DOI:10.32604/biocell.2022.018326
| BIOCELL DOI:10.32604/biocell.2022.018326 | |
| Article |
Effect of VirD4 on gastric epithelial-1 cells and its mechanism
1Clinical Laboratory Center, Jiangsu Taizhou People’s Hospital, Taizhou, 225399, China
2School of Medicine, Jiangsu University, Zhenjiang, 212013, China
*Address correspondence to: Yang Yang, princess_yanggirl@163.com; Chenglin Zhou, 18762340015@126.com
Received: 16 July 2021; Accepted: 16 September 2021
Abstract: The gene of Helicobacter pylori can encode three to four type IV secretory systems, of which a new gene region has been found in the H. pylori plasticity region. The coding products of this region can form a new T4SS named tfs3, but its function is unclear. This study investigated the effect of VirD4 recombinant protein in the tfs3 secretory system of the H. pylori clinical strain SBK on GES-1 cells. We observed changes in cell morphology after VirD4 treatment. Further analysis indicated that VirD4 increased inflammation by increasing the activation of NF-κB. VirD4 can also inhibited proliferation, and induced migration of cells. Moreover, VirD4 caused apoptosis in GES-1 cells in caspase and ERK1/2/Ras dependent signaling events. Our study laid a foundation for further research on the biological function of VirD4 and the detection and treatment of H. pylori-related diseases.
Keywords: H. pylori; tfs3; VirD4; NF-κB; Ras/Erk
Helicobacter pylori is a common pathogenic bacterium in the human digestive system (Backert et al., 2004; Chmiela and Gonciarz, 2017). H. pylori infection often causes chronic active gastritis, peptic ulcers, and can develop into malignant tumors of the digestive system (Sanders and Peura, 2002). The type IV secretory system (T4SS) is one of the secretory and transporting mechanisms of H. pylori’s pathogenic factors and causes the proliferation of host cells, promotes the secretion of cytokines, and facilitates bacterial colonization and persistent infection (Naumann et al., 2017). In addition to the cag pathogenicity island and comB locus that encode the different T4SSs of H. pylori, the plasticized zones of the H. pylori genome, of which tfs3 is one, can also encode T4SS (Hofreuter et al., 1998; Kersulyte et al., 2003; Tegtmeyer et al., 2011).
The cluster size of the tfs3 gene is 16.3 KB, and the four coding genes virB4, virB7, virB11, and VirD4 are homologous with the well-studied virB/D4 TFSS of the plant pathogen Agrobacterium tumefaciens (Alvi et al., 2007). Furthermore, it became clear that tfs3 and tfs4 genes are not restricted to the original “plasticity zones”. They are organized together with further genes as genome islands in many other genomic locations (Fischer et al., 2020). The function of the T4SS encoded by tfs3 remains unclear. A possible role of Tfs3 has been suggested in DNA transfer, but the actual function of Tfs3 still remains widely unclear (Fernandez-Gonzalez and Backert, 2014). Some studies have suggested that this system constitutes a complete set of putative T4SS channels with other homologous genes (Mcclelland et al., 2007). Alandiyjany et al. indicated a role for the tfs3 T4SS in CtkA-mediated pro-inflammatory signalling by H. pylori (Alandiyjany et al., 2017a). In addition, in the tfs3 system of strain PeCan18B, homologs VirB4, VirB11, and VirD4 jointly form an enzyme complex related to the hydrolysis of adenosine triphosphate in the cytoplasmic membrane, providing sufficient energy for the transmembrane transport of macromolecular substrates (Fernandez-Gonzalez and Backert, 2014).
With the continual production of in-depth research on the pathogenic mechanism of H. pylori, the tfs3 secretion system of H. pylori has attracted the attention of scholars. The discovery of the new transport system suggests the complexity, multiplicity, and strain-specificity of the pathogenic mechanism of H. pylori. This study investigated the role of VirD4 in the tfs3 gene cluster to examine the effect of VirD4 on gastric epithelial (GES) cells.
VirD4 caused inflammation in GES-1 cells
First, GES-1 cells were treated with VirD4 at a concentration of 10 μg/mL for 0, 12, and 36 h. Under optical microscopy, we observed that over time, the cells appeared to shrink and swell gradually, varied in size, and exhibited obvious granulation, and the number of vacuoles and dead cells increased (Fig. 1A).

Figure 1: VirD4 affects the morphology of GES-1 cells and causes inflammation. (A) White light images were observed through phase-contrast microscopy after GES-1 cells were treated with VirD4 for 0, 12, and 36 h. Images in (i), (ii), and (iii) are of the control, and those in (iv), (v), and (vi) are of the treatment groups. Magnification, ×200; scale bar = 200 μm. (B) Quantitation of cytokine levels in culture supernatants of cells treated with VirD4 for 4 h using ELISA (IL-1β, IL-6, IL-8 and TNFα). (C) Expression of major proteins in the NF-kB pathway after the GES-1 cells were treated with VirD4 was examined using western blotting. Experiments were performed in triplicate with similar results. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.001.
To explore whether VirD4 would alter the expression of inflammatory cytokines in GES-1 cells, the recombinant VirD4 protein was used to act on the cells in vitro. We then examined the effects of VirD4 on the secretion of proinflammatory cytokines in the GES-1 cells, after which the concentrations of these cytokines in cell culture supernatants were determined using an enzyme-linked immunosorbent assay. We observed that VirD4 increased the expression of interleukin (IL)-1β, IL-6, IL-8, and tumor necrosis factor (TNF)α in vitro (Fig. 1B).
Signal transduction pathways are often activated in response to external stimuli. Nuclear factor (NF)-κB plays a vital role in H. pylori infection by regulating the expression of various inflammatory genes. Therefore, we investigated whether VirD4 would modulate the activity of NF-κB under our experimental conditions. We hypothesized that the increased expression of VirD4 promotes NF-kB pathway activation.
To test this hypothesis and further confirm the role of VirD4 in the regulation of inflammatory responses, proteins were extracted. We examined key molecules in the NF-κB pathway using western blotting; after the GES-1 cells were treated with VirD4, the expression of A20, IκB kinase (IKK)α, IKKβ, NF-κB P105, NF-κB P50, P65/phosphor (P)-P65, NF of IKBα/P-IKBα, and P-glycogen synthase kinase (GSK)3β/GSK3β were significantly induced, suggesting that VirD4 can accelerate NF-κB pathway activation (Fig. 1C). These results further corroborated our previous findings and suggested that VirD4 induced proinflammatory cytokine expression by increasing the activation of NF-κB in response to inflammation.
Role of VirD4 in the proliferation of GES cells
To examine the role of VirD4 in the proliferation of GES-1 cells, Cell Counting Kit-8 (CCK8) was used to detect the changes in proliferation of GES-1 cells under different concentrations of recombinant VirD4 proteins over time. The GES-1 cells exhibited good proliferation in the control group, but VirD4 inhibited the proliferative activity of the GES-1 cells (Fig. 2A).

Figure 2: Effect of VirD4 on proliferation and migration of gastric epithelial (GES)-1. (A) Cell Counting Kit-8 assay performed on GES-1 cells indicated that VirD4 reduced cell proliferation in GES-1. (B–C) Colony formation assay performed on GES-1 cells indicated that VirD4 reduced cell proliferation. (D–E) Migratory ability of VirD4-treated GES-1 cells was evaluated using transwell migration assay. VirD4 caused the migration of the GES-1 cells. Magnification, ×100; scale bar = 10 μm. (F–G) Expression of the proliferation and migration related proteins was evaluated by immunoblotting after GES-1 cells were treated with VirD4 for 4 h. Experiments were performed in triplicate with similar results. **P < 0.01.
Then, the colony formation assay was performed on GES-1 cells both with and without VirD4 treatment. VirD4 formed smaller clones than did the control cells (Figs. 2B and 2C). The results were consistent with those of CCK8.
The expression of a series of molecular markers was then detected in the GES-1 cells by using western blotting. As Fig. 2F shows, the expression of proliferating cell nuclear antigen, cyclinD1, and c-Myc considerably decreased in the GES-1 + VirD4 group. These results demonstrate that VirD4 expression affects GES-1 cell proliferation in vitro.
Effect of VirD4 on GES cell migration
Studies have demonstrated that H. pylori can alter migration ability in gastric cancer. To test whether VirD4 is responsible for the promotion of GES-1 cell migration, we performed a transwell migration assay, which demonstrated that the number of migrated cells after VirD4 treatment had remarkably increased compared with the control group (Figs. 2D and 2E).
The expression of a series of molecular markers was then detected in the GES-1 cells through western blotting. As shown in Fig. 2G, the expression of tissue inhibitors of metalloproteinases (TIMP)-1 and TIMP-2 considerably decreased in the GES-1 + VirD4 group, whereas matrix metalloproteinases (MMP)1, MMP2, and MMP9 increased substantially compared with the control group. These results demonstrate that VirD4 expression affects GES-1 cell migration in vitro.
VirD4 caused apoptosis in GES-1 cells
To determine whether VirD4 affects GES-1 cell apoptosis, apoptotic cells were detected in situ by using annexin V-FITC/propidium iodide (PI) and Hoechst 33258 staining. First, GES-1 cells were stained with annexin V-FITC/PI after exposure to fixed concentrations of VirD4 for 4 h. The results revealed that after VirD4 treatment, intracellular green fluorescence increased (Fig. 3A). Next, we investigated apoptosis in the GES-1 cells through Hoechst 33258 staining. After the GES-1 cells were treated with VirD4, the number of cells with bright nuclear condensation or fragmented nuclei increased, which is characteristic of apoptosis (Fig. 3B).

Figure 3: VirD4 promotes apoptosis and affects the expression of the apoptosis-Caspase family and the Ras/extracellular single-regulated kinase signaling pathways in GES-1 cells. (A) Annexin V-FITC/propidium iodide was used for situ observation through fluorescence microscopy of the apoptosis level of each group. Magnification, ×200; scale bar = 200 μm. (B) DNA segmentation of GES-1 cells detected by Hoechst 33258 stain. Magnification, ×200; scale bar = 200 μm. (C) Western blotting results suggested that apoptosis increased after GES-1 cells were treated with VirD4. (D–E) The level of proteins associated with the apoptosis-caspase family and the signaling pathway was evaluated using western blotting of GES-1. Experiments were performed in triplicate with similar results.
Western blot analysis was also performed to analyze the changes in the apoptosis-promoting Bcl-2-associated X protein (BAX) and the apoptosis-inhibiting protein Bcl2 in cells from different treatment groups. Compared with the control group, the BAX index increased after treatment with VirD4, whereas the Bcl2 index decreased (Fig. 3C). These results indicated that the effect of GES-1 apoptosis induced in the GES-1 + VirD4 group was different from that of the control group, suggesting that VirD4 treatment in GES-1 cells promotes apoptosis and that the signaling molecules associated with apoptosis may change.
To further elucidate the role of VirD4 in the apoptosis of the GES-1 cell line, we detected the change in caspase-3, cleaved caspase-3, caspase-9, cleaved caspase-9, and the substrates poly ADP-ribose polymerase (PARP) and cleaved PARP. Compared with the control cells, VirD4 reduced caspase-3 and caspase-9 and increased cleaved caspase-3 and cleaved caspase-9. In addition, VirD4 was able to cleave PARP because it decreased full-length PARP and increased c-terminal-cleaved PARP (Fig. 3D). Because the activation of the extracellular single-regulated kinase (ERK)1/2 pathway is also involved in positively regulating cell apoptosis, we detected the protein expression of P-ERK1/2 and Ras to evaluate whether it could also cause apoptosis in GES-1. The data indicated that the main proteins of the Ras/ERK signaling pathway remarkably decreased in the VirD4-treated GES-1 cells (Fig. 3E). These results demonstrate that VirD4 causes apoptosis in GES-1 cells partially by activating caspase and the ERK1/2/Ras pathway.
H. pylori is a pathogenic bacterium that colonizes the human stomach, with a carrier rate of 50% in the global population. Its secretion transport system, T4SS, can contribute to various physiological and pathological reactions, such as inflammation, ulcers, and even cancer, caused by H. pylori through the transport of cell-related toxin CagA (Sue et al., 2015; Yamaoka, 2010). Tfs3 is a new pathogenic island similar to T4SS, and approximately one-fifth of all H. pylori strains carry the tfs3 gene cluster (Alm et al., 1999). Its function and pathogenic mechanism have garnered increasing attention. As one of the fundamental components of tfs3, VirD4 plays a critical role in the assembly and transfer of CagA. Mutant strains lacking Cagβ, putative ATPases corresponding to VirD4 in prototypical T4SSs, were capable of T4SS core complex assembly but defective in CagA translocation into host cells (Lin et al., 2020). However, few studies have described the function of VirD4 in tfs3 secretion system. Therefore, identifying the function of VirD4 can deepen understanding of the pathogenic mechanism of the tfs3 secretion system.
Studies have indicated that the upregulation of proinflammatory signals is a feature of certain H. pylori strains carrying tfs3, and many of the gene fragments are closely related to the expression and secretion of these inflammatory factors in GES cells (Alandiyjany et al., 2017b; Kersulyte et al., 2009). The present study demonstrated that GES-1 cells secrete small amounts of cytokines to maintain their physiological functions without stimulation, and after VirD4 treatment, the levels of proinflammatory cytokines IL-1β, IL-6, IL-8, and TNFα increased, suggesting that the VirD4 protein can cause host cells to secrete inflammatory factors. Alandiyjany et al. (2017b) demonstrated that the activation of serine/threonine kinase on gastric mucosal cells depends on the T4SS encoded by the tfs3 gene cluster to activate the NF-κB pathway and promote the release of inflammatory cytokines IL-8 and TNFα. Western blot was used to detect the major proteins of the NF-κB pathway in our study. The results indicated that the NF-κB pathway was activated after VirD4 acted on the GES-1 cells. Therefore, our results are consistent with those of other studies. VirD4 protein may play a role in potentiating H. pylori-mediated inflammatory responses in T4SS encoded by tfs3.
Studies have demonstrated that H. pylori can promote the proliferation and increase the migration of gastric mucosal epithelial cells (Alzahrani et al., 2014; Zhu et al., 2015). However, our results indicate that VirD4 proteins can inhibit the proliferation of GES-1 cells in a concentration-dependent and time-dependent manner; this result is consistent with the effect of VirD4 on the cell proliferation of human periodontal fibroblasts (Li and Liang, 2019). We also observed that VirD4 can cause a substantial migration of GES cells and that the expression of TIMP-1 and TIMP-2 decreased, whereas that of MMP1, MMP2, and MMP9 increased in the VirD4-treated cells. Ouyang et al. (2021) found that H. pylori infection caused epithelial-mesenchymal transition in GES-1, which in turn promoted the progression of gastric cancer. Our study shows an enhanced migration ability of gastric epithelial cells after H. pylori infection, which may be involved in the development and progression of H. pylori -associated gastric cancer. Therefore, the proliferation and migration in host cells changed by VirD4 in T4SS encoded by tfs3 may be involved in tumorigenesis.
Apoptosis is a process of cell contraction and autophagy, and it is one of the molecular mechanisms required to maintain homeostasis. It denotes the autonomous and orderly death of normal cells after physiological and pathological stimulation (Fan et al., 2005). The most common apoptotic pathways are the death receptor pathway and the mitochondrial pathway (Ashkenazi et al., 2017; Czabotar et al., 2014). Studies have indicated that H. pylori can also cause the apoptosis of gastric mucosa epithelial cells (Lv et al., 2014). We used annexin V-FITC/PI and Hoechst 33258 staining to analyze the apoptosis of each group. We also detected protein expressions of BAX and Bcl2, which are crucial members of the apoptotic family (Bartchewsky et al., 2010). We discovered that VirD4 can cause apoptosis in GES-1 cells. Studies have demonstrated that apoptosis is strictly controlled by the caspase family and that the ERK signaling pathway is involved in the process (Tengku Din et al., 2018; Xu et al., 2019). To reveal the possible mechanism underlying the effect of VirD4 on GES-1 cells, we tested the main molecules in the caspase family and the Ras and ERK signaling pathways. We demonstrated that VirD4 can suppress the proteins of the Caspase family and reduce levels of P-ERK1/2 and Ras to promote apoptosis in GES-1 cells.
In conclusion, we investigated the effect of recombinant protein VirD4 of the tfs3 secretion system on the inflammation, proliferation, migration, and apoptosis of GES-1 cells to elucidate the pathogenic mechanism of the tfs3 secretion system. However, the crystal structure, subcellular location, composition, and function of this protein in the T4SS complex requires further investigation. Our study provides a new perspective for H. pylori -associated disease based on tfs3 secretion system.
The VirD4 gene segment was obtained through T-A cloning method. For this part, the DNA temple was obtained from the pylori clinical strain SBK which was cultured and maintained in our laboratory culturing on Columbia agar plates containing 10% sheep blood under microaerophilic conditions at 37°C. (The expression of VirD4 in different H. pylori type strains was showed in Suppl. Fig. S1.) Then the prokaryotic expression vector pET-28a (+)-VirD4 was constructed and transformed into E. coli Rosetta for the expression by induction of IPTG. The recombinant proteins were obtained and purified by KCL dyeing with gel cutting method, and identified via SDS-PAGE analysis.
The immortalized gastric epithelial mucosa cell line GES‑1, established by the Beijing Institute for Cancer Research (Beijing, China) was cultured and maintained in our laboratory. The cells were cultured in RPMI-1640 medium (Gibco, Thermo Fisher, USA), supplemented with 10% fetal bovine serum (FBS; Life Technologies, USA) in a humidified atmosphere of 5% CO2 at 37°C. For cell studies, 10 μg/mL purified recombinant VirD4 proteins were co-cultured with GES-1 cells for 4 h.
Cellular proteins were extracted using radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime Biotechnology, Shanghai, China) containing phenylmethanesulfonyl fluoride (PMSF) and phosphatase inhibitors on ice for 30 min. The cell lysates were centrifuged at 12,000 × g for 30 min, and protein contents were determined using a BCA protein assay kit (Pierce, Rockford, IL, USA). Then, equal amounts of protein (200 µg) were resolved on 12% SDS-polyacrylamide gels by electrophoresis and transferred to PVDF membranes. The membranes were then incubated with 5% non-fat dry milk powder in tris-buffered saline (TBS) containing 0.1% Tween-20 for 2 h at room temperature, before incubating them with primary antibody at 4°C overnight. After adding the appropriate secondary antibody (1:2000) at room temperature for 1 h, membranes were visualized using an enhanced chemiluminescence (ECL) system (Image Quant LAS 4000 mini, Pittsburgh, PA, USA) according to the instructions of the manufacturer. Target protein levels were normalized to GAPDH expression by performing densitometry, and relative fold-changes in protein levels were calculated; all experiments were repeated at least three times. Primary antibodies were as follows: GAPDH (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA); Anti-A20, anti-IKKα, anti-IKKβ, anti-NF-κB P105, anti-NF-κB P50, anti-NF-κB P65, anti-P-NF-κB P65, anti-IKBα, anti-P-IKBα, anti-GSK3β, and anti-P-GSK3β, anti-PCNA, anti-CyclinD1, anti-c-Myc, anti-MMP1, anti-MMP2, anti-MMP9, anti-TIMP-1, anti-TIMP-2, anti-BAX, anti-Bcl2, anti-PARP, anti-cleaved-PARP, anti-Caspase3, anti-cleaved-Caspase3, anti-Caspase9, anti-cleaved-Caspase9, anti-P-Erk1/2, anti-Ras (1:500, all from Cell Signaling, Danvers, MA, USA); horseradish peroxidase (HRP)-conjugated secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Particulates were removed from cell culture supernatants by centrifugation at 3000 × g for 10 min and the assay was immediately performed, or samples were aliquoted and stored at ≤ −20°C. Levels of active IL-6, IL-8, and TNF-a were measured using ELISA kits (eBioscience, Inc., San Diego, CA) following the instructions of the manufacturer.
The proliferation of GES-1 cell was examined using CCK-8 kit (Tongren, Shanghai, China) according to the manufacturer’s instructions. Approximately 2 × 103 cells were seeded in 96-well plates and cultured for 24, 48, 72 and 96 h, respectively. 10 μL CCK-8 solution was then added to each well and incubated for 1 h. The absorbance at 450 nm was measured using automatic microplate reader (Bio-Rad, USA) at different time intervals.
Cells were harvested and seeded into six-well plates (1000 cells/well) and incubated at 37°C in a 5% CO2 humidified incubator for 10 days. The medium was changed at 3-day intervals. At the end of the incubation period, the cultures were fixed with 4% paraformaldehyde and stained with crystal violet.
Transwell migration assay was performed using CoStar Transwell chambers (8 μm pore size; Corning, Costar, NY, USA). Cells (1 × 105/well) were seeded in the upper chambers of the wells in 200 μL serum-free medium, while the lower chambers were filled with 600 μL medium containing 10% fetal bovine serum to induced cell migration. After incubation at 37°C in 5% CO2 for 36 h, the cells in the upper surface of the membrane were removed with a cotton swab. Cells migrated to the lower surface of the membrane were fixed with 4% paraformaldehyde and stained with crystal violet. The images were obtained, and the cells were counted under a microscope.
In situ detection of apoptosis using V-FITC/PI staining
Cells at density of 2.5 × 105 were seeded on glass coverslips in 24-well dishes. After treatment time, the prepared apoptosis staining was added to each well according the manufacturer’s protocol (Beyotime Biotechnology), removed the cover lips and place it on the slides, the slides were mounted and visualized using an inverted wide-filed fluorescence microscope (DeltaVision Elite, GE Healthcare Life Sciences, USA) to identify apoptosis.
Immunofluorescent staining was performed with cells grown on glass cover slips. Cultured cells were fixed for 30 min with 4% formaldehyde, then after rinsing in PBS, the cells were stained with Hoechst 33258 (Sigma, USA) for 5 min. The slides were mounted and visualized using an inverted wide-filed fluorescence microscope (DeltaVision Elite, GE Healthcare Life Sciences, USA).
All results are presented as the mean ± SE of three independent experiments, and analysis was performed using SPSS 22.0 (SPSS, Chicago, IL, USA). Significant differences between groups were measured by performing a Student’s t-test, and P < 0.05 was considered statistically significant. The experimental results are made by GraphPad Prism5.0.
Acknowledgement: We acknowledge the generous support of the Clinical Laboratory Center, Jiangsu Taizhou People’s Hospital; School of Medicine, Jiangsu University. This manuscript was edited by Wallace Academic Editing.
Availability of Data and Materials: All remaining data are available within the article, or available from the authors upon request.
Author Contribution: Conception and design: All authors; Administrative support: CZ; Provision of study materials or patients: CZ; Collection and assembly of data: YY, BY; Data analysis and interpretation: YY, BY; Manuscript writing: All authors; Final approval of manuscript: All authors.
Funding Statement: This work was supported by the Natural Science Foundation of Jiangsu Province (H2018072), Taizhou Social Development Project (TS201623).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Alandiyjany MN, Croxall NJ, Grove JI, Delahay RM (2017a). A role for the tfs3 ICE-encoded type IV secretion system in pro-inflammatory signalling by the Helicobacter pylori Ser/Thr kinase, CtkA. PLoS One 12: e0182144. [Google Scholar]
Alandiyjany MN, Croxall NJ, Grove JI, Delahay RM (2017b). A role for the tfs3 ICE-encoded type IV secretion system in pro-inflammatory signalling by the Helicobacter pylori Ser/Thr kinase. CtkA 12: e0182144. [Google Scholar]
Alm RA, Ling LS, Moir DT, King BL, Brown ED et al. (1999). Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397: 176–180. [Google Scholar]
Alvi A, Devi SM, Ahmed I, Hussain MA, Rizwan M et al. (2007). Microevolution of Helicobacter pylori type IV secretion systems in an ulcer disease patient over a ten-year period. Journal of Clinical Microbiology 45: 4039–4043. [Google Scholar]
Alzahrani S, Lina TT, Gonzalez J, Pinchuk IV, Beswick EJ et al. (2014). Effect of Helicobacter pylori on gastric epithelial cells. World Journal of Gastroenterology 20: 12767–12780. [Google Scholar]
Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ (2017). From basic apoptosis discoveries to advanced selective Bcl-2 family inhibitors. Nature Reviews Drug Discovery 16: 273–284. [Google Scholar]
Backert S, Schwarz T, Miehlke S, Kirsch C, Sommer C et al. (2004). Functional analysis of the cag pathogenicity island in Helicobacter pylori isolates from patients with gastritis, peptic ulcer, and gastric cancer. Infection and Immunity 72: 1043–1056. [Google Scholar]
Bartchewsky WJr., Martini MR, Squassoni AC, Alvarez MC, Ladeira MS et al. (2010). Effects of Helicobacter pylori infection on the expressions of Bax and Bcl-2 in patients with chronic gastritis and gastric cancer. Digestive Diseases and Sciences 55: 111–116. [Google Scholar]
Czabotar PE, Lessene G, Strasser A, Adams JM (2014). Control of apoptosis by the Bcl-2 protein family: Implications for physiology and therapy. Nature Reviews Molecular Cell Biology 15: 49–63. [Google Scholar]
Chmiela M, Gonciarz W (2017). Molecular mimicry in Helicobacter pylori infections. World Journal of Gastroenterology 23: 3964–3977. [Google Scholar]
Fan TJ, Han LH, Cong RS, Liang J (2005). Caspase family proteases and apoptosis. Acta Biochimica et Biophysica Sinica 37: 719–727. [Google Scholar]
Fernandez-Gonzalez E, Backert S (2014). DNA transfer in the gastric pathogen Helicobacter pylori. Journal of Gastroenterology 49: 594–604. [Google Scholar]
Fischer W, Tegtmeyer N, Stingl K, Backert S (2020). Four chromosomal type IV secretion systems in Helicobacter pylori: composition, structure and function. Frontiers in Microbiology 11: 1592. [Google Scholar]
Hofreuter D, Odenbreit S, Henke G, Haas R (1998). Natural competence for DNA transformation in Helicobacter pylori: identification and genetic characterization of the comB locus. Molecular Microbiology 28: 1027–1038. [Google Scholar]
Kersulyte D, Lee W, Subramaniam D, Anant S, Herrera P et al. (2009). Helicobacter pylori’s plasticity zones are novel transposable elements. PLoS One 4: e6859. [Google Scholar]
Kersulyte D, Velapatino B, Mukhopadhyay AK, Cahuayme L, Bussalleu A et al. (2003). Cluster of type IV secretion genes in Helicobacter pylori’s plasticity zone. Journal of Bacteriology 185: 3764–3772. [Google Scholar]
Li H, Liang D (2019). Helicobacter pylori inhibited cell proliferation in human periodontal ligament fibroblasts through the Cdc25C/CDK1/cyclinB1 signaling cascade. Journal of Periodontal & Implant Science 49: 138–147. [Google Scholar]
Lin AS, Dooyema SDR, Frick-Cheng AE, Harvey ML, Suarez G et al. (2020). Bacterial energetic requirements for Helicobacter pylori Cag type IV secretion system-dependent alterations in gastric epithelial cells. Infection and Immunity 88: e00790–00719. [Google Scholar]
Lv G, Zhu H, Zhou F, Lin Z, Lin G et al. (2014). AMP-activated protein kinase activation protects gastric epithelial cells from Helicobacter pylori-induced apoptosis. Biochemical and Biophysical Research Communications 453: 13–18. [Google Scholar]
Mcclelland RS, Sangare L, Hassan WM, Lavreys L, Mandaliya K et al. (2007). Infection with Trichomonas vaginalis increases the risk of HIV-1 acquisition. Journal of Infectious Diseases 195: 698–702. [Google Scholar]
Naumann M, Sokolova O, Tegtmeyer N, Backert S (2017). Helicobacter pylori: A paradigm pathogen for subverting host cell signal transmission. Trends in Microbiology 25: 316–328. [Google Scholar]
Ouyang Y, Liu G, Xu W, Yang Z, Li N et al. (2021). Helicobacter pylori induces epithelial-mesenchymal transition in gastric carcinogenesis via the AKT/GSK3β signaling pathway. Oncology Letters 21: 165. [Google Scholar]
Sanders MK, Peura DA (2002). Helicobacter pylori-associated diseases. Current Gastroenterology Reports 4: 448–454. [Google Scholar]
Sue S, Shibata W, Maeda S (2015). Helicobacter pylori-induced signaling pathways contribute to intestinal metaplasia and gastric carcinogenesis. BioMed Research International 2015: 737621. [Google Scholar]
Tegtmeyer N, Wessler S, Backert S (2011). Role of the cag-pathogenicity island encoded type IV secretion system in Helicobacter pylori pathogenesis. FEBS Journal 278: 1190–1202. [Google Scholar]
Tengku Din TaDaA, Abdul Jalal MI, Seeni A, Shamsuddin S, Jaafar H (2018). The differential roles of caspase family members in mediating PF4-induced breast cancer apoptosis. Malaysian Journal of Pathology 40: 303–312. [Google Scholar]
Xu JC, Zhou XP, Wang XA, Xu MD, Chen T et al. (2019). Cordycepin induces apoptosis and G2/M phase arrest through the ERK pathways in esophageal cancer cells. Journal of Cancer 10: 2415–2424. [Google Scholar]
Yamaoka Y (2010). Mechanisms of disease: Helicobacter pylori virulence factors. Nature Reviews Gastroenterology & Hepatology 7: 629–641. [Google Scholar]
Zhu Y, Chen M, Gong Y, Liu Z, Li A et al. (2015). Helicobacter pylori FKBP-type PPIase promotes gastric epithelial cell proliferation and anchorage-independent growth through activation of ERK-mediated mitogenic signaling pathway. FEMS Microbiology Letters 362: fnv023. [Google Scholar]
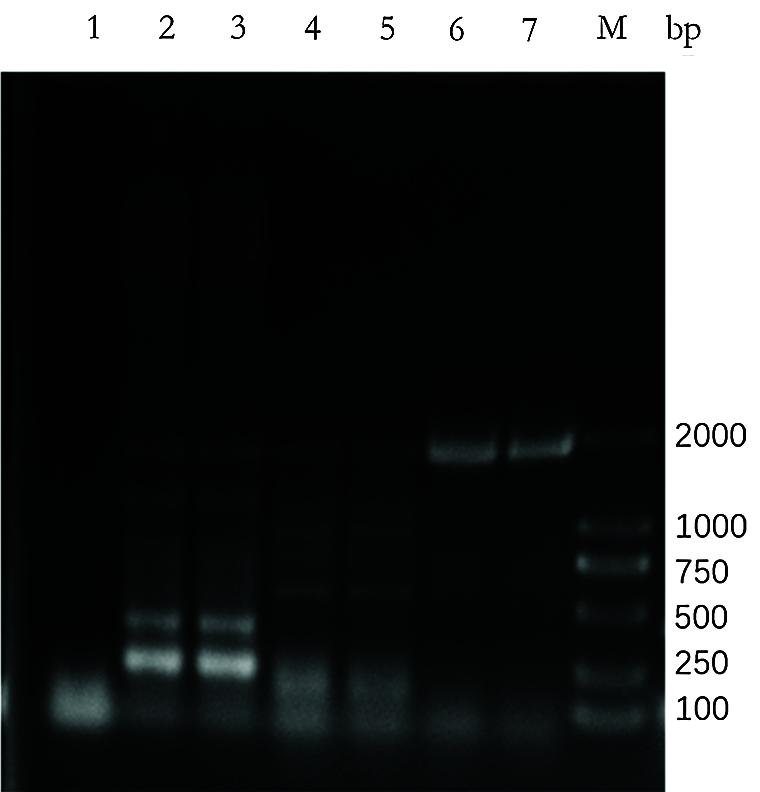
Supplementary Figure S1: Expression of VirD4 in different H. pylori type strains. The full length of VirD4 gene was about 1728 bp. VirD4 is highly expressed in the pylori clinical strain SBK (lanes 6, 7) but low expression in H. pylori type strain 11637 (lanes 2, 3) and 26695 (lanes 4, 5). Lane 1 is the negative control.
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |