
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Analysis of differentially expressed genes in Verruca vulgaris vs. adjacent normal skin by RNA-sequencing
1 School of Medicine, Nankai University, Tianjin, 300071, China
2 Department of Dermatology, First Medical Center of Chinese People’s Liberation Army General Hospital, Beijing, 100853, China
* Corresponding Authors: FANG XIE. Email: ; CHENGXIN LI. Email:
# These authors contributed equally to this work
BIOCELL 2023, 47(11), 2435-2443. https://doi.org/10.32604/biocell.2023.043126
Received 22 June 2023; Accepted 04 September 2023; Issue published 27 November 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Introduction: Verruca vulgaris is one of the most common low-risk HPV infections and is characterized by excessive proliferation of keratinocytes. Currently, very little genetic information is available regarding verruca vulgaris in the Chinese population. This study aimed to obtain comprehensive transcript information of verruca vulgaris by RNA sequencing. Methods: High-throughput sequencing was performed on three fresh verruca vulgaris samples and adjacent normal skin on the Illumina sequencing platform. The transcriptomes were analyzed using bioinformatics and the differentially expressed genes (DEGs) were verified by immunohistochemistry. Verruca vulgaris exhibited a unique molecular signature. Results: In total, 1,643 DEGs were identified in verruca vulgaris compared to normal skin. The functions of the DEGs were studies by Gene Ontology (GO) enrichment, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, DEGs Reactome analysis, disease annotation function, and STRING protein-protein interaction (PPI) network analysis. The results revealed 595 GO terms associated with the cell cycle, signal transduction, immune system, signaling molecules, and interaction. The Reactome analysis revealed enrichment in reversible hydration of carbon dioxide and BMP signaling, while the disease annotation function revealed that the enriched DEGs are involved in keratosis disorders. The STRING PPI network showed that the edges with the highest density mainly included the 2′–5′ oligoadenylate synthase (OAS) family-related proteins. Furthermore, the M-code analysis found ISG15, IRF7, and OASL were scored as significant modules and their high expression compared to the control was verified by immunohistochemistry. Conclusion: These findings contribute to the genetic information of verruca vulgaris in the Chinese population, revealing that interferon-stimulated genes may play essential roles in verruca vulgaris.Keywords
Human papillomaviruses (HPVs) are circular DNA viruses that only infect the human skin epithelium. They are classified into five genera (Alpha [α], Beta [β], Gamma [γ], Mu [µ], and Nu [ν]) based on the pairwise nucleotide sequence across the L1 open reading frame and further subdivided into species designated by a number (Bernard et al., 2010). The alpha genus is further classified based on the cancer risk, including high-risk and low-risk types. High-risk HPVs infect the mucosal epithelium and are associated with the development of cervical cancer and other human malignancies. In contrast, low-risk HPVs mostly infect the skin epithelium and cause benign hyperproliferative diseases such as verruca vulgaris, plantar warts, plane warts, and condyloma acuminatum (Doorbar et al., 2015). High-risk HPV-related diseases have been extensively researched, while low-risk HPV infection requires further study.
Verruca vulgaris is a common low-risk HPV infection that mainly manifests as excessive proliferation of keratinocytes. Clinically, the treatment methods for verruca vulgaris include cryotherapy, laser, intralesional injection, etc. However, the treatment is not specific and often places a psychological burden on the patient (Ringin, 2019). Therefore, the pathogenesis and development of low-risk HPV infections should be further explored. RNA-seq is a powerful instrument for broadly profiling the gene expression of verruca vulgaris; the technique applies next-generation sequencing to quantify mRNA levels in a tissue sample at a given moment. Messenger RNAs (mRNAs) are single-stranded RNAs that carry genetic information and guide protein synthesis. Comprehensive transcript information of samples can be obtained by mRNA transcriptome sequencing, to conduct research on transcript structure, variation, and gene expression levels. At present, only one study from Jordan investigated the differences between the gene expression profiles of common warts by RNA sequencing. The results revealed that common warts possess a unique molecular signature, including 3,140 differentially expressed genes, with the top 500 differentially expressed genes being associated with immune and autoimmune pathways (Al-Eitan et al., 2020).
In this study, a comprehensive genome-wide transcriptomic analysis of verruca vulgaris and normal skin was performed by profiling mRNA sequencing to investigate the relevant molecular biomarkers, potential mechanisms, and therapeutic markers.
Three pairs of fresh verruca vulgaris and adjacent normal skin samples were collected from discarded surgical material. This study was approved by the Ethics Committee of Chinese PLA General Hospital (S2022-724-01). Verruca vulgaris was diagnosed by an experienced dermatologist and confirmed by histopathology, showing black thrombosed capillaries on keratotic surfaces, epidermal papillomatous proliferation, and hyperkeratosis with koilocytes on the upper part of the spinous cell layer. The samples were stored in liquid nitrogen immediately and then placed on dry ice for RNA expression profiling. Table 1 shows the patient characteristics.
RNA extraction, quality control, and sequencing
Total RNA was extracted from each sample using TRIzol: RNAiso Plus (TARAKA, Beijing, China) following the manufacturer’s instructions. RNA quality was assessed by gel electrophoresis and Qubit (Thermo, Waltham, MA, USA). Libraries were constructed using VAHTS Stranded mRNA-seq Library Prep Kit for Illumina (Vazyme, Nanjing, China). 500 ng RNA from each sample was used for RNA-seq. The high-throughput sequencing of multiple samples was carried out using the pair-end (PE) sequencing mode of the Illumina Novaseq 6000 instrument (Illumina, San Diego, USA), which was performed by Genergy Biotechnology Co. Ltd. (Shanghai, China).
Skewer software (v0.2.2, Illumina, San Diego, USA) was used to delete joint sequences and poor-quality fragments from the 3′ end of the sequencing data. Clean reads were aligned to the reference genome using Spliced Transcripts Alignments to a reference, STAR (2.5.3a, Illumina, San Diego, USA). Subsequently, the expression of the transcripts was calculated in FPKM (Fragments Per Kilobase of exon model per Million mapped reads) using StringTie (v1.3.1c, Johns Hopkins University Center for Computational Biology, Washington, USA) and differentially expressed genes (DEGs) were identified by DEGseq2 (v1.16.1, Bioconductor, Boston, USA). In this study, GRCh38 was used as the reference human genome. The sequence file can be found at ftp://ftp.ensembl.org/pub/release-98/fasta/homo_sapiens/dna/ and the annotation file at ftp://ftp.ensembl.org/pub/release-98/gtf/homo_sapiens/dna/.
Analysis of differentially expressed genes
Fast QC (v0.11.5, Babraham Bioinformatics, Cambridge, UK) is a quality control analysis conduction software that calculates the base proportion of Q20 and Q30 on the preprocessed data. String Tie software was used to determine the original sequence of known genes. DESeq2 software was used to analyze and compare the genes between the verruca vulgaris samples and the normal control samples. Genes showing p < 0.05 (Paired-test) and |log2 (fold-change) | ≥ 1 were considered to be significantly differentially expressed. The DEGs were then subjected to two-way hierarchical clustering, and the clustering results were displayed in thermograms (Distance metric: Pearson correlation; Linkage rule: Average Linkage).
The rMATS software (v3.2.5, Children’s Hospital of Philadelphia, Philadelphia, USA) was used to detect alternative splicing from the RNA-seq data, and StringTie was used to assemble the transcripts of the samples. The number of alternative splicing events in each sample was calculated at the gene level. The types of alternative splicing included SE (skipped exon), RI (retained intron), A5SS (alternative 5′ splice site), A3SS (alternative 3′ splice site), and MXE (mutually exclusive exon). The rMATS algorithm was used to quantify the expression of alternative splicing. Fisher’s exact test was used to test the variability of alternative splicing between two sample groups by calculating the difference significance (p < 0.05).
Functional annotation and pathway enrichment
Functional annotation and pathway enrichment of the DEGs were performed by using the Database for Annotation, Visualization, and Integrated Discovery DAVID (version 6.8, DAVID Bioinformatics Resources, Frederick, USA) to determine the enriched GO terms (adjusted p < 0.01) and the KEGG (adjusted p < 0.05).
Disease annotation function and Reactome metabolic pathway analysis
Disease annotation function analysis was conducted on the DisGeNET disease database (http://www.disgenet.org, National Institute of General Medical Sciences of the National Institutes of Health, Bethesda, USA) for functional annotation and classification of disease types. Reactome metabolic pathway analysis was performed to annotate and classify the metabolic pathways by using the Reactome database (https://reactome.org, National Institute of General Medical Sciences of the National Institutes of Health, Bethesda, USA) (adjusted p < 0.05).
Protein-protein interaction (PPI) network and significant modules identification
The PPI network of the DEGs was constructed by the STRING database (https://string-db.org, National Institute of General Medical Sciences of the National Institutes of Health, Bethesda, USA) using a combined score >0.4 as the cut-off point. In addition, the molecular complex detection (MCODE) plugin in Cyto-scape was used to filter and identify key significant modules.
To confirm the transcriptome sequencing results, immunohistochemistry was performed to verify the expression of the significant modules genes (ISG15, IRF7, OASL). Specimens from nine patients with verruca vulgaris were excised by surgical procedures and fixed in 4% paraformaldehyde (Solarbio, Beijing, China), then embedded into paraffin. Following fixation, the samples were sliced into 4 µm-thick sections. The sections were incubated with 0.1% BSA(Solarbio, Beijing, China), then incubated overnight at 4°C with target antibodies (ISG15 dilution 1:200, Proteintech, Chicago, USA, IRF7 dilution 1:100, Proteintech, Chicago, USA; OASL dilution 1:500, Abcam, Cambridge, UK). Biotinylated IgG (Proteintech, Chicago, USA) was used as a secondary antibody. DAB kit (Proteintech, Chicago, USA) was used to react with the second antibody. Images were taken with a microscope (Nikon, Tokyo, Japan) and the staining intensity was evaluated by Image-J (Statistical comparisons were performed by Wilcoxon sign rank test; p < 0.05 was considered statistically significant).
Expression profiles of differentially expressed genes in transcriptome sequencing
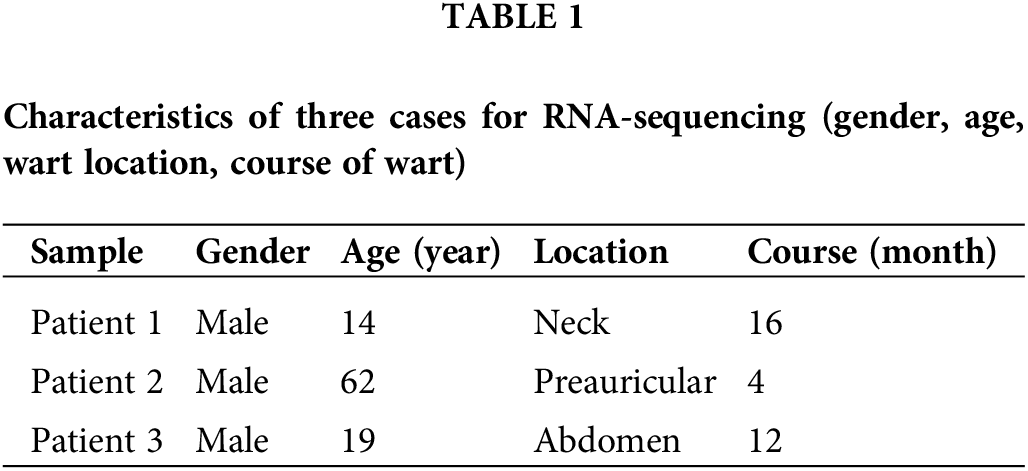
1,643 DEGs were identified by transcriptome sequencing (as displayed in Fig. 1), including 1,069 (65.1%) upregulated and 574 (34.9%) downregulated DEGs in verruca vulgaris compared with normal skin. Cluster analysis was performed on the 1,643 DEGs, as shown in Fig. 2A. Alternative splicing analysis revealed the presence of SE, RI, A5SS, A3SS, and MXE, among which SE was the most common (Fig. 2B).
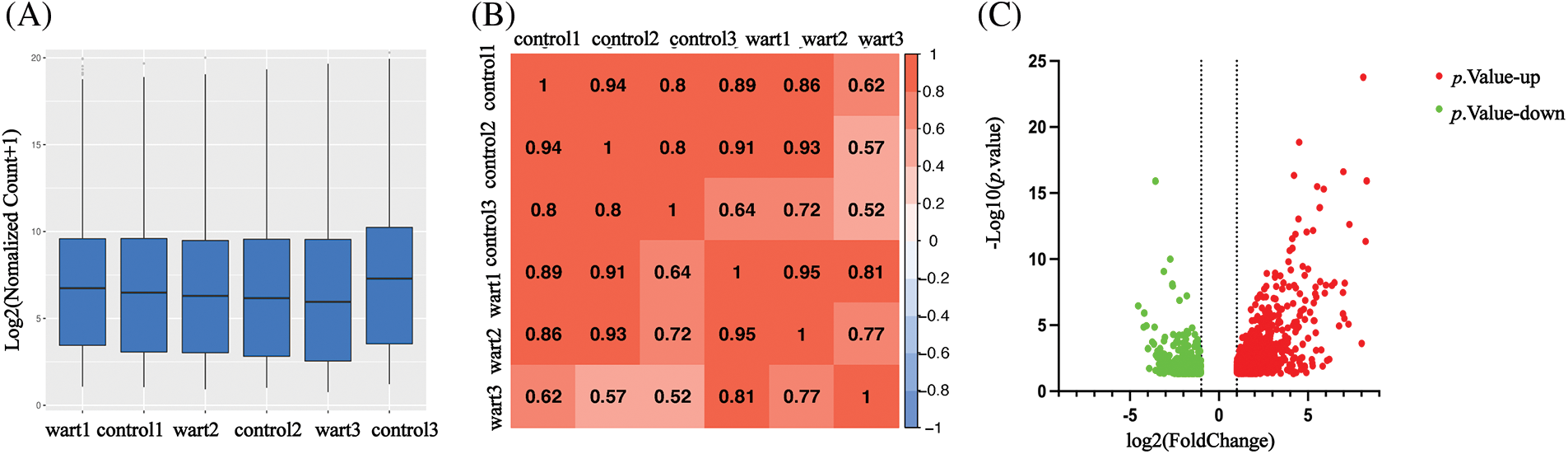
Figure 1: Gene expression abundance and sample correlation coefficient matrix heat map, and differentially expressed genes (DEGs) volcano data quality and DEG analysis: Fig. 1A gene expression abundance Boxplot; Fig. 1B sample correlation coefficient matrix heat map in warts and normal skin. The similarity in clustering patterns within each of the wart and control (normal skin) groups indicated that there are distinct expression profiles for warts. Fig. 1C DEGs between warts and normal skin (p < 0.05 and |log2 (Fold Change)| ≥ 1).
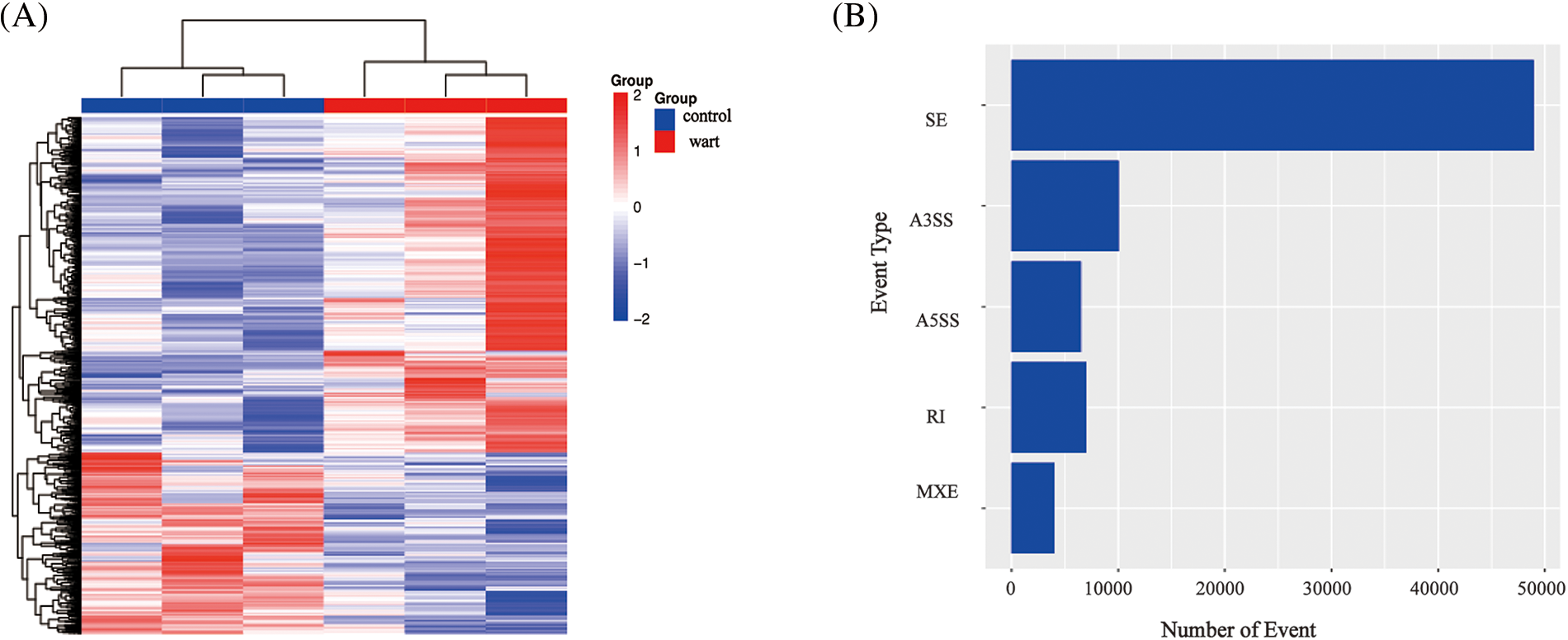
Figure 2: The cluster of differential mRNAs and alternative splicing analysis.
Fig. 2A The cluster of differential mRNAs: clustering by −logl0 (p-value), red: upregulated expression mRNA, bule: downregulated expression mRNA). Alternative splicing analysis including SE (skipped exon), RI (retained intron), A5SS (alternative 5′ splice site), A3SS (alternative 3′ splice site), MXE (mutually exclusive exons), SE was the most alternative splicing in our data (p-value < 0.05) (Fig. 2B).
Pathway and functional enrichment analysis
A total of 595 GO terms were obtained (adjusted p < 0.01). The top 60 enriched GO terms were involved in the cornification process and keratinocyte differentiation development, as shown in Fig. 3A. The most abundant functional group among the top GO terms was the regulation of the differentiation signal pathway. Pathway analysis can be used to explore the biological function of genes; the p53 signal and cell cycle signal pathways were the most prominently enriched, and the IL-17 signal pathway was also enriched (shown in Fig. 3B). These pathways are associated with the cell cycle, which could explain the hyperkeratosis in low-risk HPV infection.
Figure 3: Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis.
GO enrichment analysis (Fig. 3A): The ordinate represents the GO term, and the upper abscissa represents the number of genes compared with the GO term. The lower abscissa represents the significance level of enrichment, corresponding to the height of the column (adjusted p-value < 0.01). The top GO included the cornification process and keratinocyte differentiation development. KEGG pathway analysis (Fig. 3B): The abscissa represents the KEGG pathway, and the red column represents the number of genes compared with the GO term. The ordinate represents the significance level of enrichment, corresponding to the height of the column (adjusted p-value < 0.05). The top KEGG pathway included the p53 signal pathway, cell cycle.
Disease annotation function and Reactome metabolic pathway analysis
Disease annotation function analysis was conducted on the DisGeNET disease database for functional annotation and classification of disease types. The DEGs were enriched in palmoplantar keratosis, psoriasis, and ichthyoses with dermatologic disorders (shown in Fig. 4A). Further research is needed to determine whether HPV infection development is related to these skin diseases. Reactome metabolic pathway analysis revealed enrichment in the inhibition of signaling by overexpressed epidermal growth factor receptor (EGFR), antimicrobial protein-mediated metal sequestration, post-translational modification, and OAS antiviral response, as shown in Fig. 4B. These results suggested the complexity of HPV infection.
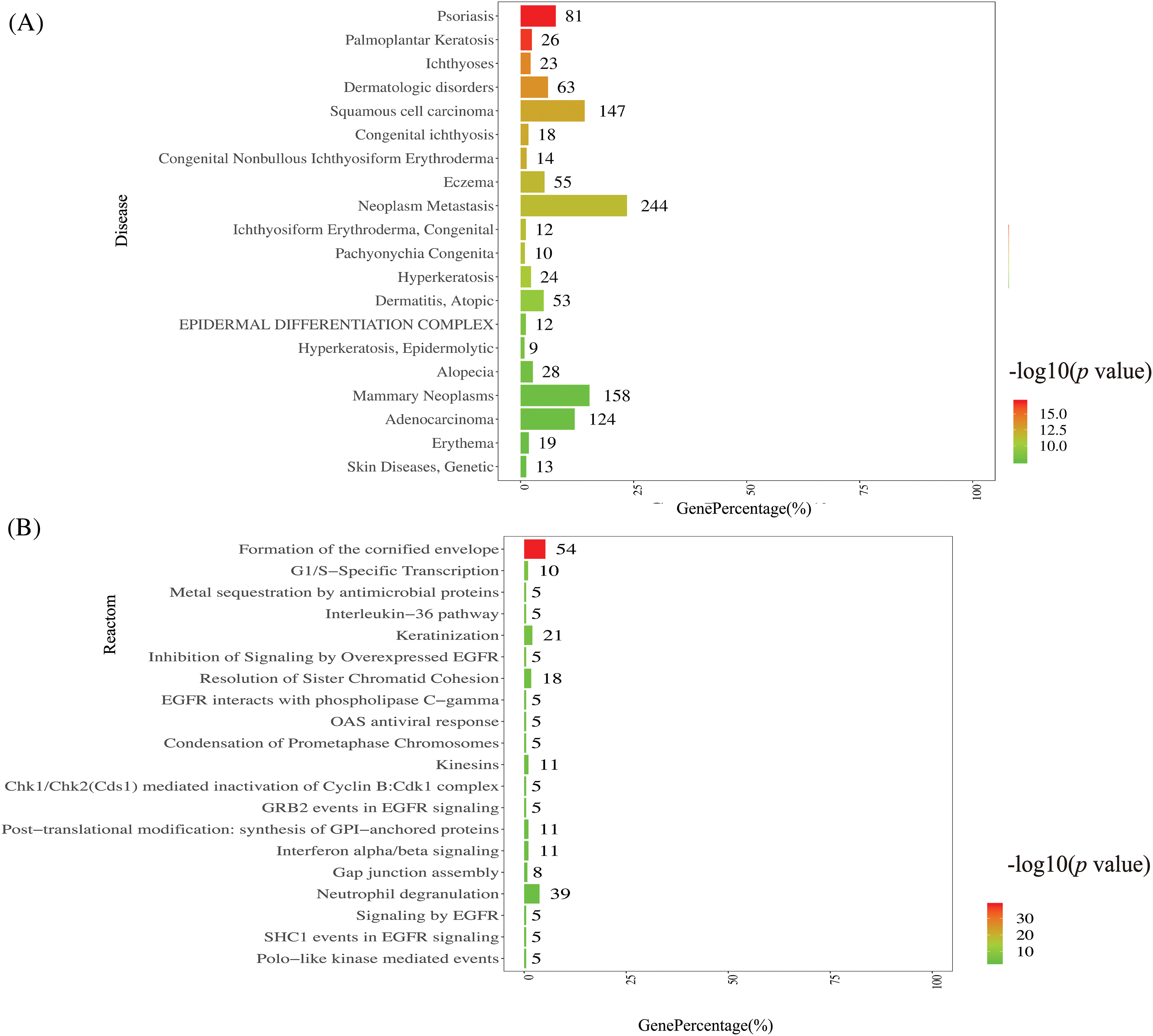
Figure 4: Disease annotation function analysis and Reactome metabolic pathway analysis.
Disease annotation function analysis (Fig. 4A): the ordinate represents the top 20 diseases, and the lower abscissa represents the percentage of genes compared with the disease data; the upper abscissa represents the significance level of enrichment, corresponding to the width of the column. DEGs were enriched in palmoplantar keratosis and psoriasis. Reactome metabolic pathway analysis is to annotate and classify the metabolic pathways by Reactome database (Fig. 4B): the ordinate represents the top 20 Reactomes, and the lower abscissa represents the percentage of genes compared with the disease data; the upper abscissa represents the significance level of enrichment, corresponding to the width of the column. The top 1 Reactome pathway is the formation of the cornified envelope.
String PPI network and significant module analysis
The String PPI network database was used to construct the protein interaction network of DEGs. Protein interaction plays an essential role in the human body. In total, 1,643 differentially expressed genes were mapped to proteins in the STRING database, and the top 50 DEGs were further screened. Finally, the protein interaction network was constructed (shown in Fig. 5). The edges with the highest density mainly included OAS family-related proteins, kinase family, and MAPK signal pathway proteins. Significant modules analysis with M-code found three significant modules, including module 1 (score of 53.633, 61 nodes, 3,218 edges), module 2 (score of 23, 23 nodes, 506 edges), and module 3 (score of 16.625, 17 nodes, 266 edges), as shown in Fig. 6A.
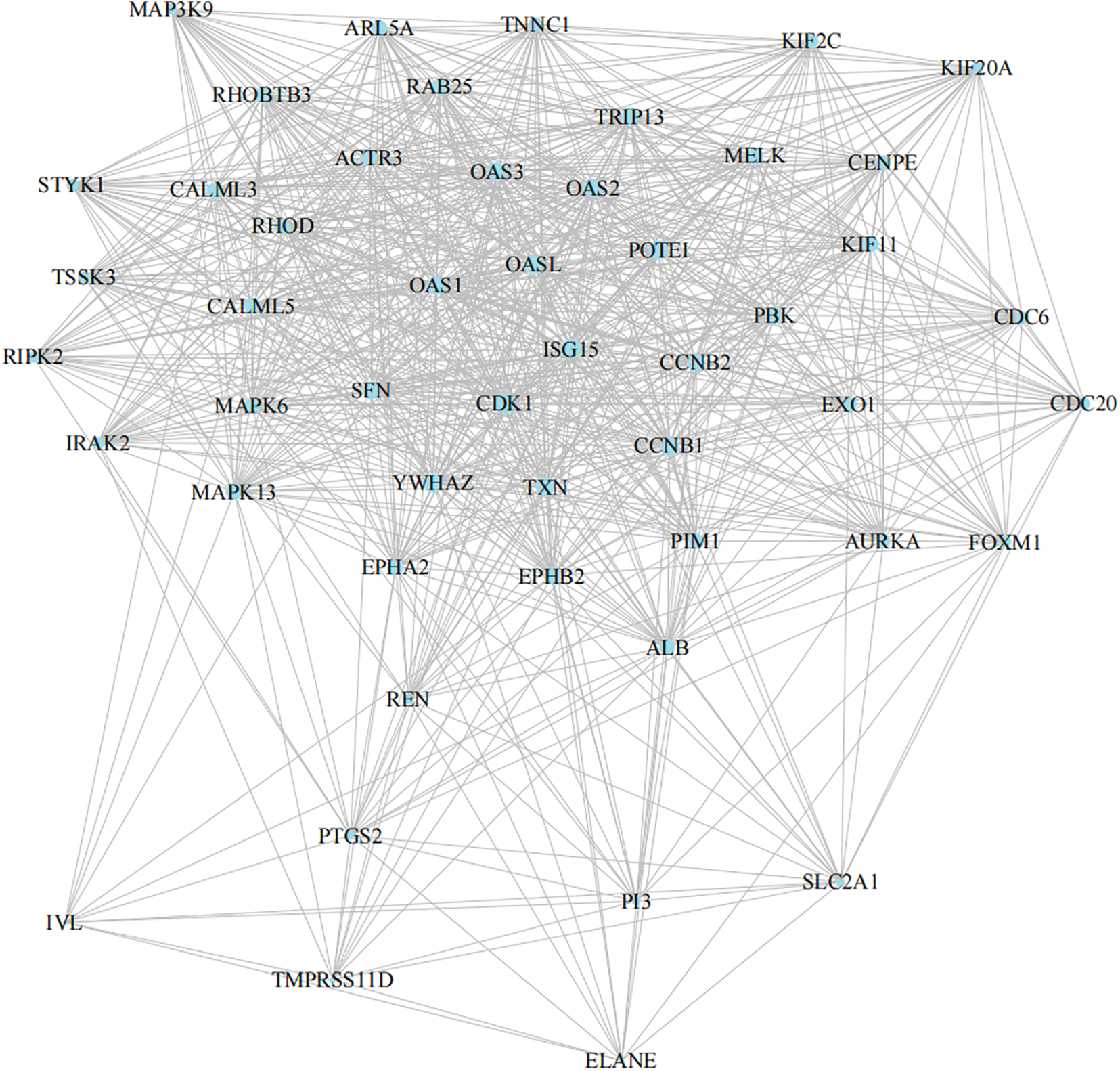
Figure 5: SignificantString protein interaction network.
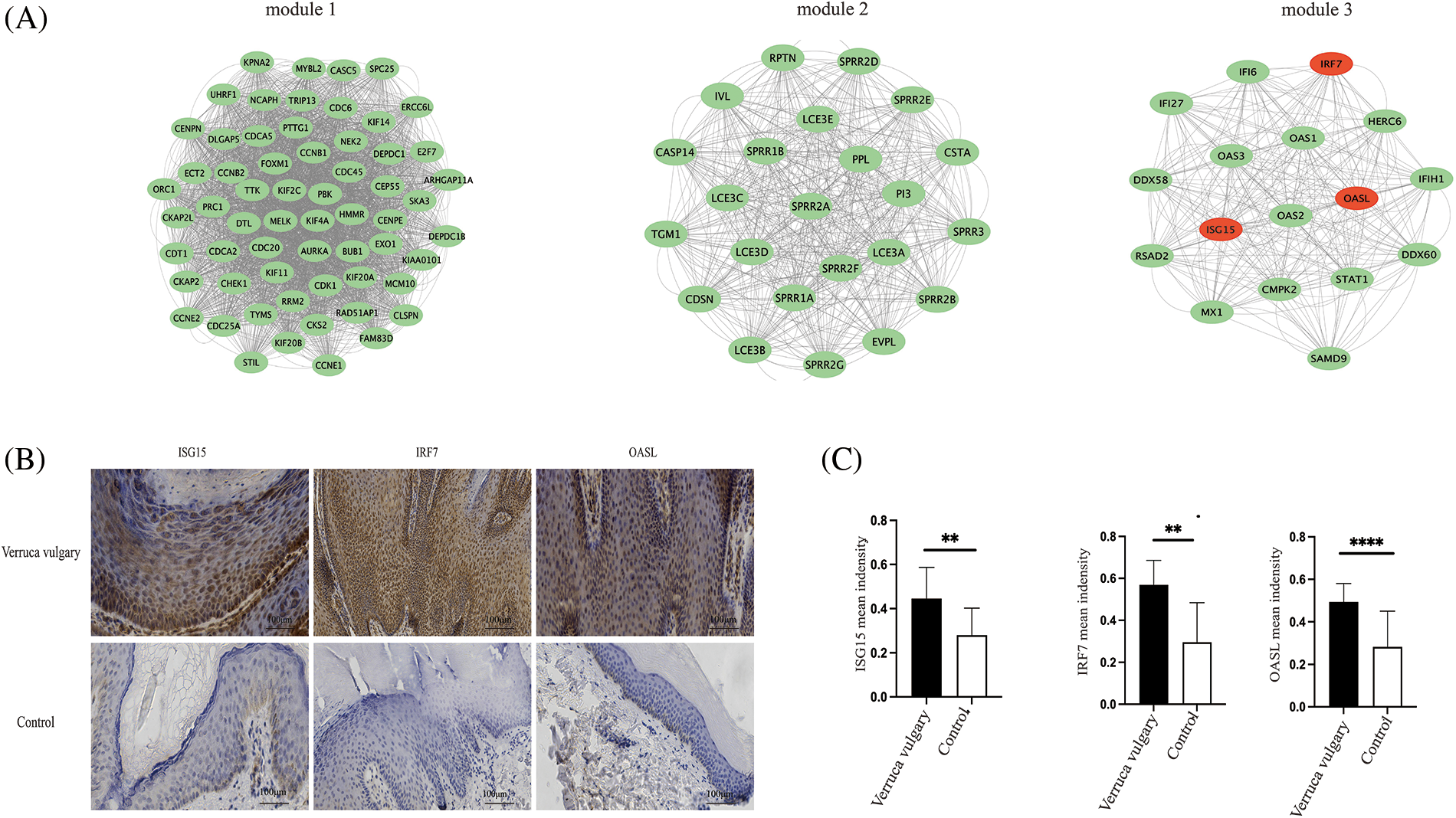
Figure 6: Significant modules analysis and verification of differentially expressed genes. Significant modules analysis with M-code found three significant modules, including module 1 (score of 53.633, 61 nodes, 3218 edges), module 2 (score of 23, 23 nodes, 506 edges), module 3 (score of 16.625, 17 nodes, 266 edges) (A). Expression of differentially expressed genes in verruca vulgaris compared with the adjacent normal skin. Immunohistochemistry staining of ISG15, IRF7, and OASL in verruca vulgaris compared with control (the adjacent normal skin). (n = 9, Magnification = 200X; scale bar represents 100 μm, Quantitation of Immunohistochemistry used Image-J, (**p < 0.05; ****p < 0.0001) (B and C).
Top 50 DEGs strings network. The densest edges mainly include OAS family-related proteins, kinase families, and MAPK signal pathway proteins.
Immunohistochemistry verification
Immunohistochemistry staining was used to explore the expression of DEGs (ISG15, IRF7, OASL) in verruca vulgaris compared with the adjacent normal skin. ISG15, IRF7, and OASL were all included in the significant module 3 of the M-code analysis. The expression of DEGs (ISG15, IRF7, OASL) was higher than in normal skin, which is in accordance with transcriptome results (n = 9) (shown in Figs. 6B and 6C).
Significant modules analysis with M-code found three significant modules (Fig. 6A), including module 1 (score of 53.633, 61 nodes, 3,218 edges), module 2 (score of 23, 23 nodes, 506 edges), module 3 (score of 16.625, 17 nodes, 266 edges). Expression of differentially expressed genes in verruca vulgaris compared with the adjacent normal skin (Figs. 6B and 6C). Immunohistochemistry staining of ISG15, IRF7, and OASL in verruca vulgaris compared with control (the adjacent normal skin).
In this study, comprehensive transcript information of verruca vulgaris was obtained by mRNA transcriptome sequencing in a Chinese population. Verruca vulgaris is a common low-risk HPV infection often manifesting as a papule or nodule with hyperkeratosis. The existing treatments for verruca vulgaris have obvious side effects, such as pain and burning sensation (Soliman et al., 2021). Moreover, treatment and recurrence place a psychological burden on the patient (Huang et al., 2020). Therefore, clarifying the mechanism and effects of HPV infection is essential (Kost et al., 2022). RNA-seq is a powerful tool for broadly profiling gene expression by applying next-generation sequencing to quantify mRNA in a tissue sample at a given moment. This technique provides information on the therapeutic strategies and pathogenesis of verruca vulgaris in the future.
High-throughput RNA deep sequencing sheds light on the mechanism of low-risk HPV infection at the transcriptome level. In total, 1,643 DEGs were identified between verruca vulgaris and adjacent normal skin tissues. However, some DEGs may be influenced by the limited sample size, and many of these DEGs were not reported to be involved in warts (Al-Eitan et al., 2020). This is also because less research has been carried out on low-risk HPV infection compared to high-risk HPV infection. Alternative splicing is widespread in eukaryotic organisms. Some studies pointed out that alternative splicing leads to structural and functional polymorphism of transcripts and proteins, which is an important transcriptional regulation mechanism in disease development (Marasco and Kornblihtt, 2023). Alternative splicing is a dynamic process that varies across different tissues or at different stages of development. Specific shear isomers are produced under specific conditions, which indicates that different isomers have various functions, thus associating variable shear with normal life activities and diseases. Many studies have found that changes in alternative splicing are related to cancer and other diseases, highlighting the importance of studying alternative splicing in different tissues and disease stages. Our study revealed alternative splicing in common warts, including SE, RI, A5SS, A3SS, MXE, and SE. Rosenberger et al. (2010) showed that the epidermal growth factor pathway regulates alternative splicing of HPV16 E6/E7 during the viral life cycle and transformation. Alternative splicing plays an important role in tumor progression, including HPV-positive cancer (Guo et al., 2017), and occurs during viral infection. The relationship between host changes and viral infection remains unelucidated, and the functions of these deformable shears require further study.
DEGs Reactome analysis novelty found the enrichment of Reversible hydration of carbon dioxide and BMP signaling in our results. Reversible hydration of carbon dioxide enrichment indicates a certain relationship between HPV infection and host metabolism. Reversible hydration of carbon dioxide influences oxidative and inflammatory processes and can also partly explain why inflammatory diseases are prone to the formation of skin warts (Radi, 2022). BMP can reduce the order of macrophage membranes, which contributes to the inhibition of TLR4 production and as a transcription factor to regulate cancer development (Cieielska et al., 2016; Lefort and Maguer, 2020). In the past, DEG Reactome analysis has rarely been performed to investigate low-risk HPV infection (Al-Eitan et al., 2020). Our study provides a new perspective on the study of low-risk HPV infection. A total of 595 GO terms were obtained, with the top terms including the cornification process and keratinocyte differentiation development, consistent with previous studies (Al-Eitan et al., 2020). In addition, the most abundant functional group in the top GO terms was the regulation of the differentiation signal pathway. Pathway analysis was used to explore the biological function of genes, revealing that the p53 signal and cell cycle signal pathway were the most enriched; the IL-17 signal pathway was also enriched. These pathways are involved in the cell cycle. Interestingly, p53 acts as a tumor suppressor and is a typical pathway in cancer development (Saha et al., 2012; Shu et al., 2022). It provides insight into exploring the mechanism of low-risk HPV infection.
Disease annotation function analysis revealed that DEGs were enriched in palmoplantar keratosis, psoriasis, ichthyoses, and dermatologic disorders. However, the relationship between HPV infection and these skin diseases remains obscure. Prior cohort studies concluded that HPV infection increased the risk of new-onset psoriasis (Tu et al., 2021; Korecka et al., 2022). Bergot confirmed that HPV 16 E7 drove the activation of TSLP and type 2 ILC infiltration to the onset of atopic dermatitis-like pathology. Chinese scholars reported a case of a 3-year-old boy with atopic dermatitis and verruca vulgaris treated with dupliumab, with an unexpected spontaneous resolution of verruca vulgaris (Shen et al., 2022). This shows a complex relationship network between inflammatory dermatosis and HPV infection. We speculate that an interestingly complex relationship between inflammatory skin diseases and HPV infection, as their etiology is related to immunity and the skin barrier. The regulation network among them needs further research. Reactome metabolic pathway analysis showed the enriched formation of the cornified envelope, inhibition of signaling by overexpressed EGFR, metal sequestration by antimicrobial proteins, post-translational modification, and OAS antiviral response. These findings indicate that HPV infection is a complex metabolic process.
The STRING PPI network database was used to build a PPI network of DEGs. The densest edges mainly included OAS family-related proteins, kinase family, and MAPK signal pathway proteins. These signal proteins play an essential role in antivirus immunity and cell cycle. Furthermore, this study reports for the first time the activation of the interferon pathway in verruca vulgaris. This contradicts the traditional view that HPV infection can only be caused by reduced immunity (Gusho and Laimins, 2021). The activation of this pathway might indicate that the human body is developing immunity to clear the virus. Further research is still needed as transcriptome sequencing only represents changes at a certain time, while HPV infection is a dynamic process that involves adsorption, invasion, replication, and transmission (Mcbride, 2022). In addition, this study investigated the expression of differentially expressed genes (ISG15, IRF7, OASL) in verruca vulgaris compared with the adjacent normal skin with immunohistochemistry staining. ISG15, IRF7, and OASL are all ISGs and play an important role in antiviral immunity (Morales and Lenschow, 2013; Tao et al., 2022). Nevertheless, few studies have investigated the expressions of these proteins in verruca vulgaris. Only one study reported a decreased expression of IRF7 in verruca vulgaris compared to normal skin (Tang et al., 2020), which is in contrast with our results. A larger sample size would be required to verify whether differences and the disease course were associated with the expression of IRF7. In addition, ISG15, IRF7, and OASL were overexpressed in low-risk HPV infection compared to normal skin (Pierangeli et al., 2011; Cannella et al., 2014; Xie et al., 2019; Zhong et al., 2022), which implies that high expression of ISGs may be universal in low-risk HPV infection and also help elucidate the role of ISGs in the clearance of low-risk HPV infection.
This study was limited by the small number of sequenced samples. Our next study will expand the sample size and integrate the data of verruca vulgaris from public databases. In addition, the role and underlying mechanism of ISG upregulation in low-risk HPV infection will be investigated.
In summary, our data provides genetic information regarding verruca vulgaris in a Chinese population. Novel DEGs were found, and alternative splicing in verruca vulgaris was reported. GO enrichment analysis included the cornification process, and keratinocyte differentiation development. KEGG pathway analysis included the p53 signal pathway and cell cycle. These results may imply that low-risk HPV infection results in the excessive proliferation of keratinocytes and hyperkeratosis. The densest edges of the PPI network mainly included OAS family-related proteins. Furthermore, ISGs (ISG15, IRF7, and OASL) were found in significant module 3 of the M-Code analysis. ISGs were overexpressed in verruca vulgaris compared to normal skin and may play a key role in the outcome of low-risk HPV infection. These data provide insights into HPV infection, which may be helpful for the treatment of low-risk HPV infection in the future.
Acknowledgement: We would like to thank the beneficial and valuable comments on our article received from our editors and reviewers.
Funding Statement: The National Natural Science Foundation of China (Grant No. 81903227) supported our study.
Author Contributions: Conceptualization and Review: Fang Xie, Chengxin Li, Qingqing Guo; Funding Acquisition: FangXie; Investigation: Qingqing Guo; Methodology: Qingqing Guo, Jiayue Qi, Xiaoqing Li; Resources: Fang Xie, Chengxin Li; Validation: all authors (equal); Software: Qingqing Guo, Jiayue Qi; Supervision: Fang Xie, Chengxin Li; Draft Preparation: Qingqing Guo.
Availability of Data and Materials: All authors confirm that the data supporting the findings of our study are available in the article.
Ethics Approval: This study was reviewed and approved by the Ethics Committee of Chinese PLA General Hospital (S2022-724-01). Informed consent was obtained from all subjects for the publication of this study.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
Al-Eitan LN, Tarkhan AH, Alghamdi MA, Al-Qarqaz FA, Al-Kofahi HS (2020). Transcriptome analysis of HPV-induced warts and healthy skin in humans. BMC Medical Genomics 13: 35. https://doi.org/10.1186/s12920-020-0700-7 [Google Scholar] [PubMed] [CrossRef]
Bernard HU, Burk RD, Chen Z, van Doorslaer K, Hausen H, de Villiers EM (2010). Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 401: 70–79. https://doi.org/10.1016/j.virol.2010.02.002 [Google Scholar] [PubMed] [CrossRef]
Cannella F, Scagnolari C, Selvaggi C, Stentella P, Recine N, Antonelli G, Pierangeli A (2014). Interferon lambda 1 expression in cervical cells differs between low-risk and high-risk human papillomavirus-positive women. Medical Microbiology & Immunology 203: 177–184. https://doi.org/10.1007/s00430-014-0330-9 [Google Scholar] [PubMed] [CrossRef]
Cieielska A, Sas-Nowosielska H, Kwiatkowska K (2016). Bis(monoacylglycero)phosphate inhibits TLR4-dependent RANTES production in macrophages. International Journal of Biochemistry & Cell Biology 83: 15–26. https://doi.org/10.1016/j.biocel.2016.12.003 [Google Scholar] [PubMed] [CrossRef]
Doorbar J, Egawa N, Griffin H, Kranjec C, Murakami I (2015). Human papillomavirus molecular biology and disease association. Reviews in Medical Virology 25: 2–23. https://doi.org/10.1002/rmv.1822 [Google Scholar] [PubMed] [CrossRef]
Guo T, Sakai A, Afsari B, Considine M, Danilova L et al. (2017). AKT3A novel functional splice variant of defined by analysis of alternative splice expression in HPV-positive oropharyngeal cancers. Cancer Research 77: 5248–5258. https://doi.org/10.1158/0008-5472.CAN-16-3106 [Google Scholar] [PubMed] [CrossRef]
Gusho E, Laimins L (2021). Human papillomaviruses target the DNA damage repair and innate immune response pathways to allow for persistent infection. Viruses 13: 1390. https://doi.org/10.3390/v13071390 [Google Scholar] [PubMed] [CrossRef]
Huang K, Li M, Xiao Y, Wu L, Chen X (2020). The application of medical scale in the treatment of plantar warts: Analysis and prospect. Journal of Dermatological Treatment 33: 637–642. https://doi.org/10.1080/09546634.2020.1781757 [Google Scholar] [PubMed] [CrossRef]
Korecka K, Anna WS, Mikiel D (2022). The impact of systemic psoriasis treatments on human papillomavirus activation and propagation. The Australasian Journal of Dermatology 63: 293–302. https://doi.org/10.1111/ajd.13865 [Google Scholar] [PubMed] [CrossRef]
Kost Y, Deutsch A, Zhu TH, Hulur I, Blasiak RC (2022). Vaccination against human papillomavirus is not associated with resolution of verruca vulgaris in immunocompetent 9- to 21-year-olds. Journal of the American Academy of Dermatology 87: 250–252. https://doi.org/10.1016/j.jaad.2021.08.016 [Google Scholar] [PubMed] [CrossRef]
Lefort S, Maguer SV (2020). Targeting BMP signaling in the bone marrow microenvironment of myeloid leukemia. Biochemical Society Transactions 48: 411–418. https://doi.org/10.1042/BST20190223 [Google Scholar] [PubMed] [CrossRef]
Marasco L, Kornblihtt A (2023). The physiology of alternative splicing. Nature Reviews Molecular Cell Biology 24: 242–254. https://doi.org/10.1038/s41580-022-00545-z [Google Scholar] [PubMed] [CrossRef]
Mcbride AA (2022). Human papillomaviruses: Diversity, infection and host interactions. Nature Reviews Microbiology 20: 95–108. https://doi.org/10.1038/s41579-021-00617-5 [Google Scholar] [PubMed] [CrossRef]
Morales DJ, Lenschow DJ (2013). The antiviral activities of ISG15. Journal of Molecular Biology 425: 4995–5008. https://doi.org/10.1016/j.jmb.2013.09.041 [Google Scholar] [PubMed] [CrossRef]
Pierangeli A, Degener AM, Ferreri ML, Riva E, Rizzo B, Turriziani O, Luciani S, Scagnolari C, Antonelli G (2011). Interferon-induced gene expression in cervical mucosa during human papillomavirus infection. International Journal of Immunopathology and Pharmacology 24: 217–223. https://doi.org/10.1177/039463201102400126 [Google Scholar] [PubMed] [CrossRef]
Radi R (2022). Interplay of carbon dioxide and peroxide metabolism in mammalian cells. The Journal of Biological Chemistry 298: 102358. https://doi.org/10.1016/j.jbc.2022.102358 [Google Scholar] [PubMed] [CrossRef]
Ringin SA (2019). The effectiveness of cutaneous wart resolution with current treatment modalities. Journal of Cutaneous and Aesthetic Surgery 13: 24–30. https://doi.org/10.4103/JCAS.JCAS_62_19 [Google Scholar] [PubMed] [CrossRef]
Rosenberger S, De-Castro Arce J, Langbein L, Steenbergen RD, Rösl F (2010). Alternative splicing of. human papillomavirus type-16 E6/E6 early mRNA is coupled to EGF signaling via Erk1/2 activation. Proceedings of the National Academy of Sciences of the United States of America 107: 7006–7011. https://doi.org/10.1073/pnas.1002620107 [Google Scholar] [PubMed] [CrossRef]
Saha B, Adhikary A, Ray P, Saha S, Chakraborty S, Mohanty S, Das K, Mukherjee S, Mazumdar M, Lahiri L (2012). Restoration of tumor suppressor p53 by differentially regulating pro- and anti-p53 networks in HPV-18-infected cervical cancer cells. Oncogene 31: 173–186. https://doi.org/10.1038/onc.2011.234 [Google Scholar] [PubMed] [CrossRef]
Shen CP, Zhao MT, Ma L (2022). Spontaneous resolution of Verruca Vulgaris in a paediatric atopic dermatitis patient treated with dupilumab. Journal of the European Academy of Dermatology and Venereology 36: 396–398. https://doi.org/10.1111/jdv.17936 [Google Scholar] [PubMed] [CrossRef]
Shu S, Li Z, Liu L, Ying X, Zhang Y, Wang T, Zhou X, Jiang P, Lv W (2022). HPV16 E6-activated OCT4 promotes cervical cancer progression by suppressing p53 expression via co-repressor NCOR1. Frontiers in Oncology 12: 900856. https://doi.org/10.3389/fonc.2022.900856 [Google Scholar] [PubMed] [CrossRef]
Soliman M, Soliman MM, Ibrahim SMA, Mohamed SKA (2021). Pulsed dye laser versus Nd:YAG laser in the treatment of recalcitrant plantar warts: An intraindividual comparative study. Journal of Cosmetic and Laser Therapy 23: 130–136. https://doi.org/10.1080/14764172.2021.2016843 [Google Scholar] [PubMed] [CrossRef]
Tang Y, Zhu X, Han R, Zhou Q, Cheng H (2020). Expression of langerhans cell and plasmacytoid dendritic cell markers, and toll-like receptor 7/9 signaling pathway proteins in verruca vulgaris lesions. Medicine 99: e19214. https://doi.org/10.1097/MD.0000000000019214 [Google Scholar] [PubMed] [CrossRef]
Tao P, Sun L, Sun Y, Wang Y, Yang Y, Yang B, Li F (2022). ISG15 is associated with cervical cancer development. Oncology Letters 24: 380. https://doi.org/10.3892/ol.2022.13500 [Google Scholar] [PubMed] [CrossRef]
Tu TY, Chang R, Lai JN, Tseng CC, Wei CC (2021). Human papillomavirus symptomatic infection associated with increased risk of new-onset alopecia areata: A nationwide population-based cohort study. Journal of Autoimmunity 119: 102618. https://doi.org/10.1016/j.jaut.2021.102618 [Google Scholar] [PubMed] [CrossRef]
Xie F, Yu HS, Wang R, Wang D, Li YM et al. (2019). Photodynamic therapy for genital warts causes activation of local immunity. Journal of Cutaneous Medicine and Surgery 23: 370–379. https://doi.org/10.1177/1203475419838548 [Google Scholar] [PubMed] [CrossRef]
Zhong Y, Wei J, Song W, Wang Q, Zhang Z, Liu H, Chen X, Huang X, Zeng K (2022). Identification of novel biomarkers and key pathways of condyloma acuminata. Genomics 114: 110303. https://doi.org/10.1016/j.ygeno.2022.110303 [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools