
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW
Spliceosome-mediated RNA trans-splicing: a strategy for Huntington’s disease gene therapy
School of Environmental and Biological Engineering, Nanjing University of Science & Technology, Nanjing, 210000, China
* Corresponding Author: DAN WENG. Email:
(This article belongs to the Special Issue: Neural Stem Cells in Health and Disease: Mechanisms and Applications)
BIOCELL 2024, 48(10), 1443-1453. https://doi.org/10.32604/biocell.2024.053794
Received 10 May 2024; Accepted 01 July 2024; Issue published 02 October 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Huntington’s disease (HD) is a debilitating neurodegenerative disorder caused by an abnormal expansion of CAG repeats (Cytosine, Adenine, Guanine) in the huntingtin gene (HTT). This mutation leads to the production of a mutant huntingtin protein, resulting in neuronal dysfunction and cell death. Current treatments primarily focus on symptomatic relief and do not address the underlying genetic cause. This review explores spliceosome-mediated RNA trans-splicing (SMaRT) therapy as an innovative and potential approach for HD treatment. SMaRT leverages the cell’s natural splicing machinery to correct mutant mRNA, thereby reducing toxic protein levels while restoring functional protein production. We compare SMaRT with other gene therapy strategies, such as antisense oligonucleotides, RNA interference, and CRISPR-based systems, highlighting SMaRT’s dual-action mechanism and its potential advantages in clinical applications. Additionally, we discuss the challenges and future directions for SMaRT therapy, emphasizing the need for further research to optimize its efficacy and safety. This review aims to provide a comprehensive overview of current and emerging therapies for HD, with a focus on the innovative potential of SMaRT.Keywords
Huntington’s disease (HD) is a neurodegenerative condition caused by an abnormal increase in (Cytosine, Adenine, Guanine) CAG repeats in the huntingtin gene (HTT) on chromosome 11. Worldwide, the disease exhibits a prevalence of approximately 3.92 cases per 100,000 and an incidence of 0.48 new cases per 100,000 each year, with 7.1% of these cases due to de novo mutations [1]. The increasing prevalence in recent decades [2] underscores the urgent need for innovative therapeutic strategies. Individuals with Huntington’s disease undergo a gradual decline in motor skills, cognitive function, and mental health [3]. Initial symptoms often include chorea, characterized by rhythmic, dance-like involuntary movements. Other motor disturbances, psychiatric conditions, and cognitive impairments can also be present [4]. Following the onset of motor symptoms, the average life expectancy is 15 to 20 years [5]. At present, there are no clinically verified treatments that can stop or reverse the progression of Huntington’s disease; available treatments primarily manage symptoms such as chorea, along with psychiatric and cognitive disturbances. The FDA has approved several drugs to address these symptoms, including tetrabenazine and deutetrabenazine, which are vesicular monoamine transporter 2 (VMAT2) inhibitors that reduce hyperkinetic movements by decreasing dopaminergic neurotransmission [6–8]. Psychiatric symptoms such as depression, anxiety, obsessive-compulsive disorder (OCD), and aggression are rare behavioral manifestations, while cognitive symptoms may manifest as memory deficits and a decline in thought processing and organization [9].
Gene therapy strategies for treating HD include various approaches aimed at targeting the genetic mutations underlying the disease, whose pros and cons will be discussed later. The various methods, in addition to FDA-approved drugs for managing symptoms, are outlined in Table 1.

We present a groundbreaking gene therapy, previously successful in other conditions, which also offers considerable promise for HD management. Spliceosome-mediated RNA trans-splicing therapy (SMaRT) focuses on pre-mRNA, employing the inherent trans-splicing machinery to amend the mutated allele. This method offers a straightforward, cost-effective strategy for addressing HD. This review sheds light on current research into Huntington’s disease, outlines the progressive developments in applying SMaRT therapy to HD and related conditions, and delves into the complexities and obstacles associated with this approach.
Literature Search and Data Collection
The compilation of this review involved a comprehensive literature search focused on HD. The primary databases used were PubMed, Google Scholar, and Web of Science, selected for their comprehensive coverage of biomedical and molecular biology research. Keywords and phrases such as “Huntington’s disease”, “HTT gene”, “molecular pathogenesis”, “genetic regulation”, “Huntingtin protein”, “trans-splicing” and “neurodegeneration” were utilized to find relevant studies. Peer-reviewed articles published in English and focusing on the molecular and genetic aspects of HD were included, while studies unrelated to HD or published in non-peer-reviewed sources were excluded to ensure the information’s reliability and relevance. The Figures and table in this review were produced with Office and Figdraw.
Molecular Pathogenesis and Genetic Regulation of Huntington’s Disease
HD arises due to a genetic anomaly: a volatile expansion of a CAG triplet sequence located within exon 1 of the HTT gene. This CAG repeat is located 17 codons downstream of the initiator ATG (Adenine, Thymine, Guanine) in exon 1 of the 67-exon gene, affecting the resulting protein product. This genetic alteration leads to the synthesis of a huntingtin protein that features an elongated polyglutamine segment [10]. The misfolded mutant huntingtin protein builds up in the striatum and cerebellum, exhibiting toxic properties and leading to protein fragmentation, which in turn triggers neuronal dysfunction and death [11]. Further exacerbating the buildup of this toxic protein is the impairment of both the ubiquitin-proteasome and autophagy systems [12,13].
Moreover, the presence of tissue-specific isoenzyme forms of respiratory chain complexes contributing to mitochondrial heterogeneity may elucidate the targeted cellular susceptibility observed in HD. Although HD stems from a nuclear DNA mutation rather than a mitochondrial one, it is plausible that the huntingtin normally aids in mitochondrial function regulation or that its mutant form actively disrupts cellular respiration [14]. This emphasizes the necessity of therapies targeting protein degradation pathways, which might offer a potential therapeutic angle.
Transcripts from both normal and expanded alleles of the HTT are present in lymphoblasts and the brain [15]. Even with the assumption, there is no evidence suggesting that homozygotic carriers of the mutated HTT allele exhibit earlier onset or faster disease progression [16], indicating that the presence of normal gene copies might not mitigate the disease’s progression. Additionally, homozygotes are known to express mRNA for the expanded HTT without apparent gene inactivation by the expanded CAG repeat [17]. Moreover, certain rare genetic mutations have led to individuals with one deleted HTT who exhibit no HD symptoms.
From the earliest disease stages, oxidative stress coupled with peripheral and central inflammation occurs, impacting the central nervous system (CNS) and plasma [18]. Mutant huntingtin also disrupts transcriptional regulation [19] and axonal transport in mice, diminishing brain-derived neurotrophic factor (BDNF)-mediated signaling relative to wild-type mice [13]. Genome-wide association studies link the age at onset and disease progression in HD to specific loci within DNA repair genes [20]. HD’s genetic pattern includes the paternal transmission of varying numbers of CAG repeats, with no analogous maternal repeat expansion. This paternal transmission suggests that mitotic divisions during spermatogenesis might be responsible for repeat instability [21], implying a connection between repeat instability and gene transcription [22]. Additionally, the severity of pathology in HD does not directly correlate with the number of CAG repeats, but instead with the extent of somatic mosaicism across different tissues [23]. The role of somatic mosaicism in disease progression is a crucial area for future research, as it may provide insights into potential tissue-targeted therapy.
HTT mRNA is ubiquitously expressed across all body tissues [24], with its expression level and the resultant protein remaining unchanged in HD conditions [25]. Within the brain, the expression of HTT is primarily observed in neurons, while it is minimally expressed in glial cells [26]. The cerebellum, hippocampus, and olfactory bulb exhibit the highest levels of HTT expression, which correspond to regional neuronal densities. On the other hand, the striatum shows modest to moderate amounts of HTT mRNA [27]. Recent studies have highlighted a heterogeneous distribution of huntingtin within normal mice striatal neurons [28], predominantly in the matrix region with scant presence in the patch regions. This pattern of distribution is associated with a notable decrease in calbindin-positive neurons within the matrix, indicating that the susceptibility of medium-sized spiny neurons in the matrix area is connected to increased levels of huntingtin expression. Conversely, the enhanced resilience of larger and medium-sized aspiny neurons in the patch regions may be attributed to lower levels of huntingtin [22].
Beyond the CAG repeats, a highly toxic HTT exon 1 protein containing the polyglutamine tract exists exclusively in mutation carriers [29]. Faulty splicing of HTT mRNA leads to the formation of a brief polyadenylated mRNA strand that includes exon 1 and the initial part of intron 1, subsequently generating the exon 1 protein fragment. This toxic protein accumulates in striatal neurons and is evident in post-mortem brains of patients with juvenile-onset HD. Mouse models expressing this toxic exon 1 protein demonstrate more severe neuronal loss and quicker neurological impairments than those expressing the full-length mutation [30]. Recent research indicates toxic activity for N-terminal mutant HTT (mHTT) oligomers produced by aberrant splicing, leading to a short mRNA that translates into a toxic N-terminal fragment also affected by the proteolytic cleavage of the abnormal protein [31]. These findings underscore the potential of highly toxic exon 1 protein as a primary pathogenic species. However, the modulation of this protein by existing therapeutic agents targeting downstream HTT mRNA, such as splicing modulators (e.g., branaplam, PTC518), antisense oligonucleotides (e.g., tominersen, WVE-120101, and WVE-120102), or RNA interference agents (e.g., VY-HTT01) currently in clinical development, remains limited. Alternatively, targeting HTT DNA or HTT exon 1 mRNA may provide a more effective strategy in lowering the expression of this deleterious protein. This perspective aligns with recent advancements in gene-editing technologies, such as CRISPR/Cas9, which offer promising avenues for directly correcting the underlying genetic defect [32,33].
Huntingtin in HD Pathophysiology
Huntingtin is a complex protein consisting of approximately 3144 amino acids, with a molecular mass of about 348 kDa, and its length varies with the number of CAG repeats [10]. This protein has unique characteristics, showing no significant homology to any known proteins. The sequence includes a series of glutamine repeats beginning at residue 18, as well as a variable CCG repeat that codes for a polyproline region. Notably, huntingtin lacks hydrophobic membrane-spanning regions, distinguishing it from many transmembrane proteins. Huntingtin protein is characterized by a structural motif referred to as the ‘HEAT repeat,’ named for its identification in four key proteins: huntingtin, elongation factor 3, the regulatory A subunit of protein phosphatase 2A, and TOR [34]. The proteins containing HEAT repeats mainly function as regulatory cytoplasmic proteins involved in critical cellular transport processes.
These HEAT-containing proteins are predominantly regulatory cytoplasmic proteins involved in essential cellular transport processes. Studies indicate that huntingtin associates with cytoplasmic granules similar to multivesicular bodies, and organelles involved in retrograde transport and protein breakdown, especially in the cortical and striatal neurons of individuals with HD [35]. Moreover, other research has shown that huntingtin interacts with microtubules, indicating a potential involvement in vesicle trafficking [36]. Additional support for this interaction comes from findings that both the mutant and normal forms of huntingtin are linked to clathrin-coated vesicles located in the Golgi apparatus, cytoplasm, and plasma membrane [37]. Despite extensive research, the full spectrum of huntingtin’s physiological activities remains elusive. Over the decades, studies have elucidated huntingtin’s involvement in numerous cellular processes including cell signaling, transcriptional regulation, energy metabolism, DNA maintenance, axonal trafficking, and antiapoptotic activity [38–40].
In the striatum, projection neurons expressing huntingtin play crucial roles in preserving synaptic connections, controlling gene activity, and supporting the longevity of neurons as organisms grow older [41,42]. The loss of functional huntingtin in these neurons recapitulates the hallmark features of Huntington’s disease, indicating the critical role of this protein in neural function [41]. Interestingly, the polyglutamine tract within huntingtin may enhance its normal activity; however, in its expanded form, it could act in a dominant negative manner, negating the protein’s typical functions and affecting the transcription from normal alleles [22]. This expanded polyglutamine (polyQ) tract impairs proper protein folding, resulting in mHTT that remains as an unfolded, soluble protein capable of forming oligomers. These oligomers can accumulate into large inclusions and fibrils within the cytoplasm and nucleus [43]. Previous insights suggest that N-terminal mHTT oligomers, produced through aberrant splicing, might generate a short mRNA that translates into a toxic N-terminal fragment. This toxicity is compounded by the proteolytic cleavage of the aberrant protein, underscoring the complex pathogenic mechanisms at play in HD [44]. The understanding of huntingtin’s multifaceted role in cellular processes and its pathogenic transformation in HD underscores the importance of targeted therapeutic strategies. Future research should aim to develop interventions that can modulate the toxic effects of mutant huntingtin while preserving the essential functions of the normal protein. This dual approach could potentially mitigate the disease’s impact more effectively and improve patient outcomes.
HD Impacts Neurogenesis and Protein Loss Progression
HD significantly affects neurogenesis and is characterized by a progressive decline in neuronal function and activity due to the accumulation of toxic protein aggregates. GABAergic medium spiny neurons (MSNs) in the striatum and cortex show heightened susceptibility to the excitotoxicity associated with HD, with a predominant impact on striatal MSNs [45]. As neuronal loss exacerbates and spreads throughout the brain, it precipitates the emergence of various signs and symptoms typical of HD. Motor symptoms notably intensify as MSN degeneration progresses in the striatum. Conversely, neuronal loss in the cortex stimulates neurogenesis, serving as a compensatory mechanism to cope with critical neuron loss [46]. This compensatory neurogenesis suggests potential therapeutic targets to enhance endogenous repair mechanisms in HD-affected regions.
Concurrently, as neuron loss originates in the striatum, neural stem cells in specific brain regions generate progenitor cells and neuroblasts [47,48]. Situated in the subventricular zone (SVZ) of the lateral ventricles and the subgranular zone of the dentate gyrus in the hippocampus, these cells develop into intermediate progenitors. In turn, these intermediate progenitors generate new neuroblasts that travel to the inner granule zone, where they mature into dentate granule cells and assimilate into the surrounding neural network [49]. Furthermore, the reduction of HTT protein in postmitotic projection neurons leads to the displacement of certain neuronal groups within the mouse neocortex [50,51]. A comparable morphological variability is noted in individuals with Huntington’s disease, wherein the division of neural progenitor cells and the migration and maturation of neurons are impaired. This suggests that Huntington’s disease involves both neurodegenerative and neurodevelopmental elements [52]. The dual nature of Huntington’s pathology highlights the importance of employing a comprehensive therapeutic strategy that integrates neuroprotective measures with interventions targeting neurodevelopmental mechanisms. This indicates that the selection of target cells for HD treatment should be contingent on the patient-specific conditions. Ideal target cells include neuronal progenitor cells, astrocytes, and microglia, among others.
Natural DNA Repair in HD cells
The rate of CAG repeat expansion varies significantly between different tissues and cell types. Neurons, especially those in striatal and cortical areas, exhibit a more pronounced expansion compared to glial cells, which are highly susceptible to degeneration in Huntington’s disease [53]. Genome-wide association studies involving thousands of HD patients have identified DNA repair mechanisms as key modifiers of the disease’s progression [20,54]. Furthermore, the genes involved in these mechanisms are also linked to other trinucleotide repeat disorders [55].
A key modifier gene in this context is Fanconi-associated nuclease 1 (FAN1), which encodes a nuclease that plays an essential role in the DNA repair process by cutting DNA during the repair of crosslinks between strands. FAN1’s functionality is protective in HD because it precisely repairs loopouts at CAG repeats [56]. When FAN1 protein is depleted in the neurons of both animal models and HD patients, there is an accelerated expansion of repeats. Variants that reduce FAN1 function have been observed to advance the onset of Huntington’s disease by over five years, while those that enhance FAN1 expression can delay the onset by a similar period [57]. Given these findings, therapeutic strategies aiming to enhance FAN1 activity could potentially stabilize CAG repeats and delay disease onset, offering a new avenue for HD treatment.
Mismatch repair stands out as the most critical DNA repair pathway influencing the pathogenesis of Huntington’s disease. This pathway is triggered by the identification of base mismatches or single-strand loopouts by a MutS complex. After detection, a MutL nuclease complex is engaged to cleave the DNA, thereby aiding in the breakdown of the strand containing errors and the subsequent rebuilding using the remaining template strand. This sequence of events culminates with the sealing of the newly synthesized strand’s ends [58]. The potential for therapeutic intervention targeting the mismatch repair pathway could also be explored, specifically by developing agents that enhance the accuracy and efficiency of this repair process in neurons.
Despite these sophisticated cellular mechanisms, studies have indicated that the inherent DNA repair capacity in HD is insufficient for completely correcting the expansions of CAG repeat and exon 1 mutations involved in protein genesis [59]. These genetic errors significantly contribute to HD pathology. Therefore, the development and application of external therapies targeting the production of mutant huntingtin (mHTT) remain critical for effectively managing the disease and mitigating its impacts on patients.
Gene Therapy Strategies for Treating HD
Addressing HD is complicated due to the dominant-negative characteristics of the mHTT gene. Despite this, the monogenic nature of HD’s symptoms makes gene editing a feasible strategy. Nevertheless, the pivotal function of HTT in neurodevelopment precludes the possibility of completely knocking out the HTT gene, as such an action would be embryonically lethal, given its vital role in developmental processes [60,61]. Research using animal models that carry the HD mutation has demonstrated that these models exhibit protein inclusions, brain atrophy, and progressive neurological decline [62]. Moreover, experiments involving the ablation of wild-type huntingtin in two-month-old mice have resulted in pancreatitis but are not harmful to the adult mouse brain [63].
Previous research has indicated that lowering the expression of mutant CAG repeat alleles in mouse models can alleviate motor and cognitive dysfunctions [64]. Studies have shown that huntingtin-lowering therapies lead to increased survival and reduced weight loss compared to untreated models, and also improve biochemical deficits, synaptic dysfunction, transcriptional dysregulation, atrophy rates, and intraneuronal protein aggregates [65–68]. Given these findings, further research should focus on optimizing huntingtin-lowering strategies to balance the reduction of toxic proteins while maintaining sufficient levels of normal huntingtin to support neurodevelopmental processes. However, reports also suggest that expansions of CAG repeats in one allele do not affect the transcription of the wild-type allele [19]. Clinical observations have demonstrated that individuals with decreased transcription of the wild-type allele exhibit earlier disease onset compared to those with unaffected transcription [69], while rare homozygous expansions show similar developmental and disease onset profiles as heterozygous carriers [16].
Antisense Oligonucleotides (ASO): ASOs are synthetic short single-stranded oligonucleotides (18–50 base pairs) designed to target and modulate messenger RNA (mRNA) function through Watson-Crick base pairing. These oligonucleotides have the ability to interact with pre-mRNA within the nucleus or mature mRNA in the cytoplasm in order to control gene expression [70]. Although ASOs are effective in silencing simple CAG repeats, allele-selective approaches face limitations, such as the inability to target all individuals affected by HD and the potential to inadvertently suppress the transcription of other genes with similar sequences [71]. In clinical applications, gapmer ASO therapies to reduce huntingtin levels have shown preliminary success. After binding, the central region of the gapmer ASO recruits the RNase H enzyme, leading to the degradation of the mRNA [71]. Initial results have shown a transient increase in neurofilament light protein levels in the cerebrospinal fluid before the full suppression of mutant huntingtin, indicating possible off-target inflammatory effects [72]. Nonetheless, studies have found that using mesenchymal stem cells (MSCs) targeting astrocytes can enhance the efficacy of ASO therapy, underscoring its potential utility in HD treatment.
RNA Interference (RNAi) technology: This method utilizes small interfering RNA (siRNA) or microRNA (miRNA) that specifically targets mature mRNA within the cytosol, facilitating its breakdown via the RNA-induced silencing complex and consequently decreasing protein synthesis. Current developments include conjugated siRNAs that can be internalized by neurons, with ongoing programs such as ALN-HTT targeting exon 1 of the HTT mRNA [73].
Gene editing technologies such as ZFNs, TALENs, and CRISPR-Cas are advanced methods that target the genetic defects associated with HD for correction. Zinc finger proteins, which include a DNA-binding domain and an effector module, selectively target mutant sequences without affecting the wild type. These proteins have proven effective at lowering the expression of mutant huntingtin in cell cultures and enhancing behavioral phenotypes in models of Huntington’s disease [74]. TALENs rely on a DNA-binding domain, known as the TALE protein, which recognizes specific nucleotides within the genome. The other component of TALENs, the FokI nuclease domain, introduces double-strand breaks (DSBs) at precise genomic locations, facilitating gene knockout, correction, and transgenic functions [75]. TALENs used for targeting CAG repeats in human HD fibroblasts have shown reduced expression of mutant HTT [76]. CRISPR-based systems offer accurate and efficient gene editing abilities, allowing for targeted deactivation of the mutant HTT gene in fibroblasts derived from Huntington’s disease patients, mouse models, and human-induced pluripotent stem cells [77].
Gene therapy aims to directly target and correct the genetic anomalies that fundamentally cause genetic disorders. For recessive disorders, this often entails introducing a functional wild-type gene to compensate for the defective one. Conversely, conditions resulting from dominant mutations, often arising from haploinsufficiency or dominant-negative effects, necessitate both the suppression of the mutant allele and the restoration of the normal gene’s function. In response, technologies for silencing genes via RNA, including antisense oligonucleotides, ribozymes, and RNA interference, have been engineered. These methods are specifically designed to selectively target and neutralize mutations that have dominant-negative effects. However, these approaches do not tackle the haploinsufficiency of the wild-type allele [78]. SMaRT technology could potentially fulfill both requirements to effectively treat disorders stemming from dominant-negative mutations [79].
Gene transcription generates pre-mRNA, encompassing both exons and introns. The spliceosome, an enzymatic complex made up of ribonucleoproteins and associated proteins, assists in removing introns and joining exons to create mature mRNA, which serves as the blueprint for protein synthesis. This mechanism relies on certain cis-elements located within the introns: the 5′ and 3′ splice sites that define intron boundaries, and the branchpoint, generally situated 18–40 nucleotides upstream of the 3′ splice site, marked by a consensus sequence of adenosine and succeeded by a polypyrimidine tract in higher eukaryotes. These elements can vary slightly among organisms. The spliceosome assembly on pre-mRNA and its regulation are tightly controlled processes. Initially, U1 small nuclear ribonucleoprotein particles (snRNP) along with several proteins attach to the 5′ splice site, facilitating the binding of U2 snRNP auxiliary factor (U2AF) to the polypyrimidine tract at the 3′ splice site. Following this, U2 snRNP and related factors adhere to the 3′ splice site and start recognizing the branchpoint adenosine, leading to the formation of the pre-spliceosome. This activity triggers a transesterification reaction that generates a lariat structure at the 5′ splice site, releasing a 3′-OH group at the intron’s 5′ end. A subsequent transesterification connects this 3'-OH group to the 3′ splice site of the following exon, thus removing the lariat [80]. In the context of trans-splicing mechanisms, several configurations can be utilized to enhance splicing efficiency and specificity. Fig. 1A illustrates 3′ trans-splicing, where a pre-trans-splicing molecule (PTM) with a binding domain (BD) aligns with the complementary intronic sequence upstream of the target exon, including branchpoint and Py splice site sequences. This setup ensures the precise removal of introns upstream of the target exon. Fig. 1B demonstrates 5′ trans-splicing, which involves a PTM with a BD matching the complementary intronic sequence downstream of the target exon, facilitating the inclusion of the target exon into the mature mRNA by introducing the 5′ splice site. Fig. 1C represents internal trans-splicing, consisting of a PTM equipped with sequences essential for both 3′ and 5′ trans-splicing. This configuration includes key elements like the polypyrimidine tract (Py) and the polyA tail (pA), enabling efficient splicing events within the internal regions of pre-mRNA. At the experimental level, enhancing the recognition of the 5′ splice site can be achieved by incorporating reporter genes into U1 snRNPs or other relevant sites. By enabling efficient evaluation of splicing efficiency across different conditions, this method enhances the understanding of core splicing mechanisms and expedites the creation of targeted treatments for Huntington’s disease [81].
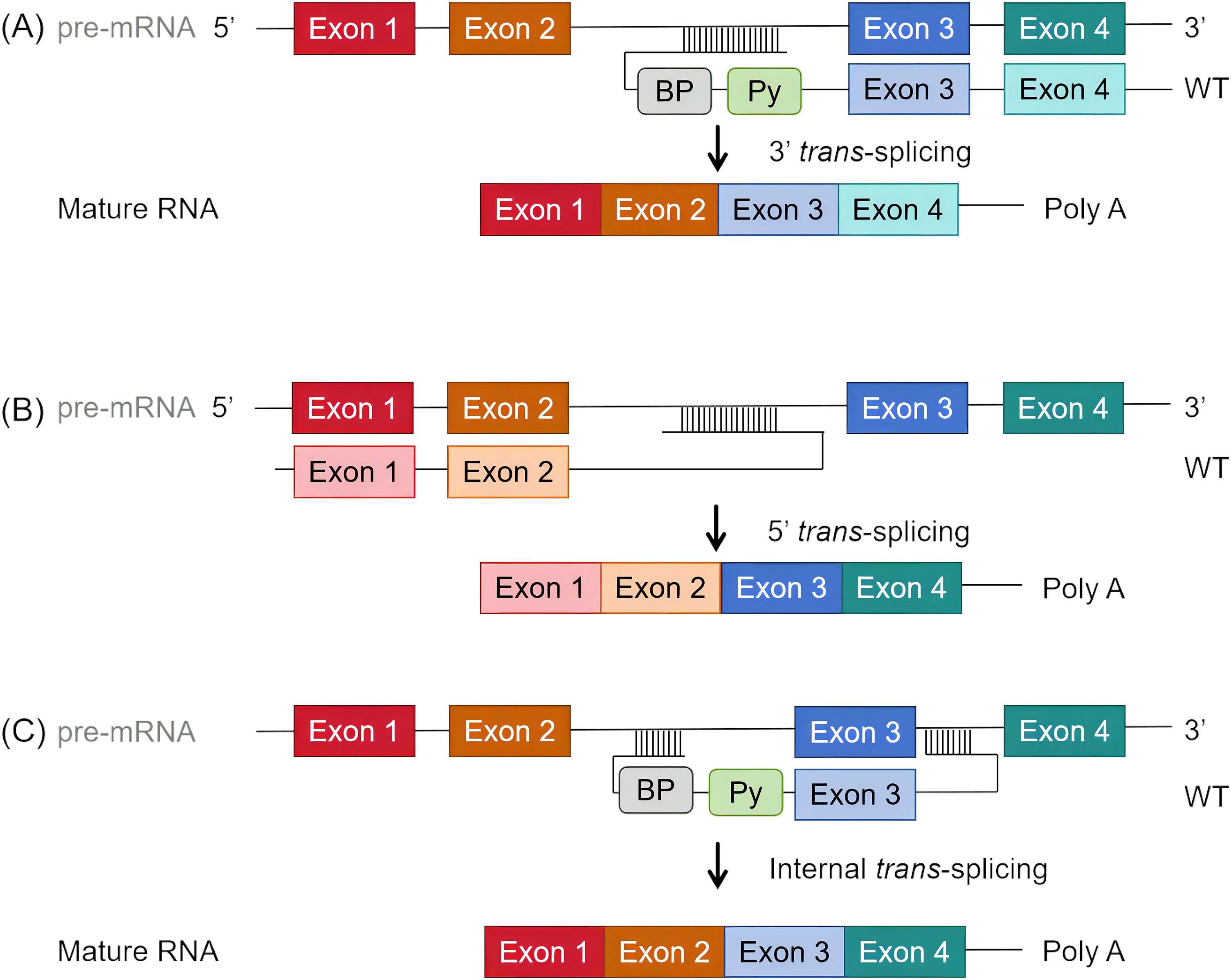
Figure 1: Schematic diagram of trans-splicing process. (A) 3′ trans-splicing: Features a pre-trans-splicing molecule (PTM) with a binding domain (BD) that aligns with the complementary intronic sequence upstream of the target exon, including branchpoint and Py splice site (ss) sequences. (B) 5′ trans-splicing: Includes a PTM with a BD matching the complementary intronic sequence downstream of the target exon, accompanied by the 5′ ss. (C) Internal trans-splicing: Consists of a PTM equipped with sequences essential for both 3′ and 5′ trans-splicing. Key elements include the Py (polypyrimidine tract) and pA (polyA tail).
The SMaRT strategy utilizes the endogenous spliceosome machinery to start trans-splicing through a custom-designed pre-trans-splicing molecule (PTM) that targets specific exons with crucial splicing elements. This method can be used to replace exons at the beginning (5′ trans-splicing) or end (3′ trans-splicing) of the pre-mRNA. Each PTM comprises a binding domain that matches the intronic sequence adjacent to the target exon and a coding domain representing the exon(s) to be joined. For 5′ trans-splicing, the binding domain includes the sequence downstream of the target exon’s first intron, including the 5' splice site. For 3′ trans-splicing, it includes the sequence upstream from the target exon’s last intron and must contain the branchpoint, the polypyrimidine tract, and the 3′ splice site. In the case of 5′ trans-splicing, the spliceosome utilizes the endogenous branchpoint and polypyrimidine tract, ensuring accurate splicing. This technology allows for the possibility of replacing internal exons, with each PTM tailored to include necessary splicing elements for both 5′ and 3′ trans-splicing [80,82].
SMaRT technology uniquely facilitates the simultaneous reduction of mutant protein levels and the synthesis of functional protein in a single reaction. However, the efficiency of trans-splicing remains challenging to predict due to its less characterized nature compared to well-documented cis-splicing. A thorough understanding of the fundamental trans-splicing mechanisms is crucial for optimizing this technology. Potential off-target effects from non-specific PTM binding might result in the production of unintended chimeric proteins. Given the scattered nature of class II missense mutations across the HTT gene, developing multiple PTM constructs targeting these mutations may be necessary.
The implementation of SMaRT in therapeutic applications involves the construction of recombinant viral plasmid vectors and the infection of neuronal progenitor cells (NPCs). Due to the vital distribution of huntingtin in the striatum, the constructed recombinant plasmid virus needs to cross the blood-brain barrier (BBB) after administration. Therefore, the preferred plasmid vector is a recombinant adeno-associated virus (rAAV) [83]. In the development phase, a recombinant AAV9 virus targeting specific mutant exons is produced. The AAV vector incorporates essential elements including the original sequence, ampicillin resistance gene, two 3′ inverted terminal repeats (ITRs), a Cytomegalovirus (CMV) enhancer, a CMV promoter, and a Multiple Cloning Site (MCS), along with the pre-trans-splicing molecule (PTM). Accompanying the AAV vector, a packaging plasmid containing replication and capping genes and an AAV protein plasmid is co-transfected into producer cells such as HEK293 cells (Fig. 2A). AAV vectors are well-established and safe gene delivery tools for the nervous system [84]. Once packaged and purified, the recombinant AAV is ready for clinical application (Fig. 2B). In the treatment phase, the recombinant adenovirus is administered to the patient via intravenous (IV) injection. The properties of AAV9 allow it to cross the BBB and reach the target cells, specifically medium spiny neurons (MSNs). Within these neurons, the trans-splicing process is completed, where the PTM aligns with the endogenous pre-mRNA of the mutant HTT gene, facilitating the correction at the RNA level (Fig. 2B) [85]. The rAAV should achieve significant expression in both the striatum and hippocampus of the brain.
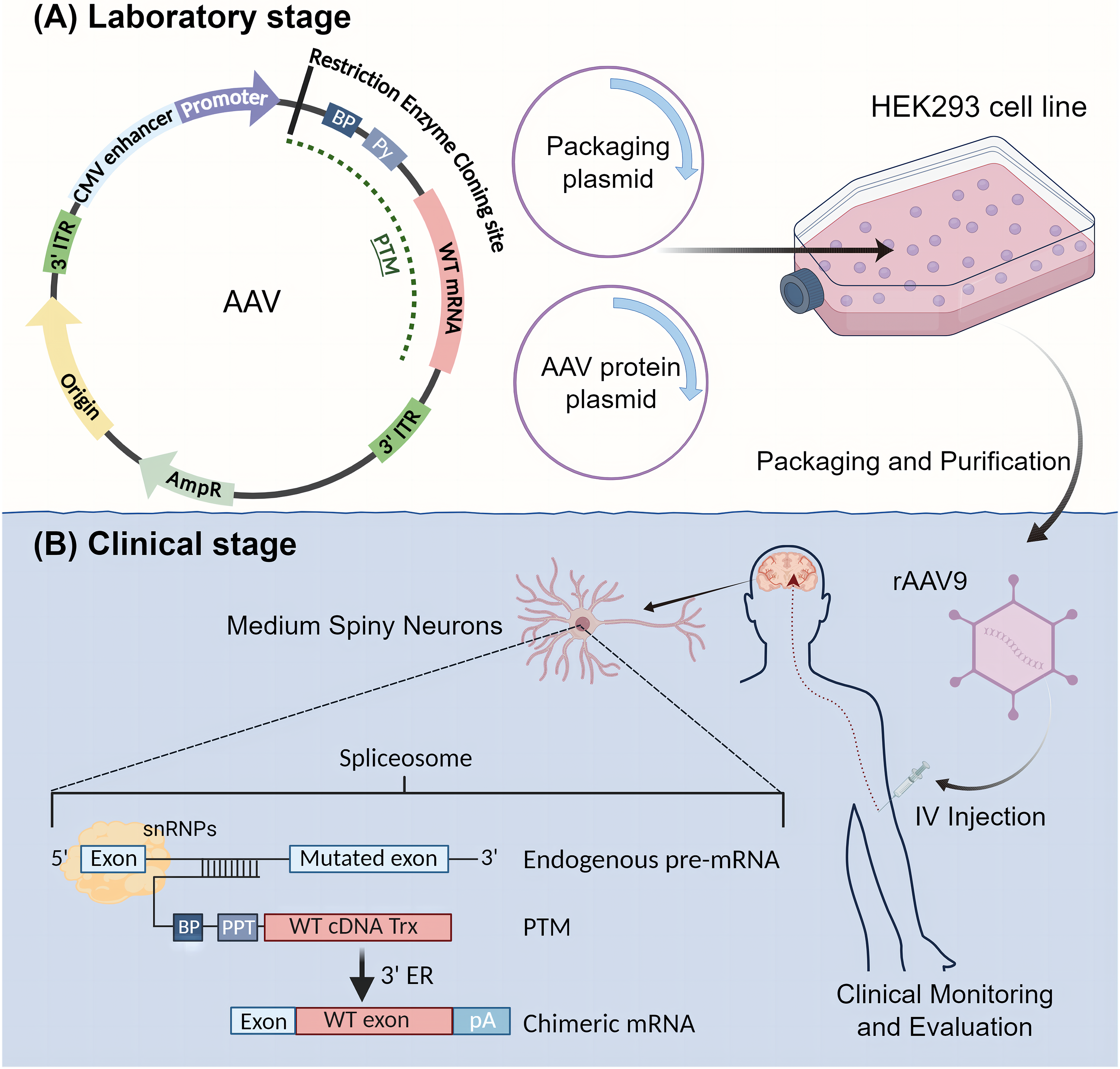
Figure 2: Schematic Diagram of SMaRT Therapy Process. SMaRT therapy includes both laboratory and clinical stages. (A) Laboratory Stage: In this stage, a recombinant AAV9 virus that targets specific mutant exons is produced. The AAV vector incorporates the original sequence, ampicillin resistance gene, two 3′ inverted terminal repeats (ITRs), a CMV enhancer, a CMV promoter, and a Multiple Cloning Site (MCS), along with the pre-trans-splicing molecule (PTM). Accompanying the AAV vector, one packaging plasmid containing replication and capping genes and one adeno-associated protein plasmid are co-transfected into producer cells (e.g., HEK293 cells). (B) Clinical Stage: The recombinant adenovirus is purified and packaged, then administered to the patient via intravenous (IV) injection. The properties of AAV9 allow it to cross the blood-brain barrier and reach the target cells, specifically medium spiny neurons (MSNs). Within these neurons, trans-splicing is completed. The treated MSNs will then express normal HTT protein, achieving the therapeutic goal.
Recent studies have shown the feasibility of SMaRT by successfully detecting the expression of wild-type HTT mRNA in induced pluripotent stem cells (iPSCs) that were transfected with lentiviral fragments containing the wild-type HTT gene [86]. SMaRT presents numerous advantages when contrasted with gene replacement and other RNA-based strategies it eliminates the need for delivering the full-length cDNA, thereby decreasing the genetic package size; it leverages the cell’s intrinsic transcriptional and regulatory mechanisms to ensure precise gene expression; and it harnesses the ubiquitous spliceosomal machinery present in nearly all cell types. Most importantly, this method transforms pathogenic mutant mRNA into beneficial, therapeutic mRNA, effectively treating diseases characterized by dominant-negative mutations like mutant HTT [87].
SMaRT technology represents a revolutionary approach in the therapeutic landscape of HD, contrasting sharply with conventional techniques like RNA interference (RNAi) or CRISPR-Cas9. SMaRT uniquely leverages the cellular spliceosomal machinery to rectify mRNA transcripts, thereby concurrently diminishing toxic protein production and reinstating functional protein synthesis. This dual action effectively addresses both the detrimental effects of pathogenic proteins and the accompanying loss of normal protein function, which are significant challenges in genetic disorder treatments.
SMaRT’s therapeutic design involves the use of recombinant adeno-associated virus (rAAV) vectors, well-established for their efficacy and safety in neurotherapeutic deliveries. These vectors facilitate the crossing of the blood-brain barrier to target neuronal progenitors in critical regions like the striatum and hippocampus. Additionally, the strategy includes utilizing PTMs tailored to specifically alter the mutated regions of the HTT mRNA, allowing for the restoration of functional protein production while bypassing the limitations of direct genetic modification techniques that do not address the loss of normal protein function.
However, SMaRT’s clinical deployment is not without challenges. The precision of the trans-splicing reaction and the targeted delivery of PTMs are important, with inaccuracies potentially leading to the production of non-functional or deleterious proteins. These challenges necessitate advanced vector technology to ensure specific, safe delivery.
Future advancements in SMaRT technology will rely on a deeper understanding of spliceosomal mechanics and the refinement of vector designs to enhance PTM delivery. Prospects include combining this approach with other therapies to augment the spliceosome’s activity and address multiple pathological aspects of HD. Ongoing research and clinical trials are essential to optimize and validate SMaRT’s efficacy, positioning it as a transformative solution for HD and potentially other disorders characterized by dominant-negative mutations. This integrated approach underscores SMaRT’s potential to significantly alter the therapeutic landscape for neurodegenerative diseases.
SMaRT offers a pioneering approach for HD treatment by concurrently reducing toxic mutant protein levels and restoring functional protein. Although the promise is there, clinical implementation is challenged by issues such as precise PTM delivery and off-target effects. Advanced vector technology and a deeper understanding of spliceosomal mechanics are essential for optimizing SMaRT’s efficacy. Future strategies may involve combining SMaRT with other therapies to enhance spliceosome activity and address multiple pathological aspects of HD. Continued research and clinical trials are critical to validate SMaRT’s effectiveness and safety, potentially revolutionizing treatment for HD and other genetic disorders caused by dominant negative mutations.
This summary provides a concise analysis of the current state and future directions of HD research, emphasizing the transformative potential of SMaRT as a therapeutic strategy.
Acknowledgement: None.
Funding Statement: This research was funded by the National Natural Science Foundation of China under Grant 22376100.
Author Contributions: The authors confirm their contribution to the paper as follows: study conception and design: Dan Weng, Qingyang Zhang; draft manuscript preparation: Qingyang Zhang, Shuxian Huang; review and editing: Dan Weng, Qingyang Zhang, Shuxian Huang; visualization: Qingyang Zhang; supervision: Dan Weng. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study. This narrative review was generated using published reviews from PubMed and other scientific databases.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Medina A, Mahjoub Y, Shaver L, Pringsheim T. Prevalence and incidence of Huntington’s disease: an updated systematic review and meta-analysis. Mov Disord. 2022;37(12):2327–35. doi:10.1002/mds.v37.12. [Google Scholar] [CrossRef]
2. Crowell V, Houghton R, Tomar A, Fernandes T, Squitieri F. Modeling manifest Huntington’s disease prevalence using diagnosed incidence and survival time. Neuroepidemiology. 2021;55(5):361–8. doi:10.1159/000516767. [Google Scholar] [PubMed] [CrossRef]
3. Bakels HS, Roos RA, van Roon-Mom WM, de Bot ST. Juvenile-onset Huntington disease pathophysiology and neurodevelopment: a review. Mov Disord. 2022;37(1):16–24. doi:10.1002/mds.v37.1. [Google Scholar] [CrossRef]
4. de la Cruz J, Hwang J. On the hunt for a cure: a guide to Huntington disease. JAAPA. 2021;34(4):26–31. doi:10.1097/01.JAA.0000735740.95438.60. [Google Scholar] [PubMed] [CrossRef]
5. Nance MA, Paulsen JS, Rosenblatt A, Wheelock V. A physician’s guide to the management of Huntington’s disease. 3rd ed. New York City: Huntingtons Disease Society of America; 2011. [Google Scholar]
6. FDA. Austedo (deutetrabenazine) approval for Huntington’s disease chorea. Available from: https://www.fda.gov/. [Accessed 2024]. [Google Scholar]
7. FDA. Xenazine (tetrabenazine) approval for Huntington’s disease chorea. Available from: https://www.fda.gov/. [Accessed 2024]. [Google Scholar]
8. FDA. Austedo XR (deutetrabenazine extended-release) approval for Huntington’s disease chorea. Available from: https://www.fda.gov/. [Accessed 2024]. [Google Scholar]
9. Mehanna R, Jankovic J. Systemic symptoms in Huntington’s disease: a comprehensive review. Mov Disord Clin Pract. 2024;11(5):453–64. doi:10.1002/mdc3.v11.5. [Google Scholar] [CrossRef]
10. MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72(6):971–83. doi:10.1016/0092-8674(93)90585-E. [Google Scholar] [PubMed] [CrossRef]
11. Mätlik K, Baffuto M, Kus L, Deshmukh AL, Davis DA, Paul MR, et al. Cell-type-specific CAG repeat expansions and toxicity of mutant Huntingtin in human striatum and cerebellum. Nat Genet. 2024;56:1–12. [Google Scholar]
12. van Well EM, Bader V, Patra M, Sánchez-Vicente A, Meschede J, Furthmann N, et al. A protein quality control pathway regulated by linear ubiquitination. EMBO J. 2019;38(9):e100730. doi:10.15252/embj.2018100730. [Google Scholar] [PubMed] [CrossRef]
13. Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, et al. Huntington disease. Nat Rev Dis Primers. 2015;1(1):1–21. [Google Scholar]
14. McColgan P, Tabrizi SJ. Huntington’s disease: a clinical review. Eur J Neurol. 2018;25(1):24–34. doi:10.1111/ene.2018.25.issue-1. [Google Scholar] [CrossRef]
15. Toneff T, Mende-Mueller L, Wu Y, Hwang SR, Bundey R, Thompson LM, et al. Comparison of huntingtin proteolytic fragments in human lymphoblast cell lines and human brain. J Neurochem. 2002;82(1):84–92. doi:10.1046/j.1471-4159.2002.00940.x. [Google Scholar] [PubMed] [CrossRef]
16. Ganesh A, Galetta S. Editors’ note: clinical manifestations of homozygote allele carriers in Huntington disease. Neurology. 2020;94(16):722–2. doi:10.1212/WNL.0000000000009305. [Google Scholar] [CrossRef]
17. Shin A, Shin B, Shin JW, Kim K-H, Atwal RS, Hope JM, et al. Novel allele-specific quantification methods reveal no effects of adult onset CAG repeats on HTT mRNA and protein levels. Hum Mol Genet. 2017;26(7):1258–67. doi:10.1093/hmg/ddx033. [Google Scholar] [PubMed] [CrossRef]
18. Politis M, Lahiri N, Niccolini F, Su P, Wu K, Giannetti P, et al. Increased central microglial activation associated with peripheral cytokine levels in premanifest Huntington’s disease gene carriers. Neurobiol Dis. 2015;83:115–21. doi:10.1016/j.nbd.2015.08.011. [Google Scholar] [PubMed] [CrossRef]
19. Langfelder P, Cantle JP, Chatzopoulou D, Wang N, Gao F, Al-Ramahi I, et al. Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat Neurosci. 2016;19(4):623–33. doi:10.1038/nn.4256. [Google Scholar] [PubMed] [CrossRef]
20. Lee J-M, Correia K, Loupe J, Kim K-H, Barker D, Hong EP, et al. CAG repeat not polyglutamine length determines timing of Huntington’s disease onset. Cell. 2019;178(4):887–900.e14. doi:10.1016/j.cell.2019.06.036. [Google Scholar] [PubMed] [CrossRef]
21. Gusella JF, MacDonald ME, Ambrose CM, Duyao MP. Molecular genetics of Huntington’s disease. Arch Neurol. 1993;50(11):1157–63. doi:10.1001/archneur.1993.00540110037003. [Google Scholar] [PubMed] [CrossRef]
22. Petersén Å, Mani K, Brundin P. Recent advances on the pathogenesis of Huntington’s disease. Exp Neurol. 1999;157(1):1–18. doi:10.1006/exnr.1998.7006. [Google Scholar] [PubMed] [CrossRef]
23. Kono Y, Agawa Y, Watanabe Y, Ohama E, Nanba E, Nakashima K. Analysis of the CAG repeat number in a patient with Huntington’s disease. Internal Med. 1999;38(5):407–11. doi:10.2169/internalmedicine.38.407. [Google Scholar] [PubMed] [CrossRef]
24. Strong TV, Tagle DA, Valdes JM, Elmer LW, Boehm K, Swaroop M, et al. Widespread expression of the human and rat Huntington’s disease gene in brain and nonneural tissues. Nat Genet. 1993;5(3):259–65. doi:10.1038/ng1193-259. [Google Scholar] [PubMed] [CrossRef]
25. Gourfinkel-An I, Cancel G, Trottier Y, Devys D, Tora L, Lutz Y, et al. Differential distribution of the normal and mutated forms of huntingtin in the human brain. Ann Neurol. 1997;42(5):712–9. doi:10.1002/ana.v42:5. [Google Scholar] [CrossRef]
26. Benraiss A, Mariani JN, Osipovitch M, Cornwell A, Windrem MS, Villanueva CB, et al. Cell-intrinsic glial pathology is conserved across human and murine models of Huntington’s disease. Cell Rep. 2021;36(1):109308. doi:10.1016/j.celrep.2021.109308. [Google Scholar] [PubMed] [CrossRef]
27. Van Raamsdonk JM, Murphy Z, Slow EJ, Leavitt BR, Hayden MR. Selective degeneration and nuclear localization of mutant huntingtin in the YAC128 mouse model of Huntington disease. Hum Mol Genet. 2005;14(24):3823–35. doi:10.1093/hmg/ddi407. [Google Scholar] [PubMed] [CrossRef]
28. Xu C, Chen S, Chen X, Ho KH, Park C, Yoo H, et al. Altered exocytosis of inhibitory synaptic vesicles at single presynaptic terminals of cultured striatal neurons in a knock-in mouse model of Huntington’s disease. Front Mol Neurosci. 2023;16:776. [Google Scholar]
29. Seto-Ohshima A, Lawson E, Emson P, Mountjoy C, Carrasco L. Loss of matrix calcium-binding protein-containing neurons in Huntington’s disease. The Lancet. 1988;331(8597):1252–5. doi:10.1016/S0140-6736(88)92073-9. [Google Scholar] [PubMed] [CrossRef]
30. Neueder A, Landles C, Ghosh R, Howland D, Myers RH, Faull RL, et al. The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Sci Rep. 2017;7(1):1307. doi:10.1038/s41598-017-01510-z. [Google Scholar] [PubMed] [CrossRef]
31. Kim H, Lenoir S, Helfricht A, Jung T, Karneva ZK, Lee Y, et al. A pathogenic proteolysis-resistant huntingtin isoform induced by an antisense oligonucleotide maintains huntingtin function. JCI Insight. 2022;7(17):135. [Google Scholar]
32. Alkanli SS, Alkanli N, Ay A, Albeniz I. CRISPR/Cas9 mediated therapeutic approach in Huntington’s disease. Mol Neurobiol. 2023;60(3):1486–98. doi:10.1007/s12035-022-03150-5. [Google Scholar] [PubMed] [CrossRef]
33. Han JY, Seo J, Choi Y, Im W, Ban J-J, Sung J-J. CRISPR-Cas9 mediated genome editing of Huntington’s disease neurospheres. Mol Biol Rep. 2023;50(3):2127–36. doi:10.1007/s11033-022-08175-6. [Google Scholar] [PubMed] [CrossRef]
34. Andrade MA, Bork P. HEAT repeats in the Huntington’s disease protein. Nat Genet. 1995;11(2):115–6. doi:10.1038/ng1095-115. [Google Scholar] [PubMed] [CrossRef]
35. Liot G, Zala D, Pla P, Mottet G, Piel M, Saudou F. Mutant Huntingtin alters retrograde transport of TrkB receptors in striatal dendrites. J Neurosci. 2013;33(15):6298–309. doi:10.1523/JNEUROSCI.2033-12.2013. [Google Scholar] [PubMed] [CrossRef]
36. Taran AS, Shuvalova LD, Lagarkova MA, Alieva IB. Huntington’s disease—An outlook on the interplay of the HTT protein, microtubules and actin cytoskeletal components. Cells. 2020;9(6):1514. doi:10.3390/cells9061514. [Google Scholar] [PubMed] [CrossRef]
37. Gutekunst C-A, Levey AI, Heilman CJ, Whaley WL, Yi H, Nash NR, et al. Identification and localization of huntingtin in brain and human lymphoblastoid cell lines with anti-fusion protein antibodies. Proc Natl Acad Sci U S A. 1995;92(19):8710–4. doi:10.1073/pnas.92.19.8710. [Google Scholar] [PubMed] [CrossRef]
38. McAdam RL, Morton A, Gordon SL, Alterman JF, Khvorova A, Cousin MA, et al. Loss of huntingtin function slows synaptic vesicle endocytosis in striatal neurons from the httQ140/Q140 mouse model of Huntington’s disease. Neurobiol Dis. 2020;134:104637. doi:10.1016/j.nbd.2019.104637. [Google Scholar] [PubMed] [CrossRef]
39. Lee H, Fenster RJ, Pineda SS, Gibbs WS, Mohammadi S, Davila-Velderrain J, et al. Cell type-specific transcriptomics reveals that mutant huntingtin leads to mitochondrial RNA release and neuronal innate immune activation. Neuron. 2020;107(5):891–908.e8. doi:10.1016/j.neuron.2020.06.021. [Google Scholar] [PubMed] [CrossRef]
40. Migazzi A, Scaramuzzino C, Anderson EN, Tripathy D, Hernández IH, Grant RA, et al. Huntingtin-mediated axonal transport requires arginine methylation by PRMT6. Cell Rep. 2021;35(2):108980. doi:10.1016/j.celrep.2021.108980. [Google Scholar] [PubMed] [CrossRef]
41. D’Egidio F, Castelli V, Lombardozzi G, Ammannito F, Cimini A, d’Angelo M. Therapeutic advances in neural regeneration for Huntington’s disease. Neural Regener Res. 2024;19(9):1991–7. doi:10.4103/1673-5374.390969. [Google Scholar] [PubMed] [CrossRef]
42. Chambon J, Komal P, Lewitus GM, Kemp GM, Valade S, Adaïdi H, et al. Early TNF-dependent regulation of excitatory and inhibitory synapses on striatal direct pathway medium spiny neurons in the YAC128 mouse model of Huntington’s disease. J Neurosci. 2023;43(4):672–80. doi:10.1523/JNEUROSCI.1655-22.2022. [Google Scholar] [PubMed] [CrossRef]
43. Castro E, Costa AR, Mysore S, Paruchuri P, Chen KY, Liu AY. PolyQ-expanded mutant Huntingtin forms inclusion body following transient cold shock in a two-step aggregation mechanism. ACS Chem Neurosci. 2022;14(2):277–88. [Google Scholar]
44. Juenemann K, Weisse C, Reichmann D, Kaether C, Calkhoven CF, Schilling G. Modulation of mutant huntingtin N-terminal cleavage and its effect on aggregation and cell death. Neurotoxic Res. 2011;20:120–33. doi:10.1007/s12640-010-9227-6. [Google Scholar] [PubMed] [CrossRef]
45. Andhale R, Shrivastava D. Huntington’s disease: a clinical review. Cureus. 2022;14(8):e28484. [Google Scholar] [PubMed]
46. Moreno-Jiménez EP, Terreros-Roncal J, Flor-García M, Rábano A, Llorens-Martín M. Evidences for adult hippocampal neurogenesis in humans. J Neurosci. 2021;41(12):2541–53. doi:10.1523/JNEUROSCI.0675-20.2020. [Google Scholar] [PubMed] [CrossRef]
47. Jurkowski MP, Bettio L, Woo K, Patten E, Yau A, Gil-Mohapel S-Y, et al. Beyond the hippocampus and the SVZ: adult neurogenesis throughout the brain. Front Cell Neurosci. 2020;14:576444. doi:10.3389/fncel.2020.576444. [Google Scholar] [PubMed] [CrossRef]
48. Cebrian-Silla A, Nascimento MA, Redmond SA, Mansky B, Wu D, Obernier K, et al. Single-cell analysis of the ventricular-subventricular zone reveals signatures of dorsal and ventral adult neurogenesis. eLife. 2021;10:e67436. doi:10.7554/eLife.67436. [Google Scholar] [PubMed] [CrossRef]
49. Abdissa D, Hamba N, Gerbi A. Review article on adult neurogenesis in humans. Transl Res Anat. 2020;20:100074. [Google Scholar]
50. Barnat M, Le Friec J, Benstaali C, Humbert S. Huntingtin-mediated multipolar-bipolar transition of newborn cortical neurons is critical for their postnatal neuronal morphology. Neuron. 2017;93(1):99–114. doi:10.1016/j.neuron.2016.11.035. [Google Scholar] [PubMed] [CrossRef]
51. Burrus CJ, McKinstry SU, Kim N, Ozlu MI, Santoki AV, Fang FY, et al. Striatal projection neurons require huntingtin for synaptic connectivity and survival. Cell Rep. 2020;30(3):642–57.e6. doi:10.1016/j.celrep.2019.12.069. [Google Scholar] [PubMed] [CrossRef]
52. Barnat M, Capizzi M, Aparicio E, Boluda S, Wennagel D, Kacher R, et al. Huntington’s disease alters human neurodevelopment. Science. 2020;369(6505):787–93. doi:10.1126/science.aax3338. [Google Scholar] [PubMed] [CrossRef]
53. Mouro Pinto R, Arning L, Giordano JV, Razghandi P, Andrew MA, Gillis T, et al. Patterns of CAG repeat instability in the central nervous system and periphery in Huntington’s disease and in spinocerebellar ataxia type 1. Hum Mol Genet. 2020;29(15):2551–67. doi:10.1093/hmg/ddaa139. [Google Scholar] [PubMed] [CrossRef]
54. Moss DJH, Pardiñas AF, Langbehn D, Lo K, Leavitt BR, Roos R, et al. Identification of genetic variants associated with Huntington’s disease progression: a genome-wide association study. The Lancet Neurology. 2017;16(9):701–11. doi:10.1016/S1474-4422(17)30161-8. [Google Scholar] [PubMed] [CrossRef]
55. Bettencourt C, Hensman-Moss D, Flower M, Wiethoff S, Brice A, Goizet C, et al. DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann Neurol. 2016;79(6):983–90. doi:10.1002/ana.v79.6. [Google Scholar] [CrossRef]
56. Porro A, Mohiuddin M, Zurfluh C, Spegg V, Dai J, Iehl F, et al. FAN1-MLH1 interaction affects repair of DNA interstrand cross-links and slipped-CAG/CTG repeats. Sci Adv. 2021;7(31):eabf7906. doi:10.1126/sciadv.abf7906. [Google Scholar] [PubMed] [CrossRef]
57. Deshmukh AL, Porro A, Mohiuddin M, Lanni S, Panigrahi GB, Caron M-C, et al. FAN1, a DNA repair nuclease, as a modifier of repeat expansion disorders. J Huntington’s Dis. 2021;10(1):95–122. doi:10.3233/JHD-200448. [Google Scholar] [PubMed] [CrossRef]
58. Iyer RR, Pluciennik A. DNA mismatch repair and its role in Huntington’s disease. J Huntington’s Dis. 2021;10(1):75–94. doi:10.3233/JHD-200438. [Google Scholar] [PubMed] [CrossRef]
59. Zhao XN, Usdin K. The repeat expansion diseases: the dark side of DNA repair. DNA Repair. 2015;32:96–105. doi:10.1016/j.dnarep.2015.04.019. [Google Scholar] [PubMed] [CrossRef]
60. Bocchi VD, Conforti P, Vezzoli E, Besusso D, Cappadona C, Lischetti T, et al. The coding and long noncoding single-cell atlas of the developing human fetal striatum. Science. 2021;372(6542):eabf5759. doi:10.1126/science.abf5759. [Google Scholar] [PubMed] [CrossRef]
61. Zeitlin S, Liu J-P, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat Genet. 1995;11(2):155–63. doi:10.1038/ng1095-155. [Google Scholar] [PubMed] [CrossRef]
62. Howland D, Ellederova Z, Aronin N, Fernau D, Gallagher J, Taylor A, et al. Large animal models of Huntington’s disease: what we have learned and where we need to go next. J Huntington’s Dis. 2020;9(3):201–16. doi:10.3233/JHD-200425. [Google Scholar] [PubMed] [CrossRef]
63. Wang G, Liu X, Gaertig MA, Li S, Li XJ. Ablation of huntingtin in adult neurons is nondeleterious but its depletion in young mice causes acute pancreatitis. Proc Natl Acad Sci U S A. 2016;113(12):3359–64. doi:10.1073/pnas.1524575113. [Google Scholar] [PubMed] [CrossRef]
64. Gilliat S, Rosa JG, Benjamin G, Sbrocco K, Lin W, Cvetanovic M. Modest reduction in CAG repeat length rescues motor deficits but not purkinje cell pathology and gliosis in spinocerebellar ataxia type 1 mice. Neuroglia. 2023;4(1):52–68. doi:10.3390/neuroglia4010005. [Google Scholar] [CrossRef]
65. Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron. 2012;74(6):1031–44. doi:10.1016/j.neuron.2012.05.009. [Google Scholar] [PubMed] [CrossRef]
66. Zeitler B, Froelich S, Marlen K, Shivak DA, Yu Q, Li D, et al. Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat Med. 2019;25(7):1131–42. doi:10.1038/s41591-019-0478-3. [Google Scholar] [PubMed] [CrossRef]
67. Wang N, Gray M, Lu XH, Cantle JP, Holley SM, Greiner E, et al. Neuronal targets for reducing mutant huntingtin expression to ameliorate disease in a mouse model of Huntington’s disease. Nat Med. 2014;20(5):536–41. doi:10.1038/nm.3514. [Google Scholar] [PubMed] [CrossRef]
68. Caron NS, Banos R, Yanick C, Aly AE, Byrne LM, Smith ED, et al. Mutant huntingtin is cleared from the brain via active mechanisms in Huntington disease. J Neurosci. 2021;41(4):780–96. doi:10.1523/JNEUROSCI.1865-20.2020. [Google Scholar] [PubMed] [CrossRef]
69. Bečanović K, Nørremølle A, Neal SJ, Kay C, Collins JA, Arenillas D, et al. A SNP in the HTT promoter alters NF-κB binding and is a bidirectional genetic modifier of Huntington disease. Nat Neurosci. 2015;18(6):807–16. doi:10.1038/nn.4014. [Google Scholar] [PubMed] [CrossRef]
70. Silva AC, Lobo DD, Martins IM, Lopes SM, Henriques C, Duarte SP, et al. Antisense oligonucleotide therapeutics in neurodegenerative diseases: the case of polyglutamine disorders. Brain. 2020;143(2):407–29. doi:10.1093/brain/awz328. [Google Scholar] [PubMed] [CrossRef]
71. Rook ME, Southwell AL. Antisense oligonucleotide therapy: from design to the Huntington disease clinic. BioDrugs. 2022;36(2):105–19. doi:10.1007/s40259-022-00519-9. [Google Scholar] [PubMed] [CrossRef]
72. Wu TT, Su FJ, Feng YQ, Liu B, Li MY, Liang FY, et al. Mesenchymal stem cells alleviate AQP-4-dependent glymphatic dysfunction and improve brain distribution of antisense oligonucleotides in BACHD mice. Stem Cells. 2020;38(2):218–30. doi:10.1002/stem.3103. [Google Scholar] [PubMed] [CrossRef]
73. McBride JL, Boudreau RL, Harper SQ, Staber PD, Monteys AM, Martins I, et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc Natl Acad Sci U S A. 2008;105(15):5868–73. doi:10.1073/pnas.0801775105. [Google Scholar] [PubMed] [CrossRef]
74. Gangwani MR, Soto JS, Jami-Alahmadi Y, Tiwari S, Kawaguchi R, Wohlschlegel JA, et al. Neuronal and astrocytic contributions to Huntington’s disease dissected with zinc finger protein transcriptional repressors. Cell Rep. 2023;42(1):111953. doi:10.1016/j.celrep.2022.111953. [Google Scholar] [PubMed] [CrossRef]
75. Ferguson MW, Kennedy CJ, Palpagama TH, Waldvogel HJ, Faull RL, Kwakowsky A. Current and possible future therapeutic options for Huntington’s disease. J Cent Nerv Syst Dis. 2022;14:11795735221092517. [Google Scholar] [PubMed]
76. Fink KD, Deng P, Gutierrez J, Anderson JS, Torrest A, Komarla A, et al. Allele-specific reduction of the mutant huntingtin allele using transcription activator-like effectors in human huntington’s disease fibroblasts. Cell Transplant. 2016;25(4):677–86. doi:10.3727/096368916X690863. [Google Scholar] [PubMed] [CrossRef]
77. Qin Y, Li S, Li XJ, Yang S. CRISPR-based genome-editing tools for Huntington’s disease research and therapy. Neurosci Bull. 2022;38(11):1397–1408. doi:10.1007/s12264-022-00880-3. [Google Scholar] [PubMed] [CrossRef]
78. Pelletier R, Caron SO, Puymirat J. RNA based gene therapy for dominantly inherited diseases. Curr Gene Ther. 2006;6(1):131–46. doi:10.2174/156652306775515592. [Google Scholar] [PubMed] [CrossRef]
79. D’Allard DL, Liu JM. Toward RNA repair of diamond blackfan anemia hematopoietic stem cells. Hum Gene Ther. 2016;27(10):792–801. doi:10.1089/hum.2016.081. [Google Scholar] [PubMed] [CrossRef]
80. Wally V, Murauer EM, Bauer JW. Spliceosome-mediated trans-splicing: the therapeutic cut and paste. J Invest Dermatol. 2012;132(8):1959–66. doi:10.1038/jid.2012.101. [Google Scholar] [PubMed] [CrossRef]
81. Wong J, Martelly W, Sharma S. A reporter based cellular assay for monitoring splicing efficiency. J Vis Exp. 2021;175:e63014. [Google Scholar]
82. Hong EM, Ingemarsdotter CK, Lever AM. Therapeutic applications of trans-splicing. Br Med Bull. 2020;136(1):4–20. doi:10.1093/bmb/ldaa028. [Google Scholar] [PubMed] [CrossRef]
83. Galvan A, Petkau TL, Hill AM, Korecki AJ, Lu G, Choi D, et al. Intracerebroventricular administration of AAV9-PHP. B SYN1-EmGFP induces widespread transgene expression in the mouse and monkey central nervous system. Hum Gene Ther. 2021;32(11–12):599–615. [Google Scholar] [PubMed]
84. Powell SK, Samulski RJ, McCown TJ. AAV capsid-promoter interactions determine CNS cell-selective gene expression in vivo. Mol Ther. 2020;28(5):1373–80. doi:10.1016/j.ymthe.2020.03.007. [Google Scholar] [PubMed] [CrossRef]
85. Maturana CJ, Verpeut JL, Engel EA. Single-cell quantification of triple-AAV vector genomes coexpressed in neurons. Curr Protocol. 2022;2(5):e430. doi:10.1002/cpz1.v2.5. [Google Scholar] [CrossRef]
86. Rindt H, Tom CM, Lorson CL, Mattis VB. Optimization of trans-splicing for Huntington’s disease RNA therapy. Front Neurosci. 2017;11:544. doi:10.3389/fnins.2017.00544. [Google Scholar] [PubMed] [CrossRef]
87. Suñé-Pou M, Prieto-Sánchez S, Boyero-Corral S, Moreno-Castro C, El Yousfi Y, Suñé-Negre J, et al. Targeting splicing in the treatment of human disease. Genes. 2017;8(3):87. doi:10.3390/genes8030087. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools