
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW
Emerging pharmaceutical therapies for targeting cholangiocarcinoma microenvironment and chemokine pathways
1 College of Osteopathic Medicine of the Pacific, Western University of Health Sciences, Pomona, CA 91766, USA
2 Department of Translational Research, Western University of Health Sciences, Pomona, CA 91766, USA
* Corresponding Author: VIKRANT RAI. Email:
BIOCELL 2024, 48(12), 1683-1702. https://doi.org/10.32604/biocell.2024.056252
Received 18 July 2024; Accepted 25 September 2024; Issue published 30 December 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Mixed cholangiocarcinoma is a rare and aggressive neoplastic proliferation of biliary tract epithelial cells, accounting for up to 20% of primary liver cancers. It is the second most common primary liver malignancy with a 5-year survivability of less than 10% at diagnosis and is associated with various inflammatory diseases. Current management involves systemic chemotherapy, targeted radiation, and surgical resection, but long-term survival remains low, especially for surgically unresectable cases. Novel discoveries and understandings of the tumor microenvironment reveal new opportunities for targeted therapies for cholangiocarcinoma. Specifically, new pharmaceuticals including cell-based vaccines, tumor-associated neutrophils, and hepatic stellate cells may make good therapeutic targets. Tumor-reactive stroma and cancer-associated fibroblasts are also heavily implicated in disease progression. This comprehensive review aims to discuss emerging pharmaceutical therapies for targeting the cholangiocarcinoma microenvironment and chemokine pathways involved in the pathogenesis of cholangiocarcinoma followed by a risk-benefit analysis of proposed pharmaceutical therapies for treatment. A literature search on PubMed, Google Scholar, and PMC was done including the terms cholangiocarcinoma, targeted therapies, chemokine pathways, microenvironment, therapeutic targets, chemotherapy, and immune cell, alone or in combination. The articles in the English language and published in the last 10–15 years were selected to discuss in this review article. Selective therapies targeting tumor microenvironment can be fruitful in inducing tumor apoptosis and suppressing cholangiocarcinoma proliferation. Immunosuppressive therapies and immune checkpoint inhibitors also demonstrate promise in improving patient outcomes, specifically in patient’s intolerance to chemotherapy.Keywords
Mixed cholangiocarcinoma is a rare and aggressive neoplastic proliferation of epithelial cells of the biliary tract accounting for up to 20% of primary liver cancers. It is the second most common primary liver malignancy, behind hepatocellular carcinoma, and can be further subclassified as extrahepatic (75% of cases) or intrahepatic (25%) [1,2]. Many patients do not possess risk factors or specific clinical presentations; therefore, conditions are often deemed terminal at the time of diagnosis with a 5-year survivability of less than 10% [3,4]. Furthermore, many intrahepatic inflammatory diseases including Hepatitis B and C infection, choledocholithiasis, liver cirrhosis, and primary sclerosing cholangitis are associated with the pathogenesis of cholangiocarcinoma (CCA) [5,6]. Further, Caroli’s disease, ulcerative colitis, Crohn’s disease, type II diabetes, obesity, fatty liver disease, alcoholism, and smoking are risk factors for CCA [7]. CCA generally arises in the presence of chronic inflammation and mutations in various protooncogenes and tumor suppressor genes including TP53, RAS, ARIDIA, GNAS, BAP1, IDH1/2, BRAF, K-ras, and SMAD4 are involved in carcinogenesis. Additionally, genetic alterations, epigenetics involving DNA methylation, histone modification, and noncoding RNAs also play a role in carcinogenesis. CCA may be intrahepatic, perihilar, and distal arising from biliary epithelium proximal to the segmental bile ducts, left or right hepatic ducts or their confluence, and distal to the biliary confluence, respectively (Fig. 1). The three different types of CCA differ histologically [3,8,9]. Chronic inflammation contributes to carcinogenesis by increased exposure of cholangiocytes to interleukin (IL)-6, tumor necrosis factor-ɑ, and cyclo-oxygenase-2, altered extracellular signal-regulated kinase (ERK)1/2, protein kinase B (Akt), and nuclear factor kappa beta (NF-κB) signaling, and growth factors effects on the cholangiocytes [10] (Fig. 1).
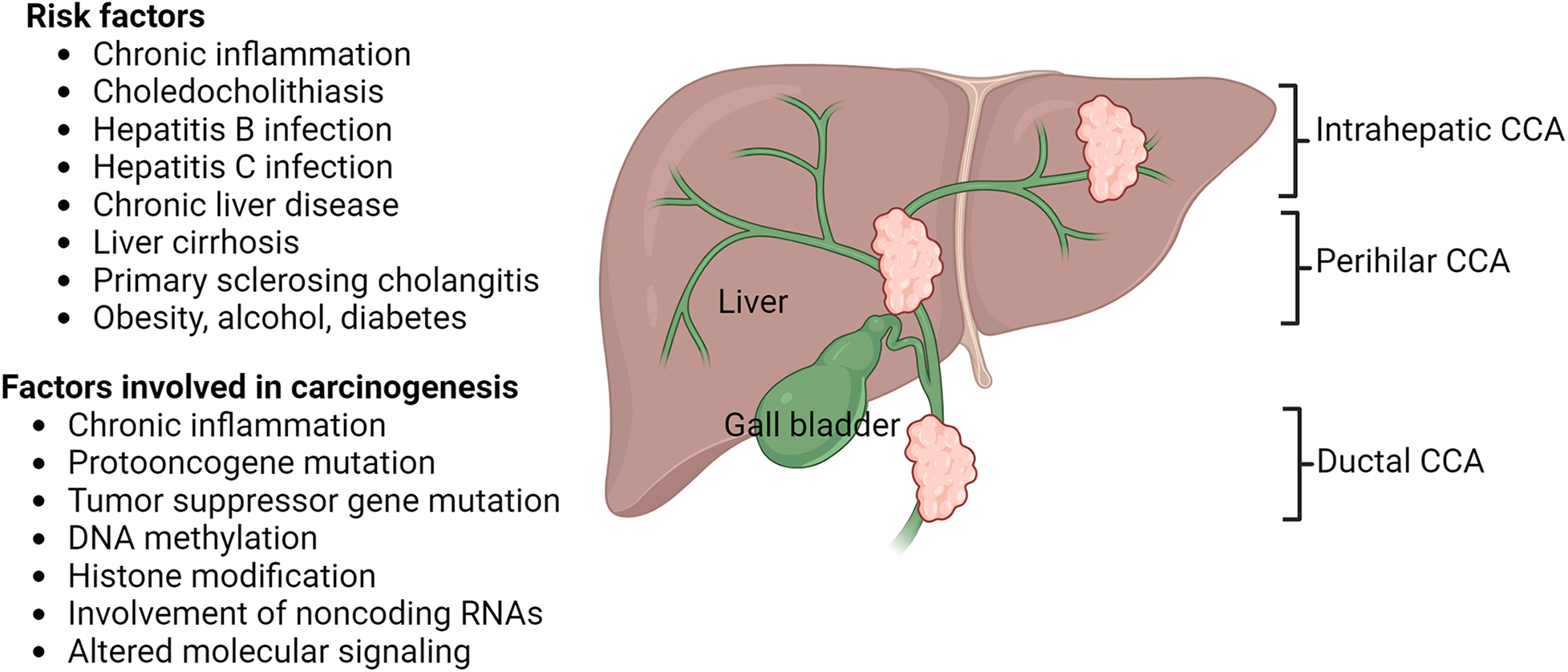
Figure 1: Risk factors, factors involved in carcinogenesis, and histologic types of cholangiocarcinoma (CCA). Cholangiocarcinoma may be intrahepatic, perihilar, and ductal originating in different parts of bile ducts connecting liver and gall bladder with small intestine. The figure is created using BioRender.
Management of Cholangiocarcinoma currently combines systemic chemotherapy, targeted radiation, and surgical resection; however, long-term survival remains low, and surgically unresectable diseases are incurable [3]. All patients with CCA should be laparoscopically staged before any surgical procedure though there may be risk of occult metastasis. Parenchymal sparing, nonanatomic liver resection in peripheral tumors, anatomic liver resection for solitary liver lesion, either left or right hepatectomy for perihilar tumors, and pancreatoduodenectomy or Whipple procedure for extrahepatic tumors are common surgical procedures [7]. Systemic therapies with neoadjuvant therapy and adjuvant therapy, radiation therapy, and locoregional therapy for nonsurgical lesions are other avenues for therapy. Fibroblast growth factor receptor 2 inhibitors, isocitrate dehydrogenase inhibitors, B-Raf Proto-Oncogene, Serine/Threonine Kinase (BRAF) inhibitor, mitogen-activated protein kinase kinase (MEK) inhibitor, anti-human epidermal growth factor receptor 2 (HER2) therapies, and immunotherapy with immune checkpoint inhibitors are emerging therapeutics for CCA [7]. Recently a new realm of research has emerged studying the role of the tumor microenvironment and chemokine pathways supporting the proliferation of cholangiocarcinoma. The tumor microenvironment including the desmoplastic stroma, hepatic stellate cells, cancer-associated fibroblasts, tumor-infiltrating immune cells, and tumor-associated macrophages have been implicated in the disease progression, metastasis, chemoresistance, and tumor-specific immune tolerance. Studies of the tumor microenvironment reveal various chemokine pathways that may serve as novel targets for cholangiocarcinoma therapy [11,12].
The primary objective of this paper is to comprehensively explore the intricate roles played by innate immunity, adaptive immunity, tumor-reactive stroma, cancer-associated fibroblasts, and immunosuppression in the pathophysiology of cholangiocarcinoma. By delving into these aspects, the aim is to shed light on how they contribute to the disease’s progression and resistance to treatment, ultimately paving the way for the development of novel therapeutic strategies. This research endeavors to enhance the current understanding of the cholangiocarcinoma tumor microenvironment by providing a detailed risk-benefit analysis of various proposed pharmaceutical therapies. In doing so, it seeks to offer valuable insights that could guide future clinical approaches, optimizing treatment outcomes for patients afflicted by this challenging malignancy.
The innate immune response, the first line of defense of the body against invading pathogens, is also referred to as the “nonspecific” immune response because the innate immune response is an antigen-independent defense mechanism and remains unable to recognize or memorize (no immunologic memory) the same pathogen again. The innate immune response is quick within hours of encountering an antigen [13]. Innate immunity comprises four types of defensive barriers including anatomic (skin and mucous membrane), physiologic, endocytic, phagocytic, and inflammatory. Macrophages and neutrophils (phagocytes), basophils, eosinophils, dendritic cells, mast cells, natural killer (NK) cells, and innate lymphoid cells are involved in innate immune response (Fig. 2).
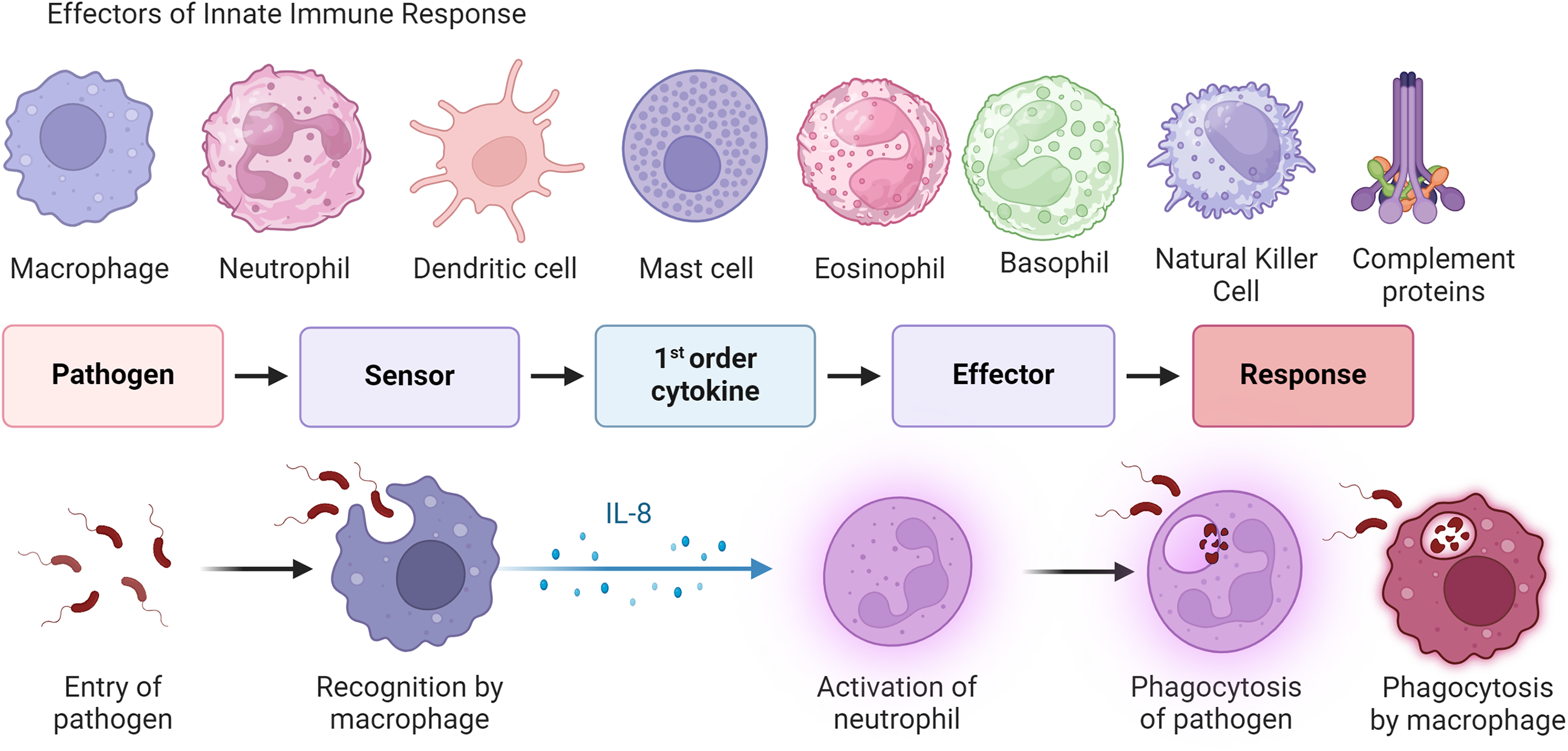
Figure 2: General principle of innate immune response. This figure displays different cells of the innate immune system, such as macrophages, neutrophils, and dendritic cells, and the process by which the innate immune system acts to recognize pathogens to create a host immune response. This process involves a sensor that leads to cell production of pro-inflammatory cytokines and the migration of effector cells (neutrophils, macrophages, T cells, and B cells) to inflammation. While this often occurs in infection, it can also occur within tumor microenvironments in response to pro-inflammatory signaling. The figure is created using BioRender.
These immune cells (Fig. 3) are involved in the tumor microenvironment and play a critical role in the inflammatory microenvironment-cholangiocarcinoma crosstalk [14,15].
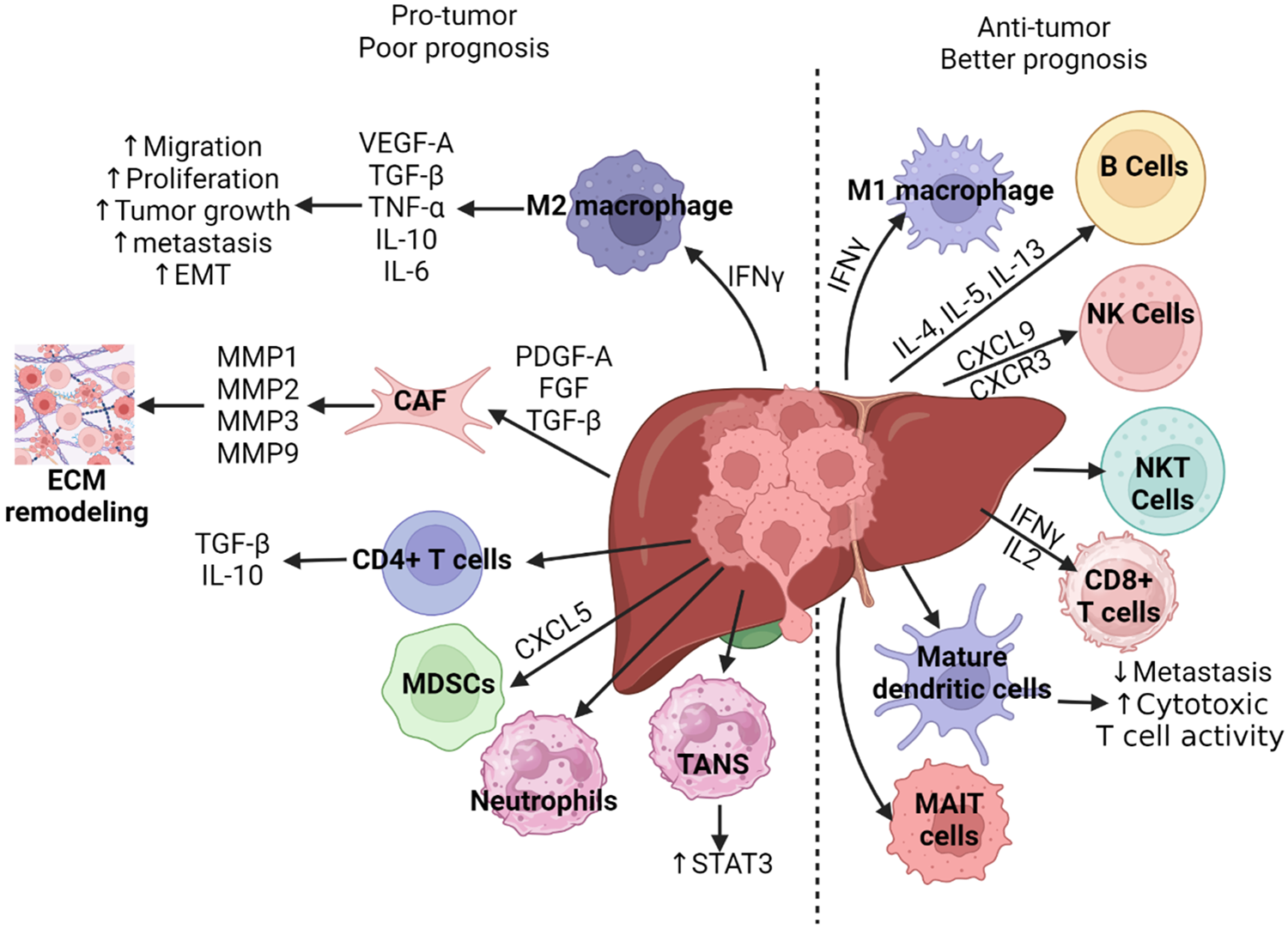
Figure 3: The role of immune cells in the pathogenesis of cholangiocarcinoma development and prognosis [11,14,15]. In cholangiocarcinoma, the tumor microenvironment is modulated by many components, both cellular and non-cellular. In some cases, these components act to promote tumor growth and progression. For example, PDGF-A, FGF, TGF-β recruit CAFs leading to the production of MMPs that result in extracellular modeling which promotes cancer cell proliferation. CD4+ T cells within the tumor stroma can secrete TGF-β and IL-10, which promote migration, proliferation, and tumor progression. Activation of M2 macrophages functions in a similar manner with secretion of TGF-β and IL-10 as well as VEGF-A, TNF-α, and IL-6, thus promoting cancer progression. MDSCs, neutrophils and TANS may similarly function to promote tumor progression within the tumor microenvironment, however, the mechanisms behind this remain unclear in cholangiocarcinoma. Alternatively, some components of the tumor microenvironment may act to suppress tumor growth. For example, CD8+ T cells, when stimulated by interferon gamma (IFN-γ) may promote apoptosis of tumor cells, thus leading to slowed progression of the tumor. Dendritic cells can function similarly by activating T cells in response to tumor associated antigens. Additionally, CXCL9 and CXCR3 promote the function of NK cells leading to tumor cell apoptosis. Similarly, B cells, M1 macrophages, MAIT cells, and NKT cells act to promote apoptosis, though these mechanisms remain unclear in cholangiocarcinoma. Cancer associated fibroblasts (CAFs), Vascular endothelial growth factor (VEGF), transforming growth factor beta (TGF-β), tumor necrosis factor-alpha (TNF-α), interleukin (IL), interferon-gamma (IFN-γ), fibroblast growth factor (FGF), platelet-derived growth factor (PDGF), matrix metalloproteinase (MMP), chemokine (C-X-C motif) ligand 5 (CXCL5), chemokine (C-X-C motif) receptor 3 (CXCR3), signal transducer and activator of transcription 3 (STAT3), extracellular matrix (ECM), epithelial-mesenchymal transition (EMT), myeloid-derived suppressor cells (MDSCs), tumor-associated neutrophils (TANs), natural killer (NK) cells, T-regulatory cells (NKT/Tregs), and mucosal-associated invariant T cells (MAIT cells). The figure is created using BioRender.
Dendritic cells are antigen-presenting cells, which stimulate T cells to invoke an immune response [16]. Within the cholangiocarcinoma (CCA) tumor microenvironment, tumor-infiltrating dendritic cells (DCs), which express CD40, are prevalent [17]. While there are immature DCs within the tumor, the majority of mature DCs remain on the tumor invasion front [18,19]. Research suggests that this might contribute to immunotolerance due to antigen presentation deficiency and maturation. This process ultimately leads to decreased T cell priming. Additionally, these DCs recruit regulatory T cells, further promoting the immunotolerance of the tumor [20,21]. Moreover, DCs within the microenvironment have been shown to decrease the local expression of human leukocyte antigen, thus lessening the response of tumor-infiltrating lymphocytes [22]. There is additional evidence that these dendritic cells may circulate and modulate the immune response systemically [23,24]. Furthermore, the CCA cells secrete IL-10 and TGFβ, further contributing to reduced antigen presentation to T cells through their effects on DCs [25,26]. Overall, the significant role of dendritic cells (Fig. 3) within the tumor microenvironment may be a vital target for the treatment of CCA.
A few studies have investigated peptide-based vaccines in CCA. Recent Phase 1 clinical trials focusing on vaccines targeting mucin protein 1 (MUC1) and Wilms tumor protein 1 (WT1) have shown promising results in the treatment of CCA. These trials have primarily aimed to assess the safety and immunogenicity of these vaccines, as both MUC1 and WT1 are frequently over-expressed in CCA tumors [27]. The results indicate that these vaccines are generally well-tolerated, with no significant adverse events reported, making them a potentially viable option for further development [27–30]. In the WT1 vaccine study, data indicated that, when combined with gemcitabine, the tumor control rate was nearly 50% [30]. In another small study, a vaccine that consisted of 4 peptides was found to be very effective in reducing CCA due to their effects on cytotoxic T lymphocytes [31]. However, these two studies were conducted with small patient populations. Overall, these results are promising for peptide-based vaccines in CCA. However, research has shown that cell-based vaccines are more effective than non-cell-based vaccines in stimulating an immune response to tumors [32].
Thus far, there have been several studies utilizing and targeting the role of dendritic cells within the CCA microenvironment. Researchers have investigated immunization as a mechanism to stimulate an immune response to CCA. For example, one group used a transmembrane cell surface protein aspartate-β-hydroxylase, which is highly conserved and expressed within CCA tumors but not in surrounding tissues [33–37]. This protein is involved in proliferation, cell migration, and invasion within tumors [38]. In their rat CCA study, they found that DCs loaded with aspartate-β-hydroxylase (ASPH) had significant anti-tumor effects through an increase in tumor-infiltrating lymphocyte recruitment [34]. However, their research indicates that multiple immunizations may be necessary to obtain optimal anti-tumor effects [34]. It has also been shown that blockage of DCs IL-10 and TGF-β receptors, using DCs loaded with Protein Kinase CAMP-Dependent Type I Regulatory Subunit Alpha (PRKAR1A), can enhance the activity of cytotoxic T cells against tumor cells [39,40]. Moreover, a retrospective study that looked at patients with advanced CCA treated with a DC vaccine indicated good tolerance of the vaccine; 15% of these patients also remained stable for 6 months [41]. Additionally, when combined with chemotherapy, the prognosis of CCA patients improved even more [25]. Further research in mice has shown that gemcitabine/cisplatin and anti-CD40/PD-1 treatments combination can activate DCs to significantly decrease the tumor burden [17]. Additionally, DCs combined with granulocyte-macrophage colony-stimulating factor (GM-CSF) could further improve outcomes in CCA patients. Thus far, data on DC vaccines is promising in CCA.
Some other studies have investigated other means of targeting DCs in CCA. More recently, studies have suggested that some antigens may be good candidates for making an mRNA vaccine. Research remains ongoing on the use of these mRNA vaccines [42]. Alternatively, other research has opted for DC vaccines and T-cell transfer after surgical resection, which may prevent recurrence and improve survival for at least 5 years [43]. Some case studies have reported similar outcomes with T-cell immunotherapies [44–46]. Moreover, some studies have used T cell antigens such as autophagosome peptides or exosomes from the tumor to pulse DCs, ultimately enhancing the cytotoxic response and suppressing tumor progression [47–49].
Neutrophils are involved in the development of cancer, though the data indicates both tumor-suppressing and tumor-promoting effects [50,51]. This occurs through two different phenotypes of tumor-associated neutrophils (TANs) depending on the presence of interferons or TGF-β [51]. In CCA, TANs are attracted to the tumor where they can promote cancer progression, in part, through the recruitment of regulatory T cells [52,53]. Alternatively, some TANs have been shown to mobilize in response to immune modulation with methotrexate-loaded tumor-derived microvesicles [54,55]. Research has also shown that high levels of TANs are associated with a worse prognosis [56–60]. Thus, TANs may be a good therapeutic target (Fig. 3); however, there are no studies published offering information on such treatments in CCA.
Myeloid-derived suppressor cells
Myeloid-derived suppressor cells (MDSCs) consist of two groups including differentiated neutrophils, basophils, mast cells, and eosinophils (polymorphonuclear MDSCs) or macrophages and DCs [61]. Both groups are triggered primarily by inflammatory cytokines and often act to suppress cytotoxic T lymphocytes through PD-L1, IL-10, and TGF-β [62,63]. This allows tumors to proliferate [62]. Moreover, patients with CCA have increased circulating levels of MDSCs compared to healthy individuals [64,65]. Furthermore, a mouse model of CCA suggested that this increase in MDSCs seems to promote tumor progression. This study also indicated that the levels of MDSCs can be affected by the gut microbiota [66]. Overall, MDSCs may be a beneficial target for therapies in CCA (Fig. 3). So far, a few studies have targeted MDSCs using immune checkpoint inhibition. Research suggests that the levels of MDSCs may correspond with patients’ response to immune checkpoint inhibition [67]. Moreover, research has indicated that inhibition of both tumor-associated macrophages (TAMs) and MDSCs may be combined with immune checkpoint inhibitors, which alone have been shown to reduce the tumor burden in CCA [17,68]. Data shows that MDSC metabolism is also altered in the presence of metformin, causing a decrease in their levels and loss of function [69]. Moreover, anti-vascular endothelial growth factor (VEGF) inhibitors may reduce their number as well as the number of regulatory T cells [70].
Some more recent studies have targeted MDSCs through alternative mechanisms in CCA. Research has shown that blockage of PD-L1 and tumor-associated macrophages (TAMs) was not enough to slow the progression of CCA because MDSCs can bypass the blockade [68]. Another study showed that using a blocking monoclonal antibody for GM-CSF can decrease the recruitment of MDSCs and thus, the progression of the tumor [65]. Moreover, targeting an ApoE MDSC subset can further increase the anti-tumor effects [68]. Overall, more data is needed to determine the best mechanism for targeting MDSCs in CCA.
Hepatic stellate cells (HSCs) are activated within the CCA tumor microenvironment through TGF-β signaling [71,72]. This signaling pathway and the activation of HSCs ultimately promote liver fibrosis, tumor progression, and metastasis of CCA [73,74]. HSCs produce PD-L1, which stabilizes the TGF-β receptors, further supporting the accumulation of HSCs [75]. Moreover, focal adhesion kinase protects the receptor from degradation [76]. HSCs may also promote progression through the induction of MDSCs and regulatory T cells [77–79]. Additionally, HSCs have been shown to inhibit the infiltration of lymphocytes into tumors and induce apoptosis of mononuclear cells [80,81]. Combined, the hepatic stellate cells and the pathways involved in their activity in CCA may make good therapeutic targets, though there are not any published studies thus far documenting HSCs as therapeutics in improving the prognosis in CCA.
Tumor-Infiltrating Leukocytes/Adaptive Immunity
Tumor-infiltrating lymphocyte (TILs) therapy has become a growing adaptive immunotherapy used for treating solid tumors. A heterogeneous mix of T cells, B cells, and natural killer (NK) cells, these intratumor lymphocytes are white blood cells that already recognize targets to kill tumor cells in the microenvironment (Fig. 3). TILs can target and inhibit immune checkpoints that create an environment to immunosuppressive microenvironment which allows for greater tumor growth. Especially in cholangiocarcinoma, natural killer cells and cytotoxic T cells are reduced, and T regulatory cells (Tregs) are increased [82]. There have been three key types of TILs that play a large role in potential immunotherapies for cholangiocarcinoma: TNF-α inhibitors, anti-CD40 and anti-PD1 therapy, and cetuximab.
Tumor necrosis factor alpha (TNF-α) inhibitors have been investigated for their potential use in treating cholangiocarcinoma. TNF-α plays an important role in tumor stroma to regulate inflammation via cytokine and chemokine release and has been linked to the development of tumor development [83]. Particularly in the gallbladder, TNF-α inhibitors such as infliximab, adalimumab, and golimumab are typically used for Crohn’s disease and ulcerative colitis [84]. Though the studies are limited to TNF-α in target therapy, TNF-α inhibitor use in gastrointestinal diseases has shed light on the ways TNF-α inhibitors can interact with the tumor microenvironment. In colon cancer, TNF-α has been found to upregulate CSF-dependent genes to increase macrophage expression and modify the extracellular matrix (ECM) to conform to the environment to support the progression of the tumor [85].
CD40 and PD1 provide tumor cells with the ability to evade immune regulation and thus aid in the continued proliferation of TAMs. Cholangiocarcinoma also expresses PD-1 at high levels, potentiating greater tumor growth and decreasing survival rates to 60% decreased survival [86]. Tumor cells rely on the PD-L1 pathway to evade immune processes as seen by the observed increase in PD-L1 expression in cholangiocarcinoma [87]. CD40 is a main driver for activating macrophage and T cell activity, which increases tumor proliferation [88]. It has been found that CD40 agonists in treating pancreatic adenocarcinoma lower regulatory T cell activation and alter tumor stroma [89]. The rationale behind combining a CD40 agonist with anti-PD1 is by altering the tumor environment with a CD40 agonist, there would be a greater improvement in immunosuppressive activity to increase tumor burden as opposed to single therapy alone [90]. This type of therapy activates myeloid cells to proliferate as well as recruit more lymphocytes. Thus, the combined anti-CD40 and anti-PD-L1 therapy showed the greatest efficacy against solid tumors [17].
Cetuximab is an epidermal growth factor receptor (EGFR) inhibitor medication indicated for colorectal and head and neck cancers [91]. Through intravenous infusion, cetuximab is a chimeric anti-EGFR monoclonal antibody that blocks the dimerization of EGFR, downregulating and stopping pathways that promote metastasis and tumor growth [92]. Cholangiocarcinoma treatment using cetuximab can have less efficacy when there are KRAS mutations [93]. The KRAS gene is an instrumental gene that translates into a G protein needed for EGFR and when mutated, EGFR inhibitors cannot function on the colorectal tumor [94]. Further studies demonstrate how a combination therapy of gemcitabine-cetuximab showed an increased progression-free survival rate at 6 months [95]. Similar studies evaluating the addition of cetuximab to the combination regimen of gemcitabine plus oxaliplatin for biliary tract cancer found no correlation between KRAS mutation status and treatment efficacy and no significant improvement in treatment outcomes compared to gemcitabine plus oxaliplatin alone [96].
In many cases of cholangiocarcinoma, the tumor stroma is desmoplastic and fibrotic [52]. Within the tumor stroma, there is abundant cancer associated fibroblasts (CAFs), natural killer cells (NK), tumor associated neutrophils and macrophages (TANS and TAMS), and regulatory T cells (Tregs) [52]. These cells modulate the environment via numerous cellular signaling pathways (Fig. 2), ultimately affecting the prognosis in cholangiocarcinoma. One significant factor which affects not only tumor progression but also the response to treatment in cholangiocarcinoma is the local microvessel density. Research has shown that greater densities of microvessels within the tumor is associated with greater likelihood of metastasis [97]. Alternatively, lower densities of microvessels within the tumor stroma has been associated with poorer response to treatment.
Vascular endothelial growth factor receptor (VEGFR)
VEGFRs, particularly VEGFR-2, are often overexpressed in tumors, which promotes local angiogenesis. Moreover, this can lead to immunosuppression [98,99]. One mechanism to target angiogenesis is through VEGFR pathways, which ultimately affect the tumor microenvironment and can increase the effectiveness of immune checkpoint inhibitor therapies [100]. Thus, VGFRs may be a good target for future therapeutics in CCA. Data on monoclonal antibodies targeting VEGFR in CCA have been mixed. One study combined pembrolizumab with ramucirumab, which is an antibody against VEGFR-2 in CCAs. The response rate was less than what researchers expected, with only 50% overall disease control rate and only 35% having stable disease [101]. Alternatively, a phase two study evaluating pembrolizumab and Lenvatinib, which acts as an anti-angiogenesis, kinase inhibitor had more promising results with a response rate of 10% without chemotherapy [102]. Another phase two study using Lenvatinib and Pembrolizumab has had good outcomes as well, with a 25% response rate [103]. Data has also shown promise with the combined use of gemcitabine and elpamotide, an epitope of VEGFR-2, in CCA patients, with a response rate of 18.5% [104]. Similarly, a phase two trial of bevacizumab, along with gemcitabine and oxaliplatin, showed a response rate of 41% [105]. Within this study, they also found that the overall survival rate was higher in patients who had injection site reactions than those who had no injection site reaction [104].
Another clinical trial investigated outcomes in patients on vandetanib, a VEGFR inhibitor, alone, in combination with gemcitabine, and gemcitabine and a placebo. However, this study found no difference in progression-free survival between these groups [106]. One study looked at cediranib with and without chemotherapy as well for CCA, however, there were no significant differences in this study either [107]. Regorafenib, which is a multi-kinase inhibitor affecting the VEGFR pathway, has also been tested in CCA; however, it also had no significant difference in overall survival [108,109] (Table 1). Yet another inhibitor of VEGFR, Sorafenib, was tested in phase two trials, however, it also failed to show improvement in CCA patients [110–112]. A multikinase inhibitor, which targets VEGFR2 and MET, cabozantinib was studied in a phase two clinical trial. This study revealed significant toxicity and limited activity [113].
Platelet-derived growth factor receptors
Platelet-derived growth factor D (PDGF-D) is secreted by CCA cells to generate cancer-associated fibroblasts [114]. Meanwhile, PDGF-BB is secreted by myofibroblasts, the activated cancer-associated fibroblasts acquiring α-smooth muscle actin expression, which induces tumor cells to become resistant to apoptosis signaling [115]. It can also increase secretion of VEGF, and thus increase CCA cell intravasation, and angiogenesis, and acts as a pro-lymphangiogenic factor [52]. Research has also shown that decreasing cancer-associated fibroblasts decreases the number of lymphatic vessels within the tumor and thus decreases lymph node metastases [116]. Thus, targeting PDGF may be beneficial in the treatment of CCA. However, there are no studies targeting this pathway in CCA to date.
c-MET is a tyrosine kinase receptor that binds hepatocyte growth factor (HGF). The MET-HGF pathway has a significant role in cell proliferation, apoptosis inhibition, angiogenesis, and cell motility [117]. In CCA, MET can often be overexpressed, with higher levels corresponding to later stages, larger tumors, and poorer prognosis [118–120]. Research has suggested that MET-HGF can reduce CCA sensitization to chemotherapy because of its ability to promote the epithelial to mesenchymal transition (EMT) [121]. Thus, it may be an important pathway to target for treatment. One study investigated tivantinib, which is a MET-inhibitor, along with gemcitabine with some positive results. 20% of the patients who received the drug had a partial response, while 46% had stable disease [122]. Other studies find that the side effect profile (ascites, neutropenia, rash, and anemia) of tivantinib outweighs its marginal benefit in reducing hepatocellular carcinoma [123]. One study finds that c-MET is overexpressed in 91.3% of human cholangiocarcinoma tissues. The inhibition of c-MET in these tissues with tivantinib results in a notable loss in cell viability and colony forming properties of the tissue via knockout of the JNK pathway. Therefore, this data demonstrates c-MET inhibition may be a viable alternative approach for the treatment of human cholangiocarcinoma, particularly when used in biomarker-selected patients [124].
Fibroblast growth factor receptors
Fibroblast growth factor receptors (FGFRs) are part of a signaling pathway that acts to promote cell proliferation, migration, angiogenesis, and inhibit apoptosis. Research has shown that mutations within this pathway can lead to tumorigenesis and malignant transformation [125–127]. Studies have also shown that some intrahepatic CCA have FGFR fusion mutations, particularly in younger patients [128]. Thus far, research suggests that FGFR2 fusions are the most sensitive to FGFR inhibition, while point mutations are less sensitive [129–131]. Overall, response rates for FGFR2 inhibition trials have ranged from 21% to 50%, with high disease control rates as well. However, these treatments have not worked well in patients who do not have FGFR mutations [132–136].
Numerous studies have been completed targeting FGFR mutations. A phase 2 study looked at pemigatinib in CCA, which showed that patients with FGFR2 fusions had significant improvement compared to patients with wild-type FGFR2. This medication did have some side effects, with 45% of patients having serious adverse events. Some of the adverse events included hyperphosphatemia, hypophosphatemia, alopecia, and diarrhea, among others [133]. This suggests that some inhibitors may be more effective in patients with mutations in their tumors. Another study, which investigated infigratinib in intrahepatic CCA showed a response rate of 15% and a disease control rate of 75% [137]. Further investigation into Infigratinib showed a response rate of 23% with similar side effects to pemigatinib [138]. Another trial, which consisted of CCA patients with FGFR2 fusion mutations, investigated derazantinib, a multi-kinase inhibitor, and had a response rate of 20% [135]. A later study of derazantinib found that there was stable disease in 65% of patients [138]. The full results of this study are still pending. Another drug, TAS-120, an irreversible FGFR inhibitor, was investigated in a phase one trial, which indicated that the response rate was 35% and had a control rate of 78.6% [139,140].
However, there has been some evidence that CCA tumors can acquire resistance to FGFR inhibition [141]. Later studies have shown that treatment of these tumors resistant to some earlier FGFR2 inhibitors with TAS-120 provided some clinical benefit [142]. Futibatinib, another irreversible FGFR inhibitor, showed a 76% disease control rate and 34% response rate after 6 months. However, 73% of patients in this study had a serious adverse event [143]. Meanwhile, another study investigating futibatinib for solid tumors with FGFR2 mutations has shown nearly 18% of patients have a response to therapy [144]. Moreover, when looking specifically at intrahepatic CCA, a more recent study showed a response rate of nearly 42%, with most of these patients responding at least 6 months [145]. Overall, more research is needed in targeting FGFR in CCA.
Epidermal growth factor receptor (EGFR)
EGFR is a group of receptors, such as BReast CAncer (BRCA), HER1, and HER2, involved in cell proliferation, migration, angiogenesis, and tumorigenesis pathways [146–148]. Overexpression of EGFRs is often seen in CCA tumors, often leading to poorer prognosis in these patients [146,149]. Research has shown that targeting multiple EGFR pathway proteins is more effective than targeting one receptor alone [150]. Additionally, tumors lacking a KRAS mutation and EGFR amplification may be more sensitive to EGFR-targeted therapies [146,151]. One mechanism of targeting EGFRs is through inhibitors. A phase 3 study of CCA investigated erlotinib, an anti-EGFR inhibitor, compared to gemcitabine and oxaliplatin, and found no significant difference in progression-free survival. However, when erlotinib was combined with the standard therapy, there was an increase in tumor response to treatment [152]. A sub-analysis of this data suggested that patients with wild-type KRAS and phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha (PIK3CA) improved more with erlotinib compared to the mutated forms [153].
Another strategy to target this pathway has been using monoclonal antibodies targeting EGFR, such as cetuximab, which has been shown to improve CCA in vitro [154]. A phase 2 study investigated cetuximab with gemcitabine and oxaliplatin which had promising anti-tumor effects [155]. However, other studies comparing standard treatment with and without cetuximab did not show any differences in overall survival [96,156]. Similarly, the trial of panitumumab, which inhibits EGFR, along with gemcitabine and oxaliplatin yielded no difference in survival between groups [157]. A later study confirmed similar results [158]. Another study looked to inhibit both VEGF and EGFR with bevacizumab and erlotinib, but there were no changes in overall survival [151]. Alternatively, another study looked at erlotinib in CCA compared to chemotherapy and found that there was a significant difference in progression-free survival in a subgroup analysis of intrahepatic cases [152].
Another mechanism of interest is using chimeric antigen receptor modified T (CAR-T) cells targeting EGFR. A case report on a patient with resistant CCA showed a partial response, though there were toxicities affecting the epidermis and endothelium [159] (Table 1). A 19-patient clinical trial was later conducted on patients with EGFR + CCA and gallbladder carcinomas, which resulted in 10 patients having stable disease and 1 having a complete response to CAR-T cell therapy [160] (Table 1). Another CAR-T cell trial was done targeting HER2, another type of EGFR, similarly revealed encouraging results [161] (Table 1). Another study investigated poly ADP ribose polymerase (PARP) therapy in CCA patients with BRCA mutations. This study indicated an improvement in overall survival with PARP therapy [162]. More research is needed to determine a safe and effective method to target EGFRs in CCA.
Transforming growth factor (TGF)-β
TGF-β is expressed in high levels by HSCs during chronic liver damage and acts to promote fibrogenesis [163]. HSCs also promote fibrogenesis by differentiation into myofibroblast-like cells. These processes ultimately lead to local immunosuppression and promote tumor formation [164]. TGF-β, along with VEGF and PDGF, can promote myofibroblast transformation into carcinoma-associated fibroblasts [165,166]. Moreover, regulatory T cells secrete TGF-β, which further suppresses cytotoxic T cells locally. TGF-β overexpression in CCA tumors is associated with poorer prognosis. Thus, TGF-β and its pathways would make a good target for CCA therapies. Recent literature identifies the use of TGF-β blockage may have utility in second-line targeted systemic therapy, particularly for its role in modulating cell signaling pathways including mitogen-activated protein kinase (MAPK)/ERK, AKT/mTOR, and Erb-B2 Receptor Tyrosine Kinase 2 (ERBB2) overexpression [167]. There are three ongoing clinical trials with the identification numbers NCT04708067, NCT03833661, and NCT04066491.
Colony-stimulating factor (CSF-1) is an important regulator of cell differentiation of macrophages [168]. It has been implicated in both the promotion and inhibition of tumor progression [169–171], including in CCA. Tumors with higher concentrations of CSF-1 and thus macrophages may have better prognoses [168]. However, other studies have suggested higher levels of CSF-1 in tumors may lead to a poorer prognosis [170,172]. Currently, there are no studies targeting CSF-1 in CCA. However, clinical trial is ongoing targeting CSF-1 (NCT04301778).
Cancer-Associated Fibroblasts (CAFs) are a group of cells that are found in the tumor stroma and thus the microenvironment. As the most represented cell population in the tumor microenvironment, CAFs help to build up the stroma which serves as a key histological marker for cholangiocarcinoma via desmoplasia [173]. An increase in CAF concentration is seen as a sign of poor prognosis and most likely diagnosis of cholangiocarcinoma [174]. CAFs are unique though in that they are comprised of highly heterogeneous groups of cells. This includes but is not limited to hepatic stellate cells, pericytes, adipocytes, mesenchymal stem cells, and portal fibroblasts. The diversity of morphology and cell origins is an important reason why CAFs play a great role in the progression of CCA, but also why understanding the direct mechanism of CAFs in CCA progression can be a challenge.
In cancer-promoting functions, CAFs promote angiogenesis and secreting cytokines, aiding in tumor growth. This includes the activation of the VEGF, PDGF, and fibroblast growth factor (FGF) [175]. When originating from hepatic stellate cells, CAFs increase the pro-apoptotic sensitivity of these cells and thus further worsen acute liver injury [176]. The expression of alpha-smooth muscle actin (α-SMA) and fibroblast activation protein alpha (FAP) are markers of CAFs [177]. α-SMA plays a significant role in the connective tissue remodeling of the stroma and has demonstrated a positive correlation with cholangiocarcinoma [178]. In addition, FAP is type II transmembrane cell surface proteinase most always expressed from mesenchymal stromal cells [179]. Both markers play a significant role in the regulation of the extracellular matrix proteins, which is part of the main scaffolding for the tumor microenvironment of CCA. The overproduction of FAP and α-SMA results in the stimulation of angiogenesis and tumor growth, which coincide with the poor progression of CCA.
In immunosuppressive functions, CAFs are instrumental in the regulation, recruitment, and maintenance of T regulatory cells [180]. CAFs have also been shown to diminish the function of cytotoxic T cells through the upregulation of IL-1 α/β [181]. Especially in mesenchymal stromal cells, depleting FAP expression established the role of chemokine ligand 12 (CXCL12) and alpha-programmed cell death 1 ligand 1 (α-PD-L1) to assist in tumor destruction [182].
When targeting CAFs in potential therapies, it becomes difficult to treat with conventional chemotherapy because targeting CAFs would necessitate targeting the proliferation of cells throughout the desmoplastic stroma [12]. Studies that shown that a possible way to control CAF is through targeting CAF pathways. When studying navitoclax’s efficacy in treating CAF, it was found that navitoclax’s ability to mimic BH3 helped to increase CAF apoptosis, which thus decreased tumor growth [183]. This finding supports the conclusion that CAF’s ability to increase apoptosis is dependent on the binding of BH3-only proteins. For FGFR and VEGFR inhibitors, nintedanib has been found to reduce the expression of a-SMA, which in turn suppressed CAF proliferation [184].
Immunosuppression and Immune Checkpoint inhibitors
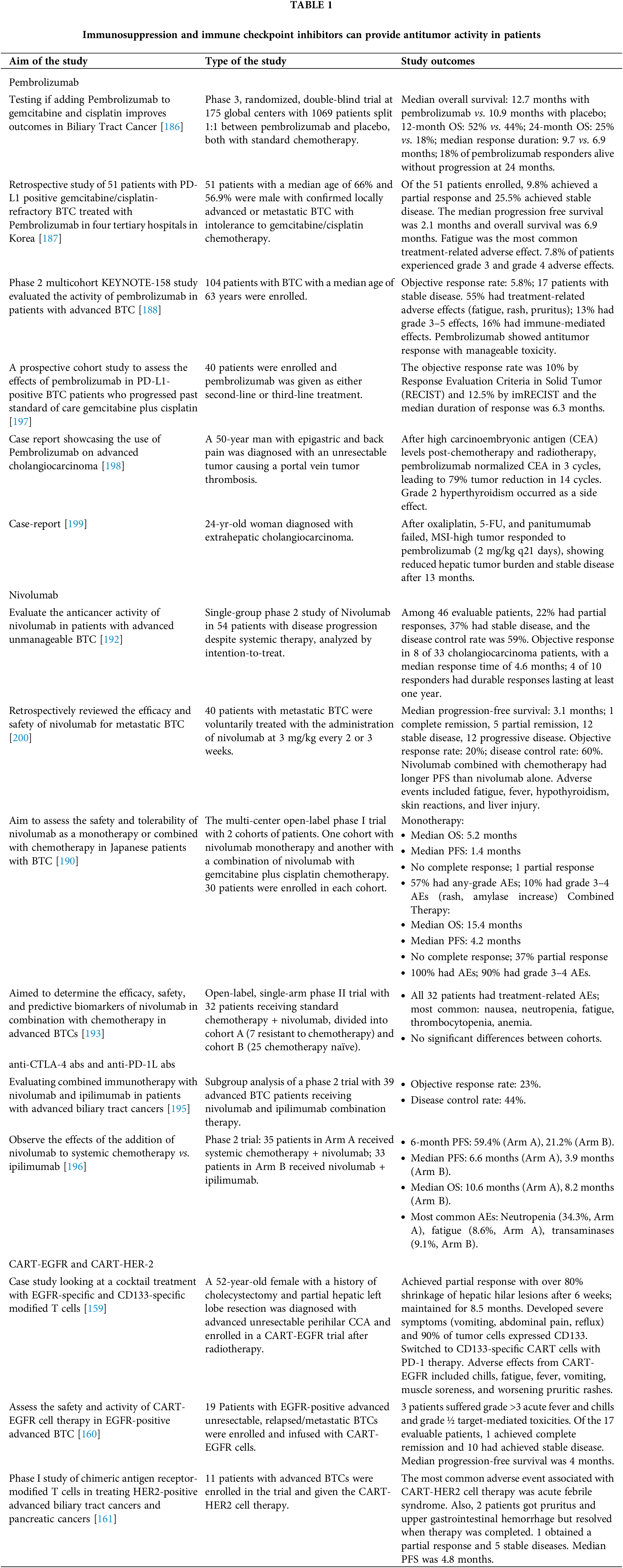
Programmed Death Receptor 1 (PD-1) is expressed on a host of immune cells including activated T and B cells and plays an essential role in regulating the magnitude of an antigen-specific immune response whether it’s against an infection or cancer. Acting as a co-inhibitory receptor, stimulation of PD-1 via the PD-1/PD-L1/PD-L2 axis negatively affects T and B cell function which prevents the formation of auto-immune, anti-tumor and anti-infectious responses [185]. Pembrolizumab is an anti-PD-1 monoclonal antibody approved by the FDA to treat patients with microsatellite instability (MSI)-high and mismatch repair deficient (dMMR) cancers and works to disrupt the PD-1/PD-L1 axis to enhance immune function against different tumors. In a randomized double-blind placebo-controlled phase 3 global trial, 1069 patients diagnosed with Biliary Tract Cancer (78% were intrahepatic and extrahepatic cholangiocarcinoma) were randomly assigned to standard-of-care chemotherapies such as gemcitabine and cisplatin or standard of care chemotherapies with the addition of Pembrolizumab [186] (Table 1). When comparing the two groups, patients in the pembrolizumab group had a 1.8-month longer median overall survival (12.7 vs. 10.9 months) and longer 12-month and 24-month overall survival rates (52% and 44% vs. 25% and 18%). The median duration of response was also longer in the Pembrolizumab group (9.7 vs. 6.9 months). These results indicate that Pembrolizumab in combination with systemic chemotherapies provides benefits that standard-of-care chemotherapy is not able to. In addition, when looking retrospectively at a group of 51 patients in Korea with Biliary Tract Cancer (82.3% Cholangiocarcinoma) that were intolerant to stand of care chemotherapies like gemcitabine and cisplatin, the addition of Pembrolizumab allowed for 9.8% to achieve partial response and 25.5% to achieve stable disease with overall survival of 6.9 months [187] (Table 1). Similarly in a phase-2 multi-cohort study, 104 patients with Biliary Tract Cancer intolerant to gemcitabine and cisplatin were given Pembrolizumab [188] (Table 1). The objective response rate was 5.8% with 16% of patients achieving a stable disease. In addition, in a prospective study, 40 patients with PD-L1 Biliary Tract Cancer who progressed past standard-of-care chemotherapies like gemcitabine and cisplatin were enrolled and given pembrolizumab as a second-line or third-line treatment. The objective response rate was 10% and the median duration was 6.3 months with a median progression-free survival of 1.5 months and overall survival of 4.3 months. These reports showcase that pembrolizumab can increase positive patient outcomes in patients intolerant to standard-of-care chemotherapy.
Similar to Pembrolizumab, Nivolumab is also an anti-PD1 monoclonal antibody used to prevent pathologic immune suppression by tumor cells [189]. In a multi-center open-label phase I trial with 2 cohorts of patients, one cohort received nivolumab monotherapy and another with the combination of nivolumab with gemcitabine plus cisplatin chemotherapy [190] (Table 1). 30 patients were enrolled in each cohort. In the monotherapy cohort, the median overall survival was 5.2 months, and the median progression-free survival was 1.4 months with no patients achieving a complete response and only one achieving a partial response. In the combined therapy cohort, median overall survival was 15.4 months and median progression-free survival was 4.2 months with no patients achieving a complete response and 37% achieving a partial response. In the monotherapy cohort, 57% reported any-grade treatment-related adverse events (AE) with 10% reporting grade 3–4 AEs. Compared to the combined therapy cohort, where all patients experienced adverse events and 90% experienced grade 3–4 adverse events. The most common adverse events in the monotherapy group were decreased appetite, malaise, pruritus, and rash with the grade 3–4 AEs being maculopapular rash and amylase increase. Compared to the combined therapy cohort, the most common treatment-related AEs decreased neutrophils, platelets, and white blood cell count. In addition, in a retrospective study, 30 patients with metastatic biliary tract cancer (BTC) were voluntarily treated with nivolumab to assess its efficacy and safety [191]. Median progression-free survival was 3.1 months with one patient achieving complete remission, 5 achieving partial remission, 12 were stable disease and 12 had progressive disease. The Objective Response Rate was 20% and the Disease Control Rate was 60%. Patients who combined nivolumab with chemotherapy had longer progression-free survival than those with Nivolumab. The only adverse events of nivolumab were fatigue, fever, hypothyroidism, skin reaction, and liver injury. These studies indicate that a combination therapy with Nivolumab could be an effective treatment. In addition, in a single-group multicenter phase 2 study of Nivolumab in 54 patients with metastatic BTC and intolerant to standard-of-care chemotherapy, 46 patients were examined for tumor response where 22% had a partial response, and 37% achieved stable disease [192] (Table 1). Objective response rate was observed in 8 of the 33 cholangiocarcinoma patients and the median time to respond was 4.6 months. Durable objective response lasted at least one year in 4 of the 10 years. This indicates that Nivolumab could be a useful treatment for patients resistant to standard-of-care chemotherapies. However, in an open-label, single-arm, phase II trial where a standard of care chemotherapy and nivolumab combination therapy was administered to 32 patients [193] (Table 1) with Cohort A resistant to standard-of-care chemotherapy (7 patients) and Cohort B was chemotherapy naïve (25 patients). All 32 patients experienced at least one treatment-related adverse event with the most frequent being nausea, neutropenia, fatigue, thrombocytopenia, and anemia. The incidence of grade 3 or higher thrombocytopenia was much higher in this study than in other studies with just chemotherapy. 15 patients among the 27 response-evaluable patients in both cohorts achieved a confirmed objective response which included 4 complete responses and 10 partial responses. Disease control was achieved in 25 patients including 10 patients who had stable disease. Median progression-free survival in the study was 6.1 months and the proportion of patients that were progression-free at 6 months and 12 months were 51.9% and 18.5%, respectively. No statistical difference between both cohorts. Median overall survival was 8.5 months and the 12-month overall survival (OS) rate, and 18-month OS rate were 3.3% and 24.7% respectively with no statistical difference among the 2 cohorts. These results could indicate that Nivolumab may not provide any additional benefit to patients resistant to stand-of-care chemotherapies.
Anti-cytotoxic T-lymphocyte associated protein 4 (CTLA-4) and anti-programmed death ligand 1 (PD-L1) antibodies
Ipilimumab is a CTLA-4 monoclonal antibody. Similarly, to PD-1, CTLA-4 is a co-inhibitory receptor on many T and B cells which gets upregulated during T-cell activation and competes with CD28 for CD80 and CD86 binding from antigen presenting cells (APCs) which prevents further T cell activation [194]. Commonly in cholangiocarcinoma, Ipilimumab is administered with a PD-1 antibody immunotherapy. In a subgroup analysis of a phase 2 nonrandomized clinical trial of 39 patients with advanced BTC who received a combination immunotherapy of nivolumab and ipilimumab, the objective response rate was 23% with a disease control rate of 44% [195] (Table 1). All responders received prior chemotherapy, and none had an unstable MSI status. Median progression-free survival was 2.9 months and overall survival was 5.7 months. Immune-related toxic events were reported in 49% of patients with 15% experiencing grade 3 or 4 events. This could indicate that a dual immuno-therapy with chemotherapy could be beneficial for patients with cholangiocarcinoma, but a study is necessary to see if it would be more beneficial than a single immuno-therapy with chemotherapy combination therapy. In addition, in a phase 2 trial for patients with advanced BTC, 35 patients in Arm A received systemic chemotherapy plus nivolumab while 33 patients in Arm B received nivolumab plus ipilimumab [196]. 6-month progression-free survival rates were 59.4% in Arm A and 21.2% in Arm B. Median progression-free survival (PFS) and overall survival were 6.6 and 10.6 months in Arm A and 3.9 and 8.2 months in Arm B. The most common hematologic adverse event was neutropenia in 34.3% in Arm A and nonhematologic adverse event was fatigue (8.6%) in Arm A and transaminases (9.1%) in Arm B. This study indicates that combination therapy between chemotherapy and immunotherapy is more effective than dual monotherapy treatment without chemotherapy.
Chimeric antigen receptor-modified T (CAR-T) cells are genetically modified T Cells that are engineered to target specific surface markers on target cells like tumor cells without the need for major histocompatibility complex (MHC)-I presentation by the said tumor cell. One such marker is EGFR, a receptor tyrosine kinase, which is commonly expressed in BTCs and nearly all intrahepatic cholangiocarcinoma and 50% of extrahepatic cholangiocarcinoma [160]. 19 Patients with EGFR-positive advanced unresectable, relapsed/metastatic BTCs were enrolled and infused with CAR-T/EGFR cells. 3 patients suffered grade >3 acute fever and chills and grade ½ target-mediated toxicities [160]. Of the 17 evaluable patients, 1 achieved complete remission and 10 had achieved stable disease. Median progression-free survival was 4 months. These results indicate that CAR T-cells could be another potential benefit to patients resistant to chemotherapies when the tumor is expressing the target marker. In a case study, a 52-year-old female with a history of cholecystectomy and partial resection of the hepatic left lobe followed by a diagnosis with advanced unresectable perihilar CCA, following radiotherapy, the patient was enrolled in CAR-T/EGFR trial [159]. She achieved a partial response in her first assessment 6 weeks later and illustrated more than 80% shrinkage of metastatic lesions in the hepatic hilar region. Partial response status was maintained for 8.5 months until she developed frequent unmanageable, vomiting, upper abdominal dull pain, and gastric acid reflux. After, 90% of tumor cells began to express CD133 protein, and the patient was switched to CD133-specific CAR-T cells with PD-1 immunotherapy. Direct adverse effects from CAR-T/EGFR were chills, fatigue, fever, vomiting, and muscle soreness. In addition, gradually worsening scattered tiny rashes appeared and became apparent and pruritic. This case study indicates that CAR-T cells are an effective treatment in chemotherapy-resistant patients and that combination with immunotherapy could enhance immune function against the tumor. In another study, 11 patients with advanced BTCs were enrolled in a trial and given CAR-T/HER2 cell therapy [161] (Table 1). The most common adverse event associated with CART-HER2 cell therapy was acute febrile syndrome. Also, 2 patients got pruritus and upper gastrointestinal hemorrhage but resolved when therapy was completed. 1 obtained partial response and 5 achieved stable disease. The median PFS was 4.8 months. These results indicate that a CAR T-Cell against HER2 could potentially benefit patients.
Recent research has shed light on the tumor microenvironment, offering new possibilities for targeted therapies. Promising approaches include cell-based vaccines, targeting specific cells like tumor-associated neutrophils and hepatic stellate cells. Cancer-associated fibroblasts are also involved in disease progression. Selective therapies aimed at the tumor microenvironment have shown the potential to induce tumor cell death and slow cancer growth. Immunosuppressive therapies and immune checkpoint inhibitors are showing promise, especially for patients who can’t tolerate chemotherapy, offering hope for improved outcomes. Given the significant role that cancer stem cells play in cholangiocarcinoma progression and resistance, targeting these cells alongside their microenvironmental niches presents a promising direction for future therapeutic strategies [201–203]. Future research should focus on refining and optimizing the efficacy of vaccines like MUC1 and WT1, particularly by exploring their use in combination with other immunotherapeutic strategies such as immune checkpoint inhibitors. Additionally, further investigation into the role of the tumor microenvironment, including tumor-associated neutrophils and cancer-associated fibroblasts, could lead to the development of more precise and effective targeted therapies that not only enhance immune response but also disrupt the supportive networks that tumors rely on for growth and survival [204]. While more research is necessary, the tumor microenvironment remains a new and exciting realm of research that may be of benefit to patients suffering from cholangiocarcinoma.
Acknowledgement: None.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: The authors confirm their contribution to the paper as follows: study conception and design: Armand N. Yazdani, Michaela Pletsch, Abraham Chorbajian, David Zitser, and Vikrant Rai; draft manuscript preparation: Armand N. Yazdani, Michaela Pletsch, Abraham Chorbajian, and David Zitser; review and editing: Vikrant Rai; visualization: Vikrant Rai; supervision: Vikrant Rai. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: As the corresponding author, I declare that this manuscript is original; that the article does not infringe upon any copyright or other proprietary rights of any third party; and that neither the text nor the figures have been reported or published previously. All the authors have no conflict of interest and have read the journal’s authorship statement.
References
1. Gad MM, Saad AM, Faisaluddin M, Gaman MA, Ruhban IA, Jazieh KA, et al. Epidemiology of cholangiocarcinoma; United States incidence and mortality trends. Clin Res Hepatol Gastroenterol. 2020;44(6):885–93. doi:10.1016/j.clinre.2020.03.024. [Google Scholar] [PubMed] [CrossRef]
2. Qurashi M, Vithayathil M, Khan SA. Epidemiology of cholangiocarcinoma. Eur J Surg Oncol. 2023;107064. doi:10.1016/j.ejso.2023.107064. [Google Scholar] [PubMed] [CrossRef]
3. Banales JM, Marin JJG, Lamarca A, Rodrigues PM, Khan SA, Roberts LR, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol. 2020;17(9):557–88. doi:10.1038/s41575-020-0310-z. [Google Scholar] [PubMed] [CrossRef]
4. Borakati A, Froghi F, Bhogal RH, Mavroeidis VK. Liver transplantation in the management of cholangiocarcinoma: evolution and contemporary advances. World J Gastroenterol. 2023;29(13):1969–81. doi:10.3748/wjg.v29.i13.1969. [Google Scholar] [PubMed] [CrossRef]
5. Zori AG, Yang D, Draganov PV, Cabrera R. Advances in the management of cholangiocarcinoma. World J Hepatol. 2021;13(9):1003–18. doi:10.4254/wjh.v13.i9.1003. [Google Scholar] [PubMed] [CrossRef]
6. Esmail A, Badheeb M, Alnahar B, Almiqlash B, Sakr Y, Khasawneh B, et al. Cholangiocarcinoma: the current status of surgical options including liver transplantation. Cancers. 2024;16(11):1946. doi:10.3390/cancers16111946. [Google Scholar] [PubMed] [CrossRef]
7. Ohaegbulam KC, Koethe Y, Fung A, Mayo SC, Grossberg AJ, Chen EY, et al. The multidisciplinary management of cholangiocarcinoma. Cancer. 2023;129(2):184–214. doi:10.1002/cncr.v129.2. [Google Scholar] [CrossRef]
8. Rodrigues PM, Olaizola P, Paiva NA, Olaizola I, Agirre-Lizaso A, Landa A, et al. Pathogenesis of cholangiocarcinoma. Annu Rev Pathol. 2021;16:433–63. doi:10.1146/pathmechdis.2021.16.issue-1. [Google Scholar] [CrossRef]
9. Rai V, Boosani CS, Agrawal DK. The promising role of epigenetic mediators and microRNAs in the early diagnosis of cholangiocarcinoma. World Acad Sci J. 2019;1(4):165–76. [Google Scholar]
10. Labib PL, Goodchild G, Pereira SP. Molecular pathogenesis of cholangiocarcinoma. BMC Cancer. 2019;19(1):185. doi:10.1186/s12885-019-5391-0. [Google Scholar] [PubMed] [CrossRef]
11. Cadamuro M, Fabris L, Zhang X, Strazzabosco M. Tumor microenvironment and immunology of cholangiocarcinoma. Hepatoma Res. 2022;8:11. doi:10.20517/2394-5079.2021.140. [Google Scholar] [PubMed] [CrossRef]
12. Montori M, Scorzoni C, Argenziano ME, Balducci D, De Blasio F, Martini F, et al. Cancer-associated fibroblasts in cholangiocarcinoma: current knowledge and possible implications for therapy. J Clin Med. 2022;11(21):6498. doi:10.3390/jcm11216498. [Google Scholar] [PubMed] [CrossRef]
13. Marshall JS, Warrington R, Watson W, Kim HL. An introduction to immunology and immunopathology. Allergy Asthma Clin Immunol. 2018;14(Suppl 2):49. [Google Scholar] [PubMed]
14. Yu X, Zhu L, Wang T, Chen J. Immune microenvironment of cholangiocarcinoma: biological concepts and treatment strategies. Front Immunol. 2023;14:1037945. doi:10.3389/fimmu.2023.1037945. [Google Scholar] [PubMed] [CrossRef]
15. Lu X, Green BL, Xie C, Liu C, Chen X. Preclinical and clinical studies of immunotherapy for the treatment of cholangiocarcinoma. JHEP Rep. 2023;5(7):100723. doi:10.1016/j.jhepr.2023.100723. [Google Scholar] [PubMed] [CrossRef]
16. Hato L, Vizcay A, Eguren I, Pérez-Gracia JL, Rodríguez J, Gállego Pérez-Larraya J, et al. Dendritic cells in cancer immunology and immunotherapy. Cancers. 2024;16(5):981. doi:10.3390/cancers16050981. [Google Scholar] [PubMed] [CrossRef]
17. Diggs LP, Ruf B, Ma C, Heinrich B, Cui L, Zhang Q, et al. CD40-mediated immune cell activation enhances response to anti-PD-1 in murine intrahepatic cholangiocarcinoma. J Hepatol. 2021;74(5):1145–54. doi:10.1016/j.jhep.2020.11.037. [Google Scholar] [PubMed] [CrossRef]
18. Katopodi T, Petanidis S, Charalampidis C, Chatziprodromidou I, Eskitzis P, Tsavlis D, et al. Tumor-infiltrating dendritic cells: decisive roles in cancer immunosurveillance, immunoediting, and tumor T cell tolerance. Cells. 2022;11(20):3183. doi:10.3390/cells11203183. [Google Scholar] [PubMed] [CrossRef]
19. Sun BY, Wang ZT, Chen KZ, Song Y, Wu JF, Zhang D, et al. Mobilization and activation of tumor-infiltrating dendritic cells inhibits lymph node metastasis in intrahepatic cholangiocarcinoma. Cell Death Discov. 2024;10(1):304. doi:10.1038/s41420-024-02079-z. [Google Scholar] [PubMed] [CrossRef]
20. Veglia F, Gabrilovich DI. Dendritic cells in cancer: the role revisited. Curr Opin Immunol. 2017;45:43–51. doi:10.1016/j.coi.2017.01.002. [Google Scholar] [PubMed] [CrossRef]
21. Xiao Z, Wang R, Wang X, Yang H, Dong J, He X, et al. Impaired function of dendritic cells within the tumor microenvironment. Front Immunol. 2023;14:1213629. doi:10.3389/fimmu.2023.1213629. [Google Scholar] [PubMed] [CrossRef]
22. Cheng JT, Deng YN, Yi HM, Wang GY, Fu BS, Chen WJ, et al. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis. 2016;5(2):e198. doi:10.1038/oncsis.2016.7. [Google Scholar] [PubMed] [CrossRef]
23. Martin-Sierra C, Martins R, Laranjeira P, Abrantes AM, Oliveira RC, Tralhao JG, et al. Functional impairment of circulating FcεRI+ monocytes and myeloid dendritic cells in hepatocellular carcinoma and cholangiocarcinoma patients. Cytometry B Clin Cytom. 2019;96(6):490–5. doi:10.1002/cyto.v96.6. [Google Scholar] [CrossRef]
24. Paillet J, Kroemer G, Pol JG. Immune contexture of cholangiocarcinoma. Curr Opin Gastroenterol. 2020;36(2):70–6. doi:10.1097/MOG.0000000000000613. [Google Scholar] [PubMed] [CrossRef]
25. Jiraviriyakul A, Songjang W, Kaewthet P, Tanawatkitichai P, Bayan P, Pongcharoen S. Honokiol-enhanced cytotoxic T lymphocyte activity against cholangiocarcinoma cells mediated by dendritic cells pulsed with damage-associated molecular patterns. World J Gastroenterol. 2019;25(29):3941–55. doi:10.3748/wjg.v25.i29.3941. [Google Scholar] [PubMed] [CrossRef]
26. Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014;2014:149185. [Google Scholar] [PubMed]
27. Marks EI, Yee NS. Immunotherapeutic approaches in biliary tract carcinoma: current status and emerging strategies. World J Gastrointest Oncol. 2015;7(11):338–46. doi:10.4251/wjgo.v7.i11.338. [Google Scholar] [PubMed] [CrossRef]
28. Mall AS, Tyler MG, Ho SB, Krige JE, Kahn D, Spearman W, et al. The expression of MUC mucin in cholangiocarcinoma. Pathol Res Pract. 2010;206(12):805–9. doi:10.1016/j.prp.2010.08.004. [Google Scholar] [PubMed] [CrossRef]
29. Takahashi R, Yoshitomi M, Yutani S, Shirahama T, Noguchi M, Yamada A, et al. Current status of immunotherapy for the treatment of biliary tract cancer. Hum Vaccin Immunother. 2013;9(5):1069–72. doi:10.4161/hv.23844. [Google Scholar] [PubMed] [CrossRef]
30. Kaida M, Morita-Hoshi Y, Soeda A, Wakeda T, Yamaki Y, Kojima Y, et al. Phase 1 trial of Wilms tumor 1 (WT1) peptide vaccine and gemcitabine combination therapy in patients with advanced pancreatic or biliary tract cancer. J Immunother. 2011;34(1):92–9. doi:10.1097/CJI.0b013e3181fb65b9. [Google Scholar] [PubMed] [CrossRef]
31. Abd-Aziz N, Poh CL. Development of peptide-based vaccines for cancer. J Oncol. 2022;2022(21):9749363. doi:10.1155/2022/9749363. [Google Scholar] [PubMed] [CrossRef]
32. Soares MM, Mehta V, Finn OJ. Three different vaccines based on the 140-amino acid MUC1 peptide with seven tandemly repeated tumor-specific epitopes elicit distinct immune effector mechanisms in wild-type versus MUC1-transgenic mice with different potential for tumor rejection. J Immunol. 2001;166(11):6555–63. doi:10.4049/jimmunol.166.11.6555. [Google Scholar] [PubMed] [CrossRef]
33. Maeda T, Taguchi K, Aishima S, Shimada M, Hintz D, Larusso N, et al. Clinicopathological correlates of aspartyl (asparaginyl) beta-hydroxylase over-expression in cholangiocarcinoma. Cancer Detect Prev. 2004;28(5):313–8. doi:10.1016/j.cdp.2004.06.001. [Google Scholar] [PubMed] [CrossRef]
34. Noda T, Shimoda M, Ortiz V, Sirica AE, Wands JR. Immunization with aspartate-beta-hydroxylase-loaded dendritic cells produces antitumor effects in a rat model of intrahepatic cholangiocarcinoma. Hepatology. 2012;55(1):86–97. doi:10.1002/hep.24629. [Google Scholar] [PubMed] [CrossRef]
35. Kanwal M, Smahel M, Olsen M, Smahelova J, Tachezy R. Aspartate beta-hydroxylase as a target for cancer therapy. J Exp Clin Cancer Res. 2020;39(1):163. doi:10.1186/s13046-020-01669-w. [Google Scholar] [PubMed] [CrossRef]
36. Nagaoka K, Bai X, Liu D, Cao K, Mulla J, Ji C, et al. Elevated 2-oxoglutarate antagonizes DNA damage responses in cholangiocarcinoma chemotherapy through regulating aspartate beta-hydroxylase. Cancer Lett. 2024;580:216493. doi:10.1016/j.canlet.2023.216493. [Google Scholar] [PubMed] [CrossRef]
37. Gonul Geyik O, Anichini G, Ulukaya E, Marra F, Raggi C. DNA damage response inhibitors in cholangiocarcinoma: current progress and perspectives. Cells. 2022;11(9):1463. doi:10.3390/cells11091463. [Google Scholar] [PubMed] [CrossRef]
38. Maeda T, Sepe P, Lahousse S, Tamaki S, Enjoji M, Wands JR, et al. Antisense oligodeoxynucleotides directed against aspartyl (asparaginyl) β-hydroxylase suppress migration of cholangiocarcinoma cells. J Hepatol. 2003;38(5):615–22. doi:10.1016/S0168-8278(03)00052-7. [Google Scholar] [PubMed] [CrossRef]
39. Thepmalee C, Panya A, Junking M, Chieochansin T, Yenchitsomanus PT. Inhibition of IL-10 and TGF-β receptors on dendritic cells enhances activation of effector T-cells to kill cholangiocarcinoma cells. Hum Vaccin Immunother. 2018;14(6):1423–31. doi:10.1080/21645515.2018.1431598. [Google Scholar] [PubMed] [CrossRef]
40. Panya A, Thepmalee C, Sawasdee N, Sujjitjoon J, Phanthaphol N, Junking M, et al. Cytotoxic activity of effector T cells against cholangiocarcinoma is enhanced by self-differentiated monocyte-derived dendritic cells. Cancer Immunol Immunother. 2018;67(10):1579–88. doi:10.1007/s00262-018-2212-2. [Google Scholar] [PubMed] [CrossRef]
41. Kobayashi M, Sakabe T, Abe H, Tanii M, Takahashi H, Chiba A, et al. Dendritic cell-based immunotherapy targeting synthesized peptides for advanced biliary tract cancer. J Gastrointest Surg. 2013;17(9):1609–17. doi:10.1007/s11605-013-2286-2. [Google Scholar] [PubMed] [CrossRef]
42. Huang X, Tang T, Zhang G, Liang T. Identification of tumor antigens and immune subtypes of cholangiocarcinoma for mRNA vaccine development. Mol Cancer. 2021;20(1):50. doi:10.1186/s12943-021-01342-6. [Google Scholar] [PubMed] [CrossRef]
43. Shimizu K, Kotera Y, Aruga A, Takeshita N, Takasaki K, Yamamoto M. Clinical utilization of postoperative dendritic cell vaccine plus activated T-cell transfer in patients with intrahepatic cholangiocarcinoma. J Hepatobiliary Pancreat Sci. 2012;19(2):171–8. doi:10.1007/s00534-011-0437-y. [Google Scholar] [PubMed] [CrossRef]
44. Higuchi R, Yamamoto M, Hatori T, Shimizu K, Imai K, Takasaki K. Intrahepatic cholangiocarcinoma with lymph node metastasis successfully treated by immunotherapy with CD3-activated T cells and dendritic cells after surgery: report of a case. Surg Today. 2006;36(6):559–62. doi:10.1007/s00595-006-3201-1. [Google Scholar] [PubMed] [CrossRef]
45. Khan JA, Yaqin S. Successful immunological treatment of gallbladder cancer in India—case report. J Zhejiang Univ Sci B. 2006;7(9):719–24. doi:10.1631/jzus.2006.B0719. [Google Scholar] [PubMed] [CrossRef]
46. Kan N, Yoshikawa K, Matsushita N, Fujii T. The case of a patient with peritoneal metastasis from cholangiocarcinoma who responded to adoptive immunotherapy and cetuximab. Gan to Kagaku Ryoho Cancer Chemother. 2013;40(12):1759–61. [Google Scholar]
47. Rao Q, Zuo B, Lu Z, Gao X, You A, Wu C, et al. Tumor-derived exosomes elicit tumor suppression in murine hepatocellular carcinoma models and humans in vitro. Hepatology. 2016;64(2):456–72. doi:10.1002/hep.28549. [Google Scholar] [PubMed] [CrossRef]
48. Su S, Zhou H, Xue M, Liu JY, Ding L, Cao M, et al. Anti-tumor efficacy of a hepatocellular carcinoma vaccine based on dendritic cells combined with tumor-derived autophagosomes in murine models. Asian Pac J Cancer Prev. 2013;14(5):3109–16. doi:10.7314/APJCP.2013.14.5.3109. [Google Scholar] [PubMed] [CrossRef]
49. Homma S, Toda G, Gong J, Kufe D, Ohno T. Preventive antitumor activity against hepatocellular carcinoma (HCC) induced by immunization with fusions of dendritic cells and HCC cells in mice. J Gastroenterol. 2001;36(11):764–71. doi:10.1007/s005350170019. [Google Scholar] [PubMed] [CrossRef]
50. Shaul ME, Fridlender ZG. Tumour-associated neutrophils in patients with cancer. Nat Rev Clin Oncol. 2019;16(10):601–20. doi:10.1038/s41571-019-0222-4. [Google Scholar] [PubMed] [CrossRef]
51. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell. 2009;16(3):183–94. doi:10.1016/j.ccr.2009.06.017. [Google Scholar] [PubMed] [CrossRef]
52. Fabris L, Perugorria MJ, Mertens J, Bjorkstrom NK, Cramer T, Lleo A, et al. The tumour microenvironment and immune milieu of cholangiocarcinoma. Liver Int. 2019;39 Suppl 1:63–78. [Google Scholar] [PubMed]
53. Zhou SL, Dai Z, Zhou ZJ, Chen Q, Wang Z, Xiao YS, et al. CXCL5 contributes to tumor metastasis and recurrence of intrahepatic cholangiocarcinoma by recruiting infiltrative intratumoral neutrophils. Carcinogenesis. 2014;35(3):597–605. doi:10.1093/carcin/bgt397. [Google Scholar] [PubMed] [CrossRef]
54. Cao H, Huang T, Dai M, Kong X, Liu H, Zheng Z, et al. Tumor microenvironment and its implications for antitumor immunity in cholangiocarcinoma: future perspectives for novel therapies. Int J Biol Sci. 2022;18(14):5369–90. doi:10.7150/ijbs.73949. [Google Scholar] [PubMed] [CrossRef]
55. Gao Y, Zhang H, Zhou N, Xu P, Wang J, Gao Y, et al. Methotrexate-loaded tumour-cell-derived microvesicles can relieve biliary obstruction in patients with extrahepatic cholangiocarcinoma. Nat Biomed Eng. 2020;4(7):743–53. doi:10.1038/s41551-020-0583-0. [Google Scholar] [PubMed] [CrossRef]
56. Fabris L, Sato K, Alpini G, Strazzabosco M. The tumor microenvironment in cholangiocarcinoma progression. Hepatology. 2021;73:75–85. doi:10.1002/hep.31410. [Google Scholar] [PubMed] [CrossRef]
57. Mao ZY, Zhu GQ, Xiong M, Ren L, Bai L. Prognostic value of neutrophil distribution in cholangiocarcinoma. World J Gastroenterol. 2015;21(16):4961–8. doi:10.3748/wjg.v21.i16.4961. [Google Scholar] [PubMed] [CrossRef]
58. Okabe H, Beppu T, Ueda M, Hayashi H, Ishiko T, Masuda T, et al. Identification of CXCL5/ENA-78 as a factor involved in the interaction between cholangiocarcinoma cells and cancer-associated fibroblasts. Int J Cancer. 2012;131(10):2234–41. doi:10.1002/ijc.v131.10. [Google Scholar] [CrossRef]
59. Gentilini A, Pastore M, Marra F, Raggi C. The role of stroma in cholangiocarcinoma: the intriguing interplay between fibroblastic component, immune cell subsets and tumor epithelium. Int J Mol Sci. 2018;19(10):2885. doi:10.3390/ijms19102885. [Google Scholar] [PubMed] [CrossRef]
60. Rimassa L, Personeni N, Aghemo A, Lleo A. The immune milieu of cholangiocarcinoma: from molecular pathogenesis to precision medicine. J Autoimmun. 2019;100:17–26. doi:10.1016/j.jaut.2019.03.007. [Google Scholar] [PubMed] [CrossRef]
61. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19(2):108–19. doi:10.1038/s41590-017-0022-x. [Google Scholar] [PubMed] [CrossRef]
62. Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, et al. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell. 2016;30(4):533–47. doi:10.1016/j.ccell.2016.09.003. [Google Scholar] [PubMed] [CrossRef]
63. Ma C, Zhang Q, Greten TF. MDSCs in liver cancer: a critical tumor-promoting player and a potential therapeutic target. Cell Immunol. 2021;361:104295. doi:10.1016/j.cellimm.2021.104295. [Google Scholar] [PubMed] [CrossRef]
64. Xu XD, Hu J, Wang M, Peng F, Tian R, Guo XJ, et al. Circulating myeloid-derived suppressor cells in patients with pancreatic cancer. Hepatobiliary Pancreat Dis Int. 2016;15(1):99–105. doi:10.1016/S1499-3872(15)60413-1. [Google Scholar] [PubMed] [CrossRef]
65. Ruffolo LI, Jackson KM, Kuhlers PC, Dale BS, Figueroa Guilliani NM, Ullman NA, et al. GM-CSF drives myelopoiesis, recruitment and polarisation of tumour-associated macrophages in cholangiocarcinoma and systemic blockade facilitates antitumour immunity. Gut. 2022;71(7):1386–98. doi:10.1136/gutjnl-2021-324109. [Google Scholar] [PubMed] [CrossRef]
66. Zhang Q, Ma C, Duan Y, Heinrich B, Rosato U, Diggs LP, et al. Gut microbiome directs hepatocytes to recruit MDSCs and promote cholangiocarcinoma. Cancer Discov. 2021;11(5):1248–67. doi:10.1158/2159-8290.CD-20-0304. [Google Scholar] [PubMed] [CrossRef]
67. Yuan ZG, Zeng TM, Tao CJ. Current and emerging immunotherapeutic approaches for biliary tract cancers. Hepatobiliary Pancreat Dis Int. 2022;21(5):440–9. doi:10.1016/j.hbpd.2022.08.015. [Google Scholar] [PubMed] [CrossRef]
68. Loeuillard E, Yang J, Buckarma E, Wang J, Liu Y, Conboy C, et al. Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments PD-1 blockade in cholangiocarcinoma. J Clin Invest. 2020;130(10):5380–96. doi:10.1172/JCI137110. [Google Scholar] [PubMed] [CrossRef]
69. Uehara T, Eikawa S, Nishida M, Kunisada Y, Yoshida A, Fujiwara T, et al. Metformin induces CD11b+-cell-mediated growth inhibition of an osteosarcoma: implications for metabolic reprogramming of myeloid cells and anti-tumor effects. Int Immunol. 2019;31(4):187–98. doi:10.1093/intimm/dxy079. [Google Scholar] [PubMed] [CrossRef]
70. Roland CL, Lynn KD, Toombs JE, Dineen SP, Udugamasooriya DG, Brekken RA. Cytokine levels correlate with immune cell infiltration after anti-VEGF therapy in preclinical mouse models of breast cancer. PLoS One. 2009;4(11):e7669. doi:10.1371/journal.pone.0007669. [Google Scholar] [PubMed] [CrossRef]
71. Shiraha H, Iwamuro M, Okada H. Hepatic stellate cells in liver tumor. Adv Exp Med Biol. 2020;1234:43–56. doi:10.1007/978-3-030-37184-5. [Google Scholar] [CrossRef]
72. Shi Y, Massague J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113(6):685–700. doi:10.1016/S0092-8674(03)00432-X. [Google Scholar] [PubMed] [CrossRef]
73. Liu C, Billadeau DD, Abdelhakim H, Leof E, Kaibuchi K, Bernabeu C, et al. IQGAP1 suppresses TβRII-mediated myofibroblastic activation and metastatic growth in liver. J Clin Invest. 2013;123(3):1138–56. doi:10.1172/JCI63836. [Google Scholar] [PubMed] [CrossRef]
74. Tu K, Li J, Verma VK, Liu C, Billadeau DD, Lamprecht G, et al. Vasodilator-stimulated phosphoprotein promotes activation of hepatic stellate cells by regulating Rab11-dependent plasma membrane targeting of transforming growth factor beta receptors. Hepatology. 2015;61(1):361–74. doi:10.1002/hep.27251. [Google Scholar] [PubMed] [CrossRef]
75. Sun L, Wang Y, Wang X, Navarro-Corcuera A, Ilyas S, Jalan-Sakrikar N, et al. PD-L1 promotes myofibroblastic activation of hepatic stellate cells by distinct mechanisms selective for TGF-β receptor I versus II. Cell Rep. 2022;38(6):110349. doi:10.1016/j.celrep.2022.110349. [Google Scholar] [PubMed] [CrossRef]
76. Chen Y, Li Q, Tu K, Wang Y, Wang X, Liu D, et al. Focal adhesion kinase promotes hepatic stellate cell activation by regulating plasma membrane localization of TGFβ receptor 2. Hepatol Commun. 2020;4(2):268–83. doi:10.1002/hep4.1452. [Google Scholar] [PubMed] [CrossRef]
77. Neaud V, Faouzi S, Guirouilh J, Le Bail B, Balabaud C, Bioulac-Sage P, et al. Human hepatic myofibroblasts increase invasiveness of hepatocellular carcinoma cells: evidence for a role of hepatocyte growth factor. Hepatology. 1997;26(6):1458–66. doi:10.1002/(ISSN)1527-3350. [Google Scholar] [CrossRef]
78. Hochst B, Schildberg FA, Sauerborn P, Gabel YA, Gevensleben H, Goltz D, et al. Activated human hepatic stellate cells induce myeloid derived suppressor cells from peripheral blood monocytes in a CD44-dependent fashion. J Hepatol. 2013;59(3):528–35. doi:10.1016/j.jhep.2013.04.033. [Google Scholar] [PubMed] [CrossRef]
79. Dunham RM, Thapa M, Velazquez VM, Elrod EJ, Denning TL, Pulendran B, et al. Hepatic stellate cells preferentially induce Foxp3+ regulatory T cells by production of retinoic acid. J Immunol. 2013;190(5):2009–16. doi:10.4049/jimmunol.1201937. [Google Scholar] [PubMed] [CrossRef]
80. Zhao W, Zhang L, Yin Z, Su W, Ren G, Zhou C, et al. Activated hepatic stellate cells promote hepatocellular carcinoma development in immunocompetent mice. Int J Cancer. 2011;129(11):2651–61. doi:10.1002/ijc.25920. [Google Scholar] [PubMed] [CrossRef]
81. Zhao W, Su W, Kuang P, Zhang L, Liu J, Yin Z, et al. The role of hepatic stellate cells in the regulation of T-cell function and the promotion of hepatocellular carcinoma. Int J Oncol. 2012;41(2):457–64. doi:10.3892/ijo.2012.1497. [Google Scholar] [PubMed] [CrossRef]
82. Zhou G, Sprengers D, Mancham S, Erkens R, Boor PPC, van Beek AA, et al. Reduction of immunosuppressive tumor microenvironment in cholangiocarcinoma by ex vivo targeting immune checkpoint molecules. J Hepatol. 2019;71(4):753–62. doi:10.1016/j.jhep.2019.05.026. [Google Scholar] [PubMed] [CrossRef]
83. Shiode Y, Kodama T, Shigeno S, Murai K, Tanaka S, Newberg JY, et al. TNF receptor-related factor 3 inactivation promotes the development of intrahepatic cholangiocarcinoma through NF-κB-inducing kinase-mediated hepatocyte transdifferentiation. Hepatology. 2023;77(2):395–410. [Google Scholar] [PubMed]
84. Gerriets V, Goyal A, Khaddour K. Tumor necrosis factor inhibitors. Treasure Island (FLStatPearls Publishing; 2024. https://www.ncbi.nlm.nih.gov/books/NBK482425/. [Accessed 2024]. [Google Scholar]
85. Zins K, Abraham D, Sioud M, Aharinejad S. Colon cancer cell–derived tumor necrosis factor-α mediates the tumor growth–promoting response in macrophages by up-regulating the colony-stimulating factor-1 pathway. Cancer Res. 2007;67(3):1038–45. doi:10.1158/0008-5472.CAN-06-2295. [Google Scholar] [PubMed] [CrossRef]
86. Gani F, Nagarajan N, Kim Y, Zhu Q, Luan L, Bhaijjee F, et al. Program death 1 immune checkpoint and tumor microenvironment: implications for patients with intrahepatic cholangiocarcinoma. Ann Surg Oncol. 2016;23(8):2610–7. doi:10.1245/s10434-016-5101-y. [Google Scholar] [PubMed] [CrossRef]
87. Fontugne J, Augustin J, Pujals A, Compagnon P, Rousseau B, Luciani A, et al. PD-L1 expression in perihilar and intrahepatic cholangiocarcinoma. Oncotarget. 2017;8(15):24644–51. doi:10.18632/oncotarget.15602. [Google Scholar] [PubMed] [CrossRef]
88. Byrne KT, Vonderheide RH. CD40 stimulation obviates innate sensors and drives T cell immunity in cancer. Cell Rep. 2016;15(12):2719–32. doi:10.1016/j.celrep.2016.05.058. [Google Scholar] [PubMed] [CrossRef]
89. Lim CY, Chang JH, Lee WS, Kim J, Park IY. CD40 agonists alter the pancreatic cancer microenvironment by shifting the macrophage phenotype toward M1 and suppress human pancreatic cancer in organotypic slice cultures. Gut Liver. 2022;16(4):645–59. doi:10.5009/gnl210311. [Google Scholar] [PubMed] [CrossRef]
90. Leblond MM, Tille L, Nassiri S, Gilfillan CB, Imbratta C, Schmittnaegel M, et al. CD40 agonist restores the antitumor efficacy of Anti-PD1 therapy in muscle-invasive bladder cancer in an IFN I/II-mediated manner. Cancer Immunol Res. 2020;8(9):1180–92. doi:10.1158/2326-6066.CIR-19-0826. [Google Scholar] [PubMed] [CrossRef]
91. Dean L, Kane M. Cetuximab therapy and RAS and BRAF genotype. In: Pratt VM, Scott SA, Pirmohamed M, editors. Medical Genetics Summaries. Bethesda (MDNational Center for Biotechnology Information (US); 2012. Available from: https://www.ncbi.nlm.nih.gov/books/NBK564547/. [Accessed 2024]. [Google Scholar]
92. Wang M, Chen Z, Guo P, Wang Y, Chen G. Therapy for advanced cholangiocarcinoma: current knowledge and future potential. J Cell Mol Med. 2021;25(2):618–28. doi:10.1111/jcmm.v25.2. [Google Scholar] [CrossRef]
93. Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360(14):1408–17. doi:10.1056/NEJMoa0805019. [Google Scholar] [PubMed] [CrossRef]
94. Chan DLH, Segelov E, Wong RS, Smith A, Herbertson RA, Li BT, et al. Epidermal growth factor receptor (EGFR) inhibitors for metastatic colorectal cancer. Cochrane Database Syst Rev. 2017;6(6):CD007047. [Google Scholar] [PubMed]
95. Borbath I, Ceratti A, Verslype C, Demols A, Delaunoit T, Laurent S, et al. Combination of gemcitabine and cetuximab in patients with advanced cholangiocarcinoma: a phase II study of the Belgian Group of Digestive Oncology. Ann Oncol. 2013;24(11):2824–9. doi:10.1093/annonc/mdt337. [Google Scholar] [PubMed] [CrossRef]
96. Chen JS, Hsu C, Chiang NJ, Tsai CS, Tsou HH, Huang SF, et al. A KRAS mutation status-stratified randomized phase II trial of gemcitabine and oxaliplatin alone or in combination with cetuximab in advanced biliary tract cancer. Ann Oncol. 2015;26(5):943–9. doi:10.1093/annonc/mdv035. [Google Scholar] [PubMed] [CrossRef]
97. Tamma R, Annese T, Ruggieri S, Brunetti O, Longo V, Cascardi E, et al. Inflammatory cells infiltrate and angiogenesis in locally advanced and metastatic cholangiocarcinoma. Eur J Clin Invest. 2019;49(5):e13087. doi:10.1111/eci.2019.49.issue-5. [Google Scholar] [CrossRef]
98. Yoshikawa D, Ojima H, Iwasaki M, Hiraoka N, Kosuge T, Kasai S, et al. Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br J Cancer. 2008;98(2):418–25. doi:10.1038/sj.bjc.6604129. [Google Scholar] [PubMed] [CrossRef]
99. Yuan Y, Shao C, Guan Y, Lu H, Wang D, Zhang S. Association between the VEGFR-2 -604T/C polymorphism (rs2071559) and type 2 diabetic retinopathy. Open Life Sci. 2023;18(1):20220081. doi:10.1515/biol-2022-0081. [Google Scholar] [PubMed] [CrossRef]
100. Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol. 2018;15(5):325–40. doi:10.1038/nrclinonc.2018.29. [Google Scholar] [PubMed] [CrossRef]
101. Arkenau HT, Martin-Liberal J, Calvo E, Penel N, Krebs MG, Herbst RS, et al. Ramucirumab plus pembrolizumab in patients with previously treated advanced or metastatic biliary tract cancer: nonrandomized, open-label, phase I trial (JVDF). Oncologist. 2018;23(12):1407–e136. doi:10.1634/theoncologist.2018-0044. [Google Scholar] [PubMed] [CrossRef]
102. Lwin Z, Gomez-Roca C, Saada-Bouzid E, Yanez E, Muñoz FL, Im S, et al. LBA41 LEAP-005: phase II study of lenvatinib (len) plus pembrolizumab (pembro) in patients (pts) with previously treated advanced solid tumours. Ann Oncol. 2020;31:S1170. doi:10.1016/j.annonc.2020.08.2271. [Google Scholar] [CrossRef]
103. Zhang Q, Liu X, Wei S, Zhang L, Tian Y, Gao Z, et al. Lenvatinib plus PD-1 inhibitors as first-line treatment in patients with unresectable biliary tract cancer: a single-arm, open-label, phase ii study. Front Oncol. 2021;11:751391. doi:10.3389/fonc.2021.751391. [Google Scholar] [PubMed] [CrossRef]
104. Matsuyama M, Ishii H, Furuse J, Ohkawa S, Maguchi H, Mizuno N, et al. Phase II trial of combination therapy of gemcitabine plus anti-angiogenic vaccination of elpamotide in patients with advanced or recurrent biliary tract cancer. Invest New Drugs. 2015;33(2):490–5. doi:10.1007/s10637-014-0197-z. [Google Scholar] [PubMed] [CrossRef]
105. Zhu AX, Meyerhardt JA, Blaszkowsky LS, Kambadakone AR, Muzikansky A, Zheng H, et al. Efficacy and safety of gemcitabine, oxaliplatin, and bevacizumab in advanced biliary-tract cancers and correlation of changes in 18-fluorodeoxyglucose PET with clinical outcome: a phase 2 study. Lancet Oncol. 2010;11(1):48–54. doi:10.1016/S1470-2045(09)70333-X. [Google Scholar] [PubMed] [CrossRef]
106. Santoro A, Gebbia V, Pressiani T, Testa A, Personeni N, Arrivas Bajardi E, et al. A randomized, multicenter, phase II study of vandetanib monotherapy versus vandetanib in combination with gemcitabine versus gemcitabine plus placebo in subjects with advanced biliary tract cancer: the VanGogh study. Ann Oncol. 2015;26(3):542–7. doi:10.1093/annonc/mdu576. [Google Scholar] [PubMed] [CrossRef]
107. Valle JW, Wasan H, Lopes A, Backen AC, Palmer DH, Morris K, et al. Cediranib or placebo in combination with cisplatin and gemcitabine chemotherapy for patients with advanced biliary tract cancer (ABC-03a randomised phase 2 trial. Lancet Oncol. 2015;16(8):967–78. doi:10.1016/S1470-2045(15)00139-4. [Google Scholar] [PubMed] [CrossRef]
108. Sun W, Patel A, Normolle D, Patel K, Ohr J, Lee JJ, et al. A phase 2 trial of regorafenib as a single agent in patients with chemotherapy-refractory, advanced, and metastatic biliary tract adenocarcinoma. Cancer. 2019;125(6):902–9. doi:10.1002/cncr.31872. [Google Scholar] [PubMed] [CrossRef]
109. Demols A, Borbath I, Van den Eynde M, Houbiers G, Peeters M, Marechal R, et al. Regorafenib after failure of gemcitabine and platinum-based chemotherapy for locally advanced/metastatic biliary tumors: rEACHIN, a randomized, double-blind, phase II trial. Ann Oncol. 2020;31(9):1169–77. doi:10.1016/j.annonc.2020.05.018. [Google Scholar] [PubMed] [CrossRef]
110. El-Khoueiry AB, Rankin CJ, Ben-Josef E, Lenz HJ, Gold PJ, Hamilton RD, et al. SWOG 0514: a phase II study of sorafenib in patients with unresectable or metastatic gallbladder carcinoma and cholangiocarcinoma. Invest New Drugs. 2012;30(4):1646–51. doi:10.1007/s10637-011-9719-0. [Google Scholar] [PubMed] [CrossRef]
111. Bengala C, Bertolini F, Malavasi N, Boni C, Aitini E, Dealis C, et al. Sorafenib in patients with advanced biliary tract carcinoma: a phase II trial. Br J Cancer. 2010;102(1):68–72. doi:10.1038/sj.bjc.6605458. [Google Scholar] [PubMed] [CrossRef]
112. Lee JK, Capanu M, O’Reilly EM, Ma J, Chou JF, Shia J, et al. A phase II study of gemcitabine and cisplatin plus sorafenib in patients with advanced biliary adenocarcinomas. Br J Cancer. 2013;109(4):915–9. doi:10.1038/bjc.2013.432. [Google Scholar] [PubMed] [CrossRef]
113. Goyal L, Zheng H, Yurgelun MB, Abrams TA, Allen JN, Cleary JM, et al. A phase 2 and biomarker study of cabozantinib in patients with advanced cholangiocarcinoma. Cancer. 2017;123(11):1979–88. doi:10.1002/cncr.30571. [Google Scholar] [PubMed] [CrossRef]
114. Cadamuro M, Nardo G, Indraccolo S, Dall’olmo L, Sambado L, Moserle L, et al. Platelet-derived growth factor-D and Rho GTPases regulate recruitment of cancer-associated fibroblasts in cholangiocarcinoma. Hepatology. 2013;58(3):1042–53. doi:10.1002/hep.26384. [Google Scholar] [PubMed] [CrossRef]
115. Fingas CD, Bronk SF, Werneburg NW, Mott JL, Guicciardi ME, Cazanave SC, et al. Myofibroblast-derived PDGF-BB promotes Hedgehog survival signaling in cholangiocarcinoma cells. Hepatology. 2011;54(6):2076–88. doi:10.1002/hep.24588. [Google Scholar] [PubMed] [CrossRef]
116. Cadamuro M, Brivio S, Mertens J, Vismara M, Moncsek A, Milani C, et al. Platelet-derived growth factor-D enables liver myofibroblasts to promote tumor lymphangiogenesis in cholangiocarcinoma. J Hepatol. 2019;70(4):700–9. doi:10.1016/j.jhep.2018.12.004. [Google Scholar] [PubMed] [CrossRef]
117. Adeva J, Sangro B, Salati M, Edeline J, La Casta A, Bittoni A, et al. Medical treatment for cholangiocarcinoma. Liver Int. 2019;39(S1):123–42. doi:10.1111/liv.14100. [Google Scholar] [PubMed] [CrossRef]
118. Heo MH, Kim HK, Lee H, Kim KM, Lee J, Park SH, et al. The clinical impact of c-MET over-expression in advanced biliary tract cancer (BTC). J Cancer. 2017;8(8):1395–9. doi:10.7150/jca.17898. [Google Scholar] [PubMed] [CrossRef]
119. Appleman LJ. MET signaling pathway: a rational target for cancer therapy. J Clin Oncol. 2011;29(36):4837–8. doi:10.1200/JCO.2011.37.7929. [Google Scholar] [PubMed] [CrossRef]
120. Wang S-C, L.T. WY, Weng S, You H, Wei Y, Eng H, et al. Amplification and overexpression of the MET gene in intrahepatic cholangiocarcinoma correlate with adverse pathological features and worse clinical outcome. Int J Clin Exp Pathol. 2017;10(6):6809–17. [Google Scholar]
121. Yu G, Jing Y, Kou X, Ye F, Gao L, Fan Q, et al. Hepatic stellate cells secreted hepatocyte growth factor contributes to the chemoresistance of hepatocellular carcinoma. PLoS One. 2013;8(9):e73312. doi:10.1371/journal.pone.0073312. [Google Scholar] [PubMed] [CrossRef]
122. Pant S, Saleh M, Bendell J, Infante JR, Jones S, Kurkjian CD, et al. A phase I dose escalation study of oral c-MET inhibitor tivantinib (ARQ 197) in combination with gemcitabine in patients with solid tumors. Ann Oncol. 2014;25(7):1416–21. doi:10.1093/annonc/mdu157. [Google Scholar] [PubMed] [CrossRef]
123. Zhao S, Wu W, Jiang H, Ma L, Pan C, Jin C, et al. Selective inhibitor of the c-MET receptor tyrosine kinase in advanced hepatocellular carcinoma: no beneficial effect with the use of tivantinib? Front Immunol. 2021;12:731527. doi:10.3389/fimmu.2021.731527. [Google Scholar] [PubMed] [CrossRef]
124. Wei K, Li M, Zoller M, Wang M, Mehrabi A, Hoffmann K. Targeting c-MET by Tivantinib through synergistic activation of JNK/c-jun pathway in cholangiocarcinoma. Cell Death Discov. 2019;10(3):231. doi:10.1038/s41419-019-1460-1. [Google Scholar] [PubMed] [CrossRef]
125. Ang C. Role of the fibroblast growth factor receptor axis in cholangiocarcinoma. J Gastroenterol Hepatol. 2015;30(7):1116–22. doi:10.1111/jgh.2015.30.issue-7. [Google Scholar] [CrossRef]
126. Hierro C, Rodon J, Tabernero J. Fibroblast growth factor (FGF) receptor/FGF inhibitors: novel targets and strategies for optimization of response of solid tumors. Semin Oncol. 2015;42(6):801–19. doi:10.1053/j.seminoncol.2015.09.027. [Google Scholar] [PubMed] [CrossRef]
127. Wesche J, Haglund K, Haugsten EM. Fibroblast growth factors and their receptors in cancer. Biochem J. 2011;437(2):199–213. doi:10.1042/BJ20101603. [Google Scholar] [PubMed] [CrossRef]
128. Aitcheson G, Mahipal A, John BV. Targeting FGFR in intrahepatic cholangiocarcinoma [iCCA]: leading the way for precision medicine in biliary tract cancer [BTC]? Expert Opin Investig Drugs. 2021;30(4):463–77. doi:10.1080/13543784.2021.1900821. [Google Scholar] [PubMed] [CrossRef]
129. Jain A, Borad MJ, Kelley RK, Wang Y, Abdel-Wahab R, Meric-Bernstam F, et al. Cholangiocarcinoma With FGFR genetic aberrations: a unique clinical phenotype. JCO Precis Oncol. 2018;2:1–12. [Google Scholar] [PubMed]
130. Arai Y, Totoki Y, Hosoda F, Shirota T, Hama N, Nakamura H, et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology. 2014;59(4):1427–34. doi:10.1002/hep.26890. [Google Scholar] [PubMed] [CrossRef]
131. Wu YM, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013;3(6):636–47. doi:10.1158/2159-8290.CD-13-0050. [Google Scholar] [PubMed] [CrossRef]
132. Javle M, Roychowdhury S, Kelley RK, Sadeghi S, Macarulla T, Weiss KH, et al. Infigratinib (BGJ398) in previously treated patients with advanced or metastatic cholangiocarcinoma with FGFR2 fusions or rearrangements: mature results from a multicentre, open-label, single-arm, phase 2 study. Lancet Gastroenterol Hepatol. 2021;6(10):803–15. doi:10.1016/S2468-1253(21)00196-5. [Google Scholar] [PubMed] [CrossRef]
133. Abou-Alfa GK, Sahai V, Hollebecque A, Vaccaro G, Melisi D, Al-Rajabi R, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 2020;21(5):671–84. doi:10.1016/S1470-2045(20)30109-1. [Google Scholar] [PubMed] [CrossRef]
134. Hoy SM. Pemigatinib: first approval. Drugs. 2020;80(9):923–9. doi:10.1007/s40265-020-01330-y. [Google Scholar] [PubMed] [CrossRef]
135. Mazzaferro V, El-Rayes BF, Droz Dit Busset M, Cotsoglou C, Harris WP, Damjanov N, et al. Derazantinib (ARQ 087) in advanced or inoperable FGFR2 gene fusion-positive intrahepatic cholangiocarcinoma. Br J Cancer. 2019;120(2):165–71. doi:10.1038/s41416-018-0334-0. [Google Scholar] [PubMed] [CrossRef]
136. Park JO, Feng Y-H, Chen Y-Y, Su W-C, Oh D-Y, Shen L, et al. Updated results of a phase IIa study to evaluate the clinical efficacy and safety of erdafitinib in Asian advanced cholangiocarcinoma (CCA) patients with FGFR alterations. J Clin Oncol. 2019;37(15_suppl). doi:10.1200/JCO.2019.37.15_suppl.4117. [Google Scholar] [CrossRef]
137. Javle M, Lowery M, Shroff RT, Weiss KH, Springfeld C, Borad MJ, et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J Clin Oncol. 2018;36(3):276–82. doi:10.1200/JCO.2017.75.5009. [Google Scholar] [PubMed] [CrossRef]
138. Javle MM, Abou-Alfa GK, Macarulla T, Personeni N, Adeva J, Bergamo F, et al. Efficacy of derazantinib in intrahepatic cholangiocarcinoma patients with FGFR2 mutations or amplifications: interim results from the phase 2 study FIDES-01. J Clin Oncol, 2022;40(4_suppl):427. doi:10.1200/JCO.2022.40.4_suppl.42. [Google Scholar] [CrossRef]
139. Kalyukina M, Yosaatmadja Y, Middleditch MJ, Patterson AV, Smaill JB, Squire CJ. TAS-120 cancer target binding: defining reactivity and revealing the first fibroblast growth factor receptor 1 (FGFR1) irreversible structure. ChemMedChem. 2019;14(4):494–500. doi:10.1002/cmdc.v14.4. [Google Scholar] [CrossRef]
140. Goyal L, Arkenau H-T, Tran B, Soria J-C, Bahleda R, Mak G, et al. Early clinical efficacy of TAS-120, a covalently bound FGFR inhibitor, in patients with cholangiocarcinoma. Ann Oncol. 2017;28:iii145. doi:10.1093/annonc/mdx262.019. [Google Scholar] [CrossRef]
141. Goyal L, Saha SK, Liu LY, Siravegna G, Leshchiner I, Ahronian LG, et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov. 2017;7(3):252–63. doi:10.1158/2159-8290.CD-16-1000. [Google Scholar] [PubMed] [CrossRef]
142. Goyal L, Shi L, Liu LY, Fece de la Cruz F, Lennerz JK, Raghavan S, et al. TAS-120 overcomes resistance to ATP-competitive FGFR inhibitors in patients with FGFR2 fusion-positive intrahepatic cholangiocarcinoma. Cancer Discov. 2019;9(8):1064–79. doi:10.1158/2159-8290.CD-19-0182. [Google Scholar] [PubMed] [CrossRef]
143. Goyal L, Meric-Bernstam F, Hollebecque A, Valle JW, Morizane C, Karasic TB, et al. FOENIX-CCA2: a phase II, open-label, multicenter study of futibatinib in patients (pts) with intrahepatic cholangiocarcinoma (iCCA) harboring FGFR2 gene fusions or other rearrangements. J Clin Oncol. 2020;38(15_suppl). doi:10.1200/JCO.2020.38.15_suppl.108. [Google Scholar] [CrossRef]
144. Meric-Bernstam F, Bahleda R, Hierro C, Sanson M, Bridgewater J, Arkenau H-T, et al. Futibatinib, an irreversible FGFR1-4 inhibitor, in patients with advanced solid tumors harboring FGF/FGFR aberrations: a phase I dose-expansion study. Cancer Discov. 2022;12(2):402–15. doi:10.1158/2159-8290.CD-21-0697. [Google Scholar] [PubMed] [CrossRef]
145. Goyal L, Meric-Bernstam F, Hollebecque A, Valle JW, Morizane C, Karasic TB, et al. Futibatinib for FGFR2-rearranged intrahepatic cholangiocarcinoma. N Engl J Med. 2023;388(3):228–39. doi:10.1056/NEJMoa2206834. [Google Scholar] [PubMed] [CrossRef]
146. Yoshikawa D, Ojima H, Kokubu A, Ochiya T, Kasai S, Hirohashi S, et al. Vandetanib (ZD6474an inhibitor of VEGFR and EGFR signalling, as a novel molecular-targeted therapy against cholangiocarcinoma. Br J Cancer. 2009;100(8):1257–66. doi:10.1038/sj.bjc.6604988. [Google Scholar] [PubMed] [CrossRef]
147. Philip PA, Mahoney MR, Allmer C, Thomas J, Pitot HC, Kim G, et al. Phase II study of erlotinib in patients with advanced biliary cancer. J Clin Oncol. 2006;24(19):3069–74. doi:10.1200/JCO.2005.05.3579. [Google Scholar] [PubMed] [CrossRef]
148. Sohal DP, Mykulowycz K, Uehara T, Teitelbaum UR, Damjanov N, Giantonio BJ, et al. A phase II trial of gemcitabine, irinotecan and panitumumab in advanced cholangiocarcinoma. Ann Oncol. 2013;24(12):3061–5. doi:10.1093/annonc/mdt416. [Google Scholar] [PubMed] [CrossRef]
149. Yang X, Wang W, Wang C, Wang L, Yang M, Qi M, et al. Characterization of EGFR family gene aberrations in cholangiocarcinoma. Oncol Rep. 2014;32(2):700–8. doi:10.3892/or.2014.3261. [Google Scholar] [PubMed] [CrossRef]
150. Zhang Z, Oyesanya RA, Campbell DJ, Almenara JA, Dewitt JL, Sirica AE. Preclinical assessment of simultaneous targeting of epidermal growth factor receptor (ErbB1) and ErbB2 as a strategy for cholangiocarcinoma therapy. Hepatology. 2010;52(3):975–86. doi:10.1002/hep.23773. [Google Scholar] [PubMed] [CrossRef]
151. Lubner SJ, Mahoney MR, Kolesar JL, Loconte NK, Kim GP, Pitot HC, et al. Report of a multicenter phase II trial testing a combination of biweekly bevacizumab and daily erlotinib in patients with unresectable biliary cancer: a phase II Consortium study. J Clin Oncol. 2010;28(21):3491–7. doi:10.1200/JCO.2010.28.4075. [Google Scholar] [PubMed] [CrossRef]
152. Lee J, Park SH, Chang HM, Kim JS, Choi HJ, Lee MA, et al. Gemcitabine and oxaliplatin with or without erlotinib in advanced biliary-tract cancer: a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2012;13(2):181–8. doi:10.1016/S1470-2045(11)70301-1. [Google Scholar] [PubMed] [CrossRef]
153. Kim ST, Jang KT, Lee J, Jang HM, Choi HJ, Jang HL, et al. Molecular subgroup analysis of clinical outcomes in a phase 3 study of gemcitabine and oxaliplatin with or without erlotinib in advanced biliary tract cancer. Transl Oncol. 2015;8(1):40–6. doi:10.1016/j.tranon.2014.12.003. [Google Scholar] [PubMed] [CrossRef]
154. Morisaki T, Umebayashi M, Kiyota A, Koya N, Tanaka H, Onishi H, et al. Combining cetuximab with killer lymphocytes synergistically inhibits human cholangiocarcinoma cells in vitro. Anticancer Res. 2012;32(6):2249–56. [Google Scholar] [PubMed]
155. Gruenberger B, Schueller J, Heubrandtner U, Wrba F, Tamandl D, Kaczirek K, et al. Cetuximab, gemcitabine, and oxaliplatin in patients with unresectable advanced or metastatic biliary tract cancer: a phase 2 study. Lancet Oncol. 2010;11(12):1142–8. doi:10.1016/S1470-2045(10)70247-3. [Google Scholar] [PubMed] [CrossRef]
156. Malka D, Cervera P, Foulon S, Trarbach T, de la Fouchardiere C, Boucher E, et al. Gemcitabine and oxaliplatin with or without cetuximab in advanced biliary-tract cancer (BINGOa randomised, open-label, non-comparative phase 2 trial. Lancet Oncol. 2014;15(8):819–28. doi:10.1016/S1470-2045(14)70212-8. [Google Scholar] [PubMed] [CrossRef]
157. Leone F, Marino D, Cereda S, Filippi R, Belli C, Spadi R, et al. Panitumumab in combination with gemcitabine and oxaliplatin does not prolong survival in wild-type KRAS advanced biliary tract cancer: a randomized phase 2 trial (Vecti-BIL study). Cancer. 2016;122(4):574–81. doi:10.1002/cncr.v122.4. [Google Scholar] [CrossRef]
158. Vogel A, Kasper S, Bitzer M, Block A, Sinn M, Schulze-Bergkamen H, et al. PICCA study: panitumumab in combination with cisplatin/gemcitabine chemotherapy in KRAS wild-type patients with biliary cancer—a randomised biomarker-driven clinical phase II AIO study. Eur J Cancer. 2018;92:11–9. doi:10.1016/j.ejca.2017.12.028. [Google Scholar] [PubMed] [CrossRef]
159. Feng KC, Guo YL, Liu Y, Dai HR, Wang Y, Lv HY, et al. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J Hematol Oncol. 2017;10(1):4. doi:10.1186/s13045-016-0378-7. [Google Scholar] [PubMed] [CrossRef]
160. Guo Y, Feng K, Liu Y, Wu Z, Dai H, Yang Q, et al. Phase I study of chimeric antigen receptor-modified T cells in patients with EGFR-positive advanced biliary tract cancers. Clin Cancer Res. 2018;24(6):1277–86. doi:10.1158/1078-0432.CCR-17-0432. [Google Scholar] [PubMed] [CrossRef]
161. Feng K, Liu Y, Guo Y, Qiu J, Wu Z, Dai H, et al. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell. 2018;9(10):838–47. doi:10.1007/s13238-017-0440-4. [Google Scholar] [PubMed] [CrossRef]
162. Golan T, Raitses-Gurevich M, Kelley RK, Bocobo AG, Borgida A, Shroff RT, et al. Overall survival and clinical characteristics of BRCA-associated cholangiocarcinoma: a multicenter retrospective study. Oncologist. 2017;22(7):804–10. doi:10.1634/theoncologist.2016-0415. [Google Scholar] [PubMed] [CrossRef]
163. Lee UE, Friedman SL. Mechanisms of hepatic fibrogenesis. Best Pract Res Clin Gastroenterol. 2011;25(2):195–206. doi:10.1016/j.bpg.2011.02.005. [Google Scholar] [PubMed] [CrossRef]
164. Kuang P, Zhao W, Su W, Zhang Z, Zhang L, Liu J, et al. 18beta-glycyrrhetinic acid inhibits hepatocellular carcinoma development by reversing hepatic stellate cell-mediated immunosuppression in mice. Int J Cancer. 2013;132(8):1831–41. doi:10.1002/ijc.v132.8. [Google Scholar] [CrossRef]
165. Torok NJ. Recent advances in the pathogenesis and diagnosis of liver fibrosis. J Gastroenterol. 2008;43(5):315–21. doi:10.1007/s00535-008-2181-x. [Google Scholar] [PubMed] [CrossRef]
166. Tacke F, Luedde T, Trautwein C. Inflammatory pathways in liver homeostasis and liver injury. Clin Rev Allergy Immunol. 2009;36(1):4–12. doi:10.1007/s12016-008-8091-0. [Google Scholar] [PubMed] [CrossRef]
167. Wheless M, Agarwal R, Goff L, Lockney N, Padmanabhan C, Heumann T. Current standards, multidisciplinary approaches, and future directions in the management of extrahepatic cholangiocarcinoma. Curr Treat Options Oncol. 2024;25(1):127–60. doi:10.1007/s11864-023-01153-5. [Google Scholar] [PubMed] [CrossRef]
168. Oishi K, Sakaguchi T, Baba S, Suzuki S, Konno H. Macrophage density and macrophage colony-stimulating factor expression predict the postoperative prognosis in patients with intrahepatic cholangiocarcinoma. Surg Today. 2015;45(6):715–22. doi:10.1007/s00595-014-0989-y. [Google Scholar] [PubMed] [CrossRef]
169. Zhu XD, Zhang JB, Zhuang PY, Zhu HG, Zhang W, Xiong YQ, et al. High expression of macrophage colony-stimulating factor in peritumoral liver tissue is associated with poor survival after curative resection of hepatocellular carcinoma. J Clin Oncol. 2008;26(16):2707–16. doi:10.1200/JCO.2007.15.6521. [Google Scholar] [PubMed] [CrossRef]
170. Toy EP, Chambers JT, Kacinski BM, Flick MB, Chambers SK. The activated macrophage colony-stimulating factor (CSF-1) receptor as a predictor of poor outcome in advanced epithelial ovarian carcinoma. Gynecol Oncol. 2001;80(2):194–200. doi:10.1006/gyno.2000.6070. [Google Scholar] [PubMed] [CrossRef]
171. Beck AH, Espinosa I, Edris B, Li R, Montgomery K, Zhu S, et al. The macrophage colony-stimulating factor 1 response signature in breast carcinoma. Clin Cancer Res. 2009;15(3):778–87. doi:10.1158/1078-0432.CCR-08-1283. [Google Scholar] [PubMed] [CrossRef]
172. Laoui D, Movahedi K, Van Overmeire E, Van den Bossche J, Schouppe E, Mommer C, et al. Tumor-associated macrophages in breast cancer: distinct subsets, distinct functions. Int J Dev Biol. 2011;55(7–9):861–7. doi:10.1387/ijdb.113371dl. [Google Scholar] [PubMed] [CrossRef]
173. Lee JI, Campbell JS. Role of desmoplasia in cholangiocarcinoma and hepatocellular carcinoma. J Hepatol. 2014;61(2):432–4. doi:10.1016/j.jhep.2014.04.014. [Google Scholar] [PubMed] [CrossRef]
174. Ying F, Chan MSM, Lee TKW. Cancer-associated fibroblasts in hepatocellular carcinoma and cholangiocarcinoma. Cell Mol Gastroenterol Hepatol. 2023;15(4):985–99. doi:10.1016/j.jcmgh.2023.01.006. [Google Scholar] [PubMed] [CrossRef]
175. Weber CE, Kuo PC. The tumor microenvironment. Surg Oncol. 2012;21(3):172–7. doi:10.1016/j.suronc.2011.09.001. [Google Scholar] [PubMed] [CrossRef]
176. Iredale JP, Benyon RC, Pickering J, McCullen M, Northrop M, Pawley S, et al. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest. 1998;102(3):538–49. doi:10.1172/JCI1018. [Google Scholar] [PubMed] [CrossRef]
177. Yin Z, Dong C, Jiang K, Xu Z, Li R, Guo K, et al. Heterogeneity of cancer-associated fibroblasts and roles in the progression, prognosis, and therapy of hepatocellular carcinoma. J Hematol Oncol. 2019;12(1):101. doi:10.1186/s13045-019-0782-x. [Google Scholar] [PubMed] [CrossRef]
178. Sirica AE, Gores GJ. Desmoplastic stroma and cholangiocarcinoma: clinical implications and therapeutic targeting. Hepatology. 2014;59(6):2397–402. doi:10.1002/hep.v59.6. [Google Scholar] [CrossRef]
179. Knopf JD, Tholen S, Koczorowska MM, De Wever O, Biniossek ML, Schilling O. The stromal cell-surface protease fibroblast activation protein-α localizes to lipid rafts and is recruited to invadopodia. Biochim Biophys Acta. 2015;1853(10):2515–25. doi:10.1016/j.bbamcr.2015.07.013. [Google Scholar] [PubMed] [CrossRef]
180. Monteran L, Erez N. The dark side of fibroblasts: cancer-associated fibroblasts as mediators of immunosuppression in the tumor microenvironment. Front Immunol. 2019;10:1835. doi:10.3389/fimmu.2019.01835. [Google Scholar] [PubMed] [CrossRef]
181. Khalili JS, Liu S, Rodriguez-Cruz TG, Whittington M, Wardell S, Liu C, et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res. 2012;18(19):5329–40. doi:10.1158/1078-0432.CCR-12-1632. [Google Scholar] [PubMed] [CrossRef]
182. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110(50):20212–7. doi:10.1073/pnas.1320318110. [Google Scholar] [PubMed] [CrossRef]
183. Mertens JC, Fingas CD, Christensen JD, Smoot RL, Bronk SF, Werneburg NW, et al. Therapeutic effects of deleting cancer-associated fibroblasts in cholangiocarcinoma. Cancer Res. 2013;73(2):897–907. doi:10.1158/0008-5472.CAN-12-2130. [Google Scholar] [PubMed] [CrossRef]
184. Yamanaka T, Harimoto N, Muranushi R, Hagiwara K, Gantumur D, Ishii N, et al. New therapy for intrahepatic cholangiocarcinoma targeted to cancer associated fibroblasts. Ann Oncol. 2019;30:v20. doi:10.1093/annonc/mdz238.068. [Google Scholar] [CrossRef]
185. Khoja L, Butler MO, Kang SP, Ebbinghaus S, Joshua AM. Pembrolizumab. J Immunother Cancer. 2015;3(1):1–13. doi:10.1186/s40425-015-0078-9. [Google Scholar] [PubMed] [CrossRef]
186. Kelley RK, Ueno M, Yoo C, Finn RS, Furuse J, Ren Z, et al. Pembrolizumab in combination with gemcitabine and cisplatin compared with gemcitabine and cisplatin alone for patients with advanced biliary tract cancer (KEYNOTE-966a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2023;401(10391):1853–65. doi:10.1016/S0140-6736(23)00727-4. [Google Scholar] [PubMed] [CrossRef]
187. Lee SH, Lee HS, Lee SH, Woo SM, Kim DU, Bang S. Efficacy and safety of pembrolizumab for gemcitabine/cisplatin-refractory biliary tract cancer: a multicenter retrospective study. J Clin Med. 2020;9(6):1769. doi:10.3390/jcm9061769. [Google Scholar] [PubMed] [CrossRef]
188. Ueno M, Chung H, Nagrial A, Marabelle A, Kelley R, Xu L, et al. Pembrolizumab for advanced biliary adenocarcinoma: results from the multicohort, phase II KEYNOTE-158 study. Ann Oncol. 2018;29:viii210. doi:10.1093/annonc/mdy282.009. [Google Scholar] [CrossRef]
189. Rendon A, Rayi A. Nivolumab. In: StatPearls. Treasure Island (FLStatPearls Publishing; 2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK567801/. [Accessed 2024]. [Google Scholar]
190. Ueno M, Ikeda M, Morizane C, Kobayashi S, Ohno I, Kondo S, et al. Nivolumab alone or in combination with cisplatin plus gemcitabine in Japanese patients with unresectable or recurrent biliary tract cancer: a non-randomised, multicentre, open-label, phase 1 study. Lancet Gastroenterol Hepatol. 2019;4(8):611–21. doi:10.1016/S2468-1253(19)30086-X. [Google Scholar] [PubMed] [CrossRef]
191. Gou M, Zhang Y, Si H, Dai G. Efficacy and safety of nivolumab for metastatic biliary tract cancer. Onco Targets Ther. 2019;12:861–7. doi:10.2147/OTT. [Google Scholar] [CrossRef]
192. Kim RD, Chung V, Alese OB, El-Rayes BF, Li D, Al-Toubah TE, et al. A Phase 2 multi-institutional study of nivolumab for patients with advanced refractory biliary tract cancer. JAMA Oncol. 2020;6(6):888–94. doi:10.1001/jamaoncol.2020.0930. [Google Scholar] [PubMed] [CrossRef]
193. Feng K, Liu Y, Zhao Y, Yang Q, Dong L, Liu J, et al. Efficacy and biomarker analysis of nivolumab plus gemcitabine and cisplatin in patients with unresectable or metastatic biliary tract cancers: results from a phase II study. J Immunother Cancer. 2020;8(1):e000367. doi:10.1136/jitc-2019-000367. [Google Scholar] [PubMed] [CrossRef]
194. Saad P, Kasi A. Ipilimumab. In: StatPearls. Treasure Island (FLStatPearls Publishing; 2023. Available from: https://pubmed.ncbi.nlm.nih.gov/32491727/. [Accessed 2024]. [Google Scholar]
195. Klein O, Kee D, Nagrial A, Markman B, Underhill C, Michael M, et al. Evaluation of combination nivolumab and ipilimumab immunotherapy in patients with advanced biliary tract cancers: subgroup analysis of a phase 2 nonrandomized clinical trial. JAMA Oncol. 2020;6(9):1405–9. doi:10.1001/jamaoncol.2020.2814. [Google Scholar] [PubMed] [CrossRef]
196. Sahai V, Griffith KA, Beg MS, Shaib WL, Mahalingam D, Zhen DB, et al. A randomized phase 2 trial of nivolumab, gemcitabine, and cisplatin or nivolumab and ipilimumab in previously untreated advanced biliary cancer: BilT-01. Cancer. 2022;128(19):3523–30. doi:10.1002/cncr.v128.19. [Google Scholar] [CrossRef]
197. Kang J, Jeong JH, Hwang HS, Lee SS, Park DH, Oh DW, et al. Efficacy and safety of pembrolizumab in patients with refractory advanced biliary tract cancer: tumor proportion score as a potential biomarker for response. Cancer Res Treat. 2020;52(2):594–603. doi:10.4143/crt.2019.493. [Google Scholar] [PubMed] [CrossRef]
198. Ikeda Y, Ono M, Ohmori G, Ameda S, Yamada M, Abe T, et al. Successful pembrolizumab treatment of microsatellite instability-high intrahepatic cholangiocarcinoma: a case report. Clin Case Rep. 2021;9(4):2259–63. doi:10.1002/ccr3.v9.4. [Google Scholar] [CrossRef]
199. Czink E, Kloor M, Goeppert B, Frohling S, Uhrig S, Weber TF, et al. Successful immune checkpoint blockade in a patient with advanced stage microsatellite-unstable biliary tract cancer. Cold Spring Harb Mol Case Stud. 2017;3(5):a001974. doi:10.1101/mcs.a001974. [Google Scholar] [PubMed] [CrossRef]
200. Elias C, Zeidan YH, Bouferraa Y, Mukherji D, Temraz S, Charafeddine M, et al. A phase II single arm study of Nivolumab with stereotactic Ablative radiation Therapy after induction chemotherapy in CHOlangiocarcinoma (NATCHO). BMC Cancer. 2022;22(1):1296. doi:10.1186/s12885-022-10373-1. [Google Scholar] [PubMed] [CrossRef]
201. McGrath NA, Fu J, Gu SZ, Xie C. Targeting cancer stem cells in cholangiocarcinoma (Review). Int J Oncol. 2020;57(2):397–408. doi:10.3892/ijo.2020.5074. [Google Scholar] [PubMed] [CrossRef]
202. Lee TK, Guan XY, Ma S. Cancer stem cells in hepatocellular carcinoma-from origin to clinical implications. Nat Rev Gastroenterol Hepatol. 2022;19(1):26–44. doi:10.1038/s41575-021-00508-3. [Google Scholar] [PubMed] [CrossRef]
203. Tamai K, Fujimori H, Mochizuki M, Satoh K. Cancer stem cells in intrahepatic cholangiocarcinoma; their molecular basis, and therapeutic implications. Front Physiol. 2021;12:824261. doi:10.3389/fphys.2021.824261. [Google Scholar] [PubMed] [CrossRef]
204. Fan T, Zhang M, Yang J, Zhu Z, Cao W, Dong C. Therapeutic cancer vaccines: advancements, challenges, and prospects. Signal Transduct Target Ther. 2023;8(1):450. doi:10.1038/s41392-023-01674-3. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools