DOI:10.32604/biocell.2022.017745
| BIOCELL DOI:10.32604/biocell.2022.017745 | |
| Article |
Murine double minute gene 2 (MDM2) promoted hepatocellular carcinoma (HCC) cell growth by targeting fructose-1,6-bisphosphatase (FBP1) for degradation
1Department of Intensive Care Unit, Changzhou No. 3 People’s Hospital, Changzhou, 213000, China
2Department of General Surgery, Changzhou No. 3 People’s Hospital, Changzhou, 213000, China
3Department of Gastroenterology, Changzhou No. 1 People’s Hospital, Changzhou, 213000, China
4Department of Liver Disease, Changzhou No. 3 People’s Hospital, Changzhou, 213000, China
*Address correspondence to: Jiakai Jiang, jiangjiakai8338@163.com; Suobao Xu, 13196797210@163.com
#These authors have contributed equally to this work
Received: 02 June 2021; Accepted: 19 July 2021
Abstract: To study the roles and association of murine double minute gene 2 (MDM2) and fructose-1,6-biphosphatase (FBP1) in human hepatocellular carcinoma (HCC), growth response of human HCC cells was assessed using proliferation and apoptosis assay. Pro-survival AKT signaling associated proteins (p-AKT, survivin and cleaved caspase 3) were assessed using western blotting. The correlation between MDM2 and FBP1 was assessed using co-immunoprecipitation combined with ubiquitination assay. Our data suggested that low expression of FBP1 was correlated with high levels of MDM2 in HCC cell lines (Huh7 and Hep3B). Overexpression of FBP1 resulted in anti-proliferation, pro-apoptosis, the up-regulation of cleaved caspase 3 while the downregulation of survivin and phosphor (p)-AKT, however, knockdown of FBP1 led to the opposite. Furthermore, overexpression of MDM2 potently reversed FBP1-induced proliferation inhibition and apoptosis, while Nutlin-3 (an MDM2 inhibitor) reversed the change trends induced by FBP1 knockdown in the aforementioned events. Lastly, but not least importantly, our data elucidated that MDM2 binds directly to FBP1 and promotes FBP1 ubiquitination. In conclusion, our data firstly suggested the involvement of FBP1 and its association with MDM2 in HCC cell growth. MDM2-FBP1-regulated HCC cell growth and the activation of AKT were mediated, at least in part, through FBP1 degradation.
Keywords: Human hepatocellular carcinoma; MDM2; FBP1; FBP1 ubiquitination
Due to a lack of treatment options, currently, hepatocellular carcinoma (HCC) has constituted the 5th most frequent and accounted for 3rd most lethal malignancies worldwide (Zender and Kubicka, 2008). According to a reported study, HCC treatment has been focused on targeted therapy rather than systemic chemotherapy that aims to inhibit pathways which are essential for tumor growth, even in the late stages of carcinogenesis (Zender and Kubicka, 2008). It is believed that, presently, murine double minute gene 2 (MDM2), also known as HDM2, appears to be the prospect target for molecular therapy of HCC (Meng et al., 2014). In HCC, MDM2 behaves as an independent oncogene and contributes to cancer process via negatively regulating tumor suppressor p53-mediated transcriptional transactivation (Cao et al., 2020; Gramantieri et al., 2020). However, the specific mechanisms, by which MDM2 regulates HCC tumorigenesis have not been fully understood. MDM2 is a substrate of pro-survival AKT pathway, a core component of PI3K/Akt/mTOR signaling that is implicated in HCC tumor growth (Zhou et al., 2011). In human HCC tissues, reduction in p-AKT significantly correlates with nuclear FBP1 accumulation (Samarin et al., 2016). Human breast cancer investigation from Xiong et al. (2016) have exposed a restriction in MDM2 expression leading to the decreased in AKT activation. However, the association between FBP1 and MDM2 in HCC remained extremely poor.
It has been reported that hepatocytes are equipped with a mechanism to control the rate of hepatic gluconeogenesis in response to energy status (Hunter et al., 2018). Fructose-1,6-bisphosphatase (FBP1) has long been recognized as a rate-limiting enzyme in gluconeogenesis (Hunter et al., 2018), and in the presence of divalent cations, FBP1 can catalyze the hydrolysis of fructose-1,6-bisphosphate (F-1,6-BP) to fructose 6-phosphate and inorganic phosphate (Dai et al., 2017). A study have shown that due to impaired gluconeogenesis, individuals with FBP1 deficiency can cause hypoglycemia and metabolic acidosis (Bijarnia-Mahay et al., 1993). Metformin could reduce liver glucose production by inhibition of fructose-1-6-bisphosphatase (Hunter et al., 2018). In addition to its function as a metabolic enzyme, FBP1 also found to be a tumor suppressor in multiple cancers (Liao et al., 2020; Lu et al., 2020), and lost in various cancers, such as HCC, kidney cancer and lung cancer (Alderton, 2014; Zhang et al., 2016), and loss of FBP1 is associated with advanced tumor stage and poor prognosis (Chen et al., 2011; Zhang et al., 2016). For example, FBP1 is found to oppose renal carcinoma progression (Li et al., 2014). Studies also have shown that FBP1 may act as a co-suppressor for multiple transcription factors to reduce downstream gene expression and control various cellular processes including proliferation and apoptosis (Chen et al., 2021; Hu et al., 2021; Liu et al., 2017). Loss of FBP1 can induce glycolysis and promote growth and apoptosis resistance in cancer cells (Dai et al., 2017; Li et al., 2013). There is also revealed that decreased expression of FBP1 is closely associates with glucose metabolism and tumor progression in HCC (Hirata et al., 2016). Mounting studies have elucidated that FBP1 is frequently lost in HCC, and strategies to restore the levels and activities of FBP1 in tumor cells may affect cellular processes in carcinogenesis (Yang et al., 2017b).
This study selected two HCC cell lines Huh7 and Hep3B to analyze the effect of FBP1 and its association with MDM2 on HCC cell growth and the underlying molecular mechanism. Our data not only identified MDM2-mediated FBP1 ubiquitination as a key mechanism to down-regulate FBP1 proteins levels in HCC, but also revealed that MDM2-regulated cancer progress of HCC is mediated, at least in part, through FBP1 degradation.
Culture medium for Huh7 and Hep3B cells (ATCC, Manassas, VA, USA) was: 90% (v/v) of Hyclone DMEM (SH30243.01), 100 U/mL of penicillin (Solarbio, Beijing, China) and 10% (v/v) of heat-inactivated fetal bovine serum (FBS) (Gibco Laboratories). Those two HCC cell lines grew in this medium at 37°C under 5% CO2 to reach the confluency above 80%.
Cell counting kit-8 (CCK-8) assay
After treatment of Vector, oeFBP1, oeMDM2 + Vector, oeMDM2 + oeFBP1, or siNC, siFBP1, Nutlin-3 + siNC, Nutlin-3 + siFBP1-1 at 12, 24, 48 h, Huh7 and Hep3B cells (3 × 103 cell/well in a 96-wellplate) was incubated with cultured medium (90 μL/well) containing CCK-8 working solution (10 mL/well) (P002, SAB) at 37°C for 1 h. Optical density (OD) value at 450 nm was recorded following the provided instruction.
For analysis of Huh7 and Hep3B cell apoptosis, an Annexin V-FITC/PI apoptosis detection kit was used (C1063, Beyotime, Shanghai, China). According to the manufacturer’s instructions, 5 × 105–1 × 106 cells were resuspended in 195 µL Annexin V-FITC binding buffer, followed by incubated in 5 µL Annexin V-FITC for 15 min at 4°C in the dark. Subsequently, the cells were incubated in 5 µL PI for 5 min at 4°C in the dark. A tube without incubation of both Annexin V-FITC and PI was a control. Cells underwent apoptosis were annexin V-FITC-positive staining while propidium iodide (PI)-negative staining, and analyzed using BD Accuri C6 Flowcytometry (Biosciences, CA, USA) with FlowJo v10 (Tree Star, Ashland, OR).
The primers of human FBP1 gene (NCBINM_000507.3) were: 5’-CGGAATTCATGGCTGACCAGGCGC-3’ (forward) and 5’- CGGGATCCTCACTGGGCAGAGTGCTTCTC-3’ (reverse). The primers of human MDM2 gene (NCBINM_002392.5) were: 5’-CGGAATTCATGGTGAGGAGCAGGCAAATG-3’ (forward) and 5’-CGGGATCCCTAGGGGAAATAAGTTAGCACAATC-3’ (reverse). Primers of human FBP1 and MDM2 were implanted into pLVX-Puro vector (Clontech), named pLVX-Puro-FBP1 and pLVX-Puro-MDM2, respectively. pLVX-Puro-FBP1, psPAX2 and pMD2G (Addgen) were packaged in 293T cells (ATCC) using Lipofectamine™ 2000 (Invitrogen, CA, USA) for 4–6 h. Then 293T cells were placed in a complete medium, and lentiviruses with FBP1 expression were harvested at 48 h and 72 h. Lentiviruses with MDM2 expression was obtained in the same way. Lentiviruses with FBP1 or MDM2 expression were transfected into Huh7 and Hep3B cells using Lipofectamine 2000 reagent. Meanwhile, Huh7 and Hep3B cells transfected with blank lentiviruses without FBP1 or MDM2 expression were used as the control.
Three siRNA targeting human FBP1 mRNA (NM_000507.3) were designed in Table 1. Hep3B cells were transfected with siRNAs using Lipofectamine® 2000 Reagent following the provided instruction, whereas transfected with non-specific siRNA (siRNA-NC) that composed the inverted sequence (Sense: 5’-CAGUACUUUUGUGUAGUACAA-3’ and antisense: 5’-UUGUACUACACAAAAGUACUG-3’) served as negative control.

Huh7 and Hep3B cells were treated with Trizol Reagent (1596-026, Invitrogen) to isolate total RNA, and 200 ng of RNA were reverse transcribed using cDNA synthesis kit (Fermentas). The primers used in this study were shown in Table 2. RT-PCR was run using SYBR Green PCR Kit (Thermo Fisher) on an ABI7300 system (Applied Biosystem). mRNA levels of MDM2 and FBP1 was normalized by β-actin using 2−ΔΔCt method (Livak and Schmittgen, 2001).

Quantitative analysis of total protein extract from Huh7 and Hep3B cells was performed using BCA protein assay kit (Thermo, MA, USA), and 25 μg of which was run on 15% SDS-PAGE for seperation. Electrophoretically pure containing FBP1, MDM2, cleaved caspase3, survivin, AKT, p-AKT and β-actin were transported onto PVDF membranes (Millipore, USA). After blocked with 5% nonfat milk, the membranes were maintained with primary antibodies at 4°C overnight followed by secondary antibodies (Beyotime, Shanghai, China) for 1 h at room temperature in the dark. Immunoreactive bands for FBP1, MDM2, cleaved caspase3, Survivin, AKT, p-AKT and β-actin were analyzed using ECL system (GE Healthcare/Amersham Biosciences) with ImageJ software (NIH, Bethesda, USA). The involved primary antibodies were: anti-FBP1 antibody (Ab109020, Abcam), anti-MDM2 antibody (Ab16895, Abcam), antibody against cleaved caspase3 (Ab13847, Abcam), anti-survivin antibody (Ab76424, Abcam), anti-AKT antibody (#4691, CST) antibody against p-AKT (#4060, CST), and antibody against β-actin (Ab8227, Abcam).
Co-Immunoprecipitation (Co-IP)
30 μL of protein G-Agarose beads (Roche) and 2 mg of protein extract from Hep3B cells were fully mixed, then underwent immune-precipitation with anti-FBP1 (Ab109732, Abcam), anti-MDM2 (Ab16895, Abcam) or control IgG antibody (1 μg) overnight at 4°C. After centrifugation for 4 min at 3000 rpm at 4°C, 1 mL of lysate were added to wash the Protein A/G Plus-Agarose beads 4 times, and appropriate protein loading buffer were added to boil for 5 min. Following centrifugation at 3000 rpm for 1 min, the supernatants were collected for western blot analysis. The content of FBP1 and MDM2 in the immune–complex was assessed using western blot method. Same amount of total protein extract before every treatment was used as an input control.
Hep3B with MDM2 overexpression was homogenized, and protein extract (100 μg) was mixed with protein A/G PLUS-Agarose (Santa Cruz Biotechnology, sc-2003) thoroughly, and then underwent immune-precipitation using 1 μg of IgG (Santa Cruz Biotechnology, sc-2027) or Anti-FBP1 antibody (ab227499). Levels of ubiquitinated FBP1 (Ub-FBP1) in immune-precipitation was evaluated using Anti-ubiquitin antibody (ab7780) by western blot.
For data analysis, GraphPad Prism®7 software (La Jolla, CA, USA) was used. Results were described as mean ± standard error of the mean (SEM), and one-way ANOVA with Bonferroni correction was conducted for multiple comparisons with P-value < 0.05 being statistically significance.
MDM2 directly interacted with FBP1 and regulated FBP1 ubiquitination
To study the interaction between MDM2 and FBP1 in cancer progress of HCC in vitro, lentiviral-mediated MDM2 overexpression (oeMDM2) was transfected into Huh7 and Hep3B cells, respectively. The results showed that in Huh7 and Hep3B cells, both MDM2 mRNA and protein were significantly increased in oeMDM2 group, in comparison to Vector (Figs. S1A and S1B). Later, expression of FBP1 in Huh7 and Hep3B cells transfected with oeMDM2 was determined. Figs. 1A and 1B showed that overexpression of MDM2 decreased mRNA expression of FBP1 within Huh7 and Hep3B cells by approximate 18% and 21%, respectively, while reduced protein expression of FBP1 within Huh7 and Hep3B cells by approximate 63% and 45%, respectively, when compared with Vector. Fig. 1C showed that there was a physical interaction between MDM2 and FBP1 within HCC cells. We then studied whether MDM2 acted as an E3 ubiquitin ligase to regulate FBP1 degradation in human HCC cells. Fig. 1D showed that Ub-FBP1 was significantly enhanced in oeMDM2 group, comparison to Vector. To confirm that MDM2 may act as a ubiquitin ligase that accelerated FBP1 ubiquitination, and subsequently favoring FBP1 degradation, Hep3B cells transfected with oeMDM2 lentivirus were treated with proteasome inhibitor MG132 (10 µmol/L). Fig. 1E showed that MG132 significantly enhanced FBP1 protein when compared with Vector or oeMDM2, substantiating that protein levels of FBP1 was determined in an ubiquitin-proteasome pathway, which is consistent with the evidence that MDM2 regulates protein ubiquitination of FBP1 (http://ubibrowser.ncpsb.org/ubibrowser/).
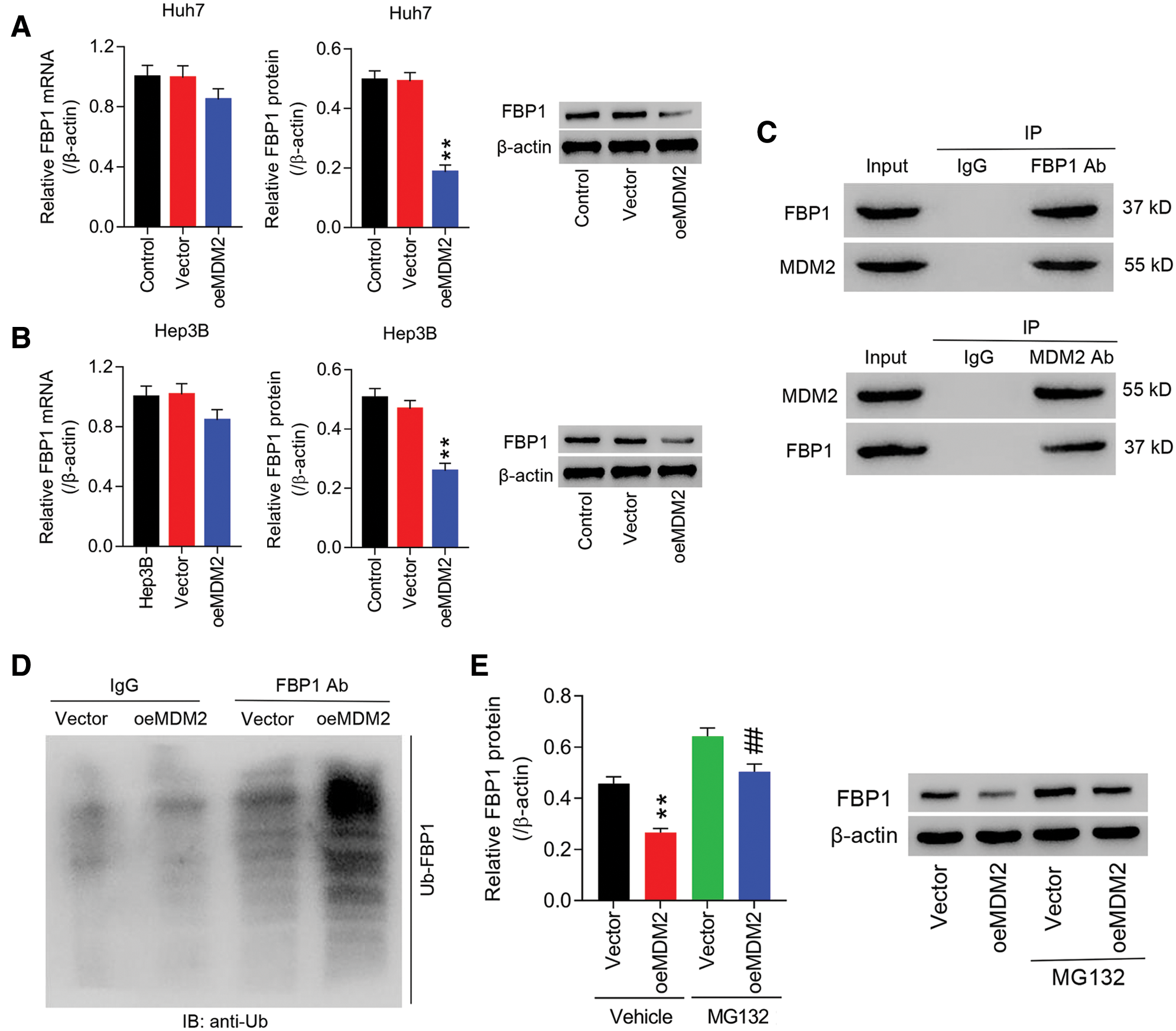
Figure 1: MDM2 was correlated with FBP1, and influence FBP1 ubiquitinoylation in human HCC cells. (A, B) oeMDM2 significantly reduced FBP1 mRNA and protein within Huh7 (A) and Hep3B (B) cells. **P < 0.01: oeMDM2 vs. Vector. (C) Following the Co-IP with anti-FBP1 antibody (or anti-MDM2 antibody), the presence of MDM2 (or FBP1) in Hep3B was detected by Western blot. (D) Ubiquitination assay demonstrating the enhanced Ub-FBP1 in Hep3B cells induced by MDM2 overexpression. (E) Hep3B cells with MDM2 overexpression was treated with MG132 (10 µmol/L) for 24 h, and then protein levels of FBP1 were assessed, using western blot. **P < 0.01: Vehicle + oeMDM2 vs. Vehicle + Vector; ##P < 0.01: MG132 + oeMDM2 vs. Vehicle + oeMDM2.
Overexpression of MDM2 ameliorated FBP1-induced the inhibition of HCC cell growth and the blockade of AKT pathway
To study the roles of FBP1 in HCC cell growth, Huh7 and Hep3B cells were treated with oeFBP1. The results showed that in Huh7 and Hep3B cells, both FBP1 mRNA and protein were significantly increased in oeFBP1 group, in comparison to Vector (Figs. S2A and S2B). Furthermore, our data showed that oeFBP1 significantly reduced the proliferation of Huh7 cells (Fig. 2A) by approximate 5.87%, 9.87% and 23.75% at 12, 24, and 48 h, respectively, while the apoptotic ratio of oeFBP1 group was evidently increased (Fig. 2B). Furthermore, oeFBP1 significantly reduced the proliferation of Hep3B cells (Fig. 2C) by approximate 6.42%, 10.49% and 25.20% at 12, 24, and 48 h, respectively, while the apoptotic ratio of oeFBP1 group was evidently enhanced (Fig. 2D) (all P < 0.01), demonstrating the anti-growth effect of FBP1 in HCC cell lines.
AKT pathway crucially regulates cell survival and apoptosis, and AKT regulates survivin expression, which is involved in tumorigenesis by modulating cell division, blocking caspase activation, and inhibiting apoptosis. To investigate the molecular mechanism of FBP1 in growth inhibition of Huh7 and Hep3B cells, protein expression of survivin, cleaved caspase 3 and p-AKT were assessed. Figs. 2E and 2F showed that both in Huh7 and Hep3B cells, overexpression of FBP1 dramatically increased cleaved caspase 3 while decreased survivin and p-AKT when compared with Vector (all P < 0.01), yet has almost no effect on total AKT expression, demonstrating the pro-apoptotic effect of FBP1 at molecular mechanism, and during which was associated with the blockade of AKT pathway.
To study the involvement of MDM2 in anti-growth effect of FBP1 on HCC cells, Huh7 and Hep3B cells were treated with oeFBP1 + oeMDM2. Fig. 2E showed that FBP1 expression in Vector + Vector or Vector + oeFBP1 group was significantly reduced by additional oeMDM2 treatment, suggesting the involvement of MDM2 in regulating FBP1 expression. Moreover, both in Huh7 and Hep3B cells, FBP1-induced proliferation inhibition and apoptosis were dramatically inhibited by MDM2 overexpression (Figs. 2A–2D). Currently, the protein expression of cleaved caspase 3 was significantly decreased, while survivin and p-AKT were increased when compared with Vector + Vector group or Vector + oeFBP1 groups (Figs. 2E and 2F) (all P < 0.01), suggesting the participation of MDM2 in regulating cell growth in HCC cells expressing the basal or enhanced level of FBP1.
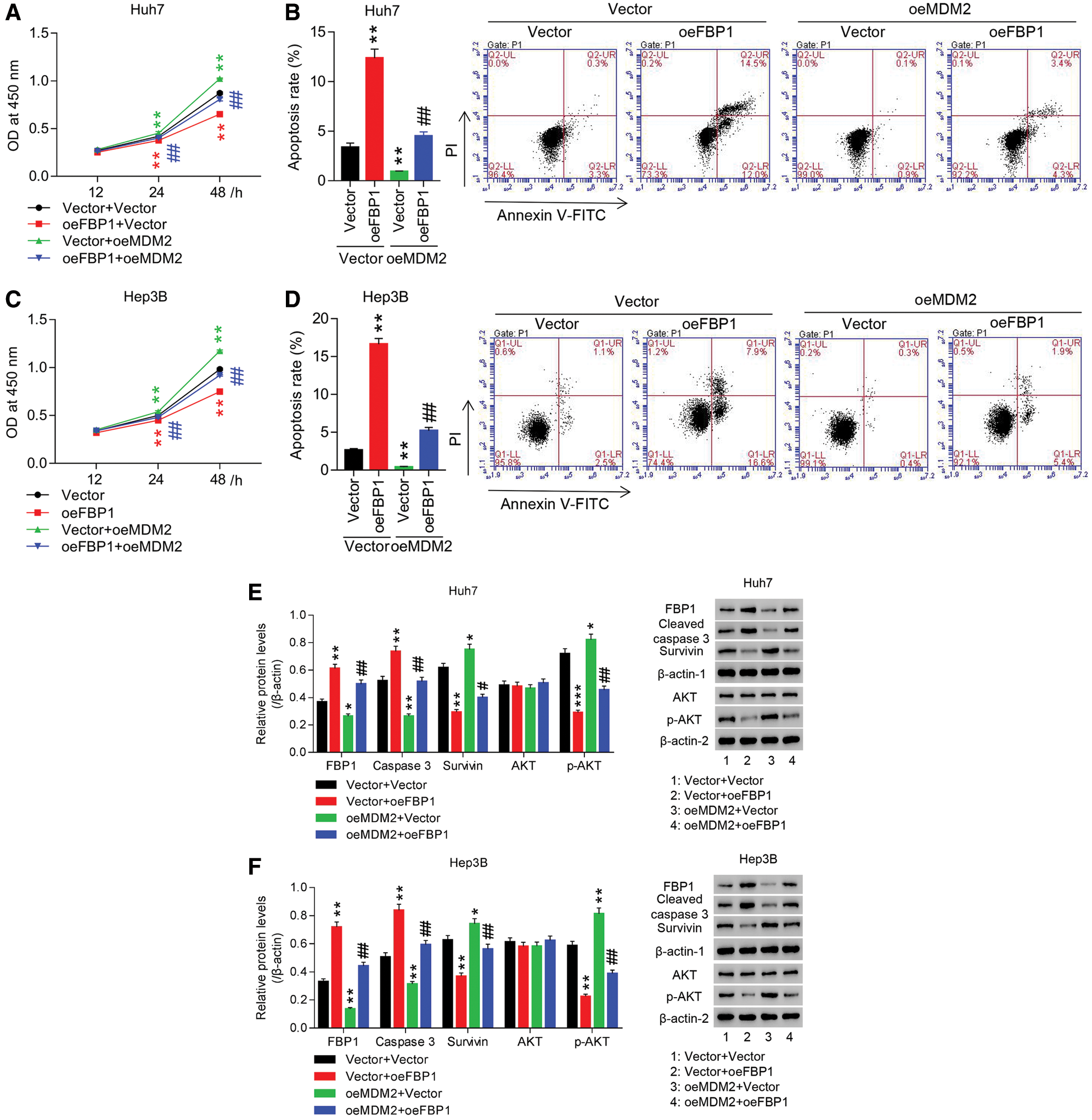
Figure 2: Overexpression of MDM2 inhibited FBP1-induced anti-growth effect and the blockade of AKT pathway in HCC cell lines. (A, C) CCK-8 analysis showed that anti-proliferative effect of oeFBP1 was significantly inhibited by oeMDM2 in a time-dependent manner in both Huh7 (A) and Hep3B (C) cells. (B, D) Flow cytometry showed that pro-apoptotic effect of oeFBP1 was significantly inhibited by oeMDM2 in both Huh7 (B) and Hep3B (D) cells. (E, F) Western blot analysis showed that oeFBP1 up-regulated FBP1 and cleaved caspase 3 while down regulated survivin and p-AKT in Huh7 (E) and Hep3B (F) cells, yet oeFBP1-induced the change treads of those proteins was significantly reversed by additional oeMDM2 treatment. FBP1, cleaved caspase 3 and survivin were normalized using β-actin-1, AKT was normalized using β-actin-2, and p-AKT was normalized using total AKT. *P < 0.05, **P < 0.01, ***P < 0.001: Vector + oeFBP1 or oeMDM2 + Vector vs. Vector + Vector; #P < 0.05, ##P < 0.01: oeMDM2 + oeFBP1 vs. Vector + oeFBP1.
Nutlin-3 ameliorated siFBP1-induced HCC cell growth and the activation of AKT pathway
To substantiate the roles of FBP1 in HCC cell growth, Hep3B cells were transfected with siRNA-FBP1 (siFBP1). Fig. S3 showed that siFBP1 (siFBP1-1, siFBP1-2, and siFBP1-3) significantly reduced FBP1 mRNA and protein expression in Hep3B cells with the maximum effect being obtained in siFBP1-1, which was chosen for the following studies. Figs. 3A and 3B showed that down-regulation of FBP1 reduced apoptotic ratio of Hep3B cells by approximate 80% at 48 h when compared with siNC, while enhanced proliferation of Hep3B cells in a time-dependent manner with the promoted rate being 21.75% at 48 h (all P < 0.01). Furthermore, Fig. 3C showed that down-regulation of FBP1 significantly decreased cleaved caspase 3 expression, while increased survivin and p-AKT expression, demonstrating an obvious anti-growth effect of FBP1 in HCC cell lines, and the activation of AKT pathway in this process.
To substantiate the involvement of MDM2 in pro-growth effect of FBP1 on HCC cells, Hep3B cells were treated with siFBP1 + Nutlin-3 (a specific MDM2 inhibitor). Fig. 3C showed that FBP1 expression in siFBP1 group was significantly enhanced by additional Nutlin-3 treatment, suggesting the involvement of Nutlin-3 in regulating FBP1 expression. Our data showed that siFBP1-inhibited apoptosis and increased proliferation were dramatically reversed by Nutlin-3 in Hep3B cells. Furthermore, the protein expression of cleaved caspase 3 was significantly increased, while survivin and p-AKT were decreased, demonstrating an obvious pro-growth effect of Nutlin-3 in HCC cell lines expressing the basal or reduced level of FBP1, and the blockade of AKT pathway in this process.
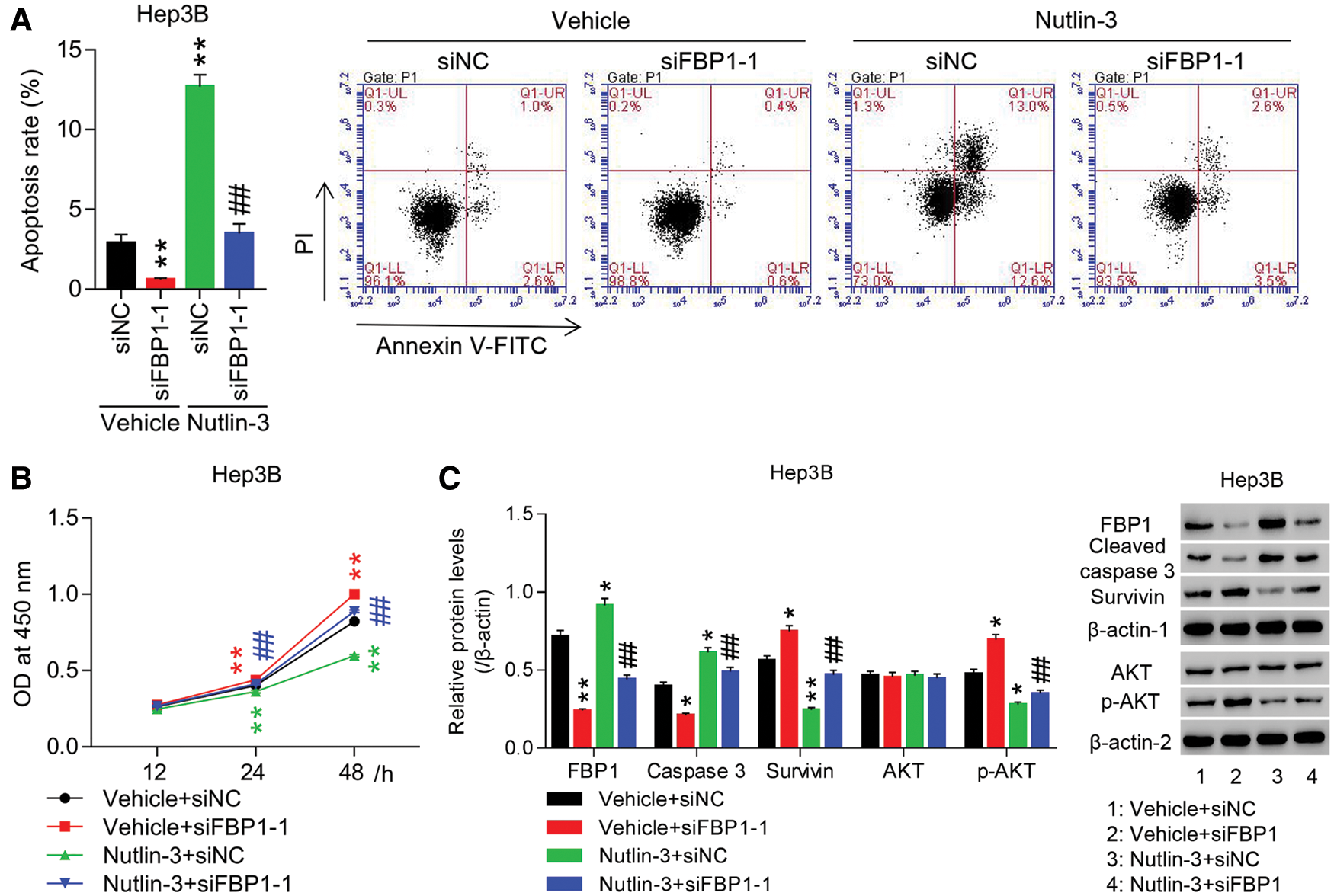
Figure 3: Knockdown of FBP1 promoted HCC cell growth and activated AKT pathway, which was significantly reversed by additional Nutin-3 treatment. HCC cells (Hep3B) transfected with siFBP1-1 were treated with Nutin-3 (an MDM2 inhibitor), and then (A) apoptosis, (B) proliferation, and (C) protein expression of FBP1, cleaved caspase 3, survivin and p-AKT were assessed. FBP1, cleaved caspase 3 and survivin were normalized using β-actin-1, AKT was normalized using β-actin-2, and p-AKT was normalized using total AKT. *P < 0.05, **P < 0.01: Vehicle + siFBP1-1 or Nutlin-3 + siNC vs. Vehicle + siNC; ##P < 0.01: Nutlin-3 + siFBP1-1 vs. Vehicle + siFBP1-1.
FBP1 is a tumor suppressor in HCC and exerts anti-growth effect on HCC cell growth, most likely by altering glucose metabolism (Hirata et al., 2016; Yang et al., 2017a). Studies have revealed the role of FBP1 in altered glucose metabolism in HCC (Hirata et al., 2016). In this study, our results confirmed the anti-proliferation of FBP1 on HCC cell lines (Yang et al., 2017a), however, firstly suggested the pro-apoptotic effect of FBP1 in parallel, which was associated with the up-regulation of cleaved caspase 3 (pro-apoptotic protein) while down-regulation of survivin (anti-apoptotic protein) (Fig. 2). Based on previous reports, hepatocyte apoptosis is associated with the early up-regulation of mitochondrial glucose metabolism (Gottschalk et al., 2012; Yang et al., 2021). Here, we speculate that it may be due to the dysregulation of glucose metabolism that leads the apoptosis of these HCC cells. In addition, AKT pathway is famous to counteract apoptosis, and enhanced p-AKT significantly diminishes FBP1 accumulation in human HCC cells (Samarin et al., 2016). In this work, our data elucidated the involvement of the inactivation of AKT pathway in the anti-growth effect of FBP1 in human HCC cells.
MDM2 is overexpressed in HCC and contributes to HCC cell growth and apoptosis, probably by influencing the expression and activity of p53 (Azer, 2018). More intriguingly, a direct correlation between MDM2 expression levels and HCC cell growth was confirmed in this study. Our data showed that overexpression of MDM2 within HCC cells promoted cell proliferation while reduced apoptosis (Fig. 2). Gastric cancer investigation from Song et al. (2018) exposes arestriction in Akt/MDM2 pathway leading to cell growth inhibition. In this study, our results showed that MDM2 enhanced the expression of p-AKT and influenced the expression of its downstream targets: cleaved caspase 3 and survivin, however, whether there was a loop between MDM2/AKT pathway in regulating HCC cell growth has been included in our future study. Nutlin-3, a MDM2 inhibitor, prevents HCC cell growth by suppressing the combination of MDM2 with p53 or p73, resulting in the activation of p53- or p73-mediated apoptosis signaling (Wang et al., 2011). In this study, our data confirmed the inhibited effect of Nutlin-3 on HCC cells (Fig. 3), which was accompanied with the inactivation of AKT pathway, yet, whether Nutlin-3 directly regulated AKT pathway to influence HCC growth remained largely unknown and needed for further study in our lab.
Our data showed that MDM2 inhibited FBP1 transcription and translation (Figs. 1A and 1B), implying that MDM2 may be a key mediator of FBP1-induced inhibited effect on HCC cell growth. Increased ubiquitination of FBP1 has been implicated in retarding cancer cell proliferation and tumorigenesis (Atanassov and Dent, 2011). MDM2 is known as an oncogenic E3 ubiquitin ligase (Nihira et al., 2017), and the UbiBrowser website predicts that MDM2 may be involved in regulating FBP1 ubiquitination (http://ubibrowser.ncpsb.org/ubibrowser/strict/networkview/networkview/name/P09467/jobId/ubibrowse-I2019-06-05-32017-1559700243). Herein, we found that in HCC in vitro, the inhibited effect of MDM2 on protein levels of FBP1, while the mRNA levels of FBP1 have no significant changes (Figs. 1A and 1B). In this work, our data firstly elucidated that, by ubiquitinating FBP1, MDM2 accelerated the degradation of FBP1 protein, and most likely to favor the subsequent growth of HCC cells (Figs. 2 and 3).
There are also certainly some limitations in this study. For example, this study was mainly performed in cells. Future studies in animal models or using clinical samples will provide more relevant data. Although further studies are needed, the current study reports a new mechanism underlying the occurrence and development of HCC.
Collectively, our results show that FBP1 and MDM2 are inversely correlated in HCC in vitro. Repression of FBP1 by MDM2 has important implications for HCC cell growth, and the underlying mechanism involved in MDM2-mediated the ubiquitination degradation of FBP1 in this process (as shown in Fig. 4).
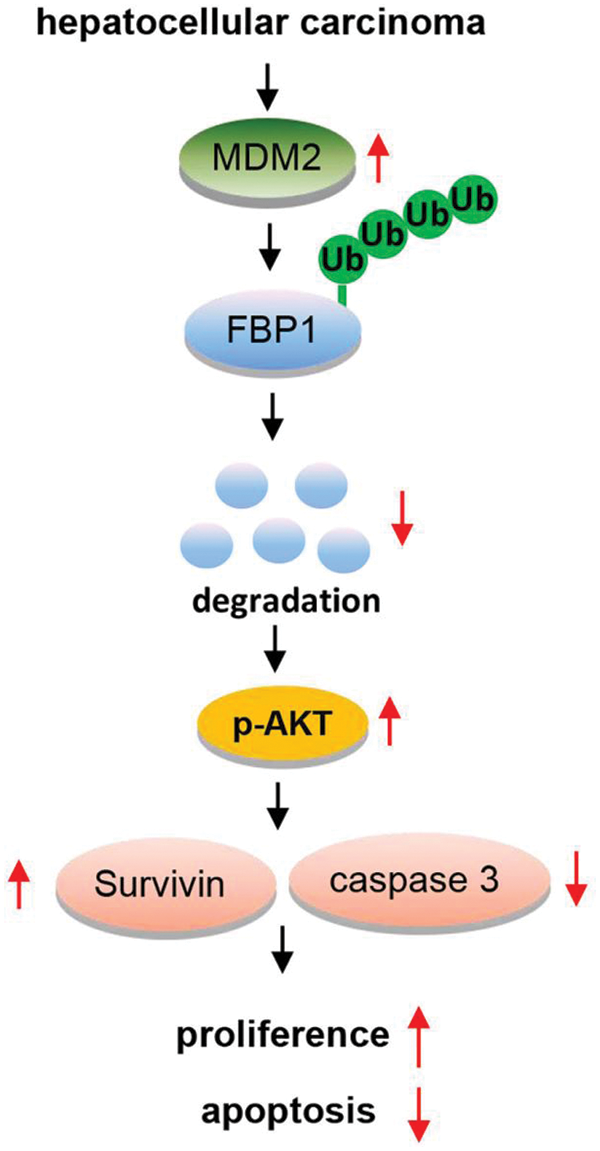
Figure 4: An overview of MDM2/FBP1 in hepatocellular carcinoma.
Authors’ Contribution: S-BX and J-KJ designed the study; YX and BW performed the experiment and made data entry; JY, SZ and L-GL made data collection/entry/analysis; J-KJ drafted the manuscript, and given final approval of the version to be published.
Availability of Data and Materials: All data generated or analyzed during this study are included in this published article (and its supplementary information files).
Funding Statement: The study was supported by the Grant QN201706 from Young Talent Training Project of Health Development Planning Commission of Changzhou City, and Grant CE20205034 from Science and Technology Supporting Project of Changzhou City.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Alderton GK (2014). Tumorigenesis: FBP1 is suppressed in kidney tumours. Nature Reviews Cancer 14: 575. DOI 10.1038/nrc3810. [Google Scholar] [CrossRef]
Atanassov BS, Dent SY (2011). USP22 regulates cell proliferation by deubiquitinating the transcriptional regulator FBP1. EMBO Reports 12: 924–930. DOI 10.1038/embor.2011.140. [Google Scholar] [CrossRef]
Azer SA (2018). MDM2-p53 interactions in human hepatocellular carcinoma: What is the role of nutlins and new therapeutic options? Journal of Clinical Medicine 7: 64. DOI 10.3390/jcm7040064. [Google Scholar] [CrossRef]
Bijarnia-Mahay S, Bhatia S, Arora V (1993). Fructose-1,6-bisphosphatase deficiency. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (eds.) GeneReviews®. Seattle, WA: University of Washington, Seattle. [Google Scholar]
Cao H, Chen X, Wang Z, Wang L, Xia Q, Zhang W (2020). The role of MDM2-p53 axis dysfunction in the hepatocellular carcinoma transformation. Cell Death Discovery 6: 53. DOI 10.1038/s41420-020-0287-y. [Google Scholar] [CrossRef]
Chen L, Yuan R, Wen C, Liu T, Feng Q et al. (2021). E3 ubiquitin ligase UBR5 promotes pancreatic cancer growth and aerobic glycolysis by downregulating FBP1 via destabilization of C/EBPα. Oncogene 40: 262–276. DOI 10.1038/s41388-020-01527-1. [Google Scholar] [CrossRef]
Chen M, Zhang J, Li N, Qian Z, Zhu M et al. (2011). Promoter hypermethylation mediated downregulation of FBP1 in human hepatocellular carcinoma and colon cancer. PLoS One 6: e25564. DOI 10.1371/journal.pone.0025564. [Google Scholar] [CrossRef]
Dai J, Ji Y, Wang W, Kim D, Fai LY et al. (2017). Loss of fructose-1,6-bisphosphatase induces glycolysis and promotes apoptosis resistance of cancer stem-like cells: An important role in hexavalent chromium-induced carcinogenesis. Toxicology and Applied Pharmacology 331: 164–173. DOI 10.1016/j.taap.2017.06.014. [Google Scholar] [CrossRef]
Gottschalk S, Zwingmann C, Raymond VA, Hohnholt MC, Chan TS, Bilodeau M (2012). Hepatocellular apoptosis in mice is associated with early upregulation of mitochondrial glucose metabolism. Apoptosis 17: 143–153. DOI 10.1007/s10495-011-0669-y. [Google Scholar] [CrossRef]
Gramantieri L, Pollutri D, Gagliardi M, Giovannini C, Quarta S et al. (2020). MiR-30e-3p influences tumor phenotype through MDM2/TP53 axis and predicts sorafenib resistance in hepatocellular carcinoma. Cancer Research 80: 1720–1734. DOI 10.1158/0008-5472.CAN-19-0472. [Google Scholar] [CrossRef]
Hirata H, Sugimachi K, Komatsu H, Ueda M, Masuda T et al. (2016). Decreased expression of fructose-1,6-bisphosphatase associates with glucose metabolism and tumor progression in hepatocellular carcinoma. Cancer Research 76: 3265–3276. DOI 10.1158/0008-5472.CAN-15-2601. [Google Scholar] [CrossRef]
Hu J, Wang J, Li C, Shang Y (2021). Fructose-1,6-bisphosphatase aggravates oxidative stress-induced apoptosis in asthma by suppressing the Nrf2 pathway. Journal of Cellular and Molecular Medicine 25: 5001–5014. DOI 10.1111/jcmm.16439. [Google Scholar] [CrossRef]
Hunter RW, Hughey CC, Lantier L, Sundelin EI, Peggie M et al. (2018). Metformin reduces liver glucose production by inhibition of fructose-1-6-bisphosphatase. Nature Medicine 24: 1395–1406. DOI 10.1038/s41591-018-0159-7. [Google Scholar] [CrossRef]
Li B, Qiu B, Lee DSM, Walton ZE, Ochocki JD et al. (2014). Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 513: 251–255. DOI 10.1038/nature13557. [Google Scholar] [CrossRef]
Li H, Wang J, Xu H, Xing R, Pan Y et al. (2013). Decreased fructose-1,6-bisphosphatase-2 expression promotes glycolysis and growth in gastric cancer cells. Molecular Cancer 12: 110. DOI 10.1186/1476-4598-12-110. [Google Scholar] [CrossRef]
Liao K, Deng S, Xu L, Pan W, Yang S et al. (2020). A feedback circuitry between polycomb signaling and fructose-1,6-bisphosphatase enables hepatic and renal tumorigenesis. Cancer Research 80: 675–688. DOI 10.1158/0008-5472.CAN-19-2060. [Google Scholar] [CrossRef]
Liu Y, Jiang Y, Wang N, Jin Q, Ji F et al. (2017). Invalidation of mitophagy by FBP1-mediated repression promotes apoptosis in breast cancer. Tumor Biology 39: 1010428317708779. DOI 10.1177/1010428317708779. [Google Scholar] [CrossRef]
Livak KJ, Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25: 402–408. DOI 10.1006/meth.2001.1262. [Google Scholar] [CrossRef]
Lu C, Ren C, Yang T, Sun Y, Qiao P et al. (2020). Fructose-1,6-bisphosphatase 1 interacts with NF-κB p65 to regulate breast tumorigenesis via PIM2 induced phosphorylation. Theranostics 10: 8606–8618. DOI 10.7150/thno.46861. [Google Scholar] [CrossRef]
Meng X, Franklin DA, Dong J, Zhang Y (2014). MDM2-p53 pathway in hepatocellular carcinoma. Cancer Research 74: 7161–7167. DOI 10.1158/0008-5472.CAN-14-1446. [Google Scholar] [CrossRef]
Nihira NT, Ogura K, Shimizu K, North BJ, Zhang J et al. (2017). Acetylation-dependent regulation of MDM2 E3 ligase activity dictates its oncogenic function. Science Signaling 10: eaai8026. DOI 10.1126/scisignal.aai8026. [Google Scholar] [CrossRef]
Samarin J, Laketa V, Malz M, Roessler S, Stein I et al. (2016). PI3K/AKT/mTOR-dependent stabilization of oncogenic far-upstream element binding proteins in hepatocellular carcinoma cells. Hepatology 63: 813–826. DOI 10.1002/hep.28357. [Google Scholar] [CrossRef]
Song J, Ma SJ, Luo JH, Zhang H, Wang RX et al. (2018). Melatonin induces the apoptosis and inhibits the proliferation of human gastric cancer cells via blockade of the AKT/MDM2 pathway. Oncology Reports 39: 1975–1983. DOI 10.3892/or.2018.6282. [Google Scholar] [CrossRef]
Wang J, Zheng T, Chen X, Song X, Meng X et al. (2011). MDM2 antagonist can inhibit tumor growth in hepatocellular carcinoma with different types of p53 in vitro. Journal of Gastroenterology and Hepatology 26: 371–377. DOI 10.1111/j.1440-1746.2010.06440.x. [Google Scholar] [CrossRef]
Xiong J, Su T, Qu Z, Yang Q, Wang Y et al. (2016). Triptolide has anticancer and chemosensitization effects by down-regulating Akt activation through the MDM2/REST pathway in human breast cancer. Oncotarget 7: 23933–23946. DOI 10.18632/oncotarget.8207. [Google Scholar] [CrossRef]
Yang J, Jin X, Yan Y, Shao Y, Pan Y et al. (2017a). Inhibiting histone deacetylases suppresses glucose metabolism and hepatocellular carcinoma growth by restoring FBP1 expression. Scientific Reports 7: 43864. DOI 10.1038/srep43864. [Google Scholar] [CrossRef]
Yang J, Wang C, Zhao F, Luo X, Qin M et al. (2017b). Loss of FBP1 facilitates aggressive features of hepatocellular carcinoma cells through the Warburg effect. Carcinogenesis 38: 134–143. [Google Scholar]
Yang W, Liu J, Hou L, Chen Q, Liu Y (2021). Shikonin differentially regulates glucose metabolism via PKM2 and HIF1α to overcome apoptosis in a refractory HCC cell line. Life Sciences 265: 118796. DOI 10.1016/j.lfs.2020.118796. [Google Scholar] [CrossRef]
Zender L, Kubicka S (2008). Molecular pathogenesis and targeted therapy of hepatocellular carcinoma. Onkologie 31: 550–555. DOI 10.1159/000151586. [Google Scholar] [CrossRef]
Zhang J, Wang J, Xing H, Li Q, Zhao Q, Li J (2016). Down-regulation of FBP1 by ZEB1-mediated repression confers to growth and invasion in lung cancer cells. Molecular and Cellular Biochemistry 411: 331–340. DOI 10.1007/s11010-015-2595-8. [Google Scholar] [CrossRef]
Zhou Q, Lui VW, Yeo W (2011). Targeting the PI3K/Akt/mTOR pathway in hepatocellular carcinoma. Future Oncology 7: 1149–1167. DOI 10.2217/fon.11.95. [Google Scholar] [CrossRef]
Supplementary Files
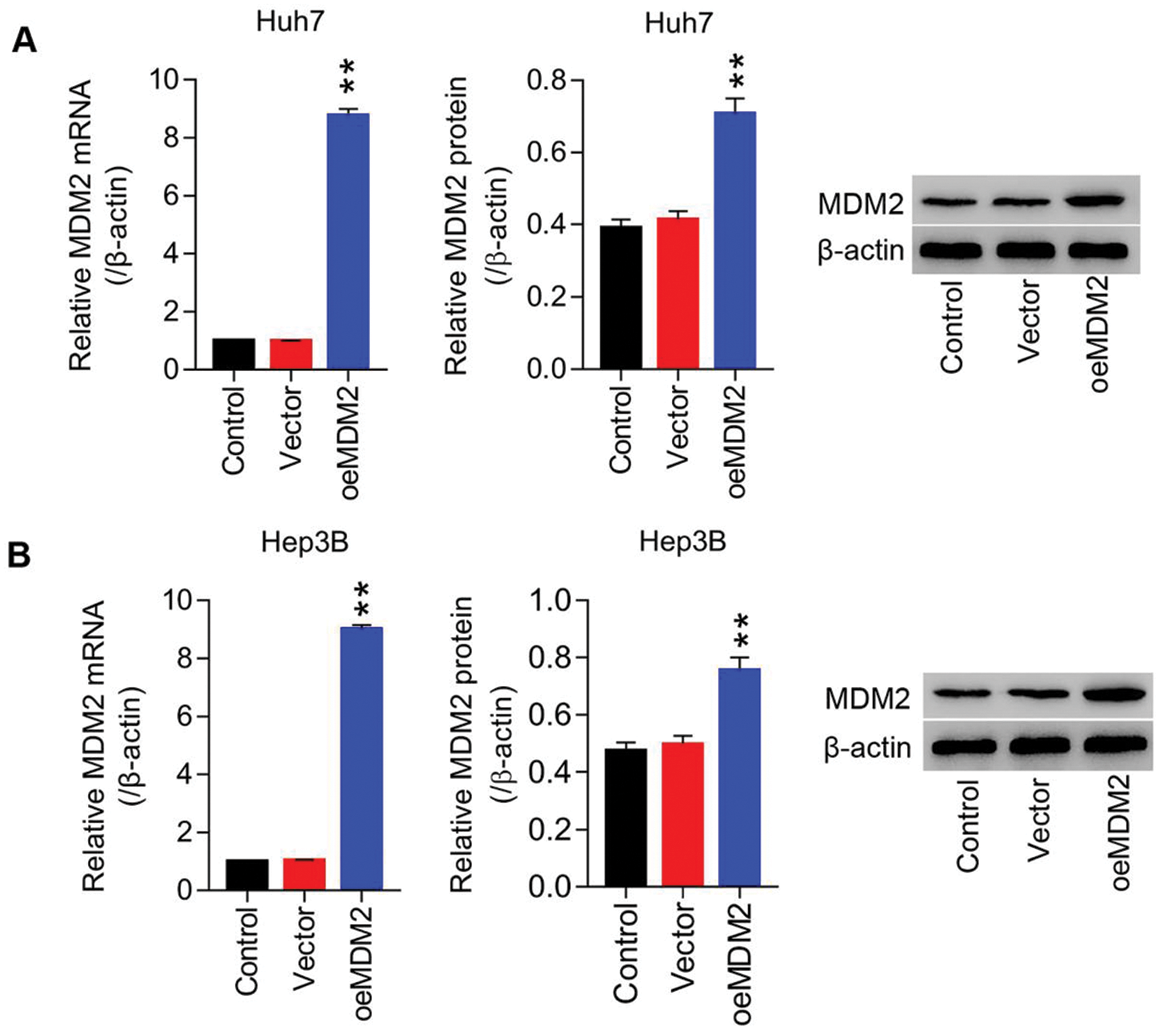
Figure S1: Lentiviruses-mediated overexpression of MDM2 within HCC cell lines (Huh7 and Hep3B) (A) mRNA and protein level of MDM2 in Huh7 cells were detected using RT-PCT and Western blot at least three repeats. (B) mRNA and protein level of MDM2 in Hep3B cells were detected using RT-PCT and Western blot at least three repeats. **P < 0.01: oeMDM2 vs. Vector.
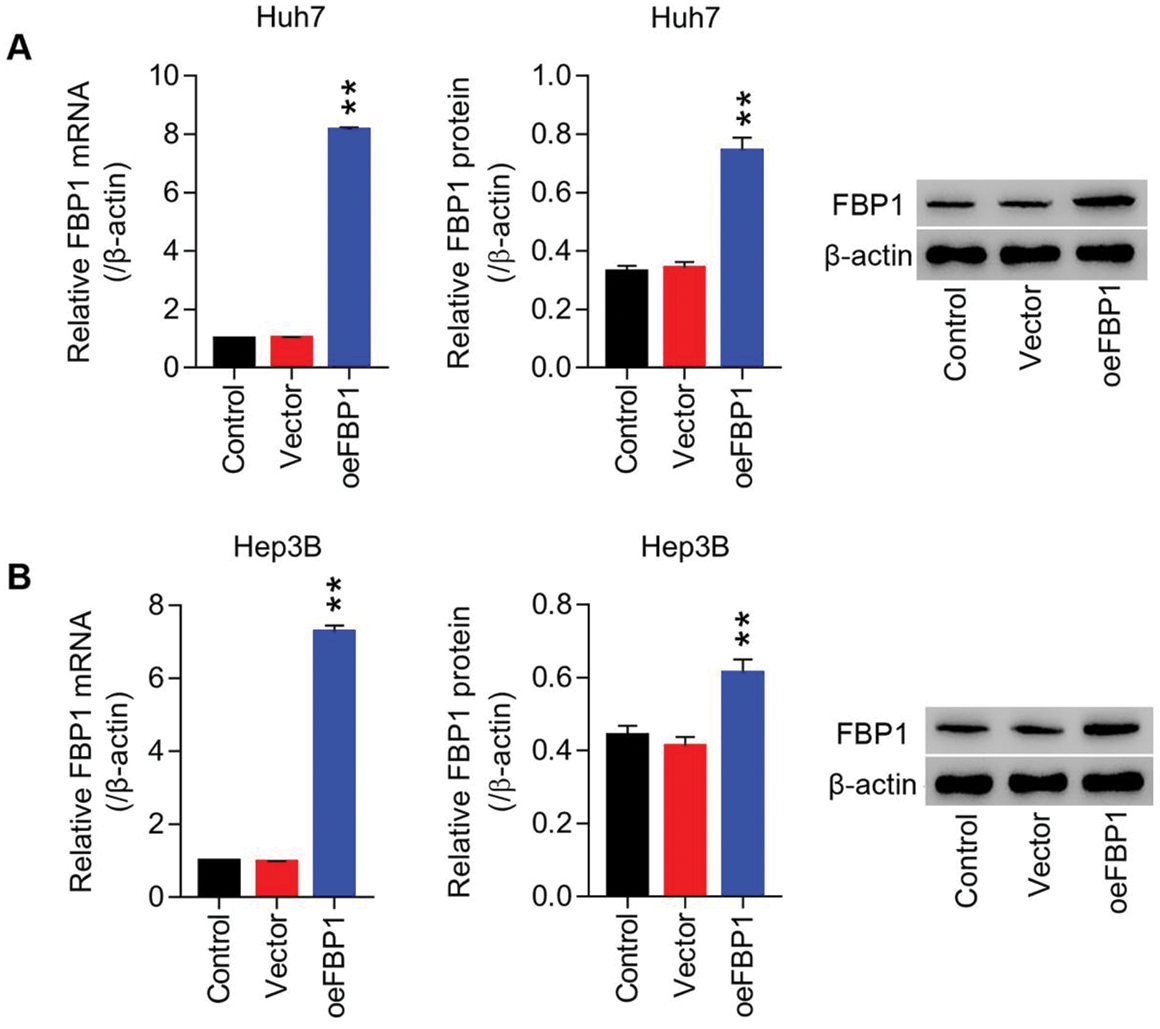
Figure S2: Lentiviruses-mediated overexpression of FBP1 within HCC cell lines (Huh7 and Hep3B) (A) mRNA and protein level of FBP1 in Huh7 cells were detected using RT-PCT and Western blot at least three repeats. (B) mRNA and protein level of FBP1 in Hep3B cells were detected using RT-PCT and Western blot at least three repeats. **P < 0.01: oeFBP1 vs. Vector.
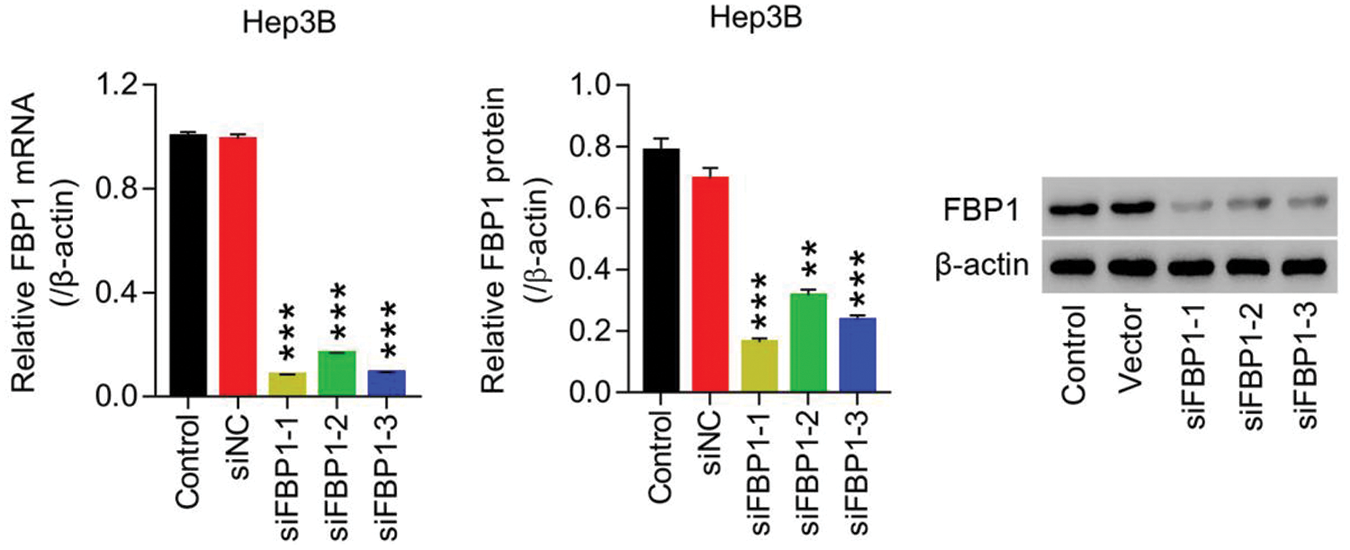
Figure S3: Lentiviruses-mediated knockdown of FBP1 within HCC cell lines (Hep3B) siRNA-FBP1 (siFBP1-1, -2 and -3) significant reduced FBP1 mRNA and protein expression, demonstrating a successful establishment of FBP1 silencing within HCC cells (Hep3B). **P < 0.01, ***P < 0.001: siFBP1-1, siFBP1-2, or siFBP1-3 vs. siNC.
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |