
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

DNA methylation as a mediator of epigenetic regulation in the pathogenesis and precision medicine of osteoarthritis: An updated review
1 Anhui University of Chinese Medicine, Hefei, 230012, China
2 Institute of Rheumatism Prevention and Treatment of Traditional Chinese Medicine, Anhui Academy of Chinese Medicine Sciences, Hefei, 230031, China
3 Department of Rheumatism Immunity, The Second Affiliated Hospital, Anhui University of Chinese Medicine, Hefei, 230061, China
4 Department of Rheumatism Immunity, The First Affiliated Hospital, Anhui University of Chinese Medicine, Hefei, 230031, China
* Corresponding Authors: Qiao Zhou, ; Jian Liu,
(This article belongs to the Special Issue: Biochemical and Epigenetics Changes in Health and Disease)
BIOCELL 2023, 47(4), 761-772. https://doi.org/10.32604/biocell.2023.026698
Received 20 September 2022; Accepted 28 November 2022; Issue published 08 March 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
The pathophysiology of osteoarthritis (OA) is multifactorial, with the primary risk factors being obesity, age, environmental variables, and genetic predisposition. The available evidence suggests that genetic diversity does not adequately account for all clinical characteristics and heterogeneity of OA. Genetics has emerged as a nascent and crucial area of research in OA. The epigenetic module presents a potential link between genetic and environmental risk factors and the susceptibility and pathogenesis of OA. As a critical epigenetic alteration, DNA methylation has been shown to have an important role in the etiology of OA and is a viable biomarker for predicting disease progression and medication response, as shown in this research. This review aims to update knowledge in the field of DNA methylation associated with OA to better identify the essential features of OA subtypes and pathological conditions, hence accelerating individualized treatment and precision medicine.Keywords
It is estimated that 250 million individuals worldwide are living with some kind of osteoarthritis (OA), the most prevalent cause of joint disability, degeneration, and pain (Wilkinson and Zeggini, 2021). The pathogenesis of OA is also uncertain, including stress stimulation, age, genetics, inflammation, obesity, injury, and environmental factors, as well as nuclear and mitochondrial genetics associated with the disease (Molnar et al., 2021). Currently, there are no disease-modifying agents that can be used to slow or reverse OA. OA treatment is limited to drug pain management and arthroplasty (Primorac et al., 2020). The discovery of diagnostic and prognostic biomarkers for OA has lately received significant attention due to its importance and economic impact.
With the rapid development of epigenetics in recent years, large-scale meta-analyses have shown that epigenetics has a role in the pathophysiology of OA (Boer et al., 2021; Styrkarsdottir et al., 2017, 2018; Tachmazidou et al., 2019; Zengini et al., 2018). Numerous genomes have been linked to OA risk by genome-wide association studies and quantitative trait loci analysis (Gari et al., 2016; Tachmazidou et al., 2019). Aberrant epigenetic modifications are associated with a number of pathological conditions and include DNA methylation, expression of non-coding RNAs, and histone modifications that regulate the gene expression at transcriptional and/or post-transcriptional levels. Here, we will briefly highlight the DNA methylation-mediated regulation of gene expression in OA. The methylation of DNA is an influential epigenetic change that regulates the expression of immune-related genes as well as the development of inflammation (Mazzone et al., 2019). DNA methylation modification involves two different processes: the addition and removal of a methyl group at the fifth position of cytosine or the sixth position of adenine in the DNA (Jiang and Guo, 2020), that is, DNA 5-methylcytosine (5 mC) and DNA 6-adenine methylation (6 mA), respectively. DNA 5-methylcytosine generally occurs in cytosine guanine dinucleotide (CpG), the most prevalent DNA methylation modification in eukaryotic genomes (Greenberg and Bourc’his, 2019). These 5 mC methylation marks are created by DNA methyltransferases (DNMTs), such as DNMT3B, DNMT3A, and DNMT1. 5 mC methylation can be oxidized and changed into 5-hydroxymethylcytosine (5 hmC) by ten-eleven translocation (TET) enzymes, TET1, TET2, and TET3 (Skvortsova et al., 2017). However, few enzymes are known to be involved in 6 mA methylation in mammals, such as N6 adenine–specific DNA methyltransferase 1 and alkylated DNA repair protein B homolog 1 (Kweon et al., 2019). Using DNA methylation data together with genotypes can identify genetic variants associated with differential methylation levels at CpGs, i.e., methylation qualitative trait loci (mQTLs) (Venkateswaran et al., 2022). The clinical value of DNA methylation changes, which are involved in the pathological process of OA, has been widely recognized (Tachmazidou et al., 2019).
This review mainly summarizes recent advances in understanding the role of DNA methylation in the pathogenesis of OA. We also have accumulated evidence indicating that methylation is a connection between genetic and environmental exposures, and a potential biomarker for subtype identification, treatment, and diagnosis. Finally, we show that epigenetic modules have the potential to be revolutionary biomarkers and precision medicine tools for the later development of personalized therapy and precision medicine.
Genome-wide methylation analysis was carried out to identify epigenetic variants associated with osteoarthritis
The methylation of DNA is a significant epigenetic modification that is referred to both the expression of genes and the splicing of transcriptional information. Identifying aberrant DNA methylation variations throughout the whole genome gives a comprehensive view of the genetic pattern alterations that occur during OA start and course. The Steinberg group established cell-specific maps of molecular QTLs in chondrocytes and synoviocytes derived from the primary joint tissue of OA patients. Combining these maps with Genome-Wide Association Study (GWAS) results, they discovered substantial evidence of the co-localization of five OA markers aldehyde dehydrogenase 1 family member A2 (ALDH1A2) and family with sequence similarity 53 member A in low-grade OA cartilage, solute carrier family 44-member 2 (SLC44A2), NADPH-cytochrome P450 reductase and SMAD Family Member 3 (SMAD3) in high-grade OA cartilage (Steinberg et al., 2021). Alvarez-Garcia et al. (2016) compared DNA from knee cartilage from 11 normal and 12 OA donors for the first time using the Illumina Infinium Human Methylation 450 array, the most comprehensive methylation array available. Methylation profiles identified 929 differentially methylated sites united with 500 distinct genes. Finally, it was revealed that normal and knee OA cartilage exhibit significantly different methylation groups. Comprehensive analysis and functional validation revealed significant hypermethylation and downregulation of a subset of six transcription factors T-box transcription factor 4, atonal BHLH transcription factor 8, zinc finger and BTB domain containing 16, MAF basic leucine zipper transcription, nuclear receptor corepressor 2 and zinc fingers and homeoboxes 2 in OA cartilage.
Differences in DNA methylation patterns may also explain the different phenotypes of OA. Genome-wide association approaches are used in studies of OA patients with distinct characteristics (77,052 cases and 378,169 controls): hip OA, knee OA, knee and/or hip OA, have identified genes enrichment in single-gene forms of skeletal developmental disorders, as well as in biological pathways for collagen formation and extracellular matrix organization (Tachmazidou et al., 2019). Simultaneously, the mechanism of ten possible effector genes, including fibroblast growth factor 18 (FGF18), cathepsin K (CTSK), transforming growth factor beta 1 (TGFB1), and interleukin 11 (IL11) supports efficacy evaluation in OA. One research investigated DNA methylation patterns of cartilage from 16 OA hip samples, 62 OA knee samples, and 19 normal hip samples. From a total of 482,421 sites, 12 OA discriminative methylation sites were chosen. These sites are linked to the Meis homeobox 1 (MEIS1), engrailed homeobox 1 (EN1) gamma-aminobutyric acid type A receptor subunit gamma3 (GABRG3), and retinoid X receptor alpha (RXRA) genes (Wu et al., 2020). Similarly, Lin et al. (2020) quantified CpG methylation in human reinforcers, including methylation profiles of 470,870 CpG probes in 108 samples of hip tissue from hip/knee OA and healthy individuals. The discovery of 8,111 differentially methylated CpGs from enhancer regions shows the importance of DNA methylation changes in both phenotypes of OA. On the side, sex-specific enhancer methylation in hip OA was revealed to be associated with OA phenotype. Subsequently, additional large-scale GWAS studies of hand OA indicate the association between the matrix Gla protein (MGP) variant and increased risk for hand OA, which may increase the burden of hand OA through decreased inhibition of cartilage calcification (den Hollander et al., 2017). Here, we summarize the characteristic genes of aberrant DNA methylation in different clinical phenotypes of osteoarthrosis, as well as the commonly shared genes (Table 1).

Methylation differences may also explain different mechanisms of action in different diseases. The results of genome-wide DNA methylation showed that the overall DNA methylation pattern of rheumatoid arthritis (RA) and OA did differ considerably, but 523 low-methylated regions were exclusive to RA. The presence of different low-methylated regions in synovial cells from RA and OA patients reflects different functional changes and supports their significance as regulatory elements for RA-specific immune function (Ham et al., 2019). Kashin-Beck disease (KBD) is a regional and disabling form of OA that causes severe alterations in the joints and has an ill-defined mechanism (Kang et al., 2022). Patients with OA demonstrated increased methylation in iodothyronine deiodinase-2, glutathione peroxidase 3, and thioredoxin reductase 1 promoter areas compared to persons with those having KBD when MALDI-TOF-MS was utilized. This suggests that distinct epigenetic alterations cause KBD and OA. Furthermore, these modifications cause a disruption in the production of selenoproteins, as well as a regulation of chondrocyte anti-oxidation and anti-apoptosis (Zhang et al., 2021a). Similarly, compared with KBD, differentially methylated genes (DMG) associated with OA are abundant in multiple ion channel pathways, hinting that ion channels are underlying biomarkers of OA. Lastly, the findings demonstrated that a great number of intriguing differentially expressed genes are associated with DMGs. SPARC-related modular calcium binding 2 and homeobox D3, two key genes that govern cartilage and skeletal physiologic and pathologic processes, are examples of differentially methylated regions (DMRs) and DMGs. These genes are also abundant in pathways associated with the skeletal system and limbs (Fan et al., 2021). These findings highlight the importance of examining alterations in DNA methylation across disorders to discover common and disease-specific components of epigenetic dysfunction. In addition, further studies have identified differentially expressed genes and differentially methylated sites for OA via the combined gene expression and bioinformatics analyses. Some intriguing biomarkers were found, such as microtubule-associated protein 1B, fibronectin type III domain containing 1 (FNDC1), anillin, potassium calcium-activated channel subfamily N member 4, sodium channel epithelial 1α subunit, and stanniocalcin 2 (Ball et al., 2022; Yi et al., 2021). The relationship between single nucleotide polymorphism changes and surrounding DMR in the proximal regions of several genes associated with OA risk has been the topic of numerous studies that have interpreted the correlation plectin (PLEC), glutamate ionotropic receptor NMDA type subunit associated protein 1 (GRINA), ALDH1A2, MGP (Rice et al., 2019; Shepherd et al., 2018; Shepherd et al., 2019). Zhao et al. (2017) first found hypomethylation of TNF receptor-associated factor 1, C-X3-C motif chemokine ligand 1, and connective tissue growth factor genes in OA chondrocytes. They also observed a consistent relationship between hypomethylation and mRNA expression, indicating that DNA methylation may impact gene and non-coding RNA expression as well as gene transcription and translation. These findings imply that DNA methylation signals might aid in the early detection of OA or risk assessment.
In this review, we summarized a list of the differentially methylated genes of OA that have previously been reported in the scientific literature. We then utilized the GeneMANIA website (Yue et al., 2021a) (http://genemania.org) to predict genes functionally comparable to hub genes and built a related PPI network (Ratnakumar et al., 2020). The PPI network can forecast the relationships between functionally similar genes and hub genes, including positive regulation of DNA-binding transcription factor activity: positive regulation of DNA-binding transcription factor activity, embryo development culminating in birth or egg hatching, shared protein domains, response to bone morphogenetic proteins, transmembrane receptor protein serine/threonine kinase signaling pathway, ossification, positive regulation of cell adhesion, skeletal system development, co-expression, co-localization, genetic interactions, and so forth (Fig. 1).
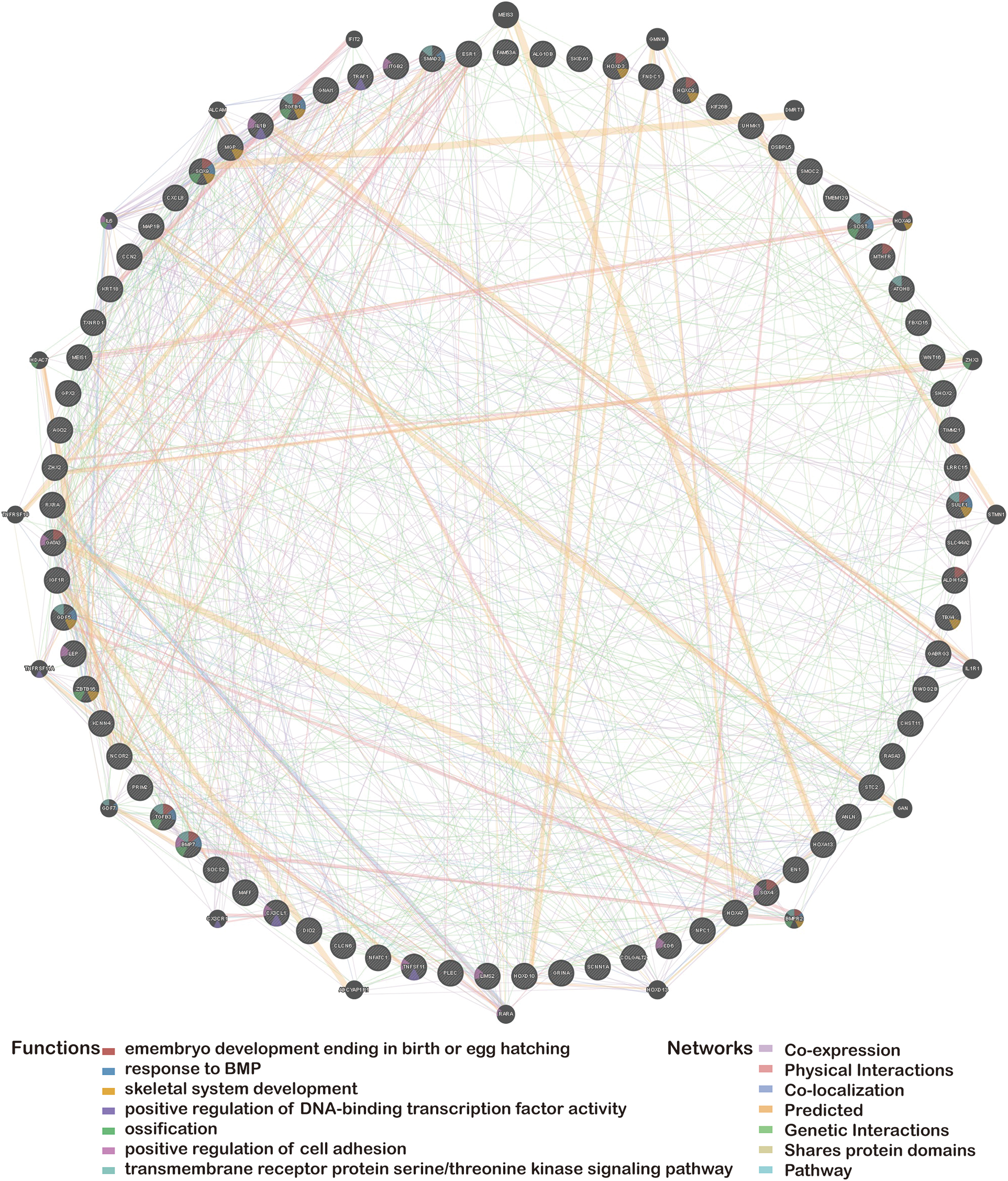
Figure 1: Protein-protein interaction network of hub genes.
Epigenetic regulation in the underlying pathogenesis of osteoarthritis
Epigenetic changes regulate gene expression by affecting gene transcription, which in turn causes variations in the amount of the protein encoded by the gene. Epigenetic processes have an effect on somatic cells that are operating normally, including articular chondrocytes. These mechanisms serve to stabilize the phenotype of the somatic cells. Epigenetic modification of chondrocytes can modify their phenotype and function, causing them to overexpress chondrodenatase and proinflammatory mediators. By attracting methyl-binding and chromatin-silencing proteins, CpG methylation promotes transcription factor binding and/or boosts gene expression. Inflammatory signals in OA joints enhance DNA CpG demethylation, and some research has revealed changes in DNA methylation of genes affecting cartilage catabolism (de Andrés et al., 2013; Hashimoto et al., 2013; Taylor et al., 2014) and anabolic metabolism (Imagawa et al., 2014; Kim et al., 2013) during OA progression. Changes in the methylation pattern of DNA seem to trigger the generation of inflammatory cytokines and matrix-degrading mediators, for example, interleukin (IL)-6 (Yang et al., 2017), matrix metalloproteinase 13 (MMP13) (Li et al., 2017) and tumor necrosis factor-α (Zhang et al., 2021b). These proteins contribute to cartilage degeneration and synovial inflammation in knee OA.
Studies of DNA methylation patterns in relation to OA have focused on candidate genes associated with the pathophysiology of OA. Singh et al. (2021) integrated transcription and epigenome data in OA cartilage and confirmed the increased expression of leucine-rich repeat containing 15 (LRCC15) in human OA cartilage samples. Additionally, they demonstrated that changes in DNA methylation might partially drive the differential expression of LRRC15 in OA cartilage. They also highlighted the probable role that LRCC15 plays in OA illness as a component of a functional network involved in chondrocyte phenotypic alterations and stress response coordination. These reactions include hypertrophic changes and extracellular matrix disintegration. The genotype rs11732213 is associated with DNA methylation at nine CpGs, which forms a DMR, which acts as an enhancer and demethylates CpGs that alter the expression of transmembrane protein 129 (TMEM129), which is a target of OA genetic risk at this locus (Brumwell et al., 2022). According to other research findings, the expression of C-terminal binding proteins is shown to be mediated by DNA methylation. Overexpressed C-terminal binding proteins form a transcriptional complex with p300 and AP1. The CtBP-p300-AP1 complex transcriptional machinery selectively stimulates the NLR family pyrin domain containing 3 (NLRP3) expression and its downstream signaling in this complex, which eventually leads to an amplification of the inflammatory response and plays a role in OA pathogenesis (Sun et al., 2020). Subsequently, based on the cross-omics analysis, other candidate methylation markers that are specific and sensitive and associated with the diagnosis are summarized, such as DNMT3B (Shen et al., 2017; Xiong et al., 2021; Zhou et al., 2019), cartilage oligomeric matrix protein (COMP), ALDH1A2 (Steinberg et al., 2018), and PLEC (Rice et al., 2019; Sorial et al., 2020). By recapitulating the mechanism of these candidate methylation markers, we could link OA genetic susceptibility with a number of cellular pathways or targets, which may provide a scope for the development of simpler OA therapeutics to target this mechanism (Table 2).

DNA methylation as a biomarker in the diagnosis of osteoarthritis
DNA methylation refers to a chemical modification process in which the base on the DNA sequence changes s-adenosine methionine into a methyl group by means of covalent bonding under the action of DNA methyltransferase without changing the genetic expression (Ming et al., 2020). DNA methylation is the most prevalent kind of epigenetic control of gene expression in mammalian cells. This alteration occurs naturally and often in eukaryotic cells and is considered normal. Although methylation can be modified in a variety of ways, the bases of the modified sites can be adenine N-6, guanine N-7, cytosine N-4, and cytosine C-5, which are catalyzed by different DNA methylases, but most occur in the gene promoter region CpG island. How DNA methylation patterns are regulated at each site in the genome and in different tissues or cells is a fundamental question that needs to be answered in epigenetics. It is diffusely accepted that dynamic DNA methylation signatures at particular CpG loci can serve as biomarkers of age-related and aging diseases. These biomarkers, which represent essential clinical goals for diagnosis, prognosis, and the prediction of treatment response, are found at specific loci (Salameh et al., 2020). The epigenetic regulation mechanism of whole blood, cartilage, and synovium in OA patients is described in Table 3.
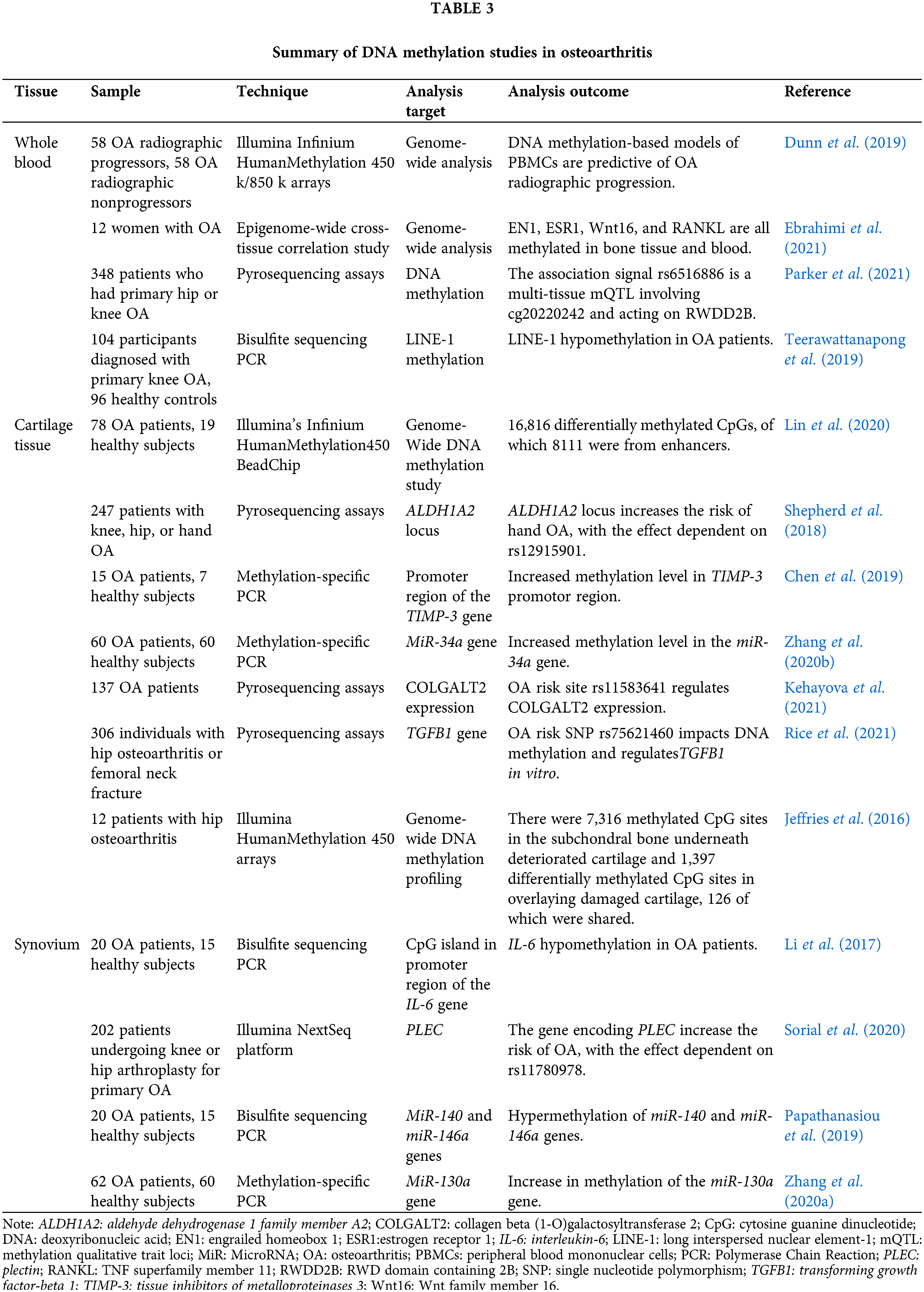
Monitoring changes in methylation might be a useful technique for predicting the progression of a disease and determining how well a patient responds to therapy. Recent research has investigated the methylation status of readily accessible tissues, such as blood and synovial fluid, to ascertain whether these biomarkers indicate synovial disease.
Patterns of DNA methylation in the blood may have a lot of promise as non-invasive biomarkers as well. Dunn et al. (2019) concluded peripheral blood epigenetic pattern of OA for the first time. DNA from peripheral blood mononuclear cells (PBMCs) was analyzed at the time of visit in 58 future radiographic progressions (joint space narrowing at 24 months, sustained at 48 months) compared to that of 58 non-progressors. The collected data was utilized to create a prediction approach based on DNA methylation to distinguish between two groups. Methylation in PBMCs might be beneficial as indicators of OA development, and bigger patient cohorts should be studied further (Dunn et al., 2019). In a different study examining DNA methylation at more than 850K locations, the researchers discovered 28,549 CpG sites that were methylated in the same way in bone and blood. This research aimed to determine whether or not blood can be deemed a viable bone-surrogate (Ebrahimi et al., 2021). Numerous key genes involved in bone metabolism are represented, such as EN1, estrogen receptor 1 (ESR1), TNF superfamily member 11 (RANKL), and Wnt family member 16 (Wnt16), and major bone-regulating pathways, such as Wnt signaling and estrogen response, are enriched among similarly methylated sites, further substantiating the feasibility of our approach. In general, our findings imply that bone methylation groups and trapping sites linked with bone modulation might be reflected in peripheral blood.
In a similar manner, as a target of the rs6516886 OA association signal, the RWD domain containing 2B (RWDD2B) is accompanied by differential methylation of Cg20242 and additional CpGs in its vicinity as intermediaries in the regulation of the gene. RWDD2B is derived from peripheral blood, cartilage, and synovium and codes for a poorly understood protein, and additional analysis of genetic risk targets for OA will provide fresh insights into this complex disease (Parker et al., 2021). Teerawattanapong et al. (2019) measured line-1 methylation levels in 104 patients with knee OA and 96 healthy controls, and the results showed that long interspersed nuclear element-1 (LINE-1) methylation in white blood cells was linked to radiographic severity of knee osteoarthritis and increased risk. Patients suffering from knee OA were found to have higher than normal amounts of 8-hydroxy-2′-deoxyguanosine (8-OHdG) in their synovial fluid, suggesting that their elevated levels can be potentially used as biomarkers for the severity of knee OA and possible involvement in its pathology.
In general, these results suggest peripheral blood can reflect bone methylation and explore sites associated with bone regulation.
Epigenetic changes in cartilage are strongly associated with OA. As part of methylation analysis, genes related to matrix metalloproteinases and leptin genes (IL1β, SOD2, NOS2,SOX9, GDF5), linked to obesity and inflammation, showed significant changes (Koike et al., 2018). In a study (Chen et al., 2019), the expression of the long non-coding RNAs (LncRNA) XIST and tissue inhibitors of metalloproteinases 3 (TIMP-3) were analyzed, and chondrocytes were separated and identified from OA cartilage tissues and normal cartilage tissues. Functional investigations involving the recruitment of DNMT3A, DNMT3B, and DNMT1 to the TIMP-3 promoter have shown that the LncRNA XIST enhances the methylation of the TIMP-3 promoter. This, in turn, stimulates collagen breakdown in the chondrocytes of OA patients after tibial plateau fracture. These two molecules can potentially be attractive therapeutic targets for people suffering from OA. Certain RNA-seq tests have revealed that the short nucleolar RNA host gene 9 is repressed in OA and inhibits chondrocytes from committing apoptosis by lowering the activity of miR-34a through methylation (Zhang et al., 2020b).
In addition, epigenetics and the genetics of alleles linked with an increased risk of OA have direct and functional interactions. Methylation quantitative trait loci, or mQTLs, are genetic polymorphisms that correspond with levels of methylation at a CpG site. There is also a possibility that up to 25% of OA at-risk loci could be mQTLs34. Kehayova et al. (2021) built functional analyzes of OA risk loci to demonstrate causal relationships between single genetic risk on nearby genes, DNA methylation, and the expression of target genes. Research utilizing cartilage samples from patients and a chondrocyte cell culture demonstrate that the OA-affected allele rs11583641 is associated with an increase in collagen beta (1-O) galactosyltransferase 2 (COLGALT2) expression. In particular, the researchers found a correlation between rs11583641 and a closed methylation site of a COLGALT2 enhancer in OA the cartilage of patients. In addition, other researchers found that the risk mutation rs75621460 impacts transforming growth factor-beta 1 (TGFB1) expression in distinct tissues. Genotyping articular cartilage samples from 319 individuals with OA revealed that the risk allele “A” of rs75621460 is related to higher methane levels at surrounding methylation sites. These locations were linked to TGFB1 expression and took on opposing effects in OA patients’ synovium and cartilage (Rice et al., 2021).
To investigate the role of enhancers in OA, 16,816 differentially methylated CpGs were identified, of which approximately half (8,111) were from enhancers, forecasting major DNA methylation changes in the enhancer region for both types of OA (Lin et al., 2020). The majority of the differentially methylated CpG sites in the subchondral bone underlying eroded cartilage were hypomethylated, according to a study that investigated the DNA methylation patterns of patients with hip OA. The researchers discovered 7,316 differentially methylated CpG sites in the subchondral bone underneath deteriorated cartilage, the vast majority of which (75%) were hypomethylated, and 1,397 sites in covering damaged cartilage, 126 of which were shared. Some original differentially methylated genes involved in the pathogenesis of OA were found, such as hypomethylated genes argonaute RISC catalytic component 2, transforming growth factor beta 1, nuclear factor of activated T cells 1, insulin-like growth factor 1 receptor and hypermethylated genes NR2F1 antisense RNA 1, homeobox A7, CD6 molecule, etc. (Jeffries et al., 2016). It seems that the ALDH1A2 locus raises the risk of hand OA via lower ALDH1A2 expression in the joint tissues; the impact relies on rs12915901. These results point to a mechanism that might now potentially be addressed to control the risk of OA (Shepherd et al., 2018). These studies uncovered new mQTLs and shed insight into the complex relationship between gene expression in cartilage, the genetic predisposition to OA, and DNA methylation.
The accumulation of inflammatory cells and the expression of inflammatory mediators in synovial tissue are also crucial factors in the pathogenesis of OA. Activated fibroblast-like synoviocytes (FLS) in the OA synovium produce, among other things, cytokines, growth factors, MMPs, and TIMPs to drive catabolic pathways in chondrocytes and contribute to macrophage activation (Haubruck et al., 2021).
Yang et al. (2017) isolated IL-6 overexpressing FLS from OA patients to examine epigenetic alterations in the IL-6 promoter region. OA patients had a pattern of hypomethylation and histone hyperacetylation, as well as decreased binding of methyl CpG binding protein 2, histone deacetylase 1, and DNMT3a, suggesting the role of epigenetic alterations in the IL-6 promoter at the synovial level in the pathogenesis of OA. Recently, small non-coding RNAs, for example, have been found in FLS, while DNA hypermethylation can silence the transcription of non-coding RNAs. Studies revealed that hypermethylation of miR-140 and miR-146a regulation region-specific CpG sites is associated with down-regulation of miR-140-5p and miR-146a in FLS and OA chondrocyte, separately. In addition, hypomethylation of the miR-146a gene leads to a decrease in the binding affinity of the nuclear factor-κB transcription factor, confirming the inflammatory role of miR-146a in OA, which is also a current strategy for treating OA (Papathanasiou et al., 2019). In another study (Zhang et al., 2020a), analysis of chondrocyte proliferation revealed that overexpression of the miR-130a gene, a promoter of chondrocyte proliferation, resulted in increased proliferation rates in chondrocytes from patients with OA. PLEC CpGs were analyzed for mQTLs in cartilage, fat pads, synovium, and peripheral blood of patients with OA, and imbalance in allelic expression in synovium was observed in the same direction as the cartilage, and methylation and PLEC expression were correlated. However, depletion of PLEC and Plec affects a wide range of pathways reported to play critical roles in cartilage biology and OA (Sorial et al., 2020).
With all things above, a series of analyses were performed to further identify DNA methylation sites among cartilage, synovium, and blood, which, as expected, had distinct methylation patterns. These differential DNA methylation genes from different tissues might be involved in the development and progression of OA, providing potential therapeutic targets for OA. Interestingly, some identical methylated genes, such as RWDD2B (Parker et al., 2021) and PLEC (Sorial et al., 2020), were found.
DNA Methylation-mediated epigenetic modules are promising targets for osteoarthritis
Epigenetic modification is dynamic, and its reversibility facilitates the development of new drugs. Smeriglio et al. (2020) revealed that cytosine hydroxymethylation regulated the activation of Wnt signaling, metalloproteinases, and mTOR through TET1. Inhibition of TET1 activity lowers the production of matrix metalloproteinases MMP3 and MMP13, as well as several inflammatory factors, which have anti-arthritic effects. By targeting myeloid cell leukemia-1 and modulating the downstream phosphoinositide-3-kinase/protein kinase B pathway, miR-34a silencing by DNMT3B may substantially decrease chondrocyte ECM breakdown and inflammatory response in OA (Xiong et al., 2021). This study demonstrated that miR-34a knockdown may result in the development of a new OA therapy (Xiong et al., 2021). The current investigation revealed that cryptotanshinone might downregulate the expression of miR-574-5p by altering its methylation, hence enhancing YY1-associated factor 2 (YAF2) expression and influencing chondrocyte apoptosis (Yue et al., 2021b). A study by Wang et al. (2018) found that Chinese medicine compound Qi-Fang-Xi-Bi-Granules (QFXBGs) promoted methylation of CCAAT enhancer-binding protein alpha 2 (C/ebpα-2) CpG island at ten bases from the promoter, which facilitates cartilage growth and regulates C/ebp expression while reducing chondrocyte apoptosis, restoring cartilage and subchondral bone.
In short, medicines that ameliorate abnormal methylation and target methylation are expected to be used in OA, and genes involved in the epigenetic regulation of OA can be used as new targets.
Current challenges and opportunities in osteoarthritis epigenetics research
OA possesses a unique methylation profile, and DNA methylation profiling has the potential to be a beneficial diagnostic tool, allowing not only the identification of OA but also the identification of potential therapeutic targets for some of the several differential methylation pathways involved in OA-specific pathways. Methylation alterations have been implicated in the development, diagnosis, therapy, and prognosis of OA, according to epigenetic research. Differential methylation genes discovered may be used as biomarkers to predict OA development and disease severity, as well as possible treatment targets for OA. As a drug target, epigenetic modification can provide a new pharmacological direction for the development of new drugs, which supplies a direction for the precise treatment of OA. While DNA methylation studies have led to significant advances in OA, some serious questions remain in current research. More studies are needed to detect the function and targets of differential methylation and to shed light on the pathogenesis and drug discovery of OA.
Acknowledgement: We would like to thank the editor and the reviewers for their useful feedback that improved this paper.
Funding Statement: This work was supported by the Anhui Famous Traditional Chinese Medicine Liu Jian Studio Construction Project (Traditional Chinese Medicine Development Secret [2018] No. 11), the Ministry of Science and Technology National Key Research and Development Program Chinese Medicine Modernization Research Key Project (No. 2018YFC1705204), Anhui Province Traditional Chinese Medicine Leading Talent Project (Traditional Chinese Medicine Development Secret [2018] No. 23), and the Anhui Key Research and Development Program Foreign Science and Technology Cooperation Project (No. 201904b11020011).
Author Contributions: The authors confirm contribution to the paper as follows:study conception and design: QIAO ZHOU and JIAN LIU; data collection: QIAO ZHOU, LING XIN, YANYAN FANG, LEI WAN, DAN HUANG, JINCHEN GUO, JIANTING WEN; draft manuscript preparation: QIAO ZHOU and JIAN LIU. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The datasets generated during the current study are available from the corresponding author on reasonable request.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
Alvarez-Garcia O, Fisch KM, Wineinger NE, Akagi R, Saito M, Sasho T, Su AI, Lotz MK (2016). Increased DNA methylation and reduced expression of transcription factors in human osteoarthritis cartilage: Differential DNA methylation in OA cartilage. Arthritis & Rheumatology 68: 1876–1886. https://doi.org/10.1002/art.39643 [Google Scholar] [PubMed] [CrossRef]
Ball HC, Alejo AL, Samson TK, Alejo AM, Safadi FF (2022). Epigenetic regulation of chondrocytes and subchondral bone in osteoarthritis. Life 12: 582. https://doi.org/10.3390/life12040582 [Google Scholar] [PubMed] [CrossRef]
Boer CG, Hatzikotoulas K, Southam L, Stefánsdóttir L, Zhang Y et al. (2021). Deciphering osteoarthritis genetics across 826,690 individuals from 9 populations. Cell 184: 4784–4818.e17. https://doi.org/10.1016/j.cell.2021.07.038 [Google Scholar] [PubMed] [CrossRef]
Brumwell A, Aubourg G, Hussain J, Parker E, Deehan DJ, Rice SJ, Loughlin J (2022). Identification of TMEM129, encoding a ubiquitin-protein ligase, as an effector gene of osteoarthritis genetic risk. Arthritis Research & Therapy 24: 189. https://doi.org/10.1186/s13075-022-02882-y [Google Scholar] [PubMed] [CrossRef]
Chen H, Yang S, Shao R (2019). Long non-coding XIST raises methylation of TIMP-3 promoter to regulate collagen degradation in osteoarthritic chondrocytes after tibial plateau fracture. Arthritis Research & Therapy 21: 271. https://doi.org/10.1186/s13075-019-2033-5 [Google Scholar] [PubMed] [CrossRef]
de Andrés MC, Imagawa K, Hashimoto K, Gonzalez A, Roach HI, Goldring MB, Oreffo ROC (2013). Loss of methylation in CpG sites in the NF-κB enhancer elements of inducible nitric oxide synthase is responsible for gene induction in human articular chondrocytes. Arthritis & Rheumatism 65: 732–742. https://doi.org/10.1002/art.37806 [Google Scholar] [PubMed] [CrossRef]
den Hollander W, Boer CG, Hart DJ, Yau MS, Ramos YFM et al. (2017). Genome-wide association and functional studies identify a role for matrix Gla protein in osteoarthritis of the hand. Annals of the Rheumatic Diseases 76: 2046–2053. https://doi.org/10.1136/annrheumdis-2017-211214 [Google Scholar] [PubMed] [CrossRef]
Dunn M, Nevitt C, MC, Lynch JA, Jeffries MA (2019). A pilot study of peripheral blood DNA methylation models as predictors of knee osteoarthritis radiographic progression: Data from the Osteoarthritis Initiative (OAI). Scientific Reports 9: 16880. https://doi.org/10.1038/s41598-019-53298-9 [Google Scholar] [PubMed] [CrossRef]
Ebrahimi P, Luthman H, McGuigan FE, Akesson KE (2021). Epigenome-wide cross-tissue correlation of human bone and blood DNA methylation—can blood be used as a surrogate for bone? Epigenetics 16: 92–105. https://doi.org/10.1080/15592294.2020.1788325 [Google Scholar] [PubMed] [CrossRef]
Fan Y, Gao D, Zhang Y, Zhu J, Zhang F, Wang L, Wen Y, Guo X, Sun S (2021). Genome-wide differentially methylated region analysis to reveal epigenetic differences of articular cartilage in kashin-beck disease and osteoarthritis. Frontiers in Cell and Developmental Biology 9: 636291. https://doi.org/10.3389/fcell.2021.636291 [Google Scholar] [PubMed] [CrossRef]
Gari MA, AlKaff M, Alsehli HS, Dallol A, Gari A et al. (2016). Identification of novel genetic variations affecting osteoarthritis patients. BMC Medical Genetics 17: 68. https://doi.org/10.1186/s12881-016-0330-2 [Google Scholar] [PubMed] [CrossRef]
Greenberg MVC, Bourc′his D (2019). The diverse roles of DNA methylation in mammalian development and disease. Nature Reviews Molecular Cell Biology 20: 590–607. https://doi.org/10.1038/s41580-019-0159-6 [Google Scholar] [PubMed] [CrossRef]
Ham S, Bae JB, Lee S, Kim BJ, Han BG, Kwok SK, Roh TY (2019). Epigenetic analysis in rheumatoid arthritis synoviocytes. Experimental & Molecular Medicine 51: 1–13. https://doi.org/10.1038/s12276-019-0215-5 [Google Scholar] [PubMed] [CrossRef]
Hashimoto K, Otero M, Imagawa K, de Andrés M C, Coico J M, Roach H I, Oreffo R O C, Marcu K B, Goldring M B (2013). Regulated transcription of human Matrix Metalloproteinase 13 (MMP13) and Interleukin-1β (IL1B) genes in chondrocytes depends on methylation of specific proximal promoter CpG sites. Journal of Biological Chemistry 288: 10061–10072. https://doi.org/10.1074/jbc.M112.421156 [Google Scholar] [PubMed] [CrossRef]
Haubruck P, Pinto MM, Moradi B, Little CB, Gentek R (2021). Monocytes, macrophages, and their potential niches in synovial joints—therapeutic targets in post-traumatic osteoarthritis? Frontiers in Immunology 12: 763702. https://doi.org/10.3389/fimmu.2021.763702 [Google Scholar] [PubMed] [CrossRef]
Imagawa K, de Andrés MC, Hashimoto K, Itoi E, Otero M, Roach HI, Goldring MB, Oreffo ROC (2014). Association of reduced type IX collagen gene expression in human osteoarthritic chondrocytes with epigenetic silencing by DNA hypermethylation: Collagen expression and DNA methylation in OA chondrocytes. Arthritis & Rheumatology 66: 3040–3051. https://doi.org/10.1002/art.38774 [Google Scholar] [PubMed] [CrossRef]
Jeffries MA, Donica M, Baker LW, Stevenson ME, Annan AC, Beth Humphrey M, James JA, Sawalha AH (2016). Genome-wide DNA methylation study identifies significant epigenomic changes in osteoarthritic subchondral bone and similarity to overlying cartilage: Genome-wide methylation in OA subchondral bone. Arthritis & Rheumatology 68: 1403–1414. https://doi.org/10.1002/art.39555 [Google Scholar] [PubMed] [CrossRef]
Jiang S, Guo Y (2020). Epigenetic clock: DNA methylation in aging. Stem Cells International 2020: 1–9. https://doi.org/10.1155/2020/1047896 [Google Scholar] [PubMed] [CrossRef]
Kang X, Liu Y, Gong Y, Huang L, Liu H et al. (2022). The status of selenium and zinc in the urine of children from endemic areas of kashin-beck disease over three consecutive years. Frontiers in Nutrition 9: 862639. https://doi.org/10.3389/fnut.2022.862639 [Google Scholar] [PubMed] [CrossRef]
Kehayova YS, Watson E, Wilkinson JM, Loughlin J, Rice SJ (2021). Genetic and epigenetic interplay within a COLGALT2 enhancer associated with osteoarthritis. Arthritis & Rheumatology 73: 1856–1865. https://doi.org/10.1002/art.4173 [Google Scholar] [PubMed] [CrossRef]
Kim KI, Park YS, Im GI (2013). Changes in the epigenetic status of the SOX-9 promoter in human osteoarthritic cartilage. Journal of Bone and Mineral Research 28: 1050–1060.https://doi.org/10.1002/jbmr.1843 [Google Scholar] [PubMed] [CrossRef]
Koike M, Nojiri H, Kanazawa H, Yamaguchi H, Miyagawa K, Nagura N, Banno S, Iwase Y, Kurosawa H, Kaneko K (2018). Superoxide dismutase activity is significantly lower in end-stage osteoarthritic cartilage than non-osteoarthritic cartilage. PLoS One 13: e0203944. https://doi.org/10.1371/journal.pone.0203944 [Google Scholar] [PubMed] [CrossRef]
Kweon SM, Chen Y, Moon E, Kvederaviciutė K, Klimasauskas S, Feldman DE (2019). An adversarial DNA N6-methyladenine-sensor network preserves polycomb silencing. Molecular Cell 74: 1138–1147.e6. https://doi.org/10.1016/j.molcel.2019.03.018 [Google Scholar] [PubMed] [CrossRef]
Li H, Wang D, Yuan Y, Min J (2017). New insights on the MMP-13 regulatory network in the pathogenesis of early osteoarthritis. Arthritis Research & Therapy 19: 248. https://doi.org/10.1186/s13075-017-1454-2 [Google Scholar] [PubMed] [CrossRef]
Lin X, Li L, Liu X, Tian J, Zheng W, Li J, Wang L (2020). Genome-wide analysis of aberrant methylation of enhancer DNA in human osteoarthritis. BMC Medical Genomics 13: 1. https://doi.org/10.1186/s12920-019-0646-9 [Google Scholar] [PubMed] [CrossRef]
Mazzone R, Zwergel C, Artico M, Taurone S, Ralli M, Greco A, Mai A (2019). The emerging role of epigenetics in human autoimmune disorders. Clinical Epigenetics 11: 34. https://doi.org/10.1186/s13148-019-0632-2 [Google Scholar] [PubMed] [CrossRef]
Ming X, Zhang Z, Zou Z, Lv C, Dong Q, He Q, Yi Y, Li Y, Wang H, Zhu B (2020). Kinetics and mechanisms of mitotic inheritance of DNA methylation and their roles in aging-associated methylome deterioration. Cell Research 30: 980–996. https://doi.org/10.1038/s41422-020-0359-9 [Google Scholar] [PubMed] [CrossRef]
Molnar V, Matišić V, Kodvanj I, Bjelica R, Jeleč Ž et al. (2021). Cytokines and chemokines involved in osteoarthritis pathogenesis. International Journal of Molecular Sciences 22: 9208. https://doi.org/10.3390/ijms22179208 [Google Scholar] [PubMed] [CrossRef]
Papathanasiou I, Trachana V, Mourmoura E, Tsezou A (2019). DNA methylation regulates miR-140-5p and miR-146a expression in osteoarthritis. Life Sciences 228: 274–284. https://doi.org/10.1016/j.lfs.2019.05.018 [Google Scholar] [PubMed] [CrossRef]
Parker E, Hofer IMJ, Rice SJ, Earl L, Anjum SA, Deehan DJ, Loughlin J (2021). Multi-tissue epigenetic and gene expression analysis combined with epigenome modulation identifies RWDD2B as a target of osteoarthritis susceptibility. Arthritis & Rheumatology 73: 100–109. https://doi.org/10.1002/art.41473 [Google Scholar] [PubMed] [CrossRef]
Primorac D, Molnar V, Rod E, Jeleč Ž., Čukelj F, Matišić V, Vrdoljak T, Hudetz D, Hajsok H, Borić I (2020). Knee osteoarthritis: A review of pathogenesis and state-of-the-art non-operative therapeutic considerations. Genes 11: 854. https://doi.org/10.3390/genes11080854 [Google Scholar] [PubMed] [CrossRef]
Ratnakumar A, Weinhold N, Mar JC, Riaz N (2020). Protein-protein interactions uncover candidate ‘core genes’ within omnigenic disease networks. PLoS Genetics 16: e1008903. 10.1371/journal.pgen.1008903. [Google Scholar] [PubMed] [CrossRef]
Rice SJ, Roberts JB, Tselepi M, Brumwell A, Falk J, Steven C, Loughlin J (2021). Genetic and epigenetic fine-tuning of TGFB1 expression within the human osteoarthritic joint. Arthritis & Rheumatology 73: 1866–1877. https://doi.org/10.1002/art.41736 [Google Scholar] [PubMed] [CrossRef]
Rice SJ, Tselepi M, Sorial AK, Aubourg G, Shepherd C et al. (2019). Prioritization of PLEC and GRINA as osteoarthritis risk genes through the identification and characterization of novel methylation quantitative trait loci. Arthritis & Rheumatology 71: 1285–1296. https://doi.org/10.1002/art.40849 [Google Scholar] [PubMed] [CrossRef]
Salameh Y, Bejaoui Y, El Hajj N (2020). DNA methylation biomarkers in aging and age-related diseases. Frontiers in Genetics 11: 171. https://doi.org/10.3389/fgene.2020.00171 [Google Scholar] [PubMed] [CrossRef]
Shen J, Wang C, Li D, Xu T, Myers J, Ashton JM, Wang T, Zuscik MJ, McAlinden A, O’Keefe RJ (2017). DNA methyltransferase 3b regulates articular cartilage homeostasis by altering metabolism. JCI Insight 2: e93612. https://doi.org/10.1172/jci.insight.93612 [Google Scholar] [PubMed] [CrossRef]
Shepherd C, Reese AE, Reynard LN, Loughlin J (2019). Expression analysis of the osteoarthritis genetic susceptibility mapping to the matrix Gla protein gene MGP. Arthritis Research & Therapy 21: 149. https://doi.org/10.1186/s13075-019-1934-7 [Google Scholar] [PubMed] [CrossRef]
Shepherd C, Zhu D, Skelton AJ, Combe J, Threadgold H, Zhu L, Vincent TL, Stuart P, Reynard LN, Loughlin J (2018). Functional characterization of the osteoarthritis genetic risk residing at ALDH1A2 identifies rs12915901 as a key target variant. Arthritis & Rheumatology 70: 1577–1587. https://doi.org/10.1002/art.40545 [Google Scholar] [PubMed] [CrossRef]
Singh P, Wang M, Mukherjee P, Lessard SG, Pannellini T, Carballo CB, Rodeo SA, Goldring MB, Otero M (2021). Transcriptomic and epigenomic analyses uncovered Lrrc15 as a contributing factor to cartilage damage in osteoarthritis. Scientific Reports 11: 21107. https://doi.org/10.1038/s41598-021-00269-8 [Google Scholar] [PubMed] [CrossRef]
Skvortsova K, Zotenko E, Luu PL, Gould CM, Nair SS, Clark SJ, Stirzaker C (2017). Comprehensive evaluation of genome-wide 5-hydroxymethylcytosine profiling approaches in human DNA. Epigenetics & Chromatin 10: 16. https://doi.org/10.1186/s13072-017-0123-7 [Google Scholar] [PubMed] [CrossRef]
Smeriglio P, Grandi FC, Davala S, Masarapu V, Indelli PF, Goodman SB, Bhutani N (2020). Inhibition of TET1 prevents the development of osteoarthritis and reveals the 5hmC landscape that orchestrates pathogenesis. Science Translational Medicine 12: eaax2332. https://doi.org/10.1126/scitranslmed.aax2332 [Google Scholar] [PubMed] [CrossRef]
Sorial AK, Hofer IM, Tselepi M, Parker E, Deehan DJ, Rice SJ, Loughlin J (2020). Multi-tissue epigenetic analysis of the osteoarthritis susceptibility locus mapping to the plectin gene PLEC. Osteoarthritis and Cartilage 28: S338–S339. https://doi.org/10.1016/j.joca.2020.02.526 [Google Scholar] [CrossRef]
Steinberg J, Brooks RA, Southam L, Bhatnagar S, Roumeliotis TI et al. (2018). Widespread epigenomic, transcriptomic and proteomic differences between hip osteophytic and articular chondrocytes in osteoarthritis. Rheumatology 57: 1481–1489. https://doi.org/10.1093/rheumatology/key101 [Google Scholar] [PubMed] [CrossRef]
Steinberg J, Southam L, Roumeliotis TI, Clark MJ, Jayasuriya RL et al. (2021). A molecular quantitative trait locus map for osteoarthritis. Nature Communications 12: 1309. https://doi.org/10.1038/s41467-021-21593-7 [Google Scholar] [PubMed] [CrossRef]
Styrkarsdottir U, Helgason H, Sigurdsson A, Norddahl GL, Agustsdottir AB et al. (2017). Whole-genome sequencing identifies rare genotypes in COMP and CHADL associated with high risk of hip osteoarthritis. Nature Genetics 49: 801–805. https://doi.org/10.1038/ng.3816 [Google Scholar] [PubMed] [CrossRef]
Styrkarsdottir U, Lund SH, Thorleifsson G, Zink F, Stefansson OA et al. (2018). Meta-analysis of Icelandic and UK data sets identifies missense variants in SMO, IL11, COL11A1 and 13 more new loci associated with osteoarthritis. Nature Genetics 50: 1681–1687. https://doi.org/10.1038/s41588-018-0247-0 [Google Scholar] [PubMed] [CrossRef]
Sun X, Xiao L, Chen J, Chen X, Chen X, Yao S, Li H, Zhao G, Ma J (2020). DNA methylation is involved in the pathogenesis of osteoarthritis by regulating CtBP expression and CtBP-mediated signaling. International Journal of Biological Sciences 16: 994–1009. https://doi.org/10.7150/ijbs.39945 [Google Scholar] [PubMed] [CrossRef]
Tachmazidou I, Hatzikotoulas K, Southam L, Esparza-Gordillo J, Haberland V et al. (2019). Identification of new therapeutic targets for osteoarthritis through genome-wide analyses of UK Biobank data. Nature Genetics 51: 230–236. https://doi.org/10.1038/s41588-018-0327-11 [Google Scholar] [PubMed] [CrossRef]
Taylor SEB, Smeriglio P, Dhulipala L, Rath M, Bhutani N (2014). A global increase in 5-hydroxymethylcytosine levels marks osteoarthritic chondrocytes: Dysregulation of 5 hmC homeostasis in OA. Arthritis & Rheumatology 66: 90–100. https://doi.org/10.1002/(ISSN)2326-5205 [Google Scholar] [CrossRef]
Teerawattanapong N, Udomsinprasert W, Ngarmukos S, Tanavalee A, Honsawek S (2019). Blood leukocyte LINE-1 hypomethylation and oxidative stress in knee osteoarthritis. Heliyon 5: e01774. https://doi.org/10.1016/j.heliyon.2019.e01774 [Google Scholar] [PubMed] [CrossRef]
Venkateswaran S, Somineni HK, Kilaru V, Katrinli S, Prince J et al. (2022). Methylation quantitative trait loci are largely consistent across disease states in Crohn’s disease. G3 Genes|Genomes|Genetics 12: jkac041. https://doi.org/10.1093/g3journal/jkac041 [Google Scholar] [PubMed] [CrossRef]
Wang X, Shen P, Tang D, Xu H, Qiu H, Wu T, Gao X (2018). Effects of Qi-Fang-Xi-Bi-Granules on cartilage morphology and C/ebp α promoter methylation in rats with knee osteoarthritis. Evidence-Based Complementary and Alternative Medicine 2018: 1–12. https://doi.org/10.1155/2018/2074976 [Google Scholar] [PubMed] [CrossRef]
Wilkinson JM, Zeggini E (2021). The genetic epidemiology of joint shape and the development of osteoarthritis. Calcified Tissue International 109: 257–276. https://doi.org/10.1007/s00223-020-00702-6 [Google Scholar] [PubMed] [CrossRef]
Wu Z, Shou L, Wang J, Huang T, Xu X (2020). The methylation pattern for knee and hip osteoarthritis. Frontiers in Cell and Developmental Biology 8: 602024. https://doi.org/10.3389/fcell.2020.602024 [Google Scholar] [PubMed] [CrossRef]
Xiong S, Zhao Y, Xu T (2021). DNA methyltransferase 3 beta mediates the methylation of the microRNA-34a promoter and enhances chondrocyte viability in osteoarthritis. Bioengineered 12: 11138–11155. https://doi.org/10.1080/21655979.2021.2005308 [Google Scholar] [PubMed] [CrossRef]
Yang F, Zhou S, Wang C, Huang Y, Li H, Wang Y, Zhu Z, Tang J, Yan M (2017). Epigenetic modifications of interleukin-6 in synovial fibroblasts from osteoarthritis patients. Scientific Reports 7: 43592. https://doi.org/10.1038/srep43592 [Google Scholar] [PubMed] [CrossRef]
Yi P, Xu X, Yao J, Qiu B (2021). Effect of DNA methylation on gene transcription is associated with the distribution of methylation sites across the genome in osteoarthritis. Experimental and Therapeutic Medicine 22: 719. https://doi.org/10.3892/etm.2021.10151 [Google Scholar] [PubMed] [CrossRef]
Yue T, Chen S, Zhu J, Guo S, Huang Z, Wang P, Zuo S, Liu Y (2021a). The aging-related risk signature in colorectal cancer. Sedentary Life and Nutrition 13: 7330–7349. https://doi.org/10.18632/aging.202589 [Google Scholar] [PubMed] [CrossRef]
Yue S, Su X, Teng J, Wang J, Guo M (2021b). Cryptotanshinone interferes with chondrocyte apoptosis in osteoarthritis by inhibiting the expression of miR‐574‐5p. Molecular Medicine Reports 23: 424. https://doi.org/10.3892/mmr.2021.12063 [Google Scholar] [PubMed] [CrossRef]
Zengini E, Hatzikotoulas K, Tachmazidou I, Steinberg J, Hartwig FP et al. (2018). Genome-wide analyses using UK Biobank data provide insights into the genetic architecture of osteoarthritis. Nature Genetics 50: 549–558. https://doi.org/10.1038/s41588-018-0079-y [Google Scholar] [PubMed] [CrossRef]
Zhang R, Guo H, Yang X, Zhang D, Zhang D, Li Q, Wang C, Yang X, Xiong Y (2021a). Patients with Osteoarthritis and kashin-beck disease display distinct CpG methylation profiles in the DIO2, GPX3, and TXRND1 promoter regions. CARTILAGE 13: 797S–808S. https://doi.org/10.1177/1947603520988165 [Google Scholar] [PubMed] [CrossRef]
Zhang H, Li J, Shao W, Shen N (2020b). LncRNA SNHG9 is downregulated in osteoarthritis and inhibits chondrocyte apoptosis by downregulating miR-34a through methylation. BMC Musculoskeletal Disorders 21: 511. https://doi.org/10.1186/s12891-020-03497-7 [Google Scholar] [PubMed] [CrossRef]
Zhang H, Li J, Shao W, Shen N (2020a). LncRNA CTBP1-AS2 is upregulated in osteoarthritis and increases the methylation of miR-130a gene to inhibit chondrocyte proliferation. Clinical Rheumatology 39: 3473–3478. https://doi.org/10.1007/s10067-020-05113-4 [Google Scholar] [PubMed] [CrossRef]
Zhang Q, Ouyang Z, Song X, Zhu W, Tang X, Liu Z, Chen X (2021b). Epigenetic modifications of tumor necrosis factor-alpha in joint cartilage tissue from osteoarthritis patients—CONSORT. Medicine 100: e27868. https://doi.org/10.1097/MD.0000000000027868 [Google Scholar] [PubMed] [CrossRef]
Zhao L, Wang Q, Zhang C, Huang C (2017). Genome-wide DNA methylation analysis of articular chondrocytes identifies TRAF1, CTGF, and CX3CL1 genes as hypomethylated in osteoarthritis. Clinical Rheumatology 36: 2335–2342. https://doi.org/10.1007/s10067-017-3667-9 [Google Scholar] [PubMed] [CrossRef]
Zhou Y, Chen M, O’Keefe RJ, Shen J, Li Z, Zhou J, Zhou X, Mao JJ (2019). Epigenetic and therapeutic implications of dnmt3b in temporomandibular joint osteoarthritis. American Journal of Translational Research 11: 1736–1747. [Google Scholar] [PubMed]
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools