
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW
Realizing the potential of exploiting human IPSCs and their derivatives in research of Down syndrome
1 Nanophotonics Research Center, Institute of Microscale Optoelectronics & State Key Laboratory of Radio Frequency Heterogeneous Integration, Shenzhen University, Shenzhen, 518060, China
2 Phil Rivers Technology, Beijing, 100871, China
3 Department of Ophthalmology, Peking University Shenzhen Hospital, Shenzhen, 518036, China
* Corresponding Author: YANXIANG NI. Email:
# These authors contributed equally to this work
(This article belongs to the Special Issue: Perspectives on Stem Cells and Regenerative Medicine)
BIOCELL 2023, 47(12), 2567-2578. https://doi.org/10.32604/biocell.2023.043781
Received 12 July 2023; Accepted 16 October 2023; Issue published 27 December 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Down syndrome (DS) is a genetic condition characterized by intellectual disability, delayed brain development, and early onset Alzheimer’s disease. The use of primary neural cells and tissues is important for understanding this disease, but there are ethical and practical issues, including availability from patients and experimental manipulability. Moreover, there are significant genetic and physiological differences between animal models and humans, which limits the translation of the findings in animal studies to humans. Advancements in induced pluripotent stem cells (iPSC) technology have revolutionized DS research by providing a valuable tool for studying the cellular and molecular pathologies associated with DS. Induced pluripotent stem cells derived from cells obtained from DS patients contain the patient’s entire genome including trisomy 21. Trisomic iPSCs as well as their derived cells or organoids can be useful for disease modeling, investigating the molecular mechanisms, and developing potential strategies for treating or alleviating DS. In this review, we focus on the use of iPSCs and their derivatives obtained from DS individuals and healthy humans for DS research. We summarize the findings from the past decade of DS studies using iPSCs and their derivatives. We also discuss studies using iPSC technology to investigate DS-associated genes (e.g., APP, OLIG1, OLIG2, RUNX1, and DYRK1A) and abnormal phenotypes (e.g., dysregulated mitochondria and leukemia risk). Lastly, we review the different strategies for mitigating the limitations of iPSCs and their derivatives, for alleviating the phenotypes, and for developing therapies.Keywords
Down syndrome (DS) is a genetic condition caused by either partial or full trisomy of human chromosome 21 (HSA21), which leads to delayed brain development, intellectual disability, and premature Alzheimer’s disease (AD) (Esposito et al., 2008; Liu et al., 2023; Ma et al., 2021; Ponroy Bally et al., 2020). Individuals with DS also have smaller whole brain volume (Giffin-Rao et al., 2022; Huo et al., 2018), specifically, they have reduced numbers of calretinin-expressing interneurons in the cortex (Giffin-Rao et al., 2022). The DS-associated intellectual disability has been linked to the various changes in early brain development, including altered proliferation and differentiation of neural progenitor cells and abnormal formation and maintenance of myelin (Klein et al., 2021). Mitochondrial dysfunction and oxidative stress are also regarded as hallmarks of DS (Omori et al., 2017; Prutton et al., 2023). Trisomy 21 alters gene dosage and expression, which leads to abnormal neurogenesis and metabolism across various cell types including neurons and astrocytes (Banno et al., 2016; Omori et al., 2017). Transcriptomic analysis showed there was altered transcription, splicing, and RNA editing in DS patients compared to healthy individuals (Cypris et al., 2020). However, what causes the abnormal phenotypes in DS remains largely unknown. Moreover, effective strategies for treating or ameliorating the deficits in DS are lacking.
Primary neural cells and tissues are useful for investigating the pathology and cellular mechanisms in DS. However, there are ethical and availability issues in using human tissues, as well as limitations in their experimental manipulability (Gough et al., 2020). In the past decades, several murine models of DS have been developed that have helped us to understand the neurobiological changes responsible for the cognitive and behavioral phenotypes associated with DS (Rueda et al., 2020). However, due to the genetic and physiological differences between animals and humans, the applicability of these preclinical models has been questioned, particularly in testing potential therapies (Rueda et al., 2020).
Induced pluripotent stem cell (iPSC) technology has emerged as a valuable tool for studying the cellular and molecular pathologies in specific cell types affected by DS (Wu et al., 2022b). These iPSCs can be derived from somatic cells such as fibroblasts from DS patients or healthy humans (Cypris et al., 2020). The fibroblasts can be reprogrammed into iPSCs through the introduction of Yamanaka factors via retroviral integration or non-integrating methods such as Sendai virus, episomal vector, or mRNA (Banno et al., 2016; Cypris et al., 2020). The iPSCs derived from DS patients contain their complete genomic sequence including disease-causing genetic mutations. Hence, these cells could be used to study the disease, investigate the molecular mechanisms, develop treatments, and test potential drugs (Banno et al., 2016). Here, we aim to utilize iPSC models to explore the molecular mechanisms underlying DS phenotypes, understand the genetic factors that contribute to pathogenesis, and discuss potential therapeutic targets for this condition.
IPSCs and Their Derivatives in DS Research
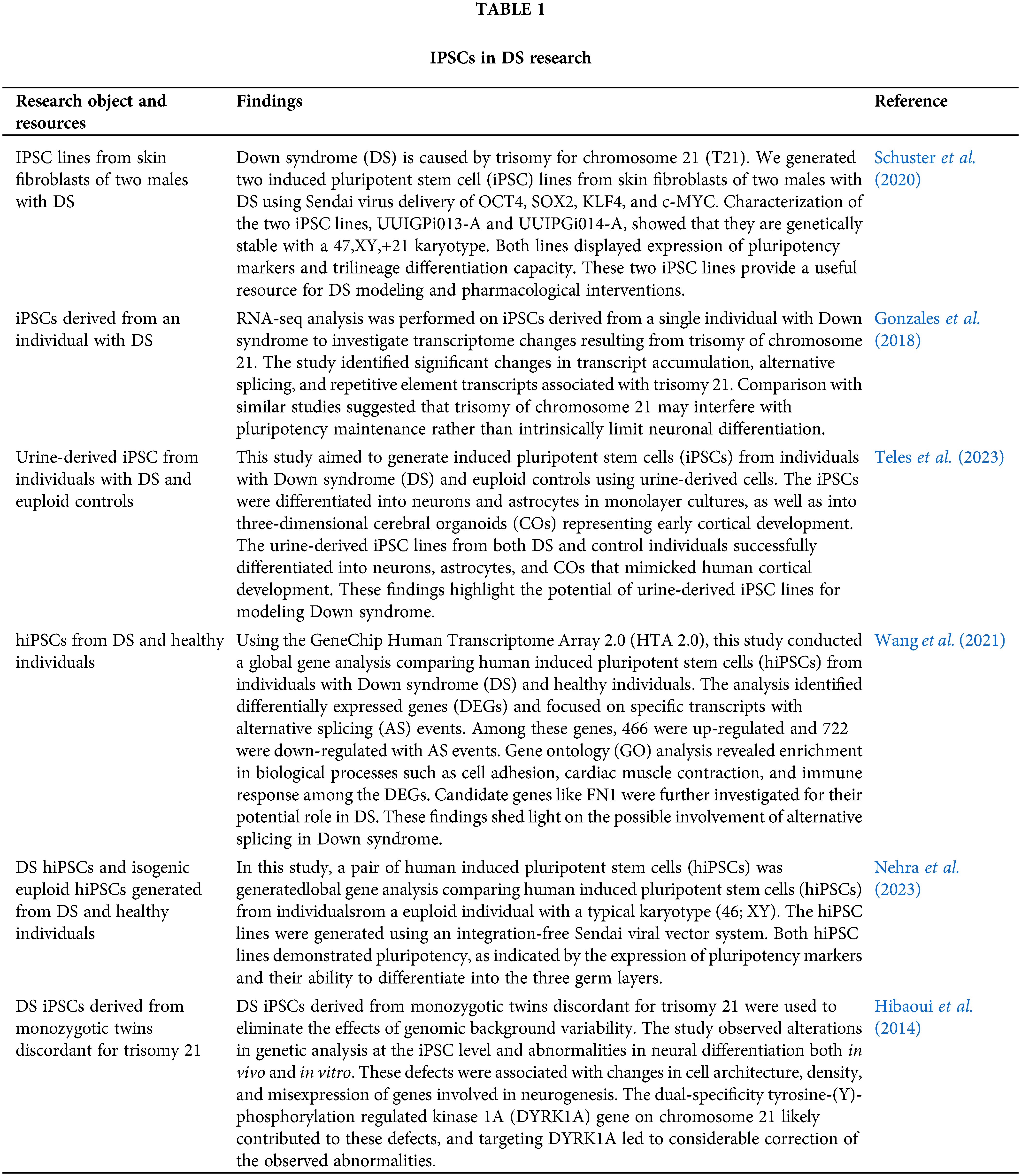
Over the past two decades, a series of iPSCs summarized in Table 1 and iPSC-derived cells or organoids summarized in Table 2 have been developed and utilized to delve into the fundamental mechanisms and characteristics of DS, thereby shedding light on the pathophysiology of this condition. DS iPSCs have undergone differentiation into various neuron types, including cortical neurons and interneurons (Sobol et al., 2019; Curtis et al., 2022). Sobol et al. (2019) conducted comprehensive transcriptomic and proteomic analyses on DS iPSCs and compared them to normal iPSCs, providing insights into DS and normal fetal brain development. These analyses revealed temporal dysregulation in genes, proteins, and pathways across 11 major functional clusters. Notable disruptions included impaired DNA replication, synaptic maturation, and neuroactive clusters during early differentiation, along with disturbances in the transition to the neural progenitor cell stage and reduced cellular growth. Additionally, other affected clusters encompassed differentiation, growth factors, extracellular matrix, oxidative phosphorylation, and glycolysis (Sobol et al., 2019).
For generating iPSCs, human somatic cells such as fibroblasts can be reprogrammed using viral vectors such as polycistronic lentiviral vectors to deliver four transcription factors OCT4, SOX2, KLF4, and c-MYC, referred to as Yamanaka factors (Gonzales et al., 2018; Grad et al., 2011; Hibaoui et al., 2014; Takahashi et al., 2007) (Table 1). To eliminate the effects of genomic background variability, Hibaoui et al. (2014) generated iPSCs from monozygotic twins discordant for trisomy 21 by transducing parental fibroblasts (Twin normal and Twin-DS) with polycistronic lentiviral vectors expressing the Yamanaka factors (Table 1). They observed the DS iPSCs had altered cell architecture, density, and misexpression of genes involved in neurogenesis in vivo and in vitro. Transcriptomic analysis of the DS iPSCs showed there were significant changes in transcript accumulation, alternative splicing, and repetitive element transcripts associated with trisomy 21 (Gonzales et al., 2018) (Table 1). These findings suggest that trisomy 21 may interfere with the maintenance of pluripotency rather than intrinsically limiting neuronal differentiation (Gonzales et al., 2018) (Table 1).
Retroviruses expressing either four reprogramming factors (OCT4, SOX2, KLF4, and c-MYC) or three factors (without c-MYC) have also been used to generate DS iPSCs from fibroblasts (Park et al., 2008; Shi et al., 2012). Subsequent differentiation of these DS iPSCs into cortical neurons revealed significant characteristics (Shi et al., 2012).
Non-integrating techniques, such as Sendai virus expressing the Yamanaka factors, have also been applied to generate iPSCs and isogenic euploid iPSCs from skin fibroblasts from DS patients (Nehra et al., 2023; Schuster et al., 2020) (Table 1). The DS iPSCs carried trisomy 21, whereas the isogenic euploid iPSCs had a typical karyotype. These DS iPSCs also displayed trilineage differentiation capacity (Nehra et al., 2023) (Table 1). Episomal plasmids have also been applied in the transient expression of OCT4, SOX2, KLF4 and MYC genes to reprogram fibroblasts into DP iPSC ACS-1003™ and normal hiPSC ATCC® ACS-1011™ (Teles et al., 2023) (Table 1). Global gene analysis of DP iPSC ACS-1003™ and hiPSC ACS-1011™ revealed differentially expressed genes (DEGs) with alternative splicing events. These DEGs were enriched in biological processes including cell adhesion and immune response. Notably, candidate genes such as FN1 were identified to potentially play a significant role in DS (Wang et al., 2021) (Table 1). Recently urine cells from DS patients and control subjects have also been reprogrammed into iPSCs in vitro (Teles et al., 2023) (Table 1). These iPSCs were generated using episomal vectors with an oriP/EBNA-1(Epstein-Barr-1 nuclear antigen) backbone for the delivery of six reprogramming factors (Oct4, Sox2, Nanog, Lin28, Klf4, and L-Myc). These iPSCs were successfully differentiated into neurons and astrocytes in the form of monolayer cultures and 3D cerebral organoids. The generated cerebral organoids exhibited early features of human cortical development, providing a valuable tool for studying DS in vitro (Teles et al., 2023) (Table 1).
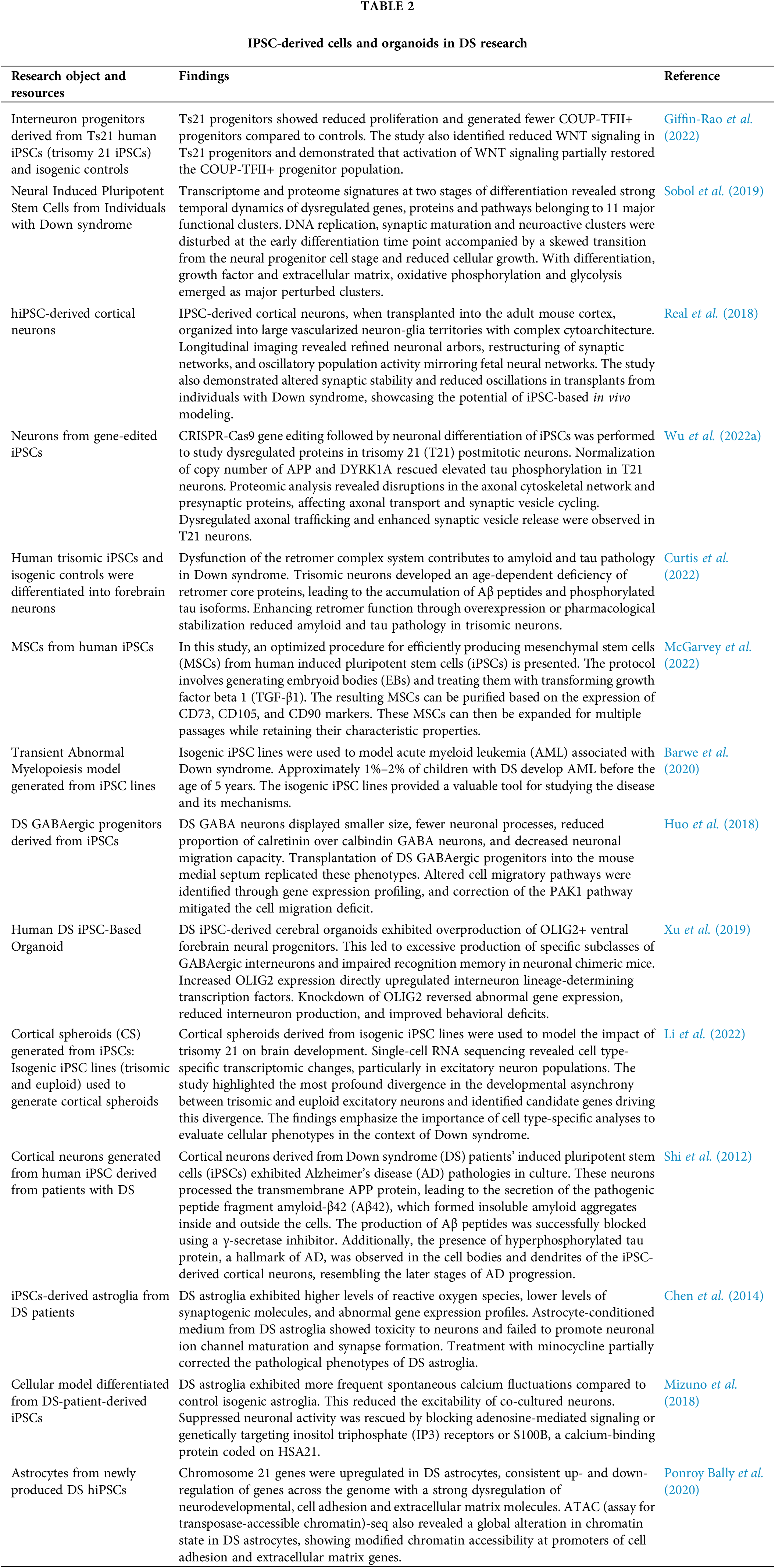
IPSC-derived cells and organoids
The generated iPSCs can be further induced into neural progenitors, which can reveal their capacity to differentiate into specific neural cells. Giffin-Rao et al. (2022) showed DS iPSCs had a reduced capacity to generate COUP-TFII+ progenitors, which has been linked to decreased WNT signaling (Table 2). Activating WNT signaling partially restored the generation of COUP-TFII+ progenitors, suggesting WNT signaling could be a target for correcting differentiation deficits (Giffin-Rao et al., 2022) (Table 2). Moreover, Curtis et al. (2022) found that DS iPSC-derived neurons developed age-dependent deficiency of retromer proteins, which regulate the retrieval and recycling of cargo from early endosomes, associated with beta-amyloid and tau accumulation (Table 2). These deficits were rescued by normalizing amyloid precursor protein (APP) and DYRK1A copy number in DS neurons via CRISPR-Cas9 editing, but not by normalizing RCAN1 and SYNJ1. They also showed that DS neurons had dysregulated axonal trafficking and enhanced synaptic retromer overexpression or stabilization, indicating that retromer dysfunction may contribute to AD pathology in DS (Curtis et al., 2022) (Table 2). Wu et al. (2022a) used DS hiPSCs to generate postmitotic neurons that showed synaptic vesicle release (Table 2). These findings provide insights into the initial changes in neurons that likely lead to neurodegeneration in DS.
Moreover, DS iPSC-derived cortical neurons transplanted in mouse cortex were found to organize into vascularized and cytoarchitecturally complex neuron-glia networks. Longitudinal imaging showed refined arbors and oscillatory activity similar to the fetal cortex. However, the DS transplants exhibited altered synaptic stability and reduced oscillations (Real et al., 2018) (Table 2).
Other studies have differentiated DS iPSCs into astrocytes (Chen et al., 2014; Mizuno et al., 2018; Ponroy Bally et al., 2020) (Table 2). Mizuno et al. (2018) found that DS iPSC-derived astrocytes had increased spontaneous calcium fluctuations and reduced neuronal excitability in neuron-astrocyte co-culture (Table 2). These abnormalities were rescued by blocking adenosine signaling, inositol triphosphate receptors, or S100B. Ponroy Bally et al. (2020) showed that DS iPSC-derived astrocytes had altered chromatin state, particularly open promoters of cell adhesion/extracellular matrix genes (Table 2). Furthermore, the conditioned media from DS iPSC-derived astrocytes was toxic to neurons and prevented channel/synapse maturation. Treatment with minocycline, an anti-oxidant and anti-inflammatory compound, partially corrected the pathological phenotypes in DS astroglia such as the decreased proliferation rate (Chen et al., 2014) (Table 2).
Organoids derived from DS iPSCs were shown to overproduce ventral forebrain neural progenitors and GABAergic interneurons. This was associated with an increase in OLIG2 that upregulated interneuron transcription factors. Knockdown of OLIG2 rescued the organoid phenotypes and improved memory in chimeric mice (Xu et al., 2019) (Table 2). Single-cell RNA sequencing of cortical spheroids derived from DS and control iPSCs revealed cell type-specific changes, particularly in excitatory neurons. A major difference between DS and controls was the asynchronous development of excitatory neurons. This highlights the need for cell type-specific analyses to fully understand DS brain phenotypes (Li et al., 2022) (Table 2).
In summary, iPSC and iPSC-derived cells or organoids have been used in a series of DS investigations, providing insights into the mechanisms underlying the abnormal development of specific cells in DS.
DS carrying the extra full or partial copy of Chr 21 shows a number of dysregulated genes including APP, OLIG1, OLIG2, RUNX1, and DYRK1A (Sobol et al., 2019). The generation of DS iPSCs through non-genomic integration showed not only the overexpression of Chr 21 genes, but also the dysregulation of thousands of other genes (Briggs et al., 2013). Therefore, iPSCs or their derivatives could provide insights into the potential roles of genes on Chr 21 and genes on other chromosomes.
• APP: The APP gene located on Chr 21 and its increased expression due to trisomy 21 is believed to contribute to the early onset of AD in DS patients. In the study by Shi et al. (2012), they found that cortical neurons derived from DS iPSCs displayed AD pathologies including the accumulation of amyloid-β42 and hyperphosphorylated tau. Deleting or overexpressing the APP gene in human iPSCs resulted in AD-related changes such as Aβ42/Aβ40 ratio and pyroglutamate aggregates in DS and control neurons, respectively (Ovchinnikov et al., 2018). Moreover, transcriptomic and proteomic profiling revealed the early upregulation of APP in DS iPSCs, supporting the role of APP in the disturbed neurogenesis in DS (Sobol et al., 2019). In a Down syndrome mouse model (Dp16), the levels of Aβ42 and Aβ40 were found to exceed those observed in AD. The products of APP processing were also increased in the Dp16 model (Sawa et al., 2022). Further study revealed that the increased dose of the APP gene, which is triplicated in DS due to trisomy 21, was necessary for the loss of vulnerable neurons, tau pathology, and the activation of astrocytes and microglia in these DS model mice (Sawa et al., 2022). In a mouse model of DS (Ts65Dn), in vivo treatment with Posiphen was well tolerated and did not show adverse effects on behavior. Posiphen treatment normalized the levels of full-length APP (fl-APP) and C-terminal fragments, slightly decreased Aβ species, restored Rab5 activity to normal levels, reduced phosphorylated tau (p-tau), and reversed the deficits in the activation of TrkB as well as Akt, ERK, and CREB signaling pathways. Remarkably, Posiphen treatment also restored the level of choline acetyltransferase protein to normal levels in disomic mice (2N) (Chen et al., 2021).
• OLIG1, OLIG2, and RUNX1: Chr 21 genes of transcription factors OLIG1, OLIG2, and RUNX1 and their dysregulation observed in transcriptome and proteome profiling of DS iPSCs were linked to the abnormal myelination and neuronal differentiation in DS (Sobol et al., 2019). Xu et al. (2019) further showed that increased OLIG2 expression in DS neural progenitors directly upregulated genes that promoted the production of specific subclasses of GABAergic interneuron lineages. Meanwhile, shRNA-mediated knockdown of OLIG2 in DS neural progenitors largely reversed the abnormal gene expression in early neural progenitors and decreased interneuron generation in DS organoids and chimeric mouse brains (Xu et al., 2019). This indicates that OLIG2 drives interneuron production in DS organoids and the behavioral deficits in DS chimeric mice (Xu et al., 2019). The expression of RUNX1 gene on Chr 21 was found to be higher in DS iPSCs compared to normal controls, which resulted in mitochondrial dysfunction in DS iPSCs via promoting apoptosis through the activation of the PI3K/Akt signaling pathway (Liu et al., 2023). Therefore, reducing RUNX1 expression could improve mitochondrial function in DS iPSCs.
• DYRK1A: The DYRK1A gene located on Chr 21 is triplicated in DS, contributing to DS-associated intellectual disability (Guimera et al., 1999). Studies using Twin-DS iPSCs-derived neural progenitor cells (NPCs) and neurons have consistently shown overexpression of DYRK1A in fetal DS (Hibaoui et al., 2014). This gene’s role in neural differentiation has been highlighted by the observation that pharmacological treatment with a DYRK1A inhibitor (EGCG) or RNA interference improved the proliferation and prevented apoptosis in Twin-DS iPSC-derived NPCs (Hibaoui et al., 2014). Deleting one copy of DYRK1A in DS iPSCs improved the development of cerebral organoids (Caglayan, 2016), suggesting that normalizing DYRK1A in DS could correct neurodevelopmental phenotypes.
• CXADR: The triplication of CXADR on Chr 21 resulted in migration deficits in neural crest cells derived from DS iPSCs (Liu et al., 2022).
• Long non-coding RNAs (lncRNAs): High-throughput sequencing and DEG screening of DS-derived iPSCs revealed the aberrant expression of lncRNAs, which are normally involved in epigenetic regulation (Qiu et al., 2017). Enrichment analysis of lncRNAs in DS indicated their target genes were mainly associated with nervous and muscle development (Ma et al., 2021).
• Fibronectin 1 (FN1): Transcriptome analysis of DS iPSCs identified FN1 gene on Chr 21 was differentially expressed, indicating it has a potential role in DS (Wang et al., 2021).
In summary, triplication of Chr 21 genes like APP, OLIG1/2, RUNX1, DYRK1A, and CXADR contributes to neurodevelopmental and mitochondrial abnormalities in DS models. Normalizing their expression could ameliorate certain DS phenotypes. Dysregulated non-Chr 21 genes like FN1 may also contribute to DS pathologies.
Insights on Mitochondrial Dysfunction and Oxidative Stress in DS
Mitochondrial dysfunction in DS
Mitochondrial dysfunction and oxidative stress observed in Down syndrome (DS), such as impaired mitochondrial dynamics, structural defects, and dysregulated bioenergetic profiles linked to OXPHOS deficiency and reduced ATP production, can be attributed to the dosage-sensitive nuclear-encoded mitochondrial genes located on HSA21 (Tan et al., 2023). In neural progenitors derived from the frontal cortex of DS patients at 19–21 weeks, genomic and functional profiling revealed dysregulated gene expression, particularly in genes related to cell death and oxidative stress (Esposito et al., 2008). In fibroblast cells from the skin of DS patients, there was mild basal mitochondrial dysfunction, but the use of a cocktail of mitochondrial stressors revealed significant mitochondrial deficits (Anderson et al., 2021). However, the underlying mechanisms of the mitochondrial dysfunction and oxidative stress in DS remain unclear. The use of iPSCs or their derivatives obtained from DS patients has provided helpful information on understanding the mitochondrial deficits in DS.
Molecular mechanisms of oxidative stress
Results from PCR-array and qRT-PCR analysis of DS iPSCs showed nearly all genes associated with mitochondria were downregulated, and a large number of lncRNAs were differentially expressed (Qiu et al., 2017). These observations provide evidence for impaired mitochondrial function in DS iPSCs, which is consistent with the observations in DS cells (Qiu et al., 2017). An RNA-seq study revealed there was significantly increased expression of RUNX1 in DS iPSCs compared to normal controls (Liu et al., 2023). Overexpression of RUNX1 in control iPSCs resulted in mitochondrial dysfunction, whereas depletion of RCAN1 or inhibition of RUNX1 expression improved mitochondrial function in DS iPSCs (Liu et al., 2023; Parra et al., 2018). Global gene expression analysis indicated the overexpression of RUNX1 could promote the induction of apoptosis in DS iPSCs by activating the PI3K/Akt signaling pathway (Liu et al., 2023). Compared to disomic iPSCs, mitochondria in trisomic iPSCs were more fused, had higher oxygen consumption, and exhibited compromised coupling efficiency and metabolic flexibility (Parra et al., 2018).
iPSC-derived cells and organoids insights into mitochondrial abnormalities
Neurons differentiated from trisomic iPSCs derived from DS fibroblasts exhibited early mitochondrial alterations. Mitochondrial dysfunction was detected as early as day 7 of neuronal differentiation in DS iPSCs, indicating the early onset of mitochondrial abnormalities (Mollo et al., 2021). Mitochondrial ROS production was also observed to impact the differentiation timing of DS iPSCs to NPCs, which could contribute to the developmental deficits in DS (Prutton et al., 2023). Notably, trisomic NPCs were observed to have greater glial-like differentiation potential compared to euploid NPCs (Mollo et al., 2021). Additionally, GABAergic interneurons and medial ganglionic eminence (MGE) organoids derived from DS iPSCs were shown to exhibit abnormal perinuclear clustering of mitochondria and impaired mitochondrial function, which was rescued by the inhibition of the DSCAM-PAK1 pathway by gene editing or treatment with a small molecule FRAX 486 (Xu et al., 2022).
In summary, impaired mitochondrial function and oxidative stress have been implicated in DS. The use of iPSCs or their derivatives could help us reveal the underlying mechanisms of the neural and cognitive deficits involving mitochondrial dysfunction.
Insights on DS-Associated Leukemogenesis and Solid Tumor Resistance
Induced pluripotent stem cell (iPSC) models have provided valuable insights into the pathogenesis of leukemia in Down syndrome (DS). Studies using iPSCs with trisomy 21 have shown perturbations in hematopoiesis, such as increased hematopoietic progenitor populations, accelerated production of aberrant cells, and upregulation of genes that promote myeloid differentiation. Stepwise introduction of mutations found in DS-associated leukemia (such as GATA1, SMC3, and MPL mutations) into DS iPSCs has allowed researchers to model the stepwise development of acute megakaryoblastic leukemia (Evans and DeGregori, 2022). These iPSC models have reproduced cellular and molecular abnormalities seen in patients and have identified mechanisms by which trisomy 21 and recurrent mutations collaborate to drive leukemogenesis. iPSC models have also been used to study other aspects of DS pathophysiology, including musculoskeletal defects and solid tumor resistance.
Enhanced hematopoietic potential and genetic factors
Maclean et al. (2012) compared hematopoietic differentiation of disomic and trisomic subclones from the same parental iPSC line, which showed cells derived from trisomic iPSCs had increased multilineage colony-forming potential and exhibited a two- to five-fold increase in CD43(+)(Leukosialin)/CD235(+) (Glycophorin A) hematopoietic cell populations. Banno et al. (2016) and McNulty and Crispino (2016) showed that iPSCs with trisomy 21 and GATA1 mutations had perturbed hematopoiesis, in which the increased early hematopoietic progenitors and upregulated mutant GATA1 accelerated aberrant cell production. These effects were mediated by the increased expression of RUNX1, ETS2, and ERG on Chr 21, which suggest multiple genetic factors contribute to leukemogenesis (Banno et al., 2016; McNulty and Crispino, 2016).
Modeling DS myeloid leukemia: altered hematopoietic pathways and proliferation
Barwe et al. (2022) developed a DS Myeloid Leukemia model via hematopoietic differentiation of gene-targeted iPSCs bearing trisomy 21. Mutations in GATA1 and STAG2, which are recurrent in this leukemia, were sequentially introduced into DS iPSCs. The introduction of GATA1 mutations downregulated megakaryocytic and erythrocytic differentiation pathways and interferon α/β signaling, and upregulated pathways promoting myeloid differentiation such as toll-like receptor cascade. Subsequent knockout of STAG2 partially reverted the expression of genes associated with myeloid differentiation, likely promoting leukemogenesis (Barwe et al., 2022). Arkoun et al. (2022) performed stepwise gene editing of GATA1s, SMC3+/−, and MPLW515K into trisomy 21 and disomic iPSCs to model megakaryocyte differentiation defects. Among the 20 different trisomic and disomic iPSC clones, they found GATA1s profoundly reshaped iPSC-derived hematopoietic architecture, with a gradual myeloid-to-megakaryocyte shift. The addition of SMC3 and MPL mutations also affected megakaryocyte differentiation, with changes in essential megakaryocyte differentiation genes, including downregulation of NFE2 (Arkoun et al., 2022). They also found that trisomy 21 enhanced the proliferative phenotype, reproducing the cellular and molecular abnormalities in DS-associated AMKL.
Galat et al. (2020) showed DS iPSC-derived progenitor stromal cells had aberrant musculoskeletal development and resistance to solid tumors. Additionally, Perepitchka et al. (2020) found that DS iPSC-derived endothelial cells exhibited decreased proliferation, migration, and inflammatory response. They also reported a set of genes potentially associated with the unfavorable solid tumor microenvironment and with the elevated leukemia risk in DS, suggesting that these genes could be potential therapeutic targets in translational cancer research (Perepitchka et al., 2020).
In summary, these studies illuminated the heightened hematopoietic potential and genetic intricacies associated with Down syndrome. Trisomic iPSCs display enhanced hematopoiesis, influenced by genetic mutations like GATA1 and STAG2, which may contribute to leukemia. Notable genes on Chr 21, including RUNX1, ETS2, and ERG, play a pivotal role. Modeling DS Myeloid Leukemia underscores the complexity of hematopoietic pathways and proliferation. Furthermore, DS iPSC-derived cells exhibit resistance to solid tumors, opening avenues for potential therapeutic targets. These findings deepen our understanding of DS-related hematological and solid tumor abnormalities, paving the way for further research and therapies.
Mechanisms of the Neural Deficits in DS
Down syndrome causes a variety of developmental deficits, such as disruptions in neural progenitor division, neurogenesis and gliogenesis, altered cortical architecture, and reduced cortical volume, leading to neural abnormalities and intellectual disability. The use of DS iPSCs and their derivatives including NPCs, neurons, astrocytes, and organoids can provide a platform to investigate the cellular mechanisms underlying these neural deficits.
Chen et al. (2014) showed that DS iPSC-derived astroglia lacked the ability to support neuronal ion channel maturation and synapse formation in vitro, and were unable to promote neurogenesis in endogenous neural stem cells in vivo. Moreover, DS astroglia exhibited aberrant calcium signaling that led to inhibited neuron excitability, which could be rescued by suppressing astrocytic calcium activity (Mizuno et al., 2018).
GABAergic neurons differentiated from DS-derived iPSCs showed cellular/migratory deficits, calretinin/calbindin imbalance, and impaired migration in mouse brain (Huo et al., 2018). Correcting the disrupted pathways mitigated the migration deficits in DS GABA progenitors in vitro (Huo et al., 2018). Real et al. (2018) transplanted DS iPSC-derived cortical neurons into adult mouse cortex, which showed increased synaptic stability and reduced oscillations. DS iPSC-derived organoids and chimeric mouse brain showed overproduction of OLIG2(+) progenitors and excess GABAergic neurons (Xu et al., 2019). The increase in OLIG2 in DS progenitors upregulated interneuron determinants, whereas suppressing OLIG2 reversed the gene expression changes, reduced excess neurons, and improved the behavior in mice (Xu et al., 2019). Neurons derived from DS iPSCs also displayed widespread transcriptomic, proteomic, and cellular changes associated with neurodevelopment (Sobol et al., 2019; Wu et al., 2022a). The DS neurons also exhibited progressive retromer deficiency together with amyloid and tau pathologies (Curtis et al., 2022). Enhancing retromer function in DS neurons alleviated these pathologies (Curtis et al., 2022).
In summary, the disruption of neural development in DS involves astroglia, progenitors, and GABAergic/other neurons. Exploiting DS iPSCs and their derivatives could help to identify dysregulated pathways and provide insights in the neuropathological mechanisms.
Potential Targets for DS Treatments
Although there has been much research focused on elucidating neurodevelopmental and neurological alterations in DS, laboratory studies on DS are still relatively challenging. Murine models have been developed and revealed many changes similar to those observed in DS individuals, which can potentially provide insights into disease mechanisms and the functions of specific genes. For example, the Ts65Dn DS model has three copies of most of the Chr 16 genes, which are homologs of human Chr 21 genes. This model has been used to investigate how triplication of genes results in abnormalities in DS patients (Murakami et al., 2023). Several phenotypes observed in this DS mouse model have yet to be confirmed in DS individuals. Nevertheless, human neurodevelopment and neuropathological processes are too complex to recapitulate in mouse model. Consequently, most of the tested drugs indicated by DS studies using animal models have not demonstrated any positive effect or very limited efficacy in clinical trials, leading to only few novel treatments for DS in the past decade (Rueda et al., 2020).
Current research using iPSCs and their derivatives obtained from DS patients revealed that the abnormalities in DS were associated with specific genes, suggesting these genes could be potential targets for treatments. Considering the presence of an extra full or partial copy of Chr 21 in DS, much effort has been made to normalize Chr 21 genes, suppress the expression of Chr 21 genes, or even completely remove this extra chromosome. For example, pharmacological targeting or use of shRNA to suppress DYRK1A on Chr 21 resulted in the significant correction of defects, such as the abnormal neural differentiation observed in DS iPSCs (Hibaoui et al., 2014). Caglayan (2016) revealed that single-copy DYRK1A knockout in DS iPSC-derived cerebral organoids could correct neurodevelopmental phenotypes such as impaired neocortical development. These findings implicate DYRK1A on Chr 21 as a strong target for ameliorating cognitive impairments in DS. Chen et al. (2014) showed that DS astroglia treated with minocycline, an FDA-approved antibiotic, suppressed the expression of S100B, GFAP, inducible nitric oxide synthase, and thrombospondins 1 and 2, leading to the partial correction of the pathological phenotypes in DS astroglia.
Recent studies have uncovered promising pathway-specific therapeutic targets for DS. Suppressing the DSCAM/PAK1 pathway in cerebral organoids formed from DS iPSCs reversed the neurogenesis deficits (Tang et al., 2021), which could be a potential therapeutic strategy. A chemical chaperone 4-phenylbutyrate was able to decrease protein aggregates, prevent premature senescence, and reduce apoptotic cell death in trisomy 21 cells (Hirata et al., 2020; Nawa et al., 2019), which suggests targeting aneuploidy-associated stress could benefit human trisomies like DS. Activating Sonic hedgehog signaling rescued early defects in neural lineage specification of neural precursors that were differentiated from DS iPSCs (Klein et al., 2021), highlighting the potential of modulating key pathways at specific developmental stages. Extracellular vesicle treatment alleviated neuropathology in a DS cortical spheroid model (Campbell et al., 2023), suggesting the manipulation of intercellular communication could provide new treatment paradigms. Activating WNT signaling partially restored COUP-TFII+ progenitors, which could be targeted to correct differentiation deficits (Giffin-Rao et al., 2022). Chang et al. (2015) showed that treatment with N-butylidenephthalide (Bdph), which was emulsified by pluronic F127, promoted Wnt signaling and attenuated AD-like pathologies including Aβ40 accumulation and total or hyperphosphorylated tau levels in DS neurons.
Targeting epigenetic dysregulation
Some studies have revealed epigenetic dysregulation as a potential therapeutic target in DS. Bioinformatic analyses showed that several microRNAs encoded on chromosome 21, including miR-99a, let-7c, miR-125b-2, miR-155 and miR-802, were overexpressed in DS fetal tissues and likely contribute to cognitive and cardiac defects (Chang et al., 2009; Kuhn et al., 2008). Additionally, epigenetic therapies like DNA methyltransferase inhibitors and HDAC inhibitors have shown promise in preclinical DS models. One study found that the combination of azacitidine and panobinostat significantly improved survival compared to chemotherapy in a DS-acute myeloid leukemia patient-derived xenograft model (Barwe et al., 2019). Another study revealed synergy between the EZH2 inhibitor GSK126 and the HDAC inhibitor romidepsin in inhibiting DS-megakaryoblastic leukemia growth in cell and xenograft models (Cicek et al., 2022). These findings highlight the potential of normalizing dysregulated epigenetic pathways through microRNA, DNA methylation, or histone modification targeting therapies. Epigenetic treatment strategies hold great promise for ameliorating diverse cellular and physiological defects in DS and warrant further evaluation. In line with this epigenetic approach, Czerminski and Lawrence (2020) used XIST RNA to silence trisomy 21, which induced neural stem cells to undergo neuronal differentiation, indicating this unique non-coding RNA could have translational potential for DS neurobiology. Furthermore, reprogramming DS fibroblasts to iPSCs resulted in the spontaneous loss of the extra chromosome in some cells, which generated disomic iPSC clones. This phenomenon could be further investigated as a potential strategy for trisomy rescue (Akutsu et al., 2022).
Cell therapy has emerged as a potential treatment option for Down syndrome (DS). Implanting murine neural stem cells (mNSCs) or murine neural progenitor cells (mNPCs) has shown promising results in enhancing cell survival and injury response in trisomic mice. Notably, these transplanted mNSCs/mNPCs exhibited migration to injury sites, offering neuroprotection and facilitating axon growth (Salamt et al., 2021). However, it is important to note that cell therapy has not yielded significant findings with regard to improvements in the physical appearance, cognitive function, social skills, or behavior of individuals with DS (Salamt et al., 2021).
In summary, the above studies elucidate promising treatment strategies for DS that are emerging from studies employing cell models derived from DS patients. These strategies aim to rectify molecular and cellular deficits associated with trisomy 21 through diverse approaches, such as normalizing the expression of chromosome 21 genes, modulating key signaling pathways disrupted in DS, reducing pathological abnormalities in DS neural cells, reversing neurodevelopmental impairments, and exploring cell therapy options. These findings hold promise, but their translation into effective clinical treatments necessitates systematic evaluation of risks, benefits, and ethical considerations through rigorous preclinical studies and clinical trials. Collectively, recent advances in DS research models are uncovering molecular details that could pave the way for tailored interventions at different developmental stages to improve outcomes for individuals with DS.
Studies employing DS iPSCs and their derivatives have revealed many insights into the cellular and molecular mechanisms of the mitochondrial dysfunction, neural deficits and leukemia risk associated with DS. Some promising strategies for ameliorating DS pathology include targeting dysregulated pathways, suppressing the effects of triplicate genes, and rescuing altered network dynamics (Akutsu et al., 2022; Caglayan, 2016; Chen et al., 2014; Czerminski and Lawrence, 2020).
However, translating the promising findings from preclinical studies to clinical trials poses substantial issues. Given the complexity of DS, including the cognitive impairment, early onset AD, and leukemia risk, and the involvement of genetic, epigenetic, and environmental factors across developmental stages (Asim et al., 2015; Sobol et al., 2019), recapitulating this disease in animal models is challenging. Mouse models with engineered trisomies and neuronal defects can inform such factors, but cannot completely mimic DS (Murakami et al., 2023). With advancements in iPSC technology, iPSC-based models that integrate multiple cell types, ages, brain regions, and genetic backgrounds can now be used to study the underlying mechanisms in DS. Using 3D organoids or assembloids to model intact neural circuitry and brain regions may yield more translatable insights (Caglayan, 2016).
In summary, although the mechanistic insights gained from in vitro modeling are invaluable, developing safe and effective treatments for DS requires long-term commitment with careful consideration of the risks and real-world contexts. Understanding the multifaceted relationships between genetic, developmental, and environment factors in DS may lead to novel therapies offering dignity and independence to those affected. Although there is still a long way to go, the first steps have already been taken.
Acknowledgement: We thank DF Guo for technical support.
Funding Statement: This work was supported by Shenzhen Science and Technology Planning Project (Grant No. JCYJ20210324093209024); and Stable Support Project of Shenzhen (Grant No. 20220812182215001).
Author Contributions: The authors confirm contribution to the paper as follows: Study conception and design: Yafei Wang, Jielei Ni, Yanxiang Ni; Manuscript preparation and organization: Yafei Wang, Jielei Ni; Review and approval of the final manuscript: Yanxiang Ni. Others contributed to the intellectual content of the review paper. All authors reviewed the manuscript and approved the final version of the manuscript.
Availability of Data and Materials: Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
Akutsu SN, Miyamoto T, Oba D, Tomioka K, Ochiai H, Ohashi H, Matsuura S (2022). iPSC reprogramming-mediated aneuploidy correction in autosomal trisomy syndromes. PLoS One 17: e0264965. https://doi.org/10.1371/journal.pone.0264965 [Google Scholar] [PubMed] [CrossRef]
Anderson CC, Marentette JO, Prutton KM, Rauniyar AK, Reisz JA, D’Alessandro A, Maclean KN, Saba LM, Roede JR (2021). Trisomy 21 results in modest impacts on mitochondrial function and central carbon metabolism. Free Radical Biology and Medicine 172: 201–212. https://doi.org/10.1016/j.freeradbiomed.2021.06.003 [Google Scholar] [PubMed] [CrossRef]
Arkoun B, Robert E, Boudia F, Mazzi S, Dufour V et al. (2022). Stepwise GATA1 and SMC3 mutations alter megakaryocyte differentiation in a Down syndrome leukemia model. Journal of Clinical Investigation 132: e156290. https://doi.org/10.1172/JCI156290 [Google Scholar] [PubMed] [CrossRef]
Asim A, Kumar A, Muthuswamy S, Jain S, Agarwal S (2015). Down syndrome: An insight of the disease. Journal of Biomedical Science 22: 41. https://doi.org/10.1186/s12929-015-0138-y [Google Scholar] [PubMed] [CrossRef]
Banno K, Omori S, Hirata K, Nawa N, Nakagawa N et al. (2016). Systematic cellular disease models reveal synergistic interaction of trisomy 21 and GATA1 mutations in hematopoietic abnormalities. Cell Reports 15: 1228–1241. https://doi.org/10.1016/j.celrep.2016.04.031 [Google Scholar] [PubMed] [CrossRef]
Barwe S, Kolb EA, Crowgey EL, Druley TE, Gopalakrishnapillai A (2019). Generation, characterization and pre-clinical drug evaluation of patient-derived xenograft models of pediatric Down syndrome AML. Blood 134: 2683. https://doi.org/10.1182/blood-2019-131050 [Google Scholar] [CrossRef]
Barwe SP, Sebastian A, Sidhu I, Kolb EA, Gopalakrishnapillai A (2022). Modeling Down syndrome myeloid leukemia by sequential introduction of GATA1 and STAG2 mutations in induced pluripotent stem cells with trisomy 21. Cells 11: 628. https://doi.org/10.3390/cells11040628 [Google Scholar] [PubMed] [CrossRef]
Barwe SP, Sidhu I, Kolb EA, Gopalakrishnapillai A (2020). Modeling transient abnormal myelopoiesis using induced pluripotent stem cells and CRISPR/Cas9 technology. Molecular Therapy Methods & Clinical Development 19: 201–209. https://doi.org/10.1016/j.omtm.2020.09.007 [Google Scholar] [PubMed] [CrossRef]
Briggs JA, Sun J, Shepherd J, Ovchinnikov DA, Chung TL et al. (2013). Integration-free induced pluripotent stem cells model genetic and neural developmental features of Down syndrome etiology. Stem Cells 31: 467–478. https://doi.org/10.1002/stem.1297 [Google Scholar] [PubMed] [CrossRef]
Caglayan ES (2016). Generation of improved human cerebral organoids from single copy DYRK1A knockout induced pluripotent stem cells in trisomy 21: Hypothetical solutions for neurodevelopmental models and therapeutic alternatives in Down syndrome. Cell Biology International 40: 1256–1270. https://doi.org/10.1002/cbin.10694 [Google Scholar] [PubMed] [CrossRef]
Campbell NB, Patel Y, Moore TL, Medalla M, Zeldich E (2023). Extracellular vesicle treatment alleviates neurodevelopmental and neurodegenerative pathology in cortical spheroid model of Down syndrome. International Journal of Molecular Sciences 24: 3477. https://doi.org/10.3390/ijms24043477 [Google Scholar] [PubMed] [CrossRef]
Chang CY, Chen SM, Lu HE, Lai SM, Lai PS et al. (2015). N-butylidenephthalide attenuates Alzheimer’s disease-like cytopathy in Down syndrome induced pluripotent stem cell-derived neurons. Scientific Reports 5: 8744. https://doi.org/10.1038/srep08744 [Google Scholar] [PubMed] [CrossRef]
Chang S, Wen S, Chen D, Jin P (2009). Small regulatory RNAs in neurodevelopmental disorders. Human Molecular Genetics 18: R18–26. https://doi.org/10.1093/hmg/ddp072 [Google Scholar] [PubMed] [CrossRef]
Chen C, Jiang P, Xue H, Peterson SE, Tran HT et al. (2014). Role of astroglia in Down’s syndrome revealed by patient-derived human-induced pluripotent stem cells. Nature Communications 5: 4430. https://doi.org/10.1038/ncomms5430 [Google Scholar] [PubMed] [CrossRef]
Chen X-Q, Salehi A, Pearn ML, Overk C, Nguyen PD, Kleschevnikov AM, Maccecchini M, Mobley WC (2021). Targeting increased levels of APP in Down syndrome: Posiphen-mediated reductions in APP and its products reverse endosomal phenotypes in the Ts65Dn mouse model. Alzheimer’s & Dementia 17: 271–292. https://doi.org/10.1002/alz.12185 [Google Scholar] [PubMed] [CrossRef]
Cicek K, Faust JR, Tavenner M, Kolb EA, Barwe SP, Gopalakrishnapillai A (2022). Preclinical validation of EZH2 and HDAC I dual inhibition as a potent therapy for refractory myeloid leukemia associated with Down syndrome. Blood 140: 5951–5952. https://doi.org/10.1182/blood-2022-170549 [Google Scholar] [CrossRef]
Curtis ME, Smith T, Blass BE, Pratico D (2022). Dysfunction of the retromer complex system contributes to amyloid and tau pathology in a stem cell model of Down syndrome. Alzheimers Dement 8: e12334. https://doi.org/10.1002/trc2.12334 [Google Scholar] [PubMed] [CrossRef]
Cypris O, Eipel M, Franzen J, Rosseler C, Tharmapalan V et al. (2020). PRDM8 reveals aberrant DNA methylation in aging syndromes and is relevant for hematopoietic and neuronal differentiation. Clinical Epigenetics 12: 125. https://doi.org/10.1186/s13148-020-00914-5 [Google Scholar] [PubMed] [CrossRef]
Czerminski JT, Lawrence JB (2020). Silencing trisomy 21 with XIST in neural stem cells promotes neuronal differentiation. Developmental Cell 52: 294–308.E3. https://doi.org/10.1016/j.devcel.2019.12.015 [Google Scholar] [PubMed] [CrossRef]
Esposito G, Imitola J, Lu J, De Filippis D, Scuderi C et al. (2008). Genomic and functional profiling of human Down syndrome neural progenitors implicates S100B and aquaporin 4 in cell injury. Human Molecular Genetics 17: 440–457. https://doi.org/10.1093/hmg/ddm322 [Google Scholar] [PubMed] [CrossRef]
EvansJr EJ, DeGregori J (2022). Dissecting stepwise mutational impairment of megakaryopoiesis in a model of Down syndrome-associated leukemia. Journal of Clinical Investigation 132: e161659. https://doi.org/10.1172/JCI161659 [Google Scholar] [PubMed] [CrossRef]
Galat Y, Perepitchka M, Elcheva I, Iannaccone S, Iannaccone PMGalat V (2020). iPSC-derived progenitor stromal cells provide new insights into aberrant musculoskeletal development and resistance to cancer in Down syndrome. Scientific Reports 10: 13252. https://doi.org/10.1038/s41598-020-69418-9 [Google Scholar] [PubMed] [CrossRef]
Giffin-Rao Y, Sheng J, Strand B, Xu K, Huang L et al. (2022). Altered patterning of trisomy 21 interneuron progenitors. Stem Cell Reports 17: 1366–1379. https://doi.org/10.1016/j.stemcr.2022.05.001 [Google Scholar] [PubMed] [CrossRef]
Gonzales PK, Roberts CM, Fonte V, Jacobsen C, Stein GHLink CD (2018). Transcriptome analysis of genetically matched human induced pluripotent stem cells disomic or trisomic for chromosome 21. PLoS One 13: e0194581. https://doi.org/10.1371/journal.pone.0194581 [Google Scholar] [PubMed] [CrossRef]
Gough G, O'Brien NL, Alic I, Goh PA, Yeap YJ, Groet J, Nizetic D, Murray A (2020). Modeling Down syndrome in cells: From stem cells to organoids. Progress in Brain Research 251: 55–90. https://doi.org/10.1016/bs.pbr.2019.10.003 [Google Scholar] [PubMed] [CrossRef]
Grad I, Hibaoui Y, Jaconi M, Chicha L, Bergstrom-Tengzelius R et al. (2011). NANOG priming before full reprogramming may generate germ cell tumours. European Cells and Materials 22: 258–274. https://doi.org/10.22203/eCM.v022a20 [Google Scholar] [PubMed] [CrossRef]
Guimera J, Casas C, Estivill X, Pritchard M (1999). Human minibrain homologue (MNBH/DYRK1Characterization, alternative splicing, differential tissue expression, and overexpression in Down syndrome. Genomics 57: 407–418. https://doi.org/10.1006/geno.1999.5775 [Google Scholar] [PubMed] [CrossRef]
Hibaoui Y, Grad I, Letourneau A, Sailani MR, Dahoun S et al. (2014). Modelling and rescuing neurodevelopmental defect of Down syndrome using induced pluripotent stem cells from monozygotic twins discordant for trisomy 21. EMBO Molecular Medicine 6: 259–277. https://doi.org/10.1002/emmm.201302848 [Google Scholar] [PubMed] [CrossRef]
Hirata K, Nambara T, Kawatani K, Nawa N, Yoshimatsu H et al. (2020). 4-Phenylbutyrate ameliorates apoptotic neural cell death in Down syndrome by reducing protein aggregates. Scientific Reports 10: 14047. https://doi.org/10.1038/s41598-020-70362-x [Google Scholar] [PubMed] [CrossRef]
Huo HQ, Qu ZY, Yuan F, Ma L, Yao L et al. (2018). Modeling Down syndrome with patient IPSCS reveals cellular and migration deficits of GABAergic neurons. Stem Cell Reports 10: 1251–1266. https://doi.org/10.1016/j.stemcr.2018.02.001 [Google Scholar] [PubMed] [CrossRef]
Klein JA, Li Z, Rampam S, Cardini J, Ayoub A, Shaw P, Rachubinski AL, Espinosa JM, Zeldich E, Haydar T F (2021). Sonic hedgehog pathway modulation normalizes expression of Olig2 in rostrally patterned NPCs with trisomy 21. Frontiers in Cellular Neuroscience 15: 794675. https://doi.org/10.3389/fncel.2021.794675 [Google Scholar] [PubMed] [CrossRef]
Kuhn DE, Nuovo GJ, Martin MM, Malana GE, Pleister AP et al. (2008). Human chromosome 21-derived miRNAs are overexpressed in Down syndrome brains and hearts. Biochemical and Biophysical Research Communications 370: 473–477. https://doi.org/10.1016/j.bbrc.2008.03.120 [Google Scholar] [PubMed] [CrossRef]
Li Z, Klein JA, Rampam S, Kurzion R, Campbell NB, Patel Y, Haydar TF, Zeldich E (2022). Asynchronous excitatory neuron development in an isogenic cortical spheroid model of Down syndrome. Frontiers in Neuroscience 16: 932384. https://doi.org/10.3389/fnins.2022.932384 [Google Scholar] [PubMed] [CrossRef]
Liu H, Huang S, Wang W, Wang H, Huang W et al. (2022). Migration deficits of the neural crest caused by CXADR triplication in a human Down syndrome stem cell model. Cell Death & Disease 13: 1018. https://doi.org/10.1038/s41419-022-05481-6 [Google Scholar] [PubMed] [CrossRef]
Liu Y, Zhang Y, Ren Z, Zeng F, Yan J (2023). RUNX1 upregulation causes mitochondrial dysfunction via regulating the PI3K-Akt pathway in iPSC from patients with Down syndrome. Molecules and Cells 46: 219–230. https://doi.org/10.14348/molcells.2023.2095 [Google Scholar] [PubMed] [CrossRef]
Ma W, Liu Y, Ma H, Ren Z, Yan J (2021). Cis-acting: A pattern of lncRNAs for gene regulation in induced pluripotent stem cells from patients with Down syndrome determined by integrative analysis of lncRNA and mRNA profiling data. Experimental and Therapeutic Medicine 22: 701. https://doi.org/10.3892/etm.2021.10133 [Google Scholar] [PubMed] [CrossRef]
Maclean GA, Menne TF, Guo G, Sanchez DJ, Park IH, Daley GQ, Orkin SH (2012). Altered hematopoiesis in trisomy 21 as revealed through in vitro differentiation of isogenic human pluripotent cells. Proceedings of the National Academy of Sciences of the United States of America 109: 17567–17572. https://doi.org/10.1073/pnas.1215468109 [Google Scholar] [PubMed] [CrossRef]
McGarvey SS, Ferreyros M, Kogut I, Bilousova G (2022). Differentiating induced pluripotent stem cells toward mesenchymal stem/stromal cells. Methods in Molecular Biology 2549: 153–167. https://doi.org/10.1007/978-1-0716-2585-9 [Google Scholar] [CrossRef]
McNulty M, Crispino JD (2016). iPSCs offer a new look at GATA1-trisomy 21 cooperation. Cell Stem Cell 18: 563–564. https://doi.org/10.1016/j.stem.2016.04.007 [Google Scholar] [PubMed] [CrossRef]
Mizuno GO, Wang Y, Shi G, Wang Y, Sun J et al. (2018). Aberrant calcium signaling in astrocytes inhibits neuronal excitability in a human Down syndrome stem cell model. Cell Reports 24: 355–365. https://doi.org/10.1016/j.celrep.2018.06.033 [Google Scholar] [PubMed] [CrossRef]
Mollo N, Esposito M, Aurilia M, Scognamiglio R, Accarino R et al. (2021). Human trisomic iPSCs from Down syndrome fibroblasts manifest mitochondrial alterations early during neuronal differentiation. Biology 10: 609. https://doi.org/10.3390/biology10070609 [Google Scholar] [PubMed] [CrossRef]
Murakami K, Hamazaki N, Hamada N, Nagamatsu G, Okamoto I et al. (2023). Generation of functional oocytes from male mice in vitro. Nature 615: 900–906. https://doi.org/10.1038/s41586-023-05834-x [Google Scholar] [PubMed] [CrossRef]
Nawa N, Hirata K, Kawatani K, Nambara T, Omori S et al. (2019). Elimination of protein aggregates prevents premature senescence in human trisomy 21 fibroblasts. PLoS One 14: e0219592. https://doi.org/10.1371/journal.pone.0219592 [Google Scholar] [PubMed] [CrossRef]
Nehra S, Sharma V, Umrani M, Singhal N (2023). Generation of integration-free Down syndrome and isogenic euploid human induced pluripotent stem cells. Stem Cell Research 67: 103041. https://doi.org/10.1016/j.scr.2023.103041 [Google Scholar] [PubMed] [CrossRef]
Omori S, Tanabe H, Banno K, Tsuji A, Nawa N et al. (2017). A pair of maternal chromosomes derived from meiotic nondisjunction in trisomy 21 affects nuclear architecture and transcriptional regulation. Scientific Reports 7: 764. https://doi.org/10.1038/s41598-017-00714-7 [Google Scholar] [PubMed] [CrossRef]
Ovchinnikov DA, Korn O, Virshup I, Wells CA, Wolvetang EJ (2018). The impact of APP on alzheimer-like pathogenesis and gene expression in Down syndrome iPSC-derived neurons. Stem Cell Reports 11: 32–42. https://doi.org/10.1016/j.stemcr.2018.05.004 [Google Scholar] [PubMed] [CrossRef]
Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ (2008). Disease-specific induced pluripotent stem cells. Cell 134: 877–886. https://doi.org/10.1016/j.cell.2008.07.041 [Google Scholar] [PubMed] [CrossRef]
Parra V, Altamirano F, Hernandez-Fuentes CP, Tong D, Kyrychenko V et al. (2018). Down syndrome critical region 1 gene, RCAN1, helps maintain a more fused mitochondrial network. Circulation Research 122: e20–e33. https://doi.org/10.1161/CIRCRESAHA.117.311522 [Google Scholar] [PubMed] [CrossRef]
Perepitchka M, Galat Y, Beletsky IP, Iannaccone PM, Galat V (2020). Down syndrome iPSC model: Endothelial perspective on tumor development. Oncotarget 11: 3387–3404. https://doi.org/10.18632/oncotarget.27712 [Google Scholar] [PubMed] [CrossRef]
Ponroy Bally B, Farmer WT, Jones EV, Jessa S, Kacerovsky JB et al. (2020). Human iPSC-derived Down syndrome astrocytes display genome-wide perturbations in gene expression, an altered adhesion profile, and increased cellular dynamics. Human Molecular Genetics 29: 785–802. https://doi.org/10.1093/hmg/ddaa003 [Google Scholar] [PubMed] [CrossRef]
Prutton KM, Marentette JO, Maclean KN, Roede JR (2023). Characterization of mitochondrial and metabolic alterations induced by trisomy 21 during neural differentiation. Free Radical Biology and Medicine 196: 11–21. https://doi.org/10.1016/j.freeradbiomed.2023.01.009 [Google Scholar] [PubMed] [CrossRef]
Qiu JJ, Liu YN, Ren ZR, Yan JB (2017). Dysfunctions of mitochondria in close association with strong perturbation of long noncoding RNAs expression in Down syndrome. International Journal of Biochemistry & Cell Biology 92: 115–120. https://doi.org/10.1016/j.biocel.2017.09.017 [Google Scholar] [PubMed] [CrossRef]
Real R, Peter M, Trabalza A, Khan S, Smith MA et al. (2018). In vivo modeling of human neuron dynamics and Down syndrome. Science 362: eaau1810. https://doi.org/10.1126/science.aau1810 [Google Scholar] [PubMed] [CrossRef]
Rueda N, Florez J, Dierssen M, Martinez-Cue C (2020). Translational validity and implications of pharmacotherapies in preclinical models of Down syndrome. Progress in Brain Research 251: 245–268. https://doi.org/10.1016/bs.pbr.2019.10.001 [Google Scholar] [PubMed] [CrossRef]
Salamt N, Idrus RH, Nasri NWM (2021). Down syndrome and cell therapy: A review. Sains Malaysiana 50: 1381–1392. https://doi.org/10.17576/jsm-2021-5005-17 [Google Scholar] [CrossRef]
Sawa M, Overk C, Becker A, Derse D, Albay R et al. (2022). Impact of increased APP gene dose in Down syndrome and the Dp16 mouse model. Alzheimer’s & Dementia 18: 1203–1234. https://doi.org/10.1002/alz.12463 [Google Scholar] [PubMed] [CrossRef]
Schuster J, Hoeber J, Sobol M, Fatima A, Anneren GDahl N (2020). Generation of two human iPSC lines (UUIGPi013-A and UUIPGi014-A) from cases with Down syndrome and full trisomy for chromosome 21 (T21). Stem Cell Research 49: 102081. https://doi.org/10.1016/j.scr.2020.102081 [Google Scholar] [PubMed] [CrossRef]
Shi Y, Kirwan P, Smith J, MacLean G, Orkin SH, Livesey FJ (2012). A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Science Translational Medicine 4: 124ra29. https://doi.org/10.1126/scitranslmed.3003771 [Google Scholar] [PubMed] [CrossRef]
Sobol M, Klar J, Laan L, Shahsavani M, Schuster J et al. (2019). Transcriptome and proteome profiling of neural induced pluripotent stem cells from individuals with Down syndrome disclose dynamic dysregulations of key pathways and cellular functions. Molecular Neurobiology 56: 7113–7127. https://doi.org/10.1007/s12035-019-1585-3 [Google Scholar] [PubMed] [CrossRef]
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131: 861–872. https://doi.org/10.1016/j.cell.2007.11.019 [Google Scholar] [PubMed] [CrossRef]
Tan KL, Lee HC, Cheah PS, Ling KH (2023). Mitochondrial dysfunction in Down syndrome: From pathology to therapy. Neuroscience 511: 1–12. https://doi.org/10.1016/j.neuroscience.2022.12.003 [Google Scholar] [PubMed] [CrossRef]
Tang XY, Xu L, Wang J, Hong Y, Wang Y et al. (2021). DSCAM/PAK1 pathway suppression reverses neurogenesis deficits in iPSC-derived cerebral organoids from patients with Down syndrome. Journal of Clinical Investigation 131: e135763. https://doi.org/10.1172/JCI135763 [Google Scholar] [PubMed] [CrossRef]
Teles ESAL, Yokota BY, Sertie AL, Zampieri BL (2023). Generation of urine-derived induced pluripotent stem cells and cerebral organoids for modeling Down syndrome. Stem Cell Reviews and Reports 19: 1116–1123. https://doi.org/10.1007/s12015-022-10497-8 [Google Scholar] [PubMed] [CrossRef]
Wang Y, Li Z, Yang G, Cai L, Yang F, Zhang Y, Zeng Y, Ma Q, Zeng F (2021). The study of alternative splicing events in human induced pluripotent stem cells from a Down’s syndrome patient. Frontiers in Cell and Developmental Biology 9: 661381. https://doi.org/10.3389/fcell.2021.661381 [Google Scholar] [PubMed] [CrossRef]
Wu CI, Vinton EA, Pearse RVII, Heo K, Aylward AJ et al. (2022a). APP and DYRK1A regulate axonal and synaptic vesicle protein networks and mediate Alzheimer’s pathology in trisomy 21 neurons. Molecular Psychiatry 27: 1970–1989. https://doi.org/10.1038/s41380-022-01454-5 [Google Scholar] [PubMed] [CrossRef]
Wu Y, West NR, Bhattacharyya A, Wiseman FK (2022b). Cell models for Down syndrome-Alzheimer’s disease research. Neuronal Signaling 6: NS20210054. https://doi.org/10.1042/NS20210054 [Google Scholar] [PubMed] [CrossRef]
Xu R, Brawner AT, Li S, Liu JJ, Kim H et al. (2019). OLIG2 Drives abnormal neurodevelopmental phenotypes in human iPSC-based organoid and chimeric mouse models of Down syndrome. Cell Stem Cell 24: 908–926.E8. https://doi.org/10.1016/j.stem.2019.04.014 [Google Scholar] [PubMed] [CrossRef]
Xu L, Huo HQ, Lu KQ, Tang XY, Hong Y et al. (2022). Abnormal mitochondria in Down syndrome iPSC-derived GABAergic interneurons and organoids. Biochimica et Biophysica Acta (BBA)–Molecular Basis of Disease 1868: 166388. https://doi.org/10.1016/j.bbadis.2022.166388 [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools