
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
VIEWPOINT

DNA polymerase θ (POLQ): A druggable DNA polymerase for homologous recombination-deficient cancer cells
Department of Biochemistry and Molecular Biology, Faculty of Medicine, University of Colombo, Colombo 08, Sri Lanka
* Corresponding Authors: Meran Keshawa Ediriweera, ;
BIOCELL 2023, 47(3), 441-444. https://doi.org/10.32604/biocell.2023.025747
Received 28 July 2022; Accepted 06 October 2022; Issue published 03 January 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Irregularities in the DNA repair pathways are frequently observed in cancer. Dysregulated DNA repair pathways support growth advantages to tumor cells. DNA polymerase-theta (POLQ) is an error-prone DNA polymerase involved in double-strand break repair through microhomology-mediated end joining (MMEJ). POLQ also mediates translesion DNA synthesis and it is largely not expressed in normal cells. POLQ is overexpressed in a range of cancer cells, including homologous recombination (HR) deficient cancer cells. In HR deficient cells, MMEJ is crucial as a backup DNA repair pathway, indicating the indispensable role of POLQ-mediated MMEJ in HR deficient cancer cells. In addition, POLQ is synthetic lethal with a number of oncogenes. This viewpoint highlights the potential role of POLQ as a new target in the treatment of HR-deficient tumors and aims to develop enthusiasm to introduce fundamental aspects of Molecular Biology and Biochemistry to basic research related to Molecular Life Sciences, Cell Biology and Genetics in Sri Lanka as Molecular Biology and Biochemistry fundamentals can still do wonders in modern research.Keywords
DNA stores genetic information in cells. The nucleotide sequences of DNA guide the synthesis of cellular RNAs and proteins, making DNA a marvelous control device necessary to maintain a number of vital functions of living cells. Collectively, the DNA metabolism of living cells comprises DNA replication, repair, and recombination. The genetic information stored in DNA is transmitted from one generation to the next. Errors in DNA can cause undesirable effects as they can affect the function of a gene. The errors in DNA are inheritable. The cellular enzymes involved in DNA replication generate thousands of copies of DNA with greater fidelity. The integrity of the genetic information stored in DNA depends on the DNA repair mechanism. Unlike RNA and protein, damaged DNA molecules are irreplaceable. Agents such as chemicals, radiations (ultraviolet, X-ray, gamma-rays), toxins, reactive oxygen species, and some viruses can damage DNA. Unrepaired DNA damage, if replicated and transmitted to other generations, can cause permanent changes in the genomic sequence, thereby causing gene mutations (Dunlop et al., 1997). DNA polymerases aid DNA replication and repair. Mammalian cells contain 15 different DNA polymerases. Given the role of DNA polymerases in DNA replication and repair, it is not surprising that any disruption or irregularity in the function of DNA polymerases can disrupt the genomic stability of an individual, thereby increasing the rate of gene mutations and an individual’s vulnerability to cancer (Lange et al., 2011). Of the different DNA polymerases, DNA polymerase θ (POLQ) has become an attractive target in anti-cancer drug discovery strategies (Chen and Pomerantz, 2021). This multi-functional DNA polymerase plays a key role in tumors with deficient homologous recombination (HR). This viewpoint aims to rationalize POLQ as a druggable target for HR-deficient tumors by providing recent research advancements related to POLQ.
In healthy cells, robust and diverse DNA repair mechanisms faithfully protect from DNA damages to maintain overall survival. The availability of diverse DNA repair systems reflects the complex nature of DNA repair mechanisms in healthy cells. Five major DNA repair pathways are seen in healthy cells: mismatch repair, base excision repair (BER), HR, non-homologous end joining (NHEJ), and nucleotide excision repair (NER) (Sancar et al., 2004).
Breast cancer type 1 (BRCA 1) and BRCA2 play a prominent role in HR to maintain genetic stability. Mutations in BRCA1 or BRCA2 are strongly associated with developing breast and ovarian cancers. Cancer cells with mutated BRCA1 and BRCA2 possess impaired HR activity and show sensitivity to poly-ADP ribose polymerase (PARP) inhibitors, and PARP inhibitors in BRCA1 and BRCA2 mutated tumors work through synthetic lethality (Murai et al., 2012). By trapping PARP on DNA, PARP inhibition by PARPi leads to the collision of unrepaired single-stranded breaks within the replication forks, thereby generating one-ended double-highly cytotoxic strand breaks (DSBs), which can be corrected by the HR machinery. In BRCA-mutated cells, the HR machinery is dysregulated (Murai et al., 2012). In these cells, PARPi traps PARP-DNA complexes, leading to the accumulation of DSBs, which eventually causes genomic instability and cell death. Normal cells with undamaged BRCA1 and BRCA2 show less sensitivity to PARP inhibitors, indicating a remarkable sensitivity of BRCA1 and BRCA2 mutated cancer cells to PARP inhibitors. Despite the interesting clinical evidence, acquired resistance to PARP inhibitors has been identified as a major clinical barrier, highlighting the importance of identification of alternative drug targets implicated in DNA repair for BRCA-deficient cancer cells (Chiappa et al., 2021).
Apart from the HR machinery, canonical NHEJ (C-NHEJ, microhomology-mediated end joining (MMEJ), and alternative end joining (alt-EJ) DNA repair pathways are involved in the DSB repairs (Yu and McVey, 2010). In the G1 phase of the cell cycle, DSBs are repaired by C-NHEJ, while HR is favored in G2 and S phases. In addition to BRCA1/2, replication protein A (RPA), c-terminal binding protein interacting protein, RAD51, partner and localizer of BRCA2, and DNA replication helicase/nuclease 2 proteins promote HR pathways (Zhao et al., 2019). Resected DNA with 3′ ssDNA bound with RPA) serves as the substrate for both MMEJ and HR, suggesting the existence of possible competition between MMEJ and HR for 3′ ssDNA (Chen and Pomerantz, 2021).
DNA polymerase θ and its role in cancer
POLQ contributes to DSB repair via MMEJ (Wood and Doublié, 2016) (Fig. 1). MMEJ is also known as polymerase theta-mediated end joining (TMEJ) as it requires POLQ. POLQ is encoded by the POLQ gene. In addition to MMEJ, POLQ performs translesion DNA synthesis (TLS) as well. Compared to HR, DSBs repair by POLQ in MMEJ is highly error-prone as there is no involvement of homologous sequences. POLQ can also reverse transcribes RNA (Chen and Pomerantz, 2021).
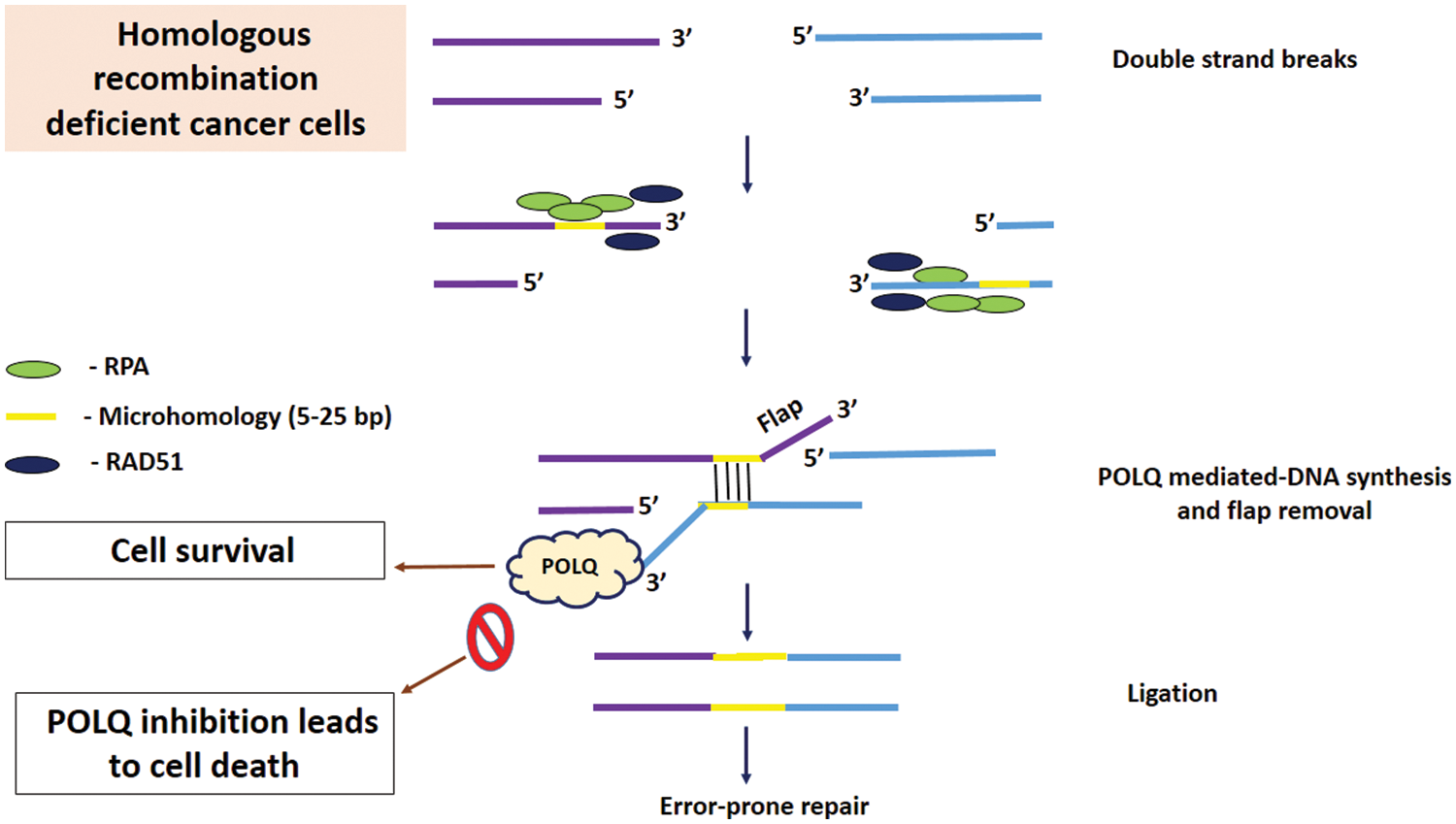
Figure 1: POLQ contributes to double strand break DNA repair through microhomology-mediated end joining (MMEJ). MMEJ is also known as polymerase theta-mediated end joining (TMEJ) due to the requirement of POLQ. In the first step of TMEJ, 5′–3′ end resection results in exposure of microhomologous regions (5-25 bp). Resected DNA with 3′ ssDNA bound to replication protein A (RPA) is a substrate for MMEJ. POLQ displaces RAD51 from ssDNA. Microhomologous regions anneal each other with the aid of POLQ, and flaps generated are removed by the endonuclease. Finally, DNA ligases complete DNA ligation. MMEJ acts as a backup DNA repair pathway in HR-deficient cancer cells. POLQ inhibition in HR-deficient cells results in cell death (Schrempf et al., 2021). Cell survival results when POLQ is not inhibited in HR-deficient cells.
POLQ is a large multi-functional DNA polymerase that comprises three main domains, namely super family 2 (SF2) helicase-like ATPase domain, central domain and DNA polymerase domain. The SF2 helicase domain of POLQ, which possesses helicase and ATPase activities, is involved in TMEJ on long ssDNA. The SF2 helicase domain comprises RAD51-binding sites, a protein that contributes to DSB repairs (Wood and Doublié, 2016).
In HR deficient cells, MMEJ functions as a backup DNA repair pathway, highlighting the essential role of POLQ mediated MMEJ in HR deficient cells (Higgins and Boulton, 2018) Fig. 1. HR deficient tumors with BRCA1 and BRCA2 mutations are exclusively dependent on POLQ-mediated DNA repair, indicating that POLQ is essential for DNA repair pathways in BRCA-deficient cancer, especially for BER and TLS (Higgins and Boulton, 2018). POLQ has been found to up-regulate in a range of human cancers including ovary, breast, lung, gastric, head and neck and colon. The up-regulation is associated with poor prognosis and clinical outcomes. The exact reason behind POLQ up-regulation in human cancer is not well understood (Chen and Pomerantz, 2021; Higgins and Boulton, 2018).
In a review, Schrempf et al. (2021) have discussed two potential mechanisms associated with POLQ up-regulation. As per the first proposed mechanism, POLQ over-expression is associated with increased replication stress which might help to improve tumor fitness. The second mechanism states that the over-expression of POLQ is associated with specific cellular signaling cascades related to DNA repair pathways (Schrempf et al., 2021). POLQ-assisted DNA repair pathways are frequently dysregulated in breast and ovarian cancer. Furthermore, it has been reported that template insertions in TMEJ are frequently found in BRCA1/2 mutated breast cancer, further indicating the dependency of POLQ-mediated repair pathways in HR deficient cancer cells. Several studies illustrate that POLQ is synthetic lethal with many HR factors and frequently mutated cancer signaling pathway genes (Schrempf et al., 2021). According to a recent investigation, POLQ not only aids MMEJ but also supports DSB repair produced by Cas9 nickase (Wang et al., 2019). Single-ended DSBs can be converted to double-ended DSBs when replication forks that arrive from the other end, meet each other. ATRs play a key role in protecting replication forks. Lack of ATR activity has been reported to cause replication fork collapse and replication stress following the accumulation of DSBs. POLQ was found responsible for repairing DSBs which are accumulated due to ATR inactivation. In addition, a synthetic lethality interaction was identified between ATR and POLQ as POLQ inactivation promoted cell death in ATR-deficient cells (Wang et al., 2019). Moreover, studies demonstrate synthetic lethal interaction between POLQ and ATM serine/threonine kinase (ATM). ATM participates as one of the key regulators in HR-mediated DSB repair (Morrison et al., 2000). Fanconi anemia group D2 protein (FANCD2) coordinates several DNA repair proteins (Nepal et al., 2017). POLQ shows a synthetic lethal relationship between FANCD2 (Schrempf et al., 2021). In addition, interesting pre-clinical investigations demonstrate synthetic lethal relationships between POLQ and BRCA1/2 (Schrempf et al., 2021).
In a study by Lemée et al. (2010), the expression of replicative (POLA, POLD, and POLE) and specialized DNA polymerases (POLH, POLL, POLM, POLN, POLK, POLB, POLI, POLQ, POLZ/REV3L, and REV1) were analyzed in breast cancer tumors. Of these, the expression of POLQ was found to be higher in tumor samples. The log-rank test predicted that up-regulation of POLQ is associated with poor survival in breast cancer patients. Ectopic expression of POLQ in MRC5-SV fibroblasts affected the MRC5-SV cell cycle and induced DNA damage. Transfection of POLQ siRNA in MRC5-SV cells and MCF-7 breast cancer cells resulted in a reduction of γ-H2AX-positive cells, which confirmed the involvement of POLQ in the induction of DNA damage response. In addition, POLQ overexpression was found to associate with the progression of defective DNA replication forks and disturb DNA replication, indicating POLQ overexpression as a promising prognostic indicator for breast cancer.
Higgins et al. (2010) demonstrated that POLQ over-expression is correlated with oestrogen receptor-negative breast cancer tumors. A seed-clustering analysis identified 97 different genes associated with POLQ overexpression related to Wnt signaling, cell cycle progression and p53 signaling. A recent study illustrated that POLQ is overexpressed in hepatocellular carcinoma and associated with its development (Pan et al., 2021). Furthermore, evidence demonstrates that POLQ confers radio-resistance in cancer cells, highlighting POLQ as an ideal target for radio-sensitization in cancer (Yousefzadeh and Wood, 2013). Tumors that receive radiotherapy show increased expression of POLQ. In a recent investigation, DNA-dependent protein kinase (DNA-PK) and POLQ dual inhibition were identified as promising strategy to overcome radiation resistance in cancer cells (Kumar et al., 2020). POLQ depletion has been reported to sensitize esophageal squamous cell carcinoma cells to genotoxic agents (Li et al., 2021).
Studies report that both the helicase domain and DNA polymerase domains of POLQ can be targeted in anti-cancer strategies. However, it is not yet clear which domain is acting as a preferred inhibitory site, revealing that more studies are necessary to elucidate the actual therapeutic functions of POLQ domains. A recent study identified the antibiotic novobiocin as a specific POLQ inhibitor. Novobiocin inhibits POLQ ATPase activity, selectively kills HR-deficient cancer cells and enhances the cytotoxic effects of PARPi (Zhou et al., 2021). Another investigation identified ART558 as a POLQ inhibitor. ART558 blocks the activity of POLQ by residing within the polymerase catalytic domain (Zatreanu et al., 2021).
To date, several biotech companies have started developing potent domain-specific POLQ inhibitors to introduce for clinical investigations for the treatment of BRCA mutated tumors. Moreover, attempts to screen POLQ inhibitors from natural sources will aid to identify novel natural-based POLQ inhibitors. Importantly, for the clinical landscape, the identification of POLQ inhibitors with less off-target toxicities and side effects is essential. Considering the recent reports, the continued development of structurally and functionally diverse POLQ inhibitors with a deep understanding of the synthetic lethal environment will create a lot of excitement in anti-cancer drug discovery platforms. According to the published reports, it is not very clear how POLQ inhibition affects the genome stability in patients undergoing POLQ inhibitor therapy. Therefore, detailed studies are necessary to elucidate whether POLQ possesses genome destabilizing effects. In addition, a better understanding of the POLQ-mediated TLS will also help to develop novel POLQ inhibitors. In summary, it is strongly suggest that POLQ inhibitors will make a breakthrough in anti-cancer drug discovery strategies. ART4215, a POLQ inhibitor is being tested in clinical trials against solid tumors (NCT04991480). However, Yoon et al. (2019) have demonstrated the occurrence of skin cancer in the absence of POLQ, indicating that targeting POLQ alone is dangerous to humans. However, more studies are needed to understand the actual function of POLQ in anti-cancer strategies. POLQ-based or DNA polymerase-based anti-cancer drug discovery strategies are new to Sri Lanka. Through this, I am trying to introduce POLQ-based anti-cancer drug discovery strategies to the Sri Lankan research landscape and encourage Sri Lankan scientists to present wonderful ideas to make POLQ-based anti-cancer strategies interesting.
Acknowledgement: I would like to thank the Department of Biochemistry and Molecular Biology, Faculty of Medicine, University of Colombo, Sri Lanka for support while writing this manuscript.
Author Contribution: The author confirms sole responsibility for the following: study conception and design, data collection, analysis and interpretation of results, and manuscript preparation.
Ethics Approval: Not applicable.
Funding Statement: The author received no specific funding for this study.
Conflicts of Interest: The author declares that they have no conflicts of interest to report regarding the present study.
References
Chen XS, Pomerantz RT (2021). DNA polymerase θ: A cancer drug target with reverse transcriptase activity. Genes 12: 1146. DOI 10.3390/genes12081146. [Google Scholar] [CrossRef]
Chiappa M, Guffanti F, Bertoni F, Colombo I, Damia G (2021). Overcoming PARPi resistance: Preclinical and clinical evidence in ovarian cancer. Drug Resistance Updates 55: 100744. DOI 10.1016/j.drup.2021.100744. [Google Scholar] [CrossRef]
Dunlop MG, Farrington SM, Carothers AD, Wyllie AH, Sharp L et al. (1997). Cancer risk associated with germline DNA mismatch repair gene mutations. Human Molecular Genetics 6: 105–110. DOI 10.1093/hmg/6.1.105. [Google Scholar] [CrossRef]
Higgins GS, Boulton SJ (2018). Beyond PARP—POLθ as an anticancer target. Science 359: 1217–1218. DOI 10.1126/science.aar5149. [Google Scholar] [CrossRef]
Higgins GS, Harris AL, Prevo R, Helleday T, McKenna WG et al. (2010). Overexpression of POLQ confers a poor prognosis in early breast cancer patients. Oncotarget 1: 175–184. DOI 10.18632/oncotarget.124. [Google Scholar] [CrossRef]
Kumar RJ, Chao HX, Simpson DA, Feng W, Cho MG et al. (2020). Dual inhibition of DNA-PK and DNA polymerase theta overcomes radiation resistance induced by p53 deficiency. NAR Cancer 2: zcaa038. DOI 10.1093/narcan/zcaa038. [Google Scholar] [CrossRef]
Lange SS, Takata KI, Wood RD (2011). DNA polymerases and cancer. Nature Reviews Cancer 11: 96–110. DOI 10.1038/nrc2998. [Google Scholar] [CrossRef]
Lemée F, Bergoglio V, Fernandez-Vidal A, Machado-Silva A, Pillaire MJ, et al. (2010). DNA polymerase θ up-regulation is associated with poor survival in breast cancer, perturbs DNA replication, and promotes genetic instability. PNAS 107: 13390–13395. DOI 10.1073/pnas.0910759107. [Google Scholar] [CrossRef]
Li J, Ko JM, Dai W, Yu VZ, Ng HY, et al. (2021). Depletion of DNA polymerase theta inhibits tumor growth and promotes genome instability through the cGAS-STING-ISG pathway in esophageal squamous cell carcinoma. Cancers 13: 3204. DOI 10.3390/cancers13133204. [Google Scholar] [CrossRef]
Morrison C, Sonoda E, Takao N, Shinohara A, Yamamoto KI, et al., Yamamoto KI, Takeda S (2000). The controlling role of ATM in homologous recombinational repair of DNA damage. The EMBO Journal 19: 463–471. DOI 10.1093/emboj/19.3.463. [Google Scholar] [CrossRef]
Murai J, Huang SY, Das BB, Renaud A, Zhang Y et al. (2012). Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Research 72: 5588–5599. DOI 10.1158/0008-5472.CAN-12-2753. [Google Scholar] [CrossRef]
Nepal M, Che R, Ma C, Zhang J, Fei P. (2017). FANCD2 and DNA damage. International Journal of Molecular Sciences. 18: 1804. DOI 10.3390/ijms18081804. [Google Scholar] [CrossRef]
Pan Q, Wang L, Liu Y, Li M, Zhang Y, et al. (2021). Knockdown of POLQ interferes the development and progression of hepatocellular carcinoma through regulating cell proliferation, apoptosis and migration. Cancer Cell International 21: 1–3. DOI 10.1186/s12935-021-02178-2. [Google Scholar] [CrossRef]
Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K, Linn S (2004). Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annual Review of Biochemistry 73: 39–85. DOI 10.1146/annurev.biochem.73.011303.073723. [Google Scholar] [CrossRef]
Schrempf A, Slyskova J, Loizou JI (2021). Targeting the DNA repair enzyme polymerase θ in cancer therapy. Trends in Cancer 7: 98–111. DOI 10.1016/j.trecan.2020.09.007. [Google Scholar] [CrossRef]
Wang Z, Song Y, Li S, Kurian S, Xiang R et al. (2019). DNA polymerase θ (POLQ) is important for repair of DNA double-strand breaks caused by fork collapse. Journal of Biological Chemistry 294: 3909–3919. DOI 10.1074/jbc.RA118.005188. [Google Scholar] [CrossRef]
Wood RD, Doublié S (2016). DNA polymerase θ (POLQdouble-strand break repair, and cancer. DNA Repair 44: 22–32. DOI 10.1016/j.dnarep.2016.05.003. [Google Scholar] [CrossRef]
Yoon JH, McArthur MJ, Park J, Basu D, Wakamiya M et al. (2019). Error-prone replication through UV lesions by DNA polymerase θ protects against skin cancers. Cell 176: 1295–1309. DOI 10.1016/j.cell.2019.01.023. [Google Scholar] [CrossRef]
Yousefzadeh MJ, Wood RD. (2013). DNA polymerase POLQ and cellular defense against DNA damage. DNA Repair 12: 1–9. DOI 10.1016/j.dnarep.2012.10.004. [Google Scholar] [CrossRef]
Yu AM, McVey M (2010). Synthesis-dependent microhomology-mediated end joining accounts for multiple types of repair junctions. Nucleic Acids Research 38: 5706–5717. DOI 10.1093/nar/gkq379. [Google Scholar] [CrossRef]
Zatreanu D, Robinson HM, Alkhatib O, Boursier M, Finch H et al. (2021). Polθ inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nature Communications 12: 1–5. DOI 10.1038/s41467-021-23463-8. [Google Scholar] [CrossRef]
Zhao W, Wiese C, Kwon Y, Hromas R, Sung P et al. (2019). The BRCA tumor suppressor network in chromosome damage repair by homologous recombination. Annual Review of Biochemistry 88: 221–245. DOI 10.1146/annurev-biochem-013118-111058. [Google Scholar] [CrossRef]
Zhou J, Gelot C, Pantelidou C, Li A, Yücel H et al. (2021). A first-in-class polymerase theta inhibitor selectively targets homologous-recombination-deficient tumors. Nature Cancer 2: 598–610. DOI 10.1038/s43018-021-00203-x. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools