference was determined by a one-sample t test. **p < 0.01, ***p < 0.001. (D) the whole protein was harvested in HT29 and CaCO2 cells treated with or without propofol using a loading buffer, and immunoblotting was applied to examine indicated protein expression. The control group was normalized as 1, the band intensity was quantified to determine the difference by one sample t test, n = 3, **p < 0.01, ***p < 0.001. (E) oil-red staining was used to detect lipid droplet in BGC823 cells treated with propofol for 24 h, the quantitation was performed as described in materials and methods. Two sample t-tests were used to determine the differences. n = 4. ***p < 0.001.
Propofol regulated DNL through SREBP1c
A large number of studies have addressed that SREBP1c initiated multiple lipogenic gene transactivation, in particular SCD1, which is critical for switching from stearic acid to OA [14,29–31], which drove us to ask whether propofol-regulated DNL is dependent of SREBP1c, as shown in Fig. 3A, propofol stimulation could attenuate SCD1 promoter activity, while overexpression of SREBP1c in HT29 could largely block the decreased SCD1 luminesce caused by propofol treatment. Moreover, ChIP analysis further indicated that, in comparison with the control group, the binding of SREBP1c to SCD1 promoter was drastically inhibited in response to propofol treatment (Fig. 3B). most importantly, a remarkable reversed phenomenon was observed in CRC cells treated with propofol after ectopic expression of SREBP1c (Fig. 3C). These findings suggested propofol-modulated DNL through SREBP1c.
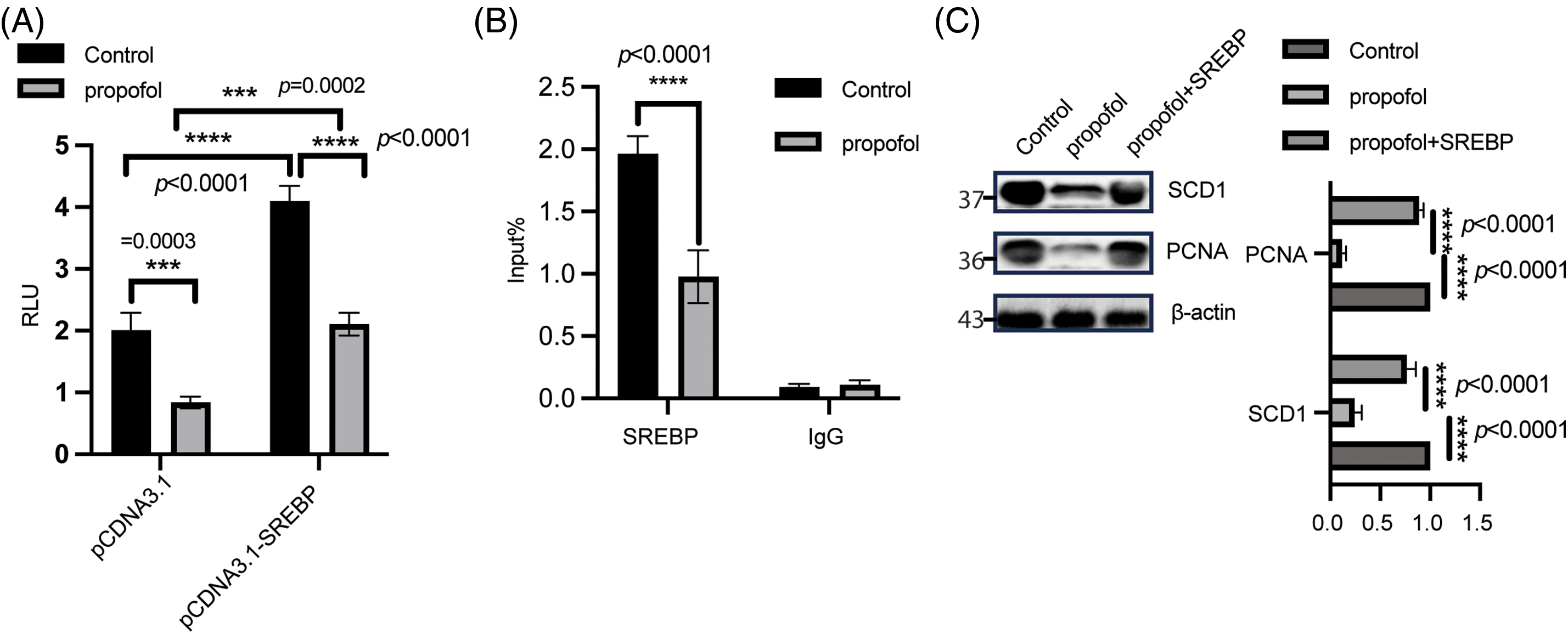
Figure 3: SREBP is critical for propofol-mediated DNL. (A) HT29 cells were transfected with a reporter gene containing SCD1 promoter and renilla plasmid for 24 h, subsequently, and treated with or without propofol (10 μg/mL) for another 24 h. The SCD1 promoter activity was determined using the Duo-Lite Luciferase Assay System. Two-way ANOVA with multiple comparisons, followed by Bonferroni post hoc test for significance was employed to determine the difference. Data was displayed as mean ± SD, n = 3, ***p < 0.001, ****p < 0.0001. (B) After treatment for 1 h, ChIP analysis was performed in HT29 cells and was fixed to isolate chromatin using an IP Kit according to the manufacturer’s protocol. Quantitative PCR of coimmunoprecipitated genomic DNA fragments was performed with indicated primers. Two-way ANOVA with multiple comparisons, followed by Bonferroni post hoc test for significance. Data was shown as mean ± SD, n = 3, ****p < 0.0001. (C) WB was used to detect indicated protein in HT29 cells transfected with SREBP plasmid for 12 h, subsequently followed by treatment with or without propofol for a further 48 h. The intensity was analyzed using one-way ANOVA, control group was normalized as 1, n = 3, ****p < 0.0001.
Propofol modulated SREBP1c mature and nuclear translocation
It is well known that SREBPs could be cleaved by site 1 and 2 proteases (S1P and S2P), which further form mature SREBPs, translocating to the nucleus to bind within target gene promoters and initiating transactivation [32]. Based on this, we further sought to determine the status of mature and nuclear translocation of SREBP1c in response to propofol stimulation. As illustrated in Fig. 4A, WB results showed that propofol treatment could induce a significant downregulation of SREBP1c mature in HT29 and CaCO2 cells, the results from a subcellular fraction analysis showed that propofol could suppress SREBP1c nuclear translocation (mature SREBP1c) in CaCO2 and HT29 cells, what’s more, the SREBPC precursor was reduced in cytosolic fraction after propofol treatment (Fig. 4B). These results implied that propofol suppressed SREBP1c mature and inhibited nuclear translocation of SREBP1c, thus repressing DNL.
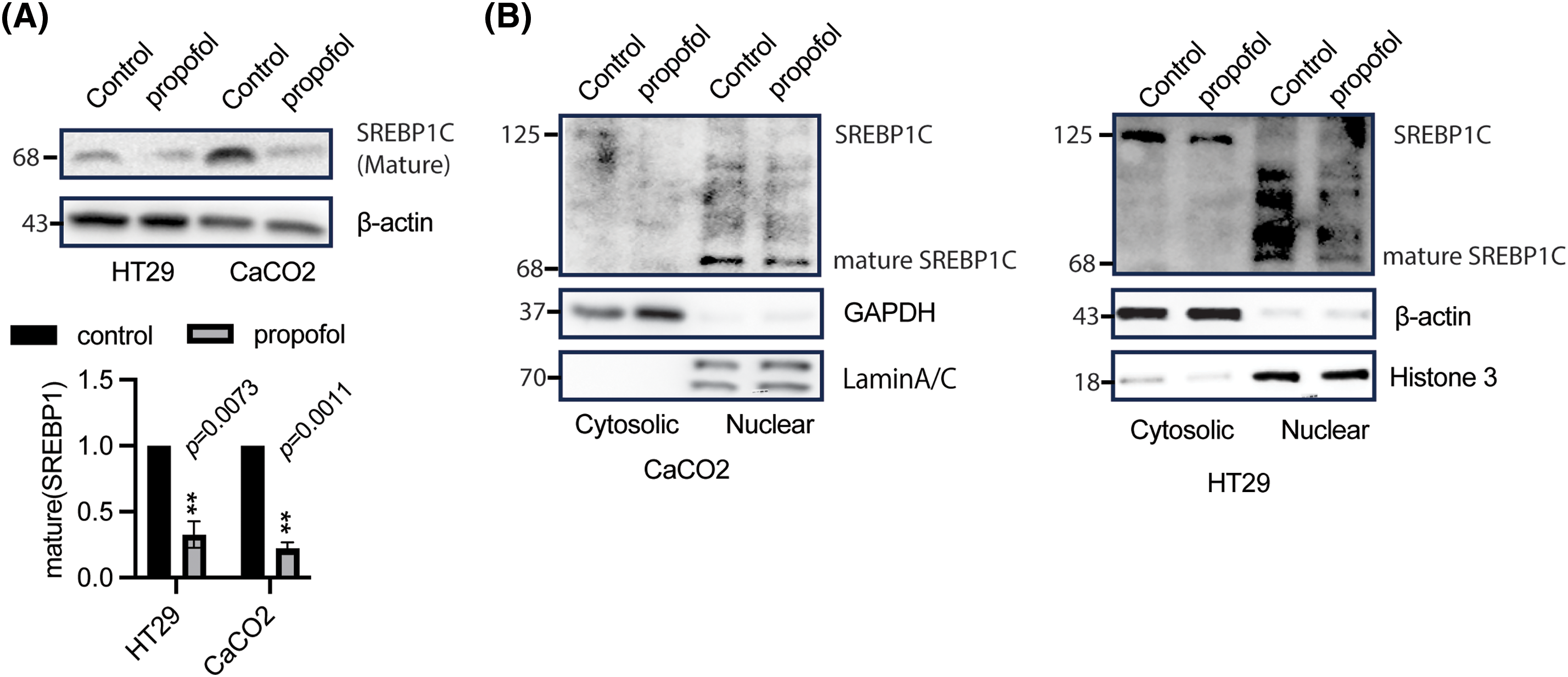
Figure 4: Propofol suppressed SREBP1c mature and nuclear translocation. (A) after treatment with or without propofol, HT29, and CaCO2 cells were harvested to detect indicated protein expression. β-actin was taken as loading control. The band of mature SREBP1c was quantified and analyzed by a one-sample t test. The control group was normalized as 1, n = 3, **p < 0.01. (B) after stimulation with or without propofol (10 μg/mL) for 1 h, CaCO2 (left panel) and HT29 (right panel) cells were harvested to separate nuclear and cytosolic fractions to perform WB to measure SREBP1c expression, histone 3/Lamin A/C and β-actin was taken as an internal control for nuclear and cytosolic fraction, respectively.
Largely number of investigations have demonstrated the antitumor properties of propofol in tumors, including proliferation, migration, invasion, apoptosis, immunology, and chemosensitivity [33–36]. To date, propofol has been shown to inhibit glycolysis reprogramming in cancer cells [17,37,38], what’s more, the polyol pathway was found to suppress gastric cancer cells in response to propofol [39]. In this work, we further demonstrated propofol suppresses CRC cell proliferation through downregulating SREBP1c-modulated DNL. Propofol treatment could result in cell proliferation inhibition in a time-dependent manner, which was attributed to the reduction of OA caused by decreased limited enzymes involved in DNL. Further results showed that propofol stimulation suppressed SREBP1c mature, thus inhibiting nuclear translocation, which further decreased the binding of SREBP1c to SCD1 promoter. Overall, this work enriched the mechanism of propofol-induced cell proliferation inhibition, and implied targeting DNL might be an alternative strategy for tumor treatment.
DNL leads to the generation of MuFAs mediated by SCD1, including OA and palmitoleic acid, which is critical for membrane phospholipids, triglycerides, and cholesterol lipids [40,41]. MuFAs could induce β-catenin stabilization and activation to trigger tumor growth [42], what’s more, MuFAs were reported to initiate lipid metabolism and inflammation through peroxisome proliferator-activated receptors (PPARs) [43]. Interestingly, in this work, MuFAs could rescue the cell proliferation inhibition caused by propofol, which implies that DNL is a critical pathway in propofol-mediated cell proliferation suppression. Further analysis demonstrated that FASN, ACCA, and SCD1 expression were largely reduced in CRC cells in response to propofol treatment. However, in addition to DNL, whether the other metabolism reprogramming involved in propofol-induced cell proliferation inhibition remained to be identified, such as sugar alcohols of the polyol pathway, Ketogenesis, tryptophan metabolism, and glutamine metabolism.
SREBP1c, a key transcription factor of lipogenesis, has been demonstrated to initiate SCD1 transactivation [12], which further led to MuFA generation. OA stimulation could enhance cell proliferation and survival in breast cancer cells and ovarian cancer cells [44,45]. In this work, propofol suppressed SREBP1c-mediated SCD1 expression, which further reduced MuFA level and cell proliferation. While addition of OA could reverse the impact of propofol on cell proliferation. Of interest, SREBP1c has been addressed to trigger NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) transactivation, leading to cell pyroptosis to regulate the cell cycle [46–48]. Based on this, we speculated propofol might regulate cell proliferation through modulation of NLRP3 inflammasome activation. However, there were several issues remained to be explored in future: whether propofol inhibited SREBP1c mature through activation of adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) or dissociation of SREBP cleavage-activating protein (SCAP)/SREBP1c complex; in addition to SREBP1c, whether there is another critical transcription factor modulated lipogenesis in propofol-mediated cell proliferation inhibition, such as Kruppel-like factor 2 (KLF2).
In conclusion, in this current study, we have revealed that propofol served as an antitumor function by suppressing DNL in CRC cells. This finding not only provided further support for the strategy for anesthetic agents used in clinics but also enriched the novel function of propofol.
Acknowledgement: None.
Funding Statement: The work is supported by Zhuhai Science and Technology Plan Project in the Field of Social Development (2320004000157), National Natural Science Foundation of China (82072215, 82272219), Shenzhen Science and Technology Program (JCYJ20210324134602006), Natural Science Foundation of Guangdong Province (2214050001873).
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Youtan Liu, Yajun Cao, Jiexian Zhou; data collection: Yajun Cao, Yidan Fang, Shuang Yin; analysis and interpretation of results: Yajun Cao, Shuang Yin; draft manuscript preparation: Yajun Cao, Shuang Yin, Jiexian Zhou, Youtan Liu. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: All data generated or analyzed during this study are included in this published article.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Oh TK, Jo H, Song IA. Propofol-based intravenous anesthesia is associated with improved survival outcomes after major cancer surgery: a nationwide cohort study in South Korea. Korean J Anesthesiol. 2023;76(5):461–70. doi:10.4097/kja.22747. [Google Scholar] [PubMed] [CrossRef]
2. Iwasaki M, Zhao H, Hu C, Saito J, Wu L, Sherwin A, et al. The differential cancer growth associated with anaesthetics in a cancer xenograft model of mice: mechanisms and implications of postoperative cancer recurrence. Cell Biol Toxicol. 2023;39(4):1561–75. doi:10.1007/s10565-022-09747-9. [Google Scholar] [PubMed] [CrossRef]
3. Glass A, McCall P, Shelley B. Comment on propofol and survival: an updated meta-analysis of randomized clinical trials. Crit Care. 2023;27(1):281. doi:10.1186/s13054-023-04550-2. [Google Scholar] [PubMed] [CrossRef]
4. Li Y, Dong W, Yang H, Xiao G. Propofol suppresses proliferation and metastasis of colorectal cancer cells by regulating miR-124-3p.1/AKT3. Biotechnol Lett. 2020;42(3):493–504. doi:10.1007/s10529-019-02787-y. [Google Scholar] [PubMed] [CrossRef]
5. Wang R, Li S, Hou Q, Zhang B, Chu H, Hou Y, et al. Propofol inhibits colon cancer cell stemness and epithelial-mesenchymal transition by regulating SIRT1, Wnt/β-catenin and PI3K/AKT/mTOR signaling pathways. Discov Oncol. 2023;14(1):137. doi:10.1007/s12672-023-00734-y. [Google Scholar] [PubMed] [CrossRef]
6. Dong H, Zhou W, Han L, Zhao Q. Propofol inhibits the proliferation, invasion, migration, and angiogenesis of oral squamous cell carcinoma through circ_0008898-mediated pathway. Chem Biol Drug Des. 2023:e14393. doi:10.1111/cbdd.14393. [Google Scholar] [PubMed] [CrossRef]
7. Chen X, Li C, Zeng R, Qiu L, Huang J, Wang N, et al. Propofol regulates HIF-1 α effect of expression of targeted SIRT1 signal pathway on kidney renal clear cell carcinoma. Cell Mol Biol. 2023;69(3):145–9. doi:10.14715/cmb/2023.69.3.21. [Google Scholar] [PubMed] [CrossRef]
8. Xiao Y, Yu TJ, Xu Y, Ding R, Wang YP, Jiang YZ, et al. Emerging therapies in cancer metabolism. Cell Metab. 2023;35(8):1283–303. doi:10.1016/j.cmet.2023.07.006. [Google Scholar] [PubMed] [CrossRef]
9. Zheng P, Lin Z, Ding Y, Duan S. Targeting the dynamics of cancer metabolism in the era of precision oncology. Metabolism. 2023;145:155615. doi:10.1016/j.metabol.2023.155615. [Google Scholar] [PubMed] [CrossRef]
10. Su F, Koeberle A. Regulation and targeting of SREBP-1 in hepatocellular carcinoma. Cancer Metastasis Rev. 2024;43:673–708. doi:10.1007/s10555-023-10156-5. [Google Scholar] [PubMed] [CrossRef]
11. Zhou Z, Guo C, Sun X, Ren Z, Tao J. Extracellular vesicles secreted by TGF-β1-treated mesenchymal stem cells promote fracture healing by SCD1-regulated transference of LRP5. Stem Cells Int. 2023;2023:4980871. [Google Scholar] [PubMed]
12. Li G, Xing Z, Wang W, Luo W, Ma Z, Wu Z, et al. Adipose-specific knockout of protein kinase D1 suppresses de novo lipogenesis in mice via SREBP1c-dependent signaling. Exp Cell Res. 2021;401(2):112548. doi:10.1016/j.yexcr.2021.112548. [Google Scholar] [PubMed] [CrossRef]
13. Sun Q, Xing X, Wang H, Wan K, Fan R, Liu C, et al. SCD1 is the critical signaling hub to mediate metabolic diseases: mechanism and the development of its inhibitors. Biomed Pharmacother. 2023;170:115586. [Google Scholar] [PubMed]
14. Sugi T, Katoh Y, Ikeda T, Seta D, Iwata T, Nishio H, et al. SCD1 inhibition enhances the effector functions of CD8+ T cells via ACAT1-dependent reduction of esterified cholesterol. Cancer Sci. 2023;115(1):48–58. doi:10.1111/cas.15999. [Google Scholar] [PubMed] [CrossRef]
15. Katoh Y, Yaguchi T, Kubo A, Iwata T, Morii K, Kato D, et al. Inhibition of stearoyl-CoA desaturase 1 (SCD1) enhances the antitumor T cell response through regulating beta-catenin signaling in cancer cells and ER stress in T cells and synergizes with anti-PD-1 antibody. J Immunother Cancer. 2022;10(7):e004616. doi:10.1136/jitc-2022-004616. [Google Scholar] [PubMed] [CrossRef]
16. Wang L, Ye G, Wang Y, Wang C. Stearoyl-CoA desaturase 1 regulates malignant progression of cervical cancer cells. Bioengineered. 2022;13(5):12941–54. doi:10.1080/21655979.2022.2079253. [Google Scholar] [PubMed] [CrossRef]
17. Qu D, Zou X, Liu Z. Propofol modulates glycolysis reprogramming of ovarian tumor via restraining circular RNA-zinc finger RNA-binding protein/microRNA-212-5p/superoxide dismutase 2 axis. Bioengineered. 2022;13(5):11881–92. doi:10.1080/21655979.2022.2063649. [Google Scholar] [PubMed] [CrossRef]
18. Chen X, Wu Q, Sun P, Zhao Y, Zhu M, Miao C. Propofol disrupts aerobic glycolysis in colorectal cancer cells via inactivation of the NMDAR-CAMKII-ERK pathway. Cell Physiol Biochem. 2018;46(2):492–504. doi:10.1159/000488617. [Google Scholar] [PubMed] [CrossRef]
19. Lu Y, Li L, Piao Z, Tan X, Su R. Sevoflurane and propofol co-affect the development of colorectal cancer by regulating TM2D1. Comb Chem High Throughput Screen. 2024. doi:10.2174/0113862073281046240527165415. [Google Scholar] [PubMed] [CrossRef]
20. Yao L, Zhai W, Jiang Z, He R, Xie W, Li Y, et al. The inhibitory effects of propofol on colorectal cancer progression through the NF-κB/HIF-1α signaling pathway. Anticancer Agents Med Chem. 2024;24(11):878–88. doi:10.2174/0118715206283884240326170501. [Google Scholar] [PubMed] [CrossRef]
21. Cigliano A, Simile MM, Vidili G, Pes GM, Dore MP, Urigo F, et al. Fatty acid synthase promotes hepatocellular carcinoma growth via S-phase kinase-associated protein 2/p27(KIP1) regulation. Medicina. 2024;60(7):1160. doi:10.3390/medicina60071160. [Google Scholar] [PubMed] [CrossRef]
22. Jiang L, Fang X, Wang H, Li D, Wang X. Ovarian cancer-intrinsic fatty acid synthase prevents anti-tumor immunity by disrupting tumor-infiltrating dendritic cells. Front Immunol. 2018;9:2927. doi:10.3389/fimmu.2018.02927. [Google Scholar] [PubMed] [CrossRef]
23. Dai L, Li S, Li X, Jiang B. Propofol inhibits the malignant development of osteosarcoma U2OS cells via AMPK/FOmicronChiO1-mediated autophagy. Oncol Lett. 2022;24(3):310. doi:10.3892/ol.2022.13430. [Google Scholar] [PubMed] [CrossRef]
24. Dong Q, Giorgianni F, Beranova-Giorgianni S, Deng X, O’Meally RN, Bridges D, et al. Glycogen synthase kinase-3-mediated phosphorylation of serine 73 targets sterol response element binding protein-1c (SREBP-1c) for proteasomal degradation. Biosci Rep. 2015;36(1):e00284. doi:10.1042/BSR20150234. [Google Scholar] [PubMed] [CrossRef]
25. Zou Z, Zeng F, Xu W, Wang C, Ke Z, Wang QJ, et al. PKD2 and PKD3 promote prostate cancer cell invasion by modulating NF-κB- and HDAC1-mediated expression and activation of uPA. J Cell Sci. 2012;125:4800–11. [Google Scholar] [PubMed]
26. Qi G, Mi Y, Shi X, Gu H, Brinton RD, Yin F. ApoE4 impairs neuron-astrocyte coupling of fatty acid metabolism. Cell Rep. 2021;34(1):108572. doi:10.1016/j.celrep.2020.108572. [Google Scholar] [PubMed] [CrossRef]
27. Qi W, Zhou L, Zhao T, Ding S, Xu Q, Han X, et al. Effect of the TYK-2/STAT-3 pathway on lipid accumulation induced by mono-2-ethylhexyl phthalate. Mol Cell Endocrinol. 2019;484:52–8. doi:10.1016/j.mce.2019.01.012. [Google Scholar] [PubMed] [CrossRef]
28. Li A, Yao L, Fang Y, Yang K, Jiang W, Huang W, et al. Specifically blocking the fatty acid synthesis to inhibit the malignant phenotype of bladder cancer. Int J Biol Sci. 2019;15(8):1610–7. doi:10.7150/ijbs.32518. [Google Scholar] [PubMed] [CrossRef]
29. Jeong DW, Park JW, Kim KS, Kim J, Huh J, Seo J, et al. Palmitoylation-driven PHF2 ubiquitination remodels lipid metabolism through the SREBP1c axis in hepatocellular carcinoma. Nat Commun. 2023;14(1):6370. doi:10.1038/s41467-023-42170-0. [Google Scholar] [PubMed] [CrossRef]
30. Hong E, Kang H, Yang G, Oh S, Kim E. The PKA-SREBP1c pathway plays a key role in the protective effects of Lactobacillus johnsonii JNU3402 against diet-induced fatty liver in mice. Mol Nutr Food Res. 2023;67(20):e2200496. doi:10.1002/mnfr.v67.20. [Google Scholar] [CrossRef]
31. Kim Y, Kim HK, Kang S, Kim H, Go GW. Rottlerin suppresses lipid accumulation by inhibiting de novo lipogenesis and adipogenesis via LRP6/mTOR/SREBP1C in 3T3-L1 adipocytes. Food Sci Biotechnol. 2023;32(10):1445–52. doi:10.1007/s10068-023-01339-5. [Google Scholar] [PubMed] [CrossRef]
32. Li Y, Wu S, Zhao X, Hao S, Li F, Wang Y, et al. Key events in cancer: dysregulation of SREBPs. Front Pharmacol. 2023;14:1130747. doi:10.3389/fphar.2023.1130747. [Google Scholar] [PubMed] [CrossRef]
33. Yao Y, Zhang F, Liu F, Xia D. Propofol-induced LINC01133 inhibits the progression of colorectal cancer via miR-186-5p/NR3C2 axis. Environ Toxicol. 2023;39(4):2265–84. doi:10.1002/tox.24104. [Google Scholar] [PubMed] [CrossRef]
34. Zhou R, Konishi Y, Zhang A, Nishiwaki K. Propofol elicits apoptosis and attenuates cell growth in esophageal cancer cell lines. Nagoya J Med Sci. 2023;85(3):579–91. [Google Scholar] [PubMed]
35. Jin Y, Xie S, Sheng B, Chen M, Zhu X. The effect of propofol on chemosensitivity of paclitaxel in cervical cancer cells. Cancer Med. 2023;12(13):14403–12. doi:10.1002/cam4.v12.13. [Google Scholar] [CrossRef]
36. Yan R, Song T, Wang W, Tian J, Ma X. Immunomodulatory roles of propofol and sevoflurane in murine models of breast cancer. Immunopharmacol Immunotoxicol. 2023;45(2):153–9. doi:10.1080/08923973.2022.2122501. [Google Scholar] [PubMed] [CrossRef]
37. Wu Z, Wang H, Shi ZA, Li Y. Propofol prevents the growth, migration, invasion, and glycolysis of colorectal cancer cells by downregulating lactate dehydrogenase both in vitro and in vivo. J Oncol. 2022;2022:8317466. [Google Scholar] [PubMed]
38. Zhao H, Wei H, He J, Wang D, Li W, Wang Y, et al. Propofol disrupts cell carcinogenesis and aerobic glycolysis by regulating circTADA2A/miR-455-3p/FOXM1 axis in lung cancer. Cell Cycle. 2020;19(19):2538–52. doi:10.1080/15384101.2020.1810393. [Google Scholar] [PubMed] [CrossRef]
39. Cao Y, Fan L, Li L, Zhou J. Propofol suppresses cell proliferation in gastric cancer cells through NRF2-mediated polyol pathway. Clin Exp Pharmacol Physiol. 2022;49(2):264–74. doi:10.1111/cep.v49.2. [Google Scholar] [CrossRef]
40. Williams KJ, Argus JP, Zhu Y, Wilks MQ, Marbois BN, York AG, et al. An essential requirement for the SCAP/SREBP signaling axis to protect cancer cells from lipotoxicity. Cancer Res. 2013;73(9):2850–62. doi:10.1158/0008-5472.CAN-13-0382-T. [Google Scholar] [PubMed] [CrossRef]
41. Mohammadzadeh F, Mosayebi G, Montazeri V, Darabi M, Fayezi S, Shaaker M, et al. Fatty acid composition of tissue cultured breast carcinoma and the effect of stearoyl-CoA desaturase 1 inhibition. J Breast Cancer. 2014;17(2):136–42. doi:10.4048/jbc.2014.17.2.136. [Google Scholar] [PubMed] [CrossRef]
42. Kim H, Rodriguez-Navas C, Kollipara RK, Kapur P, Pedrosa I, Brugarolas J, et al. Unsaturated fatty acids stimulate tumor growth through stabilization of beta-catenin. Cell Rep. 2015;13(3):495–503. doi:10.1016/j.celrep.2015.09.010. [Google Scholar] [PubMed] [CrossRef]
43. Varga T, Czimmerer Z, Nagy L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim Biophys Acta. 2011;1812(8):1007–22. doi:10.1016/j.bbadis.2011.02.014. [Google Scholar] [PubMed] [CrossRef]
44. Kado T, Kusakari N, Tamaki T, Murota K, Tsujiuchi T, Fukushima N. Oleic acid stimulates cell proliferation and BRD4-L-MYC-dependent glucose transporter transcription through PPARα activation in ovarian cancer cells. Biochem Biophys Res Commun. 2023;657:24–34. doi:10.1016/j.bbrc.2023.03.051. [Google Scholar] [PubMed] [CrossRef]
45. Hardy S, Langelier Y, Prentki M. Oleate activates phosphatidylinositol 3-kinase and promotes proliferation and reduces apoptosis of MDA-MB-231 breast cancer cells, whereas palmitate has opposite effects. Cancer Res. 2000;60(22):6353–8. [Google Scholar] [PubMed]
46. Liu MH, Lin XL, Xiao LL. SARS-CoV-2 nucleocapsid protein promotes TMAO-induced NLRP3 inflammasome activation by SCAP-SREBP signaling pathway. Tissue Cell. 2023;86:102276. doi:10.1016/j.tice.2023.102276. [Google Scholar] [PubMed] [CrossRef]
47. Varghese JF, Patel R, Yadav UCS. Sterol regulatory element binding protein (SREBP)-1 mediates oxidized low-density lipoprotein (oxLDL) induced macrophage foam cell formation through NLRP3 inflammasome activation. Cell Signal. 2019;53:316–26. doi:10.1016/j.cellsig.2018.10.020. [Google Scholar] [PubMed] [CrossRef]
48. Wang X, Yang M, Yu G, Qi J, Jia Q, Liu S, et al. Promoting the proliferation of osteoarthritis chondrocytes by resolvin D1 regulating the NLRP3/caspase-1 signaling pathway. Cell Signal. 2024;113:110960. doi:10.1016/j.cellsig.2023.110960. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools