
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW
Review on microbial metabolomics of probiotics and pathogens: Methodologies and applications
School of Pharmacy, Heilongjiang University of Chinese Medicine, Harbin, 150040, China
* Corresponding Author: XIN MENG. Email:
(This article belongs to the Special Issue: Bioinformatics Study of Diseases)
BIOCELL 2023, 47(1), 91-107. https://doi.org/10.32604/biocell.2023.024310
Received 29 May 2022; Accepted 07 July 2022; Issue published 26 September 2022
 View Full Text
View Full Text Download PDF
Download PDFAbstract
In recent years, microbial metabolomics, a new field that has attracted wide attention, provides a map of metabolic pathways and clarifies the interaction mechanism between microorganisms and hosts. Many microorganisms are found in the human intestine, oral cavity, vagina, etc. Probiotics could maintain the good health of the host, while pathogens and an imbalance of bacterial flora lead to a series of diseases of the body and mind. Metabolomics is a science for qualitative and quantitative analysis of all metabolites in an organism or biological system, which could provide key information to understand the related metabolic pathways and associated changes. This approach analyzes the final products of cellular regulatory processes, the level of which can be regarded as the ultimate response of the biological system to genetic or environmental changes. Microbial metabolomics has been widely used in different research fields, such as microbial phenotypic classification, mutant screening, metabolic pathways, microbial metabolic engineering, fermentation engineering monitoring and optimization, microbial environmental pollution, and so on. However, there are only a few reviews on microbial metabolomics of probiotics and pathogens. This review summarizes the main methodologies, including sample preparation, identification of metabolites, data processing, and analysis. Recent applications in microbial metabolomics of probiotics and pathogens are also described. This paper first summarized the research progress and application of microbial metabolomics from two aspects: probiotics and pathogenic bacteria. Probiotics and pathogenic bacteria do not exist independently most of the time; hence, these were reviewed in the research field of coexistence of probiotics and pathogenic bacteria, which was subdivided into important microbial research fields closely related to human health, including the human gut, oral cavity, food, and nutrition-related microorganisms. Then, the main problems and trends associated with microbial metabolomics are discussed.Keywords
Microbial metabolomics, as a new field, could provide a map of metabolic pathways and clarify the mechanism of interaction between microorganisms and hosts (Fobofou and Savidge, 2022; Joshua, 2019). There are many microorganisms in the human intestine, oral cavity, and so on (Moreno et al., 2022; Vlachojannis et al., 2018) (Fig. 1). Whether in quantity or gene, the microbes parasitizing on the surface and inside of the human body are far more than the human body cells (Ke et al., 2018b). Of the trillions of microorganisms, more than 1,000 species of bacteria live in the intestine and possess more than 3 million genes (Sebastian Domingo and Sanchez Sanchez, 2018; Zhu et al., 2015). The intestine carries up to 2 kg of microorganisms (Macfarlane and Macfarlane, 1997; Mondot and Lepage, 2016). Two-thirds of an individual’s intestinal flora is unique to oneself; even in identical twins, the composition of bacterial communities is not exactly the same. Probiotics could maintain the health of the host, while pathogens and imbalance of bacterial flora lead to a series of diseases of the body and mind (Lukic et al., 2017; Meng et al., 2018; Gupta et al., 2021; Manole et al., 2021). Bacteria existing in the human body possess a wide range of functions, such as affecting fat storage, angiogenesis, and immune system, regulating the nervous system and bone mineral density, protecting epithelial cells from injury and pathological tolerance, degrading food components, biosynthesis of vitamin and amino acids, and metabolism of drugs and so on (Bliss and Whiteside, 2018; Boursi et al., 2018) (Fig. 2).
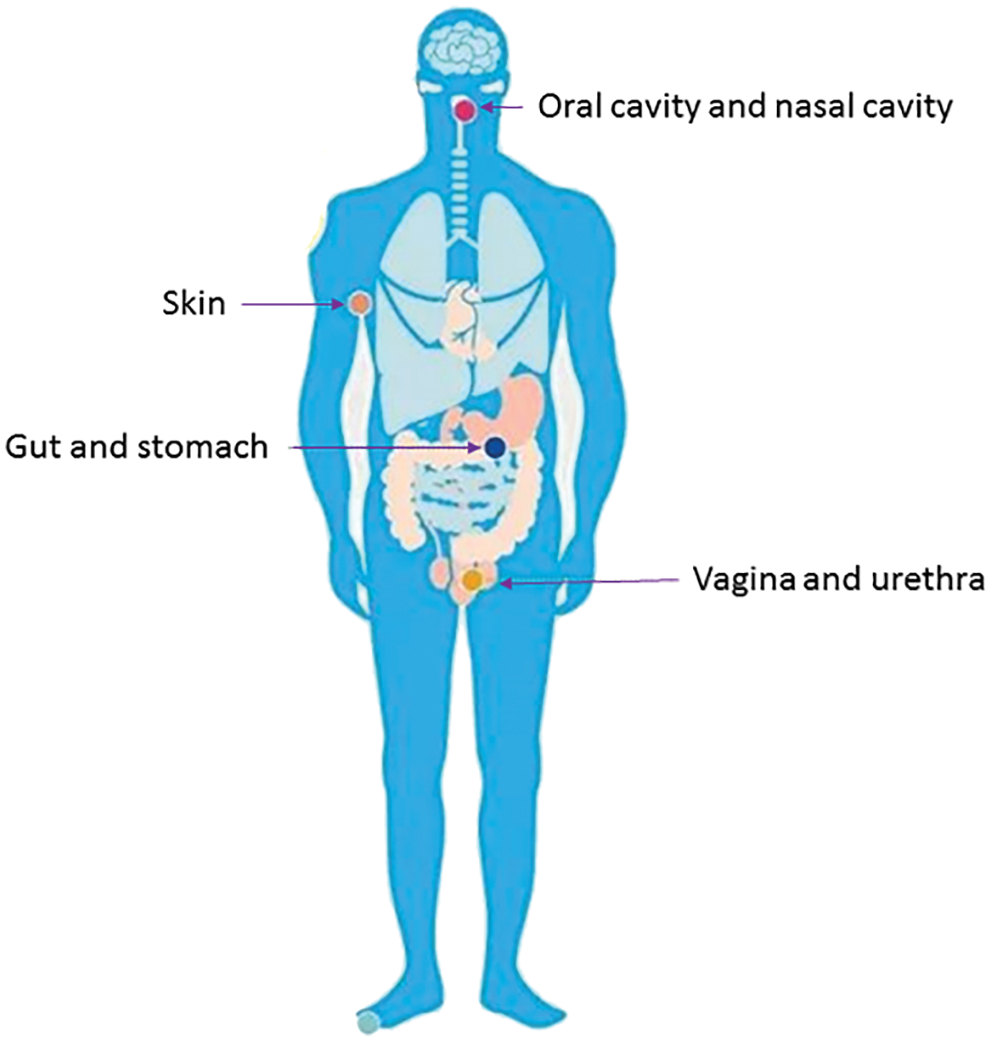
Figure 1: Distribution of microorganisms in the human body. A large number of microorganisms are distributed in the human skin, oral cavity, gastrointestinal tract, respiratory tract, genitourinary tract, etc.
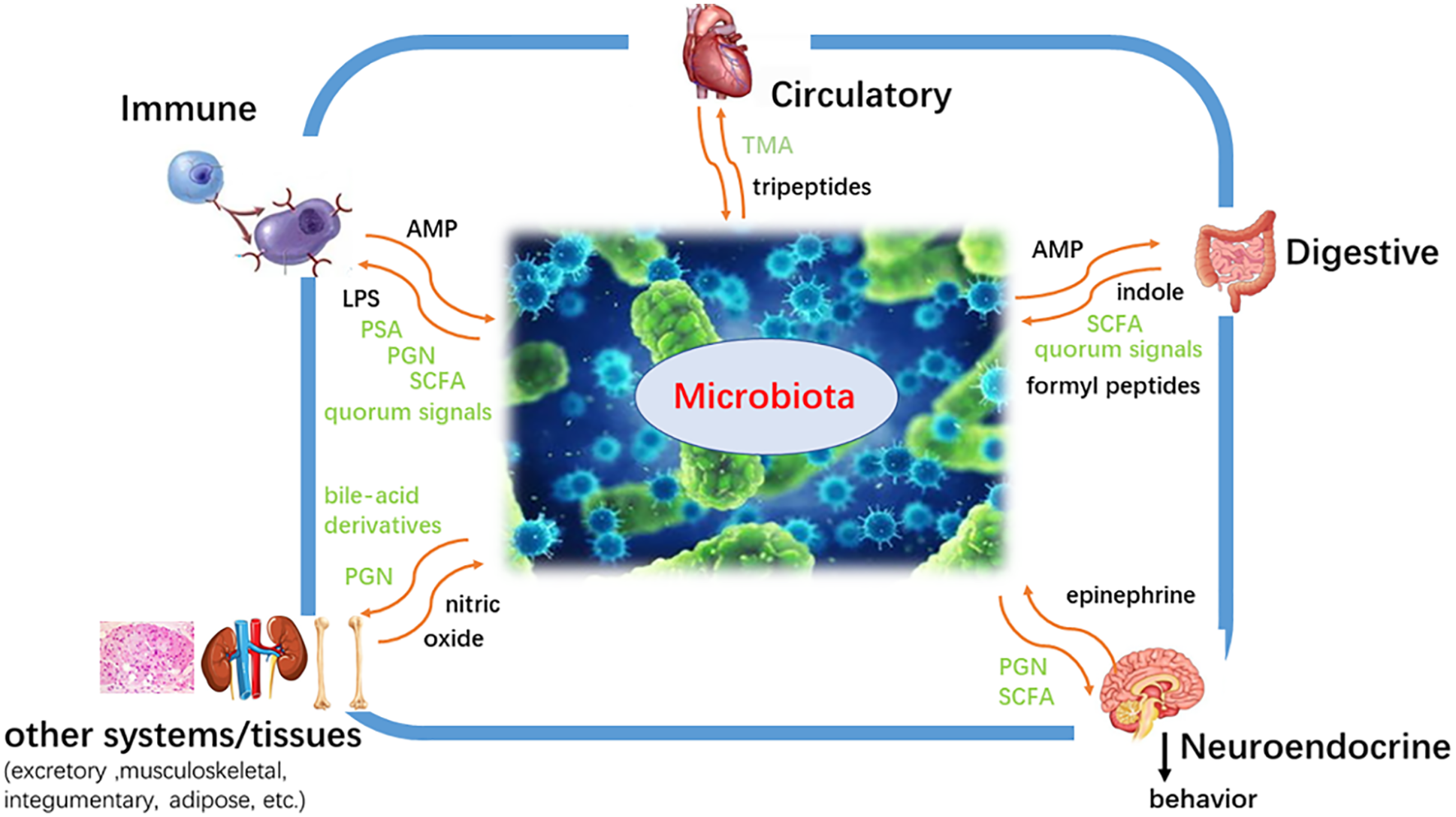
Figure 2: The effect of microorganisms on the human body. Microorganisms exert a wide range of functions, including affecting fat storage, angiogenesis, and immune system, regulating the nervous system and bone mineral density, protecting epithelial cells from injury and pathological tolerance, degrading food components, biosynthesis of vitamin and amino acid, metabolic drugs and so on.
Metabolomics is a science for qualitative and quantitative analysis of all metabolites in an organism or biological system, which could provide key information to people to understand the related metabolic pathways and their changes. This approach analyzes the final products of cellular regulatory processes, the level of which can be regarded as the ultimate response of the biological system to genetic or environmental changes. Due to the importance of microorganisms in biological systems, metabolomics technology has attracted wide attention in microbial research (Baidoo and Teixeira Benites, 2019; Joshua, 2019; Mousavi et al., 2019; Murovec et al., 2018). In 1992, Elmroth et al. (1992) assessed microbial metabolomics for the first time (Marciniec et al., 1992). Fatty acids, amino acids, and carbohydrates were detected by gas chromatography-mass spectrometry to evaluate bacterial contamination of Leuconostoc mesenteroides during their culture.
Initially, microbial metabolomics was mainly used for the biological identification of different strains (Boersma et al., 2001; Koek et al., 2006; Mashego et al., 2007; van der Werf et al., 2005). Over recent years, microbial metabolomics has been widely used in different research fields, such as microbial phenotypic classification, mutant screening, engineering of metabolic pathways and microbial metabolism, monitoring and optimization of fermentation engineering, monitoring microbial environmental pollution, and so on (Covington et al., 2017; Jacobs et al., 2016; Joshua, 2019; Sumner et al., 2015). The precise, sensitive and high-throughput research methods of microbial metabolomics are the basis of microbial metabolomics research (Mousavi et al., 2019). However, the methodology and applications of microbial metabolomics have been rarely reviewed. This review summarizes the main methodologies and applications of probiotics and pathogens in microbial metabolomics, and discusses the main problems and trends of microbial metabolomics.
Methodology of Microbial Metabolomics
In the studies of microbial metabolomics, the main research methods are roughly the same, as described below (Fig. 3).
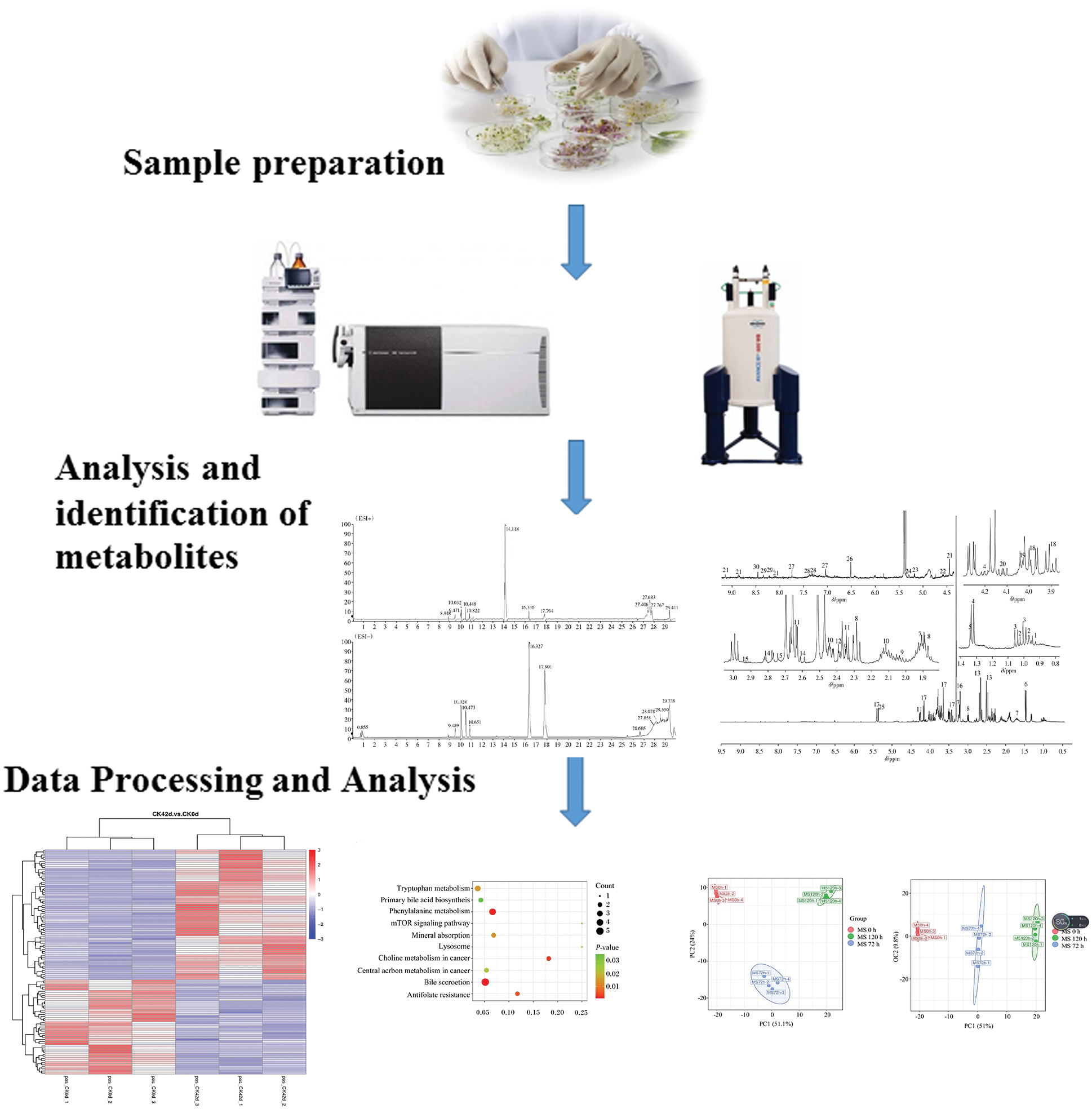
Figure 3: A broad schematic for research on microbial metabolomics. The main research workflow involves sample preparation, analysis and identification of metabolites, and data processing and analysis.
To obtain meaningful information, microbial metabolomics research needs to adopt appropriate sample preparation steps, including rapid sampling, quenching, and extraction of the metabolite. Rapid sampling prevents the dramatic change in the concentration of substrate and helps to maintain the stability of microbial metabolites. Therefore, many simple sample collection devices, such as the BioScope device and Fast Swinnex filtration (FSF) device, are often used to achieve rapid sample collection (Zhu et al., 2013).
To ensure the acquisition of the information of the sample at a specific time, it is necessary to quench the sample quickly to stop the metabolic reaction. The ideal quenching technology should quench the enzyme activity quickly and maintain the integrity of the cell or organism (Almanza-Aguilera et al., 2017; Joshua, 2019; Mayta-Apaza et al., 2018; Teoh et al., 2016). However, many quenching methods, such as organic solvent quenching, could destroy cell walls and cell membranes, leading to a significant leakage of intracellular metabolites (Mojica et al., 2008; Nandakumar et al., 2000). For example, methanol or ethanol as a quencher may cause the leakage of some metabolites (Wang et al., 2017a). At present, flow cytometry is used to evaluate the degree of cell membrane damage caused by different quenching methods. The quenching of Escherichia coli by normal saline at −80°C results only in 6% damage to the cell membrane, while conventional methanol quenching causes only 1/10 of the cell membrane damage, which reduces the leakage of metabolites (Lin et al., 2016; Wang et al., 2014).
Rapid filtration is also one of the effective methods to reduce metabolite leakage. In one study, 110 intracellular metabolites of Saccharomyces cerevisiae, four quenching methods, i.e., 60% (v/v) methanol, pure methanol (−40°C), boiling ethanol (75%, v/v), and rapid filtration, were compared (Kim et al., 2013). The results showed that rapid filtration could greatly reduce the loss of metabolites.
Metabolite extraction is an important step in microbial metabolomics. The common methods of metabolite extraction are cold methanol, hot methanol, perchloric acid or alkali, chloroform-methanol mixture, and acetonitrile (Baidoo and Teixeira Benites, 2019; Mousavi et al., 2019). A comparison of seven methods for extracting E. coli metabolites by GC-MS analysis (Chakraborty et al., 2022) showed that ammonium and ethanol were effective methods for extracting E. coli metabolites, and 289 substances were detected. However, due to the diversity of metabolites, it is usually difficult to extract all intracellular metabolites by a single extraction method. Therefore, combining different methods is conducive to improving the extraction effect of metabolites.
Analysis and identification of metabolites
Analysis and identification of metabolites is the basis of metabolomics. Mass spectrometry and nuclear magnetic resonance are two main platforms for microbial metabolomics.
Mass spectrometry
Mass spectrometry is widely used in microbial metabolomics analysis due to its high specificity and sensitivity.
Gas chromatography-mass spectrometry (GC-MS) is a mature analytical platform and the earliest analytical method used in microbial metabolomics (Elmroth et al., 1992). GC-MS can simultaneously analyze hundreds of compounds (including organic acids, amino acids, carbohydrates, glycols, aromatic amines, and fatty acids) and possess a standard metabolite spectrum database (Migne et al., 2018; Wu et al., 2018). It can analyze metabolites quickly and accurately, but the samples need to be treated by derivatization. In recent years, many studies on microbial metabolomics based on GC-MS have been conducted (Baidoo, 2019; Baidoo and Teixeira Benites, 2019; Ponnusamy et al., 2013; Yang et al., 2014). Amino acid profiles of different filamentous fungi have been obtained using chloroformate derivatization. Fatty acids in yeast samples have been analyzed by microwave derivatization. The application of GC-MS technology (GC-GC-MS) possesses significantly improved the separation efficiency and sensitivity of complex samples and has been applied to microbial metabolomics. The volatile metabolites of Pseudomonas aeruginosa were analyzed by GC-GC-time of flight (TOF)-MS, and 28 new volatile compounds were identified, including alcohols, aldehydes, ketones, functional benzenes, and aromatic molecules (Adetunji et al., 2017; Bean et al., 2012; Depke et al., 2017).
Liquid chromatography-mass spectrometry (LC-MS) is another important platform for the analysis of unstable, non-volatile, non-polar compounds without derivatization of samples (Dodda et al., 2018; Zhao and Li, 2018). Hydrophilic interaction liquid chromatography-tandem mass spectrometry (HILIC-MS) is a high throughput intracellular metabolomics analysis technology that can analyze polar and non-polar metabolites simultaneously (Teleki and Takors, 2019). Its data acquisition and analysis speed are very fast. In the metabolic extracts of Sinorhizobium meliloti, 92.2% detectable polar and lipid metabolites were obtained by HILIC-MS (Fei et al., 2014). Ion chromatography-electrospray ionization mass spectrometry, as an effective platform for quantitative analysis of microbial metabolites, could be used for simultaneous quantitative analysis of several polar metabolites, such as nucleic acid, coenzyme A ester, glyconucleotide, and diphosphate (Jerome Jeyakumar et al., 2018; Reichert et al., 2018). Although LC-MS has been often applied in many studies, the study of microbial metabolomics using this method still encounters some problems. For example, the high salt concentration in the culture medium could inhibit the ionization efficiency of ESI, block pump, and ultimately affect the validity and repeatability of quantitative analysis.
Capillary electrophoresis-mass spectrometry (CE-MS) technology has the advantages of rapid analysis, less sample requirement, and less reagent consumption (Ortiz-Villanueva et al., 2017; Sato et al., 2017). In 2003, researchers used CE-MS to analyze 1692 metabolites extracted from Bacillus subtilis, 150 of them were identified, and different CE-MS platforms were used to analyze nucleosides, acetyl coenzyme A, cationic metabolites, and anionic metabolites, which provided a map for understanding the changes of metabolites during sporogenesis of B. subtilis (Soga et al., 2003). Pressure-assisted capillary electrophoresis-mass spectrometry (PACE-MS) was used to analyze the intracellular nucleoside and coenzyme A metabolites of pykA and pykB gene-deficient strains of E. coli, and led to the conclusion that pykA gene encodes for an enzyme (Soga et al., 2007).
Nuclear magnetic resonance (NMR)
NMR can rapidly and accurately analyze samples with high throughput and non-invasiveness (Araujo et al., 2019; Swann et al., 2017). It is an important analytical technique for identifying the structure of organic compounds, which could provide complete metabolic maps of biological tissues or body fluids under certain conditions (Kiselev and Novikov, 2018; Smolyanskaya et al., 2018). The study of microbial metabolomics has many broad application prospects (Almanza-Aguilera et al., 2017; Murovec et al., 2018). Using 1H-NMR technology, the changes in metabolites during liquor fermentation under different effects of Saccharomyces cerevisiae could be monitored and the fermentation characteristics of yeast strains could be evaluated (Son et al., 2009). The intracellular metabolites of Vibrio coralliilyticus were analyzed by NMR at 27°C (highly toxic) and 24°C (non-toxic) incubation temperatures. Combined with principal component analysis (PCA) analysis, it was found that at different incubation temperatures, intracellular metabolites of V. coralliilyticus were significantly separated under PC1, PC2, and PC3 principal components, and with the increase in temperature, betaine decreased, while succinic acid and glutamic acid increased. However, due to the complex composition of microbial intracellular metabolites, including organic acids, hydrophobic substances, and complex natural products, changes in molecular concentration across several orders of magnitude (from pmol to mmol), while the sensitivity of NMR is low, it is difficult to simultaneously detect metabolites with different concentrations in biological systems (Barrilero et al., 2018). Hence, the application of NMR in microbial metabolomics is limited.
Data processing and analysis are the key processes in metabolomics research, and the main steps of microbial metabolomics data preprocessing and analysis are shown in Fig. 4. Consistent with metabolomics, data processing and analysis are briefly introduced in this review. It is necessary to pre-process the original data to eliminate the interference factors. Data processing generally includes noise elimination, baseline correction, peak alignment, binning, peak identification, normalization, and data scaling (Li et al., 2018c; Liggi et al., 2018; Montenegro-Burke et al., 2017). At present, a large number of software can pre-process the original data obtained by MS into two-dimensional data tables, such as MZmine, XCMS, and METIDEA (Myers et al., 2017). Many instrument companies have developed their proprietary software, such as Marker Lynx (Waters), Progenesis QI (Waters), Mass Profiler (Agilent), MarkerView (Applied Biosystems/MDS SCIEX), and ThermoFisher Science (Marzouki et al., 2011; Tulipani et al., 2011). Table 1 shows the main microbial metabolomics data processing and analysis software/websites. The pre-processed data need multivariate statistical analysis and bioinformatics analysis, such as PCA, and partial least squares discriminant analysis, to obtain effective information, identify biomarkers, metabolic pathways, etc. (Li et al., 2018b). Metabolic pathway analysis is helpful to understand the interaction between metabolites and exploring gene expression data to complete functional genomics research.
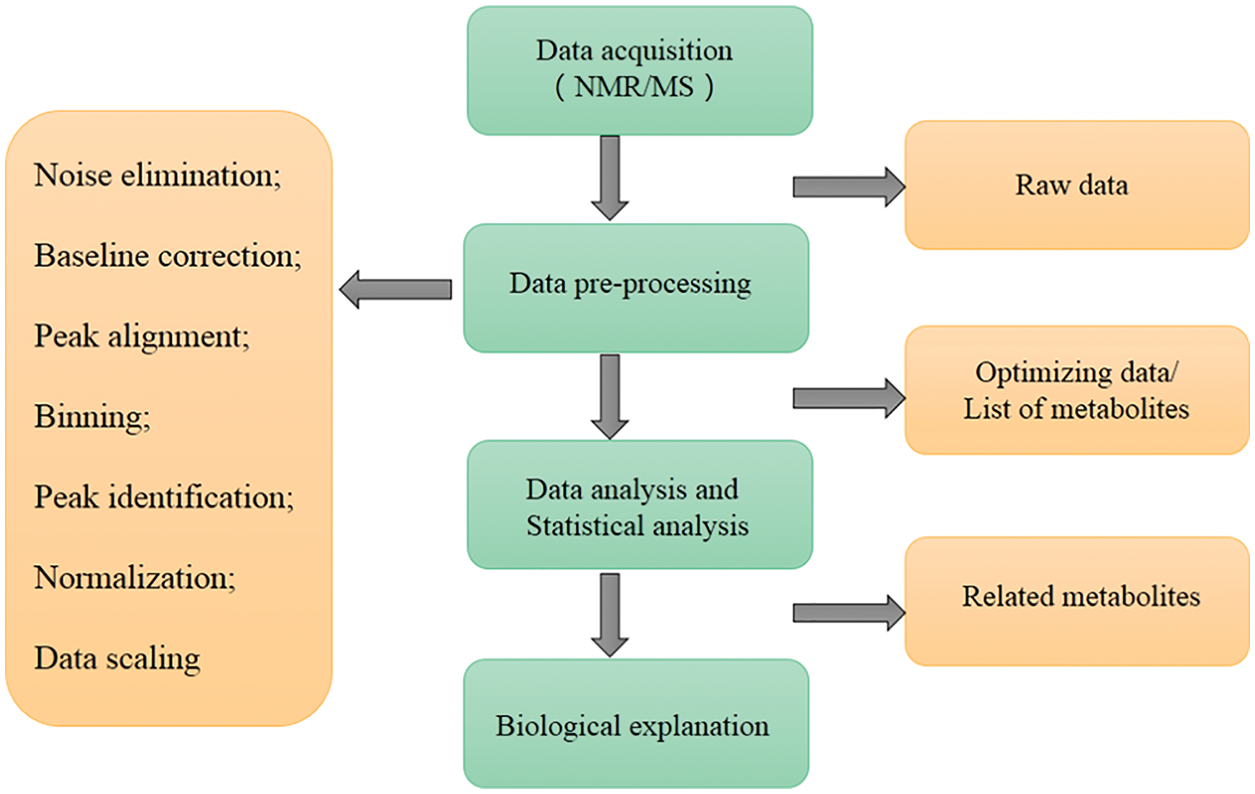
Figure 4: The main steps of microbial metabolomics data preprocessing and analysis. The main data processing procedures include: data acquisition, data pre-processing, data and statistical analysis, and biological explanation.
However, microbial metabolomics still faces serious problems in signal processing and data analysis, which pose a great challenge in its research. To effectively eliminate the undesirable signal fluctuations introduced by environmental, instrumental, and biological factors, it is necessary to develop new methods for the optimization of metabolomics signaling systems and to tailor optimal data analysis strategies for different omics studies. In response to the above problems, some researchers have established a new method for metabolomics data processing based on machine learning and parallel computing for optimizing omics signal processing strategies (Fu et al., 2021). This method can quickly optimize the best-performing omics data processing flow based on the metabolomics raw data given by the user by scanning the existing massive signal processing flow on a large scale. This approach enables data processing for “time-series” and “multi-taxonomic” metabolomics problems common to the field of microbiology.
Applications of microbial metabolomics
Microbial metabolomics is a discipline that combines bioinformatics and systematic microbiology. It is not only helpful in exploring the relationship between species but also a reliable analytical tool for the study of microbial evolution and development dynamics. As shown in Fig. 5, this review first summarized the research progress and application of microbial metabolomics from two aspects, probiotics and pathogenic bacteria. Because probiotics and pathogenic bacteria do not exist independently most of the time, we reviewed the research on the coexistence of probiotics and pathogenic bacteria. The research is subdivided into important microbial research fields closely related to human health, and the research progress and application of microbial metabolomics in the human gut, oral cavity, food and, nutrition-related microorganisms were reviewed. The recent progress and application of microbial metabolomics are discussed herein.
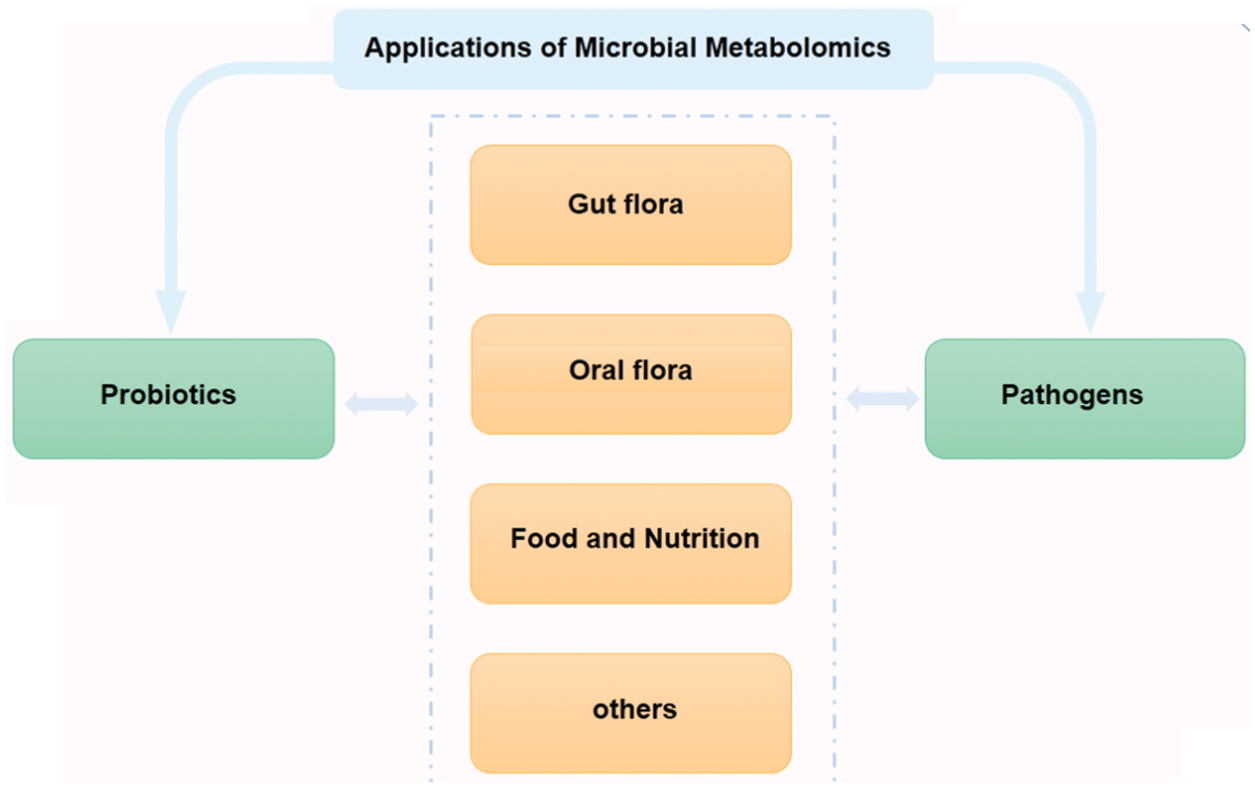
Figure 5: Main applications of probiotics and pathogenic bacteria microbial metabolomics. In recent years, research on microbial metabolomics has mainly focused on gut flora, oral flora, food, and nutrition.
Probiotics
Lactic acid bacteria (LAB) are a generic term for bacteria that produce large amounts of lactic acid from fermentable carbohydrates (Liang et al., 2020). In recent years, microbial metabolomics has made great progress and breakthroughs in the field of LAB research, such as strain screening and identification, metabolic pathway analysis, fermentation engineering, and beneficial effects (Kim et al., 2017; Zhao et al., 2016). The traditional classification of LAB is mainly based on morphological observation and biochemical experiments (Cai et al., 1999; Makela et al., 1992; Shaw et al., 1985). With the development of molecular biology techniques, genotyping methods such as microbial genome sequencing, 16S rDNA sequence analysis, polymerase chain reaction (PCR) fingerprinting, and DNA hybridization technology have been widely used (Domingos-Lopes et al., 2017; Ghodhbane et al., 2016; Ramos et al., 2018; Zhao et al., 2015). However, the genotype and phenotype of some strains are inconsistent, resulting in different classification results. Metabolic profiling analysis could identify different strains by comparing the characteristic peaks of extracellular metabolites (Bachmann et al., 2017; Filannino et al., 2018). Microbial metabolomics has gradually become an effective, fast, and high throughput method. The extracellular metabolites of Streptococcus mutans, Streptococcus haemophilus, and Lactobacillus acidophilus were studied by the 1H-NMR microbial metabolomics method (Lee et al., 2009). It was confirmed that the microbial metabolomics method could detect the differences among different strains and possess a good application prospect in microbial identification. Researchers compared the identification ability of LAB by PCR, 16S-amplified ribosomal DNA restriction analysis, and matrix-assisted laser desorption-ionization time-of-flight mass spectrometry (MALDI-TOF-MS) and found that MALDI-TOF-MS had more advantages and accuracy at the species level (Doan et al., 2012; Duskova et al., 2012).
Microbial metabolomics is also widely used to monitor the changes in components and bacterial phase during fermentation and evaluate the sensory and nutritional quality of fermented food (Castro et al., 2018; Pisano et al., 2016; Renes et al., 2019). High throughput sequencing and 1H-NMR techniques were used to monitor the changes in microbial flora and metabolites during kimchi fermentation in South Korea (Jung et al., 2012). The results showed that as a starter, Leuconostoc mesenteroides not only increased the proportion of Leuconostoc in the fermentation process but also decreased the proportion of Lactobacillus, shortened the fermentation time, and led to a higher production of organic acids and mannitol, which provided a direction for the regulation of kimchi fermentation. Meiju is a traditional fermented soybean paste in Korea. It is usually fermented by Bacillus, Aspergillus, and Mucor. Using UPLC-Q-TOF MS technology, the researchers revealed 22 markers in the Meiju fermentation process, including amino acids, small peptides, nucleic acids, ornithine circulating intermediates, and organic acids, and constructed the metabolic pathway of Meiju fermentation, which provided a theoretical reference for improving the nutrition and quality of Meiju products (Kang et al., 2011). Therefore, metabolomics technology can directly detect the changes of components in LAB fermentation products, which provides an effective tool for process optimization and quality control (Zhao et al., 2016).
In recent years, the effect of Lactobacillus on health has attracted extensive attention from researchers (Rezazadeh et al., 2018; Zhang et al., 2018). The effects of Lactobacillus on the intestinal tract by genomics, transcriptome, proteomics, and metabolomics have become a research hotspot (Awais et al., 2018; Toshimitsu et al., 2018). Microbial metabolomics can detect the changes in intestinal metabolites under the action of Lactobacillus and clarify the effects of probiotic metabolites on cytokine expression. The effects of probiotics on irritable bowel syndrome (IBS) mice were evaluated using 1H-NMR-based microbial metabolomics techniques and clinical parameters (Hong et al., 2011). IBS mice were found to have a potential energy imbalance (serum glucose) and liver dysfunction (serum tyrosine), and the symptoms improved by probiotic intake. The high-resolution magic angle spinning-NMR technique was used to evaluate the effect of probiotics on IBS patients (Hong et al., 2011). The plasma concentration of glucose, tyrosine, and lactic acid in IBS patients who drank fermented milk containing Lactobacillus and Bifidobacterium for two weeks gradually became normal and thus provided a basis for exploring the mechanism of action of probiotics on IBS. Combined with GC-GC-TOF-MS and multivariate statistical analysis, it was found that Lactobacillus rhamnosus GG (LGs) could effectively regulate the structure and metabolism of intestinal flora in mice with alcoholic fatty liver and improve the health of the host (Shi et al., 2015). Administration of LGGs increased the long-chain fatty acids in the intestine, decreased the fatty acid content in the liver, and increased the amino acid content in the liver (Castaneda-Gutierrez et al., 2014; Ivanovic et al., 2015; Ivanovic et al., 2016). Therefore, microbial metabolomics is an effective tool to study the effects of probiotics on host health too. It could provide the metabolic response of host flora under the action of probiotics and lay a foundation for exploring the regulation mechanism.
Pathogens
In recent years, microbial metabolomics technology has been widely used in the research of pathogenic bacteria, which could analyze pathogenic bacteria in many aspects in detail (Akkerman et al., 2018; Fairley et al., 2021). MS is the most commonly used microbial metabolomic analysis technology in the study of fungal plant pathogens (Sevastos et al., 2018; Sirichokchatchawan et al., 2018). It is widely used to analyze mutations in pathogens and to detect and screen secondary metabolites (Nielsen and Smedsgaard, 2003).
GC-MS analysis showed that the concentration of secondary metabolites in Stagonospora nodorum mutant strains lacking the Sch1 gene was 200 times higher than that of wild strains (Tan et al., 2009). ESI-MS/MS confirmed that the secondary metabolites were Alternaria phenol, which laid a theoretical foundation for elucidating the function and function of the Sch1 gene. The researchers used GC-MS to analyze the metabolites related to the sporogenesis of wheat pathogen Stagonospora nodorum, and found that chitosan plays an important role in sporogenesis (Lowe et al., 2009). Microbial metabolomics has also been applied to diagnose diseases caused by pathogenic bacterial infections. GC-MS analysis of volatile organic compounds in feces reveals that metabolomic techniques could be used to distinguish diarrhea caused by different pathogens. Studies have revealed that furan could be detected in diarrhoeal feces of the patient infected by Clostridium difficile, while the presence of dodecanoate compounds in feces indicates that diarrhea is a rotavirus disease (Probert et al., 2004). Infections with Rotavirus and Campylobacter lead to the increase in terpenoids in feces. The decrease in dodecanoate compounds and the increase in amino compounds are the biomarkers of other enterovirus infections. While such studies provided a theoretical basis for rapid diagnosis of diarrhea etiology and a reference for the identification of different diarrhea pathogens, other studies have shown that some volatile substances are closely related to diseases caused by fungal infections.
Some researchers studied the metabolites of three different tissues (immature leaves, leaves, and sheaths) of ryegrass infected with endophytic fungi (Neotyphodium lolii) and ryegrass not infected with N. lolii and revealed the presence of mannitol and bolamine in the infected ryegrass by LC-MS (Cao et al., 2008; Li et al., 2018a; Wiewiora et al., 2015; Zhou et al., 2014). These results fully demonstrate that microbial metabolomics can identify bacterial species through metabolites secreted by pathogenic bacteria and also be an effective tool for the diagnosis of pathogenic bacterial infections.
Probiotics and pathogenic bacteria often coexist; therefore, it is necessary to study the field where the two coexist. The recent progress and application of microbial metabolomics in the important microbial research fields closely related to human health are discussed in the following sections.
In the human body, the number of microorganisms is much larger than that of cells, and gut flora is one of the key points in systematic biology and metabolomics (Geng et al., 2018; Ke et al., 2018a, 2018b; Si et al., 2018; Liu et al., 2022). The functions and metabolism of microorganisms are closely related to the health and disease of the host (Song et al., 2018; Suzuki-Iwashima et al., 2020; Xing et al., 2018). They can prevent the infection of pathogens and provide energy for the host through their own metabolism, enhance the immunity of the host, and regulate the metabolic phenotype through their interaction with the host, and so on (Bhatnagar, 2015; Jain et al., 2012; Wang et al., 2011; Na and Lim, 2022).
The metabolic interactions between gut flora and host in mice models were studied by various techniques, including 1H-NMR analysis of metabolic profiles of the liver, plasma, urine, and ileum contents, LC-MS targeting analysis of bile acids, and GC technique detecting fatty acid fingerprints in the cecum (Martin et al., 2007). The concentration of bile acid was higher in ileum and triglyceride in the liver of sterile mice colonized with infant gut flora than that of normal mice, while the level of plasma lipoprotein was lower. These data showed that gut flora could regulate intestinal absorption and energy storage and acquisition.
Microbial metabolomics technology based on MS was used to study the plasma of aseptic mice and normal mice (Wikoff et al., 2009), and the results revealed that gut flora possessed significant effects on the plasma metabolites of mammals. For example, the antioxidant 3-indolepropionic acid only exists in the plasma of normal mice, but Clostridium was colonized in aseptic mice. After um sporogenes, 3-indolepropionic acid was also detected in plasma, indicating that the production of 3-indolepropionic acid in the host was influenced by gut flora. The water-soluble fecal metabolites of humans, mice, and rats were compared by 1H-NMR (Ganda Mall et al., 2018; Saric et al., 2008). These three metabolites were found to have unique fecal metabolite profiles. For example, beta-alanine was detected only in rat feces, while glycerol and malonate were found to be unique to human feces. It was thus confirmed that different gastrointestinal microorganisms have significant effects on fecal metabolites. Therefore, microbial metabolomics technology could directly detect the changes in host metabolic phenotype under the changes in gut flora, which provides a new research strategy and method for further elucidating the interaction mechanism between gut flora and the human body (Goodwin et al., 2015; Lyte et al., 2019) (Fig. 6).
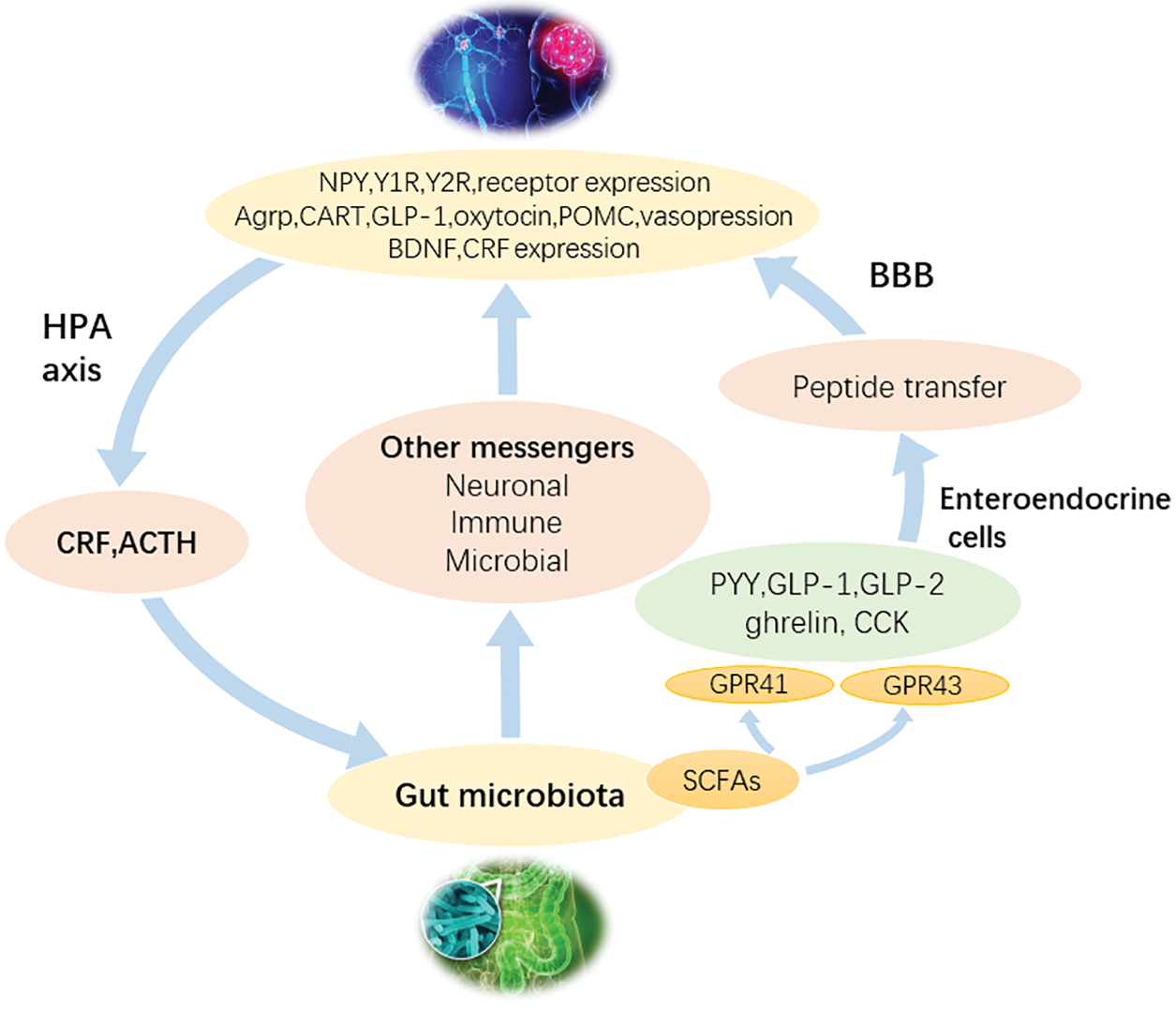
Figure 6: A flowchart to depict the mechanism of the effect of gut flora on the brain. Gut flora could interact with the brain through a variety of signal pathways.
Normally, gut flora structure plays an important role in the prevention and control of diseases, but gut flora imbalance and changes in microbial biodiversity lead to a series of adverse effects on the host, such as the occurrence of various gastrointestinal diseases, metabolic diseases, and immune diseases (Jain et al., 2012; Nagai, 2015; Wang et al., 2017b). Many researchers have used microbial metabolomics technology to study gut flora and host metabolites to explore the effects of gut flora on host health and disease and have achieved various important results (Geng et al., 2018; He et al., 2018; Ke et al., 2018b). GC-MS and pyrosequencing were used to study the changes in volatile metabolites and gut flora structure in stool samples of obese patients with non-alcoholic fatty liver (Raman et al., 2013). The results showed that the increase in esters in the stool of non-alcoholic fatty liver obesity patients was related to the changes in gut flora structure.
Researchers used GC-MS technology to study the metabolomics of fecal volatile organic compounds in IBS, Crohn’s disease, ulcerative colitis patients, and healthy people (Ahmed et al., 2013; Furnari et al., 2016; Rossi et al., 2018). The results showed that IBS occurred in patients harboring short-chain fatty acids, cyclic acetic acid, and acetic acid. Microbial metabolomics and aseptic mouse experiments showed that phosphatidylcholine metabolism in gut flora can promote the occurrence of cardiovascular diseases, and gut flora plays an important role in the production of trimethylamine and the formation of foam cells (Bhatnagar, 2015). Therefore, it is essential to analyze gut flora and host co-metabolism by microbial metabolomics technology to reveal the metabolic function of gut flora and their effects on host health and disease.
Recently, microbial metabolomics technology has gradually penetrated the field of stomatology (Shakhatreh et al., 2018); it has been reported in the study of microorganisms, caries, periodontal diseases, cleft lip, palate, and oral tumors. Most of them are used to analyze the metabolites of cariogenic bacteria and periodontal pathogens in the oral cavity. The oral cavity harbors more than 700 species of prokaryotes (Mager et al., 2003). Some bacteria could cause oral infectious diseases, such as dental caries, periodontitis, pulp disease, alveolar osteitis, and glossitis (Burczynska et al., 2017; Dong et al., 2018; Krzyzek and Gosciniak, 2018; Xu et al., 2015). Different oral structures and tissues are colonized by different microbial communities. Oral microbial identification is very important for the etiology, treatment, and prevention of infectious diseases such as dental caries, and periodontal diseases (Lira-Junior et al., 2018; Mombelli, 2018; Rafiei et al., 2018).
When using microbial metabolomics to identify Streptococcus mutans and Actinomyces viscosus, the researchers found that PCA could classify and identify the two species of bacteria, and the one-dimensional spectrum time of magnetic resonance samples was only 5 min, which was convenient and fast (Kunze et al., 2010; Ribeiro et al., 2017; Salman et al., 2018). The spectra of extracellular metabolites of Actinomyces tunica, Actinomyces neisseriae, Streptococcus sanguis, Streptococcus cousins, Porphyromonas gingivalis, Prevotella intermedia, Clostridium nucleatum, Streptococcus mutans, Lactobacillus acidophilus, Actinomyces viscosus, and Candida in a stable phase were compared by the same method (Hetrodt et al., 2018; Sanz et al., 2017). The difference between different strains could be well detected. Microbial metabolomics analysis of Streptococcus and Actinomycetes showed that the metabolic pathways of these two microorganisms were different. Researchers applied microbial metabolomics to the in vivo study of dental plaque biofilm and established dental plaque biofilm metabolomics (Keijser et al., 2018). When analyzing the effects of xylitol and fluoride on the metabolomics of plaque biofilm, fluoride was found to inhibit the glycolysis pathway of supragingival plaque but had no effect on the acid products of xylitol and the metabolic profile of supragingival plaque (Takahashi and Washio, 2011).
The analysis of the whole metabolome of the early oral cavity showed that the saliva metabolites of children with caries in deciduous dentition, mixed dentition, and permanent dentition were different. Compared with the normal group, the contents of lactic acid, acetic acid, and butyric acid increased in the infected group. The composition of metabolites in the saliva of normal children was similar, although their oral hygiene habits, socioeconomic status, and diet were different (Fidalgo et al., 2013). In addition, the salivary metabolites of children with dental caries after 3 months of composite resin repair showed a significant decrease in propionic acid, acetic acid, butyric acid, and oligosaccharide content, accompanied by a decrease in the culture level of Streptococcus mutans and Lactobacillus (Fidalgo et al., 2015). Salivary metabolites analysis in children with caries showed a prominent role of the metabolites involved in the metabolic pathway of arginine proline and the acid-base balance connecting arginine and alkali production (Edlund et al., 2015). Microbial metabolites in adult supragingival plaque were also analyzed in terms of the Embden-Meyerhof-Parnas pathway, pentose phosphate path way, and tricarboxylic acid cycle (Takahashi et al., 2010), and the main metabolic pathway was found to be the supragingival plaque. It involves all the carbohydrate metabolites except erythrose-4-phosphate in the pentose phosphate pathway. After rinsing with glucose, glucose-6-phosphate, fructose-6-phosphate, fructose-1,6-bisphosphate, dihydroxy acetone phosphate, pyruvate, and pentose phosphate pathways in the glycolysis pathway were determined. Further, 6-phosphogluconate, ribu-lose-5-phosphate, sedoheptu-lose-7-phosphate and acetyl CoA increased. Meanwhile, 3-phosphoglycerate and phosphoenolpyruvate, succinate, and malate in the tricarboxylic acid cycle decreased in the glycolysis pathway. These changes in pathways and metabolites observed in supragingival plaque are similar to those observed in Streptococcus and Actinomycetes. The composition of dental caries-related microbial communities and their relationship between them are diverse and huge. Microbial metabonomics possesses a significant space in the study of oral flora.
In recent years, food safety has received much attention. Pathogens, toxins, and by-products produced by microbial degradation of food are closely related to food safety (Putri et al., 2022). Therefore, monitoring these related metabolites is very important for food safety. Microbial metabolomics provides a new strategy for food safety assessment. It has been successfully applied to the detection of toxic substances in food, such as the fingerprint of volatile metabolites related to specific microbial contamination using GC-MS technology and the detection of microbial toxins in food by LC-MS and NMR technology (Qian et al., 2015). Researchers used GC-MS to detect toxins such as zearalenone produced by Fusarium spp. in edible vegetable oils. Studies used LC-MS/MS to detect and quantitatively analyze 23 mycotoxins in different sorghum varieties (Forero-Reyes et al., 2018; Njumbe Ediage et al., 2015).
Microbial metabolomics has also been used to assess the effects of nutrient deficiency and excess on metabolic balance, accurately monitor the effects of diet on the body, and reduce the interference of confounding factors such as age, sex, physiological status, and lifestyle (Fu and Cui, 2017). Techniques including CE, RP/UPLC and HILIC/UPLC-TOF-MS revealed the significant effect of dietary polyphenols on the anti-proliferation of human colon cancer HT29 cells (Ibanez et al., 2012). Non-targeted metabolic analysis showed that the ratio of glutathione, an antioxidant, increased after the treatment of polyphenols, while the expression of polyamines, which maintained cell proliferation and regulated gene expression, inhibited cell growth, and provides a theoretical basis for the prevention and treatment of colon cancer.
Based on the microbial metabolomics technique of NMR, researchers monitored the urine metabolic profiles of normal puppies and dietary restricted puppies (Wang et al., 2007). The results showed that dietary restriction could change the intestinal microbial activity of puppies, which was mainly manifested in the increased concentration of aromatic metabolites, creatine/creatinine, and acetic acid compounds in the urine of dietary restricted puppies. These compounds were closely related to gut flora. Analysis of the 1H-NMR metabolic profiles of piglet urine confirmed that different weaning diets produced different metabolic phenotypes, which led to changes in gut flora, and host metabolites. Studies have also found that intestinal mucosal products associated with lgA and lgM were produced by Bifidobacterium NCC2818 ingestion during the weaning diet, which provided a new research strategy for evaluating mucosal immune status. These findings fully demonstrate that microbial metabolomics can quickly and effectively make a comprehensive assessment of animal health, disease prediction and diagnosis and further understand the interaction between nutrition and metabolism in the body.
In summary, microbial metabolomics has been widely applied in microbiology research and provides a comprehensive and systematic analytical technology for microbiology research. Metabolic products in microbial cells are of great value in the analysis of cellular metabolic processes and pathogenic processes, as well as the effects of drugs or the environment on bacteria. Bacterial metabolites consist of complex mixed metabolites and are associated with a large number of metabolic pathways and signal transduction pathways. This high degree of correlation may be more meaningful than the original disturbances such as inhibition, inactivation or down-regulation of proteins. It is easier to observe the changes in metabolites.
To understand the physiology of bacteria or other microorganisms and obtain reliable results, it is necessary to identify and establish at least two reference groups for comparative analysis, such as wild type and mutant type, drug resistance type and susceptibility type, and nutritional enrichment and nutritional deficiency. Only when reference conditions are established, bacteria could be exposed to any experimental variables such as drug treatment, environmental stimuli, or gene knockout to determine any possible similarities in reference metabolites. With the continuous improvement in sample preparation methods and the rapid development of analytical techniques, great progress has been made in microbial metabolomics research. It could be used for the study of biomarkers in the microbial metabolism process and provide a comprehensive and effective evaluation method for monitoring the fermentation process, safety detection, and pathogenic bacterial infection diagnosis. It could be used to study the metabolic mechanism of intestinal flora and host and provide theoretical basis for the prevention and treatment of metabolic diseases.
Some problems in microbial metabolomics of probiotics and pathogens still need to be solved. First, there is a lack of effective methods for quenching and extracting metabolites. Microbial cell metabolism is very sensitive to changes in the surrounding environment, and both the measurement and sample preparation process could affect metabolomics. Microbial metabolomics lacks good methods for rapid inactivation of metabolic activity, comprehensive extraction of metabolites, and analysis of specific metabolites. Second, although many microbial metabolomics databases have emerged, they are limited to specific microbial populations. At present, microbial metabolomics and databases are mainly confined to specific microorganisms, mainly yeast, and E. coli, and lack integrated databases containing different microbial metabolic data, especially for bacteria and fungi. Third, microbial metabolomics still faces problems of the signal processing and data analysis, which poses a great challenge. It is necessary to develop new methods for the optimization of metabolomics signaling systems and to tailor strategies for optimal data analysis for different omics studies. Artificial intelligence and machine learning will be the trend to solve the problem of microbial metabolomics. Fourth, the study of microbial metabolomics pays much attention to metabolites rather than their sources. For example, although the chemical composition and structure of glucose from the host and microbial metabolism are identical, their biological significance and metabolic pathways involved make their regulation different. The study of microbial metabolism and host changes is of great significance to the study of the microbial-host relationship.
Compared with the application of metabolomics in drug research, disease diagnosis, and plant metabolomics, microbial metabolomics is still in its infancy. However, there are many advantages of microbial metabolomics research, such as a simple microbial system, rich genetic data, and a comprehensive understanding of microbial physiological characteristics. Concurrently, the integration of microbial metabolomics with genomics, transcriptome, and proteomics could help understand organisms more systematically by studying metabolic pathways, regulatory responses, and homeostasis mechanisms in vivo. In summary, microbial metabolomics, as a new research field with rapid development, is an important component and technical platform of systems biology, which promotes the development of systems microbiology and artificial intelligence.
Author Contribution: The authors confirm contribution to the paper as follows: study conception and design: Xin Meng; data collection: Lianrong Yang; analysis and interpretation of results: Rui Yin, Lehui Qi; draft manuscript preparation: Xin Meng, Xue Li, Qi Guo. All authors reviewed the results and approved the final version of the manuscript.
Ethics Approval: This study was approved by The Heilongjiang University of Chinese Medicine Review Board and Ethics Committee (20210711012), July 11, 2021.
Funding Statement: This work was supported by the Science Foundation of Heilongjiang Administration of Traditional Chinese Medicine (No. ZHY18-021).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
Adetunji C, Oloke J, Kumar A, Swaranjit S, Akpor B (2017). Synergetic effect of rhamnolipid from Pseudomonas aeruginosa C1501 and phytotoxic metabolite from Lasiodiplodia pseudotheobromae C1136 on Amaranthus hybridus L. and Echinochloa crus-galli weeds. Environmental Science and Pollution Research 24: 13700–13709. DOI 10.1007/s11356-017-8983-8. [Google Scholar] [CrossRef]
Ahmed I, Greenwood R, Costello Bde L, Ratcliffe NM, Probert CS (2013). An investigation of fecal volatile organic metabolites in irritable bowel syndrome. PLoS One 8: e58204. DOI 10.1371/journal.pone.0058204. [Google Scholar] [CrossRef]
Akkerman R, Faas MM, de Vos P (2018). Non-digestible carbohydrates in infant formula as substitution for human milk oligosaccharide functions: Effects on microbiota and gut maturation. Critical Reviews in Food Science and Nutrition 59: 1–12. DOI 10.1080/10408398.2017.1414030. [Google Scholar] [CrossRef]
Almanza-Aguilera E, Urpi-Sarda M, Llorach R, Vazquez-Fresno R, Garcia-Aloy M et al. (2017). Microbial metabolites are associated with a high adherence to a Mediterranean dietary pattern using a 1H-NMR-based untargeted metabolomics approach. The Journal of Nutritional Biochemistry 48: 36–43. DOI 10.1016/j.jnutbio.2017.06.001. [Google Scholar] [CrossRef]
Araujo R, Carneiro TJ, Marinho P, da Costa MM, Roque A, Silva OABDE, Fernandes MH, Vilarinho PM, Gil AM (2019). NMR metabolomics to study the metabolic response of human osteoblasts to non-poled and poled poly (L-lactic) acid. Magnetic Resonance in Chemistry 57: 919–933. DOI 10.1002/mrc.4883. [Google Scholar] [CrossRef]
Awais MM, Jamal MA, Akhtar M, Hameed MR, Anwar MI, Ullah MI (2018). Immunomodulatory and ameliorative effects of Lactobacillus and Saccharomyces based probiotics on pathological effects of eimeriasis in broilers. Microbial Pathogenesis 126: 101–108. DOI 10.1016/j.micpath.2018.10.038. [Google Scholar] [CrossRef]
Bachmann H, Molenaar D, Branco Dos Santos F, Teusink B (2017). Experimental evolution and the adjustment of metabolic strategies in lactic acid bacteria. FEMS Microbiology Reviews 41: S201–S219. DOI 10.1093/femsre/fux024. [Google Scholar] [CrossRef]
Baidoo EEK (2019). Microbial metabolomics: A general overview. Methods in Molecular Biology 1859: 1–8. [Google Scholar]
Baidoo EEK, Teixeira Benites V (2019). Mass spectrometry-based microbial metabolomics: Techniques, analysis, and applications. Methods in Molecular Biology 1859: 11–69. [Google Scholar]
Barrilero R, Gil M, Amigo N, Dias CB, Wood LG, Garg ML, Ribalta J, Heras M, Vinaixa M, Correig X (2018). LipSpin: A new bioinformatics tool for quantitative 1H NMR lipid profiling. Analytical Chemistry 90: 2031–2040. DOI 10.1021/acs.analchem.7b04148. [Google Scholar] [CrossRef]
Bean HD, Dimandja JM, Hill JE (2012). Bacterial volatile discovery using solid phase microextraction and comprehensive two-dimensional gas chromatography-time-of-flight mass spectrometry. Journal of Chromatography B, Analytical Technologies in the Biomedical and Life Sciences 901: 41–46. DOI 10.1016/j.jchromb.2012.05.038. [Google Scholar] [CrossRef]
Bhatnagar D (2015). Gut flora, diet and intestinal metabolism on cardiovascular risk. Current Opinion in Lipidology 26: 148–149. DOI 10.1097/MOL.0000000000000165. [Google Scholar] [CrossRef]
Bliss ES, Whiteside E (2018). The gut-brain axis, the human gut microbiota and their integration in the development of obesity. Frontiers in Physiology 9: 900. DOI 10.3389/fphys.2018.00900. [Google Scholar] [CrossRef]
Boersma MG, Solyanikova I, van Berkel WJ, Vervoort J, Golovleva L, Rietjens IM (2001). 19F NMR metabolomics for the elucidation of microbial degradation pathways of fluorophenols. Journal of Industrial Microbiology & Biotechnology 26: 22–34. [Google Scholar]
Boursi B, Werner TJ, Gholami S, Houshmand S, Mamtani R, Lewis JD, Wu GD, Alavi A, Yang YX (2018). Functional imaging of the interaction between gut microbiota and the human host: A proof-of-concept clinical study evaluating novel use for 18F-FDG PET-CT. PLoS One 13: e0192747. [Google Scholar]
Burczynska A, Dziewit L, Decewicz P, Struzycka I, Wroblewska M (2017). Application of metagenomic analyses in dentistry as a novel strategy enabling complex insight into microbial diversity of the oral cavity. Polish Journal of Microbiology 66: 9–15. DOI 10.5604/17331331.1234988. [Google Scholar] [CrossRef]
Cai Y, Suyanandana P, Saman P, Benno Y (1999). Classification and characterization of lactic acid bacteria isolated from the intestines of common carp and freshwater prawns. The Journal of General and Applied Microbiology 45: 177–184. DOI 10.2323/jgam.45.177. [Google Scholar] [CrossRef]
Cao M, Koulman A, Johnson LJ, Lane GA, Rasmussen S (2008). Advanced data-mining strategies for the analysis of direct-infusion ion trap mass spectrometry data from the association of perennial ryegrass with its endophytic fungus, Neotyphodium lolii. Plant Physiology 146: 1501–1514. DOI 10.1104/pp.107.112458. [Google Scholar] [CrossRef]
Castaneda-Gutierrez E, Moser M, Garcia-Rodenas C, Raymond F, Mansourian R et al. (2014). Effect of a mixture of bovine milk oligosaccharides, Lactobacillus rhamnosus NCC4007 and long-chain polyunsaturated fatty acids on catch-up growth of intra-uterine growth-restricted rats. Acta Physiologica 210: 161–173. DOI 10.1111/apha.12145. [Google Scholar] [CrossRef]
Castro RCS, David de Oliveira AP, Rodrigues de Souza EA, Correia TMA, Viana de Souza J, Dias FS (2018). Lactic acid bacteria as biological control of staphylococcus aureus in coalho goat cheese. Food Technology and Biotechnology 56: 431–440. [Google Scholar]
Chakraborty B, Kumar RS, Almansour AI, Gunasekaran P, Nayaka S (2022). Bioprospection and secondary metabolites profiling of marine Streptomyces levis strain KS46. Saudi Journal of Biological Sciences 29: 667–679. DOI 10.1016/j.sjbs.2021.11.055. [Google Scholar] [CrossRef]
Covington BC, Mclean JA, Bachmann BO (2017). Comparative mass spectrometry-based metabolomics strategies for the investigation of microbial secondary metabolites. Natural Product Reports 34: 6–24. DOI 10.1039/C6NP00048G. [Google Scholar] [CrossRef]
Depke T, Franke R, Bronstrup M (2017). Clustering of MS2 spectra using unsupervised methods to aid the identification of secondary metabolites from Pseudomonas aeruginosa. Journal of Chromatography B, Analytical Technologies in the Biomedical and Life Sciences 1071: 19–28. DOI 10.1016/j.jchromb.2017.06.002. [Google Scholar] [CrossRef]
Doan NT, van Hoorde K, Cnockaert M, de Brandt E, Aerts M, Le Thanh B, Vandamme P (2012). Validation of MALDI-TOF MS for rapid classification and identification of lactic acid bacteria, with a focus on isolates from traditional fermented foods in Northern Vietnam. Letters in Applied Microbiology 55: 265–273. DOI 10.1111/j.1472-765X.2012.03287.x. [Google Scholar] [CrossRef]
Dodda S, Makula A, Polagani SR, Kandhagatla RN (2018). High sensitive LC-MS/MS method for estimation of eluxadoline in human plasma and its application to pharmacokinetic study. Journal of Pharmaceutical and Biomedical Analysis 165: 65–72. DOI 10.1016/j.jpba.2018.11.056. [Google Scholar] [CrossRef]
Domingos-Lopes MFP, Stanton C, Ross PR, Dapkevicius MLE, Silva CCG (2017). Genetic diversity, safety and technological characterization of lactic acid bacteria isolated from artisanal Pico cheese. Food Microbiology 63: 178–190. DOI 10.1016/j.fm.2016.11.014. [Google Scholar] [CrossRef]
Dong L, Yin J, Zhao J, Ma SR, Wang HR, Wang M, Chen W, Wei WQ (2018). Microbial similarity and preference for specific sites in healthy oral cavity and esophagus. Frontiers in Microbiology 9: 1603. DOI 10.3389/fmicb.2018.01603. [Google Scholar] [CrossRef]
Duskova M, Sedo O, Ksicova K, Zdrahal Z, Karpiskova R (2012). Identification of lactobacilli isolated from food by genotypic methods and MALDI-TOF MS. International Journal of Food Microbiology 159: 107–114. DOI 10.1016/j.ijfoodmicro.2012.07.029. [Google Scholar] [CrossRef]
Edlund A, Yang Y, Yooseph S, Hall AP, Nguyen DD et al. (2015). Meta-omics uncover temporal regulation of pathways across oral microbiome genera during in vitro sugar metabolism. The ISME Journal 9: 2605–2619. DOI 10.1038/ismej.2015.72. [Google Scholar] [CrossRef]
Elmroth I, Sundin P, Valeur A, Larsson L, Odham G (1992). Evaluation of chromatographic methods for the detection of bacterial contamination in biotechnical processes. Journal of Microbiological Methods 15: 215–228. DOI 10.1016/0167-7012(92)90042-3. [Google Scholar] [CrossRef]
Fairley JK, Ferreira JA, Ziegler TR, Jones DP, Fraga LA, Ly VT, Lyon S, Collins JM (2021). High resolution metabolomics highlights differences in lipid and nutritional metabolism across the leprosy spectrum providing avenues for advances in leprosy host-pathogen research. American Journal of Tropical Medicine and Hygiene 105: 425. [Google Scholar]
Fei F, Bowdish DM, Mccarry BE (2014). Comprehensive and simultaneous coverage of lipid and polar metabolites for endogenous cellular metabolomics using HILIC-TOF-MS. Analytical and Bioanalytical Chemistry 406: 3723–3733. DOI 10.1007/s00216-014-7797-5. [Google Scholar] [CrossRef]
Fidalgo TKS, Freitas-Fernandes LB, Almeida FCL, Valente AP, Souza IPR (2015). Longitudinal evaluation of salivary profile from children with dental caries before and after treatment. Official Journal of the Metabolomic Society 11: 583–593. DOI 10.1007/s11306-014-0717-z. [Google Scholar] [CrossRef]
Fidalgo TKS, Freitas-Fernandes LB, Angeli R, Muniz AMS, Gonsalves E, Santos R, Nadal J, Almeida FCL, Valente AP, Souza IPR (2013). Salivary metabolite signatures of children with and without dental caries lesions. Metabolomics 9: 657–666. DOI 10.1007/s11306-012-0484-7. [Google Scholar] [CrossRef]
Filannino P, Di Cagno R, Gobbetti M (2018). Metabolic and functional paths of lactic acid bacteria in plant foods: Get out of the labyrinth. Current Opinion in Biotechnology 49: 64–72. DOI 10.1016/j.copbio.2017.07.016. [Google Scholar] [CrossRef]
Fobofou SA, Savidge T (2022). Microbial metabolites: Cause or consequence in gastrointestinal disease? American Journal of Physiology-Gastrointestinal and Liver Physiology 322: G535–G552. [Google Scholar]
Forero-Reyes CM, Alvarado-Fernandez AM, Ceballos-Rojas AM, Gonzalez-Carmona LC, Linares-Linares MY, Castaneda-Salazar R, Pulido-Villamarin A, Gongora-Medina ME, Cortes-Vecino JA, Rodriguez-Bocanegra MX (2018). Evaluation of Fusarium spp. pathogenicity in plant and murine models. Revista Argentina de Microbiologia 50: 90–96. [Google Scholar]
Fu, J, B., Zhang, Y., Wang, Y. X., Zhang, H. N., Liu, J. (2021). Optimization of metabolomic data processing using NOREVA. Nature Protocols 17: 129–151. [Google Scholar]
Fu ZD, Cui JY (2017). Remote sensing between liver and intestine: Importance of microbial metabolites. Current Pharmacology Reports 3: 101–113. DOI 10.1007/s40495-017-0087-0. [Google Scholar] [CrossRef]
Furnari M, Bodini G, Giannini EG, Savarino V (2016). Letter: Faecal volatile organic metabolites, promising biomarkers in inflammatory bowel disease. Alimentary Pharmacology & Therapeutics 43: 1240–1241. DOI 10.1111/apt.13581. [Google Scholar] [CrossRef]
Ganda Mall JP, Lofvendahl L, Lindqvist CM, Brummer RJ, Keita AV, Schoultz I (2018). Differential effects of dietary fibres on colonic barrier function in elderly individuals with gastrointestinal symptoms. Scientific Reports 8: 13404. DOI 10.1038/s41598-018-31492-5. [Google Scholar] [CrossRef]
Geng H, Shu S, Dong J, Li H, Xu C, Han Y, Hu J, Han Y, Yang R, Cheng N (2018). Association study of gut flora in Wilson’s disease through high-throughput sequencing. Medicine 97: e11743. DOI 10.1097/MD.0000000000011743. [Google Scholar] [CrossRef]
Gupta G, Bansal D, Sharma A, Ahmad T, Sachdev A, Ahmad A, El-Serehy HA, Kaur B (2021). GC/MS-based differential metabolic profiling of human peptic ulcer disease to study Helicobacter pylori-induced metabolic perturbations. BIOCELL 45: 1299–1311. DOI 10.32604/biocell.2021.015411. [Google Scholar] [CrossRef]
Ghodhbane H, Alessandria V, Snoussi M, Elleuch L, Trabelsi I, Abdelly C, Sabatier JM, Cocolin L, Regaya I (2016). Genetic characterization of lactic acid bacteria isolated from tunisian milk waste and their antimicrobial activity against some bacteria implicated in nosocomial infections. Infectious Disorders-Drug Targets 16: 182–191. DOI 10.2174/1871526516666160719160150. [Google Scholar] [CrossRef]
Goodwin CR, Covington BC, Derewacz DK, Mcnees CR, Wikswo JP, Mclean JA, Bachmann BO (2015). Structuring microbial metabolic responses to multiplexed stimuli via self-organizing metabolomics maps. Chemistry & Biology 22: 661–670. DOI 10.1016/j.chembiol.2015.03.020. [Google Scholar] [CrossRef]
He C, Huang L, Lei P, Liu X, Li B, Shan Y (2018). Sulforaphane normalizes intestinal flora and enhances gut barrier in mice with BBN-induced bladder cancer. Molecular Nutrition & Food Research 62: e1800427. DOI 10.1002/mnfr.201800427. [Google Scholar] [CrossRef]
Hetrodt F, Lausch J, Meyer-Lueckel H, Apel C, Conrads G (2018). Natural saliva as an adjuvant in a secondary caries model based on Streptococcus mutans. Archives of Oral Biology 90: 138–143. DOI 10.1016/j.archoralbio.2018.03.013. [Google Scholar] [CrossRef]
Hong YS, Hong KS, Park MH, Ahn YT, Lee JH, Huh CS, Lee J, Kim IK, Hwang GS, Kim JS (2011). Metabonomic understanding of probiotic effects in humans with irritable bowel syndrome. Journal of Clinical Gastroenterology 45: 415–425. DOI 10.1097/MCG.0b013e318207f76c. [Google Scholar] [CrossRef]
Ibanez C, Simo C, Garcia-Canas V, Gomez-Martinez A, Ferragut JA, Cifuentes A (2012). CE/LC-MS multiplatform for broad metabolomic analysis of dietary polyphenols effect on colon cancer cells proliferation. Electrophoresis 33: 2328–2336. DOI 10.1002/elps.201200143. [Google Scholar] [CrossRef]
Ivanovic N, Minic R, Djuricic I, Dimitrijevic L, Sobajic S, Zivkovic I, Djordjevic B (2015). Brain and liver fatty acid composition changes upon consumption of Lactobacillus rhamnosus LA68. International Journal of Food Sciences and Nutrition 66: 93–97. DOI 10.3109/09637486.2014.979313. [Google Scholar] [CrossRef]
Ivanovic N, Minic R, Djuricic I, Radojevic Skodric S, Zivkovic I, Sobajic S, Djordjevic B (2016). Active Lactobacillus rhamnosus LA68 or Lactobacillus plantarum WCFS1 administration positively influences liver fatty acid composition in mice on a HFD regime. Food & Function 7: 2840–2848. DOI 10.1039/C5FO01432H. [Google Scholar] [CrossRef]
Jacobs JP, Goudarzi M, Singh N, Tong M, Mchardy IH et al. (2016). A disease-associated microbial and metabolomics state in relatives of pediatric inflammatory bowel disease patients. Cellular and Molecular Gastroenterology and Hepatology 2: 750–766. DOI 10.1016/j.jcmgh.2016.06.004. [Google Scholar] [CrossRef]
Jain S, Marotta F, Catanzaro R, Yadav H (2012). Immune system and gut flora interactions are important episodes in metabolic diseases. Journal of Gastrointestinal and Liver Diseases 21: 347–348. [Google Scholar]
Jerome Jeyakumar JM, Zhang M, Thiruvengadam M (2018). Determination of mycotoxins by HPLC, LC-ESI-MS/MS, and MALDI-TOF MS in Fusarium species-infected sugarcane. Microbial Pathogenesis 123: 98–110. DOI 10.1016/j.micpath.2018.06.045. [Google Scholar] [CrossRef]
Joshua CJ (2019). Metabolomics: A microbial physiology and metabolism perspective. Methods in Molecular Biology 1859: 71–94. DOI 10.1007/978-1-4939-8757-3. [Google Scholar] [CrossRef]
Jung JY, Lee SH, Lee HJ, Seo HY, Park WS, Jeon CO (2012). Effects of Leuconostoc mesenteroides starter cultures on microbial communities and metabolites during kimchi fermentation. International Journal of Food Microbiology 153: 378–387. DOI 10.1016/j.ijfoodmicro.2011.11.030. [Google Scholar] [CrossRef]
Kang HJ, Yang HJ, Kim MJ, Han ES, Kim HJ, Kwon DY (2011). Metabolomic analysis of meju during fermentation by ultra performance liquid chromatography-quadrupole-time of flight mass spectrometry (UPLC-Q-TOF MS). Food Chemistry 127: 1056–1064. DOI 10.1016/j.foodchem.2011.01.080. [Google Scholar] [CrossRef]
Ke Y, Li D, Zhao M, Liu C, Liu J et al. (2018a). Erratum to Gut flora-dependent metabolite trimethylamine-N-oxide accelerates endothelial cell senescence and vascular aging through oxidative stress. Free Radical Biology & Medicine 129: 608–610. DOI 10.1016/j.freeradbiomed.2018.06.007. [Google Scholar] [CrossRef]
Ke Y, Li D, Zhao M, Liu C, Liu J et al. (2018b). Gut flora-dependent metabolite Trimethylamine-N-oxide accelerates endothelial cell senescence and vascular aging through oxidative stress. Free Radical Biology & Medicine 116: 88–100. DOI 10.1016/j.freeradbiomed.2018.01.007. [Google Scholar] [CrossRef]
Keijser BJF, van den Broek TJ, Slot DE, van Twillert L, Kool J, Thabuis C, Ossendrijver M, van der Weijden FA, Montijn RC (2018). The impact of maltitol-sweetened chewing gum on the dental plaque biofilm microbiota composition. Frontiers in Microbiology 9: 381. DOI 10.3389/fmicb.2018.00381. [Google Scholar] [CrossRef]
Kim M, Furuzono T, Yamakuni K, Li Y, Kim YI et al. (2017). 10-oxo-12(Z)-octadecenoic acid, a linoleic acid metabolite produced by gut lactic acid bacteria, enhances energy metabolism by activation of TRPV1. The FASEB Journal 31: 5036–5048. DOI 10.1096/fj.201700151R. [Google Scholar] [CrossRef]
Kim S, Lee DY, Wohlgemuth G, Park HS, Fiehn O, Kim KH (2013). Evaluation and optimization of metabolome sample preparation methods for Saccharomyces cerevisiae. Analytical Chemistry 85: 2169–2176. DOI 10.1021/ac302881e. [Google Scholar] [CrossRef]
Kiselev VG, Novikov DS (2018). Transverse NMR relaxation in biological tissues. NeuroImage 182: 149–168. DOI 10.1016/j.neuroimage.2018.06.002. [Google Scholar] [CrossRef]
Koek MM, Muilwijk B, van der Werf MJ, Hankemeier T (2006). Microbial metabolomics with gas chromatography/mass spectrometry. Analytical Chemistry 78: 1272–1281. DOI 10.1021/ac051683+. [Google Scholar] [CrossRef]
Krzyzek P, Gosciniak G (2018). Oral Helicobacter pylori: Interactions with host and microbial flora of the oral cavity. Dental and Medical Problems 55: 75–82. DOI 10.17219/dmp/81259. [Google Scholar] [CrossRef]
Kunze B, Reck M, Dotsch A, Lemme A, Schummer D, Irschik H, Steinmetz H, Wagner-Dobler I (2010). Damage of Streptococcus mutans biofilms by carolacton, a secondary metabolite from the myxobacterium Sorangium cellulosum. BMC Microbiology 10: 1–13. DOI 10.1186/1471-2180-10-199. [Google Scholar] [CrossRef]
Lee JE, Hong YS, Lee CH (2009). Characterization of fermentative behaviors of lactic acid bacteria in grape wines through 1H NMR- and GC-based metabolic profiling. Journal of Agricultural and Food Chemistry 57: 4810–4817. [Google Scholar]
Li F, Guo Y, Christensen MJ, Gao P, Li Y, Duan T (2018a). An arbuscular mycorrhizal fungus and Epichloe festucae var. lolii reduce Bipolaris sorokiniana disease incidence and improve perennial ryegrass growth. Mycorrhiza 28: 159–169. DOI 10.1007/s00572-017-0813-9. [Google Scholar] [CrossRef]
Liu S, Xu J, Liu T, Rao Z, Zhang W (2022). Co-ordinated combination of Embden-Meyerhof-Parnas pathway and pentose phosphate pathway in Escherichia coli to promote L-tryptophan production. BIOCELL 46: 2303–2313. DOI 10.32604/biocell.2022.020899. [Google Scholar] [CrossRef]
Li Y, Chen M, Liu C, Xia Y, Xu B, Hu Y, Chen T, Shen M, Tang W (2018b). Metabolic changes associated with papillary thyroid carcinoma: A nuclear magnetic resonance-based metabolomics study. International Journal of Molecular Medicine 41: 3006–3014. DOI 10.3892/ijmm.2018.3494. [Google Scholar] [CrossRef]
Li Z, Lu Y, Guo Y, Cao H, Wang Q, Shui W (2018c). Comprehensive evaluation of untargeted metabolomics data processing software in feature detection, quantification and discriminating marker selection. Analytica Chimica Acta 1029: 50–57. DOI 10.1016/j.aca.2018.05.001. [Google Scholar] [CrossRef]
Liang SX, Jiang W, Song YB, Zhou SF (2020). Improvement and metabolomics-based analysis of D-lactic acid production from agro-industrial wastes by lactobacillus delbrueckii submitted to adaptive laboratory evolution. Journal of Agricultural and Food Chemistry 68: 7660–7669. DOI 10.1021/acs.jafc.0c00259. [Google Scholar] [CrossRef]
Liggi S, Hinz C, Hall Z, Santoru ML, Poddighe S, Fjeldsted J, Atzori L, Griffin JL (2018). KniMet: A pipeline for the processing of chromatography-mass spectrometry metabolomics data. Metabolomics 14: 52. DOI 10.1007/s11306-018-1349-5. [Google Scholar] [CrossRef]
Lin Y, Bogdanov M, Tong S, Guan Z, Zheng L (2016). Substrate selectivity of lysophospholipid transporter LplT involved in membrane phospholipid remodeling in Escherichia coli. Journal of Biological Chemistry 291: 2136–2149. DOI 10.1074/jbc.M115.700419. [Google Scholar] [CrossRef]
Lira-Junior R, Akerman S, Klinge B, Bostrom EA, Gustafsson A (2018). Salivary microbial profiles in relation to age, periodontal, and systemic diseases. PLoS One 13: e0189374. DOI 10.1371/journal.pone.0189374. [Google Scholar] [CrossRef]
Lowe RGT, Lord M, Rybak K, Trengove RD, Oliver RP, Solomon PS (2009). Trehalose biosynthesis is involved in sporulation of Stagonospora nodorum. Fungal Genetics & Biology 46: 381–389. DOI 10.1016/j.fgb.2009.02.002. [Google Scholar] [CrossRef]
Lukic J, Chen V, Strahinic I, Begovic J, Lev-Tov H, Davis SC, Tomic-Canic M, Pastar I (2017). Probiotics or pro-healers: The role of beneficial bacteria in tissue repair. Wound Repair and Regeneration 25: 912–922. DOI 10.1111/wrr.12607. [Google Scholar] [CrossRef]
Lyte JM, Proctor A, Phillips GJ, Lyte M, Wannemuehler M (2019). Altered Schaedler flora mice: A defined microbiota animal model to study the microbiota-gut-brain axis. Behavioural Brain Research 356: 221–226. DOI 10.1016/j.bbr.2018.08.022. [Google Scholar] [CrossRef]
Macfarlane GT, Macfarlane S (1997). Human colonic microbiota: Ecology, physiology and metabolic potential of intestinal bacteria. Scandinavian Journal of Gastroenterology 222: 3–9. DOI 10.1080/00365521.1997.11720708. [Google Scholar] [CrossRef]
Mager DL, Ximenez-Fyvie LA, Haffajee AD, Socransky SS (2003). Distribution of selected bacterial species on intraoral surfaces. Journal of Clinical Periodontology 30: 644–654. DOI 10.1034/j.1600-051X.2003.00376.x. [Google Scholar] [CrossRef]
Manole E, Dumitrescu L, Niculițe C, Popescu BO, Ceafalan LC (2021). Potential roles of functional bacterial amyloid proteins, bacterial biosurfactants and other putative gut microbiota products in the etiopathogeny of Parkinson’s disease. BIOCELL 45: 1–16. DOI 10.32604/biocell.2021.013452. [Google Scholar] [CrossRef]
Makela P, Schillinger U, Korkeala H, Holzapfel WH (1992). Classification of ropy slime-producing lactic acid bacteria based on DNA-DNA homology, and identification of Lactobacillus sake and Leuconostoc amelibiosum as dominant spoilage organisms in meat products. International Journal of Food Microbiology 16: 167–172. DOI 10.1016/0168-1605(92)90011-Q. [Google Scholar] [CrossRef]
Marciniec B, Kujawa E, Ogrodowczyk M (1992). Evaluation of nifedipine preparations by chromatographic-spectrophotometric methods. Die Pharmazie 47: 502–504. [Google Scholar]
Martin FP, Dumas ME, Wang Y, Legido-Quigley C, Yap IK et al. (2007). A top-down systems biology view of microbiome-mammalian metabolic interactions in a mouse model. Molecular Systems Biology 3: 112. DOI 10.1038/msb4100153. [Google Scholar] [CrossRef]
Marzouki S, Ben Ahmed M, Boussoffara T, Abdeladhim M, Ben Aleya-Bouafif N, Namane A, Hamida NB, Ben Salah A, Louzir H (2011). Characterization of the antibody response to the saliva of Phlebotomus papatasi in people living in endemic areas of cutaneous leishmaniasis. The American Journal of Tropical Medicine and Hygiene 84: 653–661. DOI 10.4269/ajtmh.2011.10-0598. [Google Scholar] [CrossRef]
Mashego MR, Rumbold K, de Mey M, Vandamme E, Soetaert W, Heijnen JJ (2007). Microbial metabolomics: Past, present and future methodologies. Biotechnology Letters 29: 1–16. DOI 10.1007/s10529-006-9218-0. [Google Scholar] [CrossRef]
Mayta-Apaza AC, Pottgen E, Bodt DE, Papp J, Marasini N et al. (2018). Impact of tart cherries polyphenols on the human gut microbiota and phenolic metabolites in vitro and in vivo. The Journal of Nutritional Biochemistry 59: 160–172. DOI 10.1016/j.jnutbio.2018.04.001. [Google Scholar] [CrossRef]
Meng C, Bai C, Brown TD, Hood LE, Tian Q (2018). Human gut microbiota and gastrointestinal cancer. Genomics Proteomics Bioinformatics 16: 33–49. DOI 10.1016/j.gpb.2017.06.002. [Google Scholar] [CrossRef]
Migne C, Durand S, Pujos-Guillot E (2018). Exploratory GC/MS-based metabolomics of body fluids. Methods in Molecular Biology 1730: 239–246. DOI 10.1007/978-1-4939-7592-1. [Google Scholar] [CrossRef]
Mojica ER, Kim S, Aga DS (2008). Formation of N-ethylmaleimide (NEM)-glutathione conjugate and N-ethylmaleamic acid revealed by mass spectral characterization of intracellular and extracellular microbial metabolites of NEM. Applied and Environmental Microbiology 74: 323–326. DOI 10.1128/AEM.01407-07. [Google Scholar] [CrossRef]
Mombelli A (2018). Microbial colonization of the periodontal pocket and its significance for periodontal therapy. Periodontology 2000: 85–96. DOI 10.1111/prd.12147. [Google Scholar] [CrossRef]
Mondot S, Lepage P (2016). The human gut microbiome and its dysfunctions through the meta-omics prism. Annals of the New York Academy of Sciences 1372: 9–19. [Google Scholar]
Montenegro-Burke JR, Aisporna AE, Benton HP, Rinehart D, Fang M et al. (2017). Data streaming for metabolomics: Accelerating data processing and analysis from days to minutes. Analytical Chemistry 89: 1254–1259. DOI 10.1021/acs.analchem.6b03890. [Google Scholar] [CrossRef]
Moreno CG, Luque AT, Galvao KN, Otero MC (2022). Bacterial communities from vagina of dairy healthy heifers and cows with impaired reproductive performance. Research in Veterinary Science 142: 15–23. DOI 10.1016/j.rvsc.2021.11.007. [Google Scholar] [CrossRef]
Mousavi F, Bojko B, Pawliszyn J (2019). High-throughput solid-phase microextraction-liquid chromatography-mass spectrometry for microbial untargeted metabolomics. Methods in Molecular Biology 1859: 133–152. DOI 10.1007/978-1-4939-8757-3. [Google Scholar] [CrossRef]
Murovec B, Makuc D, Kolbl Repinc S, Prevorsek Z, Zavec D, Sket R, Pecnik K, Plavec J, Stres B (2018). (1)H NMR metabolomics of microbial metabolites in the four MW agricultural biogas plant reactors: A case study of inhibition mirroring the acute rumen acidosis symptoms. Journal of Environmental Management 222: 428–435. DOI 10.1016/j.jenvman.2018.05.068. [Google Scholar] [CrossRef]
Myers OD, Sumner SJ, Li S, Barnes S, Du X (2017). Detailed investigation and comparison of the XCMS and MZmine 2 chromatogram construction and chromatographic peak detection methods for preprocessing mass spectrometry metabolomics data. Analytical Chemistry 89: 8689–8695. DOI 10.1021/acs.analchem.7b01069. [Google Scholar] [CrossRef]
Na E, Lim SY (2022). Effect of Lycopus lucidus Turcz. supplementation on gut microflora and short chain fatty acid composition in Crj: CD-1 mice. BIOCELL 46: 2101–2109. DOI 10.32604/biocell.2022.019807. [Google Scholar] [CrossRef]
Nagai M (2015). Gut microbiota and internal diseases: Update information. Topics: V. Gut microbiota: Topics in various medical fields; 3. Does the intestinal flora relate to nervous system disorders? Nihon Naika Gakkai Zasshi 104: 75–80. DOI 10.2169/naika.104.75. [Google Scholar] [CrossRef]
Nandakumar MP, Palsson E, Gustavsson PE, Larsson PO, Mattiasson B (2000). Superporous agarose monoliths as mini-reactors in flow injection systems. On-line monitoring of metabolites and intracellular enzymes in microbial cultivation processes. Bioseparation 9: 193–202. DOI 10.1023/A:1008117827057. [Google Scholar] [CrossRef]
Nielsen KF, Smedsgaard J (2003). Fungal metabolite screening: Database of 474 mycotoxins and fungal metabolites for dereplication by standardised liquid chromatography-UV-mass spectrometry methodology. Journal of Chromatography A 1002: 111–136. DOI 10.1016/S0021-9673(03)00490-4. [Google Scholar] [CrossRef]
Njumbe Ediage E, van Poucke C, de Saeger S (2015). A multi-analyte LC-MS/MS method for the analysis of 23 mycotoxins in different sorghum varieties: The forgotten sample matrix. Food Chemistry 177: 397–404. DOI 10.1016/j.foodchem.2015.01.060. [Google Scholar] [CrossRef]
Ortiz-Villanueva E, Benavente F, Pina B, Sanz-Nebot V, Tauler R, Jaumot J (2017). Knowledge integration strategies for untargeted metabolomics based on MCR-ALS analysis of CE-MS and LC-MS data. Analytica Chimica Acta 978: 10–23. DOI 10.1016/j.aca.2017.04.049. [Google Scholar] [CrossRef]
Pisano MB, Scano P, Murgia A, Cosentino S, Caboni P (2016). Metabolomics and microbiological profile of Italian mozzarella cheese produced with buffalo and cow milk. Food Chemistry 192: 618–624. DOI 10.1016/j.foodchem.2015.07.061. [Google Scholar] [CrossRef]
Ponnusamy K, Lee S, Lee CH (2013). Time-dependent correlation of the microbial community and the metabolomics of traditional barley nuruk starter fermentation. Bioscience, Biotechnology, and Biochemistry 77: 683–690. DOI 10.1271/bbb.120665. [Google Scholar] [CrossRef]
Probert CS, Jones PR, Ratcliffe NM (2004). A novel method for rapidly diagnosing the causes of diarrhoea. Gut 53: 58–61. DOI 10.1136/gut.53.1.58. [Google Scholar] [CrossRef]
Putri SP, Ikram MMM, Sato A, Dahlan HA, Rahmawati D, Ohto Y, Fukusaki E (2022). Application of gas chromatography-mass spectrometry-based metabolomics in food science and technology. Journal of Bioscience and Bioengineering 133: 425–435. DOI 10.1016/j.jbiosc.2022.01.011. [Google Scholar] [CrossRef]
Qian M, Zhang H, Wu L, Jin N, Wang J, Jiang K (2015). Simultaneous determination of zearalenone and its derivatives in edible vegetable oil by gel permeation chromatography and gas chromatography-triple quadrupole mass spectrometry. Food Chemistry 166: 23–28. DOI 10.1016/j.foodchem.2014.05.133. [Google Scholar] [CrossRef]
Rafiei M, Kiani F, Sayehmiri K, Sayehmiri F, Tavirani M, Dousti M, Sheikhi A (2018). Prevalence of anaerobic bacteria (P.gingivalis) as major microbial agent in the incidence periodontal diseases by meta-analysis. Journal of Bioscience and Bioengineering 19: 232–242. [Google Scholar]
Raman M, Ahmed I, Gillevet PM, Probert CS, Ratcliffe NM et al. (2013). Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clinical Gastroenterology and Hepatology 11: 868–875. DOI 10.1016/j.cgh.2013.02.015. [Google Scholar] [CrossRef]
Ramos ER, Santos RA, Velazquez E, Velezmoro CE, Zuniga DE (2018). Genetic diversity and antimicrobial activity of lactic acid bacteria in the preparation of traditional fermented potato product ‘tunta’. World Journal of Microbiology and Biotechnology 34: 144. DOI 10.1007/s11274-018-2525-5. [Google Scholar] [CrossRef]
Reichert B, de Kok A, Pizzutti IR, Scholten J, Cardoso CD, Spanjer M (2018). Simultaneous determination of 117 pesticides and 30 mycotoxins in raw coffee, without clean-up, by LC-ESI-MS/MS analysis. Analytica Chimica Acta 1004: 40–50. DOI 10.1016/j.aca.2017.11.077. [Google Scholar] [CrossRef]
Renes E, Ladero V, Tornadijo ME, Fresno JM (2019). Production of sheep milk cheese with high gamma-aminobutyric acid and ornithine concentration and with reduced biogenic amines level using autochthonous lactic acid bacteria strains. Food Microbiology 78: 1–10. DOI 10.1016/j.fm.2018.09.003. [Google Scholar] [CrossRef]
Rezazadeh A, Shahabi S, Bagheri M, Nabizadeh E, Jazani NH (2018). The protective effect of Lactobacillus and Bifidobacterium as the gut microbiota members against chronic urticaria. International Immunopharmacology 59: 168–173. DOI 10.1016/j.intimp.2018.04.007. [Google Scholar] [CrossRef]
Ribeiro AA, Azcarate-Peril MA, Cadenas MB, Butz N, Paster BJ, Chen T, Bair E, Arnold RR (2017). The oral bacterial microbiome of occlusal surfaces in children and its association with diet and caries. PLoS One 12: e0180621. DOI 10.1371/journal.pone.0180621. [Google Scholar] [CrossRef]
Rossi M, Aggio R, Staudacher HM, Lomer MC, Lindsay JO, Irving P, Probert C, Whelan K (2018). Volatile organic compounds in feces associate with response to dietary intervention in patients with irritable bowel syndrome. Clinical Gastroenterology and Hepatology 16: 385–391 e381. DOI 10.1016/j.cgh.2017.09.055. [Google Scholar] [CrossRef]
Salman HA, Venkatesh S, Senthilkumar R, Gnanesh Kumar BS, Ali AM (2018). Determination of antibacterial activity and metabolite profile of Ruta graveolens against Streptococcus mutans and Streptococcus sobrinus. Journal of Laboratory Physicians 10: 320–325. DOI 10.4103/JLP.JLP_160_17. [Google Scholar] [CrossRef]
Sanz M, Beighton D, Curtis MA, Cury JA, Dige I et al. (2017). Role of microbial biofilms in the maintenance of oral health and in the development of dental caries and periodontal diseases. Consensus report of group 1 of the Joint EFP/ORCA workshop on the boundaries between caries and periodontal disease. Journal of Clinical Periodontology 44 Suppl 18: S5–S11. [Google Scholar]
Saric J, Wang Y, Li J, Coen M, Utzinger J et al. (2008). Species variation in the fecal metabolome gives insight into differential gastrointestinal function. Journal of Proteome Research 7: 352–360. DOI 10.1021/pr070340k. [Google Scholar] [CrossRef]
Sato M, Miyagi A, Yoneyama S, Gisusi S, Tokuji Y, Kawai-Yamada M (2017). CE-MS-based metabolomics reveals the metabolic profile of maitake mushroom (Grifola frondosa) strains with different cultivation characteristics. Bioscience, Biotechnology, and Biochemistry 81: 2314–2322. DOI 10.1080/09168451.2017.1387049. [Google Scholar] [CrossRef]
Sebastian Domingo JJ, Sanchez Sanchez C (2018). From the intestinal flora to the microbiome. Revista Espanola de Enfermedades Digestivas 110: 51–56. [Google Scholar]
Sevastos A, Kalampokis IF, Panagiotopoulou A, Pelecanou M, Aliferis KA (2018). Implication of Fusarium graminearum primary metabolism in its resistance to benzimidazole fungicides as revealed by 1H NMR metabolomics. Pesticide Biochemistry and Physiology 148: 50–61. DOI 10.1016/j.pestbp.2018.03.015. [Google Scholar] [CrossRef]
Shakhatreh MAK, Khabour OF, Alzoubi KH, Masadeh MM, Hussein EI, Bshara GN (2018). Alterations in oral microbial flora induced by waterpipe tobacco smoking. International Journal of General Medicine 11: 47–54. DOI 10.2147/IJGM. [Google Scholar] [CrossRef]
Shaw BG, Puckey DJ, Macfie HJ, Bolt SJ (1985). Classification of some lactic acid bacteria from vacuum-packed meats by direct probe mass spectrometry. Journal of Applied Bacteriology 59: 157–165. DOI 10.1111/j.1365-2672.1985.tb03316.x. [Google Scholar] [CrossRef]
Shi X, Wei X, Yin X, Wang Y, Zhang M, Zhao C, Zhao H, Mcclain CJ, Feng W, Zhang X (2015). Hepatic and fecal metabolomic analysis of the effects of Lactobacillus rhamnosus GG on alcoholic fatty liver disease in mice. Journal of Proteome Research 14: 1174–1182. DOI 10.1021/pr501121c. [Google Scholar] [CrossRef]
Si YC, Miao WN, He JY, Chen L, Wang YL, Ding WJ (2018). Regulating gut flora dysbiosis in obese mice by electroacupuncture. The American Journal of Chinese Medicine 46: 1–17. DOI 10.1142/S0192415X18500763. [Google Scholar] [CrossRef]
Sirichokchatchawan W, Pupa P, Praechansri P, AM-In N, Tanasupawat S, Sonthayanon P, Prapasarakul N (2018). Autochthonous lactic acid bacteria isolated from pig faeces in Thailand show probiotic properties and antibacterial activity against enteric pathogenic bacteria. Microbial Pathogenesis 119: 208–215. DOI 10.1016/j.micpath.2018.04.031. [Google Scholar] [CrossRef]
Smolyanskaya OA, Schelkanova IJ, Kulya MS, Odlyanitskiy EL, Goryachev IS, Tcypkin AN, Grachev YV, Toropova YG, Tuchin VV (2018). Glycerol dehydration of native and diabetic animal tissues studied by THz-TDS and NMR methods. Biomedical Optics Express 9: 1198–1215. DOI 10.1364/BOE.9.001198. [Google Scholar] [CrossRef]
Soga T, Ishikawa T, Igarashi S, Sugawara K, Kakazu Y, Tomita M (2007). Analysis of nucleotides by pressure-assisted capillary electrophoresis-mass spectrometry using silanol mask technique. Journal of Chromatography A 1159: 125–133. DOI 10.1016/j.chroma.2007.05.054. [Google Scholar] [CrossRef]
Soga T, Ohashi Y, Ueno Y, Naraoka H, Tomita M, Nishioka T (2003). Quantitative metabolome analysis using capillary electrophoresis mass spectrometry. Journal of Proteome Research 2: 488–494. DOI 10.1021/pr034020m. [Google Scholar] [CrossRef]
Son HS, Hwang GS, Kim KM, Kim EY, van den Berg F, Park WM, Lee CH, Hong YS (2009). 1H NMR-based metabolomic approach for understanding the fermentation behaviors of wine yeast strains. Analytical Chemistry 81: 1137–1145. DOI 10.1021/ac802305c. [Google Scholar] [CrossRef]
Song H, Wang W, Shen B, Jia H, Hou Z, Chen P, Sun Y (2018). Pretreatment with probiotic Bifico ameliorates colitis-associated cancer in mice: Transcriptome and gut flora profiling. Cancer Science 109: 666–677. DOI 10.1111/cas.13497. [Google Scholar] [CrossRef]
Sumner LW, Styczynski M, Mclean J, Fiehn O, Jander G et al. (2015). Introducing the USA plant, algae and microbial metabolomics research coordination network (PAMM-NET). Metabolomics 11: 3–5. DOI 10.1007/s11306-014-0755-6. [Google Scholar] [CrossRef]
Suzuki-Iwashima A, Matsuura H, Iwasawa A, Shiota M (2020). Metabolomics analyses of the combined effects of lactic acid bacteria and Penicillium camemberti on the generation of volatile compounds in model mold-surface-ripened cheeses. Journal of Bioscience and Bioengineering 129: 333–347. DOI 10.1016/j.jbiosc.2019.09.005. [Google Scholar] [CrossRef]
Swann JR, Garcia-Perez I, Braniste V, Wilson ID, Sidaway JE, Nicholson JK, Pettersson S, Holmes E (2017). Application of (1)H NMR spectroscopy to the metabolic phenotyping of rodent brain extracts: A metabonomic study of gut microbial influence on host brain metabolism. Journal of Pharmaceutical and Biomedical Analysis 143: 141–146. DOI 10.1016/j.jpba.2017.05.040. [Google Scholar] [CrossRef]
Takahashi N, Washio J (2011). Metabolomic effects of xylitol and fluoride on plaque biofilm in vivo. Journal of Dental Research 90: 1463–1468. DOI 10.1177/0022034511423395. [Google Scholar] [CrossRef]
Takahashi N, Washio J, Mayanagi G (2010). Metabolomics of supragingival plaque and oral bacteria. Journal of Dental Research 89: 1383–1388. DOI 10.1177/0022034510377792. [Google Scholar] [CrossRef]
Tan KC, Trengove RD, Maker GL, Oliver RP, Solomon PS (2009). Metabolite profiling identifies the mycotoxin alternariol in the pathogen Stagonospora nodorum. Metabolomics 5: 330–335. DOI 10.1007/s11306-009-0158-2. [Google Scholar] [CrossRef]
Teleki A, Takors R (2019). Quantitative profiling of endogenous metabolites using hydrophilic interaction liquid chromatography-tandem mass spectrometry (HILIC-MS/MS). Methods in Molecular Biology 1859: 185–207. DOI 10.1007/978-1-4939-8757-3. [Google Scholar] [CrossRef]
Teoh ST, Kitamura M, Nakayama Y, Putri S, Mukai Y, Fukusaki E (2016). Random sample consensus combined with partial least squares regression (RANSAC-PLS) for microbial metabolomics data mining and phenotype improvement. Journal of Bioscience and Bioengineering 122: 168–175. DOI 10.1016/j.jbiosc.2016.01.007. [Google Scholar] [CrossRef]
Toshimitsu T, Gotou A, Furuichi K, Hachimura S, Asami Y (2018). Effects of 12-wk Lactobacillus plantarum OLL2712 treatment on glucose metabolism and chronic inflammation in prediabetic individuals: A single-arm pilot study. Nutrition 58: 175–180. DOI 10.1016/j.nut.2018.07.116. [Google Scholar] [CrossRef]
Tulipani S, Llorach R, Jauregui O, Lopez-Uriarte P, Garcia-Aloy M, Bullo M, Salas-Salvado J, Andres-Lacueva C (2011). Metabolomics unveils urinary changes in subjects with metabolic syndrome following 12-week nut consumption. Journal of Proteome Research 10: 5047–5058. DOI 10.1021/pr200514h. [Google Scholar] [CrossRef]
van der Werf MJ, Jellema RH, Hankemeier T (2005). Microbial metabolomics: Replacing trial-and-error by the unbiased selection and ranking of targets. Journal of Industrial Microbiology & Biotechnology 32: 234–252. DOI 10.1007/s10295-005-0231-4. [Google Scholar] [CrossRef]
Vlachojannis C, Chrubasik-Hausmann S, Hellwig E, Vach K, AL-Ahmad A (2018). Activity of preparations from Spilanthes oleracea, propolis, Nigella sativa, and black garlic on different microorganisms involved in oral diseases and on total human salivary bacteria: A pilot study. Phytotherapy Research 32: 1992–2001. DOI 10.1002/ptr.6129. [Google Scholar] [CrossRef]
Wang HB, Feng YR, Gui SQ, Zhang Y, Lu FP (2017a). A sample pretreatment method to suit the metabolomic analysis of Bacillus licheniformis based on GC-MS. Analytical Methods 9: 2299–2304. DOI 10.1039/C7AY00008A. [Google Scholar] [CrossRef]
Wang X, Wang J, Rao B, Deng L (2017b). Gut flora profiling and fecal metabolite composition of colorectal cancer patients and healthy individuals. Experimental and Therapeutic Medicine 13: 2848–2854. DOI 10.3892/etm.2017.4367. [Google Scholar] [CrossRef]
Wang X, Xie Y, Gao P, Zhang S, Tan H, Yang F, Lian R, Tian J, Xu G (2014). A metabolomics-based method for studying the effect of yfcC gene in Escherichia coli on metabolism. Analytical Biochemistry 451: 48–55. DOI 10.1016/j.ab.2014.01.018. [Google Scholar] [CrossRef]
Wang Y, Lawler D, Larson B, Ramadan Z, Kochhar S, Holmes E, Nicholson JK (2007). Metabonomic investigations of aging and caloric restriction in a life-long dog study. Journal of Proteome Research 6: 1846–1854. DOI 10.1021/pr060685n. [Google Scholar] [CrossRef]
Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS et al. (2011). Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472: 57–63. DOI 10.1038/nature09922. [Google Scholar] [CrossRef]
Wiewiora B, Zurek G, Panka D (2015). Is the vertical transmission of Neotyphodium lolii in perennial ryegrass the only possible way to the spread of endophytes? PLoS One 10: e0117231. DOI 10.1371/journal.pone.0117231. [Google Scholar] [CrossRef]
Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, Siuzdak G (2009). Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. PNAS 106: 3698–3703. DOI 10.1073/pnas.0812874106. [Google Scholar] [CrossRef]
Wu Z, Lu X, Chen F, Dai X, Ye Y, Yan Y, Liao L (2018). Estimation of early postmortem interval in rats by GC-MS-based metabolomics. Legal Medicine 31: 42–48. DOI 10.1016/j.legalmed.2017.12.014. [Google Scholar] [CrossRef]
Xing Z, Tang W, Yang Y, Geng W, Rehman RU, Wang Y (2018). Colonization and gut flora modulation of lactobacillus kefiranofaciens ZW3 in the intestinal tract of mice. Probiotics and Antimicrobial Proteins 10: 374–382. DOI 10.1007/s12602-017-9288-4. [Google Scholar] [CrossRef]
Xu X, He J, Xue J, Wang Y, Li K et al. (2015). Oral cavity contains distinct niches with dynamic microbial communities. Environmental Microbiology 17: 699–710. DOI 10.1111/1462-2920.12502. [Google Scholar] [CrossRef]
Yang D, Fan X, Shi X, Lian S, Qiao J, Guo R (2014). Metabolomics reveals stage-specific metabolic pathways of microbial communities in two-stage anaerobic fermentation of corn-stalk. Biotechnology Letters 36: 1461–1468. DOI 10.1007/s10529-014-1508-3. [Google Scholar] [CrossRef]
Zhang J, Ma JY, Li QH, Su H, Sun X (2018). Lactobacillus rhamnosus GG induced protective effect on allergic airway inflammation is associated with gut microbiota. Cellular Immunology 332: 77–84. DOI 10.1016/j.cellimm.2018.08.002. [Google Scholar] [CrossRef]
Zhao N, Zhang C, Yang Q, Guo Z, Yang B et al. (2016). Selection of taste markers related to lactic acid bacteria microflora metabolism for chinese traditional paocai: A gas chromatography-mass spectrometry-based metabolomics approach. Journal of Agricultural and Food Chemistry 64: 2415–2422. DOI 10.1021/acs.jafc.5b05332. [Google Scholar] [CrossRef]
Zhao S, Li L (2018). Dansylhydrazine isotope labeling LC-MS for comprehensive carboxylic acid submetabolome profiling. Analytical Chemistry 90: 13514–13522. DOI 10.1021/acs.analchem.8b03435. [Google Scholar] [CrossRef]
Zhao Y, Yu X, Jia R, Yang R, Rui Q, Wang D (2015). Lactic acid bacteria protects caenorhabditis elegans from toxicity of graphene oxide by maintaining normal intestinal permeability under different genetic backgrounds. Scientific Reports 5: 17233. DOI 10.1038/srep17233. [Google Scholar] [CrossRef]
Zhou Y, Bradshaw RE, Johnson RD, Hume DE, Simpson WR, Schmid J (2014). Detection and quantification of three distinct Neotyphodium lolii endophytes in Lolium perenne by real time PCR of secondary metabolite genes. Fungal Biology 118: 316–324. DOI 10.1016/j.funbio.2014.01.003. [Google Scholar] [CrossRef]
Zhu A, Sunagawa S, Mende DR, Bork P (2015). Inter-individual differences in the gene content of human gut bacterial species. Genome Biology 16: 82. DOI 10.1186/s13059-015-0646-9. [Google Scholar] [CrossRef]
Zhu Z, Wang G, Liu J, Chen Z (2013). Fast and robust 2D-shape extraction using discrete-point sampling and centerline grouping in complex images. IEEE Transactions on Image Processing 22: 4762–4774. DOI 10.1109/TIP.2013.2277824. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools