
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

The role of 5′-adenosine monophosphate-activated protein kinase (AMPK) in skeletal muscle atrophy
Laboratory for Bone Metabolism, Xi’an Key Laboratory of Special Medicine and Health Engineering, Key Laboratory for Space Biosciences and Biotechnology, Research Center for Special Medicine and Health Systems Engineering, NPU-UAB Joint Laboratory for Bone Metabolism, School of Life Sciences, Northwestern Polytechnical University, Xi’an, 710072, China
* Corresponding Author: AIRONG QIAN. Email:
(This article belongs to the Special Issue: Biochemical and Epigenetics Changes in Health and Disease)
BIOCELL 2023, 47(2), 269-281. https://doi.org/10.32604/biocell.2023.023766
Received 13 May 2022; Accepted 12 August 2022; Issue published 18 November 2022
 View Full Text
View Full Text Download PDF
Download PDFAbstract
As a key coordinator of metabolism, AMP-activated protein kinase (AMPK) is vitally involved in skeletal muscle maintenance. AMPK exerts its cellular effects through its function as a serine/threonine protein kinase by regulating many downstream targets and plays important roles in the development and growth of skeletal muscle. AMPK is activated by phosphorylation and exerts its function as a kinase in many processes, including synthesis and degradation of proteins, mitochondrial biogenesis, glucose uptake, and fatty acid and cholesterol metabolism. Skeletal muscle atrophy is a result of various diseases or disorders and is characterized by a decrease in muscle mass. The pathogenesis and therapeutic strategies of skeletal muscle atrophy are still under investigation. In this review, we discuss the role of AMPK in skeletal muscle metabolism and atrophy. We also discuss targeting AMPK for skeletal muscle treatment, including exercise, AMPK activators including 5-amino-4-imidazolecarboxamide ribonucleoside and metformin, and low-level lasers. These studies show the important roles of AMPK in regulating muscle metabolism and function; thus, the treatment of skeletal muscle atrophy needs to take into account the roles of AMPK.Keywords
As a central coordinator of energy homeostasis, 5′-adenosine monophosphate (AMP)-activated protein kinase (AMPK) is the cellular sensor of energy and coordinates metabolism, especially of glucose and fatty acids. AMPK responds to conditions of low energy in cells, such as nutrient depletion, hypoxia, and the inhibition of mitochondrial respiratory chain complex, and regulates the synthesis and degradation of proteins (Herzig and Shaw, 2018). AMPK has been shown to play a key role in regulating muscle metabolism (Merrill et al., 1997; Winder and Hardie, 1996). Skeletal muscle atrophy is a common muscle disease attributed to muscular inactivity, caused by immobilization, denervation, unloading, aging, and starvation (Rudrappa et al., 2016). The main pathological characteristics of skeletal muscle atrophy are a decrease in protein contents, fiber, and force and providing resistance to fatigue. The molecular aspects of muscle atrophy have been well studied for over 20 years, and AMPK has been shown to play a crucial role in skeletal muscle atrophy (Thomson, 2018).
In the present review, we discuss the role of AMPK in skeletal muscle and the involved cellular processes, including protein synthesis and degradation, and autophagy. Furthermore, we also discuss the treatment of skeletal muscle atrophy through the modulation of AMPK.
5′-Adenosine Monophosphate-Activated Protein Kinase
AMPK, a serine/threonine protein kinase, is a key enzyme in coordinating cellular energy metabolism. AMPK mainly activates the uptake and oxidation of glucose and fat in conditions of energy deficiency in cells. It is a member of a eukaryotic protein family that is highly conserved from yeast to humans, and the AMPK protein consists of three subunits required for enzyme function. AMPK is widely expressed in organs, including the brain, liver, and skeletal muscle. AMPK activation is initiated upon binding ADP and AMP and leads to the stimulation of glucose uptake and fatty acid oxidation, the inhibition of cholesterol synthesis, lipogenesis and lipolysis, and the regulation of insulin secretion (Herzig and Shaw, 2018).
The structure of 5′-adenosine monophosphate-activated protein kinase
AMPK is a heterotrimeric protein complex consisting of α, β, and γ subunits. Each subunit has a specific role in the stability and activity of AMPK (Kim et al., 2016) (Fig. 1). The α subunit (α1 and α2 isoforms, encoded by PRKAA1 and PRKAA2 genes, respectively) is the catalytic subunit. Its kinase domain (KD) contains the Thr172 regulatory site, which is phosphorylated by AMPK kinase, which is required for the functioning of AMPK (Ross et al., 2016). The α subunit also has the autoinhibitory domain (AID), two α-regulatory subunit-interacting motif-2 domains that trigger a conformational change when AMP binds to the AMPK γ subunit, and a C terminal domain (α-CTD) that binds to the β subunit.

Figure 1: The structure of 5′-adenosine monophosphate-activated protein kinase (AMPK). AMPK consists of α, β, and γ subunits. The α subunit is the catalytic subunit. The kinase domain contains the predominant regulatory site, Thr172. The α subunit also has the autoinhibitory domain, two α-regulatory subunit-interacting motif-2 domains, and a C terminal domain (α-CTD). The β subunit is the scaffolding subunit; it contains an internal carbohydrate-binding motif (CBM) and a conserved C terminal domain (β-CTD). The Ser108 in the CBM is important for the effect of some AMPK activators. The γ subunit is the regulatory subunit, comprising four domains called cystathionine β-synthase domains, two of which are called Bateman domains. N terminal domains in the γ subunit are responsible for the binding of the β subunit.
The β subunit of AMPK (β1 and β2 isoforms, encoded by PRKAB1 and PRKAB2 genes, respectively) is the scaffolding subunit. It contains an internal carbohydrate-binding motif (CBM) and a conserved CTD (β-CTD). Ser108 in the CBM is necessary for the effect of some AMPK activators (Kim et al., 2016). The α subunit-binding site and the γ subunit-binding site of the β subunit are both located in β-CTD.
The γ subunit (γ1, γ2, and γ3, encoded by PRKAG1, PRKAG2, and PRKAG3 genes, respectively) is the regulatory subunit and contains four domains called cystathionine β-synthase domains, which are four binding sites for ATP, ADP or AMP. Two of them are called Bateman domains, which are sites for AMP binding, making AMPK sensitive to the change in the AMP:ATP ratio (Ross et al., 2016). The binding of AMP and one Bateman domain can increase the affinity of AMP to the other Bateman domain. When both Bateman domains are bound with AMP, the conformation of the γ subunit changes, thus exposing the catalytic domain in the α subunit. The N terminal domain in the γ subunit binds the β subunit.
Among the isoforms of α, β, and γ subunits, α1, β1, and γ1 are the most common in cells. Various combinations of the different isoforms of these three subunits result in twelve AMPK configurations. However, only α2/β2/γ1, α2/β2/γ3, and α1/β2/γ1 configurations are observed in human skeletal muscle (Birk and Wojtaszewski, 2006). In mouse muscle, only α1/β1/γ1 and α2/β1/γ1 are detected. The functional implications of different configurations are not yet clear, and a more exhaustive understanding of the functions of these trimers in different tissues and muscles is needed.
The regulation of 5′-adenosine monophosphate-activated protein kinase
AMPK is regulated structurally and by post-translational modification (Jeon, 2016). Phosphorylation of Thr172 in the AMPK α subunit is the main mechanism of AMPK activation. Blockage of the phosphatase access to Thr172 can increase AMPK activation. AMPK can be allosterically regulated by the competitive binding of its γ subunit to ATP and AMP or ADP. ATP allows the access of phosphatase to Thr172, while AMP or ADP blocks this access. Thus, AMPK is a sensitive sensor of AMP/ATP or ADP/ATP ratios, considered indicators of cell energy level (Hardie et al., 2012). The molecular mechanism of the allosteric activation of AMPK by AMP binding has been demonstrated earlier (Steinberg and Carling, 2019). The autoinhibitory domain in the AMPK α subunit interacts with the KD, leading to the inactive conformation of AMPK. Upon the binding of AMP to the AMPK γ subunit, the α-regulatory subunit-interacting motif-2 and α-CTD interact with the adenosine nucleotides, resulting in conformation changes that expose the KD to activate the AMPK subunit complex.
Many kinases and related proteins are involved in the phosphorylation and dephosphorylation of Thr172 in AMPK. The kinases that phosphorylate Thr172 include liver kinase B1 (LKB1), Ca2+/calmodulin-dependent protein kinase kinase 2 (CAMKK2), and TGF-β-activated kinase 1 (TAK1) (Neumann, 2018). Protein phosphatase 2A, protein phosphatase 2C, and Mg2+/Mn2+-dependent protein phosphatase 1E are the main phosphatases that dephosphorylate Thr172 (Yan et al., 2021). LKB1-mediated AMPK phosphorylation at Thr172 can be enhanced by AMP binding to the AMPK γ subunit (Kim et al., 2016). However, CaMKK2 responds to the increase in cellular Ca2+ and activates AMPK without influencing the ATP/ADP/AMP levels (Mairet-Coello et al., 2013). Furthermore, myristoylation of the AMPK β subunit N terminal is also involved in the effect of AMP on Thr172 phosphorylation (Ali et al., 2016).
AMPK is inhibited by insulin, leptin, and diacylglycerol (Jeon, 2016). AMPK also controls gene expression through metabolites of intermediary metabolisms, such as glucose, fatty acids, and ketogenic amino acids (Sukumaran et al., 2020). The interaction of AMPK and glycogen alters the conformation and activity of AMPK (Janzen et al., 2018). LKB1, CAMKK, and TAK1 are important kinases for AMPK activation in skeletal muscle, and LKB1 is regarded as the primary AMPK kinase (Thomson, 2018).
The physiological function of 5′-adenosine monophosphate-activated protein kinase
In response to metabolic stress, AMPK plays a pivotal role in maintaining anabolic and catabolic balance, contributing to glucose/lipid homeostasis (Trefts and Shaw, 2021). AMPK phosphorylates acetyl-CoA carboxylase 1 (ACC1) and sterol regulatory element-binding protein 1c (SREBP1c), thus inhibiting the synthesis of fatty acids and cholesterol and activating the uptake and oxidation of fatty acids (Jeon, 2016). AMPK also phosphorylates Rab-GTPase-activating proteins TBC1D1 and TCB1D4, thus inducing the fusion of glucose transporter 1 to the plasma membrane and stimulating glucose uptake (Sakamoto and Holman, 2008; Stockli et al., 2008). AMPK stimulates glycolysis by activating the phosphorylation of 6-phosphofructo-2-kinase and glycogen phosphorylase. AMPK inhibits glycogen synthesis via phosphorylation of glycogen synthase (Jeon, 2016). AMPK also inhibits transcription factors such as hepatocyte nuclear factor 4 and cAMP response element-binding (CREB) regulated transcription coactivator 2 and also inhibits gluconeogenesis in the liver (Lee et al., 2010). AMPK regulates the energy-related protein biosynthesis process through phosphorylating tuberous sclerosis complex 2 (TSC2). Upon TSC2 activation, TSC then inhibits the mechanistic target of rapamycin complex 1 (mTORC1), resulting in a decrease in protein synthesis. In energy-deficient conditions in cells, AMPK is activated; all energy-consuming pathways, such as protein synthesis are halted, and energy-producing pathways are activated (Hardie, 2011). AMPK activates autophagy by activating Unc-51 like autophagy activating kinase 1 (ULK1) directly and indirectly (Mao and Klionsky, 2011) and regulates mitochondrial biogenesis via peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1 alpha (PGC-1α), which can promote gene transcription in mitochondria (Canto and Auwerx, 2009).
AMPK is a therapeutic target in the treatment of metabolic diseases such as diabetes (Day et al., 2017) and also plays a role in tumorigenesis. The upstream regulatory kinase LKB is also a tumor suppressor (Chuang et al., 2014), and AMPK controls mTORC1 and TIF-1A, which are involved in cell proliferation (Sukumaran et al., 2020). Given its protective role in tumor cells in conditions of limited nutrients, hypoxia, and/or toxicity, AMPK activation should be considered for cancer treatment.
Skeletal muscle atrophy is the loss of skeletal muscle mass, caused by muscle disuse, malnutrition, medications, aging, injuries, or nervous system diseases, leading to muscle weakness and disability (Powers et al., 2016). Muscle disuse can lead to muscle atrophy, which often occurs after motion limitation of limb or bed-rest during related illness. Skeletal muscle atrophy induced by disuse can be fully recovered with activity if the illness is cured. Malnutrition and cachexia caused by an underlying disease can also progress to skeletal muscle atrophy, which can be reversed either completely or incompletely by nutritional therapy. Age-related skeletal muscle atrophy can be slowed down by exercise but not completely reversed. Muscle diseases (such as muscular dystrophy) and nervous system diseases (such as stroke) can result in skeletal muscle atrophy, and recovery depends on the duration and severity of the underlying diseases along with the health of the patient. The predominant symptom of skeletal muscle atrophy is increasing weakness, leading to difficulty or inability in physical tasks. Skeletal muscle atrophy can be undetected until significant loss of the muscle occurs, and symptoms are different depending on the affected muscles.
Pathologically, skeletal muscle atrophy results from the imbalance between the synthesis and degradation of the protein. The mechanisms of muscle atrophy are not yet completely understood and vary depending on the underlying reason. Skeletal muscle is a storage center for amino acids that are used for producing energy when the energy supplies are low, or energy demands are high. When the metabolic level is greater than that of protein synthesis, the loss of muscle mass will occur (Atherton and Smith, 2012). Mitochondria are vital for skeletal muscle health, and harmful changes in mitochondria may lead to skeletal muscle atrophy. Decreased density and quality of mitochondria are commonly observed in skeletal muscle atrophy when skeletal muscle is disused (Harper et al., 2021). ATP-dependent protein degradation pathway is also one of the main mechanisms for protein metabolism in muscles, and any change in this pathway might lead to muscle atrophy.
Treatment approaches for skeletal muscle atrophy include enhancing the pathways leading to muscle hypertrophy or reducing muscle breakdown. Physical exercise, especially resistance, is one of the crucial approaches to slow or reverse skeletal muscle atrophy (Lavin et al., 2019). In individuals unable to exercise due to immobilization, such as spinal cord injury, electrical stimulation of muscle can be used (Ho et al., 2014). Calories and protein supplements are also beneficial for skeletal muscles, especially for patients with malnutrition. Protein or amino acids, such as leucine, can stimulate protein synthesis in muscles and inhibit protein degradation and are, therefore, beneficial for some cases of skeletal muscle atrophy, such as cachexia (Argiles et al., 2016). A metabolite of leucine, β-hydroxy β-methylbutyrate (HMB), is effective in preventing muscle loss in patients with sarcopenia and is used as a dietary supplement (Cavallucci and Pani, 2021). HMB is also used to prevent muscle loss in the elderly. However, the detailed effect of HMB needs more investigation. Furthermore, the administration of anabolic steroids and selective androgen receptor modulators to patients with skeletal muscle atrophy are also being evaluated. Their effectiveness in promoting muscle growth and regeneration, as well as a few side effects, requires further investigation before clinical application.
The Role of 5′-Adenosine Monophosphate-Activated Protein Kinase in Skeletal Muscle Atrophy
Since skeletal muscle atrophy occurs due to the imbalance between protein synthesis and degradation and AMPK-involved pathways play important roles in protein synthesis and degradation, the role of AMPK-related mechanisms in the pathogenesis of skeletal muscle atrophy is important.
5′-adenosine monophosphate-activated protein kinase inhibits muscle protein synthesis
Skeletal muscles are the largest reservoir of proteins and amino acids in the body and are important for locomotion (Argiles et al., 2016). Muscle mass maintenance is crucial for life quality. Loss of muscle mass is the typical feature of skeletal muscle atrophy. Understanding the mechanisms of skeletal muscle atrophy, especially protein synthesis and degradation in muscle, is important for muscle rehabilitation.
The most well-recognized player in regulating muscle mass in skeletal muscle is mTOR (Yoon, 2017). Like AMPK, mTOR is also a serine/threonine kinase; it can sense intracellular and extracellular changes in nutrient and energy levels and regulates various processes in cells, such as cell growth and differentiation, autophagy, metabolism, and survival. mTORC1 and mTORC2 are two mTOR complexes with distinct functions (Grahammer et al., 2021). mTORC1 is composed of the regulatory-associated protein of mTOR (RAPTOR), the mammalian lethal with SEC13 protein 8 (mLST8), a 40 kDa proline-rich Akt substrate, and the DEP domain-containing mTOR-interacting protein (DEPTOR). mTORC2 is composed of mLST8, DEPTOR, the mammalian stress-activated map kinase-interacting protein 1, the rapamycin-insensitive companion of mTOR (RICTOR), and a protein with RICTOR 1 and 2 (PROTOR1/2). mTORC1 is sensitive to rapamycin and it is the key to regulating muscle mass in muscle contraction and loading. mTORC1 responds to cellular signals, including intracellular energy status, oxygen levels, growth factors, and amino acid availability. When activated, mTORC1 stimulates protein synthesis by phosphorylating its downstream targets, including S6 kinase 1, and inhibiting 4E-binding protein 1, promoting cell growth.
AMPK inhibits muscle protein synthesis by regulating the mTORC1 pathway. It can inhibit mTORC1 activity via the following mechanisms. a. AMPK phosphorylates mTOR at Thr2446, which can halt mTORC1 activity by preventing its phosphorylation at Ser2448; however, the impact of Ser2448 phosphorylation on mTORC1 activity has not been determined (Melick and Jewell, 2020). b. AMPK can also phosphorylate TSC2 to inhibit mTORC1 activity (Kim and Lee, 2015); TSCs can activate GTPase, which can convert GTP to GDP and can form a complex with TSC1 and TBC1D7, reducing the ability of a GTPase, Rheb, whose binding with GTP is needed for mTORC1 activity. c. AMPK can phosphorylate RAPTOR, leading to impaired mTORC1 activity mediated by 14-3-3 proteins (Thomson, 2018).
In 2002, Bolster et al. (2002) reported that the injection of 5-amino-4-imidazolecarboxamide ribonucleoside (AICAR), an AMPK activating drug, can reduce the rate of protein synthesis in skeletal muscle; this finding was considered the first evidence of the role of AMPK in protein metabolism in skeletal muscle. Since then, the negative effect of AMPK on protein synthesis has been confirmed in many other cell types, including hepatocytes (Dubbelhuis and Meijer, 2002; Reiter et al., 2005), myocytes (Chan et al., 2004), and tumor cells (Fay et al., 2009; Xiang et al., 2004). In addition to the inhibitory effect on mTORC1, AMPK can also regulate protein synthesis by inhibiting the activity of eukaryotic elongation factor 2 (eEF2) (Yamada et al., 2019). AMPK can directly phosphorylate and activate eEF2 kinase (eEF2K), which can phosphorylate eEF2 at Thr56. The phosphorylation of eEF2 leads to eEF2 inactivation, indicated as the inhibition of its binding to the ribosome, slowing down the elongation rate and the subsequent protein synthesis. AMPK also prevents the inhibitory effect of S6k, which can phosphorylate and inhibit eEF2K and lead to eEF2 activation by inhibiting the mTOR pathway (Wang et al., 2014). Thus, while AMPK acts as a negative regulator of skeletal muscle mass, the regulation of eEF2 by AMPK in skeletal muscle has not been fully unveiled and needs more in-depth investigation.
5′-adenosine monophosphate-activated protein kinase stimulates protein degradation through autophagy/ubiquitin/FoxO3a-dependent ways
In addition to the inhibitory effect on protein synthesis, AMPK is also involved in protein degradation in muscles. AMPK activator was shown to increase protein degradation in cultured mouse myoblast C2C12 cells (Das et al., 2012) And stimulate protein degradation through autophagy/ubiquitin/FoxO3a-dependent ways (Fig. 2).

Figure 2: 5′-adenosine monophosphate-activated protein kinase (AMPK) regulates protein synthesis and degradation in skeletal muscle. AMPK inhibits muscle protein synthesis by regulating the mTORC1 pathway. It also stimulates protein degradation through autophagy/ubiquitin/FoxO3a-dependent ways. TSC1, tuberous sclerosis complex 1; TSC2, tuberous sclerosis complex 2; Mtor, mammalian target of rapamycin; S6K1, ribosomal protein S6 kinase 1; ULK1, Unc-51 like autophagy activating kinase 1; FoxO3a, Forkhead box O3a; ATGs, autophagy-relatedgenes; MAFbx, muscle atrophy F-box; MuRF-1, muscle ring finger-1.
Autophagy is the process through which defective cellular components, such as organelles, are degraded and recycled in nutrition-deprived or low-energy supply conditions (Kitada and Koya, 2021). Autophagy involves the engulfment of the target into autophagosomes, followed by the fusion of the autophagosome with a lysosome and subsequent degradation of the autophagosome. Autophagy is one of the main pathways for protein degradation in skeletal muscle atrophy, especially in nutrient deprivation-induced conditions. AMPK can promote autophagy via regulating the ULK1 complex, which consists of ULK1, mTORC1, AMPK, the FAK family kinase-interacting protein of 200 kDa (FIP200), and autophagy-related 13 (ATG13) (Alers et al., 2012). ULK1, a serine/threonine kinase, plays a crucial role in the initiation of autophagy. Activation of AMPK can result in its association with the ULK1 complex, which facilitates the phosphorylation of ATG13 and FIP200, thus activating autophagy (Alers et al., 2012). AMPK also directly phosphorylates ULK1 to increase the activity of ULK1 kinase, taking part in autophagosome formation (Kim et al., 2011).
Ubiquitin-proteasome-mediated degradation is another protein degradation pathway that leads to skeletal muscle atrophy. In this process, target proteins are modified by ubiquitin in a mechanism involving three enzymes, E1, E2, and E3. The 26S proteasome then degrades the tagged proteins. In skeletal muscle atrophy, atrophy-related proteins, the muscle-specific ubiquitin ligases muscle atrophy F-box (MAFbx, also called atrogin-1) and muscle ring finger-1 (MuRF-1), are the main E3 ligases that regulate protein breakdown (Bodine and Baehr, 2014). AMPK activation can increase the mRNA content of MuRF-1 and MAFbx, leading to the initiation of the ubiquitin-proteasome-mediated degradation system (Baskin and Taegtmeyer, 2011). No evidence has shown AMPK-mediated regulation of E1 and E2 in skeletal muscle, which requires more research.
The transcription of MuRF-1 and MAFbx genes is also under the control of Forkhead box O3a (FoxO3a), which can upregulate the transcription of many genes in the autophagy pathway, such as LC3B, beclin1, ULK2, and ATG4B. FoxO proteins are evolutionarily conserved transcription factors and regulated by nuclear/cytoplasmic shuttling in a phosphorylation-dependent manner (Boccitto and Kalb, 2011). FoxO proteins play roles in the regulation of protein degradation as well as tumor suppression and development. In skeletal muscle, AMPK can activate FoxO3a via phosphorylation at Ser413/588 and regulate both autophagy and ubiquitin-proteasome degradation. The activation of FoxO3 leads to the increased expression of beclin 1 and LC3B, promoting autophagosome formation. There are debatable views on the effect of AMPK activation on FoxO3a shuttling between the nucleus and cytoplasm (Fasano et al., 2019). Nevertheless, studies have shown that AMPK can activate FoxO3a directly to increase its transcriptional activity and thus has a role in the stability of proteins (Fig. 3). AMPK can regulate the activities of class IIA histone deacetylases, such as by histone deacetylase 5 (HDAC5) phosphorylation at Ser258 and Ser498, and affect myogenesis and muscular regeneration (Fu et al., 2013).

Figure 3: The regulatory role of 5′-adenosine monophosphate-activated protein kinase (AMPK) in skeletal muscle. AMPK can exert its biological functions by regulating the phosphorylation of many kinases, some of which are listed in this figure. AMP, 5′-adenosine monophosphate; ULK1, Unc-51 like autophagy activating kinase 1; TSC2, tuberous sclerosis complex 2; eEF2K, eukaryotic elongation factor 2 kinase; GS, glycogen synthase; CREB, cAMP response element-binding; GLUT-4, glucose transporter-4; PGC-1α, peroxisome proliferator-activated receptor-gamma coactivator-1α; PPARα, peroxisome proliferator-activated receptor α; ACC2, acetyl-CoA carboxylase 2; ACC1, acetyl-CoA carboxylase 1; SREBP1c, sterol regulatory element-binding protein 1c; ChREBP, carbohydrate response element binding protein; FoxO3a, Forkhead box O3a.
The role of 5′-adenosine monophosphate-activated protein kinase in mitochondrial dysfunction
Mitochondria are the main organelles responsible for the production of energy. The fusion and fission proteins regulate the morphology and subcellular location of mitochondria (Glancy et al., 2020). The inclination to fission machinery, which involves AMPK activation, can cause muscle dysfunction. The disruption of the network in mitochondria contributes to skeletal muscle atrophy. As observed in cases of muscle atrophy, such as in disuse, aging, and diabetic amyotrophy, mitochondria dysfunction has a pivotal role in skeletal muscle atrophy, characterized by loss, morphological change, and localization of the mitochondria.
AMPK can regulate mitochondrial biogenesis, as mentioned above, by phosphorylation of PGC-1α (at Thr117 and Ser538), which is the primary controller for mitochondrial biogenesis and inhibits muscle atrophy under fasting and denervation conditions (Vainshtein et al., 2015). AMPK also directly phosphorylates FoxO3 on its regulatory sites and induces the mitochondrial fission cascade in skeletal muscle (Herzig and Shaw, 2018). The role of AMPK in regulating mitochondria function in muscle might depend on the specific conditions. By regulating ULK1, as discussed above, AMPK also modulates mitophagy, the autophagy-lysosome system in mitochondria (Fig. 4). Targeting mitochondria and improving mitochondrial function may provide a promising method for the treatment of skeletal muscle atrophy.
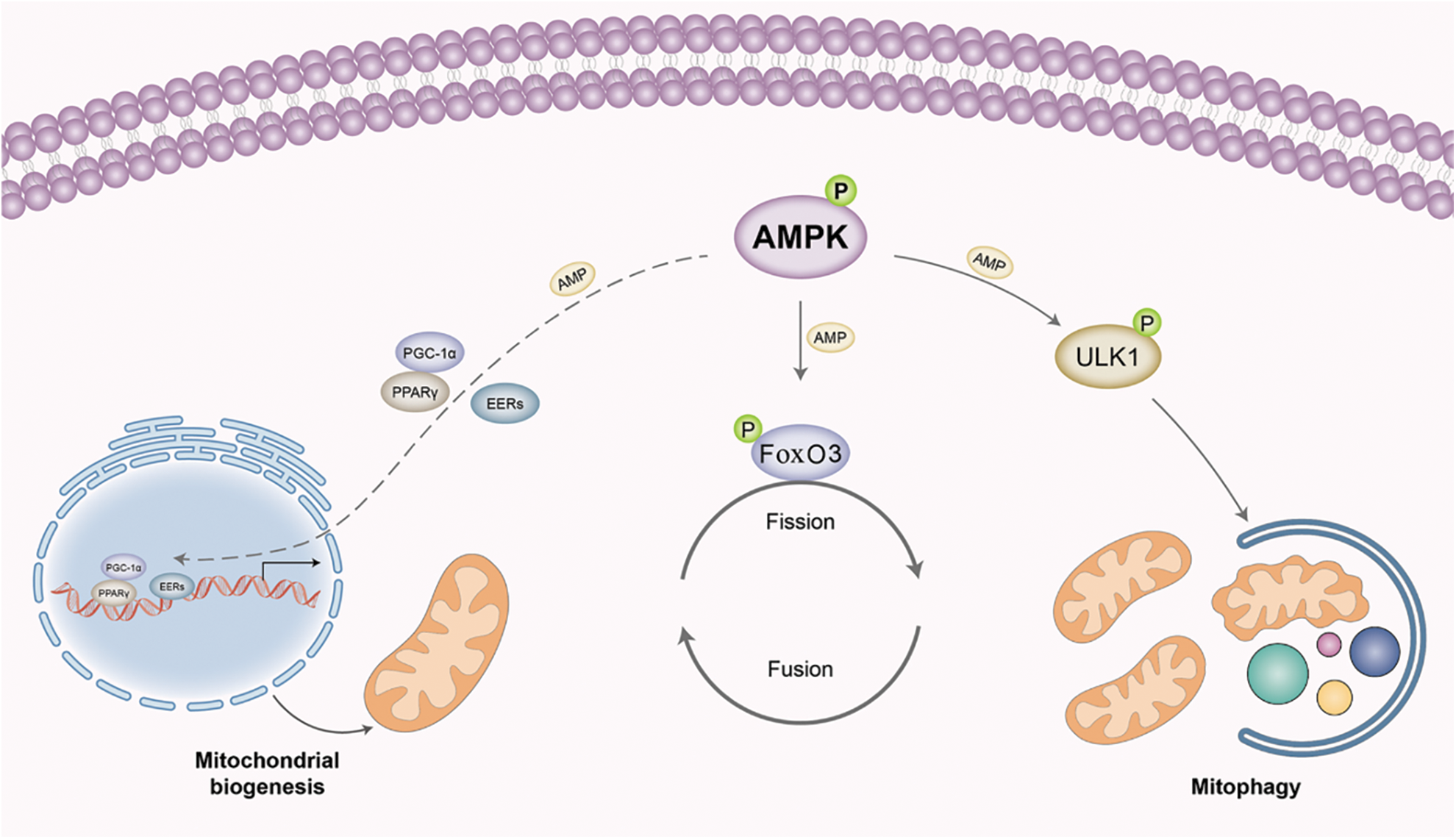
Figure 4: 5′-adenosine monophosphate-activated protein kinase (AMPK) regulates mitochondrial homeostasis in skeletal muscles. AMPK can enhance mitochondria biogenesis by phosphorylation of peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1α, which is the main controller for mitochondrial biogenesis. AMPK can also directly phosphorylate Forkhead box O3 (FoxO3) on its regulatory sites and induce the mitochondrial fission cascade. By regulating Unc-51 like autophagy activating kinase 1 (ULK1), AMPK can also modulate the autophagy-lysosome system in mitochondria, named mitophagy.
5′-adenosine monophosphate-activated protein kinase promotes glucose uptake and fatty acid and cholesterol metabolism
In the anaerobic and aerobic metabolism in skeletal muscle, insulin-dependent transport and metabolism of glucose are under the regulation of AMPK. The expression and translocation of glucose transporter-4 (GLUT-4) are induced by AMPK activation, followed by an increase in glucose uptake and oxidation (Webster et al., 2010). The effect of AMPK on GLUT-4 is thought to be mediated by AMPK-mediated phosphorylation of TBC1D1 at Ser231 (Vichaiwong et al., 2010). By phosphorylating CREB at Ser133, AMPK increases the transcription of hexokinase-2, which is responsible for the synthesis of glucose-6-phosphate (Feng et al., 2020), promoting glycolysis. Moreover, AMPK can phosphorylate and thus inactivate glycogen synthase at Ser7, resulting in the inhibition of glycogenogenesis (Janzen et al., 2018). This effect is dependent on the α2 subunit of AMPK. In skeletal muscle, AMPK activation can promote more glycolysis and inhibit glycogenolysis, resulting in more glucose uptake.
AMPK can also regulate the metabolism of fatty acid and cholesterol in skeletal muscle by phosphorylating Ser79 of ACC1, Ser871 of the hydroxymethylglutaryl-coenzyme A reductase, and Ser565 of hormone-sensitive lipase (Srivastava et al., 2012; Wang et al., 2018). AMPK also decreases the transcription of lipogenic genes by stalling the activity of SREBP1c through Ser372 phosphorylation and the activity of carbohydrate response element binding protein (ChREBP) by phosphorylation at Ser568; both these transcription factors regulate lipogenic genes, repressing the synthesis of fatty acids (Li et al., 2011; Liangpunsakul et al., 2013). AMPK can phosphorylate the Ser212 of acetyl-CoA carboxylase and inhibit its activity, leading to reduced malonyl-CoA, which can allosterically inhibit carnitine palmitoyltransferase 1 (Lee et al., 2018; Wakil and Abu-Elheiga, 2009), which is the rate-limiting enzyme in fatty acid oxidation, that transfers cytosolic long chain fatty acyl CoA into mitochondria. Thus, AMPK activation promotes the oxidation of fatty acids by downregulating malonyl-CoA in mitochondria. Furthermore, AMPK phosphorylates PGC-1α at Ser538 and Thr177 and activates PGC-1α, a crucial protein in the oxidative metabolism in skeletal muscles (McGee and Hargreaves, 2010). PGC-1α upregulates nuclear respiratory factors and other enzymes in the electron transport system, thus promoting mitochondrial respiration and biogenesis (Cheng et al., 2018). For instance, PGC-1α activates nuclear respiratory factor-1 through mitochondrial transcription factor A promoter, which regulates mitochondrial DNA replication and transcription (Scarpulla et al., 2012). During exercise, AMPK activity inhibition in skeletal muscle decreases cellular NAD+ levels, resulting in the impairment of PGC-1α deacetylation and inhibition of mitochondrial gene expression (Canto et al., 2009). Regulation of the expression of mitochondrial genes by AMPK is mostly dependent on PGC-1α.
Together, these studies indicate that AMPK promotes glucose uptake and fatty acid oxidation in skeletal muscle by direct or indirect regulation of related enzymes.
Targeting 5′-Adenosine Monophosphate-Activated Protein Kinase for the Treatment of Skeletal Muscle Atrophy
Although many studies have investigated the AMPK signaling pathway, the response of AMPK in skeletal muscle atrophy is still unclear, and conflicting results have been reported. Rodent models of disused skeletal muscle atrophy established by hindlimb unloading were reported to show increased (2–13 weeks) (Hilder et al., 2005) or decreased (3–7 d or 4 weeks) (Cannavino et al., 2015; Liu et al., 2012) AMPK phosphorylation levels. Other studies showed no effect of hindlimb unloading on AMPK activity and AMPK-related enzymes (Egawa et al., 2015). Vilchinskaya et al. (2015) analyzed signaling pathways in the proteolysis in human soleus muscle during gravitational unloading and found significant changes in AMPK phosphorylation. Later, a summary of the mechanisms for the dysregulated autophagy-mediated sarcopenic obesity indicated the involvement of AMPK and PGC1α signaling pathways in this process (Ryu et al., 2020). Increased activity of AMPK was observed in denervated mice or rats (Gao and Li, 2018; Guo et al., 2016; Ribeiro et al., 2015), but its activity was also reported to be unchanged in muscle (Dreyer et al., 2008). The difference in results in denervation models may be due to different handling times. AMPK phosphorylation was shown to increase in the early stage of denervation and decrease in later stages (Stouth et al., 2018). The activity of AMPK dynamically changes in skeletal muscle atrophy, at least in denervation models. Targeting AMPK might be an effective approach for the treatment of skeletal muscle atrophy. Furthermore, AMPK is involved in most existing treatments for skeletal muscle atrophy. The roles of AMPK in the treatment of skeletal muscle atrophy-related diseases, which mostly involve AMPK activation, are listed in Table 1.

Exercise is considered an effective recovery approach for skeletal muscle atrophy, especially atrophy caused by disuse. Exercise, especially physical exercise throughout life, plays an important role in maintaining a healthy life, including that in elderly individuals. The upregulation of skeletal muscle glucose uptake is a well-known response to exercise and is mediated by improving insulin sensitivity and maintaining glucose homeostasis in both direct and indirect methods (Parker et al., 2017). Exercise also promotes energy consumption, decreases ATP levels, and increases AMP levels in cells, which induces AMPK phosphorylation and activation, strengthening glucose uptake in skeletal muscle (Radhakrishnan et al., 2019). Nonetheless, the role of AMPK activation in stimulating glucose uptake under physiological conditions is still under investigation.
Exercise for different durations and intensities can influence the different subunits of AMPK. Under exercise of mild intensity and 1 h of duration, α1β2γ1 and α2β2γ1 are the main activated forms of AMPK (Treebak et al., 2007). Under intensive exercise for 20 min, α2β2γ3 is activated (Richter and Ruderman, 2009). The α2 subunit can be phosphorylated in prolonged exercise, and LKB1 is the primary kinase for the phosphorylation of the AMPK α2 subunit (Richter and Ruderman, 2009). The increase in intracellular Ca2+ that induces CaMKK phosphorylation is another mechanism for AMPK activation. Stress-induced reactive oxygen species increase in the endoplasmic reticulum and muscle temperature increase may also contribute to AMPK activation during physical exercise (Gariballa and Ali, 2020; Morales-Alamo and Calbet, 2016). Different modalities of exercise also show different effects on AMPK phosphorylation. Strength- and endurance-trained subjects showed different responses to AMPK phosphorylation levels in the body after cycling (Coffey et al., 2005). Furthermore, AMPK activity in exercise is dependent on the level of AMP but not on the duration and intensity of exercise, but the level of AMP in the body is higher during physical exercise than in resting conditions until exhaustion (Morales-Alamo and Calbet, 2016).
A regular habit of exercise is of great benefit to elderly individuals, since it can also induce AMPK activity. In addition to skeletal muscle atrophy, exercise-induced glucose uptake and AMPK activation can exert beneficial effects on insulin secretion and hemodynamic changes in aging people. The following cascades of AMPK activation include PGC-1α activation (for mitochondrial biogenesis, and energy homeostasis), peroxisome proliferator-activated receptor-α expression (for fatty acid oxidation), autophagy stimulation, and mTOR inhibition. However, the distinct exercise types in previous research should be considered. Especially in the recovery of skeletal muscle atrophy, one needs moderate exercise since excessive exercise can result in excessive autophagy, which can lead to skeletal muscle atrophy and related diseases (Xia et al., 2021).
The role of 5-amino-4-imidazolecarboxamide ribonucleoside (AICAR)
AICAR has long been applied as an AMPK activator in skeletal muscle and other tissues (Corton et al., 1995; Hardman et al., 2014; Thomson et al., 2008). After administration, AICAR is converted to its monophosphate form, ZMP, which is a mimic of AMP and can activate AMPK without any change in the levels of intracellular adenine nucleotide. In rat gastrocnemius, like prolonged exercise, AICAR can activate the α2 subunit of AMPK but not the α1 subunit (Bolster et al., 2002). Thus, AICAR-induced glucose uptake is diminished in AMPKα2 knockout mice but not in AMPKα1 knockout mice (Choi et al., 2019). However, AICAR can activate AMPK α2, as well as the AMPK α1 subunit in vitro (Musi et al., 2001).
Based on the role of AMPK activation (inhibiting protein synthesis, promoting protein degradation), AICAR should, in principle, accelerate the loss of muscle mass. However, in a rat denervation model, AICAR injection did not exacerbate skeletal muscle atrophy (Paulsen et al., 2001). In fact, AICAR improved muscle function in mdx mice, a mouse model of Duchenne muscular dystrophy (Ljubicic and Jasmin, 2013; Pauly et al., 2012). The mechanisms included enhanced autophagy, clearance of damaged components, and upregulation of oxidative muscle phenotype, probably through AMPK activation. More investigations of the AMPK-involved treatment for Duchenne muscular dystrophy are in progress.
Metformin is a well-known drug for the treatment of diabetes and insulin resistance. It can alleviate hyperglycemia, independent of insulin. The metabolic effect of metformin is similar to that of AMPK, and some effects of metformin are independent of AMPK activation (Zhou et al., 2001). Metformin inhibits oxidative phosphorylation in mitochondria, reduces the production of ATP, and results in energy stress in cells; all these events can indirectly activate AMPK (Rena et al., 2017). AMPK α2 (but not α1) subunit is the target for metformin in skeletal muscle, and chronic administration of metformin can increase the activity of the AMPK α2 subunit (Musi et al., 2002), an effect that can sustain even after the withdrawal of metformin. However, in other studies, both α1 and α2 subunits of AMPK were activated by metformin treatment, both in vivo and ex vivo (Oshima et al., 2015). In skeletal muscle cells, metformin has been shown to impair muscle function and induce muscle atrophy via the AMPK-FoxO3a-HDAC6 pathway (Kang et al., 2021). In burn injury patients, metformin treatment can activate AMPKα2 in skeletal muscle and lead to an increase in protein synthesis (Yousuf et al., 2020). In a tumor cachexia animal model, metformin treatment can promote protein synthesis as well as decrease protein degradation when the treatment activates AMPK (Oliveira and Gomes-Marcondes, 2016). The improvement in muscle metabolism of metformin may be attributed to its effect on insulin sensitivity, which regulates glucose uptake and protein synthesis. However, whether AMPK activation has a role in metformin-induced muscle recovery needs further investigation.
The effect of low-level lasers
Ou et al. (2021) investigated the effect of low-level laser irradiation on muscle atrophy and the related mechanism in the doxorubicin (Dox)-induced skeletal muscle atrophy model. In both animal and cultured cells, irradiation with low-level lasers alleviated Dox-induced muscle wasting; Dox-induced mitochondrial dysfunction, cell apoptosis, and oxidative stress were inhibited through the AMPK/SIRT1/PGC-1α pathway. The study not only provided evidence for the benefit of low-level lasers on Dox-induced muscle disease but also unveiled the role of AMPK activation in muscle atrophy treatment, in which the activation of the AMPK/SIRT1/PGC-1α pathway exerts protection against Dox myotoxicity through preservation of mitochondrial homeostasis and alleviation of oxidative stress and apoptosis.
AMPK is a master regulator of kinase in the metabolism of skeletal muscles. It inhibits protein synthesis in the muscle by inhibiting mTORC1 activity both directly and indirectly. AMPK can also stimulate protein degradation through the autophagy-lysosomal system, ubiquitin-proteasome system, and FoxO3a-dependent pathways, playing an important role in protein turnover. AMPK also enhances mitochondrial biogenesis and promotes glucose uptake and metabolism of fatty acids and cholesterol. Skeletal muscle is the tissue with an active metabolism, regardless of glucose and energy, making AMPK important in skeletal muscle. Modulating AMPK activity has been shown to contribute to the therapy of skeletal muscle atrophy. Therefore, it is important to understand the specific mechanisms that regulate AMPK and its downstream cascades to improve the current therapeutic strategies and develop new methods to protect against skeletal muscle atrophy. Considering the general role of AMPK activation in promoting muscle atrophy, it seems inhibition of AMPK should be a therapeutic strategy; however, many muscle atrophy treatments were shown to activate AMPK. Given the diverse effects of AMPK, inhibition of AMPK may increase the “quantity” of muscle but may result in poor “quality” of the muscle. The effect of these therapeutic means on AMPK and resultant signaling depends on the modality and duration. More detailed investigations are needed, considering the specific role of AMPK on specific types and stages of skeletal muscle atrophy. Notwithstanding, the crucial role of AMPK in skeletal muscle atrophy is incontrovertible, and targeting AMPK for developing new drugs with research on the mechanism for skeletal muscle atrophy and even other diseases, such as diabetes and cancer, is of great significance.
Author Contribution: KD designed the research and drafted the manuscript. KD, HMUF, SFJ and XD conducted the literature research. ARQ conceived the concept, participated in its design and coordination, and helped in drafting the manuscript.
Ethics Approval: Not applicable.
Funding Statement: This work was supported by the Natural Science Foundation of China (Grant No. 32071517, 82072106) and the Natural Science Basic Research Plan in Shaanxi Province of China (Grant No. 2020JM-100).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
Alers S, Loffler AS, Wesselborg S, Stork B (2012). Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Molecular and Cellular Biology 32: 2–11. DOI 10.1128/MCB.06159-11. [Google Scholar] [CrossRef]
Ali N, Ling NM, Krishnamurthy S, Oakhill JS, Scott JW, Stapleton DI, Kemp BE, Anand GS, Gooley PR (2016). Beta-subunit myristoylation functions as an energy sensor by modulating the dynamics of AMP-activated Protein Kinase. Scientific Reports 6: 39417. DOI 10.1038/srep39417. [Google Scholar] [CrossRef]
Argiles JM, Campos N, Lopez-Pedrosa JM, Rueda R, Rodriguez-Manas L (2016). Skeletal muscle regulates metabolism via interorgan crosstalk: Roles in health and disease. Journal of the American Medical Directors Association 17: 789–796. DOI 10.1016/j.jamda.2016.04.019. [Google Scholar] [CrossRef]
Atherton PJ, Smith K (2012). Muscle protein synthesis in response to nutrition and exercise. Journal of Physiology 590: 1049–1057. DOI 10.1113/jphysiol.2011.225003. [Google Scholar] [CrossRef]
Baskin KK, Taegtmeyer H (2011). AMP-activated protein kinase regulates E3 ligases in rodent heart. Circulation Research 109: 1153–U1161. DOI 10.1161/CIRCRESAHA.111.252742. [Google Scholar] [CrossRef]
Birk JB, Wojtaszewski JF (2006). Predominant α2/β2/γ3 AMPK activation during exercise in human skeletal muscle. The Journal of Physiology 577: 1021–1032. DOI 10.1113/jphysiol.2006.120972. [Google Scholar] [CrossRef]
Boccitto M, Kalb RG (2011). Regulation of foxo-dependent transcription by post-translational modifications. Current Drug Targets 12: 1303–1310. DOI 10.2174/138945011796150316. [Google Scholar] [CrossRef]
Bodine SC, Baehr LM (2014). Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. American Journal of Physiology-Endocrinology and Metabolism 307: E469–E484. DOI 10.1152/ajpendo.00204.2014. [Google Scholar] [CrossRef]
Bolster DR, Crozier SJ, Kimball SR, Jefferson LS (2002). AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. Journal of Biological Chemistry 277: 23977–23980. DOI 10.1074/jbc.C200171200. [Google Scholar] [CrossRef]
Cannavino J, Brocca L, Sandri M, Grassi B, Bottinelli R, Pellegrino MA (2015). The role of alterations in mitochondrial dynamics and PGC-1α over-expression in fast muscle atrophy following hindlimb unloading. Journal of Physiology 593: 1981–1995. DOI 10.1113/jphysiol.2014.286740. [Google Scholar] [CrossRef]
Canto C, Auwerx J (2009). PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Current Opinion in Lipidology 20: 98–105. DOI 10.1097/MOL.0b013e328328d0a4. [Google Scholar] [CrossRef]
Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J (2009). AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458: U1056–U1140. DOI 10.1038/nature07813. [Google Scholar] [CrossRef]
Cavallucci V, Pani G (2021). The leucine catabolite and dietary supplement β-hydroxy-β-methyl Butyrate (HMB) as an epigenetic regulator in muscle progenitor cells. Metabolites 11: 512. DOI 10.3390/metabo11080512. [Google Scholar] [CrossRef]
Chan AYM, Soltys CLM, Young ME, Proud CG, Dyck JRB (2004). Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. Journal of Biological Chemistry 279: 32771–32779. DOI 10.1074/jbc.M403528200. [Google Scholar] [CrossRef]
Cheng CF, Ku HC, Lin H (2018). PGC-1 as a pivotal factor in lipid and metabolic regulation. International Journal of Molecular Sciences 19: 3447. DOI 10.3390/ijms19113447. [Google Scholar] [CrossRef]
Choi RH, McConahay A, Johnson MB, Jeong HW, Koh HJ (2019). Adipose tissue-specific knockout of AMPKα1/α2 results in normal AICAR tolerance and glucose metabolism. Biochemical and Biophysical Research Communications 519: 633–638. DOI 10.1016/j.bbrc.2019.09.049. [Google Scholar] [CrossRef]
Chuang HC, Chou CC, Kulp SK, Chen CS (2014). AMPK as a potential anticancer target–friend or foe? Current Pharmaceutical Design 20: 2607–2618. DOI 10.2174/13816128113199990485. [Google Scholar] [CrossRef]
Coffey VG, Zhong ZH, Shield A, Canny BJ, Chibalin AV, Zierath JR, Hawley JA (2005). Early signaling responses to divergent exercise stimuli in skeletal muscle from well-trained humans. Faseb Journal 19: 190. DOI 10.1096/fj.05-4809fje. [Google Scholar] [CrossRef]
Corton JM, Gillespie JG, Hawley SA, Hardie DG (1995). 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? European Journal of Biochemistry 229: 558–565. DOI 10.1111/j.1432-1033.1995.tb20498.x. [Google Scholar] [CrossRef]
Das AK, Yang QY, Fu X, Liang JF, Duarte MS, Zhu MJ, Trobridge GD, Du M (2012). AMP-activated protein kinase stimulates myostatin expression in C2C12 cells. Biochemical and Biophysical Research Communications 427: 36–40. DOI 10.1016/j.bbrc.2012.08.138. [Google Scholar] [CrossRef]
Day EA, Ford RJ, Steinberg GR (2017). AMPK as a therapeutic target for treating metabolic diseases. Trends in Endocrinology and Metabolism 28: 545–560. DOI 10.1016/j.tem.2017.05.004. [Google Scholar] [CrossRef]
Dreyer HC, Glynn EL, Lujan HL, Fry CS, DiCarlo SE, Rasmussen BB (2008). Chronic paraplegia-induced muscle atrophy downregulates the mTOR/S6K1 signaling pathway. Journal of Applied Physiology 104: 27–33. DOI 10.1152/japplphysiol.00736.2007. [Google Scholar] [CrossRef]
Dubbelhuis PF, Meijer AJ (2002). Hepatic amino acid-dependent signaling is under the control of AMP-dependent protein kinase. FEBS Letters 521: 39–42. DOI 10.1016/S0014-5793(02)02815-6. [Google Scholar] [CrossRef]
Egawa T, Goto A, Ohno Y, Yokoyama S, Ikuta A et al. (2015). Involvement of AMPK in regulating slow-twitch muscle atrophy during hindlimb unloading in mice. American Journal of Physiology-Endocrinology and Metabolism 309: E651–E662. DOI 10.1152/ajpendo.00165.2015. [Google Scholar] [CrossRef]
Fasano C, Disciglio V, Bertora S, Signorile ML, Simone C (2019). FOXO3a from the nucleus to the mitochondria: A round trip in cellular stress response. Cells 8: 1110. DOI 10.3390/cells8091110. [Google Scholar] [CrossRef]
Fay JR, Steele V, Crowell JA (2009). Energy homeostasis and cancer prevention: The AMP-activated protein kinase. Cancer Prevention Research 2: 301–309. DOI 10.1158/1940-6207.CAPR-08-0166. [Google Scholar] [CrossRef]
Feng J, Li JJ, Wu LW, Yu Q, Ji J, Wu JY, Dai WQ, Guo CY (2020). Emerging roles and the regulation of aerobic glycolysis in hepatocellular carcinoma. Journal of Experimental & Clinical Cancer Research 39: 1–19. DOI 10.1186/s13046-020-01629-4. [Google Scholar] [CrossRef]
Fu X, Zhao JX, Liang J, Zhu MJ, Foretz M, Viollet B, Du M (2013). AMP-activated protein kinase mediates myogenin expression and myogenesis via histone deacetylase 5. American Journal of Physiology-Cell Physiology 305: C887–895. DOI 10.1152/ajpcell.00124.2013. [Google Scholar] [CrossRef]
Gao H, Li YF (2018). Distinct signal transductions in fast- and slow- twitch muscles upon denervation. Physiological Reports 6: e13606. DOI 10.14814/phy2.13606. [Google Scholar] [CrossRef]
Gariballa N, Ali BR (2020). Endoplasmic reticulum associated protein degradation (ERAD) in the pathology of diseases related to TGF beta signaling pathway: Future therapeutic perspectives. Frontiers in Molecular Biosciences 7: 575608. DOI 10.3389/fmolb.2020.575608. [Google Scholar] [CrossRef]
Glancy B, Kim Y, Katti P, Willingham TB (2020). The functional impact of mitochondrial structure across subcellular scales. Frontiers in Physiology 11: 541040. DOI 10.3389/fphys.2020.541040. [Google Scholar] [CrossRef]
Grahammer F, Huber TB, Artunc F (2021). Role of mTOR signaling for tubular function and disease. Physiology 36: 350–358. DOI 10.1152/physiol.00021.2021. [Google Scholar] [CrossRef]
Guo YT, Jin M, Tang YL, Wang T, Wei B, Feng R, Gong B, Wang HW, Ji GJ, Lu ZB (2016). AMP-activated kinase α2 deficiency protects mice from denervation-induced skeletal muscle atrophy. Archives of Biochemistry and Biophysics 600: 56–60. DOI 10.1016/j.abb.2016.04.015. [Google Scholar] [CrossRef]
Hardie DG (2011). AMP-activated protein kinase-an energy sensor that regulates all aspects of cell function. Genes & Development 25: 1895–1908. DOI 10.1101/gad.17420111. [Google Scholar] [CrossRef]
Hardie DG, Ross FA, Hawley SA (2012). AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nature Reviews Molecular Cell Biology 13: 251–262. DOI 10.1038/nrm3311. [Google Scholar] [CrossRef]
Hardman SE, Hall DE, Cabrera AJ, Hancock CR, Thomson DM (2014). The effects of age and muscle contraction on AMPK activity and heterotrimer composition. Experimental Gerontology 55: 120–128. DOI 10.1016/j.exger.2014.04.007. [Google Scholar] [CrossRef]
Harper C, Gopalan V, Goh J (2021). Exercise rescues mitochondrial coupling in aged skeletal muscle: A comparison of different modalities in preventing sarcopenia. Journal of Translational Medicine 19: 1–17. DOI 10.1186/s12967-021-02737-1. [Google Scholar] [CrossRef]
Herzig S, Shaw RJ (2018). AMPK: Guardian of metabolism and mitochondrial homeostasis. Nature Reviews Molecular Cell Biology 19: 121–135. DOI 10.1038/nrm.2017.95. [Google Scholar] [CrossRef]
Hilder TL, Baer LA, Fuller PM, Fuller CA, Grindeland RE, Wade CE, Graves LM (2005). Insulin-independent pathways mediating glucose uptake in hindlimb-suspended skeletal muscle. Journal of Applied Physiology 99: 2181–2188. DOI 10.1152/japplphysiol.00743.2005. [Google Scholar] [CrossRef]
Ho CH, Triolo RJ, Elias AL, Kilgore KL, DiMarco AF et al. (2014). Functional electrical stimulation and spinal cord injury. Physical Medicine and Rehabilitation Clinics of North America 25: 631–654. DOI 10.1016/j.pmr.2014.05.001. [Google Scholar] [CrossRef]
Janzen NR, Whitfield J, Hoffman NJ (2018). Interactive roles for AMPK and glycogen from cellular energy sensing to exercise metabolism. International Journal of Molecular Sciences 19: 3344. DOI 10.3390/ijms19113344. [Google Scholar] [CrossRef]
Jeon SM (2016). Regulation and function of AMPK in physiology and diseases. Experimental and Molecular Medicine 48: e245. DOI 10.1038/emm.2016.81. [Google Scholar] [CrossRef]
Kang MJ, Moon JW, Lee JO, Kim JH, Jung EJ et al. (2021). Metformin induces muscle atrophy by transcriptional regulation of myostatin via HDAC6 and FoxO3a. Journal of Cachexia Sarcopenia and Muscle 13: 605–620. DOI 10.1002/jcsm.12833. [Google Scholar] [CrossRef]
Kim J, Kundu M, Viollet B, Guan KL (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature Cell Biology 13: U132–U171. DOI 10.1038/ncb2152. [Google Scholar] [CrossRef]
Kim J, Yang G, Kim Y, Kim J, Ha J (2016). AMPK activators: Mechanisms of action and physiological activities. Experimental & Molecular Medicine 48: e224. DOI 10.1038/emm.2016.16. [Google Scholar] [CrossRef]
Kim M, Lee JH (2015). Identification of an AMPK phosphorylation site in drosophila TSC2 (gigas) that regulate cell growth. International Journal of Molecular Sciences 16: 7015–7026. DOI 10.3390/ijms16047015. [Google Scholar] [CrossRef]
Kitada M, Koya D (2021). Autophagy in metabolic disease and ageing. Nature Reviews Endocrinology 17: 647–661. DOI 10.1038/s41574-021-00551-9. [Google Scholar] [CrossRef]
Lavin KM, Roberts BM, Fry CS, Moro T, Rasmussen BB, Bamman MM (2019). The importance of resistance exercise training to combat neuromuscular aging. Physiology 34: 112–122. DOI 10.1152/physiol.00044.2018. [Google Scholar] [CrossRef]
Lee JM, Seo WY, Song KH, Chanda D, Kim YD et al. (2010). AMPK-dependent repression of hepatic gluconeogenesis via disruption of CREB center dot CRTC2 complex by orphan nuclear receptor small heterodimer partner. Journal of Biological Chemistry 285: 32182–32191. DOI 10.1074/jbc.M110.134890. [Google Scholar] [CrossRef]
Lee M, Katerelos M, Gleich K, Galic S, Kemp BE, Mount PF, Power DA (2018). Phosphorylation of Acetyl-CoA carboxylase by AMPK reduces renal fibrosis and is essential for the anti-fibrotic effect of metformin. Journal of the American Society of Nephrology 29: 2326–2336. DOI 10.1681/ASN.2018010050. [Google Scholar] [CrossRef]
Li Y, Xu SQ, Mihaylova MM, Zheng B, Hou XY et al. (2011). AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metabolism 13: 376–388. DOI 10.1016/j.cmet.2011.03.009. [Google Scholar] [CrossRef]
Liangpunsakul S, Ross RA, Crabb DW (2013). Activation of carbohydrate response element-binding protein by ethanol. Journal of Investigative Medicine 61: 270–277. DOI 10.2310/JIM.0b013e31827c2795. [Google Scholar] [CrossRef]
Liu J, Peng YH, Cui ZW, Wu ZM, Qian AR, Shang P, Qu LN, Li YH, Liu JK, Long JG (2012). Depressed mitochondrial biogenesis and dynamic remodeling in mouse tibialis anterior and gastrocnemius induced by 4-week hindlimb unloading. IUBMB Life 64: 901–910. DOI 10.1002/iub.1087. [Google Scholar] [CrossRef]
Ljubicic V, Jasmin BJ (2013). AMP-activated protein kinase at the nexus of therapeutic skeletal muscle plasticity in Duchenne muscular dystrophy. Trends in Molecular Medicine 19: 614–624. DOI 10.1016/j.molmed.2013.07.002. [Google Scholar] [CrossRef]
Mairet-Coello G, Courchet J, Pieraut S, Courchet V, Maximov A, Polleux F (2013). The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Aβ Oligomers through Tau phosphorylation. Neuron 78: 94–108. DOI 10.1016/j.neuron.2013.02.003. [Google Scholar] [CrossRef]
Mao K, Klionsky DJ (2011). AMPK activates autophagy by phosphorylating ULK1. Circulation Research 108: 787–788. DOI 10.1161/RES.0b013e3182194c29. [Google Scholar] [CrossRef]
McGee SL, Hargreaves M (2010). AMPK-mediated regulation of transcription in skeletal muscle. Clinical Science 118: 507–518. DOI 10.1042/CS20090533. [Google Scholar] [CrossRef]
Melick CH, Jewell JL (2020). Regulation of mTORC1 by upstream stimuli. Genes 11: 989. DOI 10.3390/genes11090989. [Google Scholar] [CrossRef]
Merrill GF, Kurth EJ, Hardie DG, Winder WW (1997). AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. American Journal of Physiology-Endocrinology and Metabolism 273: E1107–E1112. DOI 10.1152/ajpendo.1997.273.6.E1107. [Google Scholar] [CrossRef]
Morales-Alamo D, Calbet JAL (2016). AMPK signaling in skeletal muscle during exercise: Role of reactive oxygen and nitrogen species. Free Radical Biology and Medicine 98: 68–77. DOI 10.1016/j.freeradbiomed.2016.01.012. [Google Scholar] [CrossRef]
Musi N, Hayashi T, Fujii N, Hirshman MF, Witters LA, Goodyear LJ (2001). AMP-activated protein kinase activity and glucose uptake in rat skeletal muscle. American Journal of Physiology-Endocrinology and Metabolism 280: E677–E684. DOI 10.1152/ajpendo.2001.280.5.E677. [Google Scholar] [CrossRef]
Musi N, Hirshman MF, Nygren J, Svanfeldt M, Bavenholm P et al. (2002). Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes 51: 2074–2081. DOI 10.2337/diabetes.51.7.2074. [Google Scholar] [CrossRef]
Neumann D (2018). Is TAK1 a direct upstream kinase of AMPK? International Journal of Molecular Sciences 19: 2412. DOI 10.3390/ijms19082412. [Google Scholar] [CrossRef]
Oliveira AG, Gomes-Marcondes MCC (2016). Metformin treatment modulates the tumour-induced wasting effects in muscle protein metabolism minimising the cachexia in tumour-bearing rats. BMC Cancer 16: 418. DOI 10.1186/s12885-016-2424-9. [Google Scholar] [CrossRef]
Oshima R, Yamada M, Kurogi E, Ogino Y, Serizawa Y, Tsuda S, Ma X, Egawa T, Hayashi T (2015). Evidence for organic cation transporter-mediated metformin transport and 5′-adenosine monophosphate-activated protein kinase activation in rat skeletal muscles. Metabolism-Clinical and Experimental 64: 296–304. DOI 10.1016/j.metabol.2014.10.037. [Google Scholar] [CrossRef]
Ou HC, Chu PM, Huang YT, Cheng HC, Chou WC, Yang HL, Chen HI, Tsai KL (2021). Low-level laser prevents doxorubicin-induced skeletal muscle atrophy by modulating AMPK/SIRT1/PCG-1α-mediated mitochondrial function, apoptosis and up-regulation of pro-inflammatory responses. Cell and Bioscience 11: 200. DOI 10.1186/s13578-021-00719-w. [Google Scholar] [CrossRef]
Parker L, Shaw CS, Stepto NK, Levinger I (2017). Exercise and glycemic control: Focus on redox homeostasis and redox-sensitive protein signaling. Frontiers in Endocrinology 8: 200. DOI 10.3389/fendo.2017.00087. [Google Scholar] [CrossRef]
Paulsen SR, Rubink DS, Winder WW (2001). AMP-activated protein kinase activation prevents denervation-induced decline in gastrocnemius GLUT-4. Journal of Applied Physiology 91: 2102–2108. DOI 10.1152/jappl.2001.91.5.2102. [Google Scholar] [CrossRef]
Pauly M, Daussin F, Burelle Y, Li T, Godin R et al. (2012). AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. American Journal of Pathology 181: 583–592. DOI 10.1016/j.ajpath.2012.04.004. [Google Scholar] [CrossRef]
Powers SK, Lynch GS, Murphy KT, Reid MB, Zijdewind I (2016). Disease-induced skeletal muscle atrophy and fatigue. Medicine and Science in Sports and Exercise 48: 2307–2319. DOI 10.1249/MSS.0000000000000975. [Google Scholar] [CrossRef]
Radhakrishnan J, Baetiong A, Kaufman H, Huynh M, Leschinsky A, Fresquez A, White C, DiMario JX, Gazmuri RJ (2019). Improved exercise capacity in cyclophilin-D knockout mice associated with enhanced oxygen utilization efficiency and augmented glucose uptake via AMPK-TBC1D1 signaling nexus. The FASEB Journal 33: 11443–11457. DOI 10.1096/fj.201802238R. [Google Scholar] [CrossRef]
Reiter AK, Bolster DR, Crozier SJ, Kimball SR, Jefferson LS (2005). Repression of protein synthesis and mTOR signaling in rat liver mediated by the AMPK activator aminoimidazole carboxamide ribonucleoside. American Journal of Physiology-Endocrinology and Metabolism 288: E980–E988. DOI 10.1152/ajpendo.00333.2004. [Google Scholar] [CrossRef]
Rena G, Hardie DG, Pearson ER (2017). The mechanisms of action of metformin. Diabetologia 60: 1577–1585. DOI 10.1007/s00125-017-4342-z. [Google Scholar] [CrossRef]
Ribeiro CB, Christofoletti DC, Pezolato VA, Durigan RDM, Prestes J, Tibana RA, Pereira ECL, Neto IVD, Durigan JLQ, da Silva CA (2015). Leucine minimizes denervation-induced skeletal muscle atrophy of rats through AKT/mTOR signaling pathways. Frontiers in Physiology 6: 73. DOI 10.3389/fphys.2015.00073. [Google Scholar] [CrossRef]
Richter EA, Ruderman NB (2009). AMPK and the biochemistry of exercise: Implications for human health and disease. Biochemical Journal 418: 261–275. DOI 10.1042/BJ20082055. [Google Scholar] [CrossRef]
Ross FA, MacKintosh C, Hardie DG (2016). AMP-activated protein kinase: A cellular energy sensor that comes in 12 flavours. The FEBS Journal 283: 2987–3001. DOI 10.1111/febs.13698. [Google Scholar] [CrossRef]
Rudrappa SS, Wilkinson DJ, Greenhaff PL, Smith K, Idris I, Atherton PJ (2016). Human skeletal muscle disuse atrophy: Effects on muscle protein synthesis, breakdown, and insulin resistance—A qualitative review. Frontiers in physiology 7: 361. DOI 10.3389/fphys.2016.00361. [Google Scholar] [CrossRef]
Ryu JY, Choi HM, Yang HI, Kim KS (2020). Dysregulated autophagy mediates sarcopenic obesity and its complications via AMPK and PGC1α signaling pathways: Potential involvement of gut dysbiosis as a pathological link. International Journal of Molecular Sciences 21: 6887. DOI 10.3390/ijms21186887. [Google Scholar] [CrossRef]
Sakamoto K, Holman GD (2008). Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. American Journal of Physiology-Endocrinology and Metabolism 295: E29–E37. DOI 10.1152/ajpendo.90331.2008. [Google Scholar] [CrossRef]
Scarpulla RC, Vega RB, Kelly DP (2012). Transcriptional integration of mitochondrial biogenesis. Trends in Endocrinology and Metabolism 23: 459–466. DOI 10.1016/j.tem.2012.06.006. [Google Scholar] [CrossRef]
Srivastava RAK, Pinkosky SL, Filippov S, Hanselman JC, Cramer CT, Newton RS (2012). AMP-activated protein kinase: An emerging drug target to regulate imbalances in lipid and carbohydrate metabolism to treat cardio-metabolic diseases. Journal of Lipid Research 53: 2490–2514. DOI 10.1194/jlr.R025882. [Google Scholar] [CrossRef]
Steinberg GR, Carling D (2019). AMP-activated protein kinase: The current landscape for drug development. Nature Reviews Drug Discovery 18: 527–551. DOI 10.1038/s41573-019-0019-2. [Google Scholar] [CrossRef]
Stockli J, Davey JR, Hohnen-Behrens C, Xu AM, James DE, Ramm G (2008). Regulation of glucose transporter 4 translocation by the rab guanosine triphosphatase-activating protein AS160/TBC1D4: Role of phosphorylation and membrane association. Molecular Endocrinology 22: 2703–2715. DOI 10.1210/me.2008-0111. [Google Scholar] [CrossRef]
Stouth DW, Manta A, Ljubicic V (2018). Protein arginine methyltransferase expression, localization, and activity during disuse-induced skeletal muscle plasticity. American Journal of Physiology-Cell Physiology 314: C177–C190. DOI 10.1152/ajpcell.00174.2017. [Google Scholar] [CrossRef]
Sukumaran A, Choi K, Dasgupta B (2020). Insight on transcriptional regulation of the energy sensing AMPK and biosynthetic mTOR pathway genes. Frontiers in Cell and Developmental Biology 8: 671. DOI 10.3389/fcell.2020.00671. [Google Scholar] [CrossRef]
Thomson DM (2018). The role of AMPK in the regulation of skeletal muscle size, hypertrophy, and regeneration. International Journal of Molecular Sciences 19: 3125. DOI 10.3390/ijms19103125. [Google Scholar] [CrossRef]
Thomson DM, Fick CA, Gordon SE (2008). AMPK activation attenuates S6K1, 4E-BP1, and eEF2 signaling responses to high-frequency electrically stimulated skeletal muscle contractions. Journal of Applied Physiology 104: 625–632. DOI 10.1152/japplphysiol.00915.2007. [Google Scholar] [CrossRef]
Treebak JT, Birk JB, Rose AJ, Kiens B, Richter EA, Wojtaszewski JFP (2007). AS160 phosphorylation is associated with activation of α2β2γ1-but not α2β2γ3-AMPK-AMPK trimeric complex in skeletal muscle during exercise in humans. American Journal of Physiology-Endocrinology and Metabolism 292: E715–E722. DOI 10.1152/ajpendo.00380.2006. [Google Scholar] [CrossRef]
Trefts E, Shaw RJ (2021). AMPK: Restoring metabolic homeostasis over space and time. Molecular Cell 81: 3677–3690. DOI 10.1016/j.molcel.2021.08.015. [Google Scholar] [CrossRef]
Vainshtein A, Desjardins EMA, Armani A, Sandri M, Hood DA (2015). PGC-1α modulates denervation-induced mitophagy in skeletal muscle. Skeletal Muscle 5: 9. DOI 10.1186/s13395-015-0033-y. [Google Scholar] [CrossRef]
Vichaiwong K, Purohit S, An D, Toyoda T, Jessen N, Hirshman MF, Goodyear LJ (2010). Contraction regulates site-specific phosphorylation of TBC1D1 in skeletal muscle. Biochemical Journal 431: 311–320. DOI 10.1042/BJ20101100. [Google Scholar] [CrossRef]
Vilchinskaya NA, Mirzoev TM, Lomonosova YN, Kozlovskaya IB, Shenkman BS (2015). Human muscle signaling responses to 3-day head-out dry immersion. Journal of Musculoskeletal & Neuronal Interactions 15: 286–293. [Google Scholar]
Wakil SJ, Abu-Elheiga LA (2009). Fatty acid metabolism: Target for metabolic syndrome. Journal of Lipid Research 50: S138–S143. DOI 10.1194/jlr.R800079-JLR200. [Google Scholar] [CrossRef]
Wang Q, Liu SD, Zhai AH, Zhang B, Tian GZ (2018). AMPK-mediated regulation of lipid metabolism by phosphorylation. Biological & Pharmaceutical Bulletin 41: 985–993. DOI 10.1248/bpb.b17-00724. [Google Scholar] [CrossRef]
Wang XM, da Mota SR, Liu R, Moore CE, Xie JL et al. (2014). Eukaryotic elongation factor 2 kinase activity is controlled by multiple inputs from oncogenic signaling. Molecular and Cellular Biology 34: 4088–4103. DOI 10.1128/MCB.01035-14. [Google Scholar] [CrossRef]
Webster I, Friedrich SO, Lochner A, Huisamen B (2010). AMP kinase activation and glut4 translocation in isolated cardiomyocytes. Cardiovascular Journal of Africa 21: 10–16. [Google Scholar]
Winder WW, Hardie DG (1996). Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. American Journal of Physiology-Endocrinology and Metabolism 270: E299–304. DOI 10.1152/ajpendo.1996.270.2.E299. [Google Scholar] [CrossRef]
Xia QH, Huang XB, Huang JR, Zheng YF, March ME, Li J, Wei YJ (2021). The role of autophagy in skeletal muscle diseases. Frontiers in Physiology 12: 638983. DOI 10.3389/fphys.2021.638983. [Google Scholar] [CrossRef]
Xiang XQ, Saha AK, Wen R, Ruderman NB, Luo ZJ (2004). AMP-activated protein kinase activators can inhibit the growth of prostate cancer cells by multiple mechanisms. Biochemical and Biophysical Research Communications 321: 161–167. DOI 10.1016/j.bbrc.2004.06.133. [Google Scholar] [CrossRef]
Yamada S, Kamata T, Nawa H, Sekijima T, Takei N (2019). AMPK activation, eEF2 inactivation, and reduced protein synthesis in the cerebral cortex of hibernating chipmunks. Scientific Reports 9: 11904. DOI 10.1038/s41598-019-48172-7. [Google Scholar] [CrossRef]
Yan Y, Krecke KN, Bapat AS, Yang TY, Lopresti MW, Mashek DG, Kelekar A (2021). Phosphatase PHLPP2 regulates the cellular response to metabolic stress through AMPK. Cell Death & Disease 12: 904. DOI 10.1038/s41419-021-04196-4. [Google Scholar] [CrossRef]
Yoon MS (2017). mTOR as a key regulator in maintaining skeletal muscle mass. Frontiers in Physiology 8: 788. DOI 10.3389/fphys.2017.00788. [Google Scholar] [CrossRef]
Yousuf Y, Datu A, Barnes B, Amini-Nik S, Jeschke MG (2020). Metformin alleviates muscle wasting post-thermal injury by increasing Pax7-positive muscle progenitor cells. Stem Cell Research & Therapy 11: 1–14. DOI 10.1186/s13287-019-1480-x. [Google Scholar] [CrossRef]
Zhou GC, Myers R, Li Y, Chen YL, Shen XL et al. (2001). Role of AMP-activated protein kinase in mechanism of metformin action. Journal of Clinical Investigation 108: 1167–1174. DOI 10.1172/JCI13505. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools