
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW
Unraveling the molecular crossroads: T2DM and Parkinson’s disease interactions
Institute for Brain Sciences Research, School of Life Sciences, Henan University, Kaifeng, 475004, China
* Corresponding Author: JIANSHE WEI. Email:
# These two authors contributed equally to this work
(This article belongs to the Special Issue: Cell Death and Inflammation in Signaling and Diseases)
BIOCELL 2024, 48(12), 1735-1749. https://doi.org/10.32604/biocell.2024.056272
Received 18 July 2024; Accepted 27 September 2024; Issue published 30 December 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Type 2 diabetes mellitus (T2DM) is a chronic metabolic disorder characterized by persistent hyperglycemia. In recent times, an elevated risk of Parkinson’s disease (PD) development among individuals with T2DM has become evident. However, the molecular mechanisms that underpin the interplay between T2DM and the pathogenesis of PD remain to be elucidated. Nevertheless, recent epidemiological studies have underscored several shared molecular pathways that are crucial for normal cellular function and are also associated with the progression and etiology of both T2DM and PD. This review encapsulates some of the shared pathophysiological mechanisms, including genetic risk factors, hyperglycemia, insulin resistance (IR), inflammation, mitochondrial dysfunction, oxidative stress, autophagy, and advanced glycation end products (AGEs), which independently contribute to the onset and progression of T2DM and PD. Additionally, this review compiles studies that have investigated the relationship between T2DM and PD. This review aims to offer insights into the overlapping molecular pathways involved, which may suggest alternative therapeutic strategies for both T2DM and PD.Keywords
Type 2 diabetes mellitus (T2DM) is a chronic metabolic disease. With the continuous improvement of material living standards, T2DM has become a chronic disease that seriously threatens human health. According to the latest report from the International Diabetes Federation (IDF), the global prevalence of Type 2 Diabetes Mellitus (T2DM) among adults stood at 536.6 million individuals (10.5%) in 2021, with projections indicating that this figure will rise to 783.2 million people (12.2%) worldwide by the year 2045 [1]. Studies have shown that T2DM leads to changes in brain structure and function and increases the risk of neurological disease comorbidities. Alzheimer’s disease (AD), the most common neurodegenerative disease, is closely associated with T2DM, and patients with T2DM are at significantly increased risk of developing AD compared to healthy individuals [2]. Epidemiological studies have shown that patients with T2DM are at higher risk of developing Parkinson’s disease (PD) and have a more rapid progression of PD symptoms [3]. Diabetes can cause a variety of complications, long-term blood sugar is not well controlled can cause neuropathy and microangiopathy, can induce vascular PD, and neurological damage such as hyperosmolar coma, a symptom of diabetes, is also known as ketoacidosis, this condition is classified as a type of Parkinsonism [4]. T2DM is a complex metabolic disorder involving multiple mechanisms, such as hyperglycemia, insulin resistance (IR), disruptions in insulin receptor and insulin growth factor functions, glucose toxicity, the formation of advanced glycation end products (AGEs), and the activity of their receptors [2].
Patients with PD experience a selective loss of dopaminergic neurons in the substantia nigra (SN). It is projected that by 2030, the number of PD patients in China will surge, accounting for over half of the global PD population. Furthermore, it is estimated that the proportion of individuals aged 65 and above in China will reach 23.9%–26.9% by 2050 [5]. The typical clinical manifestations are resting tremor, myotonia, bradykinesia, and postural abnormalities, and the characteristic pathological manifestations are apoptosis of dopamine (DA) neurons and the formation of Lewy bodies [6]. Due to these morbidities, PD seriously affects the quality of life of middle-aged and elderly people in their later years. The specific pathogenic mechanisms of PD are still not fully understood, but existing studies suggest that genetic risk factors, IR, mitochondrial dysfunction, oxidative stress, autophagy, AGEs [7], and inflammation are involved in the development of PD [8] (Table 1).

Interestingly, these pathogenic mechanisms also play an important role in the development of T2DM, in which IR is the central hub of “tandem” these pathogenic mechanisms. Studies have found that in diabetic patients without PD, diabetes has motor symptoms, reduced striatum DA transporter binding, and elevated tau and α-synuclein (α-syn) levels compared with healthy controls. In the proportional hazards model (Cox model) analysis of PD patients, diabetes progressed faster with exercise and cognitive decline. Thus, diabetes may promote Parkinson-like syndrome, whereas in patients with PD, concomitant diabetes can induce a more severe phenotype [9]. Due to the loss of pancreatic β cells, a specific protein called islet amyloid misfolding may contribute to the pathogenesis of T2DM, which in turn leads to insufficient insulin and hyperglycemia, and accelerates disease progression [10]. Moreover, this islet amyloid protein is intimately linked to the pathophysiology of PD due to its interaction with α-syn, as evidenced by in vitro studies [11]. Notably, α-syn is also produced within the islets, playing a pivotal role in glucose regulation [12]. In the context of T2DM, the aggregation of islet amyloid polypeptide (IAPP) occurs within pancreatic cells [13], emphasizing the significance of maintaining a delicate balance among protein production, release, and clearance. It is crucial to note that any imbalance in the ratio of islet amyloid or α-syn may foster the development of toxic aggregates in the brain and impact pancreatic β cell function [14]. Ultimately, the potential for these two proteins to interact, either directly or indirectly, underscores their potential role in contributing to the onset of T2DM and PD when their levels shift. The currently discovered relationship between T2DM and PD is explained as follows.
Accompaniment between T2DM and PD
Both T2DM and PD are influenced by genetic and environmental factors [15,16]. Exposure to environmental factors, including heavy metals like iron, manganese, and copper, as well as pesticides such as rotenone, elevates an individual’s risk of developing PD. Notably, patients with PD often exhibit elevated concentrations of these heavy metals in their blood [17,18]. Analogously, exposure to heavy metals has been linked to insulin dysfunction and disease progression in individuals with T2DM [19]. There is growing evidence that PD and T2DM share very similar dysfunctional pathways. Genetic predisposition, lifestyle choices, and exposure to toxic environmental factors may contribute to mitochondrial dysfunction, and endoplasmic reticulum stress, inflammation, and metabolic disorders may ultimately lead to PD and/or diabetes. Other studies have shown that processes similar to peripheral IR are present in the brains of patients with PD, even without diabetes, and may lead to pathological changes in PD [20]. This finding also opens up the possibility of hypoglycemic drugs for the treatment of PD.
Exploring shared genetic risk factors
T2DM is a risk factor for the development of PD, but its effect on disease progression is unclear. Studies have evaluated the effect of T2DM on disease progression, and multi-morbidity T2DM may be associated with faster exercise and cognitive decline compared with PD patients without T2DM. Patients with T2DM are more likely to have depression, severe gait disturbance, and loss of independence than non-T2DM patients, and are less likely to have DA dysregulation [21]. The majority of T2DM cases are intricately intertwined with both genetic and environmental risk factors [22,23], along with their complex interactions [24]. Prior genome-wide association studies have uncovered numerous genetic polymorphisms and rare genetic variants that exert subtle to significant influences on T2DM, indicating that this disease arises from an intricate interplay between genetic mechanisms and environmental exposures (Fig. 1).
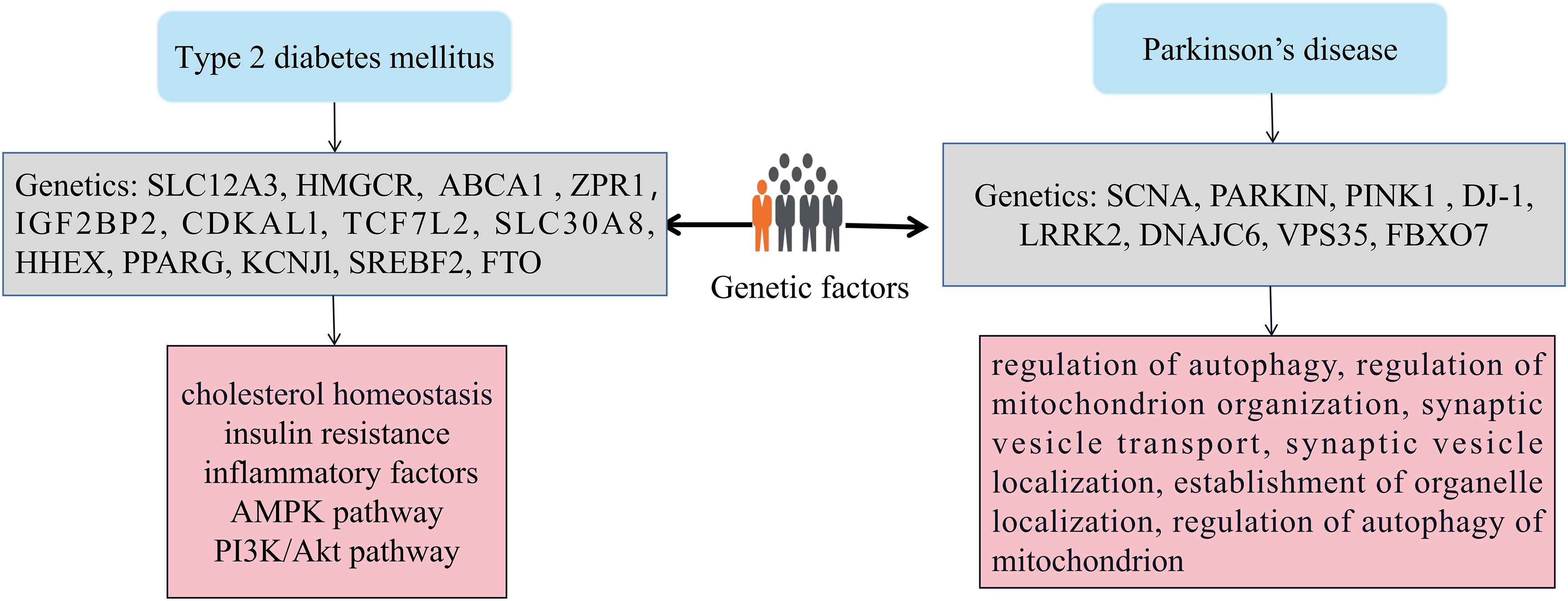
Figure 1: The impact of genetic factors on T2DM and PD. Genetic variants like SLC12A3, HMGCR, and ABCA1 are linked to T2DM, while ZPR1 is associated with PD. These genes are vital for cellular function and biological processes such as ion transport and cholesterol metabolism. Changes in these genes can increase disease risk. Chronic inflammation and abnormal AMPK signaling are associated with insulin resistance, β cell dysfunction in T2DM, and neuronal degeneration in PD. Epigenetic mechanisms, which can affect gene expression and cellular function, are significant in the development of T2DM and PD, highlighting the interaction between genetics and the environment.
As an example, Zhang et al. [25] discovered a strong correlation between the solute carrier family 12 member 3 (SLC12A3) gene and T2DM, demonstrating that the presence of a T allele within this gene modestly contributes to unfavorable lipid levels. The SLC12A3 gene encodes a sodium chloride cotransporter, also known as a urea-sensitive sodium chloride cotransporter (NCC), which primarily functions in the renal tubules. SLC12A3 participates in maintaining electrolyte balance and blood pressure regulation in the body by affecting the reabsorption of salt and water [26]. Ference et al. [27] showed that the genetic variants of the 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) gene are associated with T2DM. Maintaining cellular cholesterol homeostasis is crucial for ensuring normal cell function and viability. Accumulation of excessive cholesterol within cells is detrimental and serves as the underlying molecular mechanism for various diseases, including atherosclerosis, PD, and diabetes mellitus. HMGCR plays an important role in regulating cholesterol metabolism, lipid synthesis, and carbohydrate metabolism. It affects the cellular energy state and regulates adenosine monophosphate (AMP)--activated protein kinase (AMPK) activity, thereby impacting the insulin signaling pathway. Saturated fatty acids and insulin can activate sterol regulatory element-binding protein (SREBP), which in turn regulates the expression of HMGCR [28]. Ergen et al. [29] suggested ATP binding cassette subfamily A member 1 (ABCA1) polymorphism as a genetic marker of T2DM. ABCA1 is mainly related to the transport of cholesterol and lipids. Research has shown that the function of ABCA1 is closely related to cholesterol metabolism, and abnormal cholesterol metabolism can affect insulin sensitivity [30]; The expression of ABCA1 can be regulated by inflammatory factors, and chronic low-grade inflammation is considered to be an important pathogenic factor of T2DM. ABCA1 may affect the action of insulin by regulating inflammatory response [31]. Tokoro et al. [32] identified the genetic susceptibility of patients with a novel common variant of rs964184 in ZPR1 zinc finger (ZPR1) to T2DM. ZPR1 may participate in the formation of insulin resistance by affecting the activity of insulin receptors or downstream signaling pathways, such as the PI3K/Akt pathway [33].
The genetic architecture of PD is characterized by a unique blend of factors. Rare, highly penetrant pathogenic variants are responsible for familial forms of the disease, while genetic risk factor variants contribute significantly to the risk of PD in a minority of cases. Additionally, high-frequency, low-penetrance variants play a role in slightly elevating the risk of developing sporadic PD [34]. Notably, approximately 5%–10% of all PD patients suffer from a monogenic form of the disease, where mutations in autosomal-dominant (AD) genes such as SNCA, leucine-rich repeat kinase 2 (LRRK2), and VPS35 retromer complex component (VPS35), as well as autosomal recessive (AR) genes like PINK1, DJ-1, and Parkin, are causative. Whole-exome sequencing has further revealed AR DNAJC6 mutations in both atypical and typical PD patients [35]. Our previous research has demonstrated that the primary biological processes associated with these genes involve the regulation of autophagy, mitochondrion organization, synaptic vesicle transport, and localization, establishment of organelle localization, and regulation of mitochondrial autophagy [36]. Epigenetics plays a very important role in the occurrence and progression of T2DM and PD. Studies have shown that high glucose stimulation can cause epigenetic changes in genes, such as DNA methylation, histone acetylation, chromatin remodeling, and microRNAs, thereby regulating the signaling pathways related to the expression of inflammatory response genes and fibrotic genes in cells, and ultimately upregulate the main inflammatory response and fibrotic gene expression of target cells [37,38].
Glucose serves as virtually the sole source of energy for brain tissue. In conditions of hyperglycemia, cells undergo a series of complex reactions. Notably, hyperglycemia emerges as a significant contributor to neurodegeneration, particularly affecting the nigrostriatal pathway, the neuroanatomical foundation of motor symptoms observed in PD patients (Fig. 2). Indeed, the onset of PD stems from the loss of DA neurons in the SN. DA neurons located in the mesencephalic SN are particularly susceptible to oxidative stress and degeneration, potentially due to their inherent vulnerability to mitochondrial dysfunction. This vulnerability may be a direct consequence of hyperglycemia and the oxidative stress it induces [39]. Other pathological pathways, inducing oxidative stress response, non-enzymatic glycation of proteins, polyol pathway activation, diacylglycerol (DAG)-protein kinase C (PKC) signaling pathway activation, may all play a pivotal role in mediating the detrimental effects of hyperglycemia on nigral DA neurons.
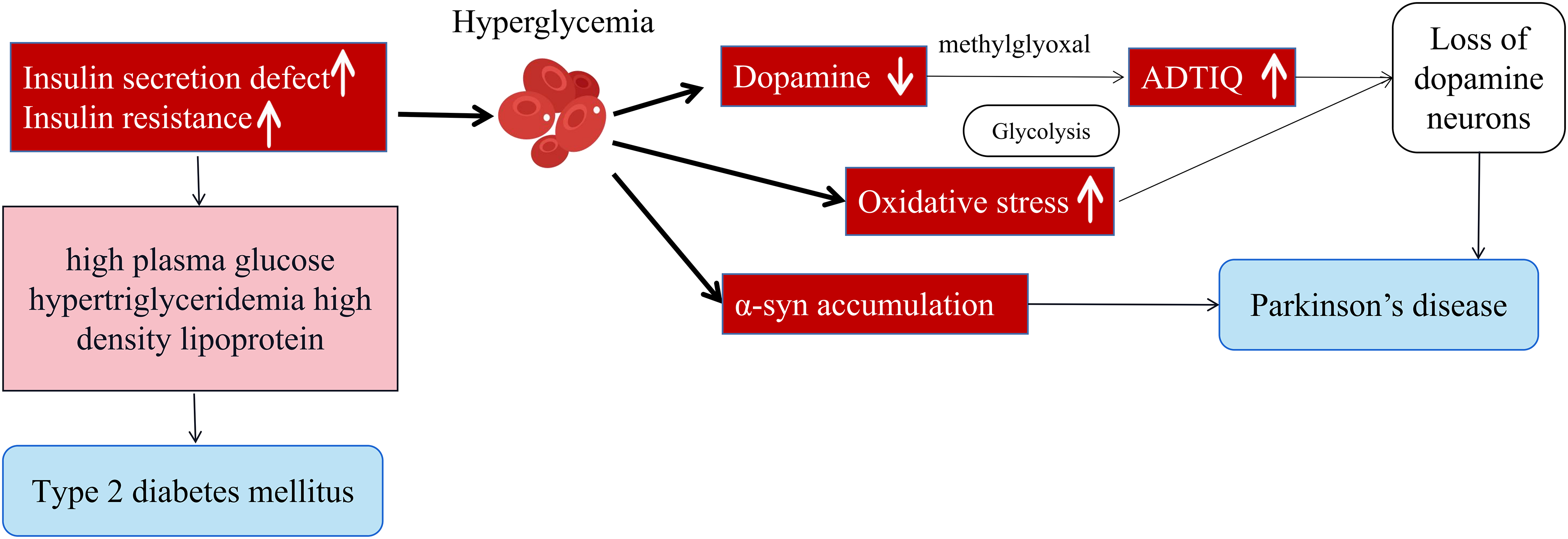
Figure 2: Hyperglycemia promotes the progression of PD, leading to a decrease in dopamine, activation of oxidative stress, and α-syn aggregation. Hyperglycemia increases oxidative stress, damaging cells and contributing to PD progression. It also promotes α-syn aggregation, a protein that misfolds and leads to neuronal death. ADTIQ may be a therapeutic agent for PD. Disruptions in glucose and lipid metabolism, common in diabetes, may also play a role in PD. Overall, the figure highlights the link between hyperglycemia and PD, emphasizing the importance of addressing high blood sugar to potentially slow the disease’s progression.
1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP) is well known to cause Parkinsonism in humans with neurotoxicity specific for DA neurons. It was found that SH-SY5Y model cells (PD cell model) are in a high-glycemic state for a long time, the DA reserves in the cells will decrease, and the concentration of methylglyoxal will increase significantly, methylglyoxal is an important metabolite in glycolysis, in the state of high sugar, cell glycolysis metabolism is vigorous, the production of methylglyoxal is further increased, methylglyoxal and DA react to generate 1-acetyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline (ADTIQ). Its chemical structure is very similar to the exogenous neurotoxin MPTP, which is often considered a neurotoxin that can cause damage to brain cells. It can be seen that hyperglycemia can affect brain cell function, damage the nervous system, and accelerate the pace of PD development [40]. In addition, in T2DM mouse brain, we observed concomitantly increased accumulation and phosphorylation of α-syn in the cortex, pre-commissural putamen, and DA neurons in the SN, which interestingly showed high correlation with levels of fasting plasma glucose (FPG), triglyceride (TG), and high-density lipoprotein (HDL) [41].
There is growing research evidence to support PD with IR (Fig. 3). Several studies have confirmed the neuroprotective effects of insulin. PD patients with low serum insulin-like growth factors (IGF-1) levels have been found to have poorer executive performance and verbal memory [42]. IR levels in the brain, especially the hypothalamus, cerebral cortex, and hippocampus, gradually decline with age. This age-related physiological insulin signaling attenuation effect appears to be amplified in PD and is accompanied by increased IR. It can be seen that IR plays a non-negligible role in the whole process of PD. Compared with normal people, the levels of insulin receptor substrate and protein kinase B (PKB/AKT) were significantly reduced, and glycogen synthase kinase-3β (GSK-3β) phosphorylation was increased in the substantia nigra pars compacta (SNpc) of PD patients [43]. There is a deletion of insulin receptor mRNA in the SN in PD patients before the death of DA neurons. In addition, 50%–80% of PD patients formally underwent glucose tolerance testing were IR [44].
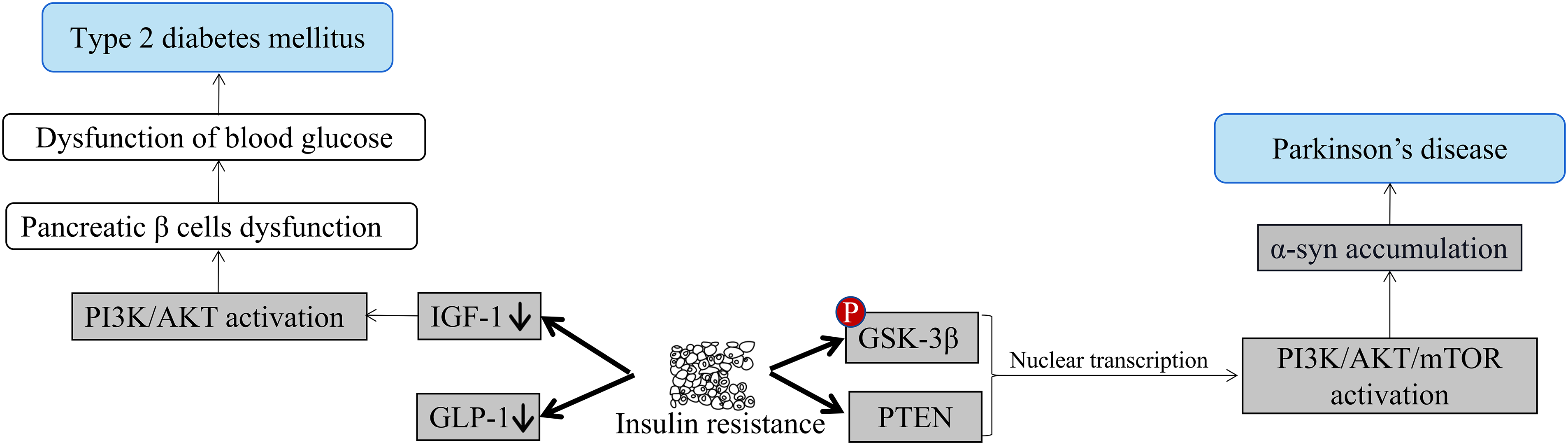
Figure 3: Insulin resistance leads to T2DM and PD. IR happens when the body’s cells stop responding properly to insulin, a hormone made by the pancreas, causing blood sugar levels to remain high. Initially, the pancreas increases insulin production to compensate, but if resistance continues, it may become exhausted and fail to produce enough insulin, leading to T2DM. The PI3K/AKT/mTOR pathway, important for cell growth, survival, and metabolism, is associated with IR and PD. IR may affect brain cells, particularly those producing dopamine, which is decreased in PD. Disruptions in the PI3K/AKT/mTOR pathway could cause α-syn accumulation, resulting in cell death and dopamine loss in PD.
T2DM and IR not only increase the possibility of developing PD but can also determine the prognosis and progression of Parkinsonian symptoms. The researchers used hypoglycemic drugs to improve IR and see if they had a positive effect on the improvement of PD. In one clinical study, metformin plus sulfonylureas reduced the incidence of PD compared with sulfonylureas alone [45]. Another clinical study showed that the glucagon-like peptide-1 (GLP-1) receptor agonist exenatide increased AKT phosphorylation levels and mammalian target of rapamycin (mTOR) levels in PD patients, thereby improving autophagy and improving clinical symptoms [46]. A large number of preclinical studies in animal models of PD have shown that the combined activation of GLP-1 and glucose-dependent insulinotropic polypeptide (GIP) pathway may have stronger neuroprotective effects, and they have been found to have neurotrophic and anti-apoptotic effects [47]. Metformin has been shown to show good neuroprotective effects in PD models in some animal experiments: in MPTP-induced PD mice, long-term metformin treatment significantly improved motor and muscle activity lowered α-syn phosphorylation, and upregulates neurotrophic factor, regulated autophagy, and mitochondrial reactive oxygen species (ROS) clearance [48,49]. In a lipopolysaccharide (LPS) induced PD mice model, metformin could inhibit microglial activation and inflammatory cytokine expression [50]. In a haloperidol-induced PD model, metformin significantly reduces memory deficits, oxidative stress, and lipid peroxidation [51].
Insulin can act as a growth factor in the brain to regulate neurons, and can also regulate DA concentration. DA neurons at the SN are highly insulin receptors, including insulin receptor β (IRβ), IR substrate-1 (IRS1), phosphoinositide 3-kinase p85 (PI3K p85), phosphatidylinositol 4,5-bisphosphate (PIP2), phosphatidylinositol (3,4,5)-trisphosphate (PIP3) [52]. DA neurons and islet β cells have similar bioenergetic properties and susceptibility to oxidative stress. Dysfunction in insulin signaling pathways, including the PI3K/Akt/mTOR pathway, can lead to IR. This disruption may trigger the synthesis and accumulation of AGEs, ROS, and reactive nitrogen species, ultimately leading to oxidative stress, protein misfolding, protein accumulation, mitochondrial dysfunction, endoplasmic reticulum stress, and dysregulation of metabolic syndrome. These processes are intimately linked to inflammation and apoptosis, playing a pivotal role in the pathogenesis of both T2DM and PD.
PD and T2DM are two progressive and devastating health conditions that affect millions of individuals globally. In recent years, the prevalence and incidence of both disorders have risen significantly due to increasing longevity and population growth. There is a growing body of evidence suggesting a connection between inflammation and central nervous system (CNS) disorders, including Alzheimer’s disease, PD, Huntington’s disease, motor neuron disease, multiple sclerosis, stroke, traumatic brain injury, and even cancers of nervous tissue. The strength of this relationship is influenced by the timing and magnitude of anti-or pro-inflammatory gene expression. Similarly, inflammation has been implicated in the pathogenesis of T2DM. A common feature of both PD and T2DM is the misfolding and fibrillization of both tissue-specific and non-specific proteins, which are both induced by and contribute to inflammation and stress (oxidative/glycation) [53] (Fig. 4). The inflammatory response is likely to contribute to the development of T2DM by causing IR, and this process is further exacerbated in the presence of hyperglycemia, ultimately promoting long-term complications of T2DM [54]. The level of inflammatory factors (such as tumor necrosis factor α (TNF-α), interleukin 6 (IL6), IL10, etc.) in the body of inflammatory patients is increased, and excess inflammatory factor promotes the differentiation of pancreatic β lymphocytes and overexpresses IgG genes, promotes the overactivation of killer T lymphocyte clones, thereby triggering the destruction and death of pancreatic β cells [55].
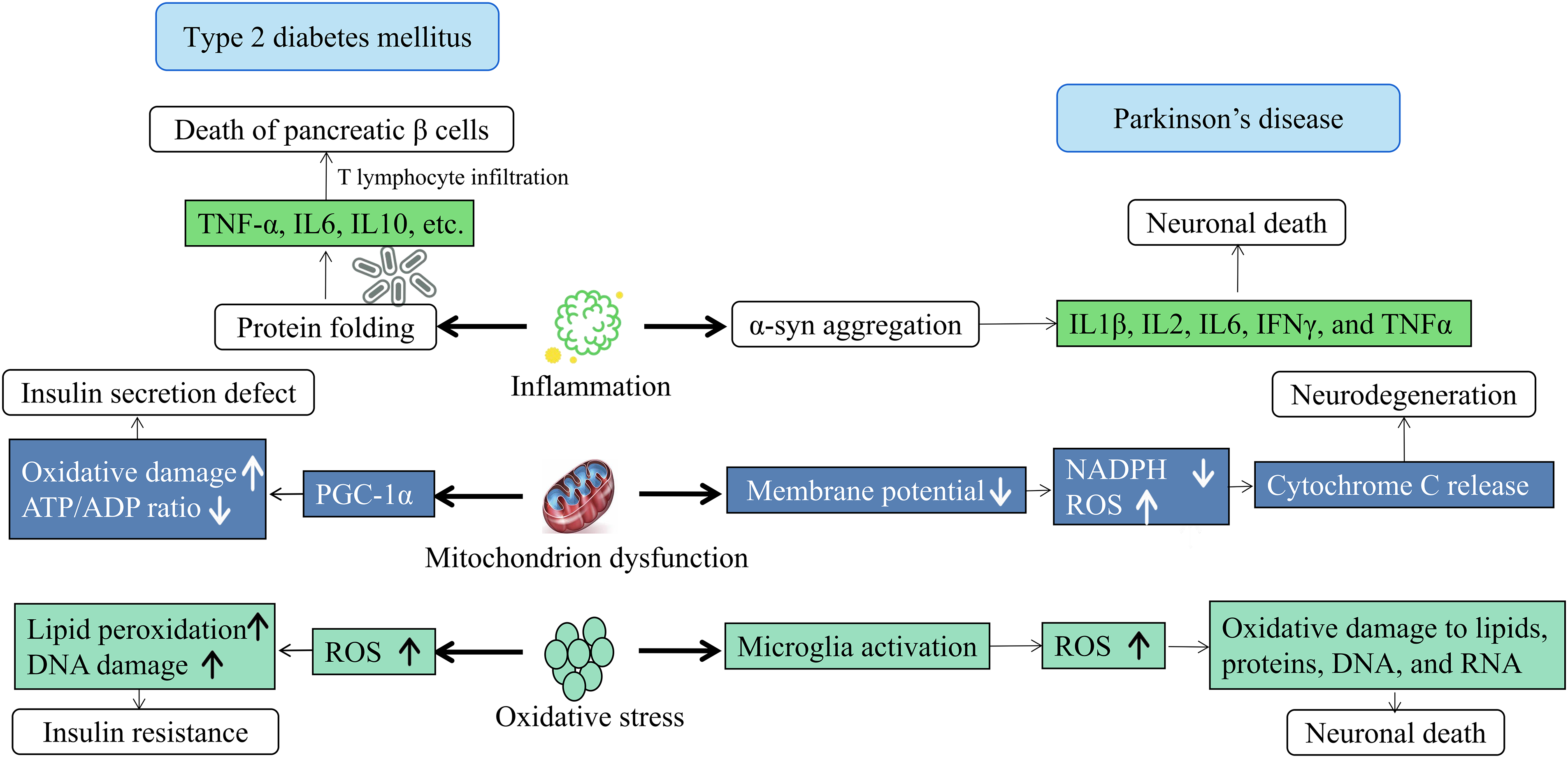
Figure 4: Inflammation, mitochondrial dysfunction, and oxidative stress lead to T2DM and PD. Mitochondrial dysfunction leads to protein misfolding and aggregation, disrupting cellular function and causing neuronal damage. It also affects energy metabolism by altering ATP/ADP ratios, worsening IR and cellular stress. Oxidative stress, characterized by the production of reactive oxygen species and lipid peroxides, damages cellular components and leads to dysfunction and death. This stress impairs insulin sensitivity and exacerbates mitochondrial dysfunction, creating a cycle of damage. T2DM and PD share common pathways such as inflammation, mitochondrial dysfunction, and oxidative stress, suggesting interconnectedness and potential shared treatment mechanisms.
Similarly, inflammation is also an indispensable and important factor in the occurrence and development of PD. The study found that PD patients had a higher proportion of pro-inflammatory factors compared with the control group, and it may affect cognition. Microglia are key to mediating neuroinflammation in PD. Positron emission tomography shows increased microglial activation in PD patients. The activation of phosphorylation of the insulin signaling pathway PI3K/AKT inhibits the activation of microglia, reduces apoptosis and neuroinflammation, and exerts neuroprotective effects [56,57]. It is noteworthy that increased cytokine levels, including IL1β, IL2, IL6, interferon γ (IFNγ), and TNFα, and higher CD4 lymphocyte counts have been detected in serum and colony-stimulating factor (CSF) from PD patients [58,59]. Neuroinflammation-mediated neurotoxicity is extremely important in the cascade of neuronal death in PD, inflammation not only appears in the brain, but peripheral inflammation also plays an important role in the occurrence and progression of PD, neuroinflammation is characterized by activated microglia and T lymphocyte infiltration near damaged neurons. Microglia maintain chronic inflammation through a self-propelling circulatory pathway that relies on the bursting of the respiratory system within microglia, thereby regulating the activity of many pro-inflammatory factors downstream and thereby promoting the progression of PD neurodegeneration.
Mitochondrial dysfunction in T2DM and PD
Mitochondrial dysfunction is an important mechanism of T2DM and PD (Fig. 4). The role of mitochondrial dysfunction in T2DM is particularly important because the relationship between mitochondrial dysfunction and IR is very clear, so T2DM exhibits typical glucose toxicity and lipid toxicity, namely high TG and low HDL cholesterol, hyperglycemia [60]. Research has revealed that fibroblasts derived from PD patients exhibit mitochondrial dysfunction, leading to the accumulation of organic and amino acids intimately linked to mitochondrial metabolism, particularly under conditions of high glucose exposure [61]. Furthermore, alterations in cerebral glucose metabolic rates within the frontal, temporal-parietal, and cingulate regions of the brain have been implicated as a crucial factor contributing to cognitive decline in prediabetic or T2DM patients. This phenomenon has been more thoroughly explored in diabetic animal models. Studies have shown that brain mitochondria from sucrose-fed T2DM mice display reduced respiratory control ratios, membrane potentials, and ATP/ADP ratios compared to their non-diabetic counterparts. The observed mitochondrial dysfunction in the brains of T2DM mice may stem from increased ROS production coupled with heightened vulnerability to oxidative damage [62].
Mesenchymal stem cells (MSCs) in the spinal cord of PD patients have mitochondrial dysfunction, including a higher base rate of mitochondrial degradation, lower levels of biogenesis, and reduced mitochondrial mass. In addition, membrane potential decreased, nicotinamide adenine dinucleotide phosphate oxidase (NADPH) dependent respiration was inhibited, ROS production and oxidative stress significantly increased, and major antioxidants glutathione (GSH) decreased [63]. The dysfunction of mitochondria can be attributed to a multitude of factors including bioenergetic defects, mutations in mitochondrial DNA, nuclear DNA gene mutations with mitochondrial connections, alterations in mitochondrial dynamics such as fusion or fission, changes in size and morphology, disruptions in trafficking or transport, impaired mitochondrial movement, compromised transcription, and the presence of mutated mitochondrial-associated proteins, has been implicated in the pathogenesis of PD [64].
Oxidative stress in T2DM and PD
Oxidative stress arises when the generation of free radicals surpasses the antioxidant defense systems, leading to the harmful effects of these radicals [65]. Biological cells possess an inherent defense mechanism comprising enzymes like superoxide dismutase (SOD), catalase (CAT), and glutathione (GLT), which shield cells from free radical assault. Free radicals, though essential for biological homeostasis [66], can induce oxidative stress when their production exceeds the body’s antioxidant capacity [67]. This oxidative stress stands as a crucial upstream event for diabetes complications and IR development [68], triggering pathophysiological molecular mechanisms and initiating a cascade of detrimental pathways that lead to IR and T2DM (Fig. 4). Oxidative stress plays a pivotal role in the pathophysiology of various diabetic complications through lipid peroxidation, DNA damage, and mitochondrial dysfunction [69]. Accumulating evidence suggests that oxidative stress is a key player in the complex cascade underpinning dopaminergic neurodegeneration in all forms of PD. This oxidative stress stems from an imbalance in cellular redox activity, where ROS production outweighs clearance by endogenous antioxidant enzymes and molecular chaperones. Thus, oxidative stress itself is not pathological; rather, it is the accumulation of ROS following redox imbalance that mediates neuronal damage.
While ROS serve as vital signaling molecules regulating physiological gene transcription and protein interactions, their accumulation can inflict oxidative damage on lipids, proteins, DNA, and RNA, depending on their subcellular location of production, thereby compromising neuronal function and structural integrity [70]. Oxidative changes are emphasized as a crucial factor in the onset of PD, with glial cell activation being the primary source of oxidative stress [71]. Key enzymes involved in the generation of oxidative species derived from oxygen and nitrogen include reduced NADPH, inducible nitric oxide synthase (iNOS), and astrocytic myeloperoxidase (MPO) [72], as well as inflammatory factors like TNF-α [73] and cyclooxygenase-2 (COX-2) [71].
The pentose phosphate pathway (PPP) plays a crucial role in cellular metabolism, particularly in its ability to counteract oxidative stress, which is a significant factor in the pathogenesis of T2DM and PD [74,75]. This metabolic pathway is responsible for generating NADPH, a critical reducing agent that participates in the biosynthesis of fatty acids and nucleotides while also serving a vital role in maintaining the redox balance within cells [76]. NADPH is integral to the functioning of major antioxidant systems, including glutathione and thioredoxin, which help neutralize ROS. In conditions characterized by IR and hyperglycemia, such as T2DM, an increase in ROS production can overwhelm the antioxidant defenses, leading to cellular damage and contributing to the progression of oxidative stress-related complications [77]. Moreover, in PD, the accumulation of α-syn and mitochondrial dysfunction further exacerbates oxidative damage, highlighting the need for robust antioxidant mechanisms [78]. The PPP, by providing NADPH, enables cells to enhance their antioxidant capacity, effectively mitigating oxidative stress and thereby potentially altering disease trajectories in both T2DM and PD. Ultimately, targeting the PPP could be a promising therapeutic strategy for ameliorating oxidative stress and its deleterious effects in these disorders.
Autophagy represents a highly conserved intracellular recycling pathway that is instrumental in regulating intracellular homeostasis. It serves as a self-protective mechanism for cells, significantly contributing to the maintenance of normal structure and function of pancreatic β cells, as well as enhancing IR. In the context of T2DM, autophagy not only supplies nutrients to sustain cellular energy during periods of fasting but also eliminates damaged organelles, lipids, and misfolded proteins [79]. The researchers found that the secretion of amylin in islet β cells is regulated by the autophagy process. In mouse models, if pancreatic β cell autophagy is lacking, oxidative damage increased, resulting in increased pancreatic β cell apoptosis. In addition, increased IR and decreased autophagy cells increase amylin toxicity and pancreatic β cell dysfunction [80]. Accumulating evidence points to the aggregation of α-syn and tau as a consequence of impaired autophagic-lysosomal degradation [81]. Conversely, α-syn and tau have been demonstrated to influence mitochondrial, autophagic, and lysosomal functions [82,83].
Dopaminergic neurons, known for their high metabolic activity and corresponding significant mitochondrial energy demand, are particularly susceptible to insufficient clearance of damaged mitochondria. Notably, a vast array of genes implicated in both familial and sporadic PD exhibits strong associations with macroautophagy, chaperone-mediated autophagy (CMA), mitophagy, and downstream lysosomal function. Macroautophagy, the primary pathway for cellular degradation, initiates with the formation of a double-membraned autophagosome from an isolated membrane (phagophore), which sequesters cytosolic material. Subsequently, the fusion of autophagosomes with lysosomes forms autolysosomes, where hydrolases mediate the degradation of their contents. In contrast, chaperone-mediated autophagy involves the cytosolic chaperone protein heat shock cognate 70 (HSC70), which targets and translocates unfolded proteins directly to lysosomes by binding to the lysosomal receptor lysosomal-associated membrane protein 2A (LAMP2A). In comparison to non-selective/bulk macroautophagy, selective mitophagy specifically tags damaged mitochondria for downstream autophagic degradation [84]. Furthermore, “loss-of-function mutations” affecting the two enzymes critical for the clearance of damaged mitochondria (mitophagy)-ubiquitin ligase Parkin and protein kinase PINK1-have been linked to familial PD. Studies have shown that ubiquitin specific peptidase 30 (USP30), a mitochondria-confined deubiquitinases (DUBs), fights mitophagy by removing ubiquitin tags placed by parkin. Inhibition of USP30 can promote mitochondrial clearance and quality control, with potential benefits for PD [85] (Fig. 5).
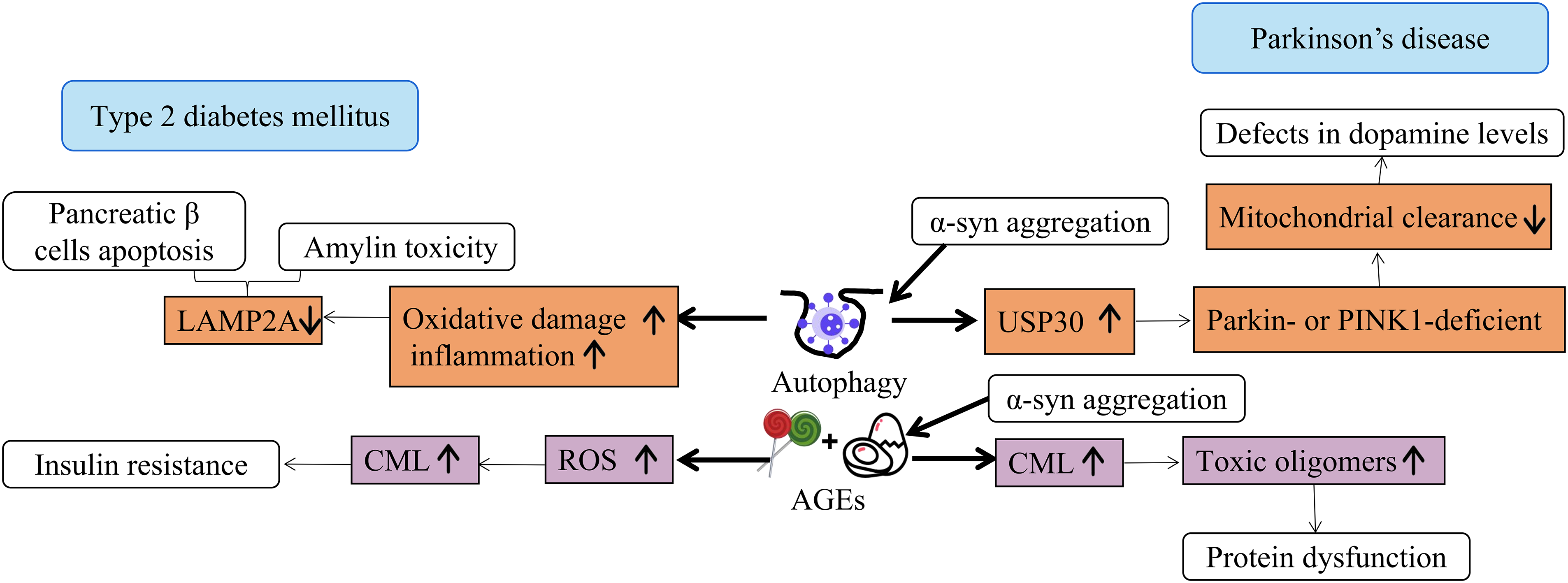
Figure 5: Autophagy and AGEs lead to T2DM and PD. AGEs accumulate with age and are associated with diseases such as T2DM and PD. They can alter proteins, leading to cellular dysfunction. AGEs can impair autophagy, leading to the accumulation of damaged proteins and organelles, exacerbating cellular stress and dysfunction. IR is a sign of T2DM and is related to AGEs and oxidative stress, which will damage cell function and lead to T2DM. Damaged autophagy may lead to IR by disrupting cellular homeostasis and allowing harmful substances to accumulate. PD is associated with autophagic damage, α-syn aggregation, and oxidative stress. Damaged autophagy may lead to PD by failing to clear damaged proteins and organelles, resulting in toxic aggregates such as α-syn, disrupting cell function, promoting neuronal death.
AGEs are hypothesized to play a pivotal role in the development and progression of both T2DM and PD (Fig. 5). The formation of AGEs, a group of potentially harmful modified proteins and/or lipids, serves as a contributing factor. On one hand, AGEs are reported to augment the generation of reactive oxygen species and impair the antioxidant system. On the other hand, the formation of some AGEs is itself induced under oxidative conditions. Consequently, AGEs contribute, at least partially, to the chronic stress state observed in diabetes. Given that AGEs arise both endogenously and exogenously (e.g., from food sources), they are considered a risk factor for T2DM [86]. Elevated concentrations of glyoxal, methylglyoxal, and 3-deoxyglucosone have been detected in the plasma of T2DM patients [87]. For instance, glyoxal promotes the formation of Nε-(carboxymethyl)lysine (CML), which is currently the most extensively characterized AGEs [88].
Studies have demonstrated that AGEs induce cell toxicity in the human neuronal cell line SH-SY5Y through oxidative stress, manifested by a dose-dependent reduction in cell viability and increase in cell toxicity in a PD model [89]. The glycation of α-syn, a process that occurs with aging, has been implicated in the progression of PD by facilitating the formation of AGEs and toxic oligomers that are not efficiently cleared from neurons [90]. α-syn is the primary constituent of Lewy bodies, where it commonly undergoes post-translational modifications, including the generation of AGEs (primarily CEL) through its reaction with methylglyoxal. Furthermore, AGEs have been non-enzymatically linked to α-syn accumulation in the chronic MPTP-induced C57BL/6 mouse model of parkinsonism [91]. The excessive production of AGEs can not only crosslink with proteins, affecting protein performance, but also change cell function by binding to specific receptors and reacting, resulting in pathological changes in the body. AGEs are closely related to the occurrence and progression of T2DM and PD.
T2DM Is Negatively Related to PD
Fig. 6 shows a negative correlation between T2DM and PD. From 1994 to 2005, the prevalence of diabetes was comparable between PD cases and a matched comparison group without PD. Notably, the risk of developing diabetes was lower among PD patients, particularly those treated with levodopa (L-dopa) [92]. Smoking, a known risk factor for diabetes, increases the risk of T2DM by 30%–40% in active smokers compared to non-smokers [93]. It predicts the progression of glucose intolerance, from normoglycemia to impaired glucose tolerance, and escalates the risk of diabetes development [94]. Smoking affects body weight and composition, peripheral insulin sensitivity, and pancreatic β cell function [95]. Smokers exhibit reduced expression of peroxisome proliferator-activated receptor-gamma (PPAR-γ), a transcription factor crucial for insulin sensitivity [96], and nicotine exposure promotes insulin receptor substrate 1 (IRS-1) Ser636 phosphorylation, activating mTOR and mitogen-activated protein kinase (MAPK) signaling [97].
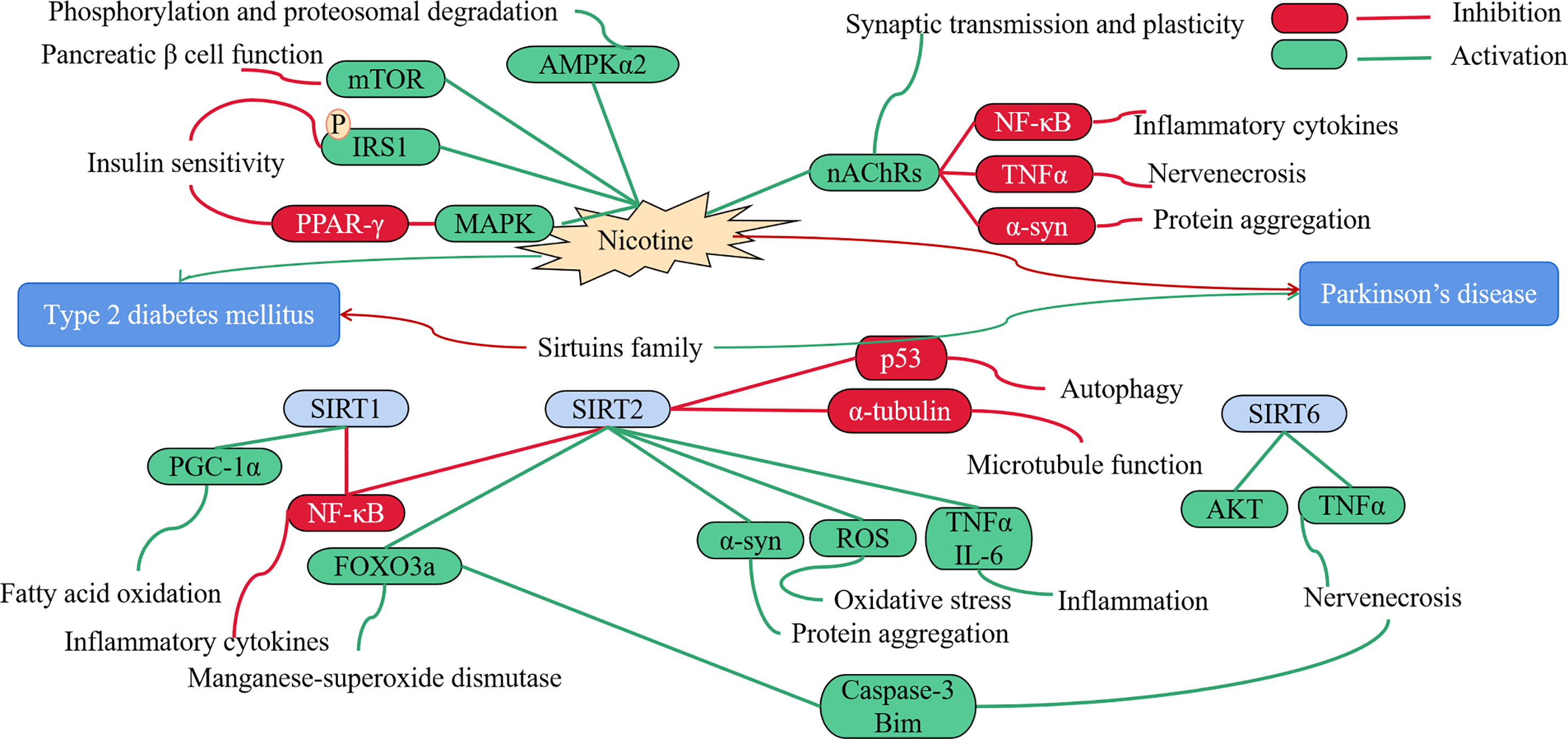
Figure 6: T2DM is negatively related to PD. Nicotine leads to dysfunction of pancreatic β cells and exacerbates IR, while inhibiting inflammatory factors reduces PD symptoms. The Sirtuins family inhibits inflammation and fatty acid oxidation, reduces T2DM, activates inflammatory factors, causes oxidative stress, and exacerbates α-syn aggregation, leading to the occurrence of PD. Red represents inhibition, green represents activation.
Although nicotine increases lipolysis in adipose tissue by activating AMP-activated protein kinase (AMPK) in adipocytes [98–100], it also possesses a definitive protective effect against PD. Nicotine or nicotine receptor ligands could serve as therapeutic agents in PD management, protecting against nigrostriatal degeneration, treating PD symptoms, or mitigating L-dopa-induced side effects [101,102]. The cholinergic receptors, which modulate the dopaminergic system and contribute to motor and cognitive functions, undergo significant losses of nicotinic acetylcholine receptors (nAChRs) in the cortex of PD patients [103]. nAChRs regulate synaptic transmission and plasticity in various brain regions, including midbrain dopamine centers. Modulators like nicotine can stimulate cholinergic anti-inflammatory pathways, such as nuclear factor kappa-B (NF-κB) and TNF pathways, yielding beneficial effects [104]. Furthermore, nicotine binds and inhibits the fibrillation of α-syn [105], inducing substantial conformational changes that prevent its toxic effects. By activating nAChRs across the blood-brain barrier (BBB), nicotine diminishes oxidative stress and neuroinflammation, enhancing synaptic plasticity and neuronal survival of dopaminergic neurons. Other studies have shown that a decrease in plasma cortisol levels, decreased blood glucose, decreased metabolic function and weight loss can be observed in the population of PD patients, and the mechanism is related to decreased activity of the sympathetic nervous system [106].
Sirtuins (SIRT) family belongs to Class III histone deacetylases (HDACs), played an opposite role in PD and diabetes. SIRT Interactions with proteins such as p53, forkhead box (FOXO), NF-κB, and Ku70 to regulate cells, possibly involving transcription, cell cycle, cell differentiation, apoptosis, oxidative stress, metabolism, and genomic stabilization [107]. SIRT1 may play a pivotal role in mitigating inflammation and oxidative stress, while enhancing mitochondrial function. This, in turn, safeguards pancreatic β cells and alleviates IR in insulin-sensitive tissues, such as skeletal muscle and adipose tissue. Consequently, SIRT1 emerges as a promising pharmacological target for the treatment of IR and T2DM [108,109]. Additionally, SIRT2 deacetylation has been shown to prevent and reverse age-related inflammation and IR [110]. It is noteworthy that SIRT2 levels escalate with aging, and mounting evidence suggests that SIRT2 activity modulates multiple processes implicated in the pathogenesis of PD, including augmenting α-syn aggregation and toxicity, diminishing microtubule function, exacerbating oxidative stress, intensifying neuroinflammation, and impairing autophagy [111–114]. Nicholatos et al. showed that brain-specific SIRT6 knockout mice are protected from MPTP-induced PD, while SIRT6 overexpressing mice develop more severe pathology and increasing neuroinflammation, neuronal apoptosis and cell death by stimulating TNFα release and suppressing AKT signaling [115].
Therapeutic Approaches to T2DM and PD
Neurological disorders rank as the second leading cause of death globally [116]. Unfortunately, current treatment strategies have fallen short in effectively addressing these conditions: the majority of drug combinations for the CNS primarily target symptoms rather than underlying causes, there is a scarcity of safe and efficacious medications, and the clinical development of drugs for neurological disorders poses significant challenges. In fact, diabetes is not the only one that increases the risk of PD, and studies have found that even in non-diabetic people, a slight increase in blood sugar may lead to an increased risk of PD. Certain glucose-lowering drugs have been found to be associated with a reduced risk of developing or progressing PD. Diabetics experienced improvement in PD symptoms after exenatide use, compared with worsening in the placebo control group [117]. The neuroprotective effects of insulin and the administration of exogenous insulin may provide potential benefits for PD patients [118]. It has been reported that intranasal insulin can improve the activation and repair of neuronal stem cells, dendritic sprouting, and the protection of nerves against inflammation and oxidative stress in rodent models [119]. Diabetes drugs, including metformin, hold potential as therapeutic agents for neurological diseases, offering novel insights into their treatment [120]. Metformin exerts a neuroprotective effect via the AMPK pathway, which concurrently serves as its anti-diabetic mechanism. AMPK activation, achieved through inhibition of mitochondrial complex I, significantly decreases ROS levels, thereby promoting neuronal survival. Metformin enhances autophagy by inhibiting mammalian target of rapamycin complex 1 (mTORC1), which is phosphorylated by AMPK. It also promotes anti-apoptosis by upregulating the anti-apoptotic protein Bcl-2, reduces DNA damage, and alleviates neuroinflammation, all contributing to neuronal protection. Furthermore, metformin diminishes α-syn aggregates by augmenting protein phosphatase 2A (PP2A) activity [121]. People with T2DM who use GLP-1RA or dipeptidyl peptidase 4 (DPP-4) inhibitors have a lower risk of PD [122]. In the brain, GLP-1 receptor (GLP-1R) is primarily expressed in the pyramidal neurons of the cortex and hippocampus, as well as in Purkinje neurons of the cerebellum and substantia nigra. GLP1R agonists also activate gene expression involved in cell growth and repair, enhancing the protective effects of neurons against stress factors such as α-syn, inflammatory mediators, and cell death through the MAPK pathway [123].
Several therapeutic approaches currently in development are aimed at targeting other mediators within the insulin signaling pathway. One such mediator is the glucose-dependent insulinotropic polypeptide (GIP). GIP, a proinsulin hormone, contributes to lowering blood glucose levels by initiating various downstream insulin pathways, thereby facilitating insulin biosynthesis and secretion [124]. A promising advancement in this field is the development of dual GLP-1/GIP receptor agonists, which demonstrate comparable affinity for both GLP-1 and GIP receptors. Both GLP-1 and GIP are crucial hormones involved in augmenting insulin signaling, and they possess the ability to traverse the blood-brain barrier (BBB), exerting neuroprotective effects [125]. Research has demonstrated that dual GLP-1/GIP receptor agonists exhibit superior efficacy in enhancing the insulin pathway and eliciting neuroprotective effects in rodent models, compared to single GLP-1R agonists, while also demonstrating a reduced incidence of adverse effects [126]. It is believed that GLP-1/GIP receptor agonists exert their effects by activating the PI3K/Akt pathway and the extracellular regulated protein kinases (Erk1/2) pathway [127]. This results in increased tyrosine hydroxylase, reduced microglial activation, promotion of dopamine production, and protection for dopaminergic neurons [128]. One study reported reduced neuroinflammation in A53T tg mouse model of PD following treatment with GLP1/GIP receptor agonists [129]. Notably, the same study also reported restored insulin sensitivity post-treatment. Currently, dual GLP-1/GIP receptor agonists are undergoing clinical trials for the treatment of T2DM, showing promising results compared to existing therapies, and further clinical trials for PD patients may be conducted in the near future.
The relationship between T2DM and PD has garnered increasing interest in recent years. Emerging evidence suggests that the metabolic dysregulation seen in T2DM may influence the pathophysiology of neurodegenerative diseases, particularly PD. Several epidemiological studies show that individuals with T2DM exhibit a higher incidence of PD compared to non-diabetic populations. Numerous cohort studies have reported that patients with T2DM have a 1.5–2 times higher risk of developing PD compared to those without diabetes [130]. For instance, a large-scale longitudinal study found that individuals with T2DM had a significantly increased risk of PD, suggesting a strong association [131,132].
In T2DM, the core pathological mechanisms include IR, beta cell dysfunction, and chronic low-grade inflammation. The key molecular pathways include abnormal insulin signaling, activation of inflammatory factors, and oxidative stress. The main pathological features of PD are progressive degeneration of dopaminergic neurons in the substantia nigra and abnormal aggregation of α-syn. The core molecular pathways include mitochondrial dysfunction, protein misfolding and aggregation, and neuroinflammation. The shared risk factors include age, genetic background (such as LRRK2, GBA gene variations), and environmental factors (such as pesticide exposure, high sugar diet), which are common risk factors for T2DM and PD, and may promote the onset of both diseases by influencing the molecular pathways mentioned above; IR in T2DM may increase the risk of PD by affecting insulin signaling in the brain, interfering with neuronal energy metabolism and synaptic plasticity. In addition, insulin also has neuroprotective effects in the brain, and its deficiency may exacerbate neuronal damage; Both T2DM and PD are accompanied by an increase in chronic low-grade inflammation and oxidative stress. These processes may mutually promote each other, forming a vicious cycle that accelerates the degeneration and death of neurons; Sugar metabolism, lipid metabolism, and energy metabolism all play important roles in T2DM and PD. The disruption of these metabolic pathways may promote the pathological progression of both diseases by affecting mechanisms such as mitochondrial function and protein folding. Understanding this common pathway can highlight potential therapeutic targets. The evidence linking T2DM and PD suggests a notable interaction that warrants further exploration. Understanding the molecular pathways involved can illuminate potential interventions to mitigate PD risk in diabetic patients.
Future research should aim to clarify these relationships and assess whether targeting metabolic dysfunction in T2DM can serve as a preventive strategy against PD. The relationship between PD and T2DM is supported by both epidemiological and experimental data. Both diseases are becoming increasingly prevalent worldwide and are closely associated with aging. Currently, many clinical trials are being conducted to repurpose existing drugs, as well as to address the need for developing vaccines and medications targeting PD related to T2DM. Established T2DM medications currently in clinical trials show potential for treating PD, including Exenatide, NLY01, PT320, Liraglutide, Semaglutide, and Lixisenatide [120]. Numerous in vivo and in vitro studies suggest a connection between T2DM and neurodegenerative diseases, enhancing the possibility of several antidiabetic drugs for preventive or therapeutic purposes in diseases such as PD. The pathophysiological links between T2DM and neurodegenerative diseases, as well as how anti-diabetic drugs affect neurodegenerative diseases, are not yet fully understood, necessitating further research to grasp these connections and their therapeutic implications.
Acknowledgement: None.
Funding Statement: This work was supported partly by National Natural Science Foundation of China (32161143021, 81271410), Henan University Graduate «Talent Program» of Henan Province (SYLYC2023092), and Henan Natural Science Foundation of China (182300410313).
Author Contributions: Study conception and design: Tingting Liu, Xiangrui Kong; draft manuscript preparation: Tingting Liu; review and editing: Tingting Liu, Xiangrui Kong, Jianshe Wei; visualization: Tingting Liu; supervision: Tingting Liu, Xiangrui Kong, Jianshe Wei. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF diabetes atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. 2022;183:109119. doi:10.1016/j.diabres.2021.109119. [Google Scholar] [PubMed] [CrossRef]
2. Hamzé R, Delangre E, Tolu S, Moreau M, Janel N, Bailbé D, et al. Type 2 diabetes mellitus and Alzheimer’s disease: shared molecular mechanisms and potential common therapeutic targets. Int J Mol Sci. 2022;23(23):15287. doi:10.3390/ijms232315287. [Google Scholar] [PubMed] [CrossRef]
3. Chohan H, Senkevich K, Patel RK, Bestwick JP, Jacobs BM, Bandres Ciga S, et al. Type 2 diabetes as a determinant of Parkinson’s disease risk and progression. Mov Disord. 2021;36(6):1420–9. doi:10.1002/mds.v36.6. [Google Scholar] [CrossRef]
4. Tao Y, Leng SX, Zhang H. Ketogenic diet: an effective treatment approach for Neurodegenerative diseases. Curr Neuropharmacol. 2022;20(12):2303–19. doi:10.2174/1570159X20666220830102628. [Google Scholar] [PubMed] [CrossRef]
5. Zheng Z, Zhu Z, Zhou C, Cao L, Zhao G. Burden of Parkinson disease in China, 1990–2019: findings from the 2019 global burden of disease study. Neuroepidemiology. 2023;57(1):51–64. doi:10.1159/000527372. [Google Scholar] [PubMed] [CrossRef]
6. Bloem BR, Okun MS, Klein C. Parkinson’s disease. Lancet. 2021;397(10291):2284–303. doi:10.1016/S0140-6736(21)00218-X. [Google Scholar] [PubMed] [CrossRef]
7. Sharma A, Weber D, Raupbach J, Dakal TC, Fließbach K, Ramirez A, et al. Advanced glycation end products and protein carbonyl levels in plasma reveal sex-specific differences in Parkinson’s and Alzheimer’s disease. Redox Biol. 2020;34:101546. doi:10.1016/j.redox.2020.101546. [Google Scholar] [PubMed] [CrossRef]
8. Liu DX, Zhao CS, Wei XN, Ma YP, Wu JK. Semaglutide protects against 6-OHDA toxicity by enhancing autophagy and inhibiting oxidative stress. Parkinsons Dis. 2022;2022:6813017. [Google Scholar] [PubMed]
9. Labandeira CM, Fraga-Bau A, Arias Ron D, Alvarez-Rodriguez E, Vicente-Alba P, Lago-Garma J, et al. Parkinson’s disease and diabetes mellitus: common mechanisms and treatment repurposing. Neural Regen Res. 2022;17(8):1652–8. doi:10.4103/1673-5374.332122. [Google Scholar] [PubMed] [CrossRef]
10. Szunerits S, Abderrahmani A, Boukherroub R. Nanoparticles and nanocolloidal carbon: will they be the next antidiabetic class that targets fibrillation and aggregation of human islet amyloid polypeptide in type 2 diabetes? Acc Chem Res. 2022;55(20):2869–81. doi:10.1021/acs.accounts.2c00415. [Google Scholar] [PubMed] [CrossRef]
11. Meng L, Li Y, Liu C, Zhang G, Chen J, Xiong M, et al. Islet amyloid polypeptide triggers α-synuclein pathology in Parkinson’s disease. Prog Neurobiol. 2023;226:102462. doi:10.1016/j.pneurobio.2023.102462. [Google Scholar] [PubMed] [CrossRef]
12. Hong CT, Chen KY, Wang W, Chiu JY, Wu D, Chao TY, et al. Insulin resistance promotes Parkinson’s disease through aberrant expression of α-synuclein, mitochondrial dysfunction, and deregulation of the polo-like kinase 2 signaling. Cells. 2020;9(3):740. doi:10.3390/cells9030740. [Google Scholar] [PubMed] [CrossRef]
13. Hsu CC, Templin AT, Prosswimmer T, Shea D, Li J, Brooks-Worrell B, et al. Human islet amyloid polypeptide-induced β-cell cytotoxicity is linked to formation of α-sheet structure. Protein Sci. 2024;33(2):e4854. doi:10.1002/pro.v33.2. [Google Scholar] [CrossRef]
14. Mezias C, Rey N, Brundin P, Raj A. Neural connectivity predicts spreading of alpha-synuclein pathology in fibril-injected mouse models: involvement of retrograde and anterograde axonal propagation. Neurobiol Dis. 2020;134:104623. doi:10.1016/j.nbd.2019.104623. [Google Scholar] [PubMed] [CrossRef]
15. Ruze R, Liu T, Zou X, Song J, Chen Y, Xu R, et al. Obesity and type 2 diabetes mellitus: connections in epidemiology, pathogenesis, and treatments. Front Endocrinol. 2023;14:1161521. doi:10.3389/fendo.2023.1161521. [Google Scholar] [PubMed] [CrossRef]
16. Simon DK, Tanner CM, Brundin P. Parkinson disease epidemiology, pathology, genetics, and pathophysiology. Clin Geriatr Med. 2020;36(1):1–12. doi:10.1016/j.cger.2019.08.002. [Google Scholar] [PubMed] [CrossRef]
17. McKnight S, Hack N. Toxin-Induced Parkinsonism. Neurol Clin. 2020;38(4):853–65. doi:10.1016/j.ncl.2020.08.003. [Google Scholar] [PubMed] [CrossRef]
18. Mezzaroba L, Alfieri DF, Colado Simão AN, Vissoci Reiche EM. The role of zinc, copper, manganese and iron in neurodegenerative diseases. Neurotoxicology. 2019;74:230–41. doi:10.1016/j.neuro.2019.07.007. [Google Scholar] [PubMed] [CrossRef]
19. Misra BB, Misra A. The chemical exposome of type 2 diabetes mellitus: opportunities and challenges in the omics era. Diabetes Metab Syndr. 2020;14(1):23–38. doi:10.1016/j.dsx.2019.12.001. [Google Scholar] [PubMed] [CrossRef]
20. Ruiz-Pozo VA, Tamayo-Trujillo R, Cadena-Ullauri S, Frias-Toral E, Guevara-Ramírez P, Paz-Cruz E, et al. The molecular mechanisms of the relationship between insulin resistance and Parkinson’s disease pathogenesis. Nutrients. 2023;15(16):3585. doi:10.3390/nu15163585. [Google Scholar] [PubMed] [CrossRef]
21. Athauda D, Evans J, Wernick A, Virdi G, Choi ML, Lawton M, et al. The impact of type 2 diabetes in Parkinson’s disease. Mov Disord. 2022;37(8):1612–23. doi:10.1002/mds.v37.8. [Google Scholar] [CrossRef]
22. American Diabetes Association Professional Practice Committee. 2. Diagnosis and classification of diabetes: standards of care in diabetes-2024. Diabetes Care. 2024;47(Suppl 1):S20–42. [Google Scholar]
23. Lyssenko V, Vaag A. Genetics of diabetes-associated microvascular complications. Diabetologia. 2023;66(9):1601–13. doi:10.1007/s00125-023-05964-x. [Google Scholar] [PubMed] [CrossRef]
24. Tinajero MG, Malik VS. An update on the epidemiology of type 2 diabetes: a global perspective. Endocrinol Metab Clin North Am. 2021;50(3):337–55. doi:10.1016/j.ecl.2021.05.013. [Google Scholar] [PubMed] [CrossRef]
25. Zhang R, Zhuang L, Li M, Zhang J, Zhao W, Ge X, et al. Arg913Gln of SLC12A3 gene promotes development and progression of end-stage renal disease in Chinese type 2 diabetes mellitus. Mol Cell Biochem. 2018;437(1–2):203–10. [Google Scholar] [PubMed]
26. Kim JH, Shin HD, Park BL, Moon MK, Cho YM, Hwang YH, et al. SLC12A3 (solute carrier family 12 member [sodium/chloride] 3) polymorphisms are associated with end-stage renal disease in diabetic nephropathy. Diabetes. 2006;55(3):843–8. doi:10.2337/diabetes.55.03.06.db05-1013. [Google Scholar] [PubMed] [CrossRef]
27. Ference BA, Robinson JG, Brook RD, Catapano AL, Chapman MJ, Neff DR, et al. Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes. N Engl J Med. 2016;375(22):2144–53. doi:10.1056/NEJMoa1604304. [Google Scholar] [PubMed] [CrossRef]
28. Fang C, Pan J, Qu N, Lei Y, Han J, Zhang J, et al. The AMPK pathway in fatty liver disease. Front Physiol. 2022;13:970292. doi:10.3389/fphys.2022.970292. [Google Scholar] [PubMed] [CrossRef]
29. Ergen HA, Zeybek U, Gök O, Karaali ZE. Investigation of ABCA1 C69T polymorphism in patients with type 2 diabetes mellitus. Biochem Med. 2012;22(1):114–20. doi:10.11613/BM.2012.013. [Google Scholar] [PubMed] [CrossRef]
30. Chen L, Zhao ZW, Zeng PH, Zhou YJ, Yin WJ. Molecular mechanisms for ABCA1-mediated cholesterol efflux. Cell Cycle. 2022;21(11):1121–39. doi:10.1080/15384101.2022.2042777. [Google Scholar] [PubMed] [CrossRef]
31. Yassine HN, Belopolskaya A, Schall C, Stump CS, Lau SS, Reaven PD. Enhanced cholesterol efflux to HDL through the ABCA1 transporter in hypertriglyceridemia of type 2 diabetes. Metabolism. 2014;63(5):727–34. doi:10.1016/j.metabol.2014.03.001. [Google Scholar] [PubMed] [CrossRef]
32. Tokoro F, Matsuoka R, Abe S, Arai M, Noda T, Watanabe S, et al. Association of a genetic variant of the ZPR1 zinc finger gene with type 2 diabetes mellitus. Biomed Rep. 2015;3(1):88–92. doi:10.3892/br.2014.379. [Google Scholar] [PubMed] [CrossRef]
33. Esteve-Luque V, Padró-Miquel A, Fanlo-Maresma M, Corbella E, Corbella X, Pintó X, et al. Implication between genetic variants from APOA5 and ZPR1 and NAFLD severity in patients with hypertriglyceridemia. Nutrients. 2021;13(2):552. doi:10.3390/nu13020552. [Google Scholar] [PubMed] [CrossRef]
34. Day JO, Mullin S. The genetics of parkinson’s disease and implications for clinical practice. Genes. 2021;12(7):1006. doi:10.3390/genes12071006. [Google Scholar] [PubMed] [CrossRef]
35. Cherian A, Divya KP. Genetics of Parkinson’s disease. Acta Neurol Belg. 2020;120(6):1297–305. doi:10.1007/s13760-020-01473-5. [Google Scholar] [PubMed] [CrossRef]
36. Liu TT, Guo DY, Wei JS. The pathogenesis of parkinson’s disease and crosstalk with other diseases. BIOCELL. 2024;48(8):1155–79. doi:10.32604/biocell.2024.051518. [Google Scholar] [CrossRef]
37. Goyal S, Rani J, Bhat MA, Vanita V. Genetics of diabetes. World J Diabetes. 2023;14(6):656–79. doi:10.4239/wjd.v14.i6.656. [Google Scholar] [PubMed] [CrossRef]
38. Song H, Chen J, Huang J, Sun P, Liu Y, Xu L, et al. Epigenetic modification in Parkinson’s disease. Front Cell Dev Biol. 2023;11:1123621. doi:10.3389/fcell.2023.1123621. [Google Scholar] [PubMed] [CrossRef]
39. Cullinane PW, de Pablo Fernandez E, König A, Outeiro TF, Jaunmuktane Z, Warner TT. Type 2 diabetes and Parkinson’s disease: a focused review of current concepts. Mov Disord. 2023;38(2):162–77. doi:10.1002/mds.v38.2. [Google Scholar] [CrossRef]
40. Deng Y, Zhang Y, Li Y, Xiao S, Song D, Qing H, et al. Occurrence and distribution of salsolinol-like compound, 1-acetyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline (ADTIQ) in parkinsonian brains. J Neural Transm. 2012;119(4):435–41. doi:10.1007/s00702-011-0724-4. [Google Scholar] [PubMed] [CrossRef]
41. Lv YQ, Yuan L, Sun Y, Dou HW, Su JH, Hou ZP, et al. Long-term hyperglycemia aggravates α-synuclein aggregation and dopaminergic neuronal loss in a Parkinson’s disease mouse model. Transl Neurodegener. 2022;11(1):14. doi:10.1186/s40035-022-00288-z. [Google Scholar] [PubMed] [CrossRef]
42. Castilla-Cortázar I, Aguirre GA, Femat-Roldán G, Martín-Estal I, Espinosa L. Is insulin-like growth factor-1 involved in Parkinson’s disease development? J Transl Med. 2020;18(1):70. doi:10.1186/s12967-020-02223-0. [Google Scholar] [PubMed] [CrossRef]
43. Wang Y, Liu W, He X, Zhou F. Parkinson’s disease-associated DJ-1 mutations increase abnormal phosphorylation of tau protein through Akt/GSK-3β pathways. J Mol Neurosci. 2013;51(3):911–8. doi:10.1007/s12031-013-0099-0. [Google Scholar] [PubMed] [CrossRef]
44. Sekar S, Taghibiglou C. Elevated nuclear phosphatase and tensin homolog (PTEN) and altered insulin signaling in substantia nigral region of patients with Parkinson’s disease. Neurosci Lett. 2018;666:139–43. doi:10.1016/j.neulet.2017.12.049. [Google Scholar] [PubMed] [CrossRef]
45. Wahlqvist ML, Lee MS, Hsu CC, Chuang SY, Lee JT, Tsai HN. Metformin-inclusive sulfonylurea therapy reduces the risk of Parkinson’s disease occurring with Type 2 diabetes in a Taiwanese population cohort. Parkinsonism Relat Disord. 2012;18(6):753–8. doi:10.1016/j.parkreldis.2012.03.010. [Google Scholar] [PubMed] [CrossRef]
46. Athauda D, Gulyani S, Karnati HK, Li Y, Tweedie D, Mustapic M, et al. Utility of neuronal-derived exosomes to examine molecular mechanisms that affect motor function in patients with Parkinson disease: a secondary analysis of the exenatide-PD trial. JAMA Neurol. 2019;76(4):420–9. doi:10.1001/jamaneurol.2018.4304. [Google Scholar] [PubMed] [CrossRef]
47. Hölscher C. Novel dual GLP-1/GIP receptor agonists show neuroprotective effects in Alzheimer’s and Parkinson’s disease models. Neuropharmacology. 2018;136:251–9. doi:10.1016/j.neuropharm.2018.01.040. [Google Scholar] [PubMed] [CrossRef]
48. Ay M, Charli A, Langley M, Jang A, Padhi P, Jin H, et al. Mito-metformin protects against mitochondrial dysfunction and dopaminergic neuronal degeneration by activating upstream PKD1 signaling in cell culture and MitoPark animal models of Parkinson’s disease. Front Neurosci. 2024;18:1356703. doi:10.3389/fnins.2024.1356703. [Google Scholar] [PubMed] [CrossRef]
49. Lu M, Su C, Qiao C, Bian Y, Ding J, Hu G. Metformin prevents dopaminergic neuron death in MPTP/P-induced mouse model of Parkinson’s disease via autophagy and mitochondrial ROS clearance. Int J Neuropsychopharmacol. 2016;19(9):pyw047. doi:10.1093/ijnp/pyw047. [Google Scholar] [PubMed] [CrossRef]
50. Taher I, El-Masry E, Abouelkheir M, Taha AE. Anti-inflammatory effect of metformin against an experimental model of LPS-induced cytokine storm. Exp Ther Med. 2023;26(3):415. doi:10.3892/etm. [Google Scholar] [CrossRef]
51. Adedeji HA, Ishola IO, Adeyemi OO. Novel action of metformin in the prevention of haloperidol-induced catalepsy in mice: potential in the treatment of Parkinson’s disease? Prog Neuropsychopharmacol Biol Psychiatr. 2014;48:245–51. doi:10.1016/j.pnpbp.2013.10.014. [Google Scholar] [PubMed] [CrossRef]
52. Wan W, Cao L, Kalionis B, Murthi P, Xia S, Guan Y. Iron deposition leads to hyperphosphorylation of tau and disruption of insulin signaling. Front Neurol. 2019;10:607. doi:10.3389/fneur.2019.00607. [Google Scholar] [PubMed] [CrossRef]
53. Delamarre A, Rigalleau V, Meissner WG. Insulin resistance, diabetes and Parkinson’s disease: the match continues. Parkinsonism Relat Disord. 2020;80:199–200. doi:10.1016/j.parkreldis.2020.10.013. [Google Scholar] [PubMed] [CrossRef]
54. Rohm TV, Meier DT, Olefsky JM, Donath MY. Inflammation in obesity, diabetes, and related disorders. Immunity. 2022;55(1):31–55. doi:10.1016/j.immuni.2021.12.013. [Google Scholar] [PubMed] [CrossRef]
55. Xxxx S, Ahmad MH, Rani L, Mondal AC. Convergent molecular pathways in type 2 diabetes mellitus and Parkinson’s disease: insights into mechanisms and pathological consequences. Mol Neurobiol. 2022;59(7):4466–87. doi:10.1007/s12035-022-02867-7. [Google Scholar] [PubMed] [CrossRef]
56. Pajares MI, Rojo A, Manda G, Boscá L, Cuadrado A. Inflammation in Parkinson’s disease: mechanisms and therapeutic implications. Cells. 2020;9(7):1687. doi:10.3390/cells9071687. [Google Scholar] [PubMed] [CrossRef]
57. Muriach M, Flores-Bellver M, Romero FJ, Barcia JM. Diabetes and the brain: oxidative stress, inflammation, and autophagy. Oxid Med Cell Longev. 2014;2014:102158. [Google Scholar] [PubMed]
58. Liu TW, Chen CM, Chang KH. Biomarker of neuroinflammation in Parkinson’s disease. Int J Mol Sci. 2022;23(8):4148. doi:10.3390/ijms23084148. [Google Scholar] [PubMed] [CrossRef]
59. Li Y, Yang Y, Zhao A, Luo N, Niu M, Kang W, et al. Parkinson’s disease peripheral immune biomarker profile: a multicentre, cross-sectional and longitudinal study. J Neuroinflamm. 2022;19(1):116. doi:10.1186/s12974-022-02481-3. [Google Scholar] [PubMed] [CrossRef]
60. Sangwung P, Petersen KF, Shulman GI, Knowles JW. Mitochondrial dysfunction, insulin resistance, and potential genetic implications. Endocrinology. 2020;161(4):bqaa017. doi:10.1210/endocr/bqaa017. [Google Scholar] [PubMed] [CrossRef]
61. Juárez-Flores DL, Ezquerra M, Gonzàlez-Casacuberta Ï., Ormazabal A, Morén C, Tolosa E, et al. Disrupted mitochondrial and metabolic plasticity underlie comorbidity between age-related and degenerative disorders as Parkinson disease and type 2 diabetes mellitus. Antioxidants. 2020;9(11):1063. doi:10.3390/antiox9111063. [Google Scholar] [PubMed] [CrossRef]
62. Pinti MV, Fink GK, Hathaway QA, Durr AJ, Kunovac A, Hollander JM. Mitochondrial dysfunction in type 2 diabetes mellitus: an organ-based analysis. Am J Physiol Endocrinol Metab. 2019;316(2):E268–85. doi:10.1152/ajpendo.00314.2018. [Google Scholar] [PubMed] [CrossRef]
63. Angelova PR, Barilani M, Lovejoy C, Dossena M, Viganò M, Seresini A, et al. Mitochondrial dysfunction in Parkinsonian mesenchymal stem cells impairs differentiation. Redox Biol. 2018;14:474–84. doi:10.1016/j.redox.2017.10.016. [Google Scholar] [PubMed] [CrossRef]
64. Moradi Vastegani S, Nasrolahi A, Ghaderi S, Belali R, Rashno M, Farzaneh M, et al. Mitochondrial dysfunction and Parkinson’s disease: pathogenesis and therapeutic strategies. Neurochem Res. 2023;48(8):2285–308. doi:10.1007/s11064-023-03904-0. [Google Scholar] [PubMed] [CrossRef]
65. Yaribeygi H, Farrokhi FR, Rezaee R, Sahebkar A. Oxidative stress induces renal failure: a review of possible molecular pathways. J Cell Biochem. 2018;119(4):2990–8. doi:10.1002/jcb.v119.4. [Google Scholar] [CrossRef]
66. Yaribeygi H, Atkin SL, Butler AE, Sahebkar A. Sodium-glucose cotransporter inhibitors and oxidative stress: an update. J Cell Physiol. 2019;234(4):3231–7. doi:10.1002/jcp.v234.4. [Google Scholar] [CrossRef]
67. Yaribeygi H, Butler AE, Barreto GE, Sahebkar A. Antioxidative potential of antidiabetic agents: a possible protective mechanism against vascular complications in diabetic patients. J Cell Physiol. 2019;234(3):2436–46. doi:10.1002/jcp.v234.3. [Google Scholar] [CrossRef]
68. Yaribeygi H, Mohammadi MT, Sahebkar A. PPAR-α agonist improves hyperglycemia-induced oxidative stress in pancreatic cells by potentiating antioxidant defense system. Drug Res. 2018;68(6):355–60. doi:10.1055/s-0043-121143. [Google Scholar] [PubMed] [CrossRef]
69. Yaribeygi H, Lhaf F, Sathyapalan T, Sahebkar A. Effects of novel antidiabetes agents on apoptotic processes in diabetes and malignancy: implications for lowering tissue damage. Life Sci. 2019;231:116538. doi:10.1016/j.lfs.2019.06.013. [Google Scholar] [PubMed] [CrossRef]
70. Trist BG, Hare DJ, Double KL. Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell. 2019;18(6):e13031. doi:10.1111/acel.v18.6. [Google Scholar] [CrossRef]
71. Araújo B, Caridade-Silva R, Soares-Guedes C, Martins-Macedo J, Gomes ED, Monteiro S, et al. Neuroinflammation and Parkinson’s disease-from neurodegeneration to therapeutic opportunities. Cells. 2022;11(18):2908. doi:10.3390/cells11182908. [Google Scholar] [PubMed] [CrossRef]
72. Percário S, da Silva Barbosa A, Varela ELP, Gomes ARQ, Ferreira MES, de Nazaré Araújo Moreira T, et al. Oxidative stress in Parkinson’s disease: potential benefits of antioxidant supplementation. Oxid Med Cell Longev. 2020;2020:2360872. [Google Scholar]
73. Rathnayake D, Chang T, Udagama P. Selected serum cytokines and nitric oxide as potential multi-marker biosignature panels for Parkinson disease of varying durations: a case-control study. BMC Neurol. 2019;19(1):56. doi:10.1186/s12883-019-1286-6. [Google Scholar] [PubMed] [CrossRef]
74. Tu D, Gao Y, Yang R, Guan T, Hong JS, Gao HM. The pentose phosphate pathway regulates chronic neuroinflammation and dopaminergic neurodegeneration. J Neuroinflamm. 2019;16(1):255. doi:10.1186/s12974-019-1659-1. [Google Scholar] [PubMed] [CrossRef]
75. Dong K, Ni H, Wu M, Tang Z, Halim M, Shi D. ROS-mediated glucose metabolic reprogram induces insulin resistance in type 2 diabetes. Biochem Biophys Res Commun. 2016;476(4):204–11. doi:10.1016/j.bbrc.2016.05.087. [Google Scholar] [PubMed] [CrossRef]
76. TeSlaa T, Ralser M, Fan J, Rabinowitz JD. The pentose phosphate pathway in health and disease. Nat Metab. 2023;5(8):1275–89. doi:10.1038/s42255-023-00863-2. [Google Scholar] [PubMed] [CrossRef]
77. He F, Huang Y, Song Z, Zhou HJ, Zhang H, Perry RJ, et al. Mitophagy-mediated adipose inflammation contributes to type 2 diabetes with hepatic insulin resistance. J Exp Med. 2021;218(3):e20201416. doi:10.1084/jem.20201416. [Google Scholar] [PubMed] [CrossRef]
78. Chen J, Gao X, Zheng C, Zhang C, Li P, He K, et al. Low-dose Cu exposure enhanced α-synuclein accumulation associates with mitochondrial impairments in mice model of Parkinson’s disease. Toxicol Lett. 2023;387:14–27. doi:10.1016/j.toxlet.2023.09.004. [Google Scholar] [PubMed] [CrossRef]
79. Zhao X, Bie LY, Pang DR, Li X, Yang LF, Chen DD, et al. The role of autophagy in the treatment of type II diabetes and its complications: a review. Front Endocrinol. 2023;14:1228045. doi:10.3389/fendo.2023.1228045. [Google Scholar] [PubMed] [CrossRef]
80. Marmentini C, Branco RCS, Boschero AC, Kurauti MA. Islet amyloid toxicity: from genesis to counteracting mechanisms. J Cell Physiol. 2022;237(2):1119–42. doi:10.1002/jcp.v237.2. [Google Scholar] [CrossRef]
81. Ding J, Hu S, Meng Y, Li C, Huang J, He Y, et al. Alpha-synuclein deficiency ameliorates chronic methamphetamine induced neurodegeneration in mice. Toxicology. 2020;438:152461. doi:10.1016/j.tox.2020.152461. [Google Scholar] [PubMed] [CrossRef]
82. Lv QK, Tao KX, Wang XB, Yao XY, Pang MZ, Liu JY, et al. Role of α-synuclein in microglia: autophagy and phagocytosis balance neuroinflammation in Parkinson’s disease. Inflamm Res. 2023;72(3):443–62. doi:10.1007/s00011-022-01676-x. [Google Scholar] [PubMed] [CrossRef]
83. Caballero B, Wang Y, Diaz A, Tasset I, Juste YR, Stiller B, et al. Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell. 2018;17(1):e12692. doi:10.1111/acel.12692. [Google Scholar] [PubMed] [CrossRef]
84. Hou X, Watzlawik JO, Fiesel FC, Springer W. Autophagy in Parkinson’s disease. J Mol Biol. 2020;432(8):2651–72. doi:10.1016/j.jmb.2020.01.037. [Google Scholar] [PubMed] [CrossRef]
85. Park GH, Park JH, Chung KC. Precise control of mitophagy through ubiquitin proteasome system and deubiquitin proteases and their dysfunction in Parkinson’s disease. BMB Rep. 2021;54(12):592–600. doi:10.5483/BMBRep.2021.54.12.107. [Google Scholar] [PubMed] [CrossRef]
86. Nowotny K, Jung T, Höhn A, Weber D, Grune T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules. 2015;5(1):194–222. doi:10.3390/biom5010194. [Google Scholar] [PubMed] [CrossRef]
87. Scheijen JL, Schalkwijk CG. Quantification of glyoxal, methylglyoxal and 3-deoxyglucosone in blood and plasma by ultra performance liquid chromatography tandem mass spectrometry: evaluation of blood specimen. Clin Chem Lab Med. 2014;52(1):85–91. doi:10.1515/cclm-2012-0878. [Google Scholar] [PubMed] [CrossRef]
88. Carvalho C, Moreira PI. MitoTempo protects against nε-carboxymethyl lysine-induced mitochondrial dyshomeostasis and neuronal cells injury. Free Radic Biol Med. 2024;220(3):192–206. doi:10.1016/j.freeradbiomed.2024.05.011. [Google Scholar] [PubMed] [CrossRef]
89. Amornrit W, Santiyanont R. Effect of amaranthus on advanced glycation end-products induced cytotoxicity and proinflammatory cytokine gene expression in SH-SY5Y cells. Molecules. 2015;20(9):17288–308. doi:10.3390/molecules200917288. [Google Scholar] [PubMed] [CrossRef]
90. Atieh TB, Roth J, Yang X, Hoop CL, Baum J. DJ-1 acts as a scavenger of α-synuclein oligomers and restores monomeric glycated α-synuclein. Biomolecules. 2021;11(10):1466. doi:10.3390/biom11101466. [Google Scholar] [PubMed] [CrossRef]
91. Choi YG, Lim S. N(ε)-(carboxymethyl)lysine linkage to α-synuclein and involvement of advanced glycation end products in α-synuclein deposits in an MPTP-intoxicated mouse model. Biochimie. 2010;92(10):1379–86. doi:10.1016/j.biochi.2010.06.025. [Google Scholar] [PubMed] [CrossRef]
92. AwadAllah Elgnainy A, Hamed MI, Osman Mohamed W, Sabri NA. Investigation of the possible correlation between idiopathic Parkinson’s disease and diabetes mellitus in egyptian patients: a pilot study. Neurol Res Int. 2021;2021:2838669. [Google Scholar] [PubMed]
93. National Center for Chronic Disease Prevention and Health Promotion (US) Office on Smoking and Health. The health consequences of smoking-50 years of progress: a report of the surgeon general. Atlanta (GACenters for Disease Control and Prevention (US); 2014. Available from: https://pubmed.ncbi.nlm.nih.gov/24455788/. [Accessed 2024]. [Google Scholar]
94. Walicka M, Russo C, Baxter M, John I, Caci G, Polosa R. Impact of stopping smoking on metabolic parameters in diabetes mellitus: a scoping review. World J Diabetes. 2022;13(6):422–33. doi:10.4239/wjd.v13.i6.422. [Google Scholar] [PubMed] [CrossRef]
95. Wei Y, Hägg S, Mak JKL, Tuomi T, Zhan Y, Carlsson S. Metabolic profiling of smoking, associations with type 2 diabetes and interaction with genetic susceptibility. Eur J Epidemiol. 2024;39(6):667–78. doi:10.1007/s10654-024-01117-5. [Google Scholar] [PubMed] [CrossRef]
96. Mukharjee S, Bank S, Maiti S. Chronic tobacco exposure by smoking develops insulin resistance. Endocr Metab Immune Disord Drug Targets. 2020;20(6):869–77. doi:10.2174/1871530320666200217123901. [Google Scholar] [PubMed] [CrossRef]
97. Hsia SH, Nisis ML, Lee ML, Goldstein C, Friedman TC. Metabolic parameters in smokers undergoing smoking reduction. J Clin Transl Endocrinol. 2021;23:100249. doi:10.1016/j.jcte.2021.100249. [Google Scholar] [PubMed] [CrossRef]
98. Espinoza-Derout J, Arambulo JML, Ramirez-Trillo W, Rivera JC, Hasan KM, Lao CJ, et al. The lipolysis inhibitor acipimox reverses the cardiac phenotype induced by electronic cigarettes. Sci Rep. 2023;13(1):18239. doi:10.1038/s41598-023-44082-x. [Google Scholar] [PubMed] [CrossRef]
99. Li GL, Ping J, Chen HJ, Zhang WX, Fan J, Peng DS, et al. Maternal nicotine exposure impairs brown adipose tissue via AMPK-SIRT1-PGC-1α signals in male offspring. Life Sci. 2021;264:118695. doi:10.1016/j.lfs.2020.118695. [Google Scholar] [PubMed] [CrossRef]
100. Wu Y, Song P, Zhang W, Liu J, Dai X, Liu Z, et al. Activation of AMPKα2 in adipocytes is essential for nicotine-induced insulin resistance in vivo. Nat Med. 2015;21(4):373–82. doi:10.1038/nm.3826. [Google Scholar] [PubMed] [CrossRef]
101. Ma C, Molsberry S, Li Y, Schwarzschild M, Ascherio A, Gao X. Dietary nicotine intake and risk of Parkinson disease: a prospective study. Am J Clin Nutr. 2020;112(4):1080–7. doi:10.1093/ajcn/nqaa186. [Google Scholar] [PubMed] [CrossRef]
102. Jung SY, Chun S, Cho EB, Han K, Yoo J, Yeo Y, et al. Changes in smoking, alcohol consumption, and the risk of Parkinson’s disease. Front Aging Neurosci. 2023;15:1223310. doi:10.3389/fnagi.2023.1223310. [Google Scholar] [PubMed] [CrossRef]
103. Soares ÉN, Costa ACDS, Ferrolho GJ, Ureshino RP, Getachew B, Costa SL, et al. Nicotinic acetylcholine receptors in glial cells as molecular target for Parkinson’s disease. Cells. 2024;13(6):474. doi:10.3390/cells13060474. [Google Scholar] [PubMed] [CrossRef]
104. Balakumar P, Kaur J. Is nicotine a key player or spectator in the induction and progression of cardiovascular disorders? Pharmacol Res. 2009;60(5):361–8. doi:10.1016/j.phrs.2009.06.005. [Google Scholar] [PubMed] [CrossRef]
105. Rose KN, Zorlu M, Fassini A, Lee H, Cai W, Xue X, et al. Neuroprotection of low dose carbon monoxide in Parkinson’s disease models commensurate with the reduced risk of Parkinson’s among smokers. npj Parkinsons Dis. 2024;10(1):152. doi:10.1038/s41531-024-00763-6. [Google Scholar] [PubMed] [CrossRef]
106. Alhowail A. Molecular insights into the benefits of nicotine on memory and cognition (Review). Mol Med Rep. 2021;23(6):398. doi:10.3892/mmr.2021.12037. [Google Scholar] [PubMed] [CrossRef]
107. Wu QJ, Zhang TN, Chen HH, Yu XF, Lv JL, Liu YY, et al. The sirtuin family in health and disease. Signal Transduct Target Ther. 2022;7(1):402. doi:10.1038/s41392-022-01257-8. [Google Scholar] [PubMed] [CrossRef]
108. Chen J, Wang Q, Li R, Li Z, Jiang Q, Yan F, et al. The role of sirtuins in the regulatin of oxidative stress during the progress and therapy of type 2 diabetes mellitus. Life Sci. 2023;333(Suppl. 1):122187. doi:10.1016/j.lfs.2023.122187. [Google Scholar] [PubMed] [CrossRef]
109. Mihanfar A, Akbarzadeh M, Ghazizadeh Darband S, Sadighparvar S, Majidinia M. SIRT1: a promising therapeutic target in type 2 diabetes mellitus. Arch Physiol Biochem. 2024 Feb;130(1):13–28. doi:10.1080/13813455.2021.1956976. [Google Scholar] [PubMed] [CrossRef]
110. He M, Chiang HH, Luo H, Zheng Z, Qiao Q, Wang L, et al. An acetylation switch of the NLRP3 inflammasome regulates aging-associated chronic inflammation and insulin resistance. Cell Metab. 2020;31(3):580–91.e5. doi:10.1016/j.cmet.2020.01.009. [Google Scholar] [PubMed] [CrossRef]
111. Singh AP, Nigam L, Yadav Y, Shekhar S, Subbarao N, Dey S. Design and in vitro analysis of SIRT2 inhibitor targeting Parkinson’s disease. Mol Divers. 2021;25(4):2261–70. doi:10.1007/s11030-020-10116-z. [Google Scholar] [PubMed] [CrossRef]
112. Guan Q, Wang M, Chen H, Yang L, Yan Z, Wang X. Aging-related 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurochemial and behavioral deficits and redox dysfunction: improvement by AK-7. Exp Gerontol. 2016;82:19–29. doi:10.1016/j.exger.2016.05.011. [Google Scholar] [PubMed] [CrossRef]
113. Zhou Z, Ma T, Zhu Q, Xu Y, Zha X. Recent advances in inhibitors of sirtuin1/2: an update and perspective. Future Med Chem. 2018;10(8):907–34. doi:10.4155/fmc-2017-0207. [Google Scholar] [PubMed] [CrossRef]
114. Liu Y, Zhang Y, Zhu K, Chi S, Wang C, Xie A. Emerging role of sirtuin 2 in Parkinson’s disease. Front Aging Neurosci. 2020;11:372. doi:10.3389/fnagi.2019.00372. [Google Scholar] [PubMed] [CrossRef]
115. Nicholatos JW, Francisco AB, Bender CA, Yeh T, Lugay FJ, Salazar JE, et al. Nicotine promotes neuron survival and partially protects from Parkinson’s disease by suppressing SIRT6. Acta Neuropathol Commun. 2018;6(1):120. doi:10.1186/s40478-018-0625-y. [Google Scholar] [PubMed] [CrossRef]
116. Feigin VL, Vos T, Nichols E, Owolabi MO, Carroll WM, Dichgans M, et al. The global burden of neurological disorders: translating evidence into policy. Lancet Neurol. 2020;19(3):255–65. doi:10.1016/S1474-4422(19)30411-9. [Google Scholar] [PubMed] [CrossRef]
117. Standaert DG. GLP-1, Parkinson’s disease, and neuroprotection. N Engl J Med. 2024;390(13):1233–4. doi:10.1056/NEJMe2401743. [Google Scholar] [PubMed] [CrossRef]
118. Shaughness M, Acs D, Brabazon F, Hockenbury N, Byrnes KR. Role of insulin in neurotrauma and neurodegeneration: a review. Front Neurosci. 2020;14:547175. doi:10.3389/fnins.2020.547175. [Google Scholar] [PubMed] [CrossRef]
119. Shen H, Gu X, Wei ZZ, Wu A, Liu X, Wei L. Combinatorial intranasal delivery of bone marrow mesenchymal stem cells and insulin-like growth factor-1 improves neurovascularization and functional outcomes following focal cerebral ischemia in mice. Exp Neurol. 2021;337:113542. doi:10.1016/j.expneurol.2020.113542. [Google Scholar] [PubMed] [CrossRef]
120. Agostini F, Masato A, Bubacco L, Bisaglia M. Metformin repurposing for parkinson disease therapy: opportunities and challenges. Int J Mol Sci. 2021;23(1):398. doi:10.3390/ijms23010398. [Google Scholar] [PubMed] [CrossRef]
121. Sabari SS, Balasubramani K, Iyer M, Sureshbabu HW, Venkatesan D, Gopalakrishnan AV, et al. Type 2 diabetes (T2DM) and Parkinson’s disease (PDa mechanistic approach. Mol Neurobiol. 2023;60(8):4547–73. doi:10.1007/s12035-023-03359-y. [Google Scholar] [PubMed] [CrossRef]
122. Jeong SH, Chung SJ, Yoo HS, Hong N, Jung JH, Baik K, et al. Beneficial effects of dipeptidyl peptidase-4 inhibitors in diabetic Parkinson’s disease. Brain. 2021;144(4):1127–37. doi:10.1093/brain/awab015. [Google Scholar] [PubMed] [CrossRef]
123. Cheong JLY, de Pablo-Fernandez E, Foltynie T, Noyce AJ. The association between type 2 diabetes mellitus and Parkinson’s disease. J Parkinsons Dis. 2020;10(3):775–89. doi:10.3233/JPD-191900. [Google Scholar] [PubMed] [CrossRef]
124. Grespan E, Mari A. Does glucose lowering restore GIP effects on insulin secretion? Nutr Metab Cardiovasc Dis. 2023;33(3):494–9. doi:10.1016/j.numecd.2022.12.021. [Google Scholar] [PubMed] [CrossRef]
125. Yang X, Feng P, Ji R, Ren Y, Wei W, Hölscher C. Therapeutic application of GLP-1 and GIP receptor agonists in Parkinson’s disease. Expert Opin Ther Targets. 2022;26(5):445–60. doi:10.1080/14728222.2022.2079492. [Google Scholar] [PubMed] [CrossRef]
126. Lv M, Xue G, Cheng H, Meng P, Lian X, Hölscher C, et al. The GLP-1/GIP dual-receptor agonist DA5-CH inhibits the NF-κB inflammatory pathway in the MPTP mouse model of Parkinson’s disease more effectively than the GLP-1 single-receptor agonist NLY01. Brain Behav. 2021;11(8):e2231. doi:10.1002/brb3.2231. [Google Scholar] [PubMed] [CrossRef]
127. Sharma MK, Jalewa J, Hölscher C. Neuroprotective and anti-apoptotic effects of liraglutide on SH-SY5Y cells exposed to methylglyoxal stress. J Neurochem. 2014;128(3):459–71. doi:10.1111/jnc.12469. [Google Scholar] [PubMed] [CrossRef]
128. Ji C, Xue GF, Cao LJ, Feng P, Li D, Li L, et al. A novel dual GLP-1 and GIP receptor agonist is neuroprotective in the MPTP mouse model of Parkinson’s disease by increasing expression of BNDF. Brain Res. 2016;1634:1–11. doi:10.1016/j.brainres.2015.09.035. [Google Scholar] [PubMed] [CrossRef]
129. Zhang Z, Shi M, Li Z, Ling Y, Zhai L, Yuan Y, et al. A dual GLP-1/GIP receptor agonist is more effective than liraglutide in the A53T mouse model of parkinson’s disease. Parkinsons Dis. 2023;2023:7427136. [Google Scholar] [PubMed]
130. Han K, Kim B, Lee SH, Kim MK. A nationwide cohort study on diabetes severity and risk of Parkinson disease. npj Parkinsons Dis. 2023;9(1):11. doi:10.1038/s41531-023-00462-8. [Google Scholar] [PubMed] [CrossRef]
131. Sánchez-Gómez A, Díaz Y, Duarte-Salles T, Compta Y, Martí MJ. Prediabetes, type 2 diabetes mellitus and risk of Parkinson’s disease: a population-based cohort study. Parkinsonism Relat Disord. 2021;89:22–7. doi:10.1016/j.parkreldis.2021.06.002. [Google Scholar] [PubMed] [CrossRef]
132. Faizan M, Sarkar A, Singh MP. Type 2 diabetes mellitus augments Parkinson’s disease risk or the other way around: facts, challenges and future possibilities. Ageing Res Rev. 2022;81:101727. doi:10.1016/j.arr.2022.101727. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools