DOI:10.32604/biocell.2022.018508
| BIOCELL DOI:10.32604/biocell.2022.018508 | |
| Article |
Divicine induces endothelial cells injury and its potential mechanism
Bio-Analytical Laboratory, Shantou University Medical College, Shantou, 51500, China
*Address correspondence to: Wenhong Luo, whluo@stu.edu.cn
#These authors contributed equally to this work
Received: 30 July 2021; Accepted: 22 October 2021
Abstract: Divicine is an active pyrimidine aglycone, generated from vicine by the enzyme β-glucosidase upon ingestion of fava beans. In this study, we investigated the effect of divicine on cultured human umbilical vein endothelial cells (HUVECs) and explored the potential mechanisms. Incubation HUVECs with 18.5–85.1 μM divicine resulted in a concentration and time dependent decrease of cell viability, followed by decrease of cellular reduced glutathione, as well as increase of reactive oxygen species (ROS), malondialdehyde (MDA), and labile iron pool. Transmission electron microscopy confirmed that the divicine treated HUVECs’ mitochondria had shrunk. Importantly, the administration of desferrioxamine, an iron chelator, to the divicine treated HUVECs significantly reduced iron overload and cell death and decreased cellular ROS and MDA. These results demonstrated that divicine could cause damaging of endothelial cells, and ferroptosis might be involved in divicine induced HUVECs injury, reminding long term ingestion of fava beans might be harmful to vascular system, especially for those suffering from glucose 6-phosphate dehydrogenase deficiency.
Keywords: Ferroptosis; Lipid oxidation; Labile iron pool
Divicine is an active pyrimidine aglycone, generated from vicine by the enzyme β-glucosidase upon ingestion of fava beans (Arbid et al., 2013). In the red blood cells (RBCs), divicine autoxidizes and forms a semiquinoid free radical (O-divicine), which subsequently produces superoxide anion and hydrogen peroxide (Baker et al., 1984). Additionally, O-divicine can be reduced to divicine in the presence of reduced glutathione (GSH), and then oxidized again back to O-divicine. This redox reaction cycle (O-divicine divicine) results in the depletion of GSH (Baker et al., 1984). Normal RBCs are well protected against the reactive oxygen species (ROS) damaging effects under normal expression of glucose 6-phosphate dehydrogenase (G6PD) due to its sufficient metabolic competence in regenerating GSH. Research showed that damaging effect to RBCs caused by divicine was not significant under normal G6PD expression (Flohe et al., 1971). Whereas under the condition of G6PD deficiency, RBCs showed a decrease in intracellular reduction potential, making it vulnerable to divicine induced membrane damaging and subsequent intravascular and extravascular hemolysis (Chevion et al., 1982).
G6PD is expressed ubiquitously in various tissues (Fagerberg et al., 2014). G6PD deficiency imparts a selective advantage against malaria infection. Survey research indicated high frequency distribution of G6PD deficiency was found in the population residing in area with long historical prevalence of endemic malaria (Sarkar et al., 2010). G6PD deficiency affects about 400 million people worldwide (Luzzatto et al., 2016). Malaria has been widespread in China before. Recent epidemiological survey showed that the G6PD deficiency frequencies was about 5.69% in Guangdong province of China (Zheng et al., 2020). Recent study showed that G6PD deficiency was associated with increased incidence of cardiovascular disease among the U.S. military personnel and civilians (Thomas et al., 2018). Another study in a Chinese population showed that G6PD-deficient females aged 20–49 had a higher risk for hypertension during pre-pregnancy and pregnancy (Zhao et al., 2019). Experimental study indicated that G6PD deficiency was involved in endothelial dysfunctions caused by higher ROS level and lower reduction potential (Parsanathan and Jain, 2020).
It has been shown that divicine could induce a favism-like response when administered to G6PD-normal rats (Mcmillan and Jollow, 1999), and could induce calcium release from rat liver mitochondria (Benatti et al., 1985), suggesting that divicine might induce cellular injury with normal G6PD levels upon ingestion of fava beans. These results suggest that cellular damaging effect of divicine to other tissues besides RBCs should not be ignored. Endothelial cells (ECs) form the inner cellular lining of all blood vessels and are vulnerable to harmful chemicals and metabolites in circulation, and the damaging of ECs contributes to the evolution of cardiovascular diseases (Ross, 1993). The present study of this article was aimed to elucidate the effects of divicine on human umbilical vein endothelial cells (HUVECs), which would be helpful for understanding the underlying mechanism of increased incidence of cardiovascular disease among G6PD-deficient population.
Vicine (4((1H)-Pyrimidinone,2,6-diamino-5-(β-D-glucopyranosyloxy)) was obtained from Chengdu Yuannuo Tiancheng Technology (Chengdu, China). Beta-glucosidase (β-GC) was obtained from Sigma. RPMI-1640 and fetal bovine serum (FBS) were from Hyclone (Logan, UT, USA). Calcein-AM, thiobarbituric acid (TBA) and desferrioxamine (DFO) were obtained from Aladdin (Shanghai, China). Cell Counting Kit-8 (CCK-8) assay kit was obtained from Dalian Meilun Biotechnology Co., Ltd. (Dalian, China). BCA protein assay kit, GSH assay kit, Cell apoptosis analysis kit, and ROS assay kit were obtained from Beyotime (Shanghai, China). All other reagents were from Energy Chemical (Shanghai, China).
Divicine was prepared by enzymatic hydrolysis according to the method of Pulkkinen with minor modification (Pulkkinen et al., 2016). Briefly, β-GC and vicine were dissolved in 0.01 M PBS (2.7 mM KCl, 2.5 mM NaH2PO4, 6 mM Na2HPO4, 137 mM NaCl; pH 5.0), and put under nitrogen to prevent divicine oxidation by oxygen and incubated at 37°C for 30 min. Then, the solution was sterile filtered and immediately added to the cell cultures. According to the reaction, vicine at concentrations of 25, 50, 100, 200, 400, and 800 μM were converted by β-GC to divicine at concentrations of 18.5, 28.1, 47.1, 85.1, 161.2, and 304.4 μM, respectively (Suppl. Fig. 1).
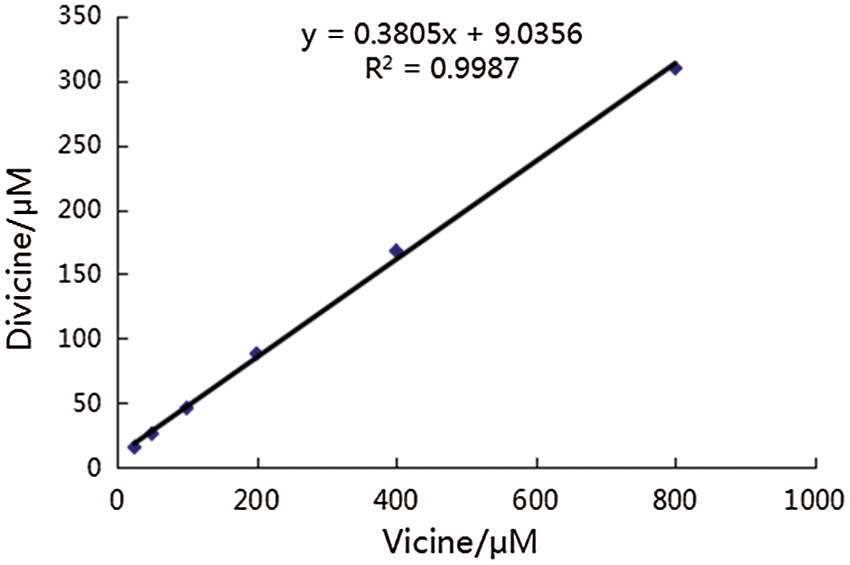
Supplemental figure 1: Linear relationship between the concentration of vicine and divicine after incubation vicine with 0.5 mg/mL β-GC. According to this reaction, divicine at concentrations of 18.5, 28.1, 47.1, 85.1, 161.2, and 304.4 μM could be derived from vicine at concentrations of 25, 50, 100, 200, 400, 800 μM at the presence of β-GC, respectively.
HUVECs (CRL-2480) were obtained from China Center for Type Culture Collection (Wuhan, China), and cultured in culture medium (RPMI-1640) supplemented with 10% FBS (v/v) in an 100% humidified incubator with 5% CO2 at 37°C.
The viability of HUVECs was determined by CCK-8 assay kit. HUVECs were seeded into 96-well microplates at a density of 5 × 104 cells/well in 100 μL culture medium and incubated for 24 h. Then the cells were treated with 100 μL culture medium containing either divicine (0, 18.5, 28.1, 47.1, 85.1 μM) or vicine (25, 50, 100, 200 μM), or β-GC (0.125 mg/mL) for 6, 12, 24, and 48 h. Duplications (N = 6) were conducted at every concentration and time points. After treatment, the cells were incubated with 100 μL medium containing 10 μL CCK-8 for 1 h. Following the incubation, the absorbance was recorded at 450 nm using a microplate reader (Multiskan spectrum, Thermo Labsystems, OH, USA). The cell viability was calculated as (ODexperiment – ODblank)/(ODcontrol – ODblank) × 100%. Cell cultures treated with 85.1 μM divicine plus 100 μM DFO together were also measured for cells viability to determine the antagonistic effect of DFO to divicine.
HUVECs were seeded into 6-well plates at a density of 4 × 105 cells/well in 2 mL of culture medium, and exposed to divicine (0, 18.5, 28.1, 47.1, 85.1 μM), or 85.1 μM divicine plus 100 μM DFO for 24 h. Then, the cells were observed for morphological changes and photographed using an inverted microscopy (Zeiss, Oberkochen, Germany).
HUVECs were grown on 25 cm2 square flask at 2 × 105 cells/mL in 5 mL of culture medium, and exposed to 85.1 μM divicine, or 85.1 μM divicine plus 100 μM DFO for 24 h. Then, the cells were scraped and collected after centrifugation at 1000×g for 5 min. The cells were fixed at 4°C in 0.01 M PBS (pH 7.4) containing 2.5% glutaraldehyde for 2 h, and then in 0.01 M PBS (pH 7.4) containing 1% oxalic acid for another 1 h in a microwave unit at 37°C. After dehydration with ethanol, the cells were embedded in LR White. Ultrathin sections (70 nm) were collected and counterstained with 5% uranyl acetate and lead citrate. Micrographs were obtained with a transmission electron microscope (JEM1400, Japan).
Determination of intracellular reduced GSH
HUVECs were seeded into 6-well plates at a density of 4 × 105 cells/well in 2 mL of culture medium. GSH assay kit was used to measure the GSH contents in HUVECs after treated with different concentrations of divicine (0, 18.5, 28.1, 47.1, 85.1 μM) or 85.1 μM divicine plus100 μM DFO for 24 h. Briefly, after treatment, the cells were harvested and fractured by ultrasonication, and the cell homogenate was then subjected to GSH determination by assay kits. The protein content of cell homogenate was determined with BCA assay kit. Assays were performed according to the manufacturer’s instructions.
Determination of intracellular ROS
Intracellular ROS levels were monitored using 2’,7’-dichlorofluorescein diacetate (DCFH-DA). Briefly, HUVECs were seeded into 6-well plates at a density of 4 × 105 cells/well in 2 mL of culture medium and incubated overnight. Then, the cells were exposed to divicine (0, 18.5, 28.1, 47.1, 85.1 μM), or 85.1 μM divicine plus100 μM DFO for 24 h. After incubation, the cells were then treated with DCFH-DA (5 μM) at 37°C for 30 min. Flow cytometry was then used to measure the fluorescence intensity of the cells. Results were expressed as the ratio of fluorescence intensity of treated groups to that of the control.
HUVECs in 6-well plates (4 × 105 cells/well) were treated with to divicine (0, 18.5, 28.1, 47.1, 85.1 μM), or 85.1 μM divicine plus100 μM DFO for 24 h. HUVECs were collected and fractured by ultrasonication, the concentration of MDA was then determined by high performance liquid chromatography (HPLC), according to the method described previously (Domijan et al., 2015). Briefly, 400 μL 0.1% phosphoric acid and 100 μL 0.6% thiobarbituric acid (TBA) were added to 60 μL cell homogenate, and incubated at 90°C for 60 min. The reaction was then stopped by placing samples on ice and kept on ice until analysis, and 20 μL of samples were injected into the HPLC column. Detector wavelengths were set at λex = 527 nm and λem = 551 nm. The protein concentration of cell homogenate was determined with BCA assay kit.
The apoptosis rate of HUVECs was evaluated by Annexin V-FITC/PI double staining assay. HUVECs in 6-well plates (4 × 105 cells/well) were treated with to divicine (0, 18.5, 28.1, 47.1, 85.1 μM), or 85.1 μM divicine plus100 μM DFO for 24 h. After incubation, the cells were collected and re-suspended in Annexin V-FITC/PI binding buffer for 20 min at room temperature in dark. The stained cells were then analyzed by flow cytometry (BD Accuri™C6, USA).
Determination of cellular labile iron pool (LIP)
LIP was determined by using Calcein-AM, a fluorescent metal-sensitive probe (Epsztejn et al., 1997). HUVECs were seeded into 96-well plates at a density of 5 × 104 cells/well in 100 μL culture medium. After 24 h, cells were treated with divicine (0, 18.5, 28.1, 47.1, 85.1 μM), or 85.1 μM divicine plus 100 μM DFO for 24 h. After incubation, the cells in each well were washed with 0.1 mL PBS (0.01 M; pH 7.4), then 0.1 mL Calcein-AM (0.125 μmol/L) was added and incubated for another 15 min. After washing with 0.1 mL PBS, 90 μL culture medium was added. Fluorescence intensity (A) was then measured at λex 490 nm and λem 530 nm with a fluorescence microplate reader (SpectraMax Gemini XS, Molecular Devices, Sunnyvale, CA, USA). Finally, 10 μL 2,2-bipyridine (1 mmol/L) was added to each well and incubated for 60 min. Then the fluorescence intensity (B) was measured. The content of cellular LIP was reflected by the relative increased fluorescence intensity (B-A) per microgram proteins. The protein concentration of cell homogenate was determined with BCA assay kit.
Data were presented as mean ± SD. The normality of results was evaluated by the Kolmogorov–Smirnov test. Comparison between groups was performed with One-way ANOVA or Mann–Whitney U-test for variables. GraphPad Prism 5.0 was used for statistical analysis, with P < 0.05 considered statistically significant.
The effect of divicine on HUVECs viability
Incubation of HUVECs with vicine or β-glucosidase did not significantly decrease the cell viability (P > 0.05) (Table 1). After treatment with 18.5 μM divicine for various time periods, the cell viability did not exhibit significant decrease compared with controls (P > 0.05) (Table 1). However, incubation of HUVECs with 28.1–85.1 μM divicine resulted in a concentration and time dependent decrease in cell viability (Table 1), with IC50 of 68.14 μM, 43.27 μM, and 39.67 μM for various time periods (12, 24, and 48 h), respectively. DFO (25–100 μM) could, in a concentration-dependent manner, alleviate the toxic effect of divicine (85.1 μM) on HUVECs (Fig. 1).
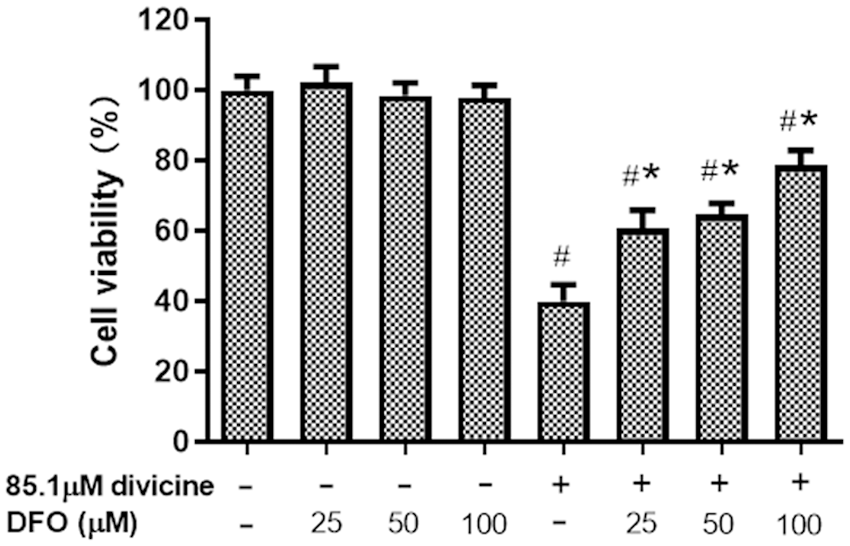
Figure 1: The effects of different concentrations of DFO on the cell viability of divicine (85.1 μM, 24 h) treated HUVECs (N = 6). #P < 0.05 vs. control group; *P < 0.05 vs. 85.1 μM divicine treatment group.
The changes of cell morphology
Confluent HUVECs exhibited a normal cobblestone-like arrangement in control group (Fig. 2A). While, after treatment with 28.1–47.1 μM divicine for 24 h, the cells became swelling, blurred boundary with refractility relatively weaker and loosely attached (Figs. 2B–2D). Cell shape became markable polymorphous after treatment with 47.1 μM divicine. A remarkable decreased cell density could be observed after 85.1 μM divicine treatment (Fig. 2E). DFO (100 μM) treatment could antagonize divicine-induced cells morphological changes including detachment of the cells (Fig. 2F).
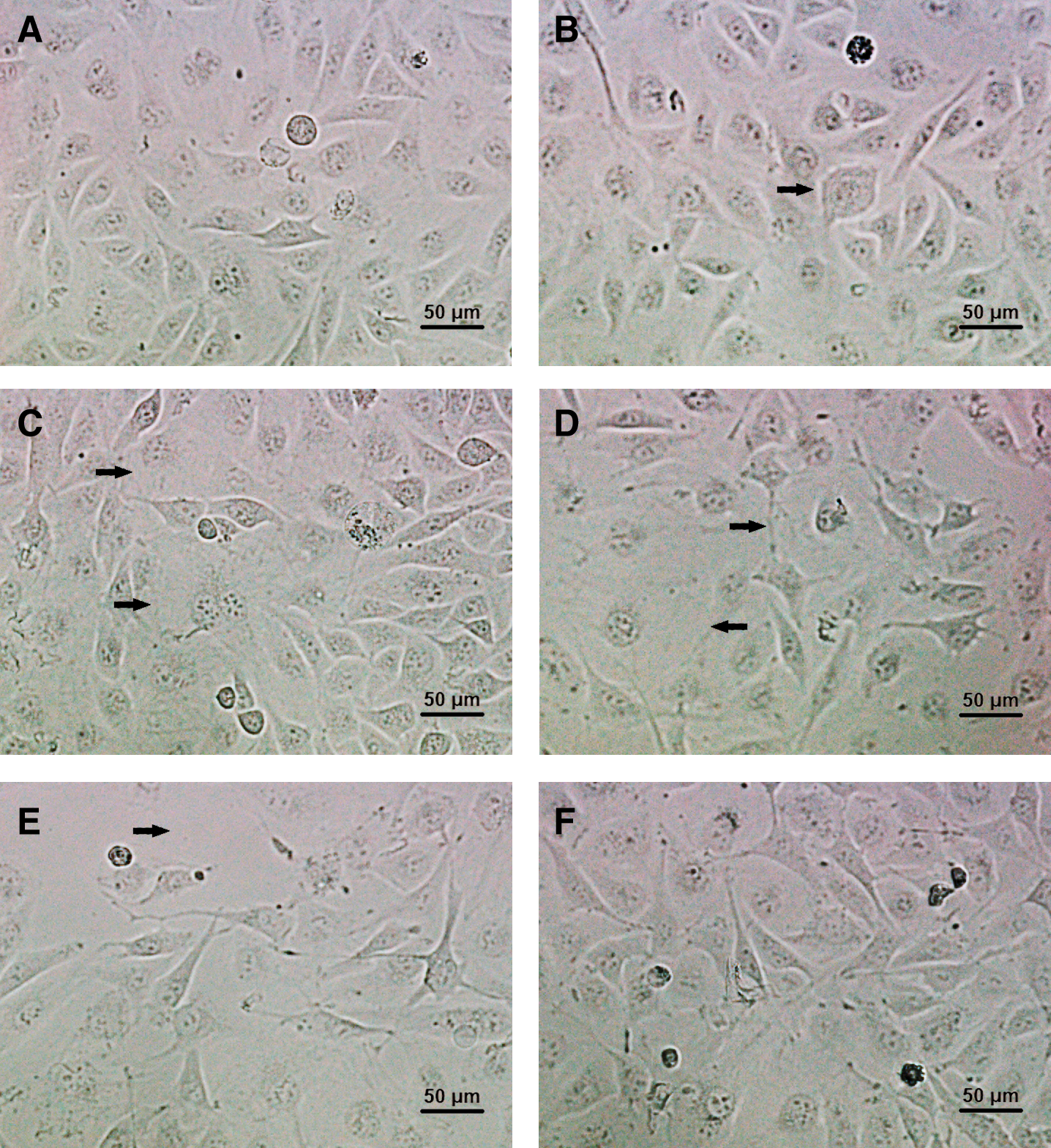
Figure 2: Morphological changes of HUVECs after incubation with different concentrations of divicine for 24 h. (A) Control. (B) 18.5 μM divicine treatment. (C) 28.1 μM divicine treatment. (D) 47.1 μM divicine treatment. (E) 85.1 μM divicine treatment. (F) 85.1 μM divicine plus 100 μM DFO treatment. The magnification of microscopic images is ×200 times. The black arrows indicate cells swelling, blurred boundary or loosely attached.
Transmission electron microscopic images of control cells showed normal intact mitochondria with bean-shaped structures and numerous transversely orientated cristae enveloped by an intact outer membrane (Fig. 3A). After treatment with divicine (85.1 μM), the cells exhibited small spherical mitochondria with disarrayed cristae and a decreased electron density of the matrix (Fig. 3B). Loss of cell nuclear membrane integrity and disintegration of organelles were also observed (Fig. 3B). DFO (100 μM) treatment could restore cell nuclear membrane integrity, but it could not restore the disintegration of organelles (Fig. 3C). Apoptotic bodies were not observed in divicine treated cells.
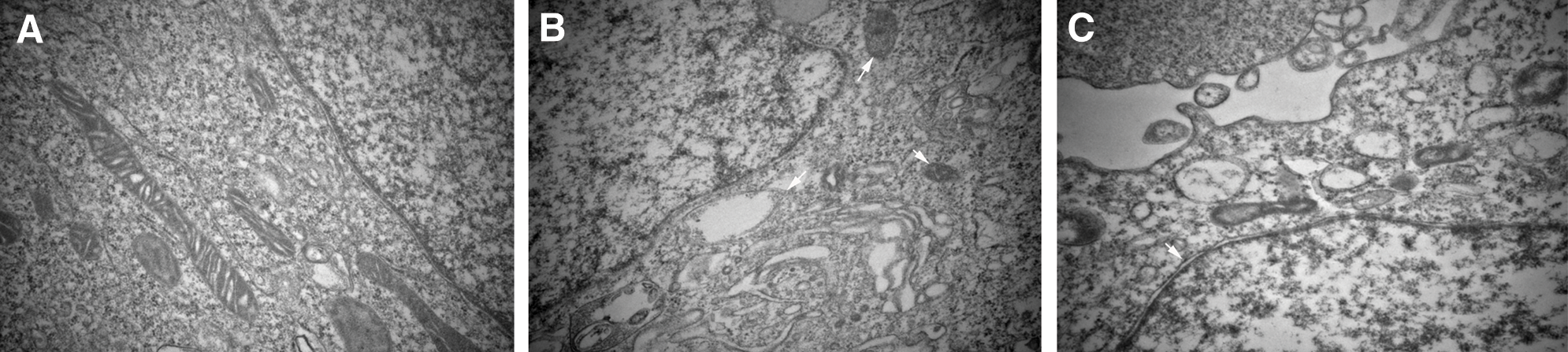
Figure 3: Divicine-induced HUVECs damaging shows ultrastructural features of mitochondria morphological changes. (A) Representative electron micrograph of control cells, showing intact mitochondria with bean-shaped structures and numerous transversely orientated cristae. (B) Representative electron micrograph of divicine treated HUVECs, showing mitochondria with membrane rupture or large vacuoles (white arrow). (C) Representative electron micrograph of DFO treated cells, showing restoration of cell nuclear membrane integrity (white arrow), without restoration of disintegration of organelles (×40000).
The effects of divicine on cellular GSH, ROS, and MDA levels in HUVECs
Divicine (18.5–85.1 μM) treatment significantly depleted the cellular GSH pool, while increase cellular ROS and MDA levels in a concentration dependent manner (Figs. 4A–4C). DFO (100 μM) treatment could significantly inhibit ROS and MDA elevation induced by divicine (P < 0.05), but DFO was not able to restore cellular GSH level (Figs. 4A–4C).
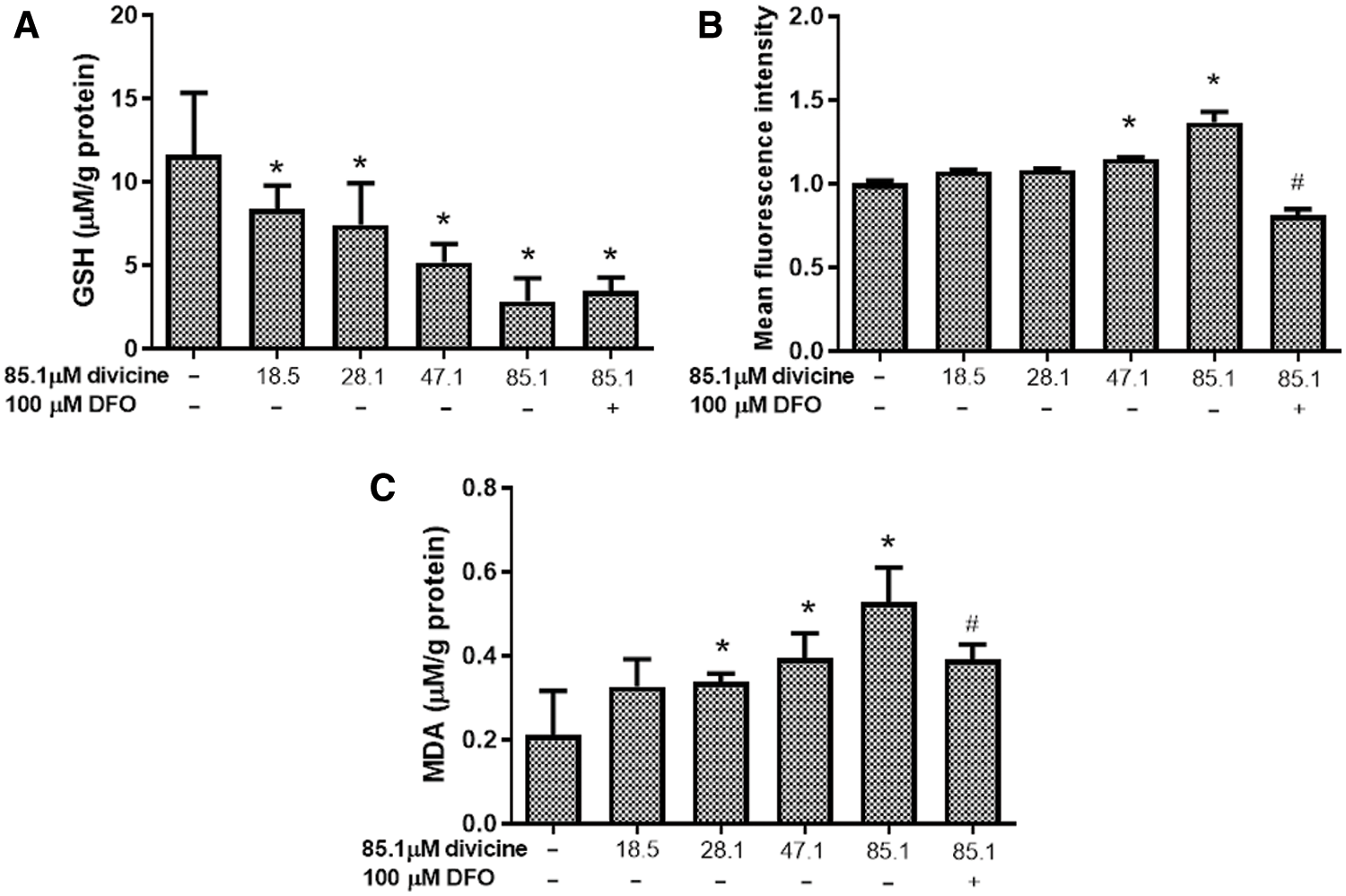
Figure 4: The effects of divicine on cellular GSH, ROS, and MDA levels in HUVECs. (A) Divicine (18.5–85.1 μM) treatment for 24 h significantly depleted the cellular GSH pool in a concentration dependent manner. DFO treatment could not significantly restore GSH level (N = 6). (B) Incubation HUVECs with 18.5–85.1 μM divicine resulted in a concentration dependent increase of cellular ROS level. DFO treatment could significantly inhibit ROS increase induced by divicine (N = 3). (C) Incubation HUVECs with 18.5–85.1 μM divicine resulted in a concentration dependent increase of cellular MDA level. DFO treatment could significantly inhibit MDA elevation induced by divicine. N = 6, *P < 0.05 vs. control; #P < 0.05 vs. 85.1 μM divicine treated group.
HUVECs apoptosis induced by divicine
The total HUVECs apoptosis rate was 0.5 ± 0.1% in control group, while 1.03 ± 0.55%, 1.20 ± 0.20%, 2.33 ± 0.12%, 3.40 ± 0.24% in divicine (18.5~85.1 μM) treatment (24 h) groups, respectively. Divicine (47.1–85.1 μM) treated groups showed a significant increase of apoptotic rate when comparing to control group (P < 0.05) (Fig. 5).
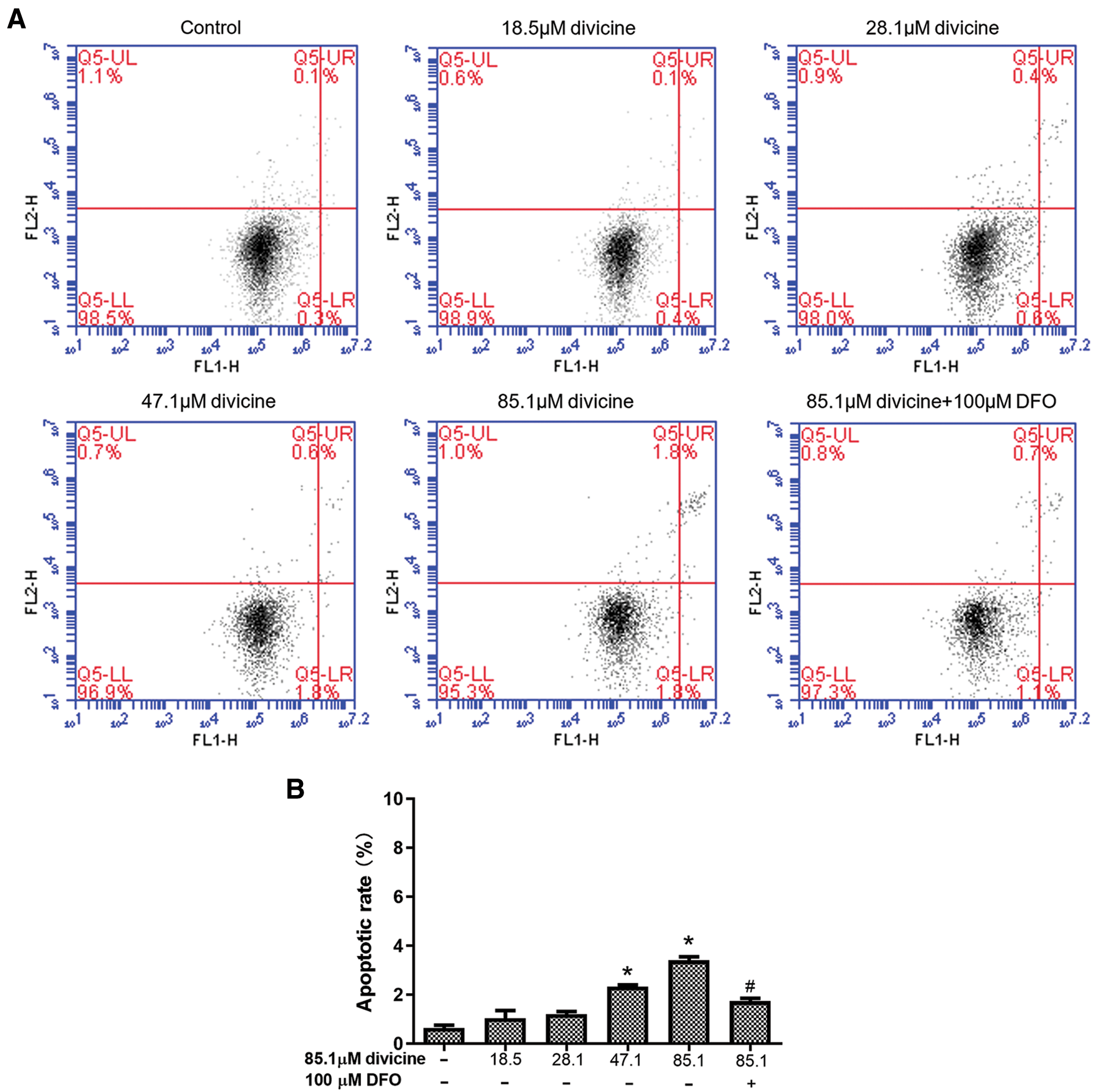
Figure 5: The effect of divicine on the apoptosis of HUVECs after 24 h treatment. (A) Representative dot plot of cells stained with Annexin V-FITC/PI following treatment with divicine (18.5–85.1 μM) for 24 h. (B) Bars represent the percentages of apoptotic cells. N = 3, *P < 0.05 vs. control; #P < 0.05 vs. 85.1 μM divicine treated group.
Divicine induced the increase of intracellular LIP
Treatment with 47.1–85.1 μM divicine for 24 h caused a significant elevation of fluorescence intensity in HUVECs (P < 0.05), with 1.00 ± 0.32 in control group and 0.92 ± 0.29, 1.57 ± 0.32, 3.36 ± 1.12, 5.03 ± 1.11 in divicine treated groups (18.5, 28.1, 47.1, 85.1 μM), respectively (Fig. 6). DFO (100 μM) could significantly inhibit the increase of LIP induced by divicine (85.1 μM) (P < 0.05) (Fig. 6).
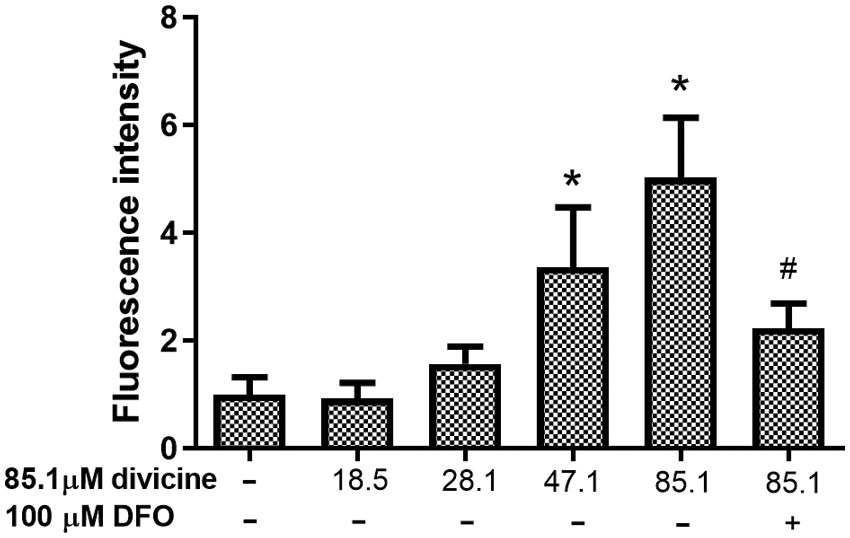
Figure 6: The alterations of LIP levels in HUVECs after treatment with different concentrations of divicine for 24 h. Divicine (18.5–85.1 μM) induced the elevation of LIP in HUVECs in a concentration dependent manner, while DFO treatment could inhibit the elevation of LIP. N = 6, *P < 0.05 vs. control; #P < 0.05 vs. 85.1 μM divicine treated group.
The present work suggested that HUVECs were sensitive to divicine, a favism-inducing agent. Divicine of concentration more than 28.1 μM could significantly reduce HUVECs viability after treatment for 12 h. The divicine cytotoxicity exhibited concentration and time dependent manner. It has been reported that myoblasts were highly sensitive to divicine induced oxidative damaging, due to intracellular low basal G6PD activity (Ninfali et al., 2000), indicating that sufficient G6PD expression is critical for cellular resistance to oxidants such as divicine. It has been demonstrated that divicine undergoes rapid auto-oxidation within cells and forms an oxidized product, a semiquinoid free radical (O-divicine), which then is reduced again by NADPH or NADH back to divicine, a reduced form. This reduction-oxidation cycle of divicine to O-divicine within cells depletes continuously the reducing power. The mechanism is a perpetuating switching between hydroquinonic and quinonic species of divicine itself (Benatti et al., 1984). This cycling process not only consumes NADPH, but also at the same time produces superoxide anion and hydrogen peroxide (Baker et al., 1984). Experimentally, oxidation of divicine within cells was responsible for about 90% depletion of GSH (Benatti et al., 1984). The depletion of NADPH and GSH would impair the cellular antioxidative capability and render cells to oxidative injury (Forman et al., 2009; Jollow and Mcmillan, 2001). In the present study, GSH depletion was observed in divicine treated HUVECs, suggesting that GSH depletion might be a driving factor for divicine-induced HUVECs injury. Consequent ROS and lipid peroxides increases were also detected in divicine-treated HUVECs. However, apoptotic rate of divicine treated HUVECs were relatively unremarkable (3.40 ± 0.24%) after treatment with 85.1 μM divicine for 24 h. The apoptotic rate of HUVECs was not consistent to the loss of cell viability (26.8 ± 9.9%, compared to control), indicating that beyond apoptosis, other mechanisms of cell death might be involved in divicine induced cell damaging and cell death.
A recent review article discussed a so-called “ferroptosis”, a form of programmed cell death, and suggested that ferroptosis could be induced by GSH depletion agents (Li and Wang, 2019). The involvement of ferroptosis in divicine induced HUVECs death was therefore proposed in this study. In addition to the observed depletion of HUVECs GSH and the increase of MDA and ROS, the shrunken mitochondria observed with transmission electron microscopy in divicine treated HUVECs, was considered to be a characteristic morphological change of HUVECs ferroptosis. Transmission electron microscopy has been an important tool for study in morphological changes in ferroptosis (Xie et al., 2019). Ferroptosis has been reported to be an iron dependent event (Dixon et al., 2012). Labile iron pool (LIP) was determined in the present study. The results showed that HUVECs LIP increased after divicine treatment, and furthermore, administration of an iron chelator DFO not only reduced iron overload and cell death but also alleviated cellular ROS and MDA alternations in HUVECs. In this study, DFO treatment did not show to restore cellular GSH level, indicating iron did not participate in the divicine to O-divicine cycle. But iron has been reported to promote ROS caused lipid peroxidation and the subsequent oxidative cellular damaging (Dixon and Stockwell, 2014).
Kagan et al. (2017) reported that oxidized arachidonoyl containing phosphatidylethanolamines navigated cells to ferroptosis. Oxidation of arachidonic acid present in cell membrane phospholipid may interfere arachidonic acid metabolism that is involved in the production of prostaglandins, an important group of vascular active factors (Luo et al., 2016). Divicine induced oxidation of HUVECs membrane phospholipid might therefore interfere HUVECs arachidonic acid metabolism and eventually affect the endothelium-derived vascular function. Divicine present in blood circulation upon ingestion of fava beans may be a risk factor of endothelial dysfunction. This may partly explain the high incidence of cardiovascular diseases in G6PD deficiency (Thomas et al., 2018; Zhao et al., 2019).
In conclusion, findings of this study suggest that endothelial cells are sensitive to divicine, and ferroptosis may be involved in divicine induced HUVECs injury, reminding long term ingestion of fava beans might be harmful to vascular system, especially for those suffering from glucose 6-phosphate dehydrogenase deficiency. More extensive research concerning endothelial function under the stress of divicine is needed.
Availability of Data and Materials: All data generated or analyzed during this study are included in this published article.
Authors’ Contribution: Long SU planned and conducted the experiments, Zhexuan LIN analyzed and interpreted the data, and wrote the manuscript. Hui LI and Hongjun LUO helped to conduct the experiments. Wenhong LUO contributed to the editing of the manuscript. Wenhong LUO took responsibility for the integrity and accuracy of the data.
Funding Statement: This work was supported by the National Natural Science Foundation of China (31671183), and the Natural Science Foundation of Guangdong Province (2018A030307005).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Arbid MS, Koriem KMM, Asaad GF, Megahed HA (2013). Effect of the antibiotic neomycin on the toxicity of the glycoside vicine in rats. Journal of Toxicology 2013: 1–8. DOI 10.1155/2013/913128. [Google Scholar] [CrossRef]
Baker MA, Bosia A, Pescarmona G, Turrini F, Arese P (1984). Mechanism of action of divicine in a cell-free system and in glucose-6-phosphate dehydrogenase-deficient red cells. Toxicologic Pathology 12: 331–336. DOI 10.1177/019262338401200405. [Google Scholar] [CrossRef]
Benatti U, Guida L, de Flora A (1984). The interaction of divicine with glutathione and pyridine nucleotides. Biochemical and Biophysical Research Communications 120: 747–753. DOI 10.1016/S0006-291X(84)80170-9. [Google Scholar] [CrossRef]
Benatti U, Guida L, Forteleoni G, Meloni T, de FA (1985). Impairment of the calcium pump of human erythrocytes by divicine. Archives of Biochemistry and Biophysics 239: 334–341. DOI 10.1016/0003-9861(85)90696-4. [Google Scholar] [CrossRef]
Chevion M, Navok T, Glaser G, Mager J (1982). The chemistry of favism-inducing compounds. The properties of isouramil and divicine and their reaction with glutathione. European Journal of Biochemistry 127: 405–409. DOI 10.1111/j.1432-1033.1982.tb06886.x. [Google Scholar] [CrossRef]
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM et al. (2012). Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149: 1060–1072. DOI 10.1016/j.cell.2012.03.042. [Google Scholar] [CrossRef]
Dixon SJ, Stockwell BR (2014). The role of iron and reactive oxygen species in cell death. Nature Chemical Biology 10: 9–17. DOI 10.1038/nchembio.1416. [Google Scholar] [CrossRef]
Domijan AM, Ralić J, Radić Brkanac S, Rumora L, Žanić-Grubišić T (2015). Quantification of malondialdehyde by HPLC-FL Application to various biological samples. Biomedical Chromatography 29: 41–46. DOI 10.1002/bmc.3361. [Google Scholar] [CrossRef]
Epsztejn S, Kakhlon O, Glickstein H, Breuer W, Cabantchik ZI (1997). Fluorescence analysis of the labile iron pool of mammalian cells. Analytical Biochemistry 248: 31–40. DOI 10.1006/abio.1997.2126. [Google Scholar] [CrossRef]
Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D et al. (2014). Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Molecular & Cellular Proteomics 13: 397–406. DOI 10.1074/mcp.M113.035600. [Google Scholar] [CrossRef]
Flohe L, Niebch G, Reiber H (1971). The effect of divicine on human erythrocytes (author’s transl). Zeitschrift fur klinische Chemie und klinische Biochemie 9: 431–437. [Google Scholar]
Forman HJ, Zhang H, Rinna A (2009). Glutathione: Overview of its protective roles, measurement, and biosynthesis. Molecular Aspects of Medicine 30: 1–12. DOI 10.1016/j.mam.2008.08.006. [Google Scholar] [CrossRef]
Jollow DJ, Mcmillan DC (2001). Oxidative stress, glucose-6-phosphate dehydrogenase and the red cell. In: Dansette PM (ed.Biological reactive intermediates VI. Advances in experimental medicine and biology. Boston, MA: Springer. [Google Scholar]
Kagan VE, Mao G, Qu F, Angeli JP, Doll S et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nature Chemical Biology 13: 81–90. DOI 10.1038/nchembio.2238. [Google Scholar] [CrossRef]
Li B, Wang Z (2019). Ferroptosis is a new form of regulatory cell death. Basic & Clinical Medicine 039: 247–251. [Google Scholar]
Luo W, Liu B, Zhou Y (2016). The endothelial cyclooxygenase pathway: Insights from mouse arteries. European Journal of Pharmacology 780: 148–158. DOI 10.1016/j.ejphar.2016.03.043. [Google Scholar] [CrossRef]
Luzzatto L, Nannelli C, Notaro R (2016). Glucose-6-phosphate dehydrogenase deficiency. Hematology/Oncology Clinics 30: 373–393. DOI 10.1016/j.hoc.2015.11.006. [Google Scholar] [CrossRef]
Mcmillan DC, Jollow DJ (1999). Favism: Divicine hemotoxicity in the rat. Toxicological Sciences: An Official Journal of the Society of Toxicology 51: 310–316. DOI 10.1093/toxsci/51.2.310. [Google Scholar] [CrossRef]
Ninfali P, Perini MP, Bresolin N, Aluigi G, Cambiaggi C et al. (2000). Iron release and oxidant damage in human myoblasts by divicine. Life Sciences 66: PL85–PL91. DOI 10.1016/S0024-3205(99)00625-6. [Google Scholar] [CrossRef]
Parsanathan R, Jain SK (2020). Glucose-6-phosphate dehydrogenase deficiency activates endothelial cell and leukocyte adhesion mediated via the TGFβ/NADPH oxidases/ROS signaling pathway. International Journal of Molecular Sciences 21: 7474. DOI 10.3390/ijms21207474. [Google Scholar] [CrossRef]
Pulkkinen M, Zhou X, Lampi AM, Piironen V (2016). Determination and stability of divicine and isouramil produced by enzymatic hydrolysis of vicine and convicine of faba bean. Food Chemistry 212: 10–19. DOI 10.1016/j.foodchem.2016.05.077. [Google Scholar] [CrossRef]
Ross R (1993). The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature 362: 801–809. DOI 10.1038/362801a0. [Google Scholar] [CrossRef]
Sarkar S, Biswas NK, Dey B, Mukhopadhyay D, Majumder PP (2010). A large, systematic molecular-genetic study of G6PD in Indian populations identifies a new non-synonymous variant and supports recent positive selection. Infection, Genetics and Evolution 10: 1228–1236. DOI 10.1016/j.meegid.2010.08.003. [Google Scholar] [CrossRef]
Thomas JE, Kang S, Wyatt CJ, Kim FS, Mangelsdorff AD, Weigel FK (2018). Glucose-6-phosphate dehydrogenase deficiency is associated with cardiovascular disease in U.S. military centers. Texas Heart Institute Journal 45: 144–150. DOI 10.14503/THIJ-16-6052. [Google Scholar] [CrossRef]
Xie BS, Wang YQ, Lin Y, Mao Q, Feng JF et al. (2019). Inhibition of ferroptosis attenuates tissue damage and improves long-term outcomes after traumatic brain injury in mice. CNS Neuroscience & Therapeutics 25: 465–475. DOI 10.1111/cns.13069. [Google Scholar] [CrossRef]
Zhao J, Zhang X, Guan T, Wang X, Zhang H, Zeng X, Dai Q, Wang Y, Zhou L, Ma X (2019). The association between glucose-6-phosphate dehydrogenase deficiency and abnormal blood pressure among prepregnant reproductive-age Chinese females. Hypertens Research 42: 75–84. DOI 10.1038/s41440-018-0118-1. [Google Scholar] [CrossRef]
Zheng Y, Wang J, Liang X, Huang H, Ma Y et al. (2020). Epidemiology, evolutionary origin, and malaria-induced positive selection effects of G6PD-deficient alleles in Chinese populations. Molecular Genetics & Genomic Medicine 8: e1540. DOI 10.1002/mgg3.1540. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |