
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Biochemical association between the prevalence of genetic polymorphism and myocardial infarction
1 Department of Pharmaceutical Chemistry, Government College University, Faisalabad, 38000, Pakistan
2 Department of Pharmacy, The Women University, Multan, 60000, Pakistan
3 Research Center for Advanced Materials Science (RCAMS), King Khalid University, Abha, 62413, Saudi Arabia
4 Department of Chemistry, Faculty of Science, King Khalid University, Abha, 62413, Saudi Arabia
* Corresponding Author: MUHAMMAD SAJID HAMID AKASH. Email:
BIOCELL 2023, 47(3), 473-484. https://doi.org/10.32604/biocell.2023.025930
Received 05 August 2022; Accepted 24 October 2022; Issue published 03 January 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Genetic polymorphism has a vital role in the pathogenesis and development of myocardial infarction (MI). Single nucleotide polymorphism at any one of the amino acid sequences can result in a diseased state. A single gene can exhibit genetic polymorphism at more than one position giving rise to different variants. Genetic polymorphism of angiotensinogen (AGT) M235T, AGT T174M, and angiotensin-1-converting enzyme (ACE) I/D, endothelial nitric oxide synthase (eNOS), and methylenetetrahydrofolate reductase (MTHFR) can be a risk factor for MI. However, it is important to study the prevalence of genetic polymorphisms of these genes among different populations. MI is influenced by genetic polymorphism of various genes, including AGT, ACE, eNOS, MTHFR, etc. However, the association of genetic polymorphism of these genes varies among different populations, but different ethnic groups could show contradictory results. These genes have shown a positive association with risks of MI in some populations, whereas the results have not been consistent with every ethnic group. In this article, we have summarized the genetic variations in the aforementioned genes and their association with MI.Keywords
The human genome shows dynamic diversity across individuals as well as between various regional, racial, and/or ethnic communities. However, only 0.1%–0.4% of the total genomic DNA or genome accounts for this genetic diversity (Karki et al., 2015). Based on the analysis of high-quality individual haplotypes from 26 different populations throughout the world, including Europe, Africa, America, East Asia, and South Asia, about 1000 Genomes Project Consortium identified more than 88 million genetic variations, including 60,000 structural variants, 3.6 million short insertions/deletions, and 84.7 million single nucleotide polymorphisms (SNPs) among these genetic differences (Aguet and Ma, 2017).
Genetic polymorphism is the most prevalent and dynamic type of genetic variation found in the human genome. It is defined as the occurrence of two or more alternative allele forms in any individual’s genome that produce different phenotypes in the same population (Cooke et al., 2012). SNPs, repeated patterns of DNA and RNA, presence or absence of specific nucleotide sequences, and exchange of genetic material are various inceptions of genetic variations (Ismail and Essawi, 2012). Mutations are the fundamental processes that give rise to almost all genetic variants. Many scientists agree that mutation is the irreversible sequence variation in DNA, which effectively covers all types of variations occurring in the human genome, whether they are spontaneous or not (McLaren et al., 2016). The interactions of many other factors, including the environment, have a significant impact on genetic variants in the genome (Landrum et al., 2018). Gene mutation occurs at a single nucleotide level, called SNP. This also leads to altered enzymatic activity as a result of changes in amino acid sequences, which leads to changes in transcription, intrinsic termination, and factor-dependent termination. In some cases, SNP or mutations do not affect the activities of the enzymes, and it is important to identify those mutations that affect the activities of the specific gene (Ding and Zhang, 2010).
Cardiovascular diseases (CVDs) are one of the major causes of mortality in the entire world. The cases of mortality due to CVDs are more in developing countries as compared to that in developed countries due to various social, psychological, and biological risk factors. The overall death rate due to CVDs is three times greater in females as compared to that in males. Developing countries like Pakistan, India, and Nepal are at high risk of coronary heart disease (CHD) (Barolia and Sayani, 2017). According to various studies, the populations of Eastern Europe and Central Asia are at a greater risk of CVDs (Thomas et al., 2018). CVDs include coronary artery disease (CAD), myocardial infarction (MI), cardiomyopathy, and congenital heart defects. Various risk factors like smoking, being overweight, raised cholesterol level, poor diet, lack of exercise, diabetes mellitus (DM), socioeconomic status, gender, and demography lead to CVDs (Chauhdary et al., 2021; Cheng et al., 2017; Rehman et al., 2020b). Other risk factors involve lipoprotein-associated phospholipase A2, C-reactive protein, fibrinogen, low-density lipoprotein particle number, lipoprotein a, triglycerides, plasminogen activator inhibitors, and interleukin-6 (Cornelis et al., 2004). MI is also known as a heart attack that occurs due to a decrease or incomplete blood supply to the heart muscle. CAD becomes the basis of MI and leads to death. Due to the blockage of the coronary artery, cardiac muscles receive less oxygen supply which leads to the death of cardiac muscles (Ojha and Dhamoon, 2022). The occurrence of MI events is high in females as compared to that in males (Nabel and Braunwald, 2012). The predisposing factor for MI includes hypertension, hyperlipidemia, DM, smoking, and obesity (Raygan et al., 2016). High blood pressure increases the shear stress of the coronary artery, which increases the production of angiotensinogen and endothelial dysfunction, leading to MI (Murphy et al., 2009). An increase in cholesterol levels also results in an increase in ischemic heart disease and MI (Nabel and Braunwald, 2012). DM is a major risk factor for the development of CAD, with an increased occurrence of MI in diabetic patients than in MI patients without DM (Leon and Maddox, 2015). Insulin resistance, hyperinsulinemia, and vascular calcification promote atherosclerosis and increase the rupturing of plaque which leads to thrombosis that ultimately leads to coronary adverse events (Yuan et al., 2019). Hyperglycemia and insulin resistance are responsible for CVDs in patients with diabetes (Rehman et al., 2020a). Other factors that contribute to MI are diabetes-induced overexpression of reactive oxygen species (ROS), secretion of inflammatory cytokines, and activation of protein kinase C (Shah and Brownlee, 2016). Augmented levels of cholesterol in plasma results in various heart diseases. Elevated plasma cholesterol leads to familial hypercholesterolemia, which results from genetic mutation; patients who are homozygous at a single locus have coronary atherosclerosis and often die in the early stages due to MI (Nabel, 2003). Various environmental factors such as diet, age, exercise, and gender are responsible for genetic mutation with myocardial infarction (Kelly and Semsarian, 2009).
Approximately 3 million populations worldwide are estimated to be affected by MI, with more than 1 million deaths annually in the United State (Nascimento et al., 2019). There are two types of acute MI. One is non-ST-segment elevation MI (NSTEMI) and ST-segment elevation MI (STEMI). Patients with STEMI need immediate treatment as compared to patients with NSTEMI (Camaro and de Boer, 2015). STEMI occurs due to complete blockage of blood vessels and is diagnosed by electrocardiography. NSTEMI occurs due to narrowing of the coronary artery, temporary blockage, and thrombosis and is diagnosed by various cardiac biomarkers (Daga et al., 2011).
Various cardiac biomarkers include cardiac troponin, creatinine kinase, myoglobin, lactate dehydrogenase, fatty acid binding proteins, myosin binding protein C, vascular endothelial growth factor, aspartate aminotransferase, and insulin-like growth factor-1 (Wu et al., 2021). In this review article, we have especially focused on the prevalence and association of genetic polymorphism of angiotensinogen (AGT) M235T, AGT T174M, and angiotensin-1-converting enzyme (ACE) insertion/deletion (I/D), endothelial nitric oxide synthase (eNOS), and methylenetetrahydrofolate reductase (MTHFR) with MI.
M235T polymorphism in the AGT gene
MI is a multifactorial disease affected by genetic mutation and various environmental factors like diet, stress, daily routine, etc. A wide range of risk factors, including smoking, hypertension, and hyperlipidemia, lead to MI (Rehman et al., 2020b). Atherosclerosis and thrombogenesis also contribute to the pathogenesis of MI. Apart from the classical risk factors, genetic mutation has been of interest to investigators. Renin-angiotensin-aldosterone system (RAAS) has genes that can be of great interest to studying the genetic susceptibility in MI. Many studies are being conducted to study the genetic risk factors in RAAS to predict the progression of MI. Several studies have shown the influence of RAAS on the development of atherosclerosis and hypertension. Coronary arteriosclerosis and its associated disorders are also seen in individuals having angiotensin II inhibition (Koh et al., 2010). Moreover, inhibition of the angiotensin-converting enzyme (ACE) also influences the development of these diseases. Both of these components of RAAS are potentially active in the initiation and development of MI. Angiotensin II can cause atherosclerotic changes and can rupture the plaque, which is triggered by different mechanisms such as vasoconstriction, thrombogenesis, antifibrinolysis, and vascular smooth muscles. Production of angiotensin II is controlled by AGT, which can be a limiting factor in the production of angiotensin II (Liang et al., 2013b; Wang and Pan, 2014).
AGT is usually found as a liver protein and interacts with renin for the production of angiotensin I, which is a precursor for angiotensin II. The AGT gene is located at 1q42-43 and consists of five exons (Sivitskaia et al., 2008). AGT is a potential factor in determining the level of angiotensin II in tissues and serum, so any mutation or polymorphism in the AGT gene may lead to thrombogenesis and atherogenesis, eventually developing into MI (Brand et al., 2002).
A homozygous variation in the gene at the molecular level can be related to a higher level of AGT in the plasma, and hence, higher risks of hypertension may occur. The polymorphism might occur when methionine at position 235 is replaced by threonine as shown in Fig. 1. Various studies have also demonstrated the role of M235T polymorphism which increases the risk of CHD (Brand et al., 2002; Ji et al., 2010). Several studies have shown the association of AGT polymorphism with the risks of MI (Xu et al., 2007; Zafarmand et al., 2008). However, some studies completely contradict these findings and do not show the association between M235T polymorphism and MI (Brenner et al., 2005; Mehri et al., 2011; Saidi et al., 2009; Um et al., 2005; Um et al., 2003).
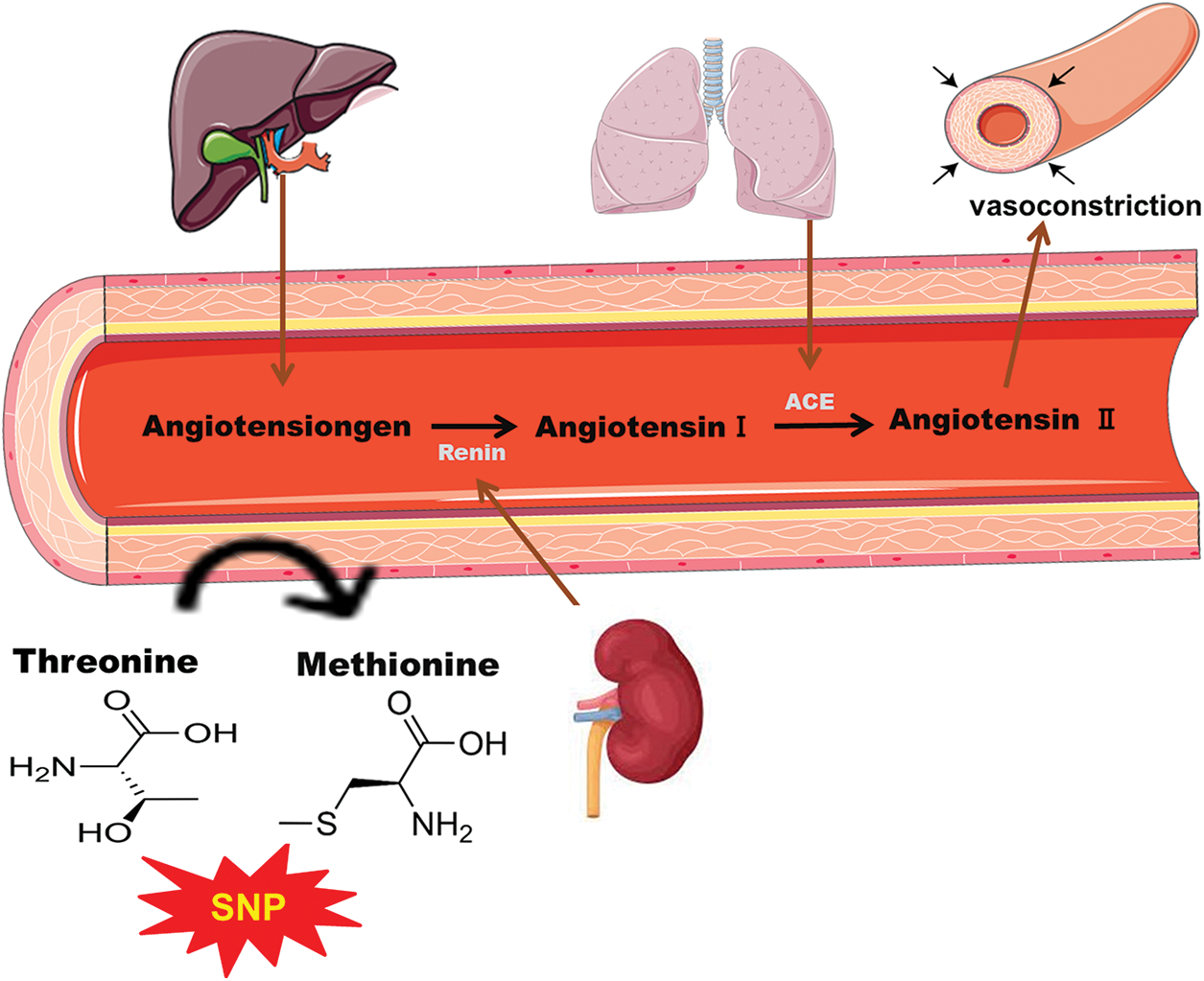
Figure 1: Schematic representation of single nucleotide polymorphism of angiotensinogen (AGT) by the substitution of threonine with methionine leading to vasoconstriction and results in coronary heart diseases.
AGT T174M polymorphism in the AGT gene
Similar to M235T polymorphism, methionine is replaced by threonine at position 174; this polymorphism is more common and is known as T174M (rs699). This genetic polymorphism can alter the functions of AGT, consequently leading to the progression of CVD (Sivitskaia et al., 2008). AGT T174M polymorphism is associated with the risk of developing CAD (Wang, 2013). Various studies conducted among different ethnic groups have shown the association of T174M polymorphism with MI. However, contradicting results are also available regarding this association (Attia et al., 2003).
Apart from AGT, RAAS has another important enzyme that has proved to be significant in regulating blood pressure and plays a vital role in various CVDs. ACE is another popular enzyme responsible for the regulation and maintenance of blood pressure through RAAS. A number of CVDs, such as MI, may arise due to the dysfunction of this enzyme (Liang et al., 2013a). The gene responsible for the encoding of this enzyme in humans is present at chromosome 17q23. The genetic polymorphism involved in this enzyme occurs in the chromosome at intron-16, where the presence or absence of 287 base pair (bp) Alu-type sequence is observed (Cambien et al., 1992). People with a homozygous deletion (DD) genotype are seen to be at higher risk of MI. Interestingly, the individuals who were considered at low risk of MI based on their data of body mass index (BMI) and the plasma concentration of apolipoprotein (Apo) B showed a significant association between MI and genetic polymorphism (Beohar et al., 1995; Schuster et al., 1995).
Various studies have confirmed that individuals with homozygous DD genotype are at the higher risk for MI. This association is even higher in people who were seen to be at low risk. However, some studies have shown contradictory results and do not support this hypothesis. The level of circulating ACE in the blood varies among individuals. This difference is attributed to I/D polymorphism is considered to be responsible for. Individuals with the homozygous DD genotype have higher levels of ACE as compared to those with the homozygous II genotype. However, its functional role is still unclear and is widely under discussion (Liang et al., 2013a).
ACE is considered a dipeptidyl carboxypeptidase. It promotes the activation of angiotensin I to angiotensin II by breaking down the carboxyterminal dipeptide. Angiotensin II is a potent vasoconstrictor. ACE not only activates angiotensin I into angiotensin II, but it also decreases the levels of bradykinin, a vasodilator (Cicoira et al., 2001; Murphey et al., 2000). It also causes the migration and adhesion of macrophages. This reduces the diameter of vessels, which results in atherosclerotic plagues (Larsson et al., 2000). Angiotensin II also has a platelet aggregating effect. It promotes the expression of plasminogen activator-inhibitor 1 and 2, which reduces the fibrinolytic activity in plasma (He et al., 2006). All these mechanisms show the possible role of ACE in MI. The I/D polymorphism in the ACE gene and its association with MI has been under consideration. It has been reported that the difference and variance in the level of ACE are majorly due to the polymorphism in I/D, which can increase the risks of MI (Dai et al., 2016; Keavney et al., 2000).
However, the results are not consistent, and various contradictions have been noted. Thirteen different polymorphisms of the ACE gene were studied among the Nigerian population to investigate the association of these polymorphisms with MI. Among all these polymorphisms, 2350G>A polymorphism showed much significant results. This polymorphism occurred at the 17th exon of chromosome and accounted for about 19% of the total variance in the plasma level of ACE among the population. This polymorphism proved to be of greater significance as compared to I/D polymorphism. The 2350G>A polymorphism was also studied among the Chinese population. Individuals with 2350G>A polymorphism showed a significant association with MI. The individuals with A allele carrier showed a higher risk of MI irrespective of their age, BMI, blood pressure, smoking status, and lipids and apolipoprotein levels in the plasma (Zhu et al., 2001). In another study, the individuals with A allele carrier in the Han Chinese population also had a high risk of MI (Jiang et al., 2013).
The eNOS is an enzyme involved in the synthesis of NO, a relaxing factor. The precursor for this synthesis is L-arginine. About three different isoforms of this enzyme are involved in the production of NO. These three isoforms are inducible nitric oxide synthase (also known as iNOS or NOS2), constitutive neuronal nitric oxide synthase (also known as nNOS or NOS1), and constitutive endothelial nitric oxide synthase (also known as eNOS or NOS3).
The eNOS is involved in the relaxation of vasculature after it moves to vascular smooth muscle cells from the endothelium. It stimulates the guanylate cyclase, which, as a result, increases the concentration of cyclic guanosine monophosphate. ENOS has a vital role in the maintenance and regulation of vascular tone. It acts as a protective agent through several mechanisms, like breaking down the superoxide radicals by reducing leukocyte adhesion and platelet aggregation. It is also involved in the proliferation of smooth muscle cells. All of these mechanisms provide a cumulative atheroprotective and, ultimately, a cardioprotective effect. However, if the synthesis of endothelial-derived NO is disturbed for any reason, it could lead to various abnormalities like thrombosis or even atherosclerosis. Despite different reasons contributing to the disturbance in the synthesis of eNOS, the genetic factor has proven to be of great importance (Dafni et al., 2010; Gluba et al., 2009; Zigra et al., 2013), as shown in Fig. 2.
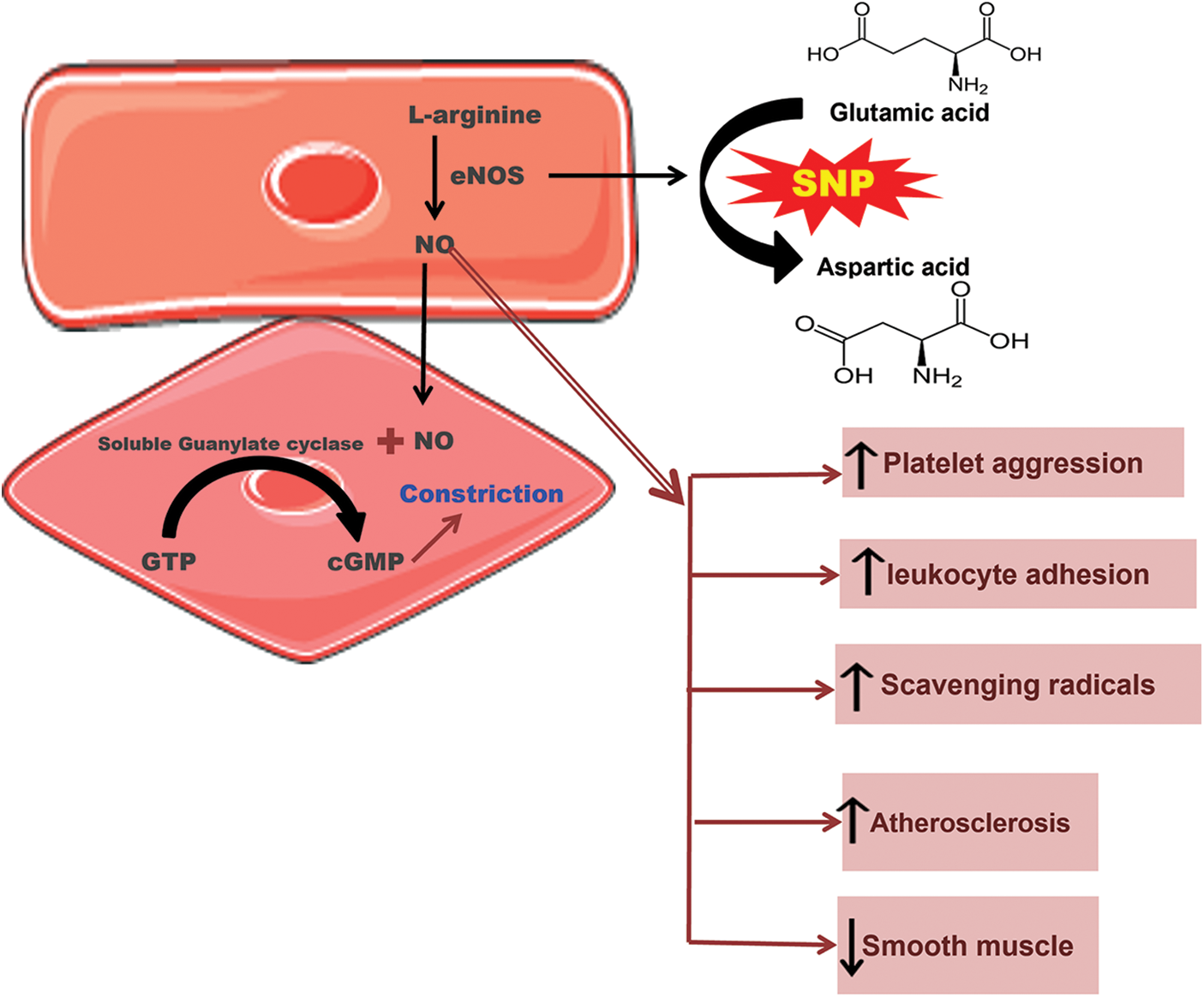
Figure 2: Schematic representation of single nucleotide polymorphism of endothelial nitric oxide synthase (eNOS) where glutamic acid is replaced with aspartic acid, leading to thrombosis and various abnormalities.
The genetic polymorphism in the gene encoding for endothelial-derived NO can seriously affect these cardioprotective effects. The gene for eNOS can be found on the chromosome 7q35-36. It has 26 exons and extends to 21 kb (21000 bp). The most common or probably the only known polymorphism of eNOS is G894T at exon 7 which results in a change in the activity of eNOS and ultimately leading to endothelial dysfunction and various CVDs. This polymorphism is also known as E298D because the amino acid glutamic acid (E) is replaced by aspartic acid (D) and is associated with various CVDs, including MI. However, the results are still contradictory. Positive association has been seen between MI and E298D polymorphism in English, German, and Japanese populations. Nonetheless, a number of studies do not support this association; no such association was found among the Austrian, Korean, Dutch, and French populations (Gardemann et al., 2002; Jo et al., 2006; Morray et al., 2007; Park et al., 2004; Schmoelzer et al., 2003).
Polymorphism in the MTHFR gene
Genetic polymorphism of MTHFR leads to the transformation of the amino acid; alanine to valine at position 226 in proteins which results in MI (Xuan et al., 2011). The gene consists of 11 exons, with the size ranging from 102 to 432 bp, which are found on chromosome 1 (1p36.3). According to the analysis of the entire genomic structure, both alternative initiation and alternative splicing may occur (Dikmen et al., 2006). MTHFR is involved in the reduction of 5, 10 methylenetetrahydrofolates to 5, methyltetrahydrofolate, which delivers a methyl group to homocysteine to produce methionine. Mutation of MTHFR results in an increase in homocysteine due to altered folic acid metabolism, resulting in MI (Shaker and Ismail, 2014). The SNPs in MTHFR are at positions 677 (MTHFR 677C>T), 1298 (MTHFR 1298A>C), 1317 (MTHFR 1317T>C), and 1793 (MTHFR 1793G>A) (Böttiger et al., 2007). The 5,10 MTHFR is an enzyme encoded by the region 1p36.3 of the chromosome. MTHFR reduces 5,10 methylenetetrahydrofolates into 5-methylenetetrahydrofolate which is involved in re-methylation (by acting as a methyl donor) of homocysteine into methionine (Biselli et al., 2010). Increased level of homocysteine has been seen to be associated with various CVDs, including MI. This factor is highly independent of other traditional risk factors for CVDs. Recent studies have provided evidence on how homocysteine leads to various CVDs. It promotes the formation of thrombus and causes platelet aggregation due to endothelial dysfunction or injury (Biselli et al., 2010; Brattström et al., 1998; Xuan et al., 2011).
How a low level of MTHFR can lead to an increase in the level of homocysteine that ultimately leads to various CVDs, can be clearly elucidated. The level of MTHFR can be altered due to various physiological reasons. However, recent developments are being made to analyze the genetic causes for the decreased level of MTHFR. Genetic polymorphism has been widely studied to investigate the alteration in the plasma level of this enzyme. A very common MTHFR polymorphism is the C677T mutation, with cysteine being replaced by thymine at the 677th position in the nucleotide sequence, consequently changing amino acid alanine to valine at the 226th position in the protein formed. This mutation changes the thermostability of the enzyme, which further affects its activity as shown in Fig. 3. This polymorphism reduces the activity of this enzyme by up to 50%, which causes an increase in the level of homocysteine and a decrease in the concentration of folic acid in the plasma. Decreased folic acid also leads to various CVDs, as folic acid improves the function of the endothelium. The polymorphism-producing homozygous genotype showed a higher level of homocysteine in the plasma than in the heterozygous condition. However, the homocysteine level in a heterozygous mutated individual is seen to be still greater than that in the control or non-mutated individuals (Doshi et al., 2001; Kerkeni et al., 2006).
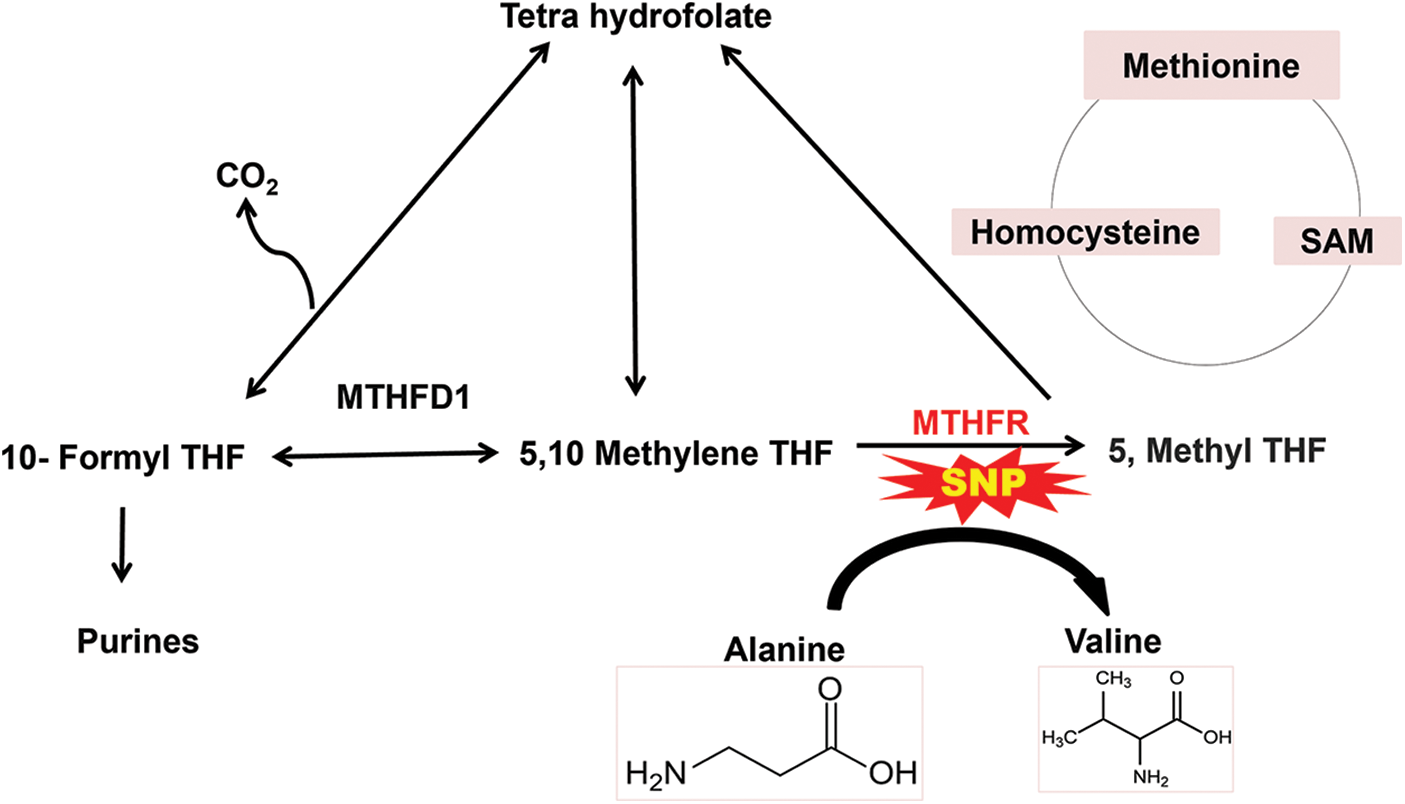
Figure 3: Schematic representation of single nucleotide polymorphism of methylenetetrahydrofolate reductase (MTHFR) where alanine is substituted by valine which results in high level of homocysteine leading to various cardiovascular diseases.
The MTHFR C677T polymorphism varies widely across the globe. The ethnic and demographic factors come into play that are not yet clearly understood. The homozygous genotype was seen to be highest among Italians and Hispanics. The lowest homozygous genotype among Europeans is found among Germans. About 13% homozygous genotype was found among the British population. About 10%–14% of homozygous polymorphism is seen in populations from America, Canada, Australia, and Brazil (Liew and Gupta, 2015).
The epidemiology and prevalence of MTHFR C677T polymorphism vary in various ethnic groups across the globe. The location and environmental factors also contribute to the polymorphism of this gene. The homozygous 677C>T was seen to be the highest among the Italians. This allele frequency was higher in Hispanics but was lower in American Blacks and those from some areas of Africa. This homozygous allele frequency ranges from less than 1% in Africans to about 20% among Americans (Botto and Yang, 2000).
In a study conducted among Europeans, Italians had the highest frequency of the C677T homozygous allele, whereas Germans were at the bottom of this list with the lowest frequency of the homozygous allele (Adams et al., 1996; Bowen et al., 1998; Markus et al., 1997). Among the British, the prevalence of the homozygous allele was about 13%. In different countries with a white population, like Canada, Australia, Brazil, and America, the C677T homozygous population ranged from 10%–14%. The Hispanic white population in California showed about 21% of homozygous allele and the rate was approximately 21% among Colombians. However, limited data is available about the Asian population. Eleven percent of the Japanese population had the homozygous allele, and the sub-Saharan Africans had zero percent (Liew and Gupta, 2015).
In Japan, an interesting study was conducted to relate age with the prevalence of the homozygous C677T allele. About 7% of people above the age of 80 years showed homozygous allele, whereas the percentage increased to 14% among the age group of 55-79 years. The homozygosity increased to 19% among individuals from the 14 to 55 years age group (Matsushita et al., 1997). However, not many elaborate studies have been conducted on the association of different age groups with the homozygous allele frequency among various populations.
Healthcare paradigm for cardiovascular diseases
Nowadays, in modern scientific research, detection and analyses of several gene SNPs are carried out to determine the molecular basics of different diseases. Genetic SNPs act as biological markers, which help to determine the possible risk factors and mechanisms for disease prevalence (Garrigós et al., 2017). With respect to the MI disease progression with genetic variants, the expression of some specific biochemical parameters is altered. Therefore, some strategies would lead to a clinical paradigm shift, moving treatment focus away from blockage and infarction treatment and toward treatment of the underlying illness process. These included drug therapy, thrombolytic therapy, percutaneous coronary interventions, and coronary artery bypass grafting.
Drug therapies include angiotensin receptor blockers (ARBs), angiotensin-converting enzyme inhibitors (ACEI), β-receptor blockers, and aldosterone receptor antagonists (Er et al., 2016; Eyuboglu, 2015; González-Cambeiro et al., 2016).
The main method for treating MI at the moment is thrombolytic therapy. According to numerous clinical investigations, the best curative outcome was reached by thrombolytic therapy administered within 6 h after the onset of MI, and the earlier treatment is initiated, the better the curative outcome (Jariwala and Chandra, 2010). Currently, the most commonly used thrombolytic drugs are urokinase, streptokinase, and tissue-type plasminogen activators. Although these thrombolytic medications are effective at breaking up blood clots, they can also cause unwanted bleeding, such as mucosal bleeding, subcutaneous bleeding, or potentially fatal intracerebral hemorrhage. However, only one-third of patients with MI satisfied the criteria for thrombolytic therapy (Burlen et al., 2017; Kunamneni and Durvasula, 2014; Omraninava et al., 2016).
Percutaneous coronary intervention (PCI)
In comparison to thrombolytic therapy, PCI eliminates the thrombus, and the post-intervention reperfusion rate ranges from 95% to 99%. However, there are some potential risks associated with PCI as well, including the possibility of bleeding or infection at the catheter insertion site, an allergic reaction to the contrast dye used, a blood clot in the blood vessel being treated, a ruptured coronary artery, and total closure of the coronary artery (Peng et al., 2017).
Coronary artery bypass grafting
Coronary artery bypass grafting (CABG) is a surgical procedure that efficiently treats CHD and myocardial ischemia. Additionally, it is a successful approach for treating patients with acute problems like restenosis. Emergency CABG will decrease myocardial damage and lower hospital mortality and unpleasant effects once the immediate problems follow surgery (Chang et al., 2016).
However, for the treatment and prevention of CVDs, more awareness and practice are required. Disparities and the understanding of cardiac illnesses would provide facilitations to the health care specialists to determine the novel disease biomarkers and precision medication for the treatment and prevention of CVD risks (Graham, 2015).
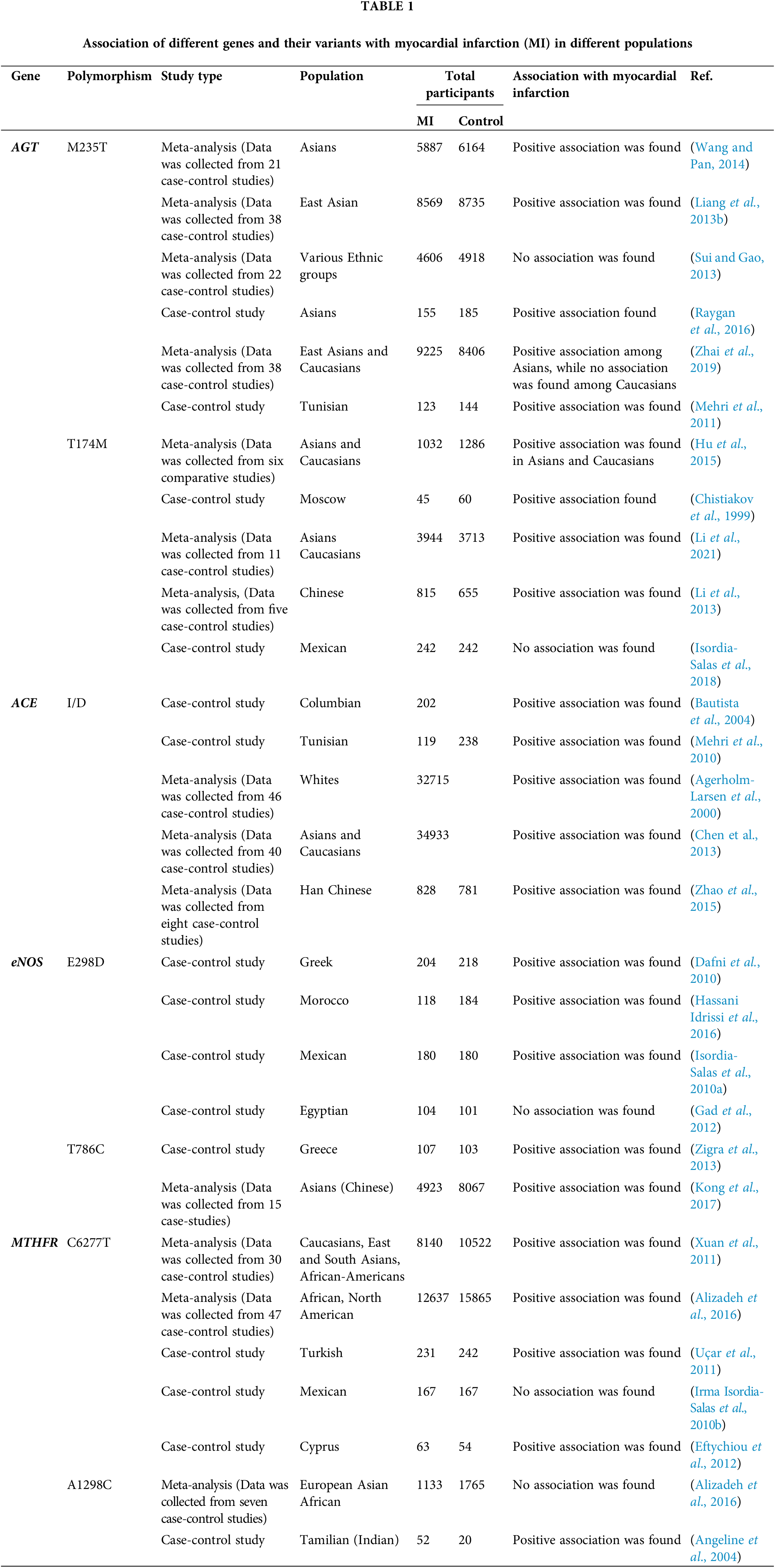
Various Genome-Wide Association Studies (GWAS) studies have revealed numerous genetic variants have a strong association with the occurrence and prevalence of MI. Several diseases, such as diabetes, might be a risk factor for the genetic polymorphism in cardioprotective genes, which then lead to the prevalence of MI. Moreover, GWAS studies have found that their association is varied among different populations due to ethnic differences, lifestyle, and environmental factors. For instance, the association between AGT T174M SNPs have found an association with the prevalence of MI among Asian, Moscow, and the Caucasian population, but not among the Mexican population. Similarly, the association of eNOS E298D polymorphism with MI was found among Greek, Moroccan, and Mexico populations but failed to show any association among the Egyptians. MTHFR C677T also showed a strong association with MI among Asians, African, Caucasian, Cyprus, and Turkish populations, but in Mexican population showed no association as shown in Table 1. This review highlights the significance of several genetic variants, with the prevalence of MI among different ethnic populations.
Availability of Data and Materials: All data generated or analyzed during this study are included in this published article.
Author Contribution: MS and KR jointly wrote this review. SS and SR drafted the manuscript. MI and MAA contributed to the in-depth discussion and conception. KR and MSHA revise and finalize the manuscript. All authors approved the final version of the manuscript.
Ethics Approval: Not applicable.
Funding Statement: Mohammed A. Assiri appreciates the support of the Research Center for Advanced Materials Science (RCAMS) at King Khalid University Abha, Saudi Arabia, through Grant (KKU/ RCAMS/22).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
Adams M, Smith P, Martin D, Thompson J, Lodwick D, Samani N (1996). Genetic analysis of thermolabile methylenetetrahydrofolate reductase as a risk factor for myocardial infarction. QJM: An International Journal of Medicine 89: 437–444. DOI 10.1093/qjmed/89.6.437. [Google Scholar] [CrossRef]
Agerholm-Larsen B, Nordestgaard BG, Tybjærg-Hansen A (2000). ACE gene polymorphism in cardiovascular disease: Meta-analyses of small and large studies in whites. Arteriosclerosis, Thrombosis, and Vascular Biology 20: 484–492. DOI 10.1161/01.ATV.20.2.484. [Google Scholar] [CrossRef]
Aguet F, Ma M (2017). Genetic effects on gene expression across human tissues. Nature 550: 204–213. DOI 10.1038/nature24277. [Google Scholar] [CrossRef]
Alizadeh S, Djafarian K, Moradi S, Shab-Bidar S (2016). C667T and A1298C polymorphisms of methylenetetrahydrofolate reductase gene and susceptibility to myocardial infarction: A systematic review and meta-analysis. International Journal of Cardiology 217: 99–108. DOI 10.1016/j.ijcard.2016.04.181. [Google Scholar] [CrossRef]
Angeline T, Jeyaraj N, Granito S, Tsongalis GJ (2004). Prevalence of MTHFR gene polymorphisms (C677T and A1298C) among Tamilians. Experimental and Molecular Pathology 77: 85–88. DOI 10.1016/j.yexmp.2004.04.006. [Google Scholar] [CrossRef]
Attia J, Thakkinstian A, D’Este C (2003). Meta-analyses of molecular association studies: Methodologic lessons for genetic epidemiology. Journal of Clinical Epidemiology 56: 297–303. DOI 10.1016/S0895-4356(03)00011-8. [Google Scholar] [CrossRef]
Barolia R, Sayani AH (2017). Risk factors of cardiovascular disease and its recommendations in Pakistani context. Journal of Pakistan Medical Association 67: 1723–1729. [Google Scholar]
Bautista LE, Ardila ME, Gamarra G, Vargas CI, Arenas IA (2004). Angiotensin-converting enzyme gene polymorphism and risk of myocardial infarction in Colombia. Medical Science Monitor 10: Cr473–479. [Google Scholar]
Beohar N, Damaraju S, Prather A, Yu QT, Raizner A, Kleiman NS, Roberts R, Marian AJ (1995). Angiotensin-I converting enzyme genotype DD is a risk factor for coronary artery disease. Journal of Investigative Medicine 43: 275–280. [Google Scholar]
Biselli PM, Guerzoni AR, de Godoy MF, Eberlin MN, Haddad R, Carvalho VM, Vannucchi H, Pavarino-Bertelli EC, Goloni-Bertollo EM (2010). Genetic polymorphisms involved in folate metabolism and concentrations of methylmalonic acid and folate on plasma homocysteine and risk of coronary artery disease. Journal of Thrombosis and Thrombolysis 29: 32–40. DOI 10.1007/s11239-009-0321-7. [Google Scholar] [CrossRef]
Böttiger AK, Hurtig-Wennlöf A, Sjöström M, Yngve A, Nilsson TK (2007). Association of total plasma homocysteine with methylenetetrahydrofolate reductase genotypes 677C> T, 1298A> C, and 1793G> A and the corresponding haplotypes in Swedish children and adolescents. International Journal of Molecular Medicine 19: 659–665. DOI 10.3892/ijmm.19.4.659. [Google Scholar] [CrossRef]
Botto LD, Yang Q (2000). 5, 10-Methylenetetrahydrofolate reductase gene variants and congenital anomalies: A HuGE review. American Journal of Epidemiology 151: 862–877. DOI 10.1093/oxfordjournals.aje.a010290. [Google Scholar] [CrossRef]
Bowen DJ, Bowley S, John M, Collins PW (1998). Factor V leiden (G1691Athe prothrombin 3’-untranslated region variant (G20210A) and thermolabile methylenetetrahydrofolate reductase (C677TA single genetic test genotypes all three loci-determination of frequencies in the S. Wales population of the UK. Thrombosis and Haemostasis 79: 949–954. DOI 10.1055/s-0037-1615100. [Google Scholar] [CrossRef]
Brand E, Chatelain N, Paillard F, Tiret L, Visvikis S, Lathrop M, Soubrier F, Demenais F (2002). Detection of putative functional angiotensinogen (AGT) gene variants controlling plasma AGT levels by combined segregation-linkage analysis. European Journal of Human Genetics 10: 715–723. DOI 10.1038/sj.ejhg.5200874. [Google Scholar] [CrossRef]
Brattström L, Wilcken DE, Öhrvik J, Brudin L (1998). Common methylenetetrahydrofolate reductase gene mutation leads to hyperhomocysteinemia but not to vascular disease: the result of a meta-analysis. Circulation 98: 2520–2526. DOI 10.1161/01.CIR.98.23.2520. [Google Scholar] [CrossRef]
Brenner D, Labreuche J, Poirier O, Cambien F, Amarenco P, Investigators G (2005). Renin-angiotensin-aldosterone system in brain infarction and vascular death. Annals of Neurology 58: 131–138. DOI 10.1002/ana.20537. [Google Scholar] [CrossRef]
Burlen J, Cholin L, Ruzieh M, Dasa O (2017). Intravenous tissue plasminogen activator as provocateur of myocardial infarction. American Journal of Therapeutics 24: e489–e490. DOI 10.1097/MJT.0000000000000494. [Google Scholar] [CrossRef]
Camaro C, de Boer M-J (2015). STEMI or non-STEMI: That is the question. Netherlands Heart Journal 23: 243–244. DOI 10.1007/s12471-015-0665-x. [Google Scholar] [CrossRef]
Cambien F, Poirier O, Lecerf L, Evans A, Cambou JP, Arveiler D, Luc G, Bard JM, Bara L, Ricard S (1992). Deletion polymorphism in the gene for angiotensin-converting enzyme is a potent risk factor for myocardial infarction. Nature 359: 641–644. DOI 10.1038/359641a0. [Google Scholar] [CrossRef]
Chang M, Lee C W, Ahn J-M, Cavalcante R, Sotomi Y et al. (2016). Coronary artery bypass grafting versus drug-eluting stents implantation for previous myocardial infarction. The American Journal of Cardiology 118: 17–22. DOI 10.1016/j.amjcard.2016.04.009. [Google Scholar] [CrossRef]
Chauhdary Z, Rehman K, Akash MSH (2021). The composite alliance of FTO locus with obesity-related genetic variants. Clinical and Experimental Pharmacology and Physiology 48: 954–965. DOI 10.1111/1440-1681.13498. [Google Scholar] [CrossRef]
Chen Y, Dong S, He M, Qi T, Zhu W (2013). Angiotensin-converting enzyme insertion/deletion polymorphism and risk of myocardial infarction in an updated meta-analysis based on 34 993 participants. Gene 522: 196–205. DOI 10.1016/j.gene.2013.03.076. [Google Scholar] [CrossRef]
Cheng J, Cai MY, Chen YN, Li ZC, Tang SS, Yang XL, Chen C, Liu X, Xiong XD (2017). Variants in ANRIL gene correlated with its expression contribute to myocardial infarction risk. Oncotarget 8: 12607–12619. DOI 10.18632/oncotarget.14721. [Google Scholar] [CrossRef]
Chistiakov DA, Turakulov RI, Moiseev VS, Nosikov VV (1999). Polymorphism of angiotensinogen T174M gene and cardiovascular diseases in the Moscow population. Genetika 35: 1160–1164. [Google Scholar]
Cicoira M, Zanolla L, Rossi A, Golia G, Franceschini L et al. (2001). Failure of aldosterone suppression despite angiotensin-converting enzyme (ACE) inhibitor administration in chronic heart failure is associated with ACE DD genotype. Journal of the American College of Cardiology 37: 1808–1812. DOI 10.1016/S0735-1097(01)01237-2. [Google Scholar] [CrossRef]
Cooke JN, Bostrom MA, Hicks PJ, Ng MC, Hellwege JN, Comeau ME, Divers J, Langefeld CD, Freedman BI, Bowden DW (2012). Polymorphisms in MYH9 are associated with diabetic nephropathy in European Americans. Nephrology Dialysis Transplantation 27: 1505–1511. DOI 10.1093/ndt/gfr522. [Google Scholar] [CrossRef]
Cornelis MC, El-Sohemy A, Campos H (2004). Genetic polymorphism of CYP1A2 increases the risk of myocardial infarction. Journal of Medical Genetics 41: 758–762. DOI 10.1136/jmg.2004.022012. [Google Scholar] [CrossRef]
Dafni C, Drakoulis N, Landt O, Panidis D, Reczko M, Cokkinos DV (2010). Association of the eNOS E298D polymorphism and the risk of myocardial infarction in the Greek population. BMC Medical Genetics 11: 133. DOI 10.1186/1471-2350-11-133. [Google Scholar] [CrossRef]
Daga LC, Kaul U, Mansoor A (2011). Approach to STEMI and NSTEMI. Journal of the Association of Physicians of India 59: 19–25. [Google Scholar]
Dai SH, Li JF, Feng JB, Li RJ, Li CB, Li Z, Zhang Y, Li DQ (2016). Association of serum levels of AngII, KLK1, and ACE/KLK1 polymorphisms with acute myocardial infarction induced by coronary artery stenosis. Journal of the Renin-Angiotensin-Aldosterone System 17: 1470320316655037. DOI 10.1177/1470320316655037. [Google Scholar] [CrossRef]
Dikmen M, Ozbabalik D, Gunes H, Degirmenci I, Bal C, Ozdemir G, Basaran A (2006). Acute stroke in relation to homocysteine and methylenetetrahydrofolate reductase gene polymorphisms. Acta Neurologica Scandinavica 113: 307–314. DOI 10.1111/j.1600-0404.2005.00556.x. [Google Scholar] [CrossRef]
Ding X, Zhang Q (2010). Enzyme regulation. In: McQueen CA (ed.Comprehensive Toxicology, vol. 10, pp. 8–44. Oxford: Elsevier B.V. DOI 10.1016/B978-0-12-801238-3.95619-8. [Google Scholar]
Doshi SN, McDowell IF, Moat SJ, Lang D, Newcombe RG, Kredan MB, Lewis MJ, Goodfellow J (2001). Folate improves endothelial function in coronary artery disease: an effect mediated by reduction of intracellular superoxide? Arteriosclerosis, Thrombosis, and Vascular Biology 21: 1196–1202. DOI 10.1161/hq0701.092000. [Google Scholar] [CrossRef]
Eftychiou C, Antoniades L, Makri L, Koumas L, Costeas PA, Kyriakou E, Nicolaides E, Papadogiannis D (2012). Homocysteine levels and MTHFR polymorphisms in young patients with acute myocardial infarction: A case control study. Hellenic Journal of Cardiology 53: 189–194. [Google Scholar]
Er F, Dahlem K M, Nia A M, Erdmann E, Waltenberger J et al. (2016). Randomized control of sympathetic drive with continuous intravenous esmolol in patients with acute ST-segment elevation myocardial infarction: The BEtA-Blocker therapy in Acute myocardial infarction (BEAT-AMI) trial. JACC: Cardiovascular Interventions 9: 231–240. DOI 10.1016/j.jcin.2015.10.035. [Google Scholar] [CrossRef]
Eyuboglu M (2015). Effects of new antiplatelet agents and aldosterone receptor antagonists on mortality in patients with myocardial infarction. Cardiovascular Drugs and Therapy 29: 495. DOI 10.1007/s10557-015-6613-6. [Google Scholar] [CrossRef]
Gad MZ, Abdel Rahman MF, Hashad IM, Abdel-Maksoud SM, Farag NM, Abou-Aisha K (2012). Endothelial nitric oxide synthase (G894T) gene polymorphism in a random sample of the Egyptian population: Comparison with myocardial infarction patients. Genetic Testing and Molecular Biomarkers 16: 695–700. DOI 10.1089/gtmb.2011.0342. [Google Scholar] [CrossRef]
Gardemann A, Lohre J, Cayci S, Katz N, Tillmanns H, Haberbosch W (2002). The T allele of the missense Glu298Asp endothelial nitric oxide synthase gene polymorphism is associated with coronary heart disease in younger individuals with high atherosclerotic risk profile. Atherosclerosis 160: 167–175. DOI 10.1016/S0021-9150(01)00554-8. [Google Scholar] [CrossRef]
Garrigós C, Espinosa M, Salinas A, Osman I, Medina R, Taron M, Molina-Pinelo S, Duran I (2017). Single nucleotide polymorphisms as prognostic and predictive biomarkers in renal cell carcinoma. Oncotarget 8: 106551–106564. DOI 10.18632/oncotarget.22533. [Google Scholar] [CrossRef]
Gluba A, Banach M, Rysz J, Piotrowski G, Fendler W, Pietrucha T (2009). Is polymorphism within eNOS gene associated with the late onset of myocardial infarction? A pilot study. Angiology 60: 588–595. DOI 10.1177/0003319709335031. [Google Scholar] [CrossRef]
González-Cambeiro MC, López-López A, Abu-Assi E, Raposeiras-Roubín S, Peña-Gil C, García-Acuña J, González-Juanatey R (2016). Mortality benefit of long-term angiotensin-converting enzyme inhibitors or angiotensin receptor blockers after successful percutaneous coronary intervention in non-ST elevation acute myocardial infarction. Revista Portuguesa de Cardiologia (English Edition) 35: 645–653. DOI 10.1016/j.repce.2016.07.004. [Google Scholar] [CrossRef]
Graham G (2015). Disparities in cardiovascular disease risk in the United States. Current Cardiology Reviews 11: 238–245. DOI 10.2174/1573403X11666141122220003. [Google Scholar] [CrossRef]
Hassani Idrissi H, Hmimech W, Diakite B, Korchi F, Baghdadi D, Habbal R, Nadifi S (2016). Association of G894T eNOS, 4G/5G PAI and T1131C APOA5 polymorphisms with susceptibility to myocardial infarction in Morocco. Meta Gene 9: 56–61. DOI 10.1016/j.mgene.2016.03.004. [Google Scholar] [CrossRef]
He M, He X, Xie Q, Chen F, He S (2006). Angiotensin II induces the expression of tissue factor and its mechanism in human monocytes. Thrombosis Research 117: 579–590. DOI 10.1016/j.thromres.2005.04.033. [Google Scholar] [CrossRef]
Hu PY, Wang YW, Pang XH, Wang HW (2015). T174M polymorphism in the angiotensinogen gene and risk of myocardial infarction: A meta-analysis. Genetics and Molecular Research 14: 3767–3774. DOI 10.4238/2015.April.22.5. [Google Scholar] [CrossRef]
Ismail S, Essawi M (2012). Genetic polymorphism studies in humans. Middle East Journal of Medical Genetics 1: 57–63. DOI 10.1097/01.MXE.0000415225.85003.47. [Google Scholar] [CrossRef]
Isordia-Salas I, Alvarado-Moreno JA, Jiménez-Alvarado RM, Hernández-Juárez J, Santiago-Germán D, Leaños-Miranda A, Majluf-Cruz A (2018). Association of renin-angiotensin system genes polymorphisms and risk of premature ST elevation myocardial infarction in young Mexican population. Blood Coagulation & Fibrinolysis 29: 267–274. DOI 10.1097/MBC.0000000000000714. [Google Scholar] [CrossRef]
Isordia-Salas I, Leaños-Miranda A, Borrayo-Sánchez G (2010a). The Glu298ASP polymorphism of the endothelial nitric oxide synthase gene is associated with premature ST elevation myocardial infarction in Mexican population. Clinica Chimica Acta 411: 553–557. DOI 10.1016/j.cca.2010.01.013. [Google Scholar] [CrossRef]
Isordia-Salas I, Trejo-Aguilar A, Valadés-Mejía MG, Santiago-Germán D, Leaños-Miranda A, Mendoza-Valdéz L, Jáuregui-Aguilar R, Borrayo-Sánchez G, Majluf-Cruz A (2010b). C677T polymorphism of the 5, 10 MTHFR gene in young Mexican subjects with ST-elevation myocardial infarction. Archives of Medical Research 41: 246–250. DOI 10.1016/j.arcmed.2010.04.008. [Google Scholar] [CrossRef]
Jariwala P, Chandra S (2010). Diagnosis and management of failed thrombolytic therapy for acute myocardial infarction. Indian Heart Journal 62: 21–28. [Google Scholar]
Ji LD, Zhang LN, Shen P, Wang P, Zhang YM, Xing WH, Xu J (2010). Association of angiotensinogen gene M235T and angiotensin-converting enzyme gene I/D polymorphisms with essential hypertension in Han Chinese population: A meta-analysis. Journal of Hypertension 28: 419–428. DOI 10.1097/HJH.0b013e32833456b9. [Google Scholar] [CrossRef]
Jiang MH, Su YM, Tang JZ, Shen YB, Deng XT, Yuan DS, Wu J, Pan M, Huang ZW (2013). Angiotensin-converting enzyme gene 2350 G/A polymorphism and susceptibility to atrial fibrillation in Han Chinese patients with essential hypertension. Clinics (Sao Paulo, Brazil) 68: 1428–1432. DOI 10.6061/clinics/2013(11)08. [Google Scholar] [CrossRef]
Jo I, Moon J, Yoon S, Kim HT, Kim E et al. (2006). Interaction between−786TC polymorphism in the endothelial nitric oxide synthase gene and smoking for myocardial infarction in Korean population. Clinica Chimica Acta 365: 86–92. DOI 10.1016/j.cca.2005.07.029. [Google Scholar] [CrossRef]
Karki R, Pandya D, Elston RC, Ferlini C (2015). Defining “mutation” and “polymorphism” in the era of personal genomics. BMC Medical Genomics 8: 1–7. DOI 10.1186/s12920-015-0115-z. [Google Scholar] [CrossRef]
Keavney B, McKenzie C, Parish S, Palmer A, Clark S, Youngman L, Delépine M, Lathrop M, Peto R, Collins R (2000). Large-scale test of hypothesised associations between the angiotensin-converting-enzyme insertion/deletion polymorphism and myocardial infarction in about 5000 cases and 6000 controls. The Lancet 355: 434–442. DOI 10.1016/S0140-6736(00)82009-7. [Google Scholar] [CrossRef]
Kelly M, Semsarian C (2009). Multiple mutations in genetic cardiovascular disease. Circulation: Cardiovascular Genetics 2: 182–190. DOI 10.1161/CIRCGENETICS.108.836478. [Google Scholar] [CrossRef]
Kerkeni M, Addad F, Chauffert M, Myara A, Gerhardt M, Chevenne D, Trivin F, Farhat MB, Miled A, Maaroufi K (2006). Hyperhomocysteinaemia, methylenetetrahydrofolate reductase polymorphism and risk of coronary artery disease. Annals of Clinical Biochemistry 43: 200–206. DOI 10.1258/000456306776865232. [Google Scholar] [CrossRef]
Koh KK, Han SH, Oh PC, Shin EK, Quon MJ (2010). Combination therapy for treatment or prevention of atherosclerosis: Focus on the lipid-RAAS interaction. Atherosclerosis 209: 307–313. DOI 10.1016/j.atherosclerosis.2009.09.007. [Google Scholar] [CrossRef]
Kong XZ, Zhang ZY, Wei LH, Li R, Yu J (2017). The endothelial nitric oxide synthase gene T-786C polymorphism increases myocardial infarction risk: A meta-analysis. Medical Science Monitor 23: 759–766. DOI 10.12659/MSM.899905. [Google Scholar] [CrossRef]
Kunamneni A, Durvasula R (2014). Streptokinase-A drug for thrombolytic therapy: A patent review. Recent Patents on Cardiovascular Drug Discovery 9: 106–121. DOI 10.2174/1574890110999150202150017. [Google Scholar] [CrossRef]
Landrum M J, Lee J M, Benson M, Brown G R, Chao C et al. (2018). ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Research 46: D1062–D1067. DOI 10.1093/nar/gkx1153. [Google Scholar] [CrossRef]
Larsson P, Schwieler J, Wallen N (2000). Platelet activation during angiotensin II infusion in healthy volunteers. Blood Coagulation & Fibrinolysis 11: 61–69. DOI 10.1097/00001721-200011010-00007. [Google Scholar] [CrossRef]
Leon BM, Maddox TM (2015). Diabetes and cardiovascular disease: Epidemiology, biological mechanisms, treatment recommendations and future research. World Journal of Diabetes 6: 1246–1258. DOI 10.4239/wjd.v6.i13.1246. [Google Scholar] [CrossRef]
Li X, Li Q, Wang Y, Li Y, Ye M, Ren J, Wang Z (2013). AGT gene polymorphisms (M235T, T174M) are associated with coronary heart disease in a Chinese population. Journal of the Renin-Angiotensin-Aldosterone System 14: 354–359. DOI 10.1177/1470320312452029. [Google Scholar] [CrossRef]
Li YY, Wang H, Wang H, Zhang YY (2021). Myocardial infarction and AGT p.Thr174Met polymorphism: A meta-analysis of 7657 subjects. Cardiovascular Therapeutics 2021: 6667934. DOI 10.1155/2021/6667934. [Google Scholar] [CrossRef]
Liang S, Pan M, Hu N, Wu YY, Chen H, Zhu JH, Guan HJ, Sang AM (2013a). Association of angiotensin-converting enzyme gene 2350 G/A polymorphism with diabetic retinopathy in Chinese Han population. Molecular Biology Reports 40: 463–468. DOI 10.1007/s11033-012-2081-2. [Google Scholar] [CrossRef]
Liang X, Qiu J, Liu X, Li X, Zhao S, Wang J, Ma Y, Gao H (2013b). Polymorphism of angiotensinogen gene M235T in myocardial infarction and brain infarction: A meta-analysis. Gene 529: 73–79. DOI 10.1016/j.gene.2013.07.095. [Google Scholar] [CrossRef]
Liew S-C, Gupta ED (2015). Methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism: Epidemiology, metabolism and the associated diseases. European Journal of Medical Genetics 58: 1–10. DOI 10.1016/j.ejmg.2014.10.004. [Google Scholar] [CrossRef]
Markus HS, Ali N, Swaminathan R, Sankaralingam A, Molloy J, Powell J (1997). A common polymorphism in the methylenetetrahydrofolate reductase gene, homocysteine, and ischemic cerebrovascular disease. Stroke 28: 1739–1743. DOI 10.1161/01.STR.28.9.1739. [Google Scholar] [CrossRef]
Matsushita S, Muramatsu T, Arai H, Matsui T, Higuchi S (1997). The frequency of the methylenetetrahydrofolate reductase-gene mutation varies with age in the normal population. The American Journal of Human Genetics 61: 1459–1460. DOI 10.1086/301640. [Google Scholar] [CrossRef]
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F (2016). The ensembl variant effect predictor. Genome Biology 17: 1–14. DOI 10.1186/s13059-016-0974-4. [Google Scholar] [CrossRef]
Mehri S, Baudin B, Mahjoub S, Zaroui A, Bénéteau-Burnat B, Mechmeche R, Hammami M, Ben Arab S (2010). Angiotensin-converting enzyme insertion/deletion gene polymorphism in a Tunisian healthy and acute myocardial infarction population. Genetic Testing and Molecular Biomarkers 14: 85–91. DOI 10.1089/gtmb.2009.0105. [Google Scholar] [CrossRef]
Mehri S, Mahjoub S, Farhati A, Bousaada R, Ben Arab S, Baudin B, Hammami M (2011). Angiotensinogen gene polymorphism in acute myocardial infarction patients. Journal of the Renin-Angiotensin-Aldosterone System 12: 42–47. DOI 10.1177/1470320310376425. [Google Scholar] [CrossRef]
Morray B, Goldenberg I, Moss AJ, Zareba W, Ryan D, McNitt S, Eberly SW, Glazko G, Mathew J (2007). Polymorphisms in the paraoxonase and endothelial nitric oxide synthase genes and the risk of early-onset myocardial infarction. The American Journal of Cardiology 99: 1100–1105. DOI 10.1016/j.amjcard.2006.12.022. [Google Scholar] [CrossRef]
Murphey LJ, Gainer JV, Vaughan DE, Brown NJ (2000). Angiotensin-converting enzyme insertion/deletion polymorphism modulates the human in vivo metabolism of bradykinin. Circulation 102: 829–832. DOI 10.1161/01.CIR.102.8.829. [Google Scholar] [CrossRef]
Murphy BP, Stanton T, Dunn FG (2009). Hypertension and Myocardial Ischemia. Medical Clinics of North America 93: 681–695. DOI 10.1016/j.mcna.2009.02.003. [Google Scholar] [CrossRef]
Nabel EG (2003). Cardiovascular disease. New England Journal of Medicine 349: 60–72. DOI 10.1056/NEJMra035098. [Google Scholar] [CrossRef]
Nabel EG, Braunwald E (2012). A tale of coronary artery disease and myocardial infarction. New England Journal of Medicine 366: 54–63. DOI 10.1056/NEJMra1112570. [Google Scholar] [CrossRef]
Nascimento BR, Brant LCC, Marino BCA, Passaglia LG, Ribeiro ALP (2019). Implementing myocardial infarction systems of care in low/middle-income countries. Heart (British Cardiac Society) 105: 20–26. DOI 10.1136/heartjnl-2018-313398. [Google Scholar] [CrossRef]
Ojha N, Dhamoon AS (2022). Myocardial infarction. https://www.ncbi.nlm.nih.gov/books/NBK537076/. [Google Scholar]
Omraninava A, Hashemian AM, Masoumi B (2016). Effective factors in door-to-needle time for streptokinase administration in patients with acute myocardial infarction admitted to the emergency department. Trauma Monthly 21: 308. DOI 10.5812/traumamon.19676. [Google Scholar] [CrossRef]
Park KW, You KH, Oh S, Chae IH, Kim HS, Oh BH, Lee MM, Park YB (2004). Association of endothelial constitutive nitric oxide synthase gene polymorphism with acute coronary syndrome in Koreans. Heart (British Cardiac Society) 90: 282–285. DOI 10.1136/hrt.2002.003616. [Google Scholar] [CrossRef]
Peng X, Zhou J, Wu X-S (2017). New strategies for myocardial infarction treatment. Journal of Cardiology and Therapy 4: 664–670. DOI 10.17554/j.issn.2309-6861.2017.04.127. [Google Scholar] [CrossRef]
Raygan F, Karimian M, Rezaeian A, Bahmani B, Behjati M (2016a). Angiotensinogen-M235T as a risk factor for myocardial infarction in Asian populations: A genetic association study and a bioinformatics approach. Croatian Medical Journal 57: 351–362. DOI 10.3325/cmj.2016.57.351. [Google Scholar] [CrossRef]
Rehman K, Jabeen K, Awan FR, Hussain M, Saddique MA, Akash MSH (2020a). Biochemical investigation of rs1801282 variations in PPAR-γ gene and its correlation with risk factors of diabetes mellitus in coronary artery disease. Clinical and Experimental Pharmacology and Physiology 47: 1517–1529. DOI 10.1111/1440-1681.13339. [Google Scholar] [CrossRef]
Rehman K, Tahir A, Niaz S, Shabbir S, Jabeen K, Faheem A, Akash MSH (2020b). Frequency of PPAR-γ, FTO and ABCC8 genetic variation in Pakistani cardiovascular smokers. Environmental Science and Pollution Research 27: 42611–42620. DOI 10.1007/s11356-020-10226-z. [Google Scholar] [CrossRef]
Saidi S, Mallat S, Almawi W, Mahjoub T (2009). Association between renin-angiotensin-aldosterone system genotypes and haplotypes and risk of ischemic stroke of atherosclerotic etiology. Acta Neurologica Scandinavica 119: 356–363. DOI 10.1111/j.1600-0404.2008.01105.x. [Google Scholar] [CrossRef]
Schmoelzer I, Renner W, Paulweber B, Malaimare L, Iglseder B, Schmid P, Schallmoser K, Wascher TC (2003). Lack of association of the Glu298Asp polymorphism of endothelial nitric oxide synthase with manifest coronary artery disease, carotid atherosclerosis and forearm vascular reactivity in two Austrian populations. European Journal of Clinical Investigation 33: 191–198. DOI 10.1046/j.1365-2362.2003.01108.x. [Google Scholar] [CrossRef]
Schuster H, Wienker TF, Stremmler U, Noll B, Steinmetz A, Luft FC (1995). An angiotensin-converting enzyme gene variant is associated with acute myocardial infarction in women but not in men. The American Journal of Cardiology 76: 601–603. DOI 10.1016/S0002-9149(99)80164-7. [Google Scholar] [CrossRef]
Shah MS, Brownlee M (2016). Molecular and cellular mechanisms of cardiovascular disorders in diabetes. Circulation Research 118: 1808–1829. DOI 10.1161/CIRCRESAHA.116.306923. [Google Scholar] [CrossRef]
Shaker OG, Ismail MF (2014). Association of genetic variants of MTHFR, ENPP1, and ADIPOQ with myocardial infarction in Egyptian patients. Cell Biochemistry and Biophysics 69: 265–274. DOI 10.1007/s12013-013-9794-2. [Google Scholar] [CrossRef]
Sivitskaia L, Kushnerevich E, Danilenko N, Novogrodskiĭ T, Davydenko O (2008). Gene polymorphism of the renin-angiotensin system in six ethnic/geographic regions of Belarus. Genetika 44: 702–709. DOI 10.1134/S1022795408050141. [Google Scholar] [CrossRef]
Sui X, Gao C (2013). The angiotensinogen gene M235T polymorphism and acute myocardial infarction risk: A meta-analysis of 22 studies. Molecular Biology Reports 40: 4439–4445. DOI 10.1007/s11033-013-2534-2. [Google Scholar] [CrossRef]
Thomas H, Diamond J, Vieco A, Chaudhuri S, Shinnar E et al. (2018). Global atlas of cardiovascular disease 2000–2016: The path to prevention and control. Global Heart 13: 143–163. DOI 10.1016/j.gheart.2018.09.511. [Google Scholar] [CrossRef]
Uçar F, Celik S, Yücel B, Sönmez M, Celep F, Erkut N (2011). MTHFR C677T polymorphism and its relationship to myocardial infarction in the Eastern Black Sea region of Turkey. Archives of Medical Research 42: 709–712. DOI 10.1016/j.arcmed.2011.12.007. [Google Scholar] [CrossRef]
Um JY, Kim HM, Park HS, Joo JC, Kim KY, Kim YK, Hong SH (2005). Candidate genes of cerebral infarction and traditional classification in Koreans with cerebral infarction. International Journal of Neuroscience 115: 743–756. DOI 10.1080/00207450590524421. [Google Scholar] [CrossRef]
Um J-Y, Moon K-S, Lee K-M, Cho K-H, Heo Y, Moon B-S, Kim H-M (2003). Polymorphism of angiotensin-converting enzyme, angiotensinogen, and apolipoprotein E genes in Korean patients with cerebral infarction. Journal of Molecular Neuroscience 21: 23–28. DOI 10.1385/JMN:21:1:23. [Google Scholar] [CrossRef]
Wang WZ (2013). Association between T174M polymorphism in the angiotensinogen gene and risk of coronary artery disease: A meta-analysis. Journal of Geriatric Cardiology 10: 59–65. DOI 10.3969/j.issn.1671-5411.2013.01.010. [Google Scholar] [CrossRef]
Wang YJ, Pan Y (2014). The M235T polymorphism in the angiotensinogen gene and myocardial infarction risk: A meta-analysis. Journal of the Renin-Angiotensin-Aldosterone System 15: 294–300. DOI 10.1177/1470320312471148. [Google Scholar] [CrossRef]
Wu Y, Pan N, An Y, Xu M, Tan L, Zhang L (2021). Diagnostic and prognostic biomarkers for myocardial infarction. Frontiers in Cardiovascular Medicine 7: 1545. DOI 10.3389/fcvm.2020.617277. [Google Scholar] [CrossRef]
Xu M-Q, Ye Z, Hu FB, He L (2007). Quantitative assessment of the effect of angiotensinogen gene polymorphisms on the risk of coronary heart disease. Circulation 116: 1356–1366. DOI 10.1161/CIRCULATIONAHA.107.728857. [Google Scholar] [CrossRef]
Xuan C, Bai XY, Gao G, Yang Q, He GW (2011). Association between polymorphism of methylenetetrahydrofolate reductase (MTHFR) C677T and risk of myocardial infarction: A meta-analysis for 8,140 cases and 10,522 controls. Archives of Medical Research 42: 677–685. DOI 10.1016/j.arcmed.2011.11.009. [Google Scholar] [CrossRef]
Yuan T, Yang T, Chen H, Fu D, Hu Y, Wang J, Yuan Q, Yu H, Xu W, Xie X (2019). New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biology 20: 247–260. DOI 10.1016/j.redox.2018.09.025. [Google Scholar] [CrossRef]
Zafarmand MH, Van Der Schouw YT, Grobbee DE, De Leeuw PW, Bots ML (2008). The M235T polymorphism in the AGT gene and CHD risk: Evidence of a Hardy-Weinberg equilibrium violation and publication bias in a meta-analysis. PLoS One 3: e2533. DOI 10.1371/journal.pone.0002533. [Google Scholar] [CrossRef]
Zhai C, Cong H, Zhang H, Hou K, Zhang Y, Zhang Y (2019). M235T polymorphism in the angiotensinogen gene and cardiovascular disease: An updated meta-analysis of 39 case-control comparisons. The Anatolian Journal of Cardiology 21: 222–232. DOI 10.14744/AnatolJCardiol.2019.75282. [Google Scholar] [CrossRef]
Zhao W, Ma ST, Cui LQ (2015). Meta-analysis of angiotensin-converting enzyme insertion/deletion polymorphism and myocardial infarction in Han Chinese. Genetics and Molecular Research 14: 8068–8076. DOI 10.4238/2015.July.17.15. [Google Scholar] [CrossRef]
Zhu X, Bouzekri N, Southam L, Cooper R S, Adeyemo A, McKenzie C A, Luke A, Chen G, Elston R C, Ward R (2001). Linkage and association analysis of angiotensin I-converting enzyme (ACE)-gene polymorphisms with ACE concentration and blood pressure. The American Journal of Human Genetics 68: 1139–1148. DOI 10.1086/320104. [Google Scholar] [CrossRef]
Zigra AM, Rallidis LS, Anastasiou G, Merkouri E, Gialeraki A (2013). eNOS gene variants and the risk of premature myocardial infarction. Disease Markers 34: 431–436. DOI 10.1155/2013/235056. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools