DOI:10.32604/biocell.2022.020109
| BIOCELL DOI:10.32604/biocell.2022.020109 | |
| Article |
PI3 kinase isoform p110δ is more important than p110α in KIT signaling in hematopoietic cells
1NHC Key Laboratory of Metabolic Cardiovascular Diseases Research, Science and Technology Center, School of Basic Medical Sciences, Ningxia Medical University, Yinchuan, 750004, China
2General Hospital of Ningxia Medical University, Yinchuan, 750004, China
*Address correspondence to: Jianmin Sun, jianmin.sun@nxmu.edu.cn, sunjm@rocketmail.com
Received: 04 November 2021; Accepted: 27 January 2022
Abstract: PI3 kinases are important for KIT signaling and KIT mutants mediated cell transformation. In order to know the difference of PI3 kinase isoforms p110α and p110δ in the signaling of wild-type KIT and the often occurred KIT mutation D816V in hematopoietic malignancy mastocytosis, the predominant PI3 kinase isoform p110δ in hematopoietic tissues was knocked out in hematopoietic cells. We found that loss of p110δ expression dramatically inhibits PI3 kinase activation mediated by both wild-type KIT and KIT/D816V. By over expression of p110α in p110δ knock out cells, wild-type KIT mediated PI3 kinase activation was not changed while over expression of p110δ increased PI3 kinase activation. Similarly, in KIT/D816V expressing cells without p110δ expression, over expression of p110δ but not p110α restored PI3 kinase activation. In agreement with the signaling results, cell proliferation, cell survival and cell cycle assay further showed that over expression of p110δ but not p110α in p110δ knock out cells increases both wild-type KIT and KIT/D816V mediated cell survival and proliferation. These results suggested that p110δ plays a more important role than p110α in KIT signaling and KIT mutant mediated cell transformation in hematopoietic cells.
Keywords: Mastocytosis; Mutation; Cell transformation
KIT is a member of type III receptor tyrosine kinase together with PDGFR, FLT3 and CSF-1R. They have an extracellular domain, a transmembrane domain and an intracellular domain with the kinase activity in common. After the binding to its ligand stem cell factor (SCF) through its extracellular domain, KIT dimerizes and the kinase activity catalyzes the phosphorylation of tyrosine residues which can bind to downstream signaling molecules and further activate a series of signaling cascades to mediate cell survival, proliferation, differentiation and others. In normal situations, KIT plays an important role in hematopoiesis, melanogenesis and gametogenesis (Cardoso et al., 2017).
Gain-of-function mutations of KIT have been identified in around 80% of gastrointestinal stromal tumors (GISTs) (Tarn et al., 2005; Steigen et al., 2007; Zhu et al., 2020) and mastocytosis (Nagata et al., 1995; Longley et al., 1999), and to a lesser extent in acute myeloid leukemia (Boissel et al., 2006) and germ cell tumors (Looijenga et al., 2003). Among all the mutations, D816V mutation in exon 17 of KIT is the most often occurred mutation which accounts for around 70% of mastocytosis but it is rarely found in GISTs. KIT mutations can confer ligand-independent activation of KIT, leading to uncontrolled downstream signaling and finally resulting in cell transformation (Mei et al., 2018; Li, 2021). Among the downstream signaling molecules, PI3 kinases play a key role in KIT mutants mediated cell transformation (Hashimoto et al., 2003; Sun et al., 2014). PI3 kinases are a family of lipid kinases that are classed into three groups according to their structures. Among all subtypes, type IA PI3 kinases are most widely distributed and best studied so far. This type of PI3 kinases have two subunits: one regulatory subunit p85α or p85β, and one catalytic subunit p110α, p110β or p110δ. Among the three catalytic subunits of PI3 kinases, p110α is widely expressed in various tissues while p110β and p110δ expression are limited in certain tissues (Hassan et al., 2013; Bauer et al., 2015).
In this study, we knocked out p110δ which is highly expressed in hematopoietic tissues and then over expressed p110δ or the widely expressed p110α to compare their function in the signaling of wild-type KIT and the most often occurred KIT mutation D816V in hematopoietic malignancy mastocytosis. We found that over expression of p110α cannot compensate the loss of p110δ expression in the signaling of both wild-type KIT and KIT/D816V.
RPMI 1640 medium was from Hyclone, antibodies against pERK, ERK, AKT, p110α, p110β, p110δ and HRP-anti-β-actin antibody were from Santa Cruz Biotechnology (USA), anti-phosphotyrosine antibody 4G10 was from Millipore (USA), anti-KIT antibody was from Biolegend (USA), anti-pAKT, anti-pp38, anti-p38, anti-pSTAT3 and anti-STAT3 antibodies were from Cell Signaling Technology (USA), HRP-anti-rabbit IgG and HRP-anti-murine IgG antibodies, fetal bovine serum, Lipofectamine™ 2000, Dynabeads™ Protein G, targeting p110δ crRNAs (AGTTAATGAGCTTCTTCACG, AGTTGGAGGATGAGCAGCGG) were from Thermo Scientific (USA), penicillin/streptomycin was from Solarbio (China), stem cell factor was from ORF Genetics (Island), Edit-R Cas9 Nuclease protein NLS and tracrRNA were from Dharmacon (USA), solution SG transfection kit was from Lonza (Switzerland).
Ba/F3 cells (DSMZ) were cultured in RPMI 1640 medium plus 10% fetal bovine serum, 10 ng/ml IL-3, 100 IU/ml penicillin and 100 μg/ml streptomycin. EcoPack cells (Clontech) were cultured in DMEM medium plus 10% fetal bovine serum, 100 IU/ml penicillin and 100 μg/ml streptomycin.
Knockout of p110δ in Ba/F3 cells
1 × 106 Ba/F3 cells were washed once with PBS and resuspended in 100 μl solution SG, 150 pmol Edit-R Cas9 Nuclease protein NLS and 300 pmol crRNA: tracrRNA were mixed and incubated for 10 min at room temperature followed by mixture with Ba/F3 cells. Cells were transfected in 4D nucleofector (Lonza, program CM150). After transfection, cells were cultured for 72 h and lysed for examination of p110δ expression. After monoclonization, each cell clone was lysed and p110δ expression was examined. p110δ knockout clone was used in further experiments.
Establishment of KIT or p110 expressing Ba/F3 cells
pMSCVpuro/wt KIT, pMSCVpuro/KIT/D816V, pMSCVneo/p110α or pMSCVneo/p110δ were transfected into EcoPack cells using Lipofectamine™ 2000 according to the manufacturer’s instructions, cell supernatants containing virus were collected to infect Ba/F3 cells followed by selection with puromycin or neomycin respectively. KIT or p110 expression was examined by western blot.
Cell stimulation, immunoprecipitation and western blot
Cell stimulation, immunoprecipitation and western blot were performed as previously described (Sun et al., 2014).
Cell proliferation, cell survival and cell cycle assay
Cell proliferation and cell survival assay were performed as previously described (Sun et al., 2014). For cell cycle analysis, cells were washed twice with PBS and resuspended in 70% ethanol. After washing with PBS twice, cells were incubated with PI/Rnase A for 30 min in the dark followed by flow cytometry analysis.
Data were presented as means ± standard deviations of three independent experiments. The difference of values was analyzed by Student’s t-test. P-values less than 0.05 were considered as statistical significance.
Establishment of p110δ knockout Ba/F3 cells
Ba/F3 cells were transfected with CRISPR-cas9 and crRNA targeting p110δ. Western blot examination showed that p110δ expression is reduced in the two crRNAs transfected Ba/F3 cells, meaning that p110δ expression might be knocked out in some cells. After monoclonization, p110δ expression in 70 clones from each crRNA transfected Ba/F3 cells were examined by western blot. The results showed that 10 clones are negative in p110δ expression, meaning that p110δ expression was knocked out in these clones (Fig. 1). In addition, all the 10 p110δ knockout clones were from one crRNA transfected Ba/F3 cells, indicating a good knockout efficiency of this crRNA (AGTTGGAGGATGAGCAGCGG).
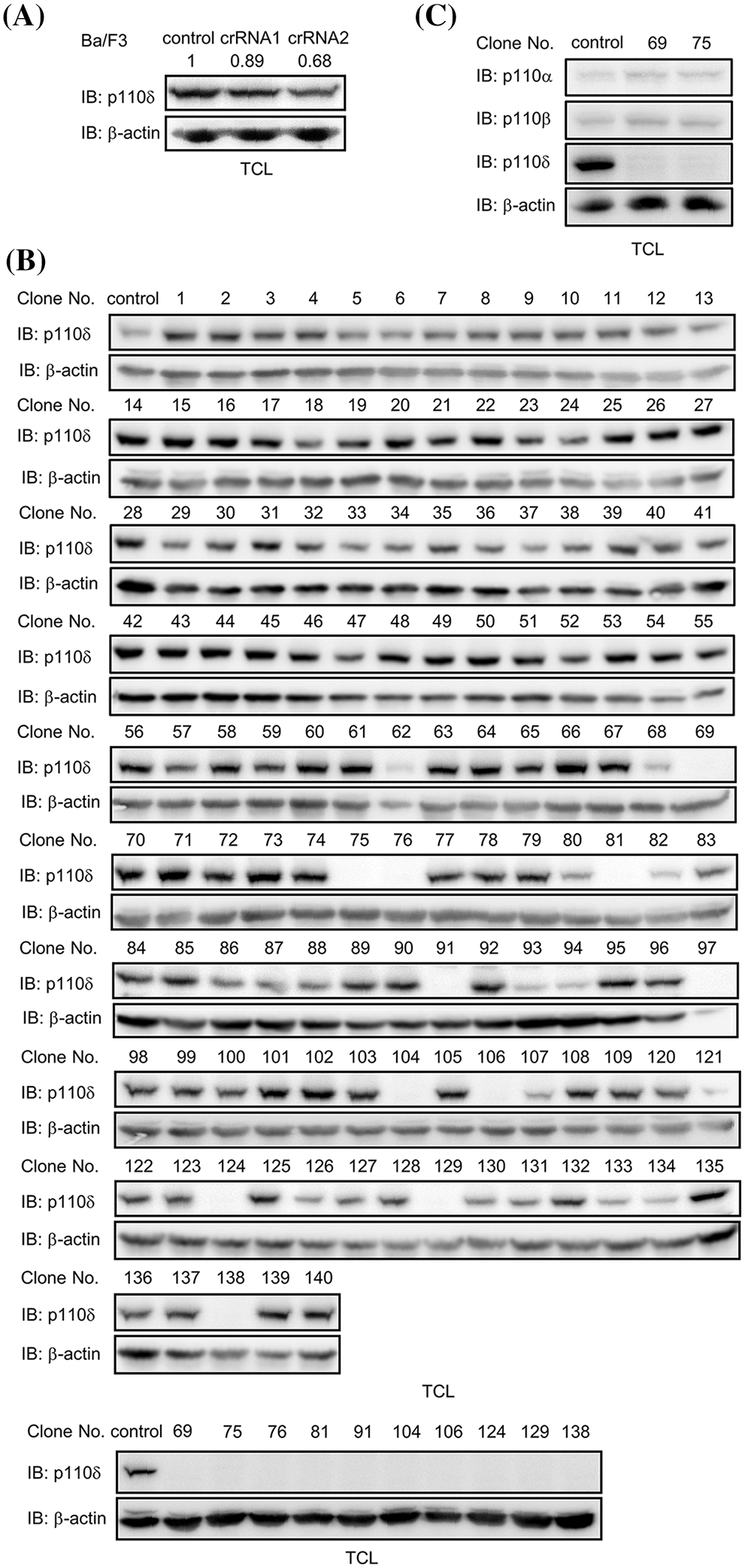
Figure 1: p110δ knockout Ba/F3 cells were established. (A) CRISPR-cas9 and crRNA targeting p110δ were transfected into Ba/F3 cells. After cell lysis and separation by SDS-PAGE, p110δ expression was examined by western blot. Signal intensity of p110δ was quantified and normalized by β-actin. (B) After monoclonization, 70 clones were lysed and p110δ expression was examined by western blot, and p110δ expression was further examined in 10 clones of p110δ knockout Ba/F3 cells. (C) The expression of p110α and p110β was examined in p110δ knockout Ba/F3 cells.
Loss of p110δ expression inhibited PI3 kinase activation mediated by both wild-type KIT and KIT/D816V
p110δ plays an important role in both wild-type KIT and KIT mutants mediated AKT activation which is important for cell survival and proliferation. In p110δ knockout Ba/F3 cells, by expression of wild-type KIT or the most often happened KIT mutation D816V in mastocytosis (Fig. 2A), we found that loss of p110δ expression inhibits both wild-type KIT and KIT/D816V mediated AKT activation, indicating the key role of p110δ in PI3 kinase activity. While loss of p110δ expression slightly inhibited wild-type KIT mediated ERK activation, and KIT/D816V mediated ERK activation was not changed. In addition, KIT/D816V mediated p38 and STAT3 activation was not altered by loss of p110δ expression (Fig. 2B).
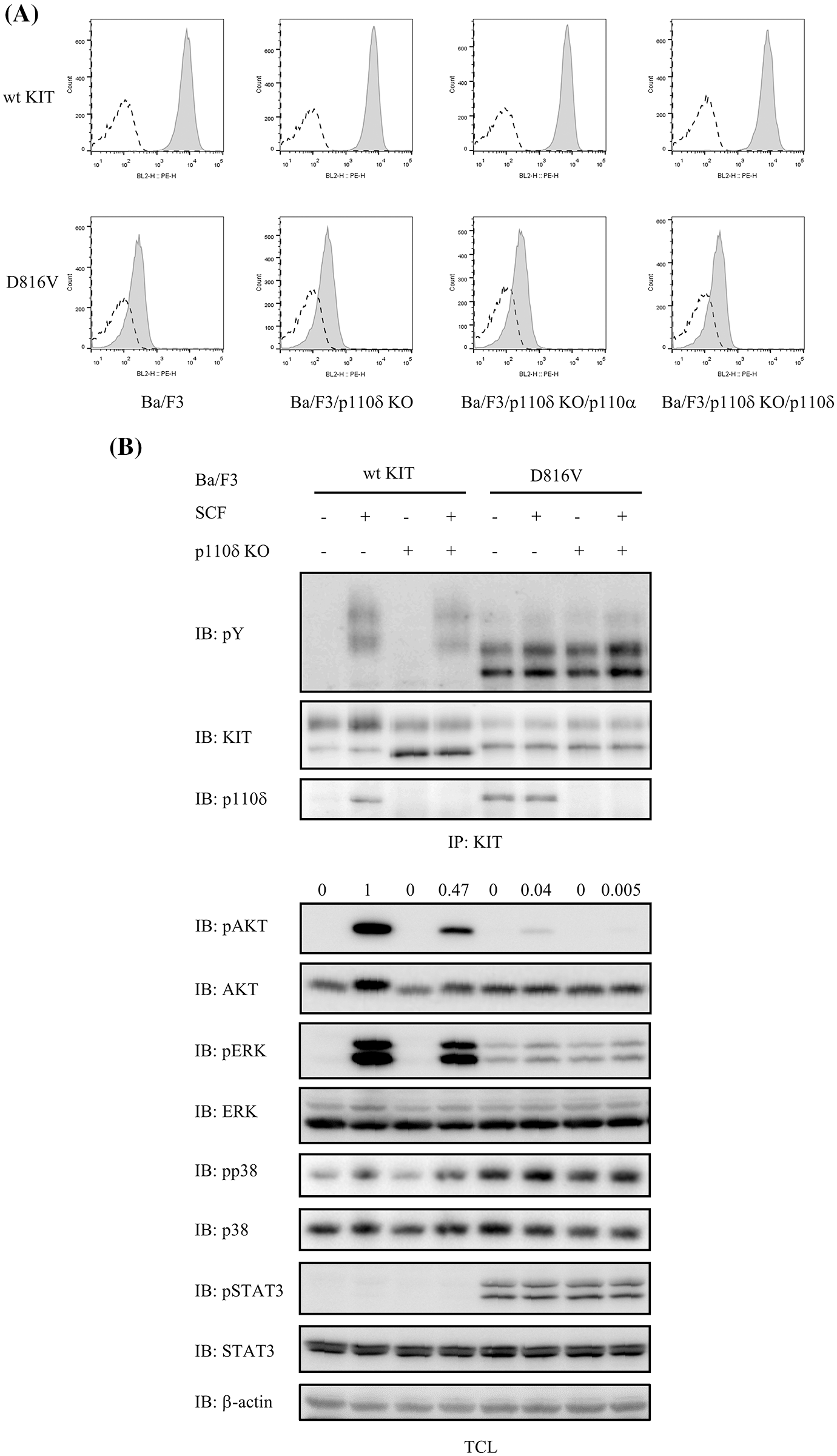
Figure 2: PI3 kinase activation mediated by both wild-type KIT and KIT/D816V was inhibited when p110δ expression was knocked out. (A) Wild-type KIT or KIT/D816V in pMSCVpuro were transfected into EcoPack cells, supernatant was collected to infect Ba/F3 cells. After selection with puromycin, expression of KIT was examined by flow cytometry. Open area: isotype control, grey area: PE-anti-KIT. (B) Ba/F3 cells were washed and starved in RPMI 1640 medium for 4 h before stimulation with 100 ng/ml SCF for 2 min, KIT was pulled down from cell lysate using its antibody, after separation by SDS-PAGE and transfer to PVDF membrane, KIT activation was detected by pY antibody 4G10. Total cell lysates were probed with antibodies against pAKT, AKT, pERK, ERK, pSTAT3, STAT3, pp38 and p38, respectively.
p110δ is more potential than p110α in both wild-type KIT and KIT/D816V mediated AKT activation
PI3 kinase isoform p110α is widely expressed in various tissues while p110δ is highly expressed in hematopoietic tissues. In order to compare the two PI3 kinase isoforms in the downstream signaling of both wild-type KIT and KIT/D816V, p110α or p110δ were transfected into p110δ knockout Ba/F3 cells. We found that p110δ but not p110α increased both wild-type KIT and KIT/D816V mediated AKT activation, suggesting a more important role of p110δ than p110α in KIT signaling (Fig. 3).
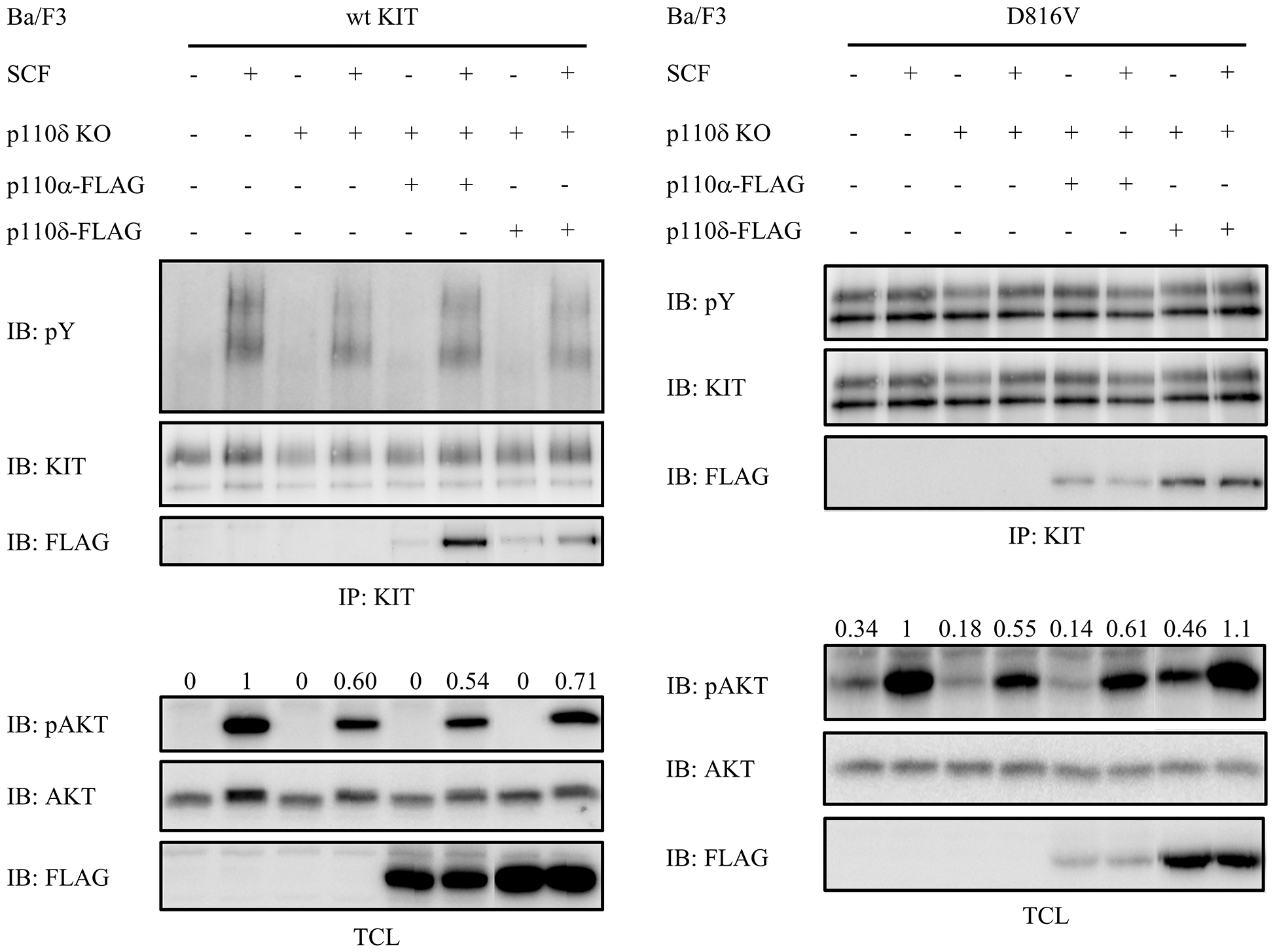
Figure 3: Compared with p110α, p110δ is more important for both wild-type KIT and KIT/D816V mediated AKT activation. Ba/F3 cells were washed and starved in RPMI 1640 medium for 4 h before stimulation with 100 ng/ml SCF for 2 min, KIT was pulled down from cell lysate using KIT antibody, after separation by SDS-PAGE and transfer to PVDF membrane, KIT activation was detected by pY antibody 4G10. Total cell lysates were probed with antibodies against pAKT and AKT, respectively.
p110δ is more potential than p110α in both wild-type KIT and KIT/D816V mediated cell proliferation and survival
Due to the important role of p110δ in both wild-type KIT and KIT/D816V mediated PI3 kinase activity, and the important role of PI3 kinase in KIT/D816V mediated cell transformation, we further compared the role of p110α and p110δ in both wild-type KIT and KIT/D816V mediated cell proliferation, survival and cell cycle. The results showed that loss of p110δ expression inhibits both wild-type KIT and KIT/D816V mediated cell proliferation and cell cycle progression, while over expression of p110δ but not p110α increases cell proliferation and cell cycle progress. Similar as that, loss of p110δ expression inhibited both wild-type KIT and KIT/D816V mediated cell survival, and which can be rescued by over expression of p110δ but not p110α (Fig. 4). These results suggested that p110δ plays a more important role than p110α in both wild-type KIT and KIT/D816V mediated cell proliferation and survival.
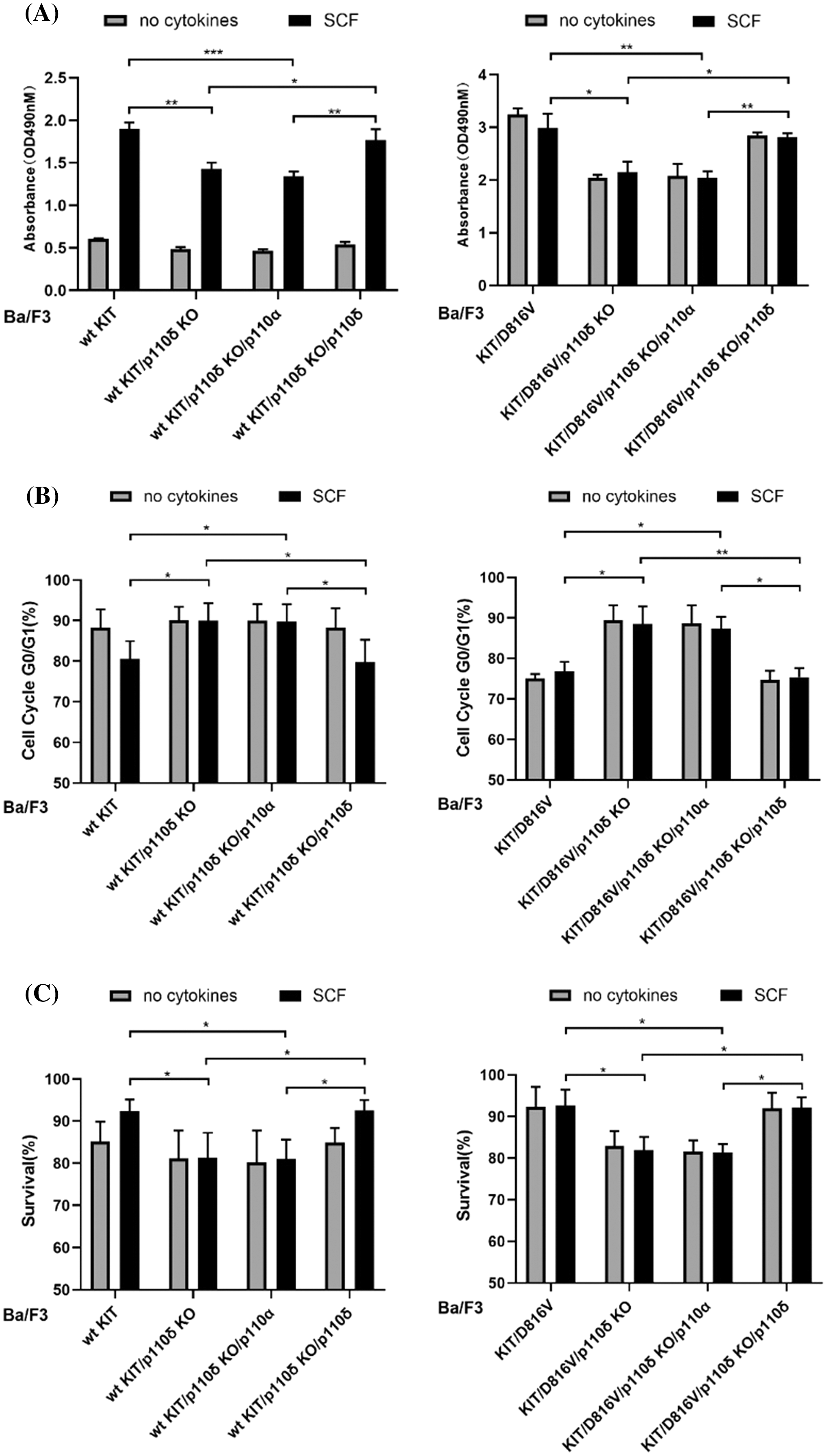
Figure 4: p110δ is more potential than p110α in both wild-type KIT and KIT/D816V mediated cell proliferation and survival. Ba/F3 cells were washed with PBS for three times, and cultured for 48 h in RPMI 1640 medium plus 10% fetal bovine serum, 100 IU/ml penicillin and 100 μg/ml streptomycin with or without 100 ng/mL SCF. (A) CCK-8 assay analyzed cell viability. (B) Cells were washed with PBS and treated with 70% ethanol, after staining with PI, cell cycle distribution was examined by flow cytometry. (C) Cells were stained with annexin V-PE and 7-AAD, apoptotic cells were examined by flow cytometry. Data were presented as mean ± SD, *p < 0.05, **p < 0.01.
Gain-of-function KIT mutations are the main gene mutations in GISTs and mastocytosis, accounting for around 70% of both malignancies. However, the distributions of these mutations in the different regions of KIT differ very much between the two malignancies. KIT mutations in GISTs mainly map to exon 11 of KIT, and to a lesser extent in exon 9, 13 and 17, and these mutations usually respond well to KIT inhibitor such as Imatinib, therefore targeted therapy of GISTs with KIT inhibitors can dramatically improve the treatment outcome of GISTs (Yan et al., 2015). Unlike that in GISTs, KIT mutations in mastocytosis are dominated by D816V mutation in exon 17. KIT/D816V is resistant to Imatinib and there is no proved targeted therapy of mastocytosis currently (Li, 2021). GISTs are mesenchymal derived tissues and mastocytosis is derived from hematopoietic tissues, different cellular context in these two tissues might play a role in the different KIT mutant distribution and KIT mutant mediated cell transformation. PI3 kinases play an important role in KIT signaling, and it has been showed that PI3 kinase isoform p110δ is necessary for KIT/D816V mediated cell transformation (Sun et al., 2014). In this study, we compared the widely expressed PI3 kinase isoform p110α and p110δ which is highly expressed in hematopoietic tissues in the signal transduction of both wild-type KIT and the most often KIT mutation D816V in mastocytosis. We found that p110δ is more potential than p110α in both wild-type KIT and KIT/D816V mediated PI3 kinase activation. Accordingly, p110δ plays a more important role than p110α in the cell proliferation and survival. Since p110δ is highly expressed in hematopoietic cells and D816V mutation of KIT mainly occur in hematopoietic malignancy mastocytosis, these results might partially explain the tissue specific distribution of KIT mutations in different malignancies.
Activity of PI3 kinases is important for cell survival and proliferation, and over activation of PI3 kinases has been widely identified in various malignancies (Mishra et al., 2021). Multiple pan-PI3 kinase inhibitors have been developed and some of them, for example copanlisib, have been proved in the clinical use of cancer treatment (Markham, 2017; Mezynski et al., 2021; Mishra et al., 2021). In addition to the pan-PI3 kinase inhibitors, subtype specific PI3 kinase inhibitors were developed to inhibit certain subtype of PI3 kinase in order to reduce the side effect (Handl et al., 2021; Mishra et al., 2021), and p110δ inhibitor delalisib has been proved for clinical use (Miller et al., 2015), therefore it is important to elucidate the key PI3 kinase isoform that contributes to the cell transformation. In the current study, the role of the wildly expressed PI3 kinase isoform p110α and p110δ that is highly expressed in hematopoietic cells were compared in the signal transduction of KIT and KIT mediated cell response, the results suggested a more important role of p110δ than p110α in the signaling of both wild-type KIT and KIT/D816V, indicating that KIT/D816V might utilize the high expressed p110δ to transform hematopoietic cells. These results make it rational to further test subtype specific PI3 kinase inhibitor in KIT related malignancies.
Availability of Data and Materials: All data generated or analyzed during this study are included in this published article.
Author Contribution: LZ run the experiments; SZ, ZF, ZJ, AL and SL analyzed the data; JS designed the project; all authors drafted and revised the manuscript.
Funding Statement: This work is supported by National Natural Science Foundation of China (82160521), Natural Science Foundation of Ningxia Province (2018A0089) and Key Research and Development Program of Ningxia Province (2019BEH03003).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Bauer TM, Patel MR, Infante JR (2015). Targeting PI3 kinase in cancer. Pharmacology & Therapeutics 146: 53–60. DOI 10.1016/j.pharmthera.2014.09.006. [Google Scholar] [CrossRef]
Boissel N, Leroy H, Brethon B, Philippe N, de Botton S et al. (2006). Incidence and prognostic impact of c-Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML). Leukemia 20: 965–970. DOI 10.1038/sj.leu.2404188. [Google Scholar] [CrossRef]
Cardoso HJ, Figueira MI, Socorro S (2017). The stem cell factor (SCF)/c-KIT signalling in testis and prostate cancer. Journal of Cell Communication and Signaling 11: 297–307. DOI 10.1007/s12079-017-0399-1. [Google Scholar] [CrossRef]
Handl SF, von Heydebrand F, Voelkl S, Oostendorp RAJ, Wilke J, Kremer AN, Mackensen A, Lutzny-Geier G (2021). Immune modulatory effects of Idelalisib in stromal cells of chronic lymphocytic leukemia. Leukemia & Lymphoma 62: 2679–2689. DOI 10.1080/10428194.2021.1927019. [Google Scholar] [CrossRef]
Hashimoto K, Matsumura I, Tsujimura T, Kim DK, Ogihara H et al. (2003). Necessity of tyrosine 719 and phosphatidylinositol 3’-kinase-mediated signal pathway in constitutive activation and oncogenic potential of c-kit receptor tyrosine kinase with the Asp814Val mutation. Blood 101: 1094–1102. DOI 10.1182/blood-2002-01-0177. [Google Scholar] [CrossRef]
Hassan B, Akcakanat A, Holder AM, Meric-Bernstam F (2013). Targeting the PI3-kinase/Akt/mTOR signaling pathway. Surgical Oncology Clinics of North America 22: 641–664. DOI 10.1016/j.soc.2013.06.008. [Google Scholar] [CrossRef]
Li Z (2021). New insights into the pathogenesis of systemic mastocytosis. International Journal of Molecular Sciences 22: 4900. DOI 10.3390/ijms22094900. [Google Scholar] [CrossRef]
Longley BJJr., Metcalfe DD, Tharp M, Wang X, Tyrrell L, Lu SZ, Heitjan D, Ma Y (1999). Activating and dominant inactivating c-KIT catalytic domain mutations in distinct clinical forms of human mastocytosis. Proceedings of the National Academy of Sciences of the United States of America 96: 1609–1614. DOI 10.1073/pnas.96.4.1609. [Google Scholar] [CrossRef]
Looijenga LH, de Leeuw H, van Oorschot M, van Gurp RJ, Stoop H et al. (2003). Stem cell factor receptor (c-KIT) codon 816 mutations predict development of bilateral testicular germ-cell tumors. Cancer Research 63: 7674–7678. [Google Scholar]
Markham A (2017). Copanlisib: First global approval. Drugs 77: 2057–2062. DOI 10.1007/s40265-017-0838-6. [Google Scholar] [CrossRef]
Mei L, Smith SC, Faber AC, Trent J, Grossman SR, Stratakis CA, Boikos SA (2018). Gastrointestinal stromal tumors: The GIST of precision medicine. Trends in Cancer 4: 74–91. DOI 10.1016/j.trecan.2017.11.006. [Google Scholar] [CrossRef]
Mezynski MJ, Farrelly AM, Cremona M, Carr A, Morgan C et al. (2021). Targeting the PI3K and MAPK pathways to improve response to HER2-targeted therapies in HER2-positive gastric cancer. Journal of Translational Medicine 19: 184. DOI 10.1186/s12967-021-02842-1. [Google Scholar] [CrossRef]
Miller BW, Przepiorka D, de Claro RA, Lee K, Nie L et al. (2015). FDA approval: Idelalisib monotherapy for the treatment of patients with follicular lymphoma and small lymphocytic lymphoma. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 21: 1525–1529. DOI 10.1158/1078-0432.CCR-14-2522. [Google Scholar] [CrossRef]
Mishra R, Patel H, Alanazi S, Kilroy MK, Garrett JT (2021). PI3K inhibitors in cancer: Clinical implications and adverse effects. International Journal of Molecular Sciences 22: 3464. DOI 10.3390/ijms22073464. [Google Scholar] [CrossRef]
Nagata H, Worobec AS, Oh CK, Chowdhury BA, Tannenbaum S, Suzuki Y, Metcalfe DD (1995). Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proceedings of the National Academy of Sciences of the United States of America 92: 10560–10564. DOI 10.1073/pnas.92.23.10560. [Google Scholar] [CrossRef]
Steigen SE, Eide TJ, Wasag B, Lasota J, Miettinen M (2007). Mutations in gastrointestinal stromal tumors—A population-based study from Northern Norway. APMIS: Acta Pathologica, Microbiologica, et Immunologica Scandinavica 115: 289–298. DOI 10.1111/j.1600-0463.2007.apm_587.x. [Google Scholar] [CrossRef]
Sun J, Mohlin S, Lundby A, Kazi JU, Hellman U, Pahlman S, Olsen JV, Ronnstrand L (2014). The PI3-kinase isoform p110delta is essential for cell transformation induced by the D816V mutant of c-Kit in a lipid-kinase-independent manner. Oncogene 33: 5360–5369. DOI 10.1038/onc.2013.479. [Google Scholar] [CrossRef]
Tarn C, Merkel E, Canutescu AA, Shen W, Skorobogatko Y et al. (2005). Analysis of KIT mutations in sporadic and familial gastrointestinal stromal tumors: Therapeutic implications through protein modeling. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 11: 3668–3677. DOI 10.1158/1078-0432.CCR-04-2515. [Google Scholar] [CrossRef]
Yan L, Zou L, Zhao W, Wang Y, Liu B, Yao H, Yu H (2015). Clinicopathological significance of c-KIT mutation in gastrointestinal stromal tumors: A systematic review and meta-analysis. Scientific Reports 5: 13718. DOI 10.1038/srep13718. [Google Scholar] [CrossRef]
Zhu G, Shi J, Zhang S, Guo Y, Huang L, Zhao H, Jiang Y, Sun J (2020). Loss of PI3 kinase association improves the sensitivity of secondary mutation of KIT to Imatinib. Cell & Bioscience 10: 16. DOI 10.1186/s13578-020-0377-9. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |