
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Comparative studies on the binding site of anesthetics to GABA a receptors using in silico docking methods
1 Department of Applied Chemistry, Dongduk Women’s University, Seoul, 02748, Korea
2 Division of Applied Chemistry and Cosmetic Science, Dongduk Women’s University, Seoul, 02748, Korea
3 Department of Anesthesiology and Pain Medicine, Anesthesia and Pain Research Institute, Yonsei University College of Medicine, Seoul, 03722, Korea
* Corresponding Author: SEYEON PARK. Email:
(This article belongs to the Special Issue: Expert Opinions and Future Trends on Stem Cells, Biomaterials and Growth Factors)
BIOCELL 2023, 47(7), 1661-1673. https://doi.org/10.32604/biocell.2023.027984
Received 24 November 2022; Accepted 28 February 2023; Issue published 21 June 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: Although the GABAA receptor (GABAAR) has been proposed as the main action site for sevoflurane, isoflurane, halothane, enflurane, propofol, and benzodiazepines (BZDs), binding of these anesthetics with high-resolution structures of the GABAAR have been rarely examined by comparative docking analyses. Moreover, various combinations of ligands on more GABAARs with various subtypes need to be analyzed to understand the elaborate action mechanism of GABAARs better because some GABAA ligands showed specificity toward the distinct subtypes of the GABAAR. Methods: We performed in silico docking analysis to compare the binding modes of sevoflurane, isoflurane, halothane, enflurane, propofol, and BZDs to the GABAAR based on one of the most recently provided 3D structures. We performed the docking analysis and the affinity-based ranking of the binding sites. Results: Our docking studies revealed that isoflurane, halothane, and enflurane docked in an extracellular domain (ECD) on GABAARs, in contrast to sevoflurane. Conclusion: Our results supported a multi-site mechanism for the allosteric modulation of propofol. Propofol was bound to the pore or favored various subsites in the transmembrane domain (TMD). Our result confirmed that different chemically related BZD ligands interact via distinct binding modes rather than by using a common binding mode, as previously suggested.Keywords
Supplementary Material
Supplementary Material FileAnesthetics can be classified into three types: general, local, and regional (Fig. 1). General anesthetics are mostly halogenated ethers, such as isoflurane, sevoflurane, desflurane, and enflurane. Inhaled anesthetics diffuse to the blood and influence the central nervous system by suppressing nicotinic acetylcholine (nACh) receptors and inhibiting spinal cord function, subsequently causing paralysis and immobility (Antognini and Schwartz, 1993; Rampil et al., 1993; Campagna et al., 2003; Grasshoff et al., 2005; Hentschke et al., 2005). General anesthetics are known to target inhibitory or excitatory ion channel receptors by prolonging the activation of inhibitory channel receptors, including glycine and γ-aminobutyric acid type A (GABAA) receptors (GABAARs), and depressing the activity of excitatory channel receptors, including serotonin, nACh, and glutamate receptors (Torri, 2010).
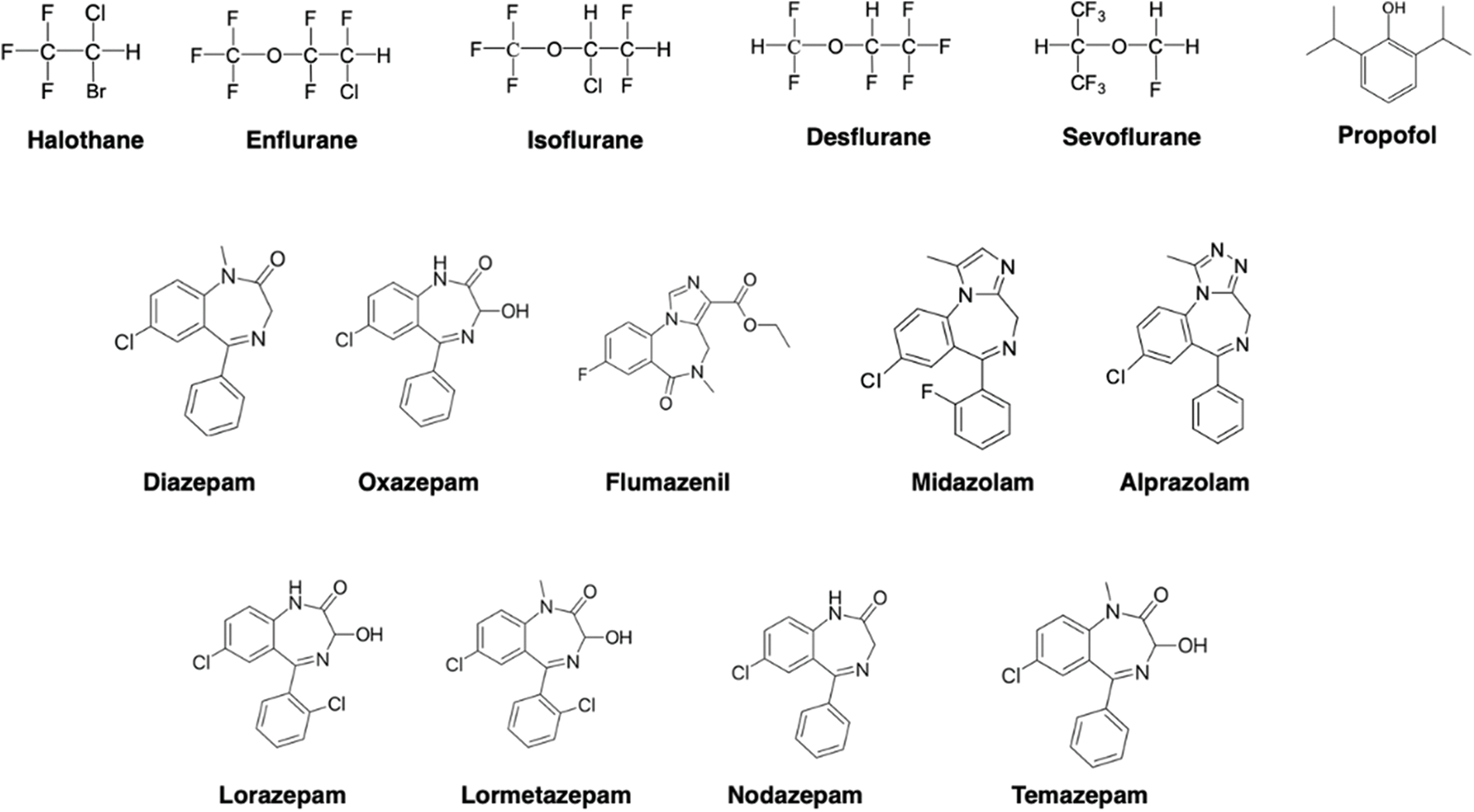
Figure 1: Structures of the anesthetics used in the docking analysis.
Although GABAARs, glycine receptors, nACh receptors, and type 3 serotonin (5-HT3) receptors are categorized as pentameric ligand-gated ion channels (pLGICs), general anesthetics generally conduct chloride ions into cells to reduce neuronal excitability in GABAA and glycine receptors but conduct cations in nACh and 5-HT3 receptors (Smart and Paoletti, 2012; Fourati et al., 2018). Moreover, in spinal motor neurons, although inhibitory glycine receptors are always stimulated by the general anesthetics, resulting in the suppression of both postsynaptic N-methyl-D-aspartate (NMDA) receptors and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors, GABAAR responses can vary among specific channel subtypes or drugs (Cheng and Kendig, 2000). Accordingly, GABAARs, are differently modulated by general anesthetics as they are inhibited rather than potentiated by anesthetics (Howard et al., 2014). Meanwhile, nACh and 5-HT3 receptors that are conventionally inhibited are activated by some anesthetics, such as ethanol (Howard et al., 2014; Fourati et al., 2018). This is a complicated but important aspect of the structure-function relationship of general anesthetics in terms of their capacity to regulate receptors in contradictory ways. Clinically relevant concentrations of enflurane and isoflurane enhanced signaling by GABA and glycine receptors (Mihic et al., 1997). Two amino acid residues buried in the transmembrane domains (TMDs) were recently discovered to be critical for the allosteric modulation of both the glycine receptors and GABAARs triggered by enflurane and isoflurane (Mihic et al., 1997). The GABAAR has also been suggested as a binding target of propofol (Krasowski et al., 2001; Kuo et al., 2005; Chiara et al., 2014). The experiment using photolabeling of a propofol analog suggested that the binding site of propofol would be located in the β subunit of the GABAAR at the boundary between the TMDs and the extracellular domain (ECD) (Yip et al., 2013). The GABAAR is one of the targets for anesthetics, including volatile and intravenous types, or neurosteroids and drugs such as benzodiazepines (BZDs) (Sieghart, 1995; Lobo and Harris, 2008; Zhu et al., 2018). Some studies have suggested that increased agonist efficacy, even though the enhanced agonist binds at the orthosteric GABA binding site, causes propofol and barbiturates to potentiate the GABAAR responses to partial agonists (O’Shea et al., 2000; Steinbach and Akk, 2001). Therefore, the interaction of anesthetic ligands and GABAARs needs to be determined to explain the anesthetic mechanism. In humans, the GABAAR has a total of 19 subunits that are encoded by nineteen different genes: α1–6, β1–3, γ1–3, δ, ε, θ, π, and ρ1–ρ3. Most physiologically predominant heteromeric synaptic isoform formats are thought to be composed of two α1-subunits, two β2-subunits, and one γ2-subunit (Sigel and Steinmann, 2012). Common synaptic GABAARs are heteropentamers with an αβγ, 2:2:1 stoichiometry (Alkire et al., 1997; Eckenhoff, 1998; Cheng and Kendig, 2000). GABAAR exists as a homopentamer (Murlidaran et al., 2019; Eckenhoff et al., 2000), and 1:2:2 heteropentamer (Bhattacharya et al., 2000; Vemparala et al., 2010). Five subunits are assembled around a pivotal pore and each subunit is composed of an ECD, including ligand binding sites, and a TMD, including M1–M4 helixes (Murlidaran et al., 2019).
The three-dimensional (3D) binding characteristics of ligands within the GABAARs should be identified using structural data to understand the action mechanism of general anesthetics. The lack of structural information has prevented a complete understanding of how anesthetics modulate the mode of action of GABAergic neurotransmission (Forman and Miller, 2011; Wei et al., 2011). Before the crystal structure of the GABAAR was discovered, other pLGICs with homology to the GABAAR and resolved 3D structures were used to determine the binding site of anesthetics (Nury et al., 2011; Chiara et al., 2014). To specify these molecular characteristics, the structures of a protein bound to anesthetics require a resolution of 2 Å (Forman and Miller, 2011). Moreover, to elucidate the conformational changes, the redundant structures of the same part need to be determined in several modes (Forman and Miller, 2011). However, crystallographic data of mammalian pLGICs with 2 Å resolution were not available until 2011 (Forman and Miller, 2011). The cryo-electron microscopy (cryo-EM) structure of the Torpedo acetylcholine receptor muscle subtype constructed in the resting mode without agonist at 4 Å resolution was the only available structural data of a vertebrate (Unwin, 2005). However, this low resolution is incapable of elucidating the elaborate binding residues of anesthetics at side chains. The other structures determined for pentameric ion-channel receptors were the superfamily of bacterial members and included the TMD without an intracellular domain (Bocquet et al., 2009; Hilf and Dutzler, 2009). Since 2012, various crystal structures of the ligand-binding domain of cysteine-loop receptor homologs such as the acetylcholine-binding protein (AChBP), the nAChR, and bacterial homologs have been solved (Richter et al., 2012). GABA is suggested to be located between β–α-subunits and BZDs are located between the α–γ subunits (Sigel and Buhr, 1997).
Furthermore, functional studies using mutagenesis have suggested the binding sites for GABA/BZD ligands and many other compounds on GABAARs. However, information on the physiological GABAAR has been rarely reported (Whiting et al., 1995; Forman and Miller, 2011; Olsen, 2015). Recently, many studies have focused on the structural determination of GABAAR, and the interaction between GABAAR and anesthetic drugs have been elucidated. Most GABAARs of the human brain in the physiological state are heteromers of two α subunits (α1–6), two β subunits (β1–3), and one γ (γ1–3) or δ subunit, out of 19 possible types (Sigel and Steinmann, 2012; Dore et al., 2014). However, further studies are needed to elucidate the previous results on the specificity of GABAA ligands toward the distinct subtypes of the GABAAR of α1, α2, α3, α4, α5, and α6. Classical BZDs, including diazepam and alprazolam, act specifically on GABAAR subtypes containing α1, α2, α3 or α5, but not α4 or α6 (Tan et al., 2011). Therefore, various combinations of ligands on more GABAARs with various subtypes need to be analyzed to understand the elaborate action mechanism of GABAARs. This explains the necessity to identify the binding site of anesthetics to GABAARs using updated and elaborate 3D structural models with more types of anesthetics. The crystallographic data of GABAAR structure determined earlier was that of β3 homopentamer in an agonist-bound state, which provided the initial guide to explain the physiological receptor (Miller and Aricescu, 2014). Photoaffinity labeling to resolve the binding site for propofol in GABAARs was performed using partially expressed GABAARs consisting of both β3 homopentamers and α1β3 heteropentamers (Yip et al., 2013). Recently, the structures of apo- and neurosteroid-bound GABAAR constructs containing an α subunit TMDs were characterized and provided (Miller et al., 2017). Later, high-resolution cryo-EM structures of the human α1β2γ2 GABAAR were provided and applied to our analysis. The α1β2γ2 isoform is a predominant form in the adult brain, and the complex of this isoform with GABA and flumazenil, the BZD antagonist, has been reported (Zhu et al., 2018). Because this α1β2γ2 isoform is a physiological GABAAR, the current high-resolution structural information finely featured the association of the heteromeric subunit and the molecular interactions of the receptor with ligands such as GABA and flumazenil. In the most up-to-date GABAA construct of β2α1β2α1γ2 form, five subunits showed a pseudo-symmetrical pattern of assembly around an integral ion channel (Zhu et al., 2018). Viewing from the extracellular space, the ring of subunits is arranged as β2α1β2α1γ2 in a counter-clockwise direction (Tretter et al., 1997; Baumann et al., 2002; Baur et al., 2006; Zhu et al., 2018). There were two ECD interfaces in α1–β2 and γ2–β2, but no ligand was bound to these clefts, which are considered putative targets for designing novel drugs (Zhu et al., 2018). According to the crystallographic model comprising the α1, β2, and γ2-subunits with GABA and flumazenil, GABA is located in the conventional neurotransmitter ligand binding site of the pLGIC superfamily at the two β2α1 interfaces in the ECD (Zhu et al., 2018). Flumazenil was found to bind at a similar form of the cleft site at the α1γ2 interface of GABARs in this model.
We used these current architectures to analyze the putative binding sites of isoflurane, enflurane, halothane, and propofol. Docking analysis of these anesthetics was performed against the entire structure and not merely the three putative ligand binding sites of recent GABAA structures. We also used these architectures to analyze the putative binding sites of various types of BZD. Docking analysis of the aesthesia was performed against a receptor construct containing ECD, TMD, and TMD hole. Docking with volatile anesthetics incurred numerous false positives because the binding mode determined by docking was unstable or short-lived (Murlidaran et al., 2019). To reduce the rate of false positives, a receptor construct containing ECD, TMD, and TMD hole was used for docking analysis in this work. Rather than being defined to the existing ligand binding site, the docking algorithm was set to predict the probability of the preferable orientation of a ligand when it was bound to the protein and to rank it by scoring (Murlidaran and Brannigan, 2018). This approach can provide locations where multiple occupancies would be considered and suggest different putative candidates of binding locations. Recently, it has been suggested that diazepam binds at the extracellular “canonical” site and also at sites in the TMD (Iorio et al., 2020). Likewise, many ligands of volatile anesthetics and BZD drugs were predicted to bind at the extracellular site and also at sites in the TMD in this docking study (Bertaccini et al., 2013).
The binding sites occupied by each anesthetic ligand under general clinical concentrations were identified and ranked by affinity scores, computational analysis was used as described previously (Murlidaran and Brannigan, 2018). Briefly, the processing includes two phases: screening or finding possible candidate binding sites occupied by anesthetics ligands at clinical concentrations and measuring and quantifying binding affinities (Murlidaran and Brannigan, 2018). All atoms contained in proteins, lipids, salt, anesthetics ligands, and water were considered to be based on the thorough physics-based Molecular Dynamics Simulation and Alchemical Free Energy Perturbation (Murlidaran and Brannigan, 2018).
High-resolution cryo-EM structures of the human α1β2γ2 GABAAR, the predominant isoform in the adult brain, co-crystalized with GABA and the BZD site antagonist flumazenil (PDB6D6U, Experimental data snapshot, Method: Electron Experimental data snapshot, Method: Electron microscopy, Resolution: 3.92 Å, Aggregation State: Particle, Reconstruction Method: Single particle, Chain A, C: 341 amino acids (AA) residues, Chain B, D: 358 AA residues, Chain F, H: 213 AA residues, Chain G, I: 454 AA residues) (Zhu et al., 2018), were used to carry out modeling. Initial models were generated depending on the lowest value of the Modeler objective function and the “Discrete Optimized Protein Energy” (DOPE) method score, accordingly, representing the largest percentage of residues in the most favored region of the Ramachandran plot (Benkert et al., 2008). The abnormalities were visually inspected for model candidates. The resulting model was slightly modified to optimize the molecular bond rotation to reduce the conflict of side chains and to optimize the protonation states for the hydrogen bond networks (Emsley et al., 2010).
GABAAR protein was considered as a system existing in a lipid membrane or water box. One of the prerequisites was that the force of the field applied to the environment, including protein, lipids, and water, was compatible with the parameters for the ligand (Murlidaran and Brannigan, 2018). The simulation was performed between a receptor and a ligand. The receptor is supposed to be inserted in a lipid bilayer hydrated with counter-ions, and the anesthetic ligands are randomly distributed in the water (Murlidaran and Brannigan, 2018). A grid box was set with the ligand randomly distributed around the receptor. In a separated docking try, a particular region of the receptor was set into a grid box as a binding candidate: whole subunit, TMD, ECD, and the TMD pore.
The receptor and the ligand in “.pdb” format were applied to the Autodock tool. The binding modes of the selected ligands were found by molecular docking simulation using a command-line prompt. In the model options, the ligand bonds were allowed to rotate. Multiple runs of docking were performed, starting from random conformations of the receptor-ligand complex. The number of these runs was represented by the value of ‘exhaustiveness,’ which was set at 15 and which depends on the flexibility of the protein side chains and the ligand. The parameter ‘nummodes,’ representing the maximum number of binding poses with multiple individual runs was set at 20 (Murlidaran and Brannigan, 2018). Software AutoDock Vina (Trott and Olson, 2010) was used as the docking algorithm. This program suggested a number of candidate binding modes between protein and ligand. The preferred orientations of the ligand were ranked by affinity-based scores (Murlidaran and Brannigan, 2018). A grid box of dimensions 25 Å × 25 Å × 25 Å was used and set with each TMD combined with ECD, TMD, ECD, and the TMD pore domain to be involved within the grid-box. When the ligands and the protein were prepared, the addition of missing hydrogens and the pairing of non-polar hydrogen atoms and computing charges were additionally inspected. Molecular docking simulations were performed with four volatile anesthetics ligands (isoflurane, sevoflurane, halothane, and enflurane), propofol, and nine BZD/BZD antagonists (flumazenil, diazepam, alprazolam, midazolam, lormetazepam, nordazepam, oxazepam, temazepam, and lorazepam). The automated docking was performed, and the preferred binding modes in a particular region of the receptor were generated. PyMOL (Schrödinger, New York, NY) was used as a software to display and explain the interaction of the receptor-ligand complex after docking analysis. The binding sites were determined by the convergence of residues in relation to anesthetic modulation on a common location in the 3D space under energy optimizations.
In this study, docking simulations of four volatile anesthetics ligands (isoflurane, sevoflurane, halothane, enflurane), propofol, and BZD/BZD antagonists (flumazenil, diazepam, alprazolam, midazolam, lormetazepam, nordazepam, oxazepam, temazepam, and lorazepam) were performed against the combined domain of TMD with ECD, TMD, ECD domains, and the TMD pore part, as shown in Fig. 2.
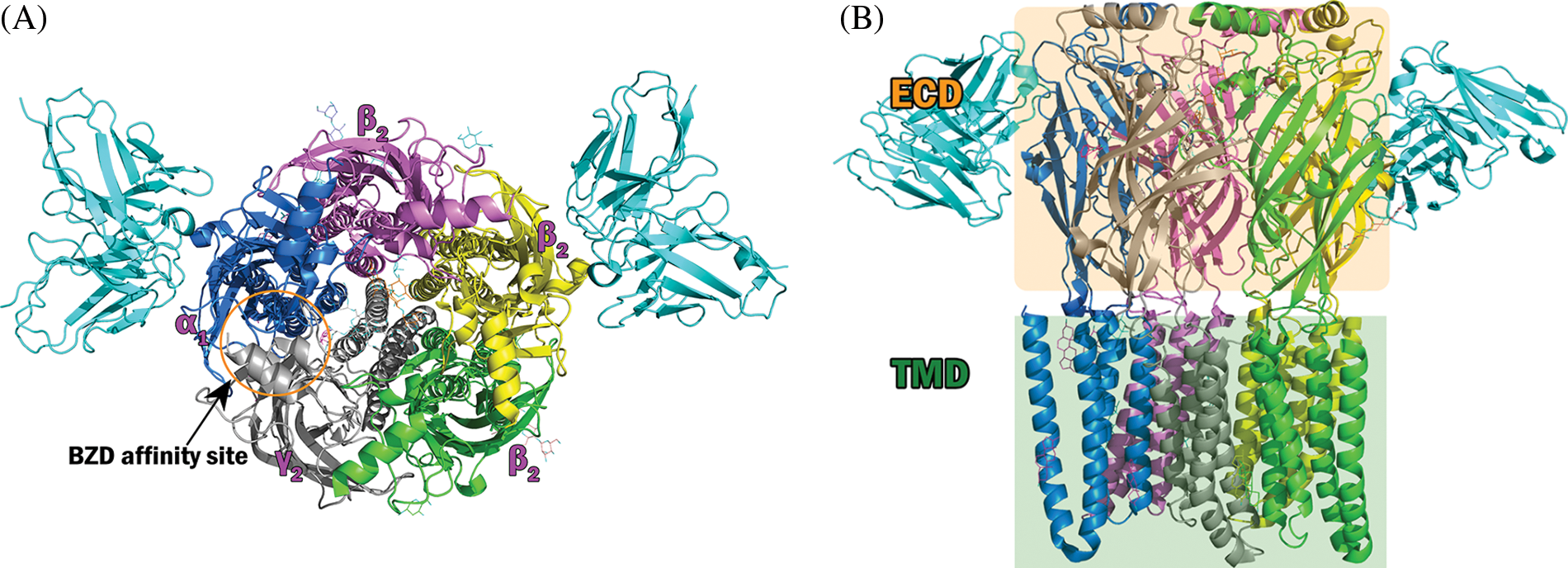
Figure 2: Structure of the γ-aminobutyric acid type A receptor (GABAAR) used in the docking experiment. Top and side views of the construction of the GABAAR-Fab fusion complex comprising subunits. α1 in marine blue (chain D), α1 in yellow (chain B), β2 in green (chain A), β2 in violet (chain C), γ2 in gray (chain E), and Fab in sky blue. (A) Side view of the extracellular and transmembrane domains of the GABAAR. (B) Front view of the receptor.
Different volatile anesthetics bind to γ-aminobutyric acid type A receptor with distinct binding modes
In the cut-off of the ninth best docking pose obtained for anesthetics within the grid box of TMD combined with ECD, the sevoflurane binding site was represented at TMD nine times (Fig. 3A), but the binding sites of isoflurane, halothane, and enflurane were represented at ECD nine times and distributed in the similar site at the best docking mode (Fig. 3B). When the docking was confined to a TMD, the binding sites of isoflurane, halothane, and enflurane were represented at the similar α1 domain but did not coincide with the sevoflurane binding site (Fig. 3C). In the best docking mode obtained for isoflurane, halothane, and enflurane (Fig. 3B), the ligand interacts with γ2Pro175 and γ2Glu178 through hydrophobic interactions (Fig. 3D). Recently, the binding sites of isoflurane and sevoflurane on GABAARs were suggested using each photoreactive ligand analog such as AziISO and AziSEVO, respectively (Woll et al., 2018). Putative binding sites identified by photolabeling were located in the TMDs in either subunit interfaces or in the interface between the ECD and the TMD (Woll et al., 2018). In agreement with these previous study results, our results suggested that sevoflurane and isoflurane did not always share binding sites, which might be related to an unexpected degree of selectivity.
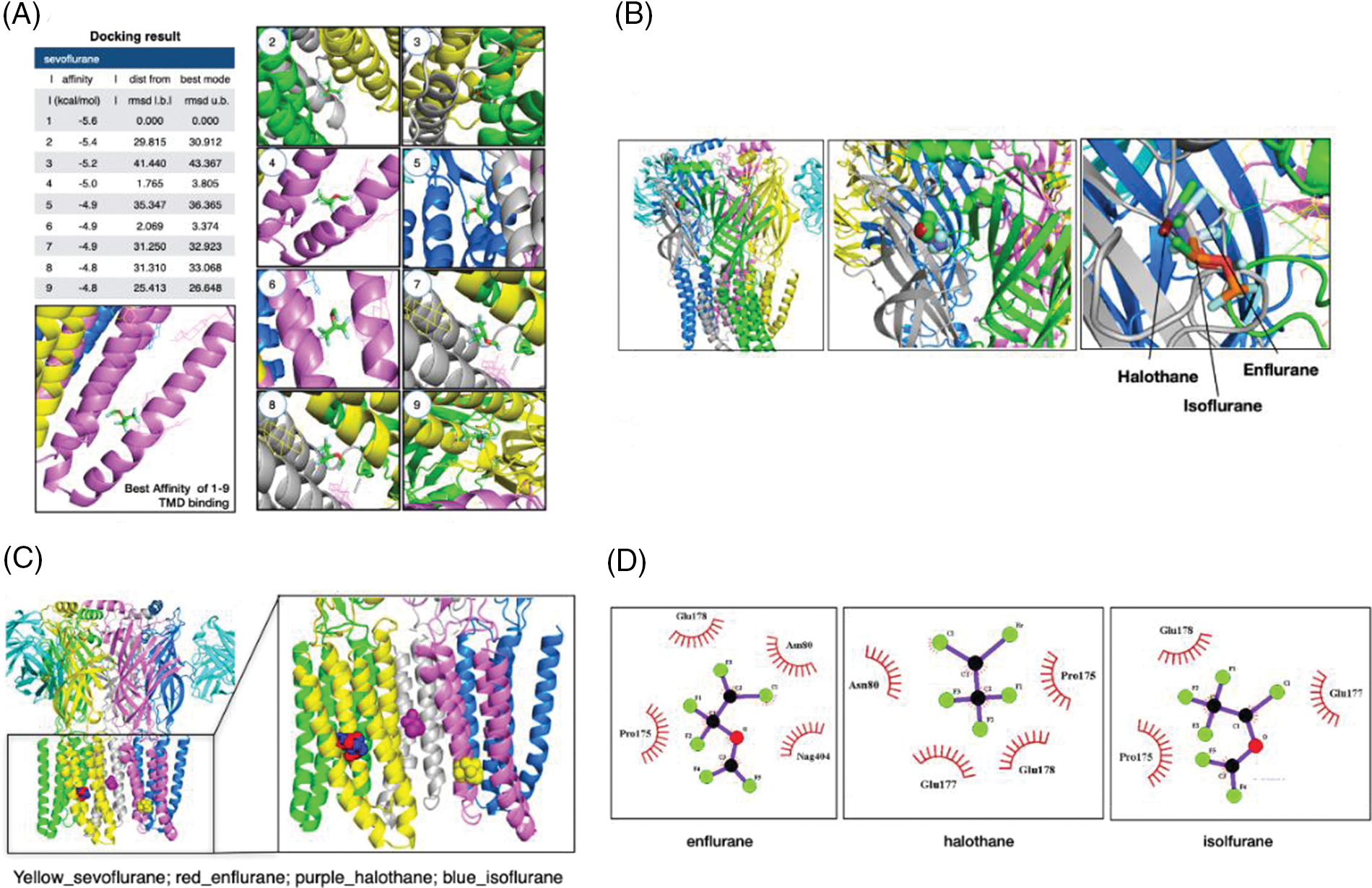
Figure 3: Binding modes of the inhalation anesthetics on the γ-aminobutyric acid type A receptor (GABAAR). (A) Nine highest-rated docking poses of sevoflurane on the GABAAR within the grid box of TMD combined with extracellular domain (ECD). The configurations are represented according to the degree of affinity. (B) The binding sites of isoflurane, halothane, and enflurane. The three anesthetics are distributed in ECD. (C) The binding sites of isoflurane, halothane, and enflurane in the case of the docking are confined to a transmembrane domain. (D) The interactions of ligands and the GABAAR in the best docking mode were obtained for isoflurane, halothane, and enflurane by 2D Ligplot analysis.
The result of the docking of propofol (Fig. 4A) within the grid box of TMD combined with ECD shows that propofol was widely distributed in TMD. The carboxyl group of ß2Trp237 interacted with propofol via hydrogen bonding (3.18 Å: weak-middle interaction) in the best docking mode (Fig. 4B). However, the binding sites of propofol were also represented in ECD and pore (Fig. 4C). This mode agrees partially with previous models obtained from GLIC showing that propofol allosterically favored closed channels (Fourati et al., 2018) by binding in the pore or favored open channels via various subsites in the TMD. Our results support a multi-site mechanism for the allosteric modulation of propofol. As previously reported, a propofol molecule was located in the lower ion pore when it was analyzed within the grid box of the TMD pore (Fig. 4D). Furthermore, propofol was located with its long axis parallel to that of the pore, making non-polar interactions with pore interface residues at Thr256 and Ile270 (Fig. 4D), in agreement with a previous report (Fourati et al., 2018).
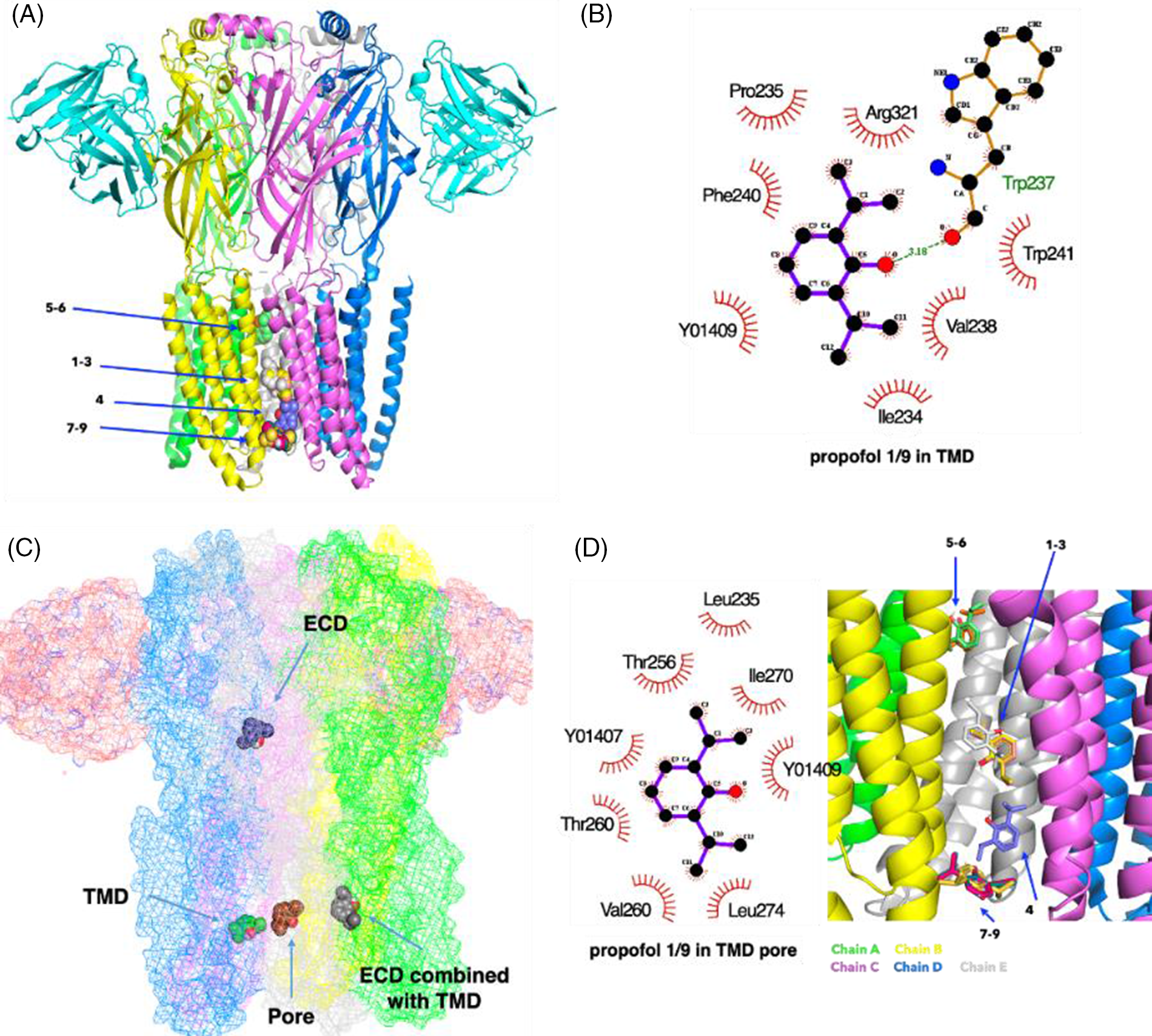
Figure 4: The binding sites of propofol on the γ-aminobutyric acid type A receptor (GABAAR). (A) Nine highest-rated docking poses of propofol on the GABAAR within the grid box of the transmembrane domain (TMD) pore. (B) The interactions of ligands and the GABAAR in the best docking mode obtained for propofol within the grid box of the TMD pore. (C) The best docking pose of propofol on the GABAAR in the case of the docking being confined to a TMD or an ECD or a TMD combined with the ECD or TMD pore. (D) The interactions of propofol on the GABAAR in the best docking pose within the grid box of a TMD pore (Y01 409 and 407 are cholesterol hemisuccinates. They are co-crystalized ligands of GABA-AR (PDB: 6D6U)).
Flumazenil, an imidazobenzodiazepine (i-BZD), is a competitive BZD antagonist which blocks diazepam potentiation of the GABA response (Zhu et al., 2018). BZD derivatives with a phenyl substituent showed a different binding mode from flumazenil with an imidazole ring (Masiulis et al., 2019). In the cryo-EM map of the α1β3γ2 GABAAR in complex with diazepam, the BZD-binding site at the α1+/γ2− interface, and sites in the β3+/α1− TMD interfaces were observed (Masiulis et al., 2019). In another cryo-EM map of the human α1β2γ2 GABAAR bound with GABA and flumazenil, flumazenil was distributed at a similar position at the α1γ2 interface (Zhu et al., 2018). Because crystallography data of the α2β2γ1 heteropentamer have not yet been proposed, the characterization of the binding mode of BZD in this specific subtype receptor cavity was difficult. In this result, the BZD binding sites were comparatively suggested using computational docking. A strong probability for flumazenil distribution was observed at analogous positions suggested for diazepam and flumazenil. In the best docking pose obtained for flumazenil from our docking result, the ligand was distributed in the well-known BZD binding site at the ECD α1+/γ2− interface and interacted with the side chain amine group of Lys106 through hydrogen bonding (3.11 Å: weak-middle interaction) (Figs. 5A and 5B). In this work, the probability for flumazenil distribution was observed at both the ECD α1+/γ2− interface and the TMD α1/ß2 interface. The result of the docking of flumazenil in TMD showed that flumazenil interacted with the hydroxyl group of ß2Thr237 via hydrogen bonding (3.15 Å: weak-middle) (Fig. 5C). Flumazenil binding sites with nine highest-ranked affinities were distributed in both TMDs and ECDs, which supports a possible multi-site mechanism for allosteric modulation such as propofol. Different BZDs bound to distinct binding sites of GABAARs. Diazepam, midazolam, nordazepam, temazepam, lorazepam, and lormetazepam were widely distributed at TMD but the best docking sites of alprazolam and oxazepam were observed at ECD (Fig. 6A). Classical BZDs, such as diazepam, predominantly exert their action via GABAARs composed of α1βγ2, α2βγ2, α3βγ2, and α5βγ2 subunits and are known to bind at the extracellular α-γ interface (Richter et al., 2012; Sigel, 2002). However, in the recently solved cryo-EM structure of the α1β3γ2L receptor in complex with GABA, and although lorazepam and lormetazepam have a very similar skeleton to diazepam, they have a 5-phenyl substituent with chloride, in contrast to nordazepam, temazepam, and oxazepam.
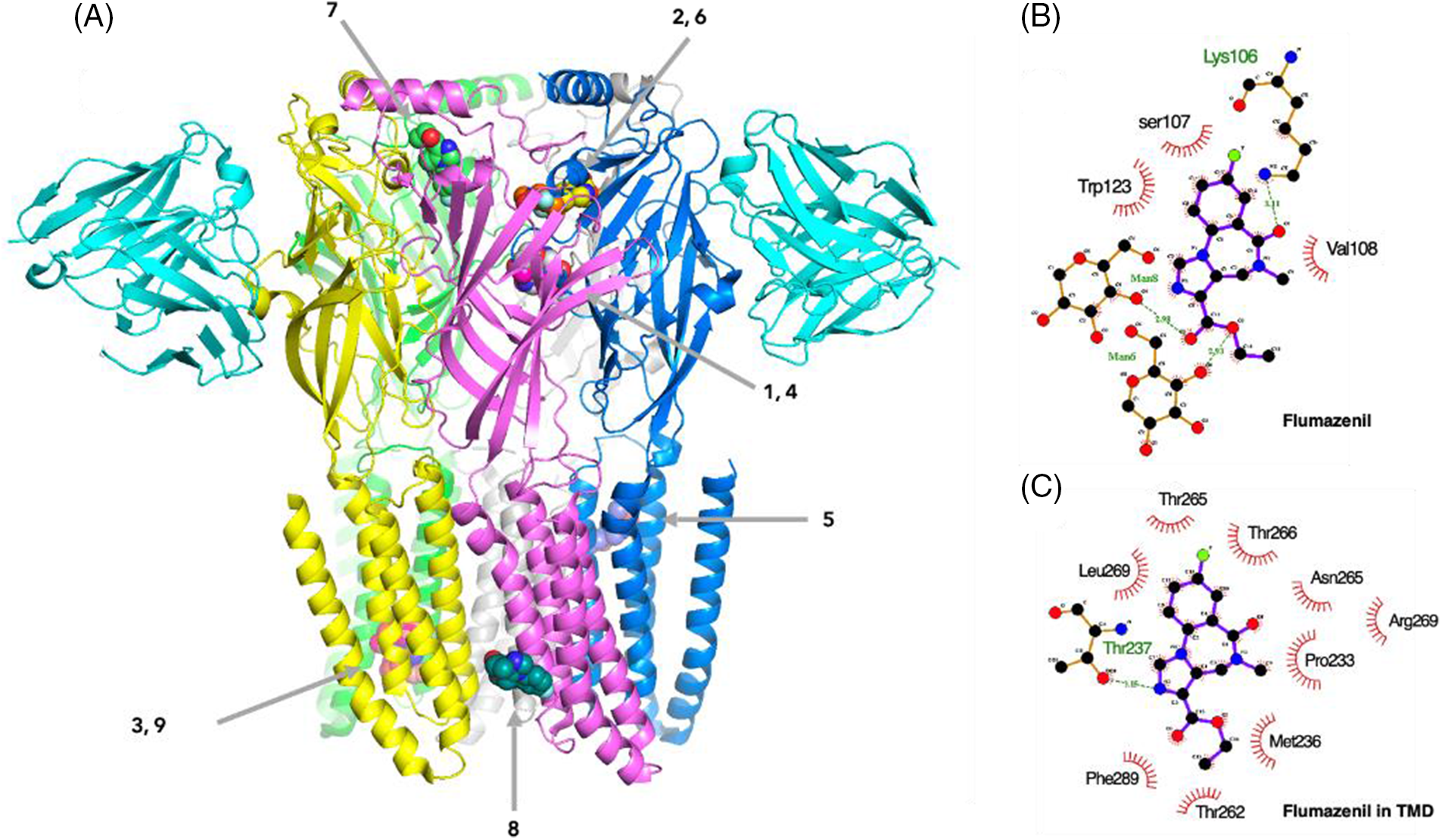
Figure 5: The binding sites of flumazenil on the γ-aminobutyric acid type A receptor (GABAAR). (A) Nine highest-rated docking poses of flumazenil on the GABAAR within the grid box of a transmembrane domain (TMD) combined with extracellular domain (ECD). (B) The interactions of ligands and the GABAAR in the best docking pose for flumazenil. (C) Three highest-rated docking poses of nordazepam, temazepam, lorazepam, lormetazapam, alprazolam, and midazolam.
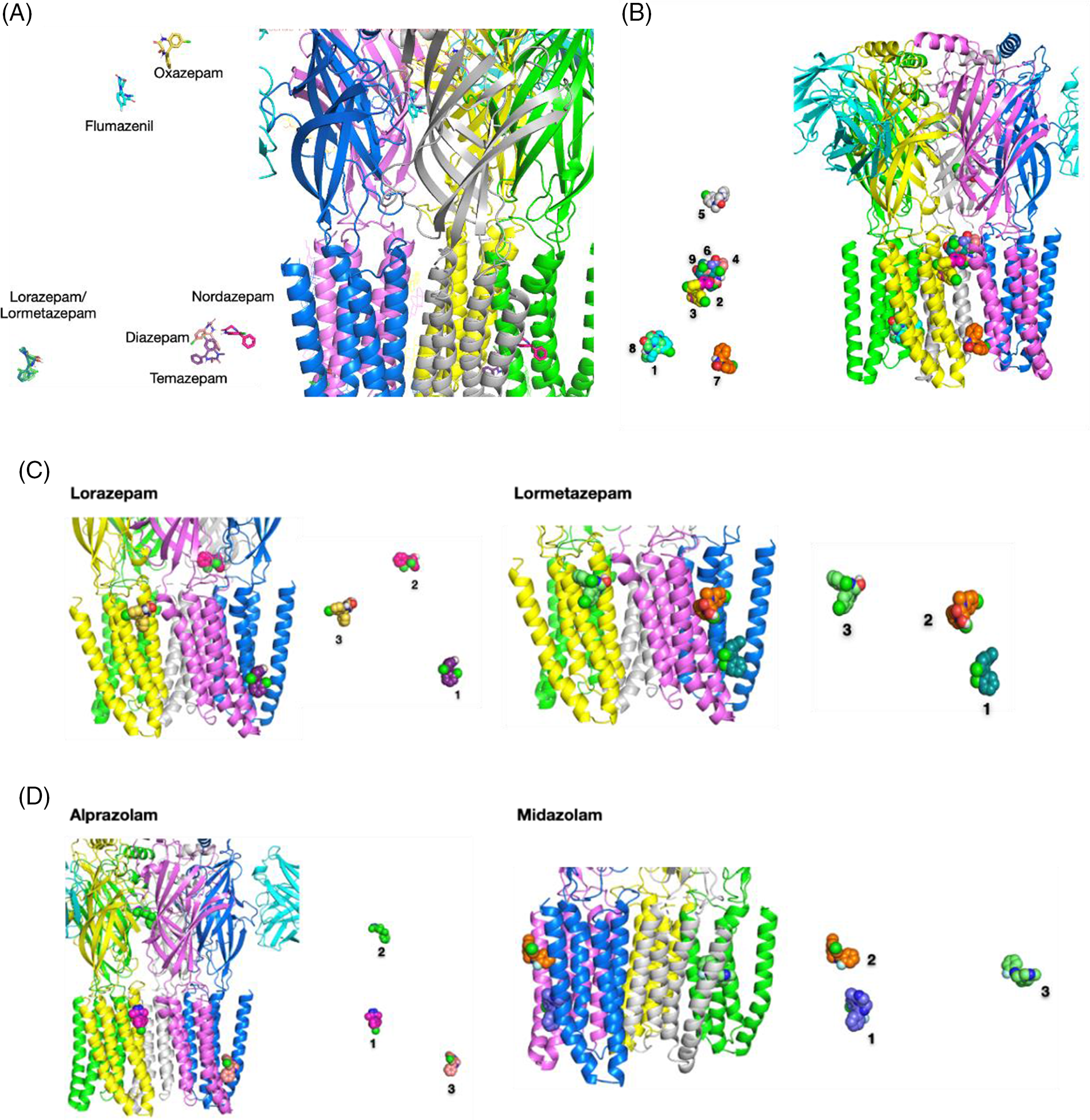
Figure 6: The binding sites of BZDs on the γ-aminobutyric acid type A receptor (GABAAR). (A) The best docking pose of benzodiazepines (BZDs) on the GABAAR. (B) Nine highest-rated docking poses of diazepam on the GABAAR. (C) Three highest-rated docking poses of lorazepam and lormetazepam on the GABAAR. (D) Three highest-rated docking poses of alprazolam and midazolam on the GABAAR.
The docking poses of lorazepam and lormetazepam displayed a high overlap bound state (Fig. 6C). One of the alprazolam binding poses on the TMD pore was overlapped with one of the propofol binding poses in an interaction of Ile232, Leu235, Y01401, and Phe306, as shown in Figs. 4D and 6D. If the ligands were docked to the ECD, flumazenil, and isoflurane were bound to the α1γ2side (chains D–E).
Docking parameters and affinity
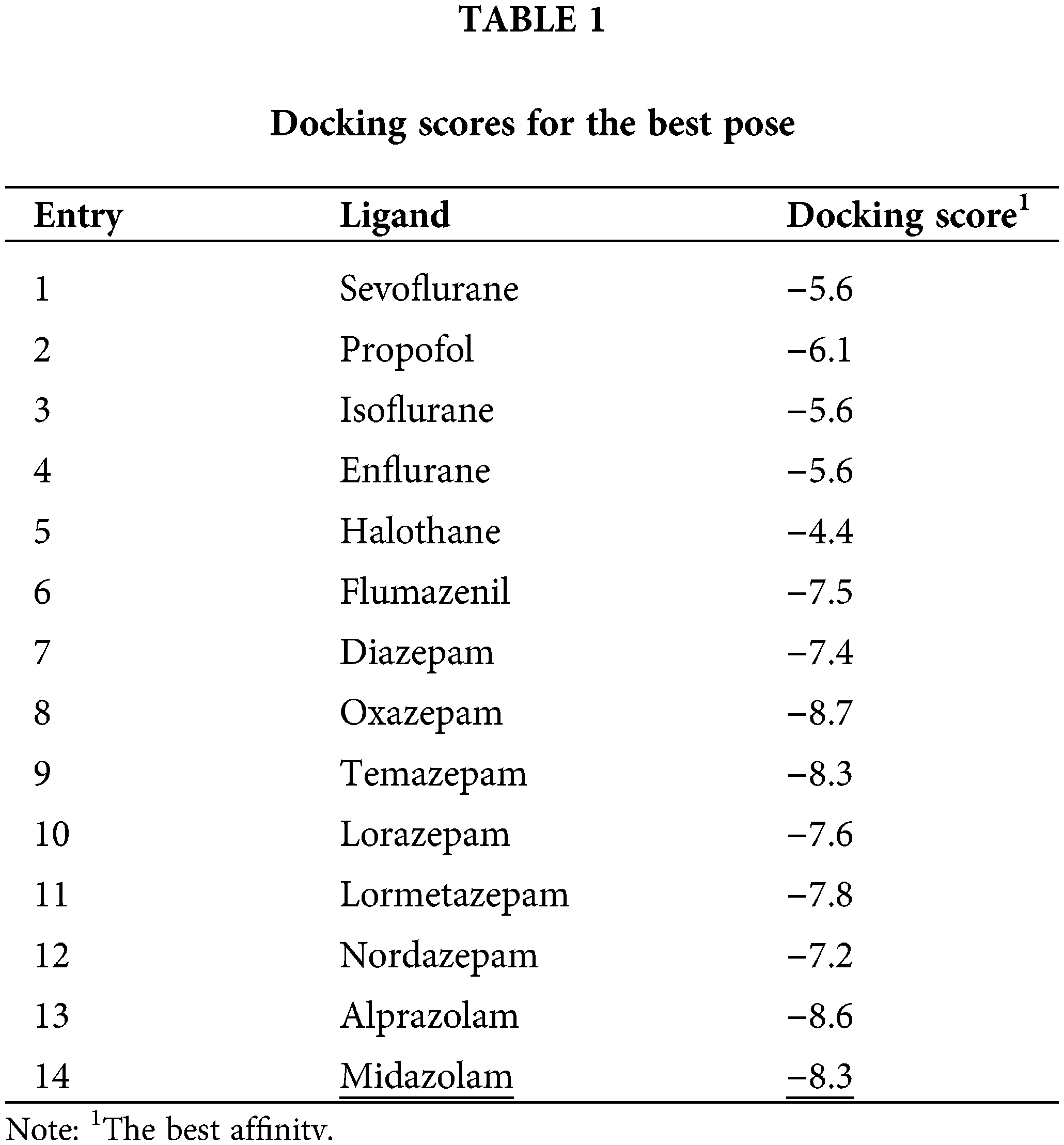
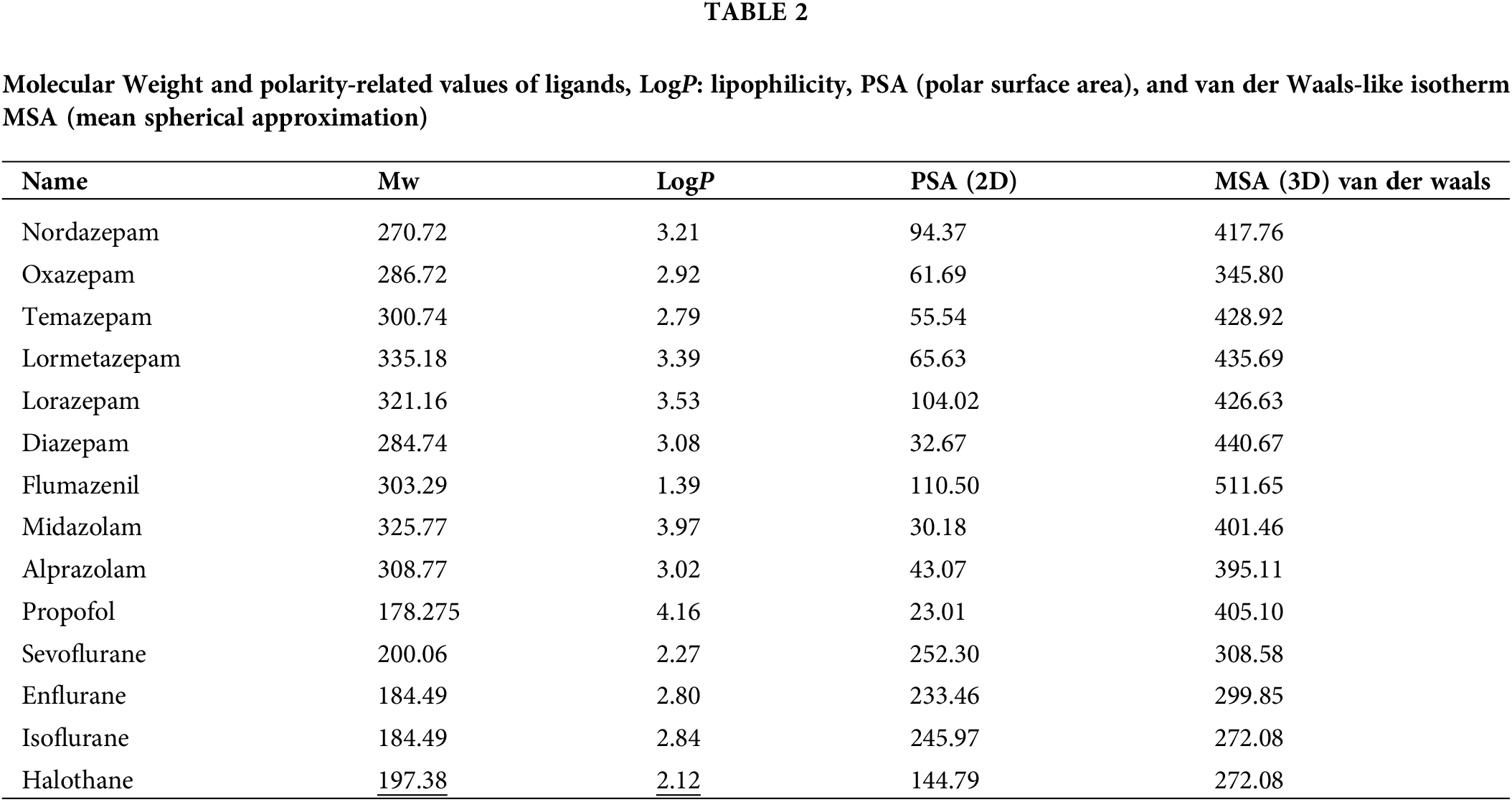
Affinity scores for the docking simulation and docking scores for the best pose are shown in Suppl. Table S1 and Table 1. Polarity-related values of ligands like logP representing lipophilicity, polar surface area (PSA), and van der Waals-like isotherm mean spherical approximation (MSA) are represented in Table 2. The scores determined from automated docking are merely function outputs and are not directly meaningful as absolute values (Murlidaran and Brannigan, 2018). Nevertheless, the relative values can be used to determine the favorable configuration and binding mode of the ligand. In the tables, we have suggested the ten most favorable ligand configurations bound to the receptor with their scoring functions. Considering the chemical properties of the ligands, the results indicated that propofol, with the highest logP value and the lowest PSA value, was distributed mostly in TMD. Meanwhile, flumazenil, with the lowest logP value and a larger PSA value than propofol, was mostly bound to ECD. Because inhalation anesthetics and propofol are low-solubility molecules, it is likely that these anesthetics are not prone to be distributed in receptor pores or intersubunit and intrasubunit spaces. However, converging evidence and our docking results demonstrated the binding of anesthetics to the pores or intersubunit and intrasubunit spaces. In the present study of the best pose, propofol interacted with residues of Phe77(E), Phe100(D), Met130(E), Tyr160(D), Val203(D), and Thr207(D) through hydrophobic interaction and Ser205(D) through hydrogen bonding. In a previous report, the degree of hydrogen bonding was not closely connected to the affinity of general anesthetics to interacting residues (Murlidaran et al., 2019). Instead, they suggested that affinity was more closely connected to the number of water molecules in inter-subunit cavities displaced by sevoflurane or propofol. Our affinity data obtained for sevoflurane and propofol determined by docking score exhibited the same trend with the results of water displacement simulation as a criterion for affinity. Although propofol is smaller than sevoflurane, propofol seems to replace more water molecules from any site because it has a smaller polar surface area than sevoflurane (Table 2). Propofol is suggested to have a larger binding affinity than that of sevoflurane (Elgarf et al., 2018).
To identify binding sites and understand molecular mechanisms of anesthetics’ action on GABAARs, docking analysis has been used by employing structures of bound proteins and ligands. Since 2007, updated protein engineering techniques and crystallography analyses have provided a greatly increased number of solved protein structures (Venkatakrishnan et al., 2013). Although diverse pLGICs, including nAChR, GABAAR, glycine receptor, and NMDA receptor, are known to bind to a variety of anesthetics, docking studies to identify binding sites were not performed in many cases because of limited protein structures (Kim et al., 2018). Crystal structures of several LGICs have been recently solved. Crystal structures of a human GABAAR and a glycine receptor were obtained in 2014 and 2015, respectively (Miller and Aricescu, 2014; Huang et al., 2015; Kim et al., 2018). Docking analyses not previously performed because of unavailable structures are now possible, especially for those on the GABAAR with a variety of anesthetics. Crystalized structures of different kinds of GABAARs in complex with various ligands, including pharma-cores, drugs, peptides, antibodies, and G-proteins, have been reported (Sieghart, 1995; Bertaccini et al., 2013; Fourati et al., 2018; Murlidaran and Brannigan, 2018; Emsley et al., 2010). However, recent studies have revealed that when an anesthetic interacts with receptors to a multi-binding site, the influence on the GABAARs varies according to distinct subtypes. Therefore, more information needs to be obtained on the structure of GABAARs bound to anesthetic ligands.
Our docking studies revealed that isoflurane, halothane, and enflurane docked in a similar way to that observed for GABAARs by diazepam, except for an absence of interaction in the same residues in the diazepam binding site on GABAARs. Furthermore, these three anesthetics were majorly bound to an ECD of GABAARs, in contrast to sevoflurane, distributed in the TMD at the nine most highly ranked affinities. This differentiation in the docking mechanism might be related to sevoflurane’s different clinical effects, such as recovery from anesthesia, preconditioning, liver, and renal toxicity. In clinical practice, sevoflurane is more frequently used in pediatric patients for induction of anesthesia than other anesthetics due to its mild airway irritation (TerRiet et al., 2000). Moreover, recovery from anesthesia with sevoflurane is more rapid than with isoflurane (Arain et al., 2005). Although solubility influences the duration of anesthesia, the approach to different binding sites with membrane receptors also seems to affect anesthesia duration because these inhalation anesthetics have poor aqueous solubility. Within the grid box of the TMD pore, sevoflurane and propofol were located in the pore face of ß2. This result agrees with a previous report showing that all sevoflurane molecules are observed to be partitioned into membrane or protein sites, especially to the intrasubunit cavity of both ß subunits (but not α or γ subunits) in the flooding of ligands rarely soluble in the aqueous phase (sevoflurane and propofol) (Murlidaran et al., 2019). In this flooding simulation, amounts of sevoflurane partitioning into the water fraction, the membrane, and protein cavities were measured, and a high portion of molecules was introduced into the cytosolic phase and spontaneously partitioned into the lipid bilayer and/or bound to receptor protein cavities (Murlidaran et al., 2019). In this flooding model, sevoflurane was reported to bind spontaneously to pore sites and inter-subunit/intra-subunit sites, in agreement with our docking results. The report suggests that sevoflurane can penetrate the pore through the ECD vestibule over 1.3 µs and transfer from the pore to the α+-β− inter-subunit site (Murlidaran et al., 2019). In our docking results, interacting residues for sevoflurane on the GABAAR existed between β2 and γ2 at the best pose (1/9) or between α1 and β2 (2/9) at the second-best pose. Contacted residues for propofol on GABAARs were between α1β2 (1/9) or between β2 and γ2 (3/9). Although inhalation anesthetics and propofol had low solubility, our results showed that they were distributed in the pores.
The competition between flumazenil and isoflurane for the binding site might be related to the clinical role of flumazenil. Flumazenil is a competitive inhibitor of isoflurane or sevoflurane. The BZD antagonist flumazenil can restore the effects of BZD drugs bound to GABARs. It could also restore the clinical effect of isoflurane (Griffiths et al., 1984a). In clinical anesthesia, extubation and early anesthetic recovery are important to decrease morbidity and incidence of complications (Tvete et al., 2016).
BZD pharmacophore models based on SAR data have predicted a lipophilic pharmacophoric feature to be an essential part of BZDs (Rikke et al., 2013).
Considering that diazepam had a similar molecular weight, oxazepam showing the lowest docking score, might have the best affinity to the receptor at the best docking mode. At the best docking mode, flumazenil and oxazepam are bound to ECD, while diazepam is bound to TMD. These differences are related to lipophilicity in that flumazenil and oxazepam are less lipophilic than diazepam. In clinical reports, diazepam is considered to be less additive to dependency than oxazepam (Griffiths et al., 1984b). Further, diazepam is more rapidly absorbed, followed by a fast distribution phase (distribution half-life of about 1 h) with a half-life of 20–200 h, whereas oxazepam is absorbed more slowly, with a half-life of 4–15 h (Tvete et al., 2016). Classified in terms of BZD elimination half-life, diazepam is a long-acting BZD, whereas alprazolam, oxazepam, lorazepam, and temazepam are short to intermediate-acting BZDs (Fox et al., 2011). However, the clinical difference between diazepam and oxazepam is likely due to these pharmacokinetic differences and also the binding site and mode to the receptor. These differences suggest that relative potency is another way to characterize BZDs. Initially used BZDs were defined as having low to medium potency. However, alprazolam, lorazepam, and clonazepam were categorized as high-potency BZDs (Griffin et al., 2013). Of three BZD receptor agonists, namely diazepam, midazolam, and lorazepam, commonly used in anesthesia in clinical treatment, midazolam is the most lipid-soluble (Griffin et al., 2013). This agrees with our result that midazolam has the highest logP value among BZD agonists. It is distributed mainly in TMD. However, overall, our results are in line with a previous report proposing at least two different “common” binding modes rather than a single one for BZDs in general (Elgarf et al., 2018). Not all BZDs bind with the same type of BZD receptor or with equivalent binding affinity to the same receptor (Griffin et al., 2013).
Each anesthetic interacts with receptors to a multi-binding site, and its influence on the GABAARs varies according to the distinct subtypes. This necessitates a better understanding of the structure of GABAARs bound to anesthetic ligands. Our study results for the computational docking of volatile anesthetics such as isoflurane, sevoflurane, halothane, and enflurane into GABAARs suggested that sevoflurane binds with a mode that is distinct from that of isoflurane, halothane, and enflurane. Comparing the inhalation anesthetics, sevoflurane was mostly distributed in TMD, whereas isoflurane, enflurane, and halothane were bound to ECD.
In the best docking pose acquired for isoflurane, halothane, and enflurane, the ligand molecule binds to γ2Pro175 through the N terminal amine group and binds to γ2Glu178 through the side carboxyl group, conforming to the previously known experimental information. Because inhalation anesthetics and propofol are low solubility molecules, these anesthetics are unlikely to be distributed in receptor pores or intersubunit and intrasubunit spaces. However, converging evidence and our docking results demonstrated the binding of anesthetics to the pores or intersubunit and intrasubunit spaces. In the best docking pose obtained for flumazenil from our docking result, the ligand was observed in a well-known BZD binding site at both the ECD α1+/γ2− interface and the TMD α1/ß2 interface. From our docking result, diazepam was mostly distributed in TMD. The docking results of these ligands showed a broad diversity of high-score poses. Overall, our results reveal at least two different “common” binding modes, rather than a single one, for BZDs in general.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: The authors confirm their contribution to the paper as follows: study conception and design: SYP; data collection: SHA, JYL; Investigation: SHA; Methodology: SHA, JYL; Project administration: SYP; Resources: SYP, HJK; Supervision: SYP, HJK; Validation: JHS, SYP; Visualization: SHA, JYL; analysis and interpretation of results: SHA, SYP; Draft manuscript preparation: Writing–original draft: SHA, SYP; Writing–review & editing: JHS, HJK.
Availability of Data and Materials: The datasets generated during analyzed study are available from the corresponding author on reasonable request.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Supplementary Materials: The supplementary material is available online at https://doi.org/10.32604/biocell.2023.027984.
References
Alkire MT, Haier RJ, Shah NK, Anderson CT (1997). Positron emission tomography study of regional cerebral metabolism in humans during isoflurane anesthesia. Anesthesiology 86: 549–557. https://doi.org/10.1097/00000542-199703000-00006 [Google Scholar] [PubMed] [CrossRef]
Antognini JF, Schwartz K (1993). Exaggerated anesthetic requirements in the preferentially anesthetized brain. Anesthesiology 79: 1244–1249. https://doi.org/10.1097/00000542-199312000-00015 [Google Scholar] [PubMed] [CrossRef]
Arain SR, Barth CD, Shankar H, Ebert TJ (2005). Choice of volatile anesthetic for the morbidly obese patient: Sevoflurane or desflurane. Journal of Clinical Anesthesia 17: 413–419. https://doi.org/10.1016/j.jclinane.2004.12.015 [Google Scholar] [PubMed] [CrossRef]
Baumann SW, Baur R, Sigel E (2002). Forced subunit assembly in α1β2γ2 GABAA receptors. Insight into the absolute arrangement. Journal of Biological Chemistry 277: 46020–46025. https://doi.org/10.1074/jbc.M207663200 [Google Scholar] [PubMed] [CrossRef]
Baur R, Minier F, Sigel E (2006). A GABAA receptor of defined subunit composition and positioning: Concatenation of five subunits. FEBS Letters 580: 1616–1620. https://doi.org/10.1016/j.febslet.2006.02.002 [Google Scholar] [PubMed] [CrossRef]
Benkert P, Tosatto SC, Schomburg D (2008). QMEAN: A comprehensive scoring function for model quality assessment. Proteins 71: 261–277. https://doi.org/10.1002/prot.21715 [Google Scholar] [PubMed] [CrossRef]
Bertaccini EJ, Yoluk O, Lindahl ER, Trudell JR (2013). Assessment of homology templates and an anesthetic binding site within the gamma-aminobutyric acid receptor. Anesthesiology 119: 1087–1095. https://doi.org/10.1097/ALN.0b013e31829e47e3 [Google Scholar] [PubMed] [CrossRef]
Bhattacharya AA, Curry S, Franks NP (2000). Binding of the general anesthetics propofol and halothane to human serum albumin. High resolution crystal structures. Journal of Biological Chemistry 275: 38731–38738. https://doi.org/10.1074/jbc.M005460200 [Google Scholar] [PubMed] [CrossRef]
Bocquet N, Nury H, Baaden M, Le Poupon C, Changeux JP, Delarue M, Corringer PJ (2009). X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature 457: 111–114. https://doi.org/10.1038/nature07462 [Google Scholar] [PubMed] [CrossRef]
Campagna JA, Miller KW, Forman SA (2003). Mechanisms of actions of inhaled anesthetics. New England Journal of Medicine 348: 2110–2124. https://doi.org/10.1056/NEJMra021261 [Google Scholar] [PubMed] [CrossRef]
Cheng G, Kendig JJ (2000). Enflurane directly depresses glutamate AMPA and NMDA currents in mouse spinal cord motor neurons independent of actions on GABAA or glycine receptors. Anesthesiology 93: 1075–1084. https://doi.org/10.1097/00000542-200010000-00032 [Google Scholar] [PubMed] [CrossRef]
Chiara DC, Gill JF, Chen Q, Tillman T, Dailey WP, Eckenhoff RG, Xu Y, Tang P, Cohen JB (2014). Photoaffinity labeling the propofol binding site in GLIC. Biochemistry 53: 135–142. https://doi.org/10.1021/bi401492k [Google Scholar] [PubMed] [CrossRef]
Dore AS, Okrasa K, Patel JC, Serrano-Vega M, Bennett K et al. (2014). Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature 511: 557–562. https://doi.org/10.1038/nature13396 [Google Scholar] [PubMed] [CrossRef]
Eckenhoff RG (1998). Do specific or nonspecific interactions with proteins underlie inhalational anesthetic action? Molecular Pharmacology 54: 610–615. [Google Scholar] [PubMed]
Eckenhoff RG, Petersen CE, Ha CE, Bhagavan NV (2000). Inhaled anesthetic binding sites in human serum albumin. Journal of Biological Chemistry 275: 30439–30444. https://doi.org/10.1074/jbc.M005052200 [Google Scholar] [PubMed] [CrossRef]
Elgarf AA, Siebert DCB, Steudle F, Draxler A, Li G, Huang S, Cook JM, Ernst M, Scholze P (2018). Different benzodiazepines bind with distinct binding modes to GABAA receptors. ACS Chemical Biology 13: 2033–2039. https://doi.org/10.1021/acschembio.8b00144 [Google Scholar] [PubMed] [CrossRef]
Emsley P, Lohkamp B, Scott WG, Cowtan K (2010). Features and development of Coot. Acta Crystallographica Section D: Structural Biology 66: 486–501. https://doi.org/10.1107/S0907444910007493 [Google Scholar] [PubMed] [CrossRef]
Forman SA, Miller KW (2011). Anesthetic sites and allosteric mechanisms of action on Cys-loop ligand-gated ion channels. Canadian Journal of Anesthesia 58: 191–205. https://doi.org/10.1007/s12630-010-9419-9 [Google Scholar] [PubMed] [CrossRef]
Fourati Z, Howard RJ, Heusser SA, Hu H, Ruza RR, Sauguet L, Lindahl E, Delarue M (2018). Structural basis for a bimodal allosteric mechanism of general anesthetic modulation in pentameric ligand-gated ion channels. Cell Reports 23: 993–1004. https://doi.org/10.1016/j.celrep.2018.03.108 [Google Scholar] [PubMed] [CrossRef]
Fox C, Liu H, Kaye AD, Manchikanti L, Trescot AM, Christo PJ (2011). Antianxiety Agents, Clinical Aspects of Pain Medicine and Interventional Pain Management: A Comprehensive Review, pp. 543–552. Paducah, KY: ASIP Publishing. [Google Scholar]
Grasshoff C, Rudolph U, Antkowiak B (2005). Molecular and systemic mechanisms of general anaesthesia: The ‘multi-site and multiple mechanisms’ concept. Current Opinion in Anaesthesiology 18: 386–391. https://doi.org/10.1097/01.aco.0000174961.90135.dc [Google Scholar] [PubMed] [CrossRef]
Griffin CEIII, Kaye AM, Bueno FR, Kaye AD (2013). Benzodiazepine pharmacology and central nervous system-mediated effects. Ochsner Journal 13: 214–223. [Google Scholar] [PubMed]
Griffiths RR, McLeod DR, Bigelow GE, Liebson IA, Roache JD (1984a). Relative abuse liability of diazepam and oxazepam: Behavioral and subjective dose effects. Psychopharmacology 84: 147–154. https://doi.org/10.1007/BF00427437 [Google Scholar] [PubMed] [CrossRef]
Griffiths RR, McLeod DR, Bigelow GE, Liebson IA, Roache JD, Nowowieski P (1984b). Comparison of diazepam and oxazepam: Preference, liking and extent of abuse. Journal of Pharmacology and Experimental Therapuetics 229: 501–508. [Google Scholar] [PubMed]
Hentschke H, Schwarz C, Antkowiak B (2005). Neocortex is the major target of sedative concentrations of volatile anesthetics: Strong depression of firing rates and increase of GABAA receptor-mediated inhibition. European Journal of Neuroscience 21: 93–102. https://doi.org/10.1111/j.1460-9568.2004.03843.x [Google Scholar] [PubMed] [CrossRef]
Hilf RJ, Dutzler R (2009). Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature 457: 115–118. https://doi.org/10.1038/nature07461 [Google Scholar] [PubMed] [CrossRef]
Howard RJ, Trudell JR, Harris RA (2014). Seeking structural specificity: Direct modulation of pentameric ligand-gated ion channels by alcohols and general anesthetics. Pharmacological Reviews 66: 396–412. https://doi.org/10.1124/pr.113.007468 [Google Scholar] [PubMed] [CrossRef]
Huang X, Chen H, Michelsen K, Schneider S, Shaffer PL (2015). Crystal structure of human glycine receptor-α3 bound to antagonist strychnine. Nature 526: 277–280. https://doi.org/10.1038/nature14972 [Google Scholar] [PubMed] [CrossRef]
Iorio MT, Vogel FD, Koniuszewski F, Scholze P, Rehman S, Simeone X, Schnurch M, Mihovilovic MD, Ernst M (2020). GABAA receptor ligands often interact with binding sites in the transmembrane domain and in the extracellular domain-can the promiscuity code be cracked? International Journal of Molecular Sciences 21: 334–354. https://doi.org/10.3390/ijms21010334 [Google Scholar] [PubMed] [CrossRef]
Kim D, Kim HJ, Ahn S (2018). Anesthetics mechanisms: A review of putative target proteins at the cellular and molecular level. Current Drug Targets 19: 1333–1343. https://doi.org/10.2174/1389450119666180502112029 [Google Scholar] [PubMed] [CrossRef]
Krasowski MD, Jenkins A, Flood P, Kung AY, Hopfinger AJ, Harrison NL (2001). General anesthetic potencies of a series of propofol analogs correlate with potency for potentiation of gamma-aminobutyric acid (GABA) current at the GABAA receptor but not with lipid solubility. Journal of Pharmacology and Experimental Therapeutics 297: 338–351. [Google Scholar] [PubMed]
Kuo A, Domene C, Johnson LN, Doyle DA, Venien-Bryan C (2005). Two different conformational states of the KirBac3.1 potassium channel revealed by electron crystallography. Structure 13: 1463–1472. https://doi.org/10.1016/j.str.2005.07.011 [Google Scholar] [PubMed] [CrossRef]
Lobo IA, Harris RA (2008). GABAA receptors and alcohol. Pharmacology Biochemistry and Behavior 90: 90–94. https://doi.org/10.1016/j.pbb.2008.03.006 [Google Scholar] [PubMed] [CrossRef]
Masiulis S, Desai R, Uchanski T, Serna Martin I, Laverty D et al. (2019). GABAA receptor signaling mechanisms revealed by structural pharmacology. Nature 565: 454–459. https://doi.org/10.1038/s41586-018-0832-5 [Google Scholar] [PubMed] [CrossRef]
Mihic SJ, Ye Q, Wick MJ, Koltchine VV, Krasowski MD et al. (1997). Sites of alcohol and volatile anaesthetic action on GABAA and glycine receptors. Nature 389: 385–389. https://doi.org/10.1038/38738 [Google Scholar] [PubMed] [CrossRef]
Miller PS, Aricescu AR (2014). Crystal structure of a human GABAA receptor. Nature 512: 270–275. https://doi.org/10.1038/nature13293 [Google Scholar] [PubMed] [CrossRef]
Miller PS, Scott S, Masiulis S, de Colibus L, Pardon E, Steyaert J, Aricescu AR (2017). Structural basis for GABAA receptor potentiation by neurosteroids. Nature Structural & Molecular Biology 24: 986–992. https://doi.org/10.1038/nsmb.3484 [Google Scholar] [PubMed] [CrossRef]
Murlidaran S, Brannigan G (2018). Physical accuracy leads to biological relevance: Best practices for simulating ligand-gated ion channels interacting with general anesthetics. Methods in Enzymology 602: 3–24. https://doi.org/10.1016/bs.mie.2018.02.001 [Google Scholar] [PubMed] [CrossRef]
Murlidaran S, Hénin J, Brannigan G (2019). Competitive dewetting underlies site-specific binding of general anesthetics to GABAA receptors. Biophysical Journal 1–15. https://doi.org/10.1101/694612 [Google Scholar] [CrossRef]
Nury H, van Renterghem C, Weng Y, Tran A, Baaden M, Dufresne V, Changeux JP, Sonner JM, Delarue M, Corringer PJ (2011). X-ray structures of general anesthetics bound to a pentameric ligand-gated ion channel. Nature 469: 428–431. https://doi.org/10.1038/nature09647 [Google Scholar] [PubMed] [CrossRef]
O’Shea SM, Wong LC, Harrison NL (2000). Propofol increases agonist efficacy at the GABAA receptor. Brain Research 852: 344–348. https://doi.org/10.1016/S0006-8993(99)02151-4 [Google Scholar] [PubMed] [CrossRef]
Olsen RW (2015). Allosteric ligands and their binding sites define gamma-aminobutyric acid (GABA) type A receptor subtypes. Advances in Pharmacology 73: 167–202. https://doi.org/10.1016/bs.apha.2014.11.005 [Google Scholar] [PubMed] [CrossRef]
Rampil IJ, Mason P, Singh H (1993). Anesthetic potency (MAC) is independent of forebrain structures in the rat. Anesthesiology 78: 707–712. https://doi.org/10.1097/00000542-199304000-00014 [Google Scholar] [PubMed] [CrossRef]
Richter L, de Graaf C, Sieghart W, Varagic Z, Morzinger M, de Esch IJ, Ecker GF, Ernst M (2012). Diazepam-bound GABAA receptor models identify new benzodiazepine binding-site ligands. Nature Chemical Biology 8: 455–464. https://doi.org/10.1038/nchembio.917 [Google Scholar] [PubMed] [CrossRef]
Rikke B, Kristine K, Pernille LS, Tommy S, Thomas B (2013). A unified moedel of the GABAA receptor comprising agonist and benzodiazepine binding sites. PLoS One 8: e52323. https://doi.org/10.1371/journal.pone.0052323 [Google Scholar] [PubMed] [CrossRef]
Sieghart W (1995). Structure and pharmacology of gamma-aminobutyric acid_A receptor subtypes. Pharmacology Reviews 47: 181–234. [Google Scholar]
Sigel E (2002). Mapping of the benzodiazepine recognition site on GABAA receptors. Current Topics in Medicinal Chemistry 2: 833–839. https://doi.org/10.2174/1568026023393444 [Google Scholar] [PubMed] [CrossRef]
Sigel E, Buhr A (1997). The benzodiazepine binding site of GABAA receptors. Trends in Pharmacological Sciences 18: 425–429. https://doi.org/10.1016/S0165-6147(97)90675-1 [Google Scholar] [CrossRef]
Sigel E, Steinmann ME (2012). Structure, function, and modulation of GABAA receptors. Journal of Biology Chemistry 287: 40224–40231. https://doi.org/10.1074/jbc.R112.386664 [Google Scholar] [PubMed] [CrossRef]
Smart TG, Paoletti P (2012). Synaptic neurotransmitter-gated receptors. Cold Spring Harber Perspectives in Biology 4. https://doi.org/10.1101/cshperspect.a009662 [Google Scholar] [PubMed] [CrossRef]
Steinbach JH, Akk G (2001). Modulation of GABAA receptor channel gating by pentobarbital. Journal of Physiology 537: 715–733. https://doi.org/10.1113/jphysiol.2001.012818 [Google Scholar] [CrossRef]
Tan KR, Rudolph U, Luscher C (2011). Hooked on benzodiazepines: GABAA receptor subtypes and addiction. Trends in Neurosciences 34: 188–197. https://doi.org/10.1016/j.tins.2011.01.004 [Google Scholar] [PubMed] [CrossRef]
TerRiet MF, DeSouza GJ, Jacobs JS, Young D, Lewis MC, Herrington C, Gold MI (2000). Which is most pungent: Isoflurane, sevoflurane or desflurane? British Journal of Anaesthesia 85: 305–307. https://doi.org/10.1093/bja/85.2.305 [Google Scholar] [PubMed] [CrossRef]
Torri G (2010). Inhalation anesthetics: A review. Minerva Anestesiologica 76: 215–228. [Google Scholar] [PubMed]
Tretter V, Ehya N, Fuchs K, Sieghart W (1997). Stoichiometry and assembly of a recombinant GABAA receptor subtype. Journal of Neuroscience 17: 2728–2737. https://doi.org/10.1523/JNEUROSCI.17-08-02728.1997 [Google Scholar] [PubMed] [CrossRef]
Trott O, Olson AJ (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry 31: 455–461. https://doi.org/10.1002/jcc.21334 [Google Scholar] [PubMed] [CrossRef]
Tvete IF, Bjorner T, Skomedal T (2016). A 5-year follow-up study of users of benzodiazepine: Starting with diazepam versus oxazepam. British Journal of General Practice 66: e241–247. https://doi.org/10.3399/bjgp16X684385 [Google Scholar] [PubMed] [CrossRef]
Unwin N (2005). Refined structure of the nicotinic acetylcholine receptor at 4A resolution. Journal of Molecular Biology 346: 967–989. https://doi.org/10.1016/j.jmb.2004.12.031 [Google Scholar] [PubMed] [CrossRef]
Vemparala S, Domene C, Klein ML (2010). Computational studies on the interactions of inhalational anesthetics with proteins. Accounts of Chemical Research 43: 103–110. https://doi.org/10.1021/ar900149j [Google Scholar] [PubMed] [CrossRef]
Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM (2013). Molecular signatures of G-protein-coupled receptors. Nature 494: 185–194. https://doi.org/10.1038/nature11896 [Google Scholar] [PubMed] [CrossRef]
Wei W, Hamby AM, Zhou K, Feller MB (2011). Development of asymmetric inhibition underlying direction selectivity in the retina. Nature 469: 402–406. https://doi.org/10.1038/nature09600 [Google Scholar] [PubMed] [CrossRef]
Whiting PJ, McKernan RM, Wafford KA (1995). Structure and pharmacology of vertebrate GABAA receptor subtypes. International Review of Neurobiology 38: 95–138. https://doi.org/10.1016/S0074-7742(08)60525-5 [Google Scholar] [PubMed] [CrossRef]
Woll KA, Zhou X, Bhanu NV, Garcia BA, Covarrubias M, Miller KW, Eckenhoff RG (2018). Identification of binding sites contributing to volatile anesthetic effects on GABA type A receptors. Federation of American Societies for Experimental Biology 32: 4172–4189. https://doi.org/10.1096/fj.201701347R [Google Scholar] [PubMed] [CrossRef]
Yip GM, Chen ZW, Edge CJ, Smith EH, Dickinson R et al. (2013). A propofol binding site on mammalian GABAA receptors identified by photolabeling. Nature Chemical Biology 9: 715–720. https://doi.org/10.1038/nchembio.1340 [Google Scholar] [PubMed] [CrossRef]
Zhu S, Noviello CM, Teng J, Walsh RMJr, Kim JJ, Hibbs RE (2018). Structure of a human synaptic GABAA receptor. Nature 559: 67–72. https://doi.org/10.1038/s41586-018-0255-3 [Google Scholar] [PubMed] [CrossRef]
Table S1. Affinity scores and docking parameters
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools