
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

The preventive mechanisms and research progress of sulforaphane in relation to prostate cancer
1 Department of Urology, The First People’s Hospital of Xiushui, Jiujiang, 332400, China
2 Department of Urology, The First Affiliated Hospital of Gannan Medical University, Ganzhou, 341000, China
* Corresponding Author: HUANGLIN DUAN. Email:
# Baisheng Xu and Tianpeng Xie are listed as co-authors
BIOCELL 2024, 48(12), 1703-1719. https://doi.org/10.32604/biocell.2024.054873
Received 10 June 2024; Accepted 20 September 2024; Issue published 30 December 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Prostate cancer is one of the most common tumors in urology. Dietary prophylaxis can effectively reduce prostate cancer incidence and progression. A growing body of research has shown that natural food ingredients such as Sulforaphane (SFN) can reduce the incidence of prostate cancer. It has a significant inhibitory effect on the progression from local prostate cancer to more aggressive prostate cancer. This article mainly expounds on the prevention mechanism and research progress of sulforaphane in various ways for prostate cancer and provides a reference for its future clinical application. In this review, ‘SFN’, ‘Prostate Cancer’, and ‘PCa’ were searched through PubMed, Embase, Web of Science, and other databases. SFN inhibits the occurrence and development of prostate cancer mainly through anti-oxidation, inhibition of fatty acid metabolism, inhibition of glycolysis, inhibition of pro-inflammatory factors, inhibition of cell proliferation and promotion of apoptosis, reduction of androgen receptors, and influence of epigenetics. Therefore, SFN is a natural compound with great potential for the prevention and treatment of prostate cancer, but the key factors such as effective chemoprevention dose, bioavailability, toxic dose, and response of sulforaphane in the human body need to be further studied in the future.Keywords
Abbreviations: Full Name of the Technical Term
| SFN | Sulforaphane |
| PCa | Prostate cancer |
| CRPC | Castration-Resistant Prostate Cancer |
| ARTA | Androgen receptor-targeted drugs |
| PARP | Poly ADP-ribose polymerase |
| nmCRPC | Nonmetastatic castration-resistant disease |
| mCRPC | Metastatic castration-resistant disease |
| AR | Androgen receptor |
| ADT | Androgen Deprivation Therapy |
| MFS | Metastasis-free survival |
| OS | Overall survival |
| PFS | Progression Free Survival |
| ROS | Reactive oxygen species |
| GST | Glutathione S-transferase |
| NQO | NAD (P)H quinone oxidoreductase |
| Nrf2 | Nuclear transcription factor-E2-related factor 2 |
| Keap1 | Kelpin-like ECH-associated protein 1 |
| MtDNA | Mitochondrial DNA |
| HO-1 | Heme oxygenase 1 |
| UGT | UDP-glucuronosyltransferases |
| SOD | Superoxide dismutase Modifications completed |
| ARE | Antioxidant response element defense system |
| sMaf | Small molecule myofascial fibrosarcoma |
| TRAMP | Ttransgenic adenocarcinoma mouse prostate |
| FASN | Fatty acid synthase |
| PIN | Prostate intraepithelial neoplasia |
| ACC1A | Acetyl coenzyme A carboxylase 1 |
| CPT1A | Carnitine palmitoyltransferase 1A |
| OXPHOS | Oxidative phosphorylation |
| HK | Hexokinase |
| PFK | Phosphofructokinase |
| PK | Pyruvate kinase |
| LDH | Lactate dehydrogenase |
| PKM2 | Pyruvate kinase M2 |
| LDHA | Lactate dehydrogenase A |
| VHL | Von Hippel-Lindau Disease Tumor Suppressor |
| HREs | Hypoxia response elements |
| TME | Tumor microenvironment |
| IL-6 | Interleukin-6 |
| TNF-α | Tumor necrosis factor α |
| IL-1 | Interleukin-1 |
| LPS | Lipopolysaccharide |
| UV | Ultraviolet |
| VEGF | Vascular endothelial growth factor |
| NO | Nitric oxide |
| PGE2 | Prostaglandin E2 |
| iNOS | Inducible NO synthase |
| COX-2 | Cyclooxygenase 2 |
| TLR | Toll-like receptors |
| HIF-1 | Hypoxia-inducible factor-1 |
| STAT-3 | Signal transducer and activator of transcription 3 |
| cdc25C | Cytokine cycle 25C |
| ChK2 | Checkpoint kinase 2 |
| PTEN | Phosphatase and tensin homolog |
| hTERC | Human telomerase RNA component |
| TEP1 | Telomerase associated protein 1 |
| DRs | Ddeath receptors |
| MEF | Mouse fibroblast |
| SFN-Cys | SFN-cysteine |
| SFN-NAC | SFN-N-acetylcysteine |
| HATs | Histone acetyltransferases |
| HDACs | Histone deacetyltransferases |
| CpG | Cytosine-phosphate-guanine |
| LncRNAs | Long non-coding RNAs |
Prostate cancer (PCa) is a type of malignant tumor that develops within the prostate tissue, Its pathogenesis is very complex and related to many factors [1,2], For example, diet [3,4], genetic or epigenetic [5–7], microbiota [8,9], inflammation [10], obesity [11] and race [12]. As reported in the Global Cancer Statistics 2020, PCa ranks as the second most prevalent solid tumor among men. Each year, millions of men worldwide succumb to PCa, making it the fifth leading cause of cancer-related fatalities [13,14]. Prostate, lung, and colorectal cancers account for almost half (48%) of all male cancer cases in the US in 2023, and prostate cancer alone accounts for 29% of diagnosed cases [15]. The incidence of prostate cancer is mainly in Europe and the United States, and the incidence of prostate cancer in China is increasing year by year [16,17]. Prostate cancer that is localized may be managed through radical surgery or radiation therapy. The five-year survival rate is nearly 100%, while the recurrence rate is approximately 30% to 40% [18–20]. If prostate cancer (PCa) has already spread beyond its original site, the preferred treatment is drug therapy aimed at providing relief [21,22]. The advancement and progression of PCa heavily rely on androgen receptor (AR) signaling, with androgen deprivation therapy (ADT) being the gold standard for treating metastatic PCa. Although there is a favorable initial response from the tumor to ADT, progression typically resumes after approximately 2 to 3 years, leading to the emergence of incurable castration-resistant prostate cancer (CRPC). Nonetheless, the five-year survival rate for patients diagnosed with metastatic PCa and CRPC is only about 30% [18,19].
When the patient’s disease develops into CRPC, the treatment mainly includes taxane chemotherapeutic drugs (such as docetaxel, and cabazitaxel); androgen receptor-targeted drugs (ARTA), such as enzalutamide and abiraterone acetate, and the recent poly ADP-ribose polymerase (PARP) inhibitor [23]. CRPC includes nonmetastatic castration-resistant disease (nmCRPC) and metastatic castration-resistant disease (mCRPC) [24]. In the past few years, Rosellini et al. showed that androgen receptor (AR) axis receptor inhibitors have significantly benefited from the results of nmCRPC. The administration of enzalutamide alongside androgen deprivation therapy (ADT) is linked to extended metastasis-free survival (MFS) and a favorable safety profile. The addition of apalutamide to the ongoing ADT significantly improved MFS and symptom progression time in patients with high-risk nmCRPC, showing an increased OS (25% lower risk of death compared to placebo). The addition of darolutamide to ADT ensures prolonged MFS and OS high-risk non-metastatic diseases. In addition, when darolutamide is analyzed alongside the previously mentioned enzalutamide and apalutamide, it appears to be associated with improved safety and a reduced incidence of adverse events [24]. In the early 2000s, mitoxantrone emerged as the initial cytotoxic chemotherapy for advanced diseases resistant to ADT. While overall survival did not show significant differences between men treated with mitoxantrone and those who were not, it enhanced palliative treatment for symptomatic cases of mCRPC [25]. Since 2004, taxane chemotherapy drugs have been widely studied to prolong the OS of mCRPC patients. Although prostate cancer is in a castration state, the AR axis continues to be a significant factor in the advancement of prostate cancer. In patients who have had prior treatment with docetaxel, abiraterone acetate has been shown to enhance OS and progression-free survival (PFS) when compared to treatment with prednisone alone. Recent studies have also validated the possible advantages of PARP inhibitors, like olaparib, in the context of prostate cancer [24,26].
In addition, antibody-drug conjugates of cytotoxic drugs (also known as payloads), are linked to specific antibodies that can recognize antigens expressed on the surface of cancer cells [24,27]. Regarding prostate cancer, scientists are exploring the possibility of applying this treatment to the condition [28–30]. Nevertheless, the existing mCRPC therapy has notably extended the lifespan of patients. Currently, there are several treatments available for metastatic castration-resistant prostate cancer (mCRPC), including PARP inhibitors (PARPi) [31–33], androgen receptor signaling inhibitors (ARSI) [33,34], taxane chemotherapeutic agents [35,36], and radium-223 [37,38]. These treatments have significantly improved the survival lifetime of prostate cancer patients [39–42]. The lasting survival advantages provided by these medications remain constrained, necessitating further investigation into more effective options.
Isothiocyanate represents one of the numerous investigations demonstrating that dietary choices may help mitigate both the onset and advancement of prostate cancer [43–45]. Sulforaphane (SFN, 1-isothiocyanato-4-(methanesulfinyl)butane) is a phytochemical belonging to the isothiocyanate family, and it is present in consumable cruciferous vegetables like broccoli, cauliflower, and cabbage [46–50], It exists in form of glucoraphanin (glucosinolate conjugate) and is produced in the catalytic reaction of thioglycosidase (myrosinase). There is no endogenous activity in mammalian cells and myrosinase enzymes are present in plants or intestinal biota, where it is physically separated through the plant cell wall and thioglucoside and is released after damage to the plant caused by cutting or chewing [51]. Talalay and Zhang were the first to isolate it from broccoli and prove its anticancer properties [52]. Its biological precursor glucoraphanin was subsequently found in large quantities in broccoli buds, and SFN was confirmed to be active in animal carcinogenic models [53]. Posner et al. evaluated the structural activity of more than 100 synthetic analogues and found no more effective phase II detoxification enzyme inducer than SFN. SFN is still one of the most effective natural inducers found so far [54]. Subsequently, studies have found that this molecule has a variety of pathways and metabolisms in mammalian cells, tissues, and humans, and has strong anticancer properties [55]. Preventing cancer can be achieved by hindering the proliferation of cancer cells, obstructing the cell cycle, and promoting apoptosis. SFN provides cancer protection by altering several epigenetic and non-epigenetic mechanisms. SFN can block the activity of histone deacetylase deacetylases are important in the prevention of cancer because they enhance multiple mechanisms such as apoptosis and cell cycle arrest. In addition, SFN also prevents histone phosphorylation by enhancing phosphatases [56]. The anticancer properties of SFN emphasize its potential as a versatile and potent agent against various malignant tumors. Its impact on the proliferation, migration, and drug resistance of cancer cells presents a potential avenue for developing innovative treatment strategies and enhancing patient prognosis. As early as 2000, studies have shown that SFN can reduce the risk of prostate cancer [57].
Studies have shown that SFN prevents prostate cancer mainly through antioxidants [58], inhibition of fatty acid metabolism [59], inhibition of glycolysis [60], inhibition of pro-inflammatory factors [61], inhibition of cell proliferation and induction of apoptosis [62,63], reduction of androgen receptors [64], and influence of epigenetics [49,65].
Prostate Cancer-Preventive Effects of SFN
Oxidative stress plays a crucial role in the onset and development of prostate cancer [66–69]. It typically denotes the disruption between the production of reactive oxygen species and the antioxidant defenses, or the capability to oxidize beyond the existing antioxidant capacity. Reactive oxygen species (ROS) are characterized as unstable and highly reactive molecules, commonly existing as superoxide anion, hypochlorous acid, hydrogen peroxide, singlet oxygen, hypochlorite, hydroxyl radicals, and lipid peroxides, which are involved in cell processes such as growth, differentiation, and death [70]. Under physiological conditions, the human body has a solid antioxidant system, including antioxidant enzymes and some small molecules, the main antioxidant enzymes are phase II detoxifying enzymes: for example, glutathione S-transferase (GST), NAD (P)H quinone oxidoreductase(NQO), epoxide hydrolase, heme oxygenase 1 (HO-1), UDP-glucuronosyltransferases (UGT), superoxide dismutase (SOD), phase II detoxification enzymes are redox-sensitive stress-inducing proteins downstream of the Nrf2-Keap1 axis that reduces various oxidative stress and inflammation-derived cytotoxicity [71,72]. Numerous elements, both inherent to the cells (Nrf2 deficiency; DNA) and from the external environment (chronic; radiation), can result in elevated ROS generation in the prostate. Elevated levels of ROS can cause prostate dysfunction, which subsequently leads to even greater ROS production [73]. An antioxidant defense system, which can be enzymatic or non-enzymatic, helps to counteract and regulate the levels of reactive oxygen species (ROS) in order to sustain physiological homeostasis. Reducing ROS levels below the homeostatic threshold might disrupt both proliferation and the host defense mechanisms. Conversely, an accumulation of ROS in the prostate can disturb its normal functioning, resulting in a decreased antioxidant capacity by interfering with the Nrf2-antioxidant response element axis (ARE), increasing mitochondrial DNA (mtDNA) mutation and aggressive phenotypes, and causing DNA damage [73]. SFN is an antioxidant that, once in the cell from the blood, reacts spontaneously with glutathione and other thiols, enhancing the oxidative state of the cell and leading to a transient increase in reactive oxygen species. This change in oxidation status led to rapid activation of the Nrf2-antioxidant response element defense system (ARE), leading to increased transcription of many genes involved in restoring the redox state of cells and preventing ROS damage [74,75]. In homeostasis, Nrf2 is associated with cytoplasmic protein KEAP1, which is continuously ubiquitous by Cul3 E3 ubiquitin ligase and subsequently degraded by proteases. In the presence of oxidative stress, conformational changes KEAP1 can occur directly with Sulfhydryl KEAP1 or indirectly through changes in the cellular redox state, leading to Nrf2 separation from KEAP1, metastasis to the nucleus, heterogeneity with the small molecule myofascial fibrosarcoma (sMaf) protein, and binding to ARE, leading to a large number of gene transcription and antioxidant responses [76,77].
SFN activates Nrf2, leading to structural changes in KEAP1, disrupting the KEAP1-Nrf2 complex, blocking Nrf2 ubiquitination from being degraded, and promoting Nrf2 nuclear translocation [77], which enhances the expression of phase II detoxification enzymes GST, HO-1, and NQO-1, causing an enhanced antioxidant effect (Fig. 1). Excitingly, the effect of adding SFN to human prostate cells and the increased activity of phase II detoxification enzymes (GST, NQO1) after tube feeding of SFN in F-344 rats [72,78]. Significantly enhanced protein expression of Nrf2 and NOQ1 was similarly observed in transgenic adenocarcinoma mouse prostate (TRAMP)-C1 cell lines after SFN treatment [79].
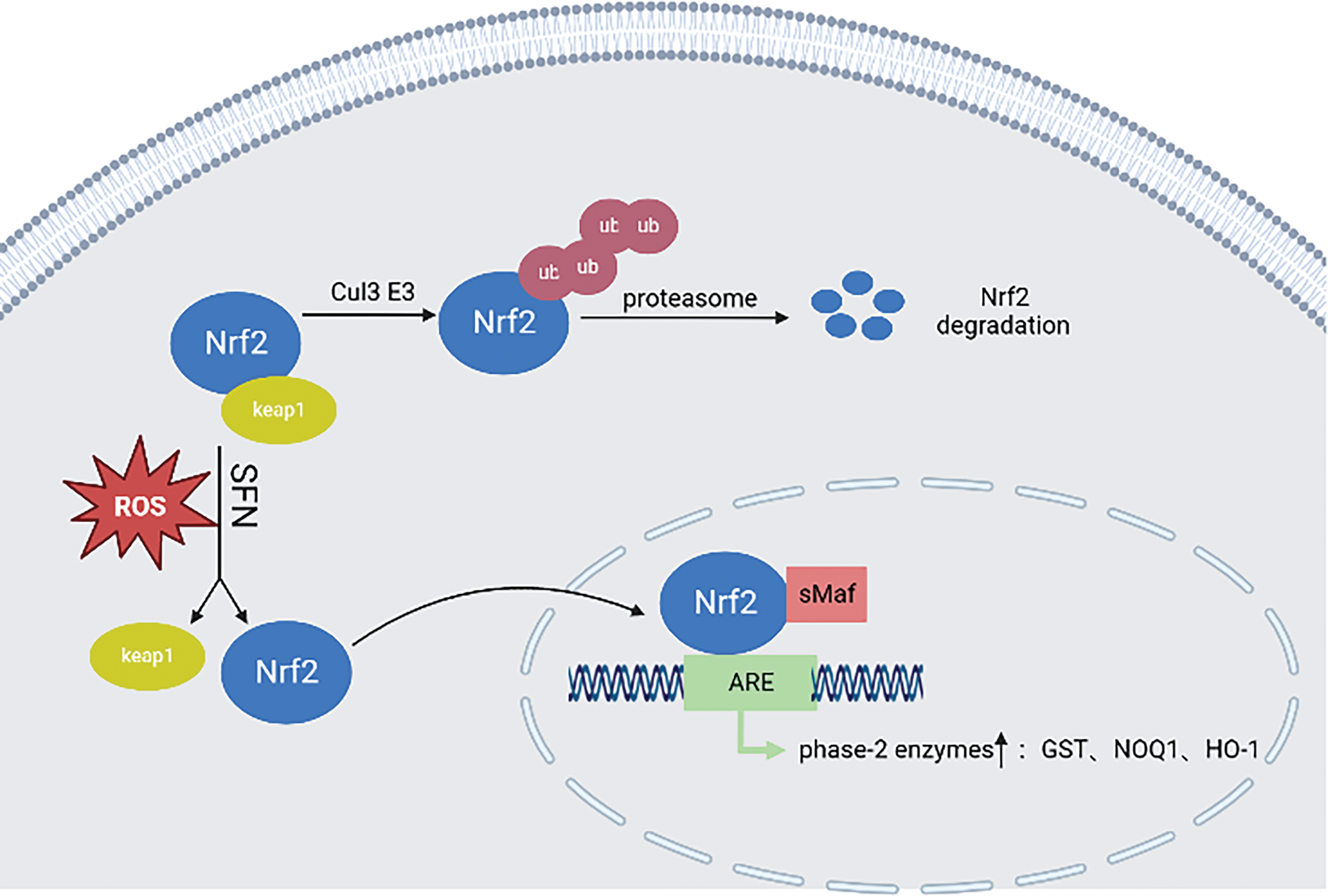
Figure 1: In the steady state, Nrf2 is linked to KEAP1, continuously ubiquitinated by Cul3 E3 ubiquitin ligase, and then degraded by proteasome. In the presence of oxidative stress, the conformational change of KEAP1 will be induced, which will lead to the separation of Nrf2 and KEAP1, translocation to the nucleus, and small molecule myofascial fibrosarcoma (sMaf) protein heterodimerization and binding to ARE, leading to the transcription and antioxidant response of a large number of genes. SFN can activate Nrf2, lead to the change of KEAP1 structure, destroy the KEAP1-Nrf2 complex, block the degradation of Nrf2 ubiquitination, and promote Nrf2 nuclear translocation, thereby enhancing the expression of phase II detoxification enzymes GST, HO-1, NQO-1, etc. This illustration was generated utilizing resources accessible on BioRender.com.
Inhibition of fatty acid metabolism
It has been shown that ab initio synthesis of fatty acids is necessary for prostate cancer growth and that lipid metabolism and genes related to lipid metabolism can play an important role in the progression of prostate cancer through metabolic pathways and anti-apoptotic effects [80–83]. The key enzyme for fatty acid synthesis is fatty acid synthase (FASN), a 250–270 kDa cytoplasmic protein, and fatty acid synthase is involved in the final step in the ab initio synthesis of fatty acids (i.e., condensation of acetyl coenzyme-A and malonyl coenzyme-A to produce palmitate), which in turn generates more types of fatty acids, which undergo β-oxidation catalyzed by the key enzyme carnitine palmitoyltransferase 1A (CPT1A). It also plays an important function in energy homeostasis, converting excess carbon into fatty acid stores and supplying energy through β-oxidation when necessary. The mechanisms regulating the expression of fatty acid synthases are complex and not fully understood, and their expression and activity are regulated by growth factors, hormones, and dietary factors, among others [84]. The expression of FASN is low in most human tissues and high in liver and adipose tissue [85]. It has been shown that FASN gene expression is upregulated in approximately one-quarter of human prostate cancer patients [86]. Migita et al. showed that FASN overexpression leads to prostate intraepithelial neoplasia (PIN) progression [87]. However, the regulation of fatty acid synthase mechanisms is unknown, cytotoxicity can be produced on tumor cells through the use of FASN inhibitors [88], and because of this, FSAN is considered a potential new target for the treatment of many cancer cell line (22Rv1), Singh et al. found that SFN could reduce the protein and mRNA levels of acetyl-CoA carboxylase 1 (ACC1A), FASN and CPT1A, and the expression of FASN and ACC1A protein in TRAMP mouse model treated with SFN was also significantly reduced chemoprevention of prostate cancer is associated with the inhibition of fatty acid synthesis and its β-oxidation [59] (Fig. 2).
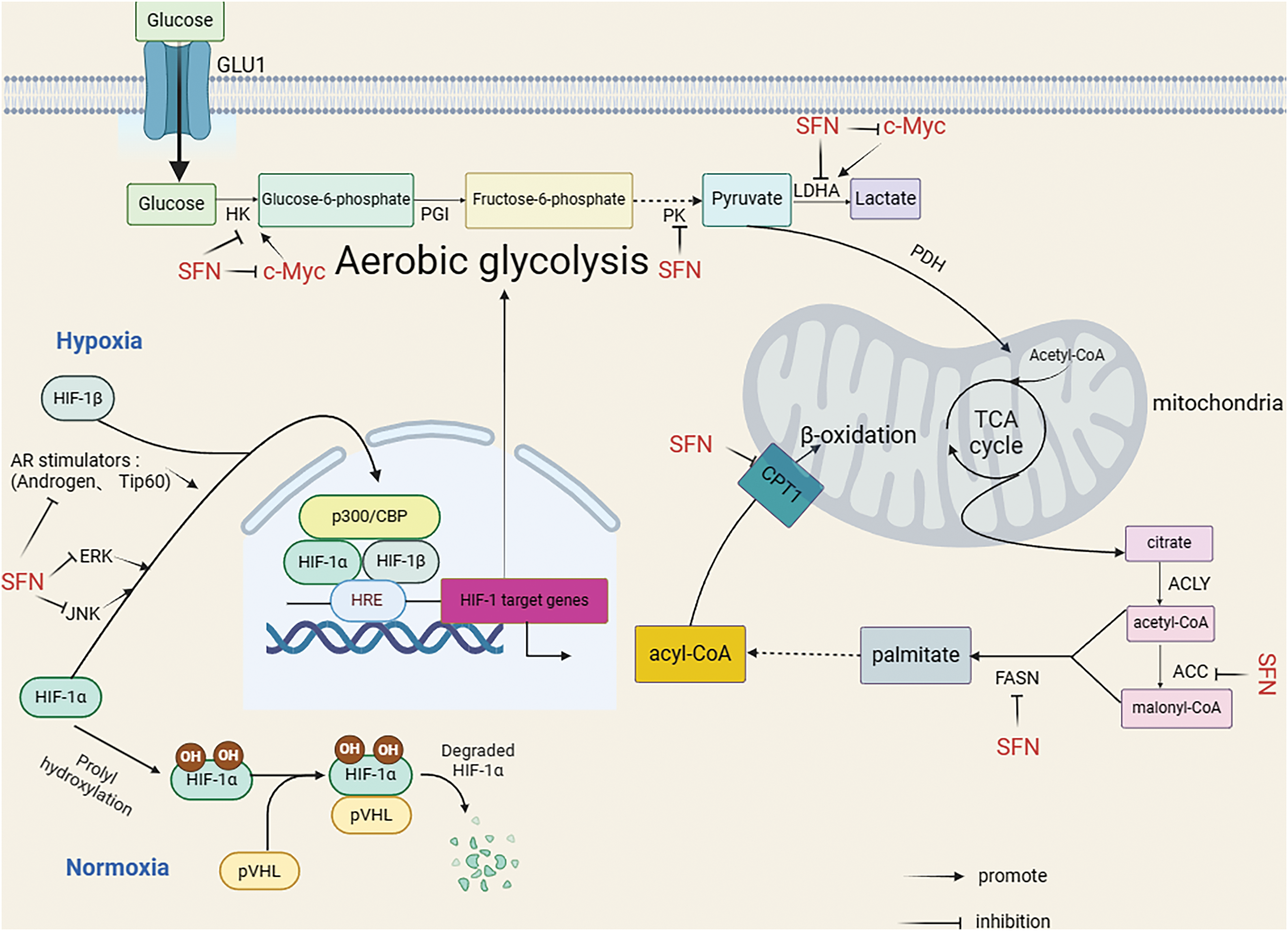
Figure 2: SFN indirectly inhibited HK and LDHA or directly inhibited HK and LDHA by inhibiting c-Myc, and also directly inhibited PK. SFN inhibits hypoxia-induced HIF-1α expression by inhibiting JNK, ERK pathways, and AR stimulators (Androgen, Tip60). In addition, SFN can inhibit ACC, FASN, and CPT1 to inhibit β-oxidation. This illustration was generated utilizing resources accessible on BioRender.com.
In normal cells, glucose undergoes initial metabolism to pyruvate within the cytoplasm; under aerobic circumstances, pyruvate is further converted to carbon dioxide through the process of oxidative phosphorylation (OXPHOS). In contrast, when oxygen is absent, pyruvate is transformed into lactate through glycolysis, thereby generating ATP [89]. In cancer cells, the energy supply comes mainly from aerobic glycolysis and tumor cells convert the major ATP pathway OXPHOS to aerobic glycolysis also known as the Warburg effect [89]. The conversion from glucose to lactate requires the involvement of several enzymes such as hexokinase (HK), phosphofructokinase (PFK), pyruvate kinase (PK), and lactate dehydrogenase (LDH) [90]. In Singh et al.’s study, it was found that SFN significantly downregulated the expression of hexokinase 2 (HK2), pyruvate kinase M2 (PKM2) and/or lactate dehydrogenase A (LDHA) in mouse TRAMP model and prostate tumor lesions in Hi-Myc mice in-vitro and in-vivo, and significantly inhibited glycolysis in the prostate of Hi-Myc mice, reversing Warburg phenomenon in prostate cancer [60]. Warburg effect is considered to be a central component of tumor metabolic recoding and is mainly associated with overexpression of the transcription factor hypoxia-inducible factor HIF-1. HIF-1 is mainly composed of two subunits, HIF-1α and HIF-1β. Under normoxic conditions, HIF-1α is hydroxylated by proline and recognized with the Von Hippel-Lindau Disease Tumor Suppressor (pVHL) complex, which exhibits ubiquitin ligase activity that ubiquitinates HIF-1α, causing its degradation through the proteasomal pathway. Under hypoxic conditions, proline hydroxylation is inhibited, resulting in the accumulation of HIF-α translocated into the nucleus and HIF-1β forming a dimer with the hypoxia response elements (HREs) binding and recruitment of transcriptional co-activators (e.g., the histone acetyltransferases CBP/P300) thus obtaining complete transcriptional activity [91]. SFN can affect directly and/or indirectly affect glycolysis in prostate cancer (Fig. 2). Findings from a study conducted on the DU145 cell line related to human prostate cancer indicated that SFN may reduce the expression of HIF-1α induced by hypoxia by inhibiting the pathways of Jun N-terminal kinase (JNK) and extracellular regulated protein kinase (ERK) [92]. In addition, Carrasco et al. showed that SFN blocked androgen receptor agonist (androgen and Tip60)-induced glycolysis in human prostate cancer LNCaP cells, and that hexokinase and acetone kinase activities increased to reduce HIF-1α stability [93]. In addition, Myc genes can upregulate genes of the glycolytic pathway (HK2, LDHA) to enhance glucose metabolism [94], and one study reported SFN-mediated inhibition of c-Myc protein levels in human prostate cancer cell lines (LNCaP, PC-3, C4-2) and prostate adenocarcinoma of a Hi-Myc transgenic mouse(Myc-CaP cell line) [95].
Inhibition of pro-inflammatory factors
Inflammation has a role in cancer in regulating the tumor microenvironment (TME) and promoting tumor proliferation and migration [96–99]. For example, the cytokine interleukin-6 (IL-6) can promote prostate cancer cell proliferation and inhibit apoptosis through a variety of cellular signaling pathways [100]. Nuclear transcription factor-κB (NF-κB) is an inducible protein transcription factor, mainly formed as a heterodimer by p50 and p65 of the Rel protein family. NF-κB can be activated by a variety of stimuli, such as tumor necrosis factor α (TNF-α), interleukin-1 (IL-1), lipopolysaccharide (LPS), ultraviolet (UV) and oxidative stress, etc. Activation of NF-κB by extracellular stimuli leads to phosphorylation, ubiquitination, and protein degradation of IκB kinase, which exposes the nuclear localization signal on NF-κB, resulting in nuclear translocation of the NF-κB complex and phosphorylation of p65. NF-κB regulates different inflammatory responses and immune responses and can also promote tumor progression by controlling tumor angiogenesis through upregulation of vascular endothelial growth factor (VEGF) and receptors [101,102], and Huang et al. have also shown that transfection of the highly metastatic prostate cancer cell line PC-3M with mutated IκBα (blocking NF-κB activity) injected into the prostate of nude mice revealed significant inhibition of major pro-angiogenic molecules such as VEGF, IL-8, and Matrix metalloproteinase 9 (MMP-9), as well as downregulation of MMP-9 mRNA and collagenase activity, and in general, blocking NF-κB signaling inhibited tumor invasion, angiogenesis, and metastasis [103]. In conclusion, NF-κB activation is common in cancer and is thought to be a key link between inflammation and cancer, with inflammatory factors in the tumor microenvironment again being the most common factors that enable NF-κB activation. Interestingly, SFN is known for its anti-inflammatory effects. Following stress, bacterial, viral, and pro-inflammatory cytokine-related cellular stimulation, IκB kinase is phosphorylated and then the kinase is degraded, which allows free translocation of NF-κB dimers into the nucleus and induces transcription of pro-inflammatory cytokines (IL-6, IL-10, TNF-α) [104]. It was shown that SFN could reduce inflammation by inhibiting the binding of NF-κB to DNA [105]. In addition, it has also been shown that SFN inhibits IκB kinase complex (IKK) phosphorylation in human prostate cancer PC-3 cells, specifically inhibiting IKKβ, leading to IKKβ-mediated inhibition of IκBα phosphorylation. This in turn leads to reduced ubiquitination and protein degradation of IκBα, with subsequent retention of NF-κB in the cytoplasm and reduced nuclear translocation of p65 and consequent attenuation of NF-κB-regulated VEGF, cyclin D1, and B-cell lymphoma/leukemia-2 protein X-linked (Bcl-XL) gene expression [106]. The stimulation of tumor cell proliferation, apoptosis, or mutations in cancer cells can be prompted by the activation of NF-κB and the subsequent cascade of inflammatory cytokines or chemokines [107]. Consequently, blocking the activation of NF-κB plays a crucial role in mitigating detrimental effects. SFN notably reduces the levels of several inflammatory mediators, including IL-6, IL-1β, TNF-α, nitric oxide (NO), and prostaglandin E2 (PGE2), along with inflammatory enzymes such as inducible NO synthase (iNOS) and cyclooxygenase 2 (COX-2), through the suppression of the NF-κB signaling pathway [61,105].
In addition, Toll-like receptors (TLR) are key pattern recognition receptors (PRR) that induce innate or adaptive immune responses, whereas TLR-4 is a key signaling receptor that triggers inflammation. LPS recognizes CD14/TLR-4/MD-2 heterotrimers and activates myeloid differentiation factor 88 (MyD88)-dependent pathways and TRIF (MyD88-independent pathway) to trigger inflammation.TLR4 activation also increases the expression of VEGF and transforming growth factor-β1 (TGF-β1) in prostate cancer cells, thus promoting tumor development [108]. It has been shown that SFN inhibits the TLR4/MyD88 pathway and reduces TNF-α and IL-6 levels [109]. In macrophages, HIF-1 can upregulate TLR4 expression, while SFN can inhibit hypoxia and CoCl2-induced TLR4 expression upregulation by inhibiting phosphatidylinositol 3 kinase/protein kinase B (PI3K/AKT) pathway and HIF-1α activation [110]. It has also been shown that the anti-inflammatory mechanism of action of SFN is closely related to the inhibition of TLR4 response by directly and indirectly targeting the TLR4-md2 receptor complex [111,112]. In addition, signal transducer and activator of transcription 3 (STAT-3) is closely associated with inflammation, and tumor promoters, lipopolysaccharide, and cigarette smoke can activate the STAT-3 signaling pathway, and STAT-3 binds competitively with NF-κB at overlapping DNA binding sites [113], and studies have shown that SFN reduces STAT-3 in the prostate cancer cell lines DU145 and LNCap expression [114]. In conclusion, SFN can inhibit pro-inflammatory factors through various targets to disrupt the tumor microenvironment in prostate cancer (Fig. 3).
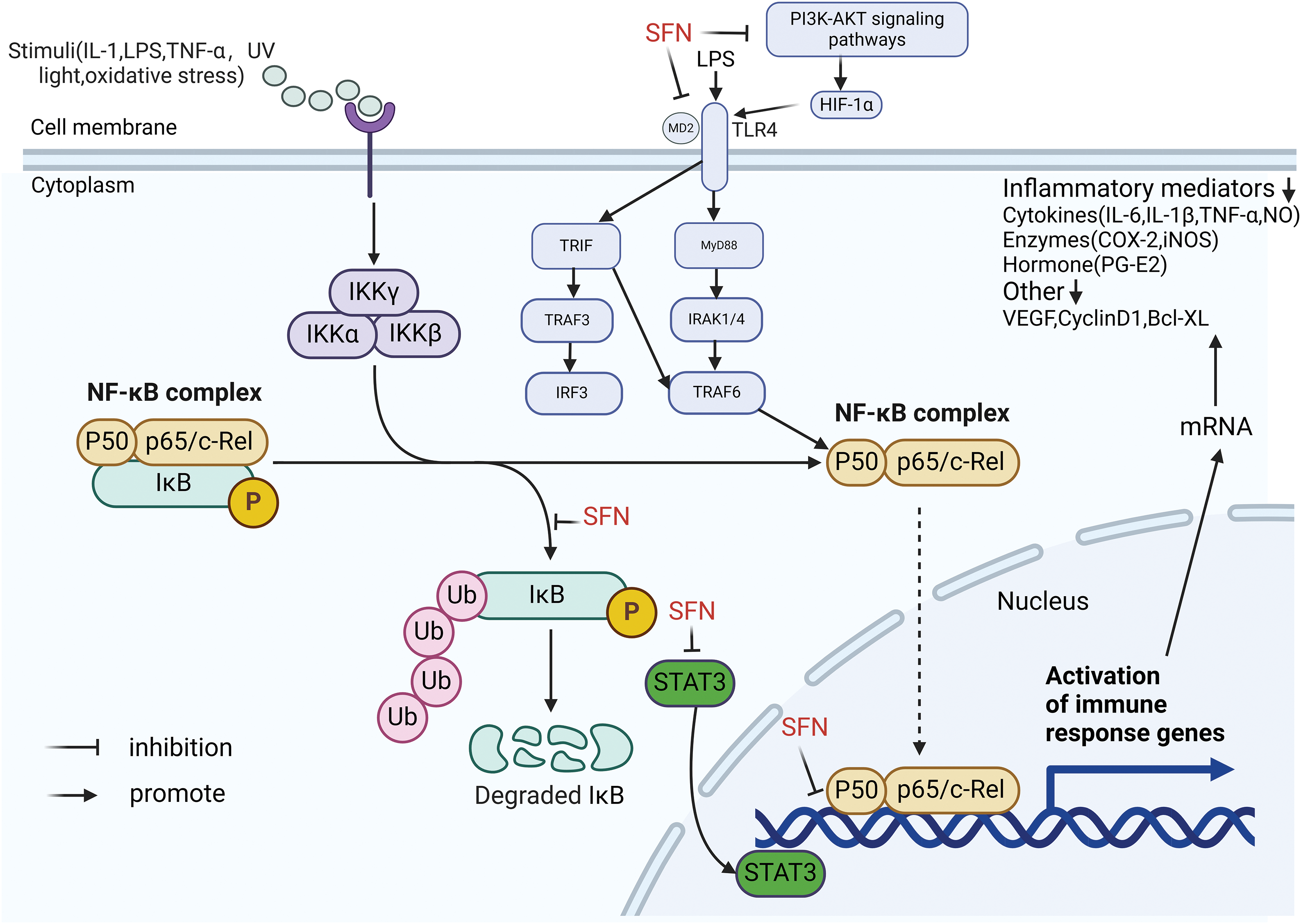
Figure 3: SFN can inhibit the phosphorylation of IκB kinase and the expression of STAT3; SFN inhibited hypoxia and CoCl2-induced upregulation of TLR4 expression by inhibiting PI3K/AKT pathway and HIF-1α activation; SFN inhibits TLR4-md2 receptor complex by direct and indirect targeting. Inhibiting the activation of the NF-κB signaling pathway by the above three methods significantly attenuated various inflammatory mediators, such as IL-6, IL-1β, TNF-α, nitric oxide (NO), and prostaglandin E2 (PGE2), as well as inflammatory enzymes: inducible NO synthase (iNOS) and cyclooxygenase 2 (COX-2). This illustration was generated utilizing resources accessible on BioRender.com.
Inhibition of cell proliferation and induction of apoptosis in cancer cells
Numerous experimental studies, both in vivo and in vitro, have demonstrated that SFN and its metabolites can inhibit cell proliferation and induce apoptosis in prostate cancer (Fig. 4). Induction of G2/M phase cell cycle arrest in SFN-treated human prostate cancer cell line (PC-3) involves checkpoint kinase 2 (ChK2) checkpoint activation leading to phosphorylation of cell division cycle 25C (cdc25C), resulting in its segregation in the cytoplasm [115]. SFN was also found to upregulate CD44 variants v4, v5, and v7 to slow down the proliferative activity of tumor cells in human prostate cancer cell lines (DU-145 and PC-3 cell lines) in-vitro, and also upregulate the tumor suppressor p19 in blocking cell cycle and apoptosis [116]. Administration of SFN in a PTEN gene-deficient mouse model reversed the effects of early prostate cancer development due to PTEN deficiency and served to inhibit cell proliferation [117].
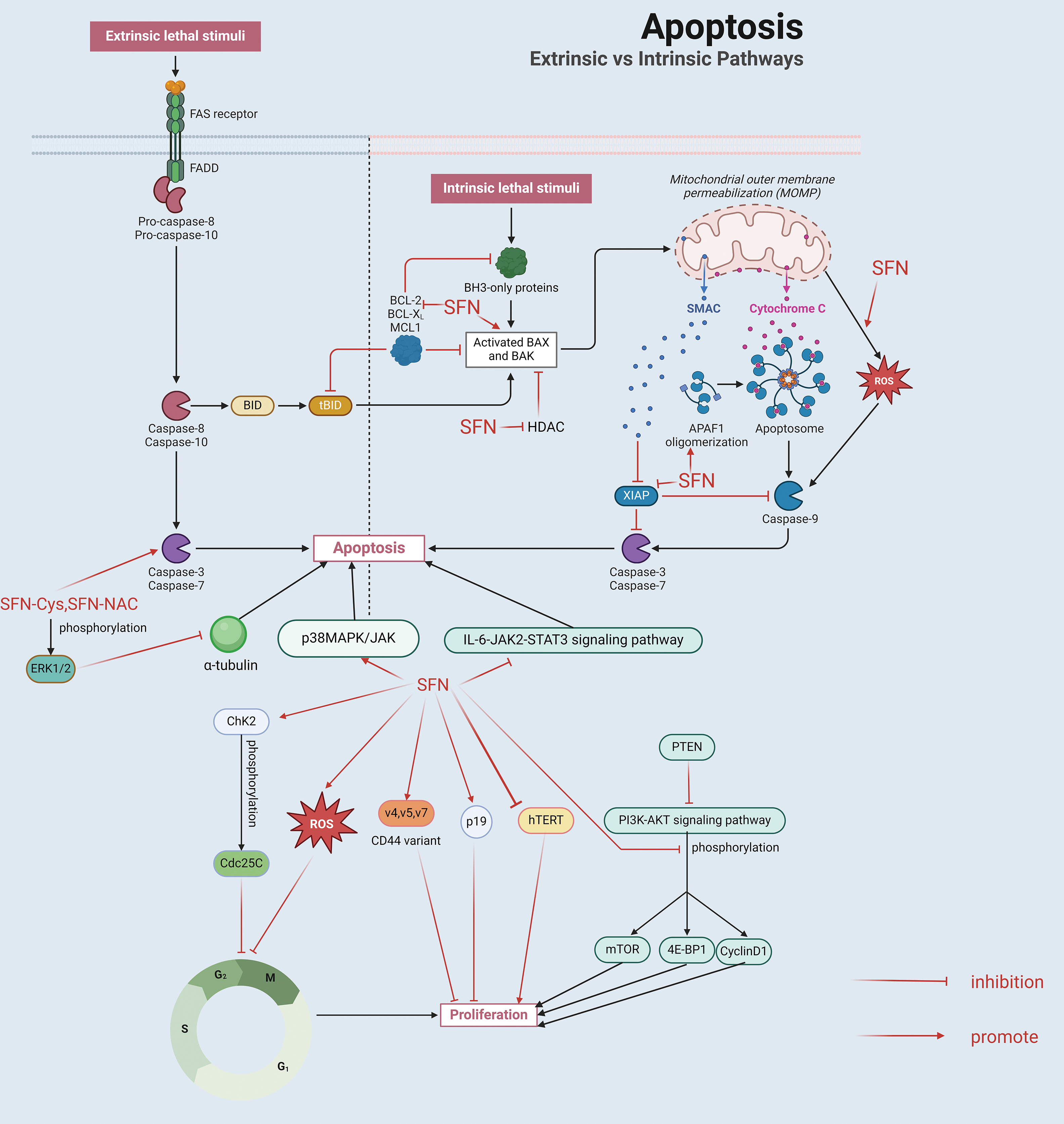
Figure 4: 1. Inhibition of cell proliferation: (1) SFN involves checkpoint kinase 2 (ChK2) checkpoint activation leading to phosphorylation of cell division cycle 25C (cdc25C), (2) SFN up-regulates CD44 variants v4, v5 and v7, (3) SFN inhibits the phosphorylation of AKT and its downstream kinases (mTOR, 4E-BP1) and target protein (cyclin D1), (4) SFN induces ROS production, (5) SFN inhibits hTERT expression. 2. Promoting apoptosis: (1) The production of ROS induced by SFN is associated with the disruption of mitochondrial membrane potential and the subsequent release of cytochrome C from the mitochondria into the cytoplasm, which results in the activation of caspase 9 and ultimately causes cell death; (2) SFN enhances the expression of the pro-apoptotic protein Bax while diminishing the levels of the anti-apoptotic protein Bcl-2 within the Bcl-2 protein family. This change results in a higher Bax to Bcl-2 ratio, which in turn triggers the activation of caspase-9. Additionally, SFN stimulates the activity of caspase-8 and caspase-9, leading to the activation of caspase-3, which subsequently cleaves the DNA repair enzyme PARP; (3) SFN can mediate the activation of caspase by inducing Apaf and down-regulating XIAP; (4) SFN promotes apoptosis by inhibiting the IL-6/JAK2/STAT3 signaling pathway; SFN metabolites SFN-cysteine (SFN-Cys) and SFN-N-acetylcysteine (SFN-NAC) activate caspase 3 and induce phosphorylation of protein kinase 1/2 (ERK1/2), which down-regulates α-tubulin; (5) SFN can activate p38 MAPK and JNK to induce apoptosis. This illustration was generated utilizing resources accessible on BioRender.com.
Phosphorylated expression of AKT protein (cell cycle protein D1) was reduced after feeding high doses of broccoli sprouts in a TRAMP mouse model, thereby inhibiting cell proliferation [118]. SFN was also found to cause G2/M phase cell cycle arrest in wild-type LNCap cell lines, but not in its variant Rh-0 cells (mitochondrial DNA deletion), thus SFN causes G2/M phase cell cycle arrest in human prostate cancer cells due to mitochondria-derived ROS-mediated [119]. Moreover, telomerase consists of a complex of ribonucleoproteins in eukaryotes that includes six distinct subunits: heat shock protein 90, the human telomerase RNA component (hTERC), dyskerin, the telomerase-associated protein 1 (TEP1), p23, and the human telomerase reverse transcriptase (hTERT). The activity of the hTERT gene is absent in normal human cells; however, it is frequently expressed in different forms of cancers (such as:prostate cancer), signifying a necessary role in the unlimited proliferation of tumor cells [120]. It has been shown that SFN can inhibit hTERT expression in prostate cancer cells in two prostate cancer cell lines, LNCap and PC-3 [121].The apoptosis triggered by SFN is primarily facilitated through intrinsic mitochondrial pathways as well as extrinsic death receptors (DRs) mechanisms [122]. Intrinsic mitochondrial-mediated apoptosis begs the question of mitochondrial autophagy, a catabolic process in which the autophagic system targets damaged mitochondria and delivers them to lysosomes for degradation. Mitochondrial autophagy helps control the quality and quantity of mitochondria [123]. Mitochondrial autophagy is one of the organelle-specific autophagic pathways that are used to maintain cell structure and function [124]. Mitochondria are believed to be the primary location for the production of ROS, primarily via the electron transport chain and various localized proteins, and overproduction of ROS occurs when mitochondrial dysfunction occurs, and overproduction of ROS further damages mitochondria, creating a vicious cycle [125]. Excessive intracellular ROS production and imbalance of antioxidant capacity cause oxidative stress, which not only leads to mitochondrial dysfunction, but excessive accumulation of damaged mitochondria may lead to apoptosis. In turn, mitochondrial autophagy and tumor cell suppression are closely related [126].
Extrinsic death receptors mainly include Tumor necrosis factor-related apoptosis-inducing ligand 1 (TNFR1), Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand 1 (TRAIL1) receptors (DR-4, DR-5), and Fas (Apo-1; CD95) mediated [122]. Research indicates that the cell death induced by SFN in human prostate cancer cells is attributed to the generation of ROS. This ROS production triggered by SFN is associated with a disruption of the mitochondrial membrane potential, which in turn facilitates the release of cytochrome C from the mitochondria into the cytoplasm. This process culminates in the activation of caspase 9, ultimately resulting in cell death [127]. It has also been reported that in human prostate cancer cell line PC-3, SFN-induced apoptosis is associated with upregulation of B-cell lymphoma-2 Associated X Protein (Bax), downregulation of B-cell lymphoma-2 Bcl-2) and activation of caspase-3, caspase-8 and caspase-9. SFN causes upregulation of the pro-apoptotic protein Bax and downregulation of the anti-apoptotic protein Bcl-2 in the Bcl-2 protein family will lead to an increase in the Bax to Bcl-2 ratio, making activation of the mitochondrial pathway for cytochrome c release, which activates caspase-9; and then caspase-8 and caspase-9 activate caspase 3 to shear the DNA repair enzyme PARP, leading to apoptosis [128]. In addition, research involving mouse embryonic fibroblasts (MEFs) revealed that SFN plays a role in triggering caspase activation, which in turn facilitates apoptosis by enhancing the expression of Apoptotic protease activating factor (Apaf) and decreasing levels of X-linked inhibitor of apoptosis protein (XIAP) [129].
In addition, in BPH1, PC3 and LNCap prostate cancer cells, SFN induced apoptosis by inhibiting HDAC activity, inducing cell cycle arrest through upregulation of mRNA and protein levels of p21 and Bax, and activating caspase [130]. Hahm et al. in DU145 and LNCap human prostate cancer cell lines found that SFN induced apoptosis by inhibiting IL-6/JAK2/STAT3 signaling pathway to promote apoptosis [114]. It has also been shown in DU-145 and PC-3 cells that the SFN metabolites SFN-cysteine (SFN-Cys) and SFN-N-acetylcysteine (SFN-NAC) cause apoptosis by activation of caspase 3 and induction of protein kinase 1/2 (ERK1/2) phosphorylation, resulting in downregulation of α-microtubulin [131]. SFN was found to activate p38 Mitogen-activated protein kinase (p38MAPK), JNK leading to apoptosis in a PC-3 prostate cancer cell line [132]. In studies on androgen-independent prostate cancer cell lines (PC-3, DU145) and androgen-dependent prostate cancer cell lines (VcaP), SFN was found to have cytotoxic and pro-apoptotic effects on prostate cancer cell lines [62].
Prostate cancer initially develops in a manner dependent on androgens, driven by the signaling pathway of the androgen receptor (AR), which is critical not only for normal prostate development and physiological function, but also for the proliferation, survival, invasion, and clonogenic capacity of PCa cells [133–135]. SFN has been shown to reduce AR protein in LNCap and C4-2 cell lines by inhibiting transcription of androgen receptor mRNA, leading to Ser210/213 phosphorylation and reduced total androgen receptor [64] and increasing proteasomal degradation of androgen receptor protein [136]. SFN was shown in LNCap cells and Vcap cells to increase HSP90 acetylation and lead to AR and HSP90 dissociation by inhibiting HDAC6 activity in cells, decreasing AR protein levels and reducing AR target gene expression [136]. In 22Rv1 cell line (a CRPC cell line expressing AR-FL and multiple AR splice variants), it was demonstrated that SFN can reduce AR-FL and AR-V7 levels, and this reduction in AR protein levels may result in diminished cell proliferation, migration and cloning capacity [137]. SFN was found to reduce AR protein levels through AR protein degradation and inhibition of AR gene expression in androgen-dependent cell lines (LNCap) and androgen-independent cell lines (C4-2B) [138]. Another interesting study in a TRAMP mouse model found that SFN treatment with Nrf2 levels similar to ADT treatment reduced ROS levels in prostate cancer cells and caused prostate cancer cells to show similar sensitivity to radiotherapy as with ADT treatment [139].
Influence on epigenetic inheritance
In epigenetics, histone modifications, DNA methylation, RNA regulation and nucleosome remodeling are considered to be important influential mechanisms that are dysregulated in cancer [140–142], as well as in prostate cancer [143–145]. Modifications of histones include acetylation, methylation, phosphorylation, and ubiquitination, among which histone acetylation plays the most prominent role. Histone acetyltransferases (HATs) and histone deacetyltransferases (HDACs) are in balance in normal cells, and when histone acetyltransferase expression is decreased and/or histone deacetyltransferase expression is increased is closely related to the process of tumorigenesis and progression, while SFN inhibits histone deacetylases (HDACs), histone deacetylase (HDAC) overexpression will lead to histone deacetylation, and deacetylation causes DNA to wrap around histones too tightly, thus inhibiting gene expression, which may lead to cancer development if the affected genes are oncogenes [146]. Zhang et al. treating TRAMP-C1 cell lines with 1.0, 2.5 μM SFN respectively found that SFN decreased HDAC1, HDAC4, HDAC5, HDAC7 proteins were reduced in a dose-dependent manner [79]. SFN inhibited the epigenetic regulator HDAC3 in PC-3 cells and decreased HDAC3 expression in TRAMP mice [147]. In prostate cancer cells PC3 and LnCap, as well as in BPH1, SFN similarly suppressed HDAC activity, which resulted in heightened global histone acetylation. This enhancement promoted the interaction between acetylated histone H4 and the P21 and Bax promoters, leading to an upregulation of both mRNA and protein levels of p21 and Bax [130].
DNA methylation occurs mainly at cytosine residues of cytosine-phosphate-guanine (CpG) dinucleotides and is regulated by DNA methylation transferase, and DNA hypermethylation leads to gene silencing. In prostate cancer, overall hypomethylation of tumor-associated genes and site-specific hypermethylation influence tumorigenesis and progression [148], with hypomethylation associated with genomic instability, transposons, and proto-oncogene activation, and hypermethylation can silence genes involved in cancer protection, with targets including those involved in DNA repair, detoxification, and apoptosis [148]. In previous studies treating TRAMP-C1 cell lines with 1.0 and 2.5 μM SFN respectively found that SFN reduced the protein levels of DNMT1 and DNMT3a in DNA methylation transferase (DNMT) in a dose-dependent manner [79]. It was also shown that SFN treatment of LNCap cell lines inhibited DNMT1 and DNMT3b expression, and SFN treatment of benign prostatic hyperplasia cells (BPH-1) and PC-3 cells significantly inhibited DNMT1 and DNMT3a mRNA expression, while protein expression only showed a downward trend. The expression of cell cycle protein D2 mRNA increased after SFN treatment in LNCap cell lines, which served to inhibit prostate cancer growth [149]. Furthermore, SFN in both LNCap and PC-3 prostate cancer cell lines by regulating and reversing aberrant promoter DNA methylation in chemokine-related gene targets in cancer cells [150].
Long non-coding RNAs (lncRNAs) are closely associated with prostate cancer development and progression [151,152]. Treatment with SFN in a study of LNCap and PC-3 cell lines was found to inhibit the expression of the long non-coding RNA (lncRNA) LINC01116 [65].
In addition to the above pathways through which SFN inhibits prostate cancer, it has been found that the SFN metabolite SFN-Cys causes sustained phosphorylation of ERK1/2 and triggers downregulation of galectin-1 (an invasion-associated protein) in DU145 and PC-3 cells, which in turn inhibits invasion [153]. SFN inhibits invasion by regulating E-calcine mucin (an invasion inhibitor), CD44v6 (invasion promoter) and MMP-2 (invasion promoter) to inhibit invasion [154]. In the advancement of lung metastasis, by diminishing cell proliferation and boosting the lytic activity of NK cells [155].
Singh et al. found that SFN treatment of TRAMP mice led to increased IL-12 production by dendritic cells. This effect leads to increased cytotoxicity of NK cells to prostate cancer cell lines, and improves the activation efficiency of dendritic cells with cytotoxic function of NK cells after co-culture. Aside from the effects observed in-vitro regarding NK cells and dendritic cells due to SFN administration, researchers noted a rise in T cell infiltration in the prostate tumors of TRAMP mice treated with SFN when contrasted with control mice. This research demonstrates that SFN has the potential to boost the immune response in prostate tumors by prompting dendritic cells to secrete IL-12, which leads to heightened NK cell cytotoxicity and, consequently, an increase in T cell presence within prostate tumors, ultimately contributing to a decrease in tumor burden and metastasis [155]. As we described earlier that the c-Myc gene is associated with cancer stem cells in addition to glycolysis [156], SFN also inhibits c-Myc protein levels to impair prostate cancer stem cell (pCSC) capacity in LNCaP, PC-3 and Myc-CaP cells [95]. It has also been observed in PC-3 cell lines that SFN inhibits protein synthesis and is accompanied by a reduction in mTOR substrate phosphorylation [157].
The value of SFN in the prevention of prostate cancer is positive, but its dose, bioavailability and safety still need further study. A study showed that the average concentration of glucoraphanin in broccoli seeds was 0.38 μmol/g. In clinical trials, the dose of glucoraphanin supplementation was 25–800 μmol broccoli [158]. However, it is not realistic to eat such a large amount of broccoli every day. Other ways to supplement SFN can be considered, such as using dried broccoli buds and seed extracts to make capsules or beverage preparations.
Secondly, in terms of bioavailability, the rapid and unstable metabolism of SFN in the body requires cryopreservation, which poses a challenge for research use in animals and humans [159]. Its precursor glucoraphanin is relatively inert and water-soluble, which is converted into sulforaphane by intestinal bacteria and myrosinase of the plant itself. The results showed that the conversion rate of SFN was only 10% when broccoli sprout extract was given. When myrosinase and glucoraphanin were administered simultaneously, the bioavailability was up to 35%–40% [160]. Unfortunately, glucoraphanin seems to be absorbed and excreted faster in the human body than SFN, and its inter-individual variability is smaller. This means that the SFN supplement study may be easier to accurately assess the dose [158].
However, as a disease prevention substance, its safety is also a problem that we must consider. In another clinical study, BSE containing 200 μmol SFN was supplemented every day, and no adverse events above grade 3 were found in the subjects [49]. At present, the exact effective and toxic doses of SFN remain undetermined. Whether there will be safety problems when the daily supplemental dose is greater than the above dose needs more tests to explore.
Viewed through an economic lens, SFN, being a natural food item, is both safer and more affordable compared to other cancer-fighting medications. Future research on SFN certainly merits further investigation.
As the second largest solid tumor in men, the treatment cost and time of PCa have become a huge burden for patients and a major burden on the global economy. At present, the treatment of PCa is progressing very rapidly, but there are still many deficiencies, especially in the prognosis of patients with mPCa and CRPC, which is still a major problem in treatment. Therefore, there is an urgent need for a safe, environmentally friendly, and effective natural compound to treat PCa on multiple potential targets. In addition, as a natural compound, the prevention of PCa is particularly critical. More and more studies have shown that SFN can play a huge advantage in the prevention and treatment of Pca. We summarized the research mechanism of prostate cancer in SFN, as shown in Table 1.
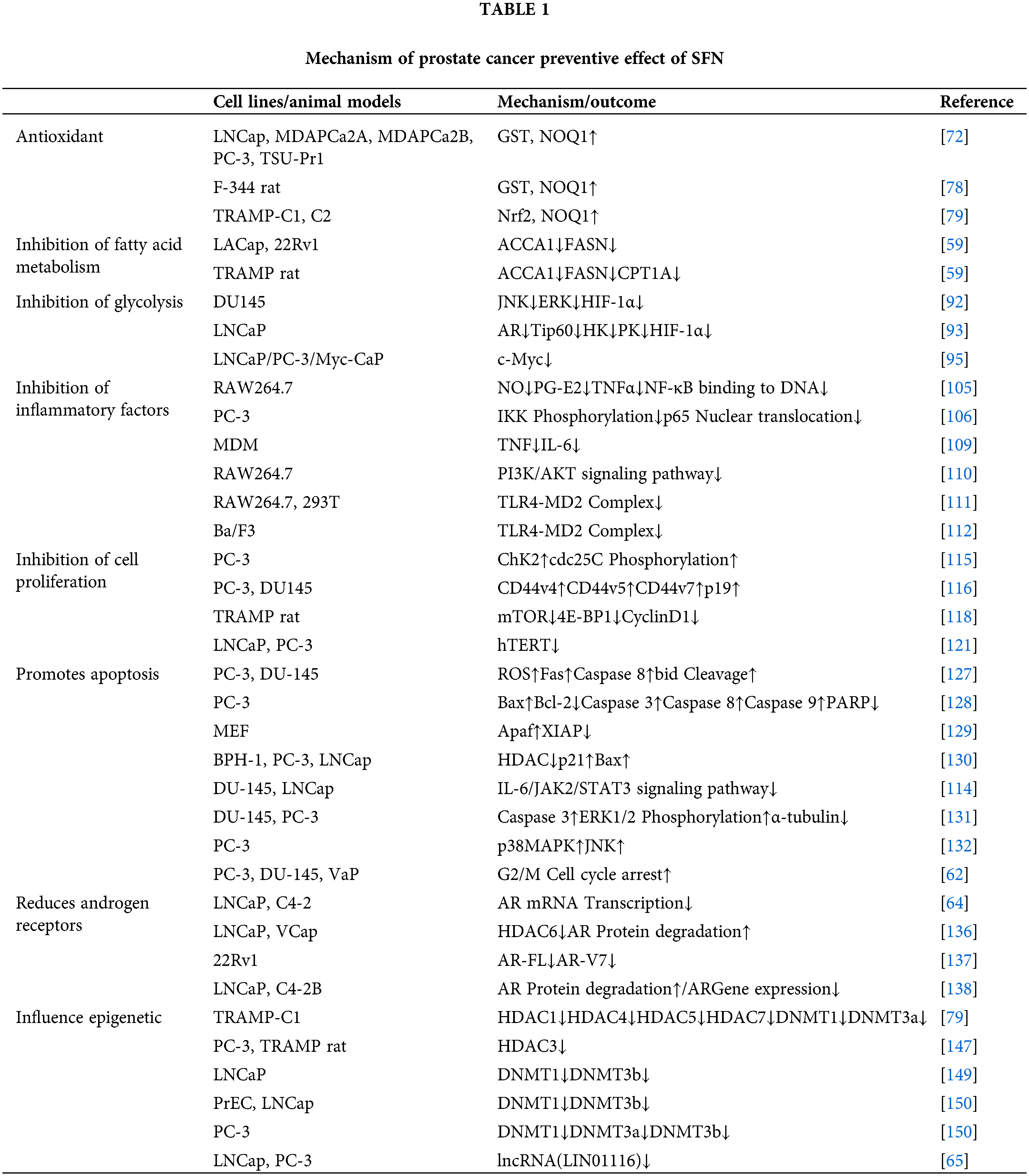
SFN can enhance the expression of phase II detoxification enzymes GST, HO-1, NQO-1, etc., thus playing an antioxidant role. Lipid metabolism and its related genes can play an important role in the progression of PCa through metabolic pathways and anti-apoptotic effects. However, SFN can inhibit the metabolism of fatty acids, which is critical for the inhibition of PCa. More excitingly, SFN can significantly down-regulate a variety of key enzymes in aerobic glycolysis hypoxia-induced HIF-1α expression. SFN significantly attenuated various inflammatory mediators signaling pathways, thereby destroying the tumor microenvironment. SFN can inhibit cell proliferation by affecting the phosphorylation of cdc25C, and lead to apoptosis of tumor cells through PI3K-AKT-mTOR signaling pathway, IL-6/JAK2/STAT3 signaling pathway, p38MAPK and JNK pathways.
In cell and animal experiments, SFN has a significant effect on the prevention of PCa. In recent years, more and more clinical trials have explored the relationship between SFN and PCa, and many studies have confirmed its preventive mechanism. However, unfortunately, the current research on the key factors such as effective chemopreventive dose, bioavailability, toxic dose, and response of SFN in the human body is still insufficient. Whether as a natural substance for prevention or treatment, these factors are particularly important. Therefore, we look forward to more clinical trials to clarify these issues in order to achieve greater therapeutic effect.
Acknowledgement: None.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: Study conception and design: Huanglin Duan, Baisheng Xu; draft manuscript preparation: Huanglin Duan; review and editing: Huanglin Duan, Baisheng Xu, Tianpeng Xie; visualization: Baisheng Xu, Tianpeng Xie; supervision: Huanglin Duan. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The datasets generated during and/or analysed during the current study are available in the PubMed repository.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Wasim S, Lee SY, Kim J. Complexities of prostate cancer. Int J Mol Sci. 2022;23(22):14257. doi:10.3390/ijms232214257. [Google Scholar] [PubMed] [CrossRef]
2. Sekhoacha M, Riet K, Motloung P, Gumenku L, Adegoke A, Mashele S. Prostate cancer review: genetics, diagnosis treatment options, and alternative approaches. Molecules. 2022;27(17):5730. doi:10.3390/molecules27175730. [Google Scholar] [PubMed] [CrossRef]
3. Matsushita M, Fujita K, Nonomura N. Influence of diet and nutrition on prostate cancer. Int J Mol Sci. 2020;21(4):1447. doi:10.3390/ijms21041447. [Google Scholar] [PubMed] [CrossRef]
4. Wang M, Jian Z, Yuan C, Jin X, Li H, Wang K. Coffee consumption and prostate cancer risk: results from national health and nutrition examination survey 1999–2010 and mendelian randomization analyses. Nutrients. 2021;13(7):2317. doi:10.3390/nu13072317. [Google Scholar] [PubMed] [CrossRef]
5. Vietri MT, D’Elia G, Caliendo G, Resse M, Casamassimi A, Passariello L, et al. Hereditary prostate cancer: genes related, target therapy and prevention. Int J Mol Sci. 2021;22(7):3753. doi:10.3390/ijms22073753. [Google Scholar] [PubMed] [CrossRef]
6. Rebello RJ, Oing C, Knudsen KE, Loeb S, Johnson DC, Reiter RE, et al. Prostate cancer. Nat Rev Dis Primers. 2021;7(1):9. doi:10.1038/s41572-020-00243-0. [Google Scholar] [PubMed] [CrossRef]
7. Ge R, Wang Z, Montironi R, Jiang Z, Cheng M, Santoni M, et al. Epigenetic modulations and lineage plasticity in advanced prostate cancer. Ann Oncol. 2020;31(4):470–9. doi:10.1016/j.annonc.2020.02.002. [Google Scholar] [PubMed] [CrossRef]
8. Rizzo A, Santoni M, Mollica V, Fiorentino M, Brandi G, Massari F. Microbiota and prostate cancer. Semin Cancer Biol. 2022;86:1058–65. doi:10.1016/j.semcancer.2021.09.007. [Google Scholar] [PubMed] [CrossRef]
9. Pernigoni N, Guo C, Gallagher L, Yuan W, Colucci M, Troiani M, et al. The potential role of the microbiota in prostate cancer pathogenesis and treatment. Nat Rev Urol. 2023;20(12):706–18. doi:10.1038/s41585-023-00795-2. [Google Scholar] [PubMed] [CrossRef]
10. Mughees M, Kaushal JB, Sharma G, Wajid S, Batra SK, Siddiqui JA. Chemokines and cytokines: axis and allies in prostate cancer pathogenesis. Semin Cancer Biol. 2022;86:497–512. doi:10.1016/j.semcancer.2022.02.017. [Google Scholar] [PubMed] [CrossRef]
11. Wilson RL, Taaffe DR, Newton RU, Hart NH, Lyons-Wall P, Galvao DA. Obesity and prostate cancer: a narrative review. Crit Rev Oncol Hematol. 2022;169:103543. doi:10.1016/j.critrevonc.2021.103543. [Google Scholar] [PubMed] [CrossRef]
12. Arenas-Gallo C, Owiredu J, Weinstein I, Lewicki P, Basourakos SP, Vince RJr., et al. Race and prostate cancer: genomic landscape. Nat Rev Urol. 2022;19(9):547–61. doi:10.1038/s41585-022-00622-0. [Google Scholar] [PubMed] [CrossRef]
13. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics 2021. CA Cancer J Clin. 2021;71(1):7–33. doi:10.3322/caac.v71.1. [Google Scholar] [CrossRef]
14. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209–49. doi:10.3322/caac.v71.3. [Google Scholar] [CrossRef]
15. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73(1):17–48. doi:10.3322/caac.v73.1. [Google Scholar] [CrossRef]
16. Gandaglia G, Leni R, Bray F, Fleshner N, Freedland SJ, Kibel A, et al. Epidemiology and prevention of prostate cancer. Eur Urol Oncol. 2021;4(6):877–92. doi:10.1016/j.euo.2021.09.006. [Google Scholar] [PubMed] [CrossRef]
17. Liu J, Dong L, Zhu Y, Dong B, Sha J, Zhu HH, et al. Prostate cancer treatment–China’s perspective. Cancer Lett. 2022;550:215927. doi:10.1016/j.canlet.2022.215927. [Google Scholar] [PubMed] [CrossRef]
18. Miller KD, Nogueira L, Devasia T, Mariotto AB, Yabroff KR, Jemal A, et al. Cancer treatment and survivorship statistics, 2022. CA Cancer J Clin. 2022;72(5):409–36. doi:10.3322/caac.v72.5. [Google Scholar] [CrossRef]
19. Beier AK, Puhr M, Stope MB, Thomas C, Erb HHH. Metabolic changes during prostate cancer development and progression. J Cancer Res Clin Oncol. 2023;149(5):2259–70. doi:10.1007/s00432-022-04371-w. [Google Scholar] [PubMed] [CrossRef]
20. Tilki D, Chen MH, Wu J, Huland H, Graefen M, Wiegel T, et al. Adjuvant versus early salvage radiation therapy for men at high risk for recurrence following radical prostatectomy for prostate cancer and the risk of death. J Clin Oncol. 2021;39(20):2284–93. doi:10.1200/JCO.20.03714. [Google Scholar] [PubMed] [CrossRef]
21. Achard V, Putora PM, Omlin A, Zilli T, Fischer S. Metastatic prostate cancer: treatment options. Oncology. 2022;100(1):48–59. doi:10.1159/000519861. [Google Scholar] [PubMed] [CrossRef]
22. Lowrance W, Dreicer R, Jarrard DF, Scarpato KR, Kim SK, Kirkby E, et al. Updates to advanced prostate cancer: AUA/SUO guideline. 2023 J Urol. 2023;209(6):1082–90. [Google Scholar] [PubMed]
23. Mollica V, Marchetti A, Rosellini M, Nuvola G, Rizzo A, Santoni M, et al. An insight on novel molecular pathways in metastatic prostate cancer: a focus on DDR, MSI and AKT. Int J Mol Sci. 2021;22(24):13519. doi:10.3390/ijms222413519. [Google Scholar] [PubMed] [CrossRef]
24. Rosellini M, Santoni M, Mollica V, Rizzo A, Cimadamore A, Scarpelli M, et al. Treating prostate cancer by antibody-drug conjugates. Int J Mol Sci. 2021;22(4):1551. doi:10.3390/ijms22041551. [Google Scholar] [PubMed] [CrossRef]
25. Tannock IF, Osoba D, Stockler MR, Ernst DS, Neville AJ, Moore MJ, et al. Chemotherapy with mitoxantrone plus prednisone or prednisone alone for symptomatic hormone-resistant prostate cancer: a Canadian randomized trial with palliative end points. J Clin Oncol. 1996;14(6):1756–64. doi:10.1200/JCO.1996.14.6.1756. [Google Scholar] [PubMed] [CrossRef]
26. de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. 2020;382(22):2091–102. doi:10.1056/NEJMoa1911440. [Google Scholar] [PubMed] [CrossRef]
27. Sardinha M, Palma Dos Reis AF, Barreira JV, Fontes Sousa M, Pacey S, Luz R. Antibody-drug conjugates in prostate cancer: a systematic review. Cureus. 2023;15(2):e34490. [Google Scholar] [PubMed]
28. Khongorzul P, Ling CJ, Khan FU, Ihsan AU, Zhang J. Antibody-drug conjugates: a comprehensive review. Mol Cancer Res. 2020;18(1):3–19. doi:10.1158/1541-7786.MCR-19-0582. [Google Scholar] [PubMed] [CrossRef]
29. Danila DC. Antibody-drug conjugates in prostate cancer. Clin Adv Hematol Oncol. 2020;18(8):447–9. [Google Scholar] [PubMed]
30. Mollica V, Rizzo A, Montironi R, Cheng L, Giunchi F, Schiavina R, et al. Current strategies and novel therapeutic approaches for metastatic urothelial carcinoma. Cancers. 2020;12(6):1449. doi:10.3390/cancers12061449. [Google Scholar] [PubMed] [CrossRef]
31. Taylor AK, Kosoff D, Emamekhoo H, Lang JM, Kyriakopoulos CE. PARP inhibitors in metastatic prostate cancer. Front Oncol. 2023;13:1159557. doi:10.3389/fonc.2023.1159557. [Google Scholar] [PubMed] [CrossRef]
32. Longoria O, Beije N, de Bono JS. PARP inhibitors for prostate cancer. Semin Oncol. 2024;51(1–2):25–35. [Google Scholar] [PubMed]
33. Messina C, Giunta EF, Signori A, Rebuzzi SE, Banna GL, Maniam A, et al. Combining PARP inhibitors and androgen receptor signalling inhibitors in metastatic prostate cancer: a quantitative synthesis and meta-analysis. Eur Urol Oncol. 2024;7(2):179–88. doi:10.1016/j.euo.2023.07.013. [Google Scholar] [PubMed] [CrossRef]
34. Ai J, Jian L, Wen X, Huo X, Yang X, Jiang J, et al. Comparative effectiveness of first-line systemic treatments for metastatic castration-resistant prostate cancer: a systematic review and network meta-analysis. Clin Transl Oncol. 2024;26(10):2559–71. doi:10.1007/s12094-024-03506-4. [Google Scholar] [PubMed] [CrossRef]
35. Thomas C. Role of chemotherapy in metastatic castration-resistant prostate cancer (mCRPC) treatment: still standard or exception? Urologie. 2023;62(12):1281–8. [Google Scholar] [PubMed]
36. Mellgard G, Saffran N, Chakrani Z, McCroskery S, Taylor N, Patel M, et al. Performance status and end-of-life outcomes in patients with metastatic castration-resistant prostate cancer treated with androgen receptor targeted therapy. Am J Clin Oncol. 2024;47(10):459–64. doi:10.1097/COC.0000000000001115. [Google Scholar] [PubMed] [CrossRef]
37. Frantellizzi V, Monari F, Mascia M, Costa R, Rubini G, Spanu A, et al. A national multicenter study on overall survival in elderly metastatic castrate-resistant prostate cancer patients treated with radium-223. Aging Clin Exp Res. 2021;33(3):651–8. doi:10.1007/s40520-020-01573-5. [Google Scholar] [PubMed] [CrossRef]
38. Konig F, Strauss A, Johannsen M, Mommsen C, Fricke E, Klier J, et al. Radium-223 for the treatment of metastatic castration-resistant prostate cancer (mCRPCthe androgen receptor-independent active agent in the therapeutic sequence. Urologe A. 2020;59(1):53–64. [Google Scholar] [PubMed]
39. Cai M, Song XL, Li XA, Chen M, Guo J, Yang DH, et al. Current therapy and drug resistance in metastatic castration-resistant prostate cancer. Drug Resist Updat. 2023;68:100962. doi:10.1016/j.drup.2023.100962. [Google Scholar] [PubMed] [CrossRef]
40. Henriquez I, Roach M, Morgan TM3rd, Bossi A, Gomez JA, Abuchaibe O, et al. Current and emerging therapies for metastatic castration-resistant prostate cancer (mCRPC). Biomedicines. 2021;9(9):1247. doi:10.3390/biomedicines9091247. [Google Scholar] [PubMed] [CrossRef]
41. Hatano K, Nonomura N. Systemic therapies for metastatic castration-resistant prostate cancer: an updated review. World J Mens Health. 2023;41(4):769–84. doi:10.5534/wjmh.220200. [Google Scholar] [PubMed] [CrossRef]
42. Yan K, Balijepalli C, Gullapalli L, Joshy J, Kotum S, Druyts E. Efficacy and safety of interventions for metastatic castration resistant prostate cancer (mCRPC) patients progressing on androgen receptor-axis-targeted (ARAT) therapy: a systematic literature review. Curr Med Res Opin. 2024:1–12. [Google Scholar]
43. Habib TN, Altonsy MO, Ghanem SA, Salama MS, Hosny MA. Optimizing combination therapy in prostate cancer: mechanistic insights into the synergistic effects of Paclitaxel and Sulforaphane-induced apoptosis. BMC Mol Cell Biol. 2024;25(1):5. doi:10.1186/s12860-024-00501-z. [Google Scholar] [PubMed] [CrossRef]
44. Zhang F, Wan X, Zhan J, Shen M, Li R. Sulforaphane inhibits the growth of prostate cancer by regulating the microRNA-3919/DJ-1 axis. Front Oncol. 2024;14:1361152. doi:10.3389/fonc.2024.1361152. [Google Scholar] [PubMed] [CrossRef]
45. Livingstone TL, Saha S, Bernuzzi F, Savva GM, Troncoso-Rey P, Traka MH, et al. Accumulation of Sulforaphane and alliin in human prostate tissue. Nutrients. 2022;14(16):3263. doi:10.3390/nu14163263. [Google Scholar] [PubMed] [CrossRef]
46. Janczewski L. Sulforaphane and its bifunctional analogs: synthesis and biological activity. Molecules. 2022;27(5):1750. doi:10.3390/molecules27051750. [Google Scholar] [PubMed] [CrossRef]
47. Schepici G, Bramanti P, Mazzon E. Efficacy of sulforaphane in neurodegenerative diseases. Int J Mol Sci. 2020;21(22):8637. doi:10.3390/ijms21228637. [Google Scholar] [PubMed] [CrossRef]
48. Zhang Y, Wu Q, Liu J, Zhang Z, Ma X, Zhang Y, et al. Sulforaphane alleviates high fat diet-induced insulin resistance via AMPK/Nrf2/GPx4 axis. Biomed Pharmacother. 2022;152:113273. doi:10.1016/j.biopha.2022.113273. [Google Scholar] [PubMed] [CrossRef]
49. Zhang Z, Garzotto M, Davis EW, Mori M, Stoller WA, Farris PE, et al. Sulforaphane bioavailability and chemopreventive activity in men presenting for biopsy of the prostate gland: a randomized controlled trial. Nutr Cancer. 2020;72(1):74–87. doi:10.1080/01635581.2019.1619783. [Google Scholar] [PubMed] [CrossRef]
50. Bousquet J, Anto JM, Czarlewski W, Haahtela T, Fonseca SC, Iaccarino G, et al. Cabbage and fermented vegetables: from death rate heterogeneity in countries to candidates for mitigation strategies of severe COVID-19. Allergy. 2021;76(3):735–50. doi:10.1111/all.v76.3. [Google Scholar] [CrossRef]
51. Vanduchova A, Anzenbacher P, Anzenbacherova E. Isothiocyanate from broccoli, sulforaphane, and its properties. J Med Food. 2019;22(2):121–6. doi:10.1089/jmf.2018.0024. [Google Scholar] [PubMed] [CrossRef]
52. Zhang Y, Talalay P, Cho CG, Posner GH. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc Natl Acad Sci U S A. 1992;89(6):2399–403. doi:10.1073/pnas.89.6.2399. [Google Scholar] [PubMed] [CrossRef]
53. Fahey JW, Zhang Y, Talalay P. Broccoli sprouts: an exceptionally rich source of inducers of enzymes that protect against chemical carcinogens. Proc Natl Acad Sci U S A. 1997;94(19):10367–72. doi:10.1073/pnas.94.19.10367. [Google Scholar] [PubMed] [CrossRef]
54. Posner GH, Cho CG, Green JV, Zhang Y, Talalay P. Design and synthesis of bifunctional isothiocyanate analogs of sulforaphane: correlation between structure and potency as inducers of anticarcinogenic detoxication enzymes. J Med Chem. 1994;37(1):170–6. doi:10.1021/jm00027a021. [Google Scholar] [PubMed] [CrossRef]
55. Otoo RA, Allen AR. Sulforaphane’s multifaceted potential: from neuroprotection to anticancer action. Molecules. 2023;28(19):6902. doi:10.3390/molecules28196902. [Google Scholar] [PubMed] [CrossRef]
56. Asif Ali M, Khan N, Kaleem N, Ahmad W, Alharethi SH, Alharbi B, et al. Anticancer properties of sulforaphane: current insights at the molecular level. Front Oncol. 2023;13:1168321. doi:10.3389/fonc.2023.1168321. [Google Scholar] [PubMed] [CrossRef]
57. Cohen JH, Kristal AR, Stanford JL. Fruit and vegetable intakes and prostate cancer risk. J Natl Cancer Inst. 2000;92(1):61–8. doi:10.1093/jnci/92.1.61. [Google Scholar] [PubMed] [CrossRef]
58. Ruhee RT, Suzuki K. The integrative role of sulforaphane in preventing inflammation, oxidative stress and fatigue: a review of a potential protective phytochemical. Antioxidants. 2020;9(6):521. doi:10.3390/antiox9060521. [Google Scholar] [PubMed] [CrossRef]
59. Singh KB, Kim SH, Hahm ER, Pore SK, Jacobs BL, Singh SV. Prostate cancer chemoprevention by sulforaphane in a preclinical mouse model is associated with inhibition of fatty acid metabolism. Carcinogenesis. 2018;39(6):826–37. doi:10.1093/carcin/bgy051. [Google Scholar] [PubMed] [CrossRef]
60. Singh KB, Hahm ER, Alumkal JJ, Foley LM, Hitchens TK, Shiva SS, et al. Reversal of the Warburg phenomenon in chemoprevention of prostate cancer by sulforaphane. Carcinogenesis. 2019;40(12):1545–56. doi:10.1093/carcin/bgz155. [Google Scholar] [PubMed] [CrossRef]
61. Ruhee RT, Ma S, Suzuki K. Sulforaphane protects cells against lipopolysaccharide-stimulated inflammation in murine macrophages. Antioxidants. 2019;8(12):577. doi:10.3390/antiox8120577. [Google Scholar] [PubMed] [CrossRef]
62. Dogan Sigva ZO, Balci Okcanoglu T, Biray Avci C, Yilmaz Susluer S, Kayabasi C, Turna B, et al. Investigation of the synergistic effects of paclitaxel and herbal substances and endemic plant extracts on cell cycle and apoptosis signal pathways in prostate cancer cell lines. Gene. 2019;687(4):261–71. doi:10.1016/j.gene.2018.11.049. [Google Scholar] [PubMed] [CrossRef]
63. Tuttis K, Machado ART, Santos P, Antunes LMG. Sulforaphane combined with vitamin D induces cytotoxicity mediated by oxidative stress, DNA damage, autophagy, and JNK/MAPK pathway modulation in human prostate tumor cells. Nutrients. 2023;15(12):2742. doi:10.3390/nu15122742. [Google Scholar] [PubMed] [CrossRef]
64. Kim SH, Singh SV. D, L-Sulforaphane causes transcriptional repression of androgen receptor in human prostate cancer cells. Mol Cancer Ther. 2009;8(7):1946–54. doi:10.1158/1535-7163.MCT-09-0104. [Google Scholar] [PubMed] [CrossRef]
65. Beaver LM, Kuintzle R, Buchanan A, Wiley MW, Glasser ST, Wong CP, et al. Long noncoding RNAs and sulforaphane: a target for chemoprevention and suppression of prostate cancer. J Nutr Biochem. 2017;42:72–83. doi:10.1016/j.jnutbio.2017.01.001. [Google Scholar] [PubMed] [CrossRef]
66. Ahmed Amar SA, Eryilmaz R, Demir H, Aykan S, Demir C. Determination of oxidative stress levels and some antioxidant enzyme activities in prostate cancer. Aging Male. 2019;22(3):198–206. doi:10.1080/13685538.2018.1488955. [Google Scholar] [PubMed] [CrossRef]
67. Miao M, Song Y, Jin M, Du Y, Xin P, Jiang Y, et al. Single-cell RNA combined with bulk RNA analysis to explore oxidative stress and energy metabolism factors and found a new prostate cancer oncogene MXRA8. Aging. 2024;16(5):4469–502. [Google Scholar] [PubMed]
68. Rago V, Di Agostino S. Novel insights into the role of the antioxidants in prostate pathology. Antioxidants. 2023;12(2):289. doi:10.3390/antiox12020289. [Google Scholar] [PubMed] [CrossRef]
69. Dickinson K, Case AJ, Kupzyk K, Saligan L. Exploring biologic correlates of cancer-related fatigue in men with prostate cancer: cell damage pathways and oxidative stress. Biol Res Nurs. 2020;22(4):514–9. doi:10.1177/1099800420933347. [Google Scholar] [PubMed] [CrossRef]
70. Tan BL, Norhaizan ME, Liew WP. Nutrients and oxidative stress: friend or foe? Oxid Med Cell Longev. 2018;2018:9719584. doi:10.1155/omcl.v2018.1. [Google Scholar] [CrossRef]
71. Fahey JW, Talalay P. Antioxidant functions of sulforaphane: a potent inducer of Phase II detoxication enzymes. Food Chem Toxicol. 1999;37(9–10):973–9. [Google Scholar] [PubMed]
72. Brooks JD, Paton VG, Vidanes G. Potent induction of phase 2 enzymes in human prostate cells by sulforaphane. Cancer Epidemiol Biomarkers Prev. 2001;10(9):949–54. [Google Scholar] [PubMed]
73. Khandrika L, Kumar B, Koul S, Maroni P, Koul HK. Oxidative stress in prostate cancer. Cancer Lett. 2009;282(2):125–36. doi:10.1016/j.canlet.2008.12.011. [Google Scholar] [PubMed] [CrossRef]
74. Zhang Y, Li J, Tang L. Cancer-preventive isothiocyanates: dichotomous modulators of oxidative stress. Free Radic Biol Med. 2005;38(1):70–7. doi:10.1016/j.freeradbiomed.2004.09.033. [Google Scholar] [PubMed] [CrossRef]
75. Li D, Shao R, Wang N, Zhou N, Du K, Shi J, et al. Sulforaphane activates a lysosome-dependent transcriptional program to mitigate oxidative stress. Autophagy. 2021;17(4):872–87. doi:10.1080/15548627.2020.1739442. [Google Scholar] [PubMed] [CrossRef]
76. Tonelli C, Chio IIC, Tuveson DA. Transcriptional regulation by Nrf2. Antioxid Redox Signal. 2018;29(17):1727–45. doi:10.1089/ars.2017.7342. [Google Scholar] [PubMed] [CrossRef]
77. Kensler TW, Egner PA, Agyeman AS, Visvanathan K, Groopman JD, Chen JG, et al. Keap1–Nrf2 signaling: a target for cancer prevention by sulforaphane. Top Curr Chem. 2013;329:163–77. doi:10.1007/128_2012_339. [Google Scholar] [PubMed] [CrossRef]
78. Jones SB, Brooks JD. Modest induction of phase 2 enzyme activity in the F-344 rat prostate. BMC Cancer. 2006;6(1):62. doi:10.1186/1471-2407-6-62. [Google Scholar] [PubMed] [CrossRef]
79. Zhang C, Su ZY, Khor TO, Shu L, Kong AN. Sulforaphane enhances Nrf2 expression in prostate cancer TRAMP C1 cells through epigenetic regulation. Biochem Pharmacol. 2013;85(9):1398–404. doi:10.1016/j.bcp.2013.02.010. [Google Scholar] [PubMed] [CrossRef]
80. De Schrijver E, Brusselmans K, Heyns W, Verhoeven G, Swinnen JV. RNA interference-mediated silencing of the fatty acid synthase gene attenuates growth and induces morphological changes and apoptosis of LNCaP prostate cancer cells. Cancer Res. 2003;63(13):3799–804. [Google Scholar] [PubMed]
81. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7(10):763–77. doi:10.1038/nrc2222. [Google Scholar] [PubMed] [CrossRef]
82. Chen L, Guo Y, Wu Z, Zhao S, Zhang Z, Zheng F, et al. Epicatechin gallate prevents the de novo synthesis of fatty acid and the migration of prostate cancer cells. Acta Biochim Biophys Sin. 2021;53(12):1662–9. doi:10.1093/abbs/gmab144. [Google Scholar] [PubMed] [CrossRef]
83. Chen L, Xu YX, Wang YS, Zhou JL. Lipid metabolism, amino acid metabolism, and prostate cancer: a crucial metabolic journey. Asian J Androl. 2024;26(2):123–34. doi:10.4103/aja202363. [Google Scholar] [PubMed] [CrossRef]
84. Heemers HV, Verhoeven G, Swinnen JV. Androgen activation of the sterol regulatory element-binding protein pathway: current insights. Mol Endocrinol. 2006;20(10):2265–77. doi:10.1210/me.2005-0479. [Google Scholar] [PubMed] [CrossRef]
85. Kusakabe T, Maeda M, Hoshi N, Sugino T, Watanabe K, Fukuda T, et al. Fatty acid synthase is expressed mainly in adult hormone-sensitive cells or cells with high lipid metabolism and in proliferating fetal cells. J Histochem Cytochem. 2000;48(5):613–22. doi:10.1177/002215540004800505. [Google Scholar] [PubMed] [CrossRef]
86. Shah US, Dhir R, Gollin SM, Chandran UR, Lewis D, Acquafondata M, et al. Fatty acid synthase gene overexpression and copy number gain in prostate adenocarcinoma. Hum Pathol. 2006;37(4):401–9. doi:10.1016/j.humpath.2005.11.022. [Google Scholar] [PubMed] [CrossRef]
87. Migita T, Ruiz S, Fornari A, Fiorentino M, Priolo C, Zadra G, et al. Fatty acid synthase: a metabolic enzyme and candidate oncogene in prostate cancer. J Natl Cancer Inst. 2009;101(7):519–32. doi:10.1093/jnci/djp030. [Google Scholar] [PubMed] [CrossRef]
88. Rae C, Fragkoulis GI, Chalmers AJ. Cytotoxicity and radiosensitizing activity of the fatty acid synthase inhibitor C75 is enhanced by blocking fatty acid uptake in prostate cancer cells. Adv Radiat Oncol. 2020;5(5):994–1005. doi:10.1016/j.adro.2020.06.022. [Google Scholar] [PubMed] [CrossRef]
89. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33. doi:10.1126/science.1160809. [Google Scholar] [PubMed] [CrossRef]
90. Fukushi A, Kim HD, Chang YC, Kim CH. Revisited metabolic control and reprogramming cancers by means of the warburg effect in tumor cells. Int J Mol Sci. 2022;23(17):10037. doi:10.3390/ijms231710037. [Google Scholar] [PubMed] [CrossRef]
91. Carroll VA, Ashcroft M. Targeting the molecular basis for tumour hypoxia. Expert Rev Mol Med. 2005;7(6):1–16. doi:10.1017/S1462399405009117. [Google Scholar] [PubMed] [CrossRef]
92. Yao H, Wang H, Zhang Z, Jiang BH, Luo J, Shi X. Sulforaphane inhibited expression of hypoxia-inducible factor-1α in human tongue squamous cancer cells and prostate cancer cells. Int J Cancer. 2008;123(6):1255–61. doi:10.1002/ijc.23647. [Google Scholar] [PubMed] [CrossRef]
93. Carrasco-Pozo C, Tan KN, Rodriguez T, Avery VM. The molecular effects of sulforaphane and capsaicin on metabolism upon androgen and Tip60 activation of androgen receptor. Int J Mol Sci. 2019;20(21):5384. doi:10.3390/ijms20215384. [Google Scholar] [PubMed] [CrossRef]
94. Zhang Y, Li J, Huang Y, Chen Y, Luo Z, Huang H, et al. Improved antitumor activity against prostate cancer via synergistic targeting of Myc and GFAT-1. Theranostics. 2023;13(2):578–95. doi:10.7150/thno.76614. [Google Scholar] [PubMed] [CrossRef]
95. Vyas AR, Moura MB, Hahm ER, Singh KB, Singh SV. Sulforaphane inhibits c-Myc-Mediated prostate cancer stem-like traits. J Cell Biochem. 2016;117(11):2482–95. doi:10.1002/jcb.25541. [Google Scholar] [PubMed] [CrossRef]
96. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–7. doi:10.1038/nature01322. [Google Scholar] [PubMed] [CrossRef]
97. Ene CV, Nicolae I, Geavlete B, Geavlete P, Ene CD. IL-6 signaling link between inflammatory tumor microenvironment and prostatic tumorigenesis. Anal Cell Pathol. 2022;2022:5980387. [Google Scholar]
98. Saydullaeva I, Butuner BD, Korkmaz KS. NKX3.1 expression contributes to epithelial-mesenchymal transition of prostate cancer cells. ACS Omega. 2023;8(36):32580–92. doi:10.1021/acsomega.3c03127. [Google Scholar] [PubMed] [CrossRef]
99. Dai YN, Yi-Wen Yu E, Zeegers MP, Wesselius A. The association between dietary inflammatory potential and urologic cancers: a meta-analysis. Adv Nutr. 2024;15(1):100124. doi:10.1016/j.advnut.2023.09.012. [Google Scholar] [PubMed] [CrossRef]
100. Nguyen DP, Li J, Tewari AK. Inflammation and prostate cancer: the role of interleukin 6 (IL-6). BJU Int. 2014;113(6):986–92. doi:10.1111/bju.2014.113.issue-6. [Google Scholar] [CrossRef]
101. Yoshida A, Yoshida S, Ishibashi T, Kuwano M, Inomata H. Suppression of retinal neovascularization by the NF-κB inhibitor pyrrolidine dithiocarbamate in mice. Invest Ophthalmol Vis Sci. 1999;40(7):1624–9. [Google Scholar] [PubMed]
102. Xie TX, Xia Z, Zhang N, Gong W, Huang S. Constitutive NF-κB activity regulates the expression of VEGF and IL-8 and tumor angiogenesis of human glioblastoma. Oncol Rep. 2010;23(3):725–32. [Google Scholar] [PubMed]
103. Huang S, Pettaway CA, Uehara H, Bucana CD, Fidler IJ. Blockade of NF-κB activity in human prostate cancer cells is associated with suppression of angiogenesis, invasion, and metastasis. Oncogene. 2001;20(31):4188–97. doi:10.1038/sj.onc.1204535. [Google Scholar] [PubMed] [CrossRef]
104. Ruhee RT, Roberts LA, Ma S, Suzuki K. Organosulfur compounds: a review of their anti-inflammatory effects in human health. Front Nutr. 2020;7:64. doi:10.3389/fnut.2020.00064. [Google Scholar] [PubMed] [CrossRef]
105. Heiss E, Herhaus C, Klimo K, Bartsch H, Gerhauser C. Nuclear factor κB is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. J Biol Chem. 2001;276(34):32008–15. doi:10.1074/jbc.M104794200. [Google Scholar] [PubMed] [CrossRef]
106. Xu C, Shen G, Chen C, Gelinas C, Kong AN. Suppression of NF-κB and NF-κB-regulated gene expression by sulforaphane and PEITC through IκBα, IKK pathway in human prostate cancer PC-3 cells. Oncogene. 2005;24(28):4486–95. doi:10.1038/sj.onc.1208656. [Google Scholar] [PubMed] [CrossRef]
107. Baldwin AS. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-αB. J Clin Invest. 2001;107(3):241–6. doi:10.1172/JCI11991. [Google Scholar] [PubMed] [CrossRef]
108. Zhao S, Zhang Y, Zhang Q, Wang F, Zhang D. Toll-like receptors and prostate cancer. Front Immunol. 2014;5(1):352. doi:10.3389/fimmu.2014.00352. [Google Scholar] [PubMed] [CrossRef]
109. Zeng X, Liu X, Bao H, Zhang Y, Wang X, Shi K, et al. Effects of sulforaphane on toll-like receptor 4/myeloid differentiation factor 88 pathway of monocyte-derived macrophages from patients with chronic obstructive pulmonary disease. Zhonghua Jie He He Hu Xi Za Zhi. 2014;37(4):250–4 (In Chinese). [Google Scholar] [PubMed]
110. Kim SY, Jeong E, Joung SM, Lee JY. PI3K/Akt contributes to increased expression of Toll-like receptor 4 in macrophages exposed to hypoxic stress. Biochem Biophys Res Commun. 2012;419(3):466–71. doi:10.1016/j.bbrc.2012.02.015. [Google Scholar] [PubMed] [CrossRef]
111. Youn HS, Kim YS, Park ZY, Kim SY, Choi NY, Joung SM, et al. Sulforaphane suppresses oligomerization of TLR4 in a thiol-dependent manner. J Immunol. 2010;184(1):411–9. doi:10.4049/jimmunol.0803988. [Google Scholar] [PubMed] [CrossRef]
112. Koo JE, Park ZY, Kim ND, Lee JY. Sulforaphane inhibits the engagement of LPS with TLR4/MD2 complex by preferential binding to Cys133 in MD2. Biochem Biophys Res Commun. 2013;434(3):600–5. doi:10.1016/j.bbrc.2013.03.123. [Google Scholar] [PubMed] [CrossRef]
113. Aggarwal BB, Kunnumakkara AB, Harikumar KB, Gupta SR, Tharakan ST, Koca C, et al. Signal transducer and activator of transcription-3, inflammation, and cancer: how intimate is the relationship? Ann N Y Acad Sci. 2009;1171(1):59–76. doi:10.1111/j.1749-6632.2009.04911.x. [Google Scholar] [PubMed] [CrossRef]
114. Hahm ER, Singh SV. Sulforaphane inhibits constitutive and interleukin-6-induced activation of signal transducer and activator of transcription 3 in prostate cancer cells. Cancer Prev Res. 2010;3(4):484–94. doi:10.1158/1940-6207.CAPR-09-0250. [Google Scholar] [PubMed] [CrossRef]
115. Singh SV, Herman-Antosiewicz A, Singh AV, Lew KL, Srivastava SK, Kamath R, et al. Sulforaphane-induced G2/M phase cell cycle arrest involves checkpoint kinase 2-mediated phosphorylation of cell division cycle 25C. J Biol Chem. 2004;279(24):25813–22. doi:10.1074/jbc.M313538200. [Google Scholar] [PubMed] [CrossRef]
116. Rutz J, Thaler S, Maxeiner S, Chun FK, Blaheta RA. Sulforaphane reduces prostate cancer cell growth and proliferation in vitro by modulating the Cdk-Cyclin axis and expression of the CD44 variants 4, 5, and 7. Int J Mol Sci. 2020;21(22):8724. doi:10.3390/ijms21228724. [Google Scholar] [PubMed] [CrossRef]
117. Traka MH, Spinks CA, Doleman JF, Melchini A, Ball RY, Mills RD, et al. The dietary isothiocyanate sulforaphane modulates gene expression and alternative gene splicing in a PTEN null preclinical murine model of prostate cancer. Mol Cancer. 2010;9:189. doi:10.1186/1476-4598-9-189. [Google Scholar] [PubMed] [CrossRef]
118. Keum YS, Khor TO, Lin W, Shen G, Kwon KH, Barve A, et al. Pharmacokinetics and pharmacodynamics of broccoli sprouts on the suppression of prostate cancer in transgenic adenocarcinoma of mouse prostate (TRAMP) mice: implication of induction of Nrf2, HO-1 and apoptosis and the suppression of Akt-dependent kinase pathway. Pharm Res. 2009;26(10):2324–31. [Google Scholar] [PubMed]
119. Xiao D, Powolny AA, Antosiewicz J, Hahm ER, Bommareddy A, Zeng Y, et al. Cellular responses to cancer chemopreventive agent D, L-sulforaphane in human prostate cancer cells are initiated by mitochondrial reactive oxygen species. Pharm Res. 2009;26(7):1729–38. doi:10.1007/s11095-009-9883-5. [Google Scholar] [PubMed] [CrossRef]
120. Taheri M, Ghafouri-Fard S, Najafi S, Kallenbach J, Keramatfar E, Atri Roozbahani G, et al. Hormonal regulation of telomerase activity and hTERT expression in steroid-regulated tissues and cancer. Cancer Cell Int. 2022;22(1):258. doi:10.1186/s12935-022-02678-9. [Google Scholar] [PubMed] [CrossRef]
121. Abbas A, Hall JA, Patterson WL, Ho E3rd, Hsu A, Al-Mulla F, et al. Sulforaphane modulates telomerase activity via epigenetic regulation in prostate cancer cell lines. Biochem Cell Biol. 2016;94(1):71–81. doi:10.1139/bcb-2015-0038. [Google Scholar] [PubMed] [CrossRef]
122. Kalkavan H, Green DR. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018;25(1):46–55. doi:10.1038/cdd.2017.179. [Google Scholar] [PubMed] [CrossRef]
123. Pickles S, Vigie P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018;28(4):R170–R85. doi:10.1016/j.cub.2018.01.004. [Google Scholar] [PubMed] [CrossRef]
124. Okamoto K. Organellophagy: eliminating cellular building blocks via selective autophagy. J Cell Biol. 2014;205(4):435–45. doi:10.1083/jcb.201402054. [Google Scholar] [PubMed] [CrossRef]
125. Chen C, Han X, Zou X, Li Y, Yang L, Cao K, et al. 4-methylene-2-octyl-5-oxotetrahydrofuran-3-carboxylic acid (C75an inhibitor of fatty-acid synthase, suppresses the mitochondrial fatty acid synthesis pathway and impairs mitochondrial function. J Biol Chem. 2014;289(24):17184–94. doi:10.1074/jbc.M114.550806. [Google Scholar] [PubMed] [CrossRef]
126. Vara-Perez M, Felipe-Abrio B, Agostinis P. Mitophagy in cancer: a tale of adaptation. Cells. 2019;8(5):493. doi:10.3390/cells8050493. [Google Scholar] [PubMed] [CrossRef]
127. Singh SV, Srivastava SK, Choi S, Lew KL, Antosiewicz J, Xiao D, et al. Sulforaphane-induced cell death in human prostate cancer cells is initiated by reactive oxygen species. J Biol Chem. 2005;280(20):19911–24. doi:10.1074/jbc.M412443200. [Google Scholar] [PubMed] [CrossRef]
128. Singh AV, Xiao D, Lew KL, Dhir R, Singh SV. Sulforaphane induces caspase-mediated apoptosis in cultured PC-3 human prostate cancer cells and retards growth of PC-3 xenografts in vivo. Carcinogenesis. 2004;25(1):83–90. doi:10.1093/carcin/bgg178. [Google Scholar] [PubMed] [CrossRef]
129. Choi S, Singh SV. Bax and Bak are required for apoptosis induction by sulforaphane, a cruciferous vegetable-derived cancer chemopreventive agent. Cancer Res. 2005;65(5):2035–43. doi:10.1158/0008-5472.CAN-04-3616. [Google Scholar] [PubMed] [CrossRef]
130. Myzak MC, Hardin K, Wang R, Dashwood RH, Ho E. Sulforaphane inhibits histone deacetylase activity in BPH-1, LnCaP and PC-3 prostate epithelial cells. Carcinogenesis. 2006;27(4):811–9. doi:10.1093/carcin/bgi265. [Google Scholar] [PubMed] [CrossRef]
131. Zhou Y, Yang G, Tian H, Hu Y, Wu S, Geng Y, et al. Sulforaphane metabolites cause apoptosis via microtubule disruption in cancer. Endocr Relat Cancer. 2018;25(3):255–68. doi:10.1530/ERC-17-0483. [Google Scholar] [PubMed] [CrossRef]
132. Pei Y, Wu B, Cao Q, Wu L, Yang G. Hydrogen sulfide mediates the anti-survival effect of sulforaphane on human prostate cancer cells. Toxicol Appl Pharmacol. 2011;257(3):420–8. doi:10.1016/j.taap.2011.09.026. [Google Scholar] [PubMed] [CrossRef]
133. Desai K, McManus JM, Sharifi N. Hormonal therapy for prostate cancer. Endocr Rev. 2021;42(3):354–73. doi:10.1210/endrev/bnab002. [Google Scholar] [PubMed] [CrossRef]
134. Aurilio G, Cimadamore A, Mazzucchelli R, Lopez-Beltran A, Verri E, Scarpelli M, et al. Androgen receptor signaling pathway in prostate cancer: from genetics to clinical applications. Cells. 2020;9(12):2653. doi:10.3390/cells9122653. [Google Scholar] [PubMed] [CrossRef]
135. Liu X, Zhang P, Song H, Tang X, Hao Y, Guan Y, et al. Unveiling a pH-responsive dual-androgen-blocking magnetic molecularly imprinted polymer for enhanced synergistic therapy of prostate cancer. ACS Appl Mater Interfaces. 2024;16(4):4348–60. doi:10.1021/acsami.3c13732. [Google Scholar] [PubMed] [CrossRef]
136. Gibbs A, Schwartzman J, Deng V, Alumkal J. Sulforaphane destabilizes the androgen receptor in prostate cancer cells by inactivating histone deacetylase 6. Proc Natl Acad Sci U S A. 2009;106(39):16663–8. doi:10.1073/pnas.0908908106. [Google Scholar] [PubMed] [CrossRef]
137. Khurana N, Kim H, Chandra PK, Talwar S, Sharma P, Abdel-Mageed AB, et al. Multimodal actions of the phytochemical sulforaphane suppress both AR and AR-V7 in 22Rv1 cells: advocating a potent pharmaceutical combination against castration-resistant prostate cancer. Oncol Rep. 2017;38(5):2774–86. doi:10.3892/or.2017.5932. [Google Scholar] [PubMed] [CrossRef]
138. Khurana N, Talwar S, Chandra PK, Sharma P, Abdel-Mageed AB, Mondal D, et al. Sulforaphane increases the efficacy of anti-androgens by rapidly decreasing androgen receptor levels in prostate cancer cells. Int J Oncol. 2016;49(4):1609–19. doi:10.3892/ijo.2016.3641. [Google Scholar] [PubMed] [CrossRef]
139. Liu M, Yao XD, Li W, Geng J, Yan Y, Che JP, et al. Nrf2 sensitizes prostate cancer cells to radiation via decreasing basal ROS levels. Biofactors. 2015;41(1):52–7. doi:10.1002/biof.v41.1. [Google Scholar] [CrossRef]
140. Li Y. Modern epigenetics methods in biological research. Methods. 2021;187:104–13. doi:10.1016/j.ymeth.2020.06.022. [Google Scholar] [PubMed] [CrossRef]
141. Nozaki T, Kanai M. Chemical catalysis intervening to histone epigenetics. Acc Chem Res. 2021;54(9):2313–22. doi:10.1021/acs.accounts.1c00144. [Google Scholar] [PubMed] [CrossRef]
142. Xie SS, Duan CG. Epigenetic regulation of plant immunity: from chromatin codes to plant disease resistance. aBIOTECH. 2023;4(2):124–39. doi:10.1007/s42994-023-00101-z. [Google Scholar] [PubMed] [CrossRef]
143. Lopez J, Anazco-Guenkova AM, Monteagudo-Garcia O, Blanco S. Epigenetic and epitranscriptomic control in prostate cancer. Genes. 2022;13(2):378. doi:10.3390/genes13020378. [Google Scholar] [PubMed] [CrossRef]
144. Rafikova G, Gilyazova I, Enikeeva K, Pavlov V, Kzhyshkowska J. Prostate cancer: genetics, epigenetics and the need for immunological biomarkers. Int J Mol Sci. 2023;24(16):12797. doi:10.3390/ijms241612797. [Google Scholar] [PubMed] [CrossRef]
145. Samija I, Frobe A. Genomics of prostate cancer: clinical utility and challenges. Acta Clin Croat. 2022;61(Suppl 3):86. [Google Scholar] [PubMed]
146. Abbas A, Gupta S. The role of histone deacetylases in prostate cancer. Epigenetics. 2008;3(6):300–9. doi:10.4161/epi.3.6.7273. [Google Scholar] [PubMed] [CrossRef]
147. Beaver LM, Löhr CV, Clarke JD, Glasser ST, Watson GW, Wong CP, et al. Broccoli sprouts delay prostate cancer formation and decrease prostate cancer severity with a concurrent decrease in HDAC3 protein expression in transgenic adenocarcinoma of the mouse prostate (TRAMP) mice. Curr Dev Nutr. 2018;2(3):nzy002. doi:10.1093/cdn/nzy002. [Google Scholar] [PubMed] [CrossRef]
148. Pardo JC, Ruiz de Porras V, Gil J, Font A, Puig-Domingo M, Jorda M. Lipid metabolism and epigenetics crosstalk in prostate cancer. Nutrients. 2022;14(4):851. doi:10.3390/nu14040851. [Google Scholar] [PubMed] [CrossRef]
149. Hsu A, Wong CP, Yu Z, Williams DE, Dashwood RH, Ho E. Promoter de-methylation of cyclin D2 by sulforaphane in prostate cancer cells. Clin Epigenetics. 2011;3(1):3. doi:10.1186/1868-7083-3-3. [Google Scholar] [PubMed] [CrossRef]
150. Wong CP, Hsu A, Buchanan A, Palomera-Sanchez Z, Beaver LM, Houseman EA, et al. Effects of sulforaphane and 3,3′-diindolylmethane on genome-wide promoter methylation in normal prostate epithelial cells and prostate cancer cells. PLoS One. 2014;9(1):e86787. doi:10.1371/journal.pone.0086787. [Google Scholar] [PubMed] [CrossRef]
151. Weiss M, Plass C, Gerhauser C. Role of lncRNAs in prostate cancer development and progression. Biol Chem. 2014;395(11):1275–90. doi:10.1515/hsz-2014-0201. [Google Scholar] [PubMed] [CrossRef]
152. Ramalho-Carvalho J, Fromm B, Henrique R, Jeronimo C. Deciphering the function of non-coding RNAs in prostate cancer. Cancer Metastasis Rev. 2016;35(2):235–62. doi:10.1007/s10555-016-9628-y. [Google Scholar] [PubMed] [CrossRef]
153. Tian H, Zhou Y, Yang G, Geng Y, Wu S, Hu Y, et al. Sulforaphane-cysteine suppresses invasion via downregulation of galectin-1 in human prostate cancer DU145 and PC3 cells. Oncol Rep. 2016;36(3):1361–8. doi:10.3892/or.2016.4942. [Google Scholar] [PubMed] [CrossRef]
154. Peng X, Zhou Y, Tian H, Yang G, Li C, Geng Y, et al. Sulforaphane inhibits invasion by phosphorylating ERK1/2 to regulate E-cadherin and CD44v6 in human prostate cancer DU145 cells. Oncol Rep. 2015;34(3):1565–72. doi:10.3892/or.2015.4098. [Google Scholar] [PubMed] [CrossRef]
155. Singh SV, Warin R, Xiao D, Powolny AA, Stan SD, Arlotti JA, et al. Sulforaphane inhibits prostate carcinogenesis and pulmonary metastasis in TRAMP mice in association with increased cytotoxicity of natural killer cells. Cancer Res. 2009;69(5):2117–25. doi:10.1158/0008-5472.CAN-08-3502. [Google Scholar] [PubMed] [CrossRef]
156. Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5(1):8. doi:10.1038/s41392-020-0110-5. [Google Scholar] [PubMed] [CrossRef]
157. Wiczk A, Hofman D, Konopa G, Herman-Antosiewicz A. Sulforaphane, a cruciferous vegetable-derived isothiocyanate, inhibits protein synthesis in human prostate cancer cells. Biochim Biophys Acta. 2012;1823(8):1295–305. doi:10.1016/j.bbamcr.2012.05.020. [Google Scholar] [PubMed] [CrossRef]
158. Yagishita Y, Fahey JW, Dinkova-Kostova AT, Kensler TW. Broccoli or sulforaphane: is it the source or dose that matters? Molecules. 2019;24(19):3593. doi:10.3390/molecules24193593. [Google Scholar] [PubMed] [CrossRef]
159. Fahey JW, Kensler TW. The challenges of designing and implementing clinical trials with broccoli sprouts and turning evidence into public health action. Front Nutr. 2021;8:648788. doi:10.3389/fnut.2021.648788. [Google Scholar] [PubMed] [CrossRef]
160. Fahey JW, Holtzclaw WD, Wehage SL, Wade KL, Stephenson KK, Talalay P. Sulforaphane bioavailability from glucoraphanin-rich broccoli: control by active endogenous myrosinase. PLoS One. 2015;10(11):e0140963. doi:10.1371/journal.pone.0140963. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools