
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE
Lovastatin modulation of YAP/TAZ signaling on cardiomyocyte autophagy and mitochondrial damage in myocardial I/R injury
1 Department of Cardiovascular Surgery, The People’s Hospital of Gaozhou, Gaozhou, 525200, China
2 Department of Pharmacy, The People’s Hospital of Gaozhou, Gaozhou, 525200, China
3 Department of Pharmacology, School of Pharmacy, Jinan University, Guangzhou, 511436, China
* Corresponding Author: MINGZHU LI. Email:
# Represented equally contribution to this work
(This article belongs to the Special Issue: Cell Death and Inflammation in Signaling and Diseases)
BIOCELL 2024, 48(10), 1489-1501. https://doi.org/10.32604/biocell.2024.053930
Received 14 May 2024; Accepted 27 August 2024; Issue published 02 October 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Objective: Studies have demonstrated that administering statins promptly following myocardial ischemia/reperfusion (MI/R) can confer cardioprotective benefits. This study investigates whether Lovastatin can modulate the Yes-associated protein/Transcriptional co-activator with PDZ-binding motif (YAP/TAZ) signaling pathway to mitigate cardiomyocyte injury caused by hypoxia/reoxygenation (H/R). Methods: The in vitro MI/R model was established by H/R in rat myocardial H9c2 cells, and the cells were pretreated with varying doses of Lovastatin before reoxygenation. The extent of cellular injury was evaluated by measuring the myocardial enzyme content and cell viability. The levels of oxidative stress and inflammatory factors were quantified by enzyme-linked immunosorbent assay (ELISA). Mitochondrial function was evaluated by detecting mitochondrial reactive oxide species (ROS), oxygen consumption rate (OCR), mitochondrial permeability transition pore (MPTP), adenosine 5′-triphosphate (ATP), mitochondrial membrane potential (MMP), and Ca2+. Western blotting (WB) and immunofluorescence assays were applied to detect the proteins related to apoptosis, autophagy, and YAP/TAZ signaling. YAP overexpression plasmid (Ov-YAP) was constructed for mechanism verification. Results: H/R leads to a reduction in H9c2 cell viability, and an elevation in the contents of myocardial enzymes, inflammatory factors, and oxidative stress. The apoptosis and autophagy were increased, and yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ) expression levels were up-regulated in the H/R group. Nevertheless, all these changes were dose-dependently reversed by Lovastatin. Meanwhile, Lovastatin restored the levels of MMP, ATP, MPTP opening, and Ca2+, and reduced OCR in H9c2 cells exposed to H/R. Nevertheless, Ov-YAP markedly attenuated the function of Lovastatin on apoptosis, autophagy, inflammation, and oxidative stress in H9c2 cells exposed to H/R. Conclusion: The result indicated that Lovastatin played a protective role in H/R-induced H9c2 cells by inhibiting YAP/TAZ signaling.Graphic Abstract
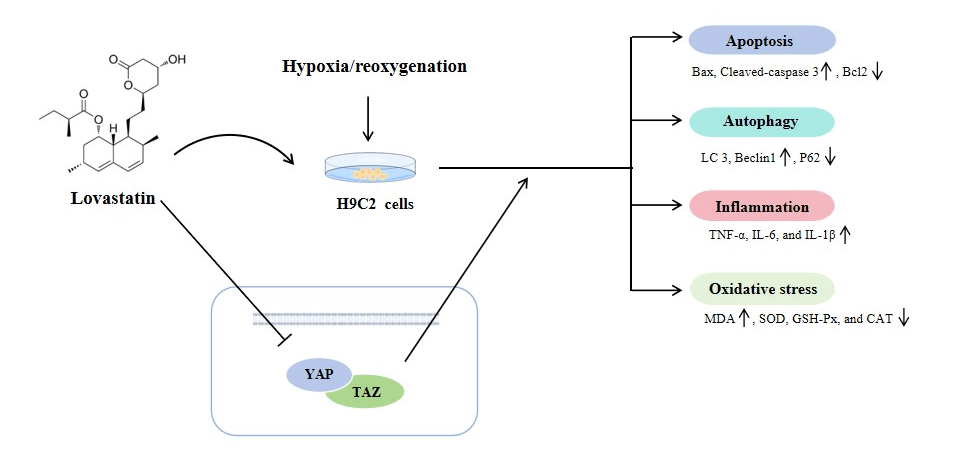
Keywords
Abbreviations
| AMI | Acute myocardial infarction |
| ATP | Adenosine triphosphate |
| BCA | Bicinchoninic acid |
| CAT | Catalase |
| CCK-8 | Cell counting kit-8 |
| CK | Creatine kinase |
| CK-MB | Creatine kinase-MB |
| FBS | Fetal bovine serum |
| GSH-Px | Glutathione peroxidase |
| H/R | Hypoxia/reoxygenation |
| IL-1β | Interleukin-1 beta |
| IL-6 | Interleukin-6 |
| LC-3 | Microtubule-associated protein 1 light chain 3 |
| LDH | Lactate dehydrogenase |
| MDA | Malonaldehyde |
| MI/R | Myocardial ischemia/reperfusion |
| MMP | Mitochondrial membrane potential |
| MPTP | Mitochondrial permeability transition pore |
| OCR | Oxygen consumption rate |
| PBS | Phosphate buffer solution |
| PVDF | Polyvinylidene difluoride |
| RIPA | Radio immunoprecipitation assay |
| ROS | Reactive oxide species |
| SOD | Superoxide dismutase |
| TAZ | Transcriptional co-activator with PDZ-binding motif |
| TNF-α | Tumor necrosis factor-alpha |
| YAP | Yes-associated protein |
Myocardial ischemia/reperfusion (MI/R) injury represents a prevalent cardiovascular complication, resulting in significant global morbidity and mortality [1]. Acute myocardial infarction (AMI) is a principal contributor to these outcomes. Current standard therapies for AMI, which emphasize reperfusion strategies, can paradoxically precipitate cardiomyocyte dysfunction, known as MI/R injury [2]. Manifestations of MI/R injury include arrhythmias triggered by reperfusion, reversible myocardial impairment, microvascular dysfunction, and fatal myocardial reperfusion damage [3]. The etiologies of MI/R injury are complex, involving molecular, cellular, and tissue changes such as oxidative stress, inflammation, etc., [4,5]. Although considerable improvements have been made in the management of MI/R, the intricate pathophysiological mechanisms remain elusive [6]. As such, a deeper exploration of the underlying mechanisms of MI/R injury is pivotal in counteracting its detrimental impacts.
MI/R injury is a major factor exacerbating myocardial disfunction and cardiomyocyte death after cardiac surgery and myocardial infarction [7]. While autophagy helps eliminate damaged proteins and organelles to support cell survival, irreversible damage can result in cell death within the intrafollicular microenvironment [8]. It has been shown that exosomes sourced from bone marrow mesenchymal stem cells reduced IR-induced cardiomyocyte apoptosis and also regulated autophagy activity, which in turn ameliorates cardiomyocyte injury [9]. Wu et al. [10] explored the effect of miR-199a-3p in MI/R injury and determined that reducing miR-199a-3p expression hastened the rate of apoptosis and autophagy in cardiomyocytes. Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ) are integral components of the Hippo signaling pathway, also referred to as YAP/TAZ signaling, which exerts a pivotal role in modulating cell growth and differentiation [11]. Previous studies have reported that the YAP/TAZ signaling pathway plays a regulatory role in autophagy and mitochondrial function [12]. In MI/R injury, modulation of this signal ameliorates MI/R-induced necroptosis [13]. However, it is still unknown whether YAP/TAZ has a role in regulating myocardial autophagy and mitochondrial function in MI/R injury.
Lovastatin, a member of the statin drug class, has been demonstrated in previous studies to confer protection to mitochondrial and renal functions in the context of rat kidney I/R injury [14]. Furthermore, lovastatin exhibits the capability to ameliorate chronic cardiac damage resulting from azithromycin treatment, serving as a myocardial protector [15]. Moreover, additional research has indicated that Lovastatin possesses the capacity to inhibit autophagy and is implicated in the pathogenesis of cerebral hemorrhage [16]. More importantly, it has been reported that Lovastatin alleviates cardiovascular fibrosis induced by angiotensin II by suppressing the YAP/TAZ signaling [17]. Nevertheless, the precise mechanism underlying Lovastatin’s impact on MI/R injury remains insufficiently elucidated, necessitating further investigation.
Therefore, this study induced a cell model of MI/R injury and observed the indicators of myocardial damage, apoptosis, inflammation, oxidative stress, autophagy, and mitochondrial function to investigate whether Lovastatin could ameliorate H9c2 cell injury caused by hypoxia/reoxygenation (H/R) via YAP/TAZ signaling.
The rat myocardial cell line H9c2 (CRL-1446, ATCC, Manassas, VA, USA) was cultivated in dulbecco’s modified eagle medium/nutrient mixture F-12 (DMEM/F12) medium (Gibco, Grand Island, NY, USA) with 10% fetal bovine serum (FBS, Gibco) and an antibiotic combination (100 U/mL streptomycin and penicillin) (C0222, Beyotime, Shanghai, China) at 37°C with 5% CO2. All cells have been identified by short tandem repeat (STR) and tested for mycoplasma, and all experiments were performed under sterile conditions.
H/R model establishment and treatment
The H/R model was adopted using H9c2 cells. Briefly, to induce in vitro ischemic conditions, H9c2 cells were maintained in glucose-free and FBS-free DMEM/F12 medium and exposed to a mixture of 94% N2, 1% O2, and 5% CO2 for 4 h. Then, H9c2 cells were subsequently placed under normoxic conditions containing 21% O2, 5% CO2, and 74% N2 for reoxygenation for a duration of 6 h. Following 24 h cultivation under normal culture conditions, the cells were harvested at the end of reoxygenation. During reoxygenation, the cells were pretreated with Lovastatin (HY-N0504, MCE, NJ, USA) at various concentrations (25, 50, 100 nmol/L).
The YAP overexpression vector (ov-YAP) and its control vector (ov-NC) were obtained from GenePharma Technology (Shanghai, China). A total of 2 × 105 H9c2 cells were plated in a 6-well plate. H9c2 cells were transfected with 50 nM ov-YAP or ov-NC with the pre-prepared transfection reagent (2 mL) after the cell confluency reached 30% to 50%. The cells were incubated for 6 h, after which the medium was replaced. Following a 48 h transfection period, the cells were harvested for the subsequent experiments.
Following the incubation period, the treated H9c2 cells were moved to a fresh 96-well plate (3 × 103 cells/well). Then, cells were treated with 10 µL of cell counting kit-8 (CCK-8, C0038, Beyotime) for 2 h at 37°C. Cell viability was evaluated by detecting the absorbance at 450 nm by a microplate reader (F50, TECAN, Männedorf, Switzerland).
Enzyme-linked immunosorbent assay (ELISA)
The tumor necrosis factor-alpha (TNF-α, H052-1, Nanjing Jiancheng, Nanjing, China), interleukin-6 (IL-6, H007-1-1, Nanjing Jiancheng), interleukin-1 beta (IL-1β, H002, Nanjing Jiancheng), and creatine kinase-MB (CK-MB, SEKR-0059, Solarbio, Beijing, China), lactate dehydrogenase (LDH, C0017, Beyotime), and creatine kinase (CK, BC1145, Solarbio) contents in the H9c2 cells were evaluated by ELISA kits in accordance with the producer’s guidelines. Cells were cultured in a 96-well plate (1 × 104 cells/well) and incubated with corresponding ELISA kits for 20 or 30 min. After washing, 3, 3′, 5, 5′-Tetramethylbenzidine (TMB) substrate solution was added and incubated for 30 min to terminate the reaction. The absorbance at 450 nm was detected by a microplate reader (F50, TECAN).
The proteins were lysed with the radioimmunoprecipitation assay (RIPA) buffer (P0013B, Beyotime) and the protein concentration was measured by a bicinchoninic acid (BCA) protein assay kit (P0012, Beyotime). A total of 30 μg of protein was loaded into sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE, P0012A, Beyotime) gel for separation and transferred onto polyvinylidene difluoride (PVDF) membranes (IPVH00010, Merck Millipore, MA, USA). The membrane was blocked with 5% skim milk for 1 h at room temperature (RT) and incubated with primary antibodies (Cleaved caspase3, 1:2000, ab214430, Abcam, MA, USA; Bax, 1:2500, CST, #2772, Danvers, MA, USA; Bcl-2, 1:3000, #3498, CST; caspase3, 1:1000, #9662, CST; Beclin1, 1:2500, 33738, CST; LC3, 1:2000, #4108, CST; P62, 1:2000, #5114, CST; YAP, 1:3000, #4912, CST; TAZ, 1:2000, #4883, CST; glyceraldehyde-3-phosphate dehydrogenase, GAPDH, 1:1000, ab9485, Abcam) overnight at 4°C. Then, incubate the membrane for 2 h at RT with the secondary antibody (1:5000, ab7090, Abcam). The membrane was developed in darkness and the blots were made visible by a gel imaging system (ChampGelTM 5000, sagecreation, Beijing, China).
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining
TUNEL Staining Kit (C1086, Beyotime) was used to investigate the apoptosis of H9c2 cells. Initially, cells were fixed with 4% paraformaldehyde (P0099, Beyotime) and permeabilized by 0.1% TritonX-100 (BL934B, Biosharp, Hefei, China) for 20 min, respectively. Then, H9c2 cells were incubated with a mixture containing terminal deoxynucleotidyl transferase (TDT) enzyme, dUTP, and buffer at a ratio of 1:5:50. After 4′,6-diamidino-2-phenylindole (DAPI, C1005, Beyotime) counterstaining, the images were captured using a fluorescence microscope (IX53, Olympus, Tokyo, Japan).
H9c2 cells were treated with 4% paraformaldehyde and 0.1% Triton X-100 (BL934B, Biosharp) for 20 min at RT. Subsequently, H9c2 cells were blocked using a 1% bovine serum albumin (BSA) solution for 30 min at RT. Then, the cells were treated with anti-LC3 (1:200, #4108, CST) or Mito-Tracker red CMXRos (C1049B, Beyotime) and anti-LC3B (1:200, ab192890, Abcam) overnight at 4°C. Then, the cells were treated with secondary antibodies (1:1000, ab7090, Abcam) at 37°C for 1 h in darkness and stained with DAPI for 10 min. The fluorescent images were captured by a microscope (CKX53, Olympus).
H9c2 cells were grown to 70%–80% confluence following H/R treatment. Then, the cells were treated with 5 μM MitoSOX Red reagent (S0061M, Beyotime) at 37°C for 15 min. Cells were rinsed with PBS and the images were captured under a fluorescence microscope (IX53, Olympus).
Assessment of mitochondrial membrane potential (MMP)
MMP was evaluated through JC-1 assay (T3168, Thermo Fisher Scientific, MA, USA). H9c2 cells were rinsed with PBS and then treated with the JC-1 probe in the dark at 37°C for 30 min. Afterward, the free probe was washed out with PBS, and results were observed under a confocal microscope (TCS SP8, Leica, Wetzlar, Germany). Changes in MMP were evaluated based on the red-to-green fluorescence ratio. For immunofluorescence analysis, the green/red immunosignals were converted into average grayscale intensity and subjected to analysis using Image-Pro Plus 6.0 (Media Cybernetics, Rockville, MD, USA).
Detection of adenosine triphosphate (ATP) content
H9c2 cells (1 × 104 cells) grown in 6-well plates were lysed using ATP lysis buffer on ice and then exposed to the ATP substrate solution. Cellular ATP level was quantified using a bioluminescent assay kit (ab113849, Abcam).
Evaluation of mitochondrial permeability transition pore (MPTP) opening
An MPTP kit (C2009S, Beyotime) was employed to assess the MPTP opening. 100 μL of Calcein AM solution was added into H9c2 cells at 37°C for 30 min in darkness, then centrifuged and resuspended in a preheated culture medium. Subsequently, cells were incubated at 37°C for 30 min in a dark environment, and the results were observed under a fluorescence microscope (IX53, Olympus).
Detection of oxygen consumption rate (OCR)
The Seahorse XF Cell Mito Stress Test Kit on a Seahorse XFp analyzer (Agilent, CA, USA) was employed for OCR assessment. H9c2 cells were seeded in XFe96 plates at a density of 5 × 104 cells per well. Then, 1 μM oligomycin, 1 μM carbonyl cyanide-4- (trifluoromethoxy) phenylhydrazone, and 1 μM rotenone were automatically added to test OCR.
Detection of Ca2+ concentration
H9c2 cells were seeded in a 96-well plate (1 × 104 cells/well) and treated with a calcium assay Kit (S1061S, Beyotime) for 5 min. The absorbance at 610 nm was measured by a microplate reader (Infinite F50, TECAN, Beijing, China). Additionally, the mitochondrial Ca2+ was detected using the Rhod-2 (40776ES50, Yeasen, Shanghai, China) and MitoTracker Green (40742ES50, Yeasen). Briefly, Rhod-2 and MitoTracker Green were solubilized in dimethyl sulfoxide and diluted to a working solution, respectively. H9c2 cells were treated with Rhod-2 (5 μM) and MitoTracker Green (20 nM) away from light for 30 min at 37°C, followed by the added tyrode’s solution for 30 min of incubation in darkness. Afterward, the cells were washed to eliminate any excess or non-specific probes in the mitochondria. Rhod-2 fluorescence (Ex/Em: 549/578 nm) and MitoTracker green fluorescence (Ex/Em: 488/523 nm) were measured under a confocal microscope (TCS SP8, Leica).
Measurement of oxidative stress
H9c2 cells were grown in a 96-well plate (1 × 103 cells/well). Subsequently, the levels of malonaldehyde (MDA, A003-4-1, Nanjing Jiancheng), superoxide dismutase (SOD, A001-3-2, Nanjing Jiancheng), glutathione peroxidase (GSH-Px, S0056, Beyotime) and catalase (CAT, S0051, Beyotime) levels were quantified using assay kits. The absorbance at 532 nm was recorded by a microplate reader (Infinite F50, TECAN).
The data was utilized by IBM SPSS 26.0 (IBM SPSS, Watson, NY, USA) and were exhibited as mean ± standard deviation (SD). A One-way ANOVA with Tukey’s post hoc test was used to compare the differences among the experimental groups. Kruskal-Wallis test was employed for statistical analysis when n < 3. Statistical significance was established when the p-value < 0.05.
Lovastatin ameliorates H/R-induced injury and apoptosis in H9c2 cells
Initially, H9c2 cells were treated with varying doses of Lovastatin (25, 50, 100 nmol/L), and the results revealed no impact on cell viability (Fig. 1A). Lovastatin dose-dependently increased the viability of H9c2 cells in contrast with the H/R group (Fig. 1B). Concurrently, a reduction was exhibited in the levels of intracellular enzymes including LDH, CK-MB, and CK as the dosage of Lovastatin was increased (Fig. 1C). Next, the apoptotic level was assessed. TUNEL staining revealed that Lovastatin intervention at varying doses reduced the apoptosis in H9c2 cells when compared to the H/R group, with the highest dose exerting the most significant effect (Fig. 1D). WB analysis demonstrated that Lovastatin reduced the expression of Cleaved caspase-3, and Bax dose-dependently, while up-regulating the level of Bcl2 (Fig. 1E). These findings indicated that Lovastatin could mitigate injury and apoptosis in the H/R-induced cellular model dose-dependently.
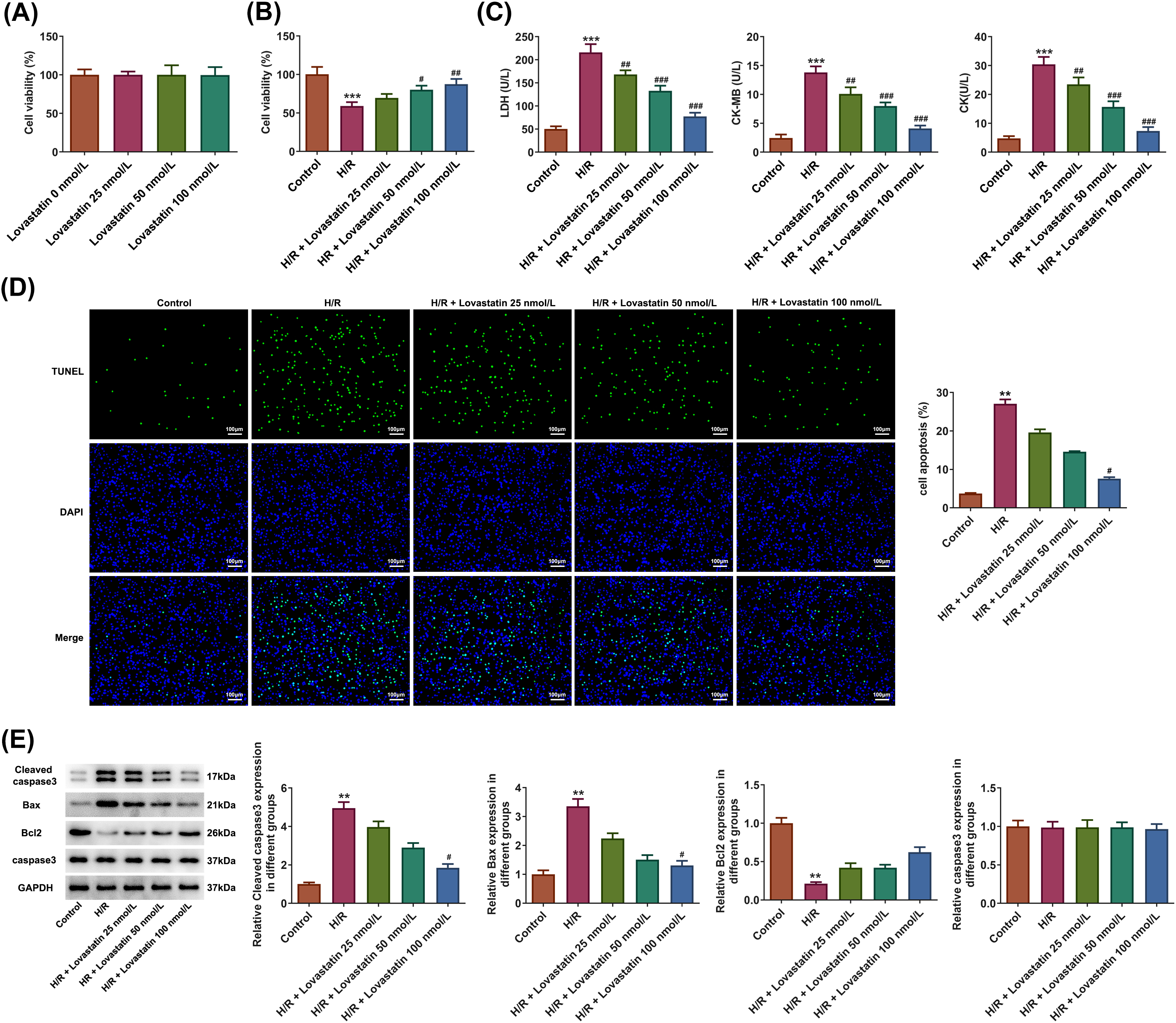
Figure 1: Lovastatin ameliorates H9c2 cell damage caused by hypoxia/reoxygenation (H/R). Effect of varying doses of Lovastatin on the H9c2 cell viability (A), (n = 5). Lovastatin up-regulated the viability of H9c2 cells (B), (n = 5). Lovastatin reduced the contents of LDH, CK-MB, and CK (C), (n = 5). Lovastatin decreased the TUNEL fluorescence intensity in H9c2 cells (D), (n = 3). Lovastatin was observed to reduce pro-apoptotic proteins and increase anti-apoptotic protein expression in H9c2 cells (E), (n = 3). **p < 0.01, ***p < 0.001 vs. Control; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. H/R. CK-MB, creatine kinase-MB; LDH, lactate dehydrogenase; CK, creatine kinase.
Lovastatin attenuates H/R-induced autophagy in H9c2 cells
Immunofluorescence assay was utilized to measure the levels of the autophagic marker microtubule-associated protein 1 light chain 3 (LC3). Lovastatin treatment effectively reduced the fluorescence intensity of LC3, displaying a dose-dependent manner (Fig. 2A). Additionally, a similar trend was observed in the level of autophagy-related proteins, with lower expression of Beclin1 and LC3-II/LC3I in the Lovastatin group relative to the H/R group, while P62 expression was increased (Fig. 2B). Meanwhile, the fluorescence intensity of LC3 was detected in mitochondria. The findings displayed that Lovastatin decreased the fluorescence intensity of LC3B in the mitochondria of H9C2 cells induced by H/R (Fig. 2C). These findings suggested that Lovastatin might suppress autophagy in H9c2 cells treated by H/R dose-dependently.
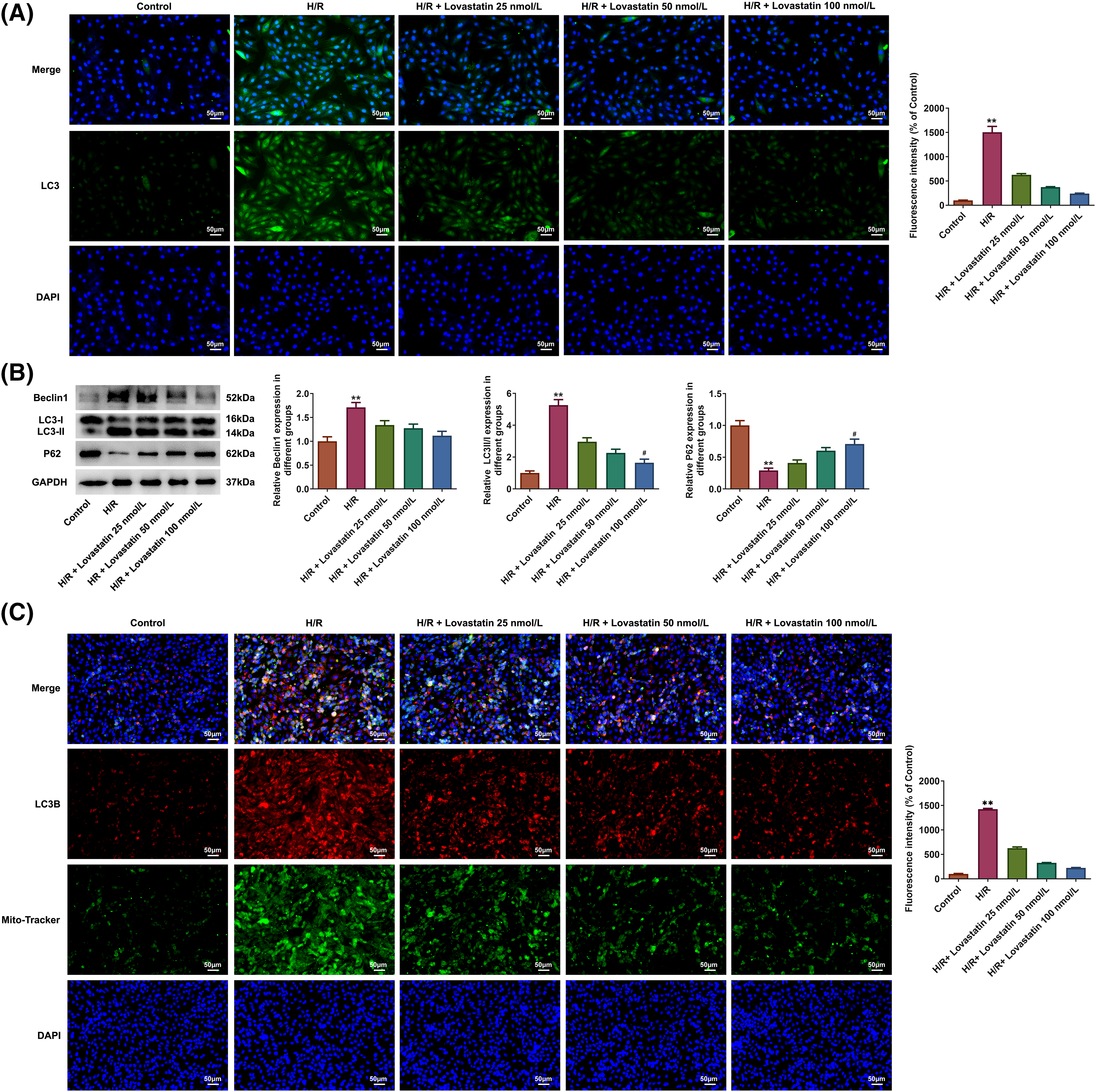
Figure 2: Lovastatin attenuates autophagy in H9c2 cells caused by H/R. Lovastatin decreased the fluorescence intensity of LC3 in cardiomyocytes (A), (n = 3). Lovastatin regulated autophagy-related proteins expression in H9c2 cells (B), (n = 3). Lovastatin decreased mitochondrial LC3B fluorescence intensity in H9c2 cells (C), (n = 3). **p < 0.01 vs. Control; #p < 0.01 vs. H/R. LC-3, microtubule-associated protein 1 light chain 3.
Lovastatin attenuates H/R-induced inflammation, oxidative stress injury and mitochondrial dysfunction in H9c2 cells
Then, the inflammatory response, oxidative stress, and mitochondrial function were further determined following myocardial injury. ELISA results showed that the levels of TNF-α, IL-6, and IL-1β were accelerated in H/R group (Fig. 3A). However, the concentrations of TNF-α, IL-6, and IL-1β were declined with the increasing doses of Lovastatin. Likewise, biochemical assay kits were used to detect oxidative stress markers. In comparison with the H/R group, Lovastatin resulted in the decreased MDA levels, and increased contents of SOD, GSH-Px, and CAT (Fig. 3B). Next, the mitochondrial function in H9c2 cells were detected after different treatments. The result demonstrated that the mitochondrial ROS level was upregulated after H/R injury and decreased following Lovastatin intervention (Fig. 3C). The results of JC-1 staining exhibited that Lovastatin dose-dependently enhanced the red fluorescence and diminished the green fluorescence in H/R-induced H9c2 cells (Fig. 3D). Additionally, Lovastatin restored the levels of MPTP opening (Fig. 4A), ATP (Fig. 4B) and OCR (Fig. 4C) in H9c2 cells subjected to H/R. Finally, the intracellular and mitochondrial Ca2+ concentrations were separately measured. The outcomes exhibited that Lovastatin dose-dependently reduced the levels of Ca2+ in H9c2 cells (Fig. 4D) and in the mitochondria of H9c2 cells (Fig. 4E). This implied that Lovastatin might mitigate inflammation, oxidative stress, and mitochondrial damage in H/R-induced H9c2 cells in a concentration-dependent manner.
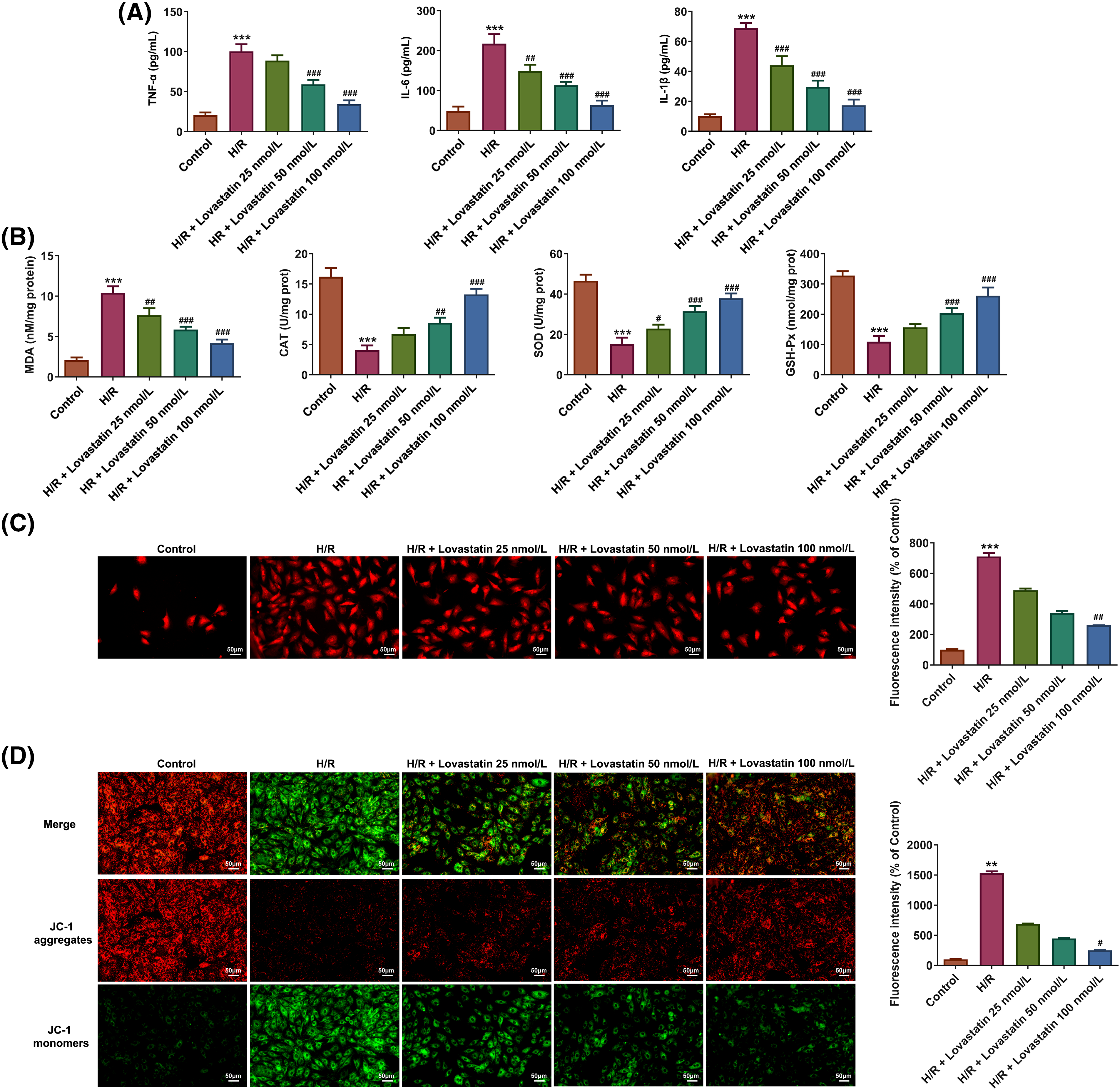
Figure 3: Lovastatin attenuates inflammation, oxidative stress and mitochondrial damage in H9c2 cells subjected to H/R. Lovastatin reduced the levels of proinflammatory cytokines in H9c2 cells (A), (n = 5). Lovastatin lowered MDA level and up-regulated SOD, GSH-Px and CAT contents in H9c2 cells (B), (n = 5). Lovastatin reduced mitochondrial ROS level in H9c2 cells (C), (n = 3). Lovastatin regulated MMP in H9c2 cells (D), (n = 3). **p < 0.001, ***p < 0.001 vs. Control; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. H/R. MDA, malonaldehyde; SOD, superoxide dismutase; GSH-Px, glutathione peroxidase; CAT, catalase; ROS, reactive oxygen species; MMP, mitochondrial membrane potential.
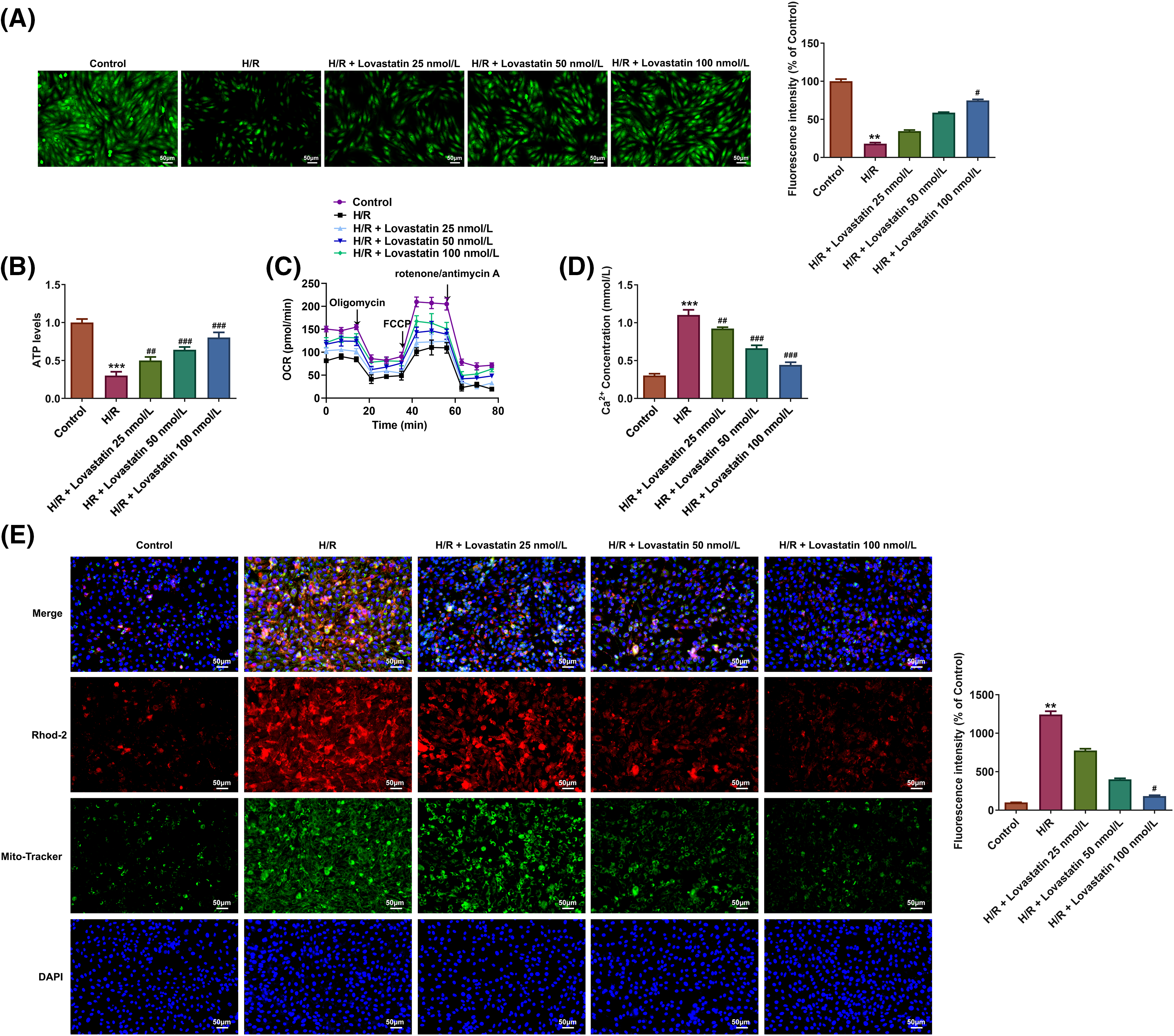
Figure 4: Lovastatin attenuates mitochondrial damage in H9c2 cells caused by H/R. Lovastatin increased mitochondrial permeability transition pore (MPTP) in H9c2 cells (A), (n = 3). Lovastatin increased mitochondrial ATP levels in H9c2 cells (B), (n = 5). Lovastatin increased oxygen consumption rate (OCR) in H9c2 cells (C), (n = 3). Lovastatin decreased the concentrations of Ca2+ in H9c2 cells (D), (n = 5). Lovastatin decreased the levels of mitochondrial Ca2+ in H9c2 cells (E), (n = 3). **p < 0.01, ***p < 0.001 vs. Control; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. H/R.
Lovastatin ameliorates H/R-induced injury in H9c2 cells through YAP/TAZ signaling
In the context of YAP/TAZ signaling, YAP was overexpressed by plasmid transfection, aiming to validate the impacts of Lovastatin on YAP/TAZ signaling-related proteins and determine whether the protective role of Lovastatin was linked to the signaling pathway. Initially, the expression of YAP and TAZ proteins involved in the signaling pathway were detected. The result illustrated that H/R treatment up-regulated the level of YAP and TAZ proteins, which were subsequently declined after Lovastatin intervention (Fig. 5A). Subsequently, Ov-YAP and Ov-NC plasmids were established and the transfection efficiency was assessed. As a result, YAP protein expression was significantly elevated after transfection of Ov-YAP (Fig. 5B). CCK-8 assay demonstrated that Ov-YAP impaired the effect of Lovastatin on H9c2 cell viability (Fig. 5C), and the contents of LDH, CK-MB, and CK were increased after overexpression of YAP (Fig. 5D). Similarly, TUNEL staining exhibited that the fluorescence intensity in the H/R + Lovastatin + Ov-YAP group was notably enhanced compared with the H/R + Lovastatin + Ov-NC group (Fig. 5E). Moreover, the H/R + Lovastatin + Ov-YAP group demonstrated the increased expression of Cleaved caspase-3, Bax, and the reduced Bcl2 expression compared with the H/R + Lovastatin + Ov-NC group (Fig. 5F). These research findings suggested that the protective role of Lovastatin in the myocardium might potentially be achieved via the mediation of the YAP/TAZ pathway.
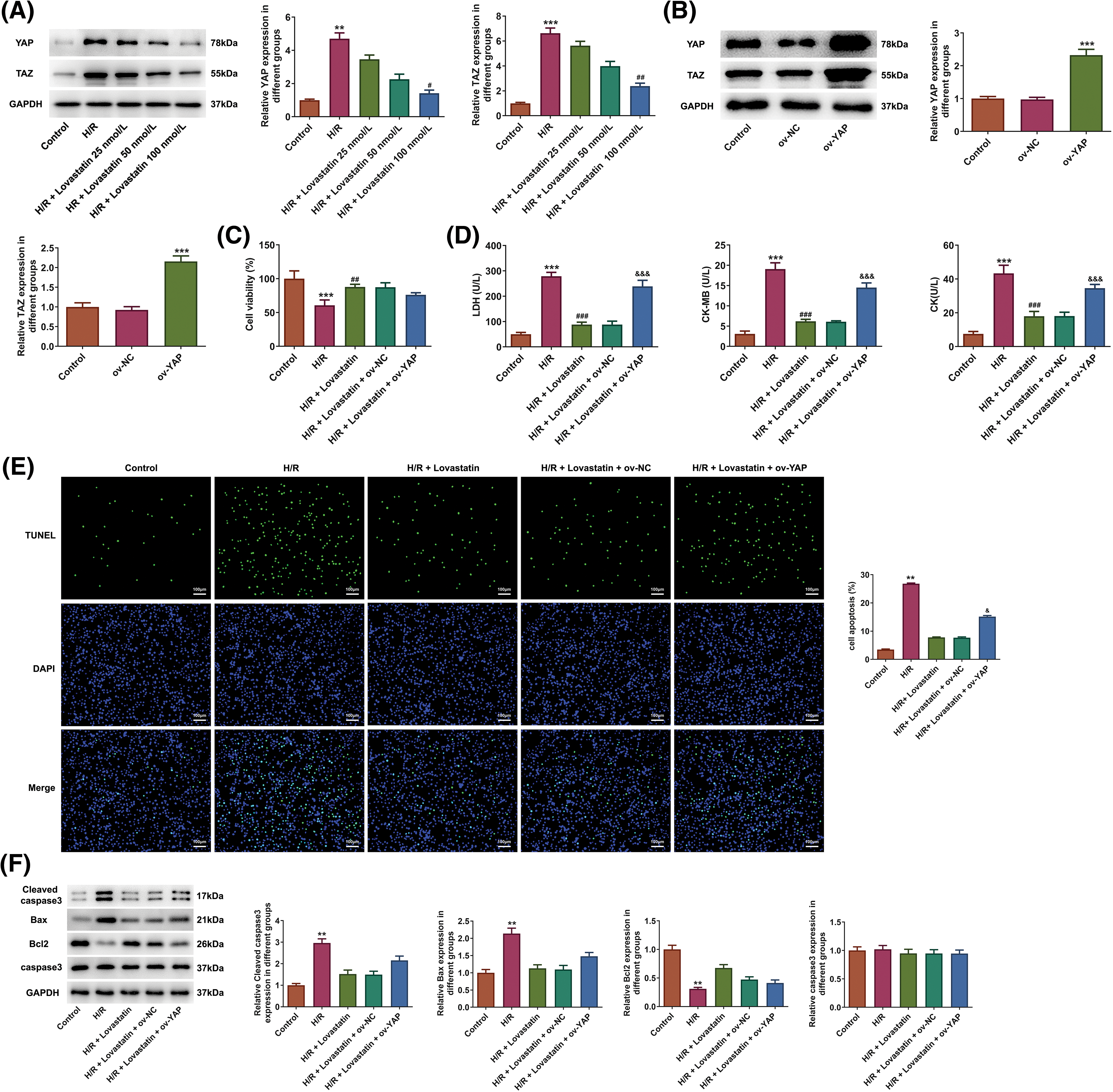
Figure 5: Lovastatin ameliorates H/R induced H9c2 cell injury through YAP/TAZ signaling. Lovastatin reduced the level of YAP and TAZ in H9c2 cells (A), (n = 3). WB detected the transfection efficacy of Ov-YAP (B), (n = 3). Ov-YAP impaired the effect of Lovastatin on H9c2 cell viability (C), (n = 5). Ov-YAP reversed effect of Lovastatin on myocardial enzymes in H9c2 cells (D), (n = 5). Ov-YAP enhanced the TUNEL fluorescence intensity in H9c2 cells treatment with H/R and Lovastatin (E), (n = 3). Ov-YAP increased pro-apoptotic proteins expression and decreased anti-apoptotic proteins expression in H9c2 cells in contrast with the H/R + Lovastatin + Ov-NC group (F), (n = 3). **p < 0.01, ***p < 0.001 vs. Control or Ov-NC; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. H/R; &p < 0.001, &&&p < 0.001 vs. H/R + Lovastain or H/R + Lovastain + Ov-NC.
Lovastatin attenuates autophagy in H9c2 cells caused by H/R via YAP/TAZ signaling
Next, to ascertain whether the impacts of Lovastatin on autophagy might be partially mediated by the YAP/TAZ signaling pathway. The result displayed that the fluorescence intensity of LC3 (Fig. 6A) and the level of Beclin1 and LC3-II/LC3I were up-regulated in the H/R + Lovastatin + Ov-YAP group in comparison with the H/R + Lovastatin + Ov-NC group, whereas P62 expression was reduced (Fig. 6B). Also, LC3B fluorescence intensity was detected in mitochondria. The results exhibited that Ov-YAP increased the fluorescence intensity of LC3B in the mitochondria of H9c2 cells, in contrast with the H/R + Lovastatin + Ov-NC group (Fig. 6C). The results illustrated that the inhibitory role of Lovastatin in H9c2 cell autophagy might be related to the YAP/TAZ signaling pathway.
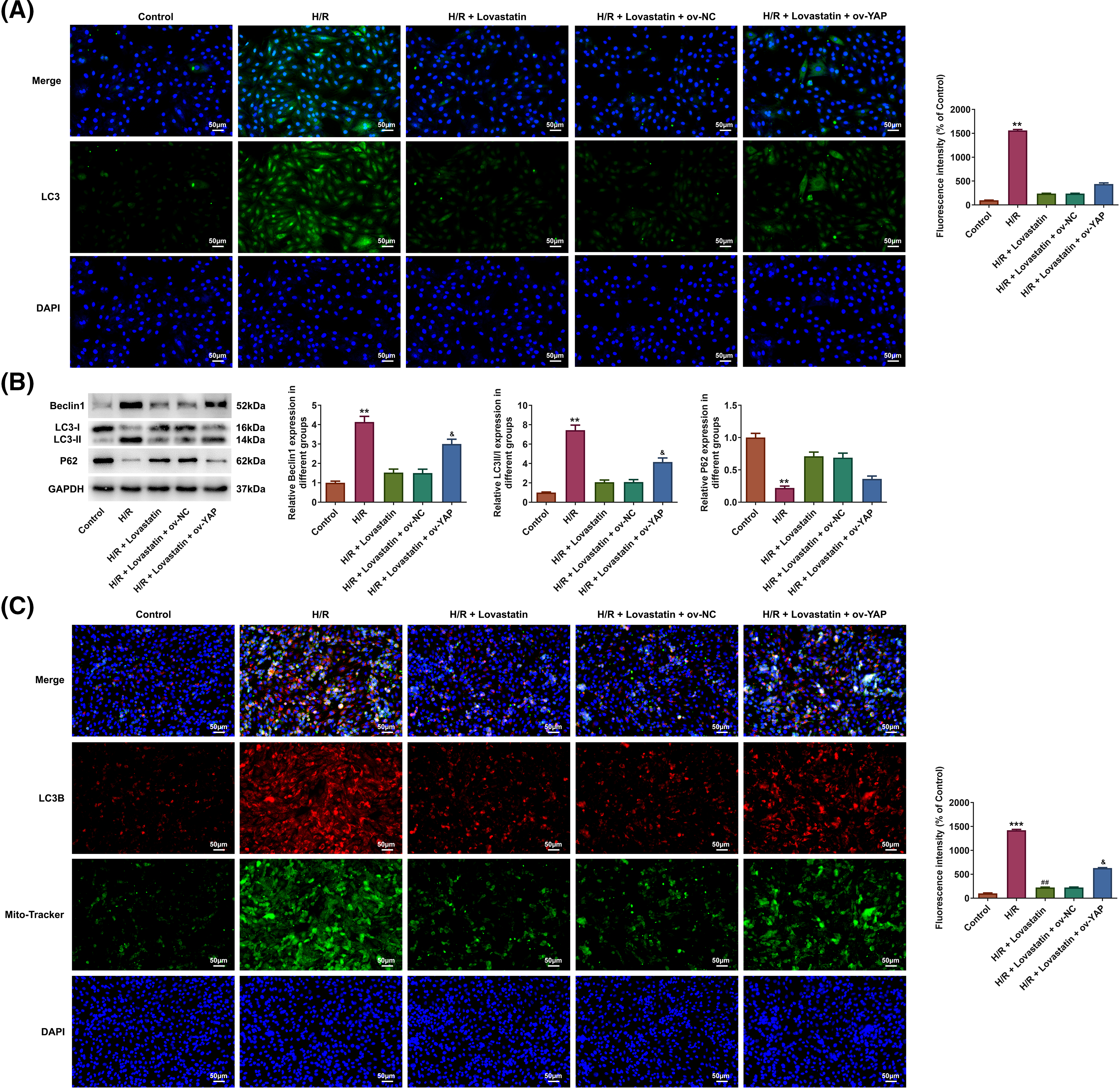
Figure 6: Lovastatin attenuates autophagy in H9c2 cells caused by H/R through YAP/TAZ pathway. Ov-YAP enhanced the expression of LC3 in H9c2 cells in contrast with the H/R + Lovastatin + Ov-NC group (A), (n = 3). Ov-YAP partially inhibited the role of Lovastatin in autophagy-related proteins expression in H9c2 cells (B), (n = 3). Ov-YAP increased the expression of LC3B in the mitochondria of H9c2 cells in contrast with the H/R + Lovastatin + Ov-NC group (C), (n = 3). **p < 0.01, ***p < 0.001 vs. Control; ##p < 0.01 vs. H/R; &p < 0.05 vs. H/R + Lovastain.
Lovastatin attenuates inflammation, oxidative stress and mitochondrial injure in H/R-induced H9c2 cells via YAP/TAZ signaling
Ultimately, whether Lovastatin exerted its influence on inflammation, oxidative stress, and mitochondrial dysfunction in H9c2 cells through mediating the YAP/TAZ signaling was explored. In contrast to the H/R + Lovastatin + Ov-NC group, the pro-inflammatory cytokines (Fig. 7A) and MDA contents were increased in H/R + Lovastatin + Ov-YAP group, and the contents of SOD, GSH-Px, and CAT were declined (Fig. 7B). Concurrently, through detecting mitochondrial ROS, the results affirmed that mitochondrial ROS level was elevated in the H/R + Lovastatin + Ov-YAP group compared with the H/R + Lovastatin + Ov-NC group (Fig. 7C). Through detecting MMP, it was discovered that the red fluorescence intensity was diminished and the green fluorescence intensity was enhanced in the H/R + Lovastatin + Ov-YAP group in comparison with the H/R + Lovastatin + Ov-NC group (Fig. 7D), Moreover, the levels of MPTP opening (Fig. 8A), ATP (Fig. 8B) and OCR (Fig. 8C) in H/R + Lovastatin + Ov-YAP group were decreased by contrast with the H/R + Lovastatin + Ov-NC group. Lastly, Ov-YAP up-regulated the concentrations of Ca2+ in H9c2 cells (Fig. 8D) and in mitochondria of H9c2 cells (Fig. 8E).
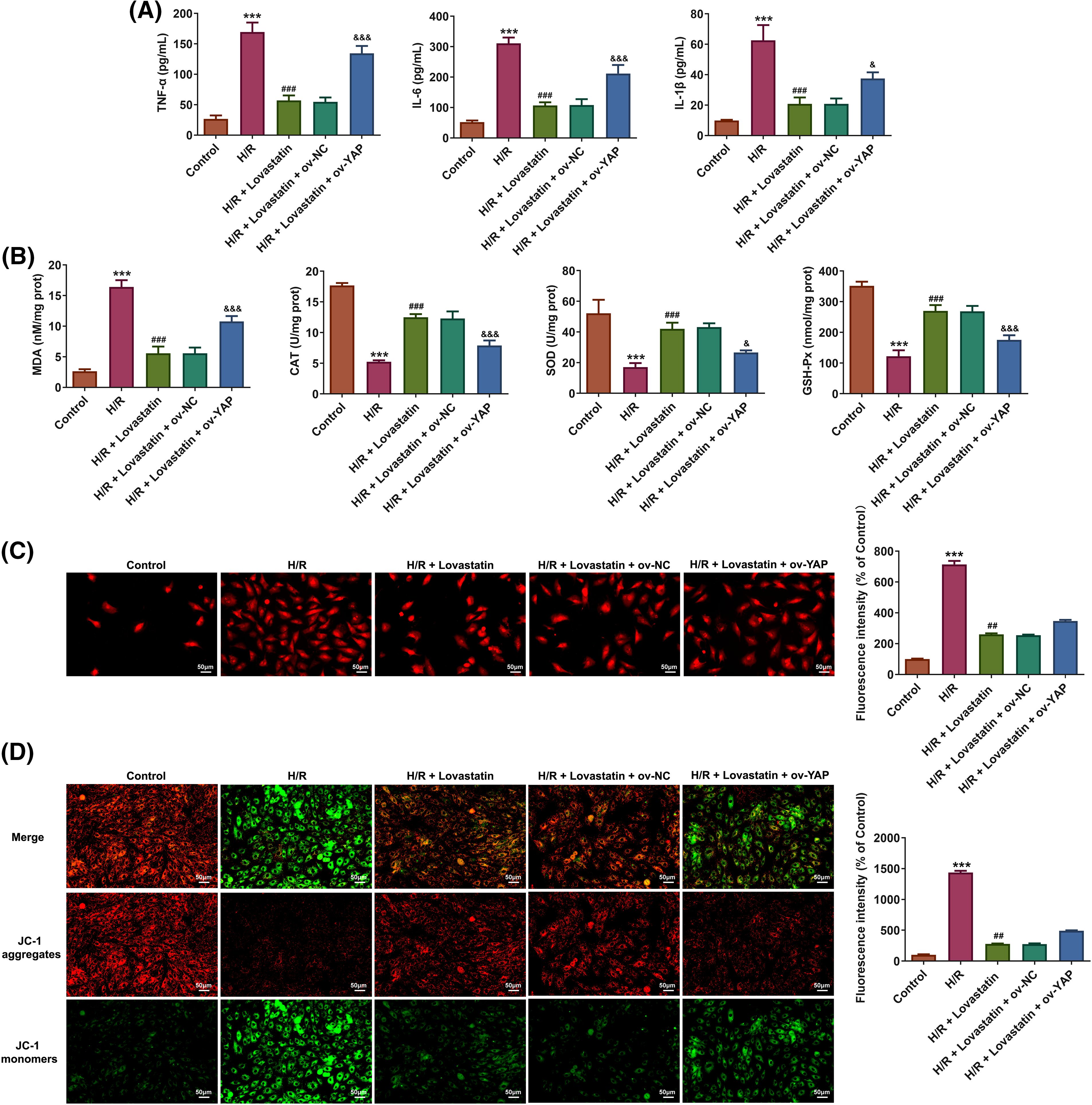
Figure 7: Lovastatin attenuates inflammation, oxidative stress and mitochondrial dysfunction in H/R-induced H9c2 cells via YAP/TAZ signaling. Ov-YAP up-regulated the contents of pro-inflammatory cytokines in H9c2 cells in contrast with the H/R + Lovastatin + Ov-NC group (A), (n = 5). Ov-YAP reversed the effect of Lovastatin on oxidative stress in H9c2 cells (B), (n = 5). Ov-YAP increased ROS level in mitochondria of H9c2 cells compared with the H/R + Lovastatin + Ov-NC group (C), (n = 3). Ov-YAP upregulated MMP in H9c2 cells compared the H/R + Lovastatin + Ov-NC group (D), (n = 3). ***p < 0.001 vs. Control; ##p < 0.01, ###p < 0.001 vs. H/R; &p < 0.05, &&&p < 0.001 vs. H/R + Lovastain.
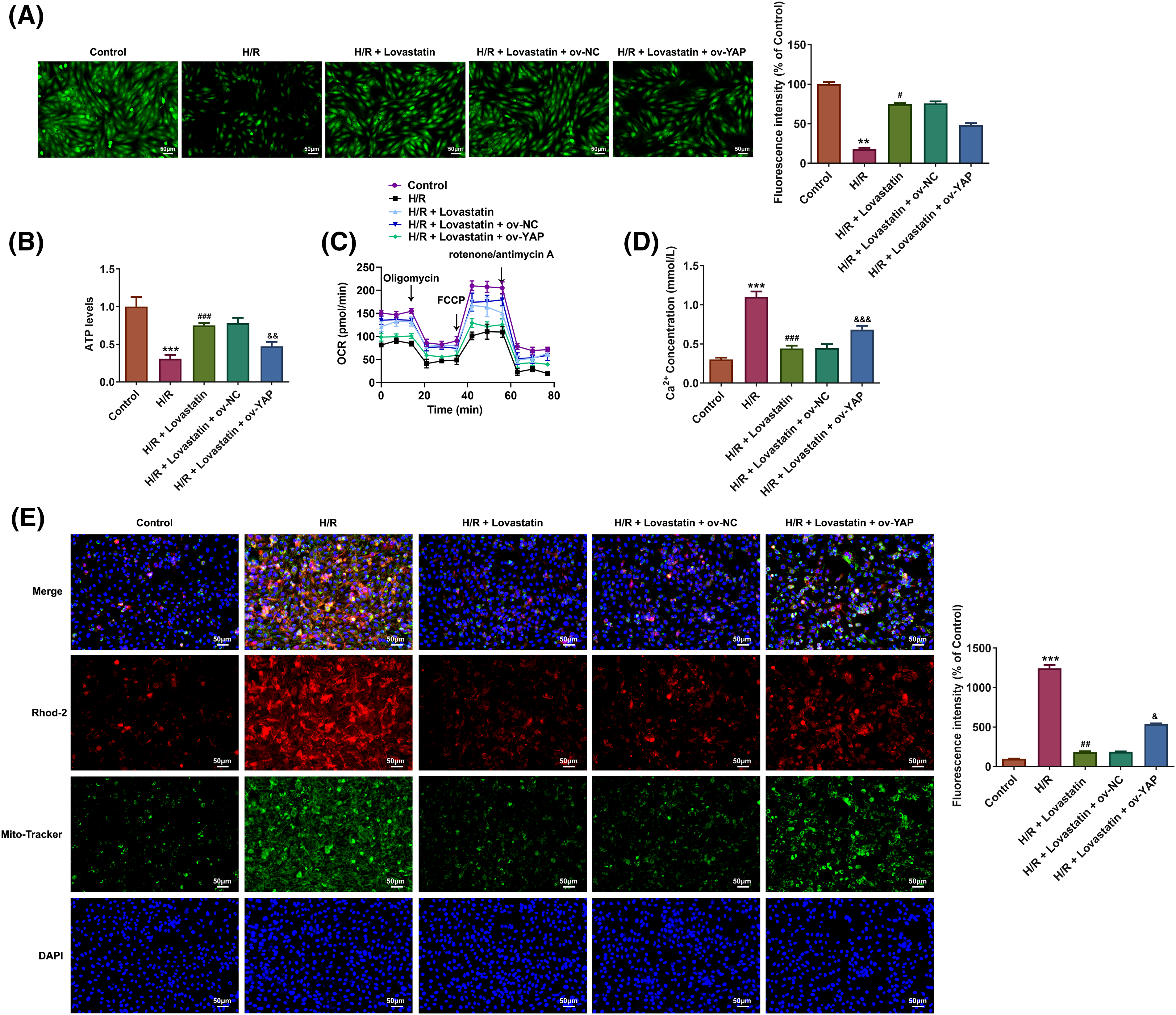
Figure 8: Lovastatin attenuates H/R-induced mitochondrial damage via YAP/TAZ signaling. Ov-YAP decreased the MPTP opening in H9c2 cells compared with the H/R + Lovastatin + Ov-NC group (A), (n = 3). Ov-YAP reduced the mitochondrial ATP levels in H9c2 cells compared with the H/R + Lovastatin + Ov-NC group (B), (n = 5). Ov-YAP decreased the OCR in H9c2 cells compared with the H/R + Lovastatin + Ov-NC group (C), (n = 3). Ov-YAP increased the levels of Ca2+ in H9c2 cells compared with the H/R + Lovastatin + Ov-NC group (D), (n = 5). Ov-YAP up-regulated the levels of Ca2+ in mitochondria of H9c2 cells compared with the H/R + Lovastatin + Ov-NC group (E), (n = 3). **p < 0.01, ***p < 0.001 vs. Control; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. H/R; &p < 0.05, &&p < 0.01, &&&p < 0.001 vs. H/R + Lovastain.
MI/R represents a predominant pathological process in the development of myocardial infarction and the subsequent heart failure, presenting a significant global health threat. Considering that, the MI/R cell model was created by H/R in H9c2 cells and treated by varying concentrations of Lovastatin. The results demonstrated that Lovastatin exerted inhibitory effects on cell injury, apoptosis, autophagy, inflammation, mitigated oxidative stress, and mitochondrial damage. Furthermore, through the construction of YAP overexpression plasmids, we have unveiled that the protective role of Lovastatin in H9c2 cells might be correlated with the YAP signaling pathway.
During the MI/R process, ROS is significantly produced [18]. Excessive ROS accumulation within cells can cause severe oxidative damage to cellular membranes, proteins, as well as RNA and DNA molecules [19]. Consequently, an excess of ROS production may lead to mitochondrial dysfunction, which may substantially impair both mitochondrial and cellular function [20]. Our research additionally revealed that H/R induction led to myocardial cell injury, elevated ROS levels, the impaired mitochondrial function, as evidenced by the conversion of the JC-1 staining fluorescent signal and changes of MPTP opening, OCR and Ca2+. In response to mitochondrial and organelle impairment, autophagy may be activated in cells [21]. During the initial phases of damage, autophagosomes can degrade compromised organelles to restore cellular equilibrium. Nevertheless, under I/R conditions, autophagy may be excessively activated, further aggravating cell death [22]. Due to mitochondrial impairment, cells are directed towards a programmed cell death pathway known as apoptosis. In this process, signaling pathways including the caspase pathway, become activated, leading to the breakdown of cellular structure [23]. In our experimental investigations, we identified the presence of both apoptosis and autophagy and verified that autophagy might occurs via this pathway, substantiated by the up-regulated expression of Cleaved caspase-3, Bax, and the down-regulated expression of Bcl2. Eventually, the accumulation of ROS, the apoptosis factors released from damaged cells, and the remnants of deceased cells might provoke an inflammatory response that exacerbates the damage, ultimately resulting in more severe myocardial impairment.
As previously discussed, our research findings were consistent with earlier research, confirming an increase in apoptosis and autophagy in myocardial cells following MI/R injury [24]. Furthermore, we explored the activation of the YAP/TAZ signaling pathway in MI/R injury, which was in line with previous research studies [13]. The YAP/TAZ signaling pathway has the capacity to regulate autophagy and mitochondrial function [25]. A study has also revealed a correlation between YAP/TAZ signaling pathway and autophagy. Autophagy inhibits the activity of YAP/TAZ signaling when the levels of α-catenin are low, while elevated α-catenin levels enhance YAP/TAZ activity [26]. This suggests that the role of YAP/TAZ signaling pathway in the initiation and progression of MI/R injury may be related to the regulation of autophagy and mitochondrial function. Lovastatin, a medication belonging to the statin class, has been demonstrates to exert protective effects in a range of organ models of I/R injury [14]. Our research results further substantiated that Lovastatin significantly reduced apoptosis, autophagy, inflammatory responses, and oxidative stress in H9c2 cells following H/R injury. This finding was consistent with earlier research on the protective properties of Lovastatin against tissue damage [16,27,28], indicating its ability to mitigate a series of pathological responses following myocardial injury. Furthermore, we have also found that Lovastatin significantly inhibits vascular fibrosis via inhibition of YAP/TAZ signaling [17]. This offers new insights into the cardioprotective mechanisms of Lovastatin, suggesting that it may suppress apoptosis, autophagy, oxidative and inflammatory damage in myocardial cells by modulating the YAP/TAZ signaling pathway, thus ameliorating myocardial injury. In addition, this study investigated the mechanism of myocardial protection by Lovastatin and focused more comprehensively on the effects of Lovastatin on a range of biological processes, including inflammation, apoptosis, oxidative stress, autophagy, and mitochondrial damage in cardiomyocytes. It was preliminarily demonstrated that the regulation of inflammation, oxidative stress, apoptosis, autophagy and mitochondrial damage in H/R-induced H9c2 cells by Lovastatin was related to the suppression of YAP/TAZ signaling. This might bring out a new insight for exploring the cardioprotective mechanism of Lovastatin and the relevance of YAP/TAZ signaling in inflammation, apoptosis, oxidative stress and autophagy.
The current study focused on the protection of Lovastatin in H/R-induced H9c2 cells through YAP/TAZ signaling, especially in terms of autophagy and mitochondrial damage. However, the limitations of the current research should not be overlooked. For example, maintaining the equilibrium between mitochondrial fusion and fission is crucial for preserving mitochondrial morphology and function [29]. Further research is required to illustrate the regulatory mechanisms governing mitochondrial function in H/R-induced cardiomyocytes in the presence of Lovastatin. Therefore, in future studies, the fusion and fission of mitochondria should be fully considered, through investigating key regulatory factors such as mitofusin 1 and dynamin-related protein 1, to ascertain the impact of Lovastatin on mitochondrial fusion/fission in H/R-induced myocardial cells. Furthermore, the present study demonstrated that the protection of Lovastatin on H9c2 cells induced by was influenced by the YAP/TAZ signaling, and the regulation of YAP/TAZ signaling was observed at an overall level. The study indicates that YAP phosphorylation can inhibit YAP transcriptional activity [30]. Therefore, how Lovastatin affects the YAP/TAZ signaling, whether it affects YAP phosphorylation or transcriptional activity, is worth further investigation. Additionally, we observed that Ov-YAP did not completely reverse the regulatory effects of Lovastatin on autophagy, apoptosis, and mitochondrial function in H9c2 cells stimulated by H/R. This suggested that the protective effect of Lovastatin on H9c2 cells might not solely depend on the YAP/TAZ pathway. Moreover, the current study is still preliminary. On one hand, the current results and conclusions were drawn from H9c2 cells. On the other hand, in vitro experiments cannot fully simulate the complex in vivo environment. Therefore, to clarify the role of Lovastatin in myocardial damage caused by I/R, more myocardial cell lines need to be included for a comprehensive study of Lovastatin’s role in I/R injury and further validation in animal MI/R models. The present results observed in H9c2 cells may provide a reference and general direction for future research. Lastly, statins work by competitively restraining the active site of HMG-CoA reductase, which in turn prevents cholesterol synthesis and decreases the risk of cardiovascular disease [31]. Other statins such as simvastatin, atorvastatin have also been shown to attenuate MI/R injury by inhibiting inflammation [32] and endoplasmic reticulum stress-related apoptosis [33], improving mitochondrial ultrastructure [34]. These studies focus on the regulation of inflammation, apoptosis, mitochondrial function, and other processes in myocardial cells by statin drugs. The findings are consistent with some of the results imposed by Lovastatin in this study. Therefore, the investigation into the role of Lovastatin and the exploration of mechanisms may serve as a reference for studying the role of other statin drugs in MI/R injury. Further research is warranted to systematically comprehend the role of statin drugs and to advance the development and utilization of such medications.
To summarize, our study has produced innovative mechanistic evidence regarding the cardioprotective properties of Lovastatin in MI/R injury. The consequences showed that Lovastatin played a protective effect on H/R-induced H9c2 cell damage by inhibiting YAP/TAZ signaling. These findings may offer new insights and references for a comprehensive understanding of the effect and mechanism of Lovastatin in MI/R injury.
Acknowledgement: None.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Mingzhu Li, Kaitian Zhang; data collection: Jianping Zhang, Jinfeng Li; analysis and interpretation of results: Kunlang Li, Huanqian Lu, Jinyan Lv; draft manuscript preparation: Kaitian Zhang, Mingzhu Li. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The analyzed data sets generated during the present study are available from the corresponding author on reasonable request.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Papatheodorou I, Makrecka-Kuka M, Kuka J, Liepinsh E, Dambrova M, Lazou A. Pharmacological activation of PPARbeta/delta preserves mitochondrial respiratory function in ischemia/reperfusion via stimulation of fatty acid oxidation-linked respiration and PGC-1alpha/NRF-1 signaling. Front Endocrinol. 2022;13:941822. [Google Scholar]
2. Yoshioka G, Tanaka A, Nishihira K, Shibata Y, Node K. Prognostic impact of serum albumin for developing heart failure remotely after acute myocardial infarction. Nutrients. 2020;12(9):2637. [Google Scholar] [PubMed]
3. Yeh KC, Lee CJ, Song JS, Wu CH, Yeh TK, Wu SH, et al. Protective effect of CXCR4 antagonist DBPR807 against ischemia-reperfusion injury in a rat and porcine model of myocardial infarction: potential adjunctive therapy for percutaneous coronary intervention. Int J Mol Sci. 2022;23(19):11730. [Google Scholar] [PubMed]
4. Boteanu RM, Suica VI, Uyy E, Ivan L, Cerveanu-Hogas A, Mares RG, et al. Short-term blockade of pro-inflammatory alarmin s100a9 favorably modulates left ventricle proteome and related signaling pathways involved in post-myocardial infarction recovery. Int J Mol Sci. 2022;23(9):5289. [Google Scholar] [PubMed]
5. Wang Y, Sheng Y, Ji N, Zhang H. Gentiopicroside enhances the protective effect of trimetazidine against myocardial ischemia-reperfusion injury via the AMPK/NLRP3 inflammasome signaling. J Biochem Mol Toxicol. 2023;37(7):e23366. [Google Scholar] [PubMed]
6. Wu J, Cai W, Du R, Li H, Wang B, Zhou Y, et al. Sevoflurane alleviates myocardial ischemia reperfusion injury by inhibiting P2X7-NLRP3 mediated pyroptosis. Front Mol Biosci. 2021;8:768594. doi:10.3389/fmolb.2021.768594. [Google Scholar] [PubMed] [CrossRef]
7. Ge L, Cai Y, Ying F, Liu H, Zhang D, He Y, et al. miR-181c-5p exacerbates hypoxia/reoxygenation-induced cardiomyocyte apoptosis via targeting PTPN4. Oxid Med Cell Longev. 2019;2019(1):1957920. doi:10.1155/2019/1957920. [Google Scholar] [PubMed] [CrossRef]
8. Ni Z, Li Y, Song D, Ding J, Mei S, Sun S, et al. Iron-overloaded follicular fluid increases the risk of endometriosis-related infertility by triggering granulosa cell ferroptosis and oocyte dysmaturity. Cell Death Dis. 2022;13(7):579. doi:10.1038/s41419-022-05037-8. [Google Scholar] [PubMed] [CrossRef]
9. Fu M, Xie D, Sun Y, Pan Y, Zhang Y, Chen X, et al. Exosomes derived from MSC pre-treated with oridonin alleviates myocardial IR injury by suppressing apoptosis via regulating autophagy activation. J Cell Mol Med. 2021;25(12):5486–96. doi:10.1111/jcmm.16558. [Google Scholar] [PubMed] [CrossRef]
10. Wu W, Chen X, Hu Q, Wang X, Zhu J, Li Q. Improvement of myocardial cell injury by miR-199a-3p/mTOR axis through regulating cell apoptosis and autophagy. J Immunol Res. 2022;2022(1):1642301. doi:10.1155/2022/1642301. [Google Scholar] [PubMed] [CrossRef]
11. Pan WW, Moroishi T, Koo JH, Guan KL. Cell type-dependent function of LATS1/2 in cancer cell growth. Oncogene. 2019;38(14):2595–610. doi:10.1038/s41388-018-0610-8. [Google Scholar] [PubMed] [CrossRef]
12. Zhou W, Shen Q, Wang H, Yang J, Zhang C, Deng Z, et al. Knockdown of YAP/TAZ inhibits the migration and invasion of fibroblast synovial cells in rheumatoid arthritis by regulating autophagy. J Immunol Res. 2020;2020(1):9510594. doi:10.1155/2020/9510594. [Google Scholar] [PubMed] [CrossRef]
13. Tian H, Xiong Y, Xia Z. Resveratrol ameliorates myocardial ischemia/reperfusion induced necroptosis through inhibition of the Hippo pathway. J Bioenerg Biomembr. 2023;55(1):59–69. doi:10.1007/s10863-022-09954-3. [Google Scholar] [PubMed] [CrossRef]
14. Tucci Junior S, Molina CA, Cassini MF, Leal DM, Schineider CA, Martins AC. Lovastatin protects mithochondrial and renal function in kidney ischemia-reperfusion in rats. Acta Cir Bras. 2012;27(7):477–81. doi:10.1590/S0102-86502012000700008. [Google Scholar] [PubMed] [CrossRef]
15. Henninger C, Huelsenbeck S, Wenzel P, Brand M, Huelsenbeck J, Schad A, et al. Chronic heart damage following doxorubicin treatment is alleviated by lovastatin. Pharmacol Res. 2015;91:47–56. doi:10.1016/j.phrs.2014.11.003. [Google Scholar] [PubMed] [CrossRef]
16. Deng X, Yang J, Qing R, Yuan H, Yue P, Tian S. Suppressive role of lovastatin in intracerebral hemorrhage through repression of autophagy. Metab Brain Dis. 2023;38(1):361–72. doi:10.1007/s11011-022-01101-6. [Google Scholar] [PubMed] [CrossRef]
17. Wu P, Liu Z, Zhao T, Xia F, Gong L, Zheng Z, et al. Lovastatin attenuates angiotensin II induced cardiovascular fibrosis through the suppression of YAP/TAZ signaling. Biochem Biophys Res Commun. 2019;512(4):736–41. doi:10.1016/j.bbrc.2019.03.158. [Google Scholar] [PubMed] [CrossRef]
18. Bae S, Park M, Kang C, Dilmen S, Kang TH, Kang DG, et al. Hydrogen peroxide-responsive nanoparticle reduces myocardial ischemia/reperfusion injury. J Am Heart Assoc. 2016;5(11):e003697. doi:10.1161/JAHA.116.003697. [Google Scholar] [PubMed] [CrossRef]
19. Yu B, Liu J, Wu D, Liu Y, Cen W, Wang S, et al. Weighted gene coexpression network analysis-based identification of key modules and hub genes associated with drought sensitivity in rice. BMC Plant Biol. 2020;20(1):478. doi:10.1186/s12870-020-02705-9. [Google Scholar] [PubMed] [CrossRef]
20. He D, Ma Z, Xue K, Li H. Juxtamembrane 2 mimic peptide competitively inhibits mitochondrial trafficking and activates ROS-mediated apoptosis pathway to exert anti-tumor effects. Cell Death Dis. 2022;13(3):264. doi:10.1038/s41419-022-04639-6. [Google Scholar] [PubMed] [CrossRef]
21. Lin Y, Yang Y, Yuan K, Yang S, Zhang S, Li H, et al. Multi-omics analysis based on 3D-bioprinted models innovates therapeutic target discovery of osteosarcoma. Bioact Mater. 2022;18:459–70. doi:10.1016/j.bioactmat.2022.03.029. [Google Scholar] [PubMed] [CrossRef]
22. Duan X, Kong Z, Mai X, Lan Y, Liu Y, Yang Z, et al. Autophagy inhibition attenuates hyperoxaluria-induced renal tubular oxidative injury and calcium oxalate crystal depositions in the rat kidney. Redox Biol. 2018;16:414–25. doi:10.1016/j.redox.2018.03.019. [Google Scholar] [PubMed] [CrossRef]
23. Yao R, Yang Y, Lian S, Shi H, Liu P, Liu Y, et al. Effects of acute cold stress on liver O-GlcNAcylation and glycometabolism in mice. Int J Mol Sci. 2018;19(9):2815. doi:10.3390/ijms19092815. [Google Scholar] [PubMed] [CrossRef]
24. Qiu Z, Wang Y, Liu W, Li C, Zhao R, Long X, et al. CircHIPK3 regulates the autophagy and apoptosis of hypoxia/reoxygenation-stimulated cardiomyocytes via the miR-20b-5p/ATG7 axis. Cell Death Discov. 2021;7(1):64. doi:10.1038/s41420-021-00448-6. [Google Scholar] [PubMed] [CrossRef]
25. Park SJ, Frake RA, Karabiyik C, Son SM, Siddiqi FH, Bento CF, et al. Vinexin contributes to autophagic decline in brain ageing across species. Cell Death Differ. 2022;29(5):1055–70. doi:10.1038/s41418-021-00903-y. [Google Scholar] [PubMed] [CrossRef]
26. Pavel M, Park SJ, Frake RA, Son SM, Manni MM, Bento CF, et al. α-Catenin levels determine direction of YAP/TAZ response to autophagy perturbation. Nat Commun. 2021;12(1):1703. doi:10.1038/s41467-021-21882-1. [Google Scholar] [PubMed] [CrossRef]
27. Xu R, Chen J, Cong X, Hu S, Chen X. Lovastatin protects mesenchymal stem cells against hypoxia- and serum deprivation-induced apoptosis by activation of PI3K/Akt and ERK1/2. J Cell Biochem. 2008;103(1):256–69. doi:10.1002/jcb.21402. [Google Scholar] [PubMed] [CrossRef]
28. Chen SF, Hung TH, Chen CC, Lin KH, Huang YN, Tsai HC, et al. Lovastatin improves histological and functional outcomes and reduces inflammation after experimental traumatic brain injury. Life Sci. 2007;81(4):288–98. doi:10.1016/j.lfs.2007.05.023. [Google Scholar] [PubMed] [CrossRef]
29. Daste F, Sauvanet C, Bavdek A, Baye J, Pierre F, Le Borgne R, et al. The heptad repeat domain 1 of Mitofusin has membrane destabilization function in mitochondrial fusion. EMBO Rep. 2018;19(6):e43637. doi:10.15252/embr.201643637. [Google Scholar] [PubMed] [CrossRef]
30. Wang T, Qin ZY, Wen LZ, Guo Y, Liu Q, Lei ZJ, et al. Epigenetic restriction of Hippo signaling by MORC2 underlies stemness of hepatocellular carcinoma cells. Cell Death Differ. 2018;25(12):2086–100. doi:10.1038/s41418-018-0095-6. [Google Scholar] [PubMed] [CrossRef]
31. Zahedipour F, Butler AE, Rizzo M, Sahebkar A. Statins and angiogenesis in non-cardiovascular diseases. Drug Discov Today. 2022;27(10):103320. doi:10.1016/j.drudis.2022.07.005. [Google Scholar] [PubMed] [CrossRef]
32. Wei T, Li J, Fu G, Zhao H, Huang C, Zhu X, et al. Simvastatin improves myocardial ischemia reperfusion injury through KLF-regulated alleviation of inflammation. Dis Markers. 2022;2022:7878602. doi:10.1155/2022/7878602. [Google Scholar] [PubMed] [CrossRef]
33. Xia JG, Xu FF, Qu Y, Song DG, Shen H, Liu XH. Atorvastatin post-conditioning attenuates myocardial ischemia reperfusion injury via inhibiting endoplasmic reticulum stress-related apoptosis. Shock. 2014;42(4):365–71. doi:10.1097/SHK.0000000000000224. [Google Scholar] [PubMed] [CrossRef]
34. Godoy JC, Niesman IR, Busija AR, Kassan A, Schilling JM, Schwarz A, et al. Atorvastatin, but not pravastatin, inhibits cardiac Akt/mTOR signaling and disturbs mitochondrial ultrastructure in cardiac myocytes. FASEB J. 2019;33(1):1209–25. doi:10.1096/fj.201800876R. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools