DOI:10.32604/biocell.2022.017544
| BIOCELL DOI:10.32604/biocell.2022.017544 | |
| Article |
The function of ubiquitin-specific protease 31 in intracerebral hemorrhage
1Department of Neurology, Shanghai Xuhui District Dahua Hospital, Shanghai, 200237, China
2Department of Endocrinology and Metabolism, Minhang Hospital, Fudan University, Shanghai, 201199, China
*Address correspondence to: Shunjun Li, liaoone04@sina.com
#These authors have contributed equally to this work
Received: 19 May 2021; Accepted: 04 August 2021
Abstract: Intracerebral hemorrhage (ICH) is the most serious type of stroke. High level of thrombin is found in the ICH. Ubiquitin-specific protease (USP) 31, a member of deubiquitinating enzymes family, has been found to negatively regulate the NF-κB pathway. However, the function of USP31 in ICH remains largely unknown. In the present study, the mRNA and protein expression levels of USP31 were measured by real-time PCR and western blot. Flow cytometry was used to measure cell apoptosis and the level of reactive oxygen species (ROS). In the current study, we found the mRNA level of USP31 was decreased in peripheral blood mononuclear cells (PBMCs) from the patients with ICH. Thrombin stimulated cell apoptosis, and increased the ROS level, the productions of interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), and monocyte chemoattractant protein-1 (MCP-1), but it decreased the expression level of USP31 in BV-2 cells. In agreement, USP31 overexpressing could alleviate these effects caused by thrombin. USP31 partially reversed the thrombin induced increase of nuclear factor kappa B (NF-κB) localization. Further, NF-κB inhibitor could alleviate the effects induced by USP31 knockdown. In addition, USP31 decreased the ubiquitination level of IκBα, which might contribute to the depression of NF-κB activation. In conclusions, USP31 played a role in the ICH might via regulating NF-κB signaling pathway.
Keywords: Intracerebral hemorrhage; ROS; USP31
Intracerebral hemorrhage (ICH) is considered as the most serious type of stroke, with one-month mortality up to 40%. For survivors, it usually leaves patients severely disabled (van Asch et al., 2010). As reported, hypertension, older age, oral anticoagulant treatment, cerebral amyloid angiopathy, and alcohol intake have been proposed as the most important risk factors for ICH (O’donnell et al., 2010; van Asch et al., 2010). Currently, the mainstays of ICH treatment are blood pressure management, hemostatic treatment and intracranial pressure management (Morotti and Goldstein, 2016). However, these treatments fail to satisfy patients so that novel treatment is urgently needed.
Thrombin, a serine protease, plays a pivotal role in the coagulation cascade (Keep et al., 2012). High concentration of thrombin has been found in the brain of patients with ICH (Xi et al., 2003). In addition, animal experiments show that thrombin may be neurotoxic and thrombin inhibitor decreases edema formation in the hematoma site (Xi et al., 2003). Microglia cells, the resident immune cells in the brain, normally stay as resting microglia. But they rapidly transform into activated microglia upon brain damage and increase the release of interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) (Hua et al., 2006; Wang and Dore, 2007). Therefore, thrombin has been considered as a trigger of microglia activation (Moller et al., 2000).
Nuclear factor kappa B (NF-κB), a well-known transcription factor, plays a critical regulatory role in various cellular processes including inflammation, cell survival, proliferation, and apoptosis (Hayden and Ghosh, 2004). Usually, NF-κB remains inactive by binding with IκB which is a NF-κB inhibitor. When cells are stimulated, NF-κB is released by the degradation of IκB. Active NF-κB enters into nucleus and regulates the expression of downstream genes (Shapira et al., 2004). The phosphorylation of IκB, mediated by IκB kinase (IKK), causes its degradation by the ubiquitin–proteasome pathway (Saccani et al., 2004) which plays a key role in all aspects of cell biology (Ehlers, 2003). Ubiquitin-specific proteases (USP) belong to the deubiquitinating enzymes family. As reported, there are approximately 58 putative USPs (Nijman et al., 2005). For example, USP11 is found to participate in promoting the brain secondary damage after ICH (Xu et al., 2016). USP4 is reported to play a role in neuronal apoptosis following ICH (Liu et al., 2017). USP31, a new member of the USP family, is identified in 2004 (Lockhart et al., 2004). Recently, USP31 has been found as a negative regulator of the NF-κB signaling (Tzimas et al., 2006). Inhibition of USP31 promotes sarcomagenesis by enhancing the NF-κB signaling (Ye et al., 2018). However, the function of USP31 in ICH is poorly understood.
In addition, the capacity of the immortalized BV-2 microglia cell line as a substitute for primary microglia in experimental studies has been investigated further (Burguillos et al., 2011; Henn et al., 2009). To explore the role of USP31 in ICH, thrombin was used to treat BV-2 cells. We found that thrombin induced cell apoptosis and the release of inflammation cytokines, and these effects could be partially reversed by USP31 overexpressing. In addition, downregulation of USP31 was observed in ICH patients. This study implied that USP31 might play a potential protective role in ICH.
Thirty ICH patients from Shanghai Xuhui District Dahua Hospital and 30 healthy volunteers were involved in the study. The exclusion criteria for the ICH patients were as follows: (1) patient age <18 or >80 years; (2) surgical history within the last 6 months; (3) coma or death within 48 h after admission; (4) obvious inflammatory disease (e.g., infectious disease, systemic lupus erythematosus (SLE), or rheumatoid arthritis; (5) presence of a hospital-acquired infection; and (6) lack of agreement with the study protocol. Peripheral blood samples from these patients were collected within 12 h after ICH. This study was approved by Shanghai Xuhui Dahua Hospital Ethics Committee (LLW-FO-401, Date 2019-8-28) and written informed consent were signed by all participants.
Peripheral Blood Mononuclear Cells (PBMCs) were isolated using the EasySep™ Direct Human PBMC Isolation Kit (Stem cell, Cambridge, USA).
Mouse BV-2 microglial cell line was purchased from ACCEGEN biotechnology (Fairfield, USA) and cultured in Dulbecco’s modified Eagle medium (Gibco, Carlsbad, USA) containing 10% heat-inactivated fetal bovine serum (Gibco) at 37°C in a 5% CO2 humidifier.
First, BV-2 cell line was treated with thrombin (Molecular Innovation, Novi, USA) at different concentration (0, 10, 20, and 40 U/mL) for 24 h. Then, BV-2 cells were treated with 20 U/mL thrombin for various time points (0, 6, 12, and 24 h). For co-treatment, BV-2 cell line was transfected with vector or USP31 overexpression plasmid and then treated with 20 U/mL thrombin. Next, BV-2 cell line was transfected with siUSP31 or siNC and then treated with 10 μM of PDTC (Selleck, Radnor, USA), a NF-κB inhibitor or 18 μM of SN50 (Selleck), a NF-κB competitor.
Three small interference RNAs (siRNA) targeted USP31 and one USP31 overexpression plasmid were purchased from Genepharma Company (Shanghai, China). Three siRNA sequences used in this study were listed below: siUSP31-1, GCTGTGTAGTCTCACCAAA; siUSP31-2, GATGATGATTGCACGACTA; siUSP31-3, GGACTTTGTTGGAGGTTAA.
BV-2 cells were seeded into six-well plate and cultured for 24 h. For silencing of USP31, siUSP31 and siNC (the control siRNA) were transfected into BV-2 cells using Lipofectamine 2000 (Invitrogen, Carlsbad, USA) following the users’ instructions. For overexpressing USP31, USP31 overexpression and vector plasmid were transfected into BV-2 cells.
Total RNA of all treated cells was isolated using TRIzol reagent (Invitrogen) and reverse transcribed into cDNA using a Reverse Transcription System (Invitrogen). Real-time PCR reactions were performed using SYBR Green PCR mix (Invitrogen) on an ABI 7300 system (Applied Biosystems, Foster City, USA). The primer sequences were as follows:
Usp31 Primer F 5’-ACATCTTGCC TTGGTAATC-3’
Primer R 5’-TAGCACACCACAAATACTC-3’
Gapdh Primer F 5’-AATCCCATCACCATCTTC-3’
Primer R 5’-AGGCTGTTGTCATACTTC-3’
Different treated cells were harvested using RIPA buffer (Beyotime, Shanghai, China) and sonicated. BCA protein kit (Bio-Rad Laboratories, Inc., Hercules, USA) was used to quantify protein concentration. 30 μg protein from each sample was separated according to their molecular weight by SDS-PAGE and transferred to Hybond-C pure nitrocellulose membrane (Millipore, Bedford, USA). Then the blots were incubated with the following primary antibodies over night: anti-USP31 (1:1000; PA5-68355, Thermo, Grand Island, USA), anti-NF-κB (1:1000; Ab16502, Abcam, St. Louis, USA), anti-IκBα (1:800; #9242, Cell Signaling Technology, Danvers, USA), anti-H3 (1:1500; Ab1791, Abcam) and anti-GAPDH (1:2500; #5174, Cell Signaling Technology, Danvers). On the second day, the blots were incubated with HRP-conjugated secondary antibody (Beyotime). Then, these brands were visualized with an enhanced chemiluminescence kit (Bio-Rad Laboratories, Inc., Hercules, USA) and analyzed with Gel Imaging System (Bio-Rad Laboratories, Inc., Hercules, USA).
Treated BV-2 cells and culture medium were collected, centrifuged, washed, and resuspended. Annexin V and propidium iodide (PI) cell apoptosis kit (Abcam) was used to analyze cell apoptosis. The staining cell were collected and analyzed by a flow cytometry (BD Biosciences, San Jose, USA).
Reactive Oxygen Species (ROS) assay
DCFH-DA Cellular ROS Detection Assay Kit (Abcam) was used to measure the ROS level of treated cells. Treated BV-2 cells were harvested and resuspended with working buffer provided in the kit. Then, the cells were incubated with 20 μM of DCFH-DA probe for 30 min. Flow cytometry (BD Biosciences) was used to detect fluorescence at 535 nm.
Enzyme-linked immunoassay (ELISA)
All the ELISA kits used in this study were purchased from XeY Biotechnology (Shanghai, China). The concentrations of IL-1β, TNF-α and MCP-1 were quantified according to the manufacturer’s protocol.
Immunoprecipitation (IP) assay
Treated BV-2 cells were lysed with IP lysis buffer for 10 min on ice. Cell lysis was harvested and incubated with anti-USP31, anti-IκBα IP primary antibodies or control IgG (Abcam) for 2 h at 4°C. Then the compound was incubated with protein A/G agarose beads (Abcam) at 4°C overnight, followed by SDS-PAGE.
All data were given as mean ± SD. Statistical analysis was carried out using GraphPad Prism software (version 6.0, San Diego, USA). ANOVA and Student’s t-tests were used to analyze statistical significance. P < 0.05 was considered significant.
Thrombin inhibits the expression of USP31to induce cell apoptosis and the production of inflammatory cytokines in BV-2 cells
To explore the function of USP31 in ICH, BV-2 cells were treated with thrombin to establish ICH cell model (Aronowski and Zhao, 2011). Firstly, different concentrations of thrombin were used to treat cells for 24 h and the expression level of USP31 was measured. As shown in Fig. 1A, thrombin induced the decrease of USP31 in a dosage-dependent manner. Because 20 U/mL thrombin exhibited the significant decrease of USP31 and the cell morphology remained normal, BV-2 cells were approached to 20 U/mL thrombin for different time points. The level of USP31 was time dependently decreased after treatment with thrombin (Fig. 1B) and 24 h showed the best inhibition of USP31.
To study the function of thrombin in BV-2 cells, flow cytometry was performed. As illustrated in Figs. 1C and 1D, thrombin induced cell apoptosis in a dosage dependent manner. As reported, ROS relates to various neurodegenerative diseases by oxidizing major cellular components and resulting in cell apoptosis (Gilgun-Sherki et al., 2001). Thus, we measured the ROS level after treatment with thrombin and found that thrombin exposure did dependently increase cellular ROS level (Fig. 1E). Furthermore, thrombin promoted the release of some inflammatory cytokines including IL-1β, TNF-α, and MCP-1 (Fig. 1F). As reported, ROS plays a crucial role in regulating NF-κB signaling pathway, so that we measured the activation of NF-κB signaling pathway by western blot. After treatment with thrombin, the level of nuclear NF-κB was upregulated whereas the level in cytoplasm was downregulated (Fig. 1G). In agreement, the protein levels of p-NF-κB and cleavage caspase 3 increased in response to the thrombin treatment (Fig. 1G). These results indicate that thrombin played as a positive regulator of NF-κB signaling pathway in BV-2 cells upon thrombin treatment.
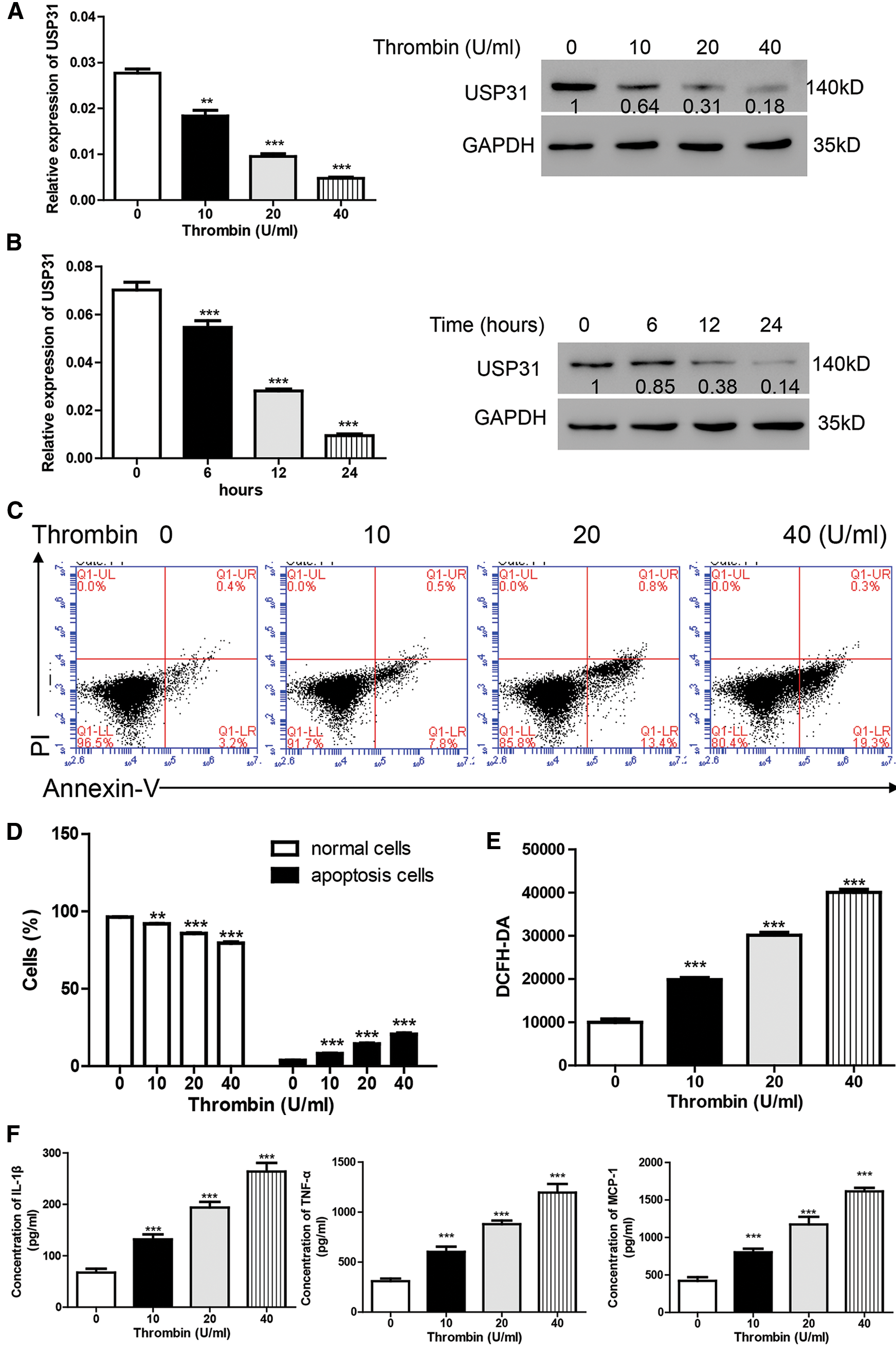
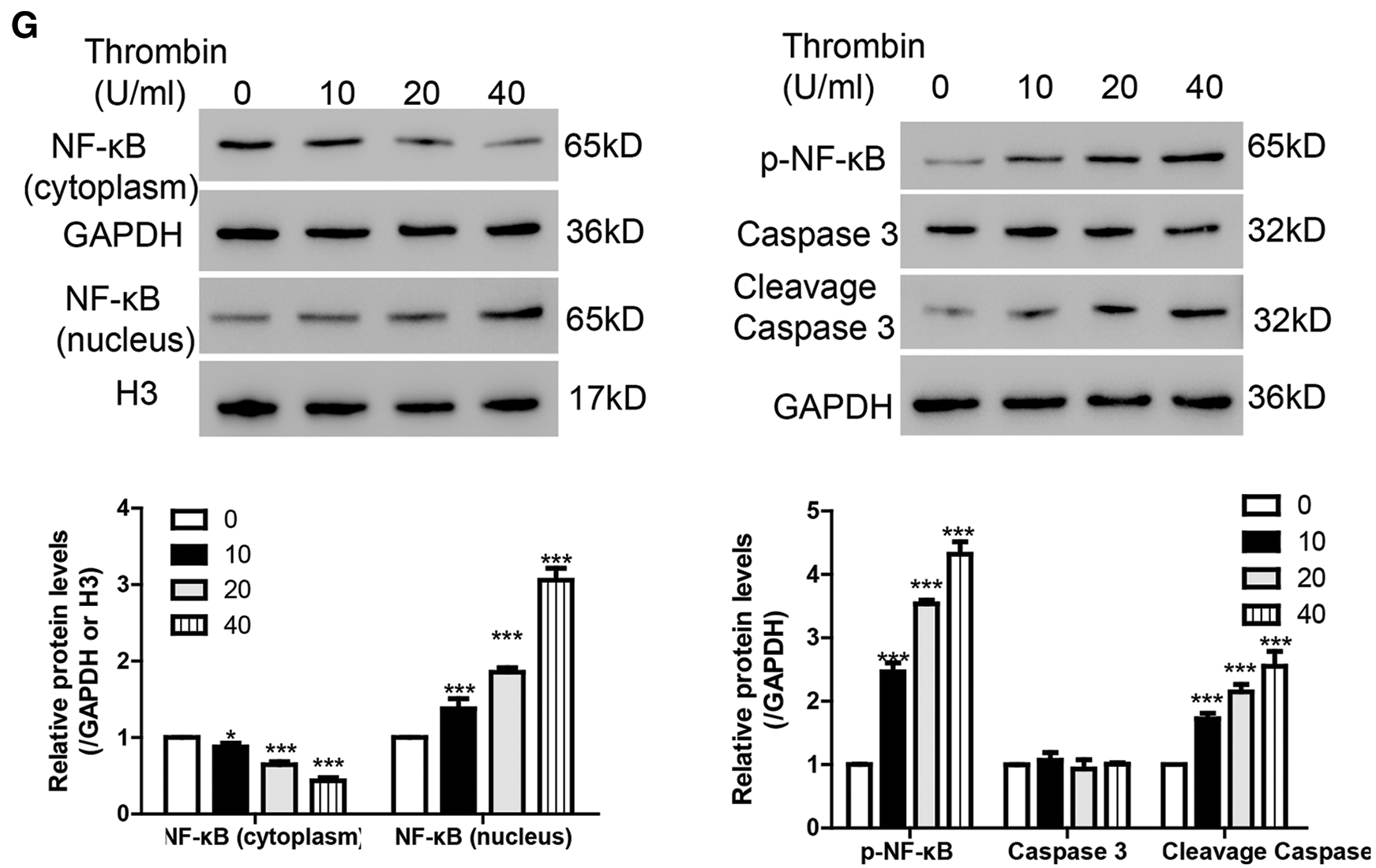
Figure 1: Thrombin inhibits the expression of USP31 and induces cell apoptosis and the productions of inflammatory cytokines in BV-2 cells. (A) Realtime PCR and western blot assay were performed to quantify the expression level of USP31 after treatment with different concentrations of thrombin. (B) Realtime PCR and western blot assay were performed to measure the expression level of USP31 after treatment with 20 U/mL thrombin for different time points. (C) BV-2 cells were treated with thrombin (0, 10, 20, or 40 U/mL) for 24 h. Cell apoptosis (C), ROS (E) and release of inflammatory cytokines (F) including IL-1β, TNF-α and MCP-1 were analyzed by flow cytometry, DCFH-DA and ELISA, respectively. (D) Apoptosis rates of different groups. (G) Western blot was used to detect the protein levels of USP31, NF-κB, p- NF-κB, caspase 3, and cleavage caspase 3. **P < 0.01; ***P < 0.001 vs. the treatment with 0 U/mL thrombin.
As previously described, the expression level of USP31 observed in the PBMCs was examined for this particular research rather than that in the brain tissue (Moore et al., 2005; Sang et al., 2017). Based on previous results that thrombin inhibited USP31, we sought to investigate whether thrombin inhibited USP31 through regulating its transcription. To this end, we analyzed the mRNA and protein levels of USP31 in the PBMCs from ICH patients and normal persons. As shown in Figs. 2A and 2B, USP31 was downregulated in ICH patients compared with normal persons.
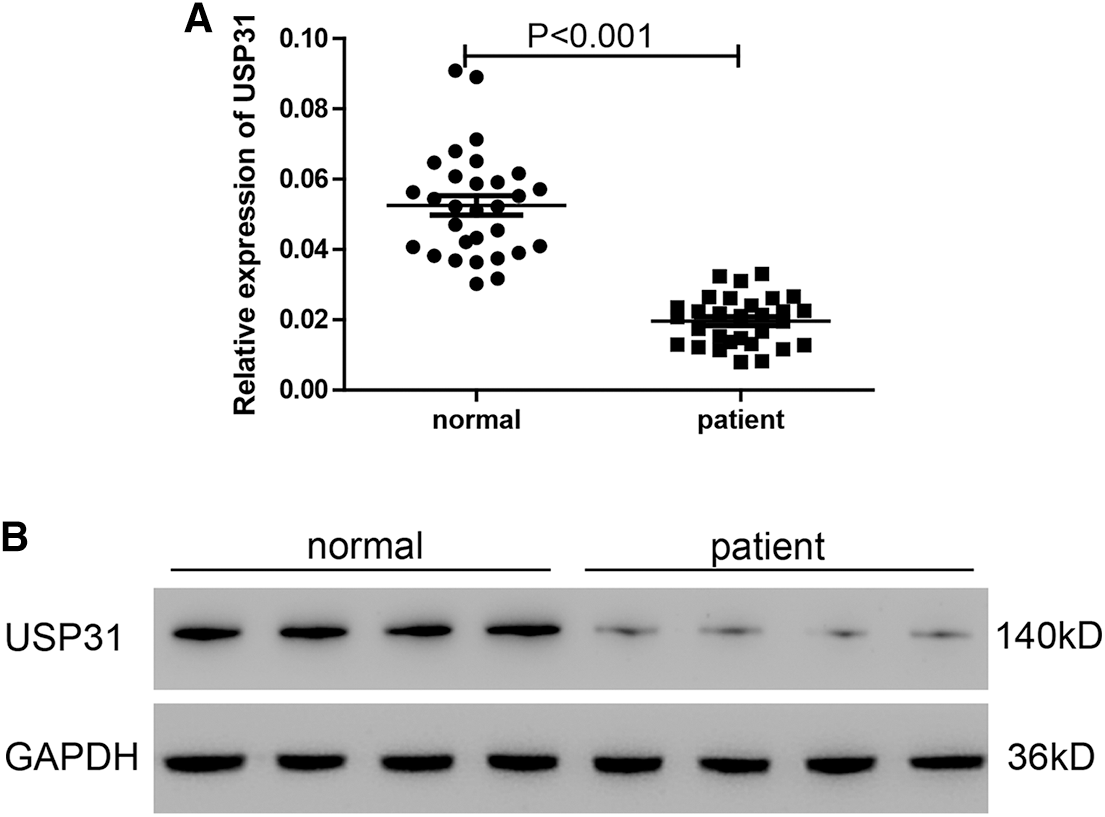
Figure 2: USP31 is downregulated in ICH patients and the transfection efficiencies of USP31 silencing and overexpressing. (A) Relative mRNA level of USP31 was measured in PBMCs form ICH patients and normal persons (N = 30). (B) Protein level of USP31 was measured in PBMCs form ICH patients and normal persons (N = 15).
UPS31 alleviates the effects induced by thrombin
To study the role of USP31 in ICH, we overexpressed the expression level of USP31 by transfection with USP31 overexpression plasmid and found that USP31 was markedly upregulated (Fig. 3A). Then, BV-2 cells overexpressing USP31 were treated with thrombin. As shown in Fig. 3B, single treatment with thrombin markedly induced cell apoptosis. Intriguingly, overexpression of USP31 exhibited lower apoptosis rate than that with thrombin treatment. Furthermore, we found that thrombin increased ROS level and the release of IL-1β, TNF-α and MCP-1, and these effects could be reversed by USP31 overexpressing (Figs. 3C–3F). In addition, upon thrombin treatment, the nuclear level of NF-κB in the group overexpressing USP31 was lower than that in the control group but still higher than that in the thrombin single treatment group (Fig. 3G). Additionally, the protein levels of p-NF-κB and cleavage caspase 3 increased in response to the thrombin treatment and USP31 overexpression could partially attenuate the increase of p-NF-κB and cleavage caspase 3 induced by thrombin treatment (Fig. 3G). These results indicate that UPS31 could alleviate the effects induced by thrombin in BV-2 cells.
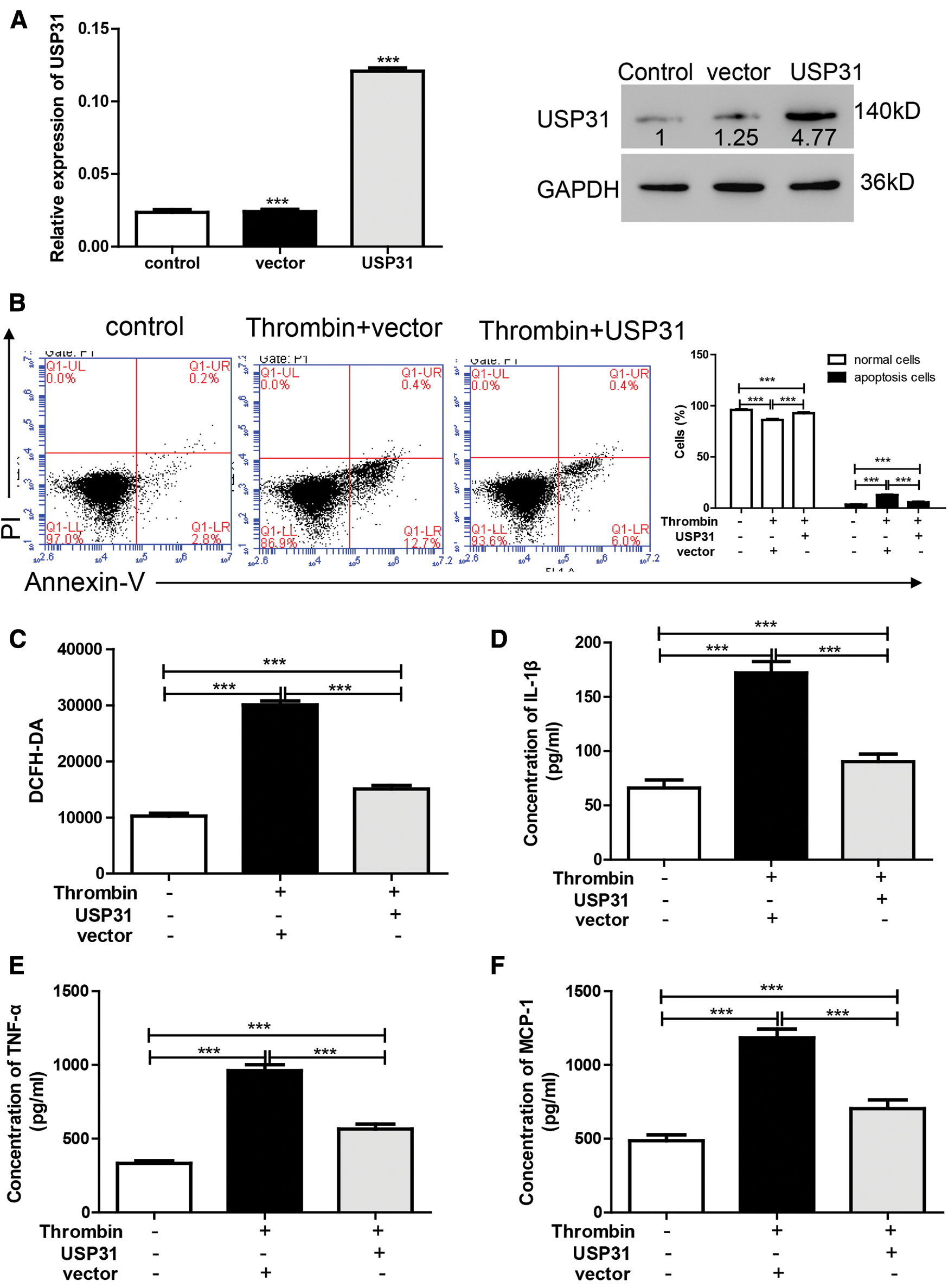
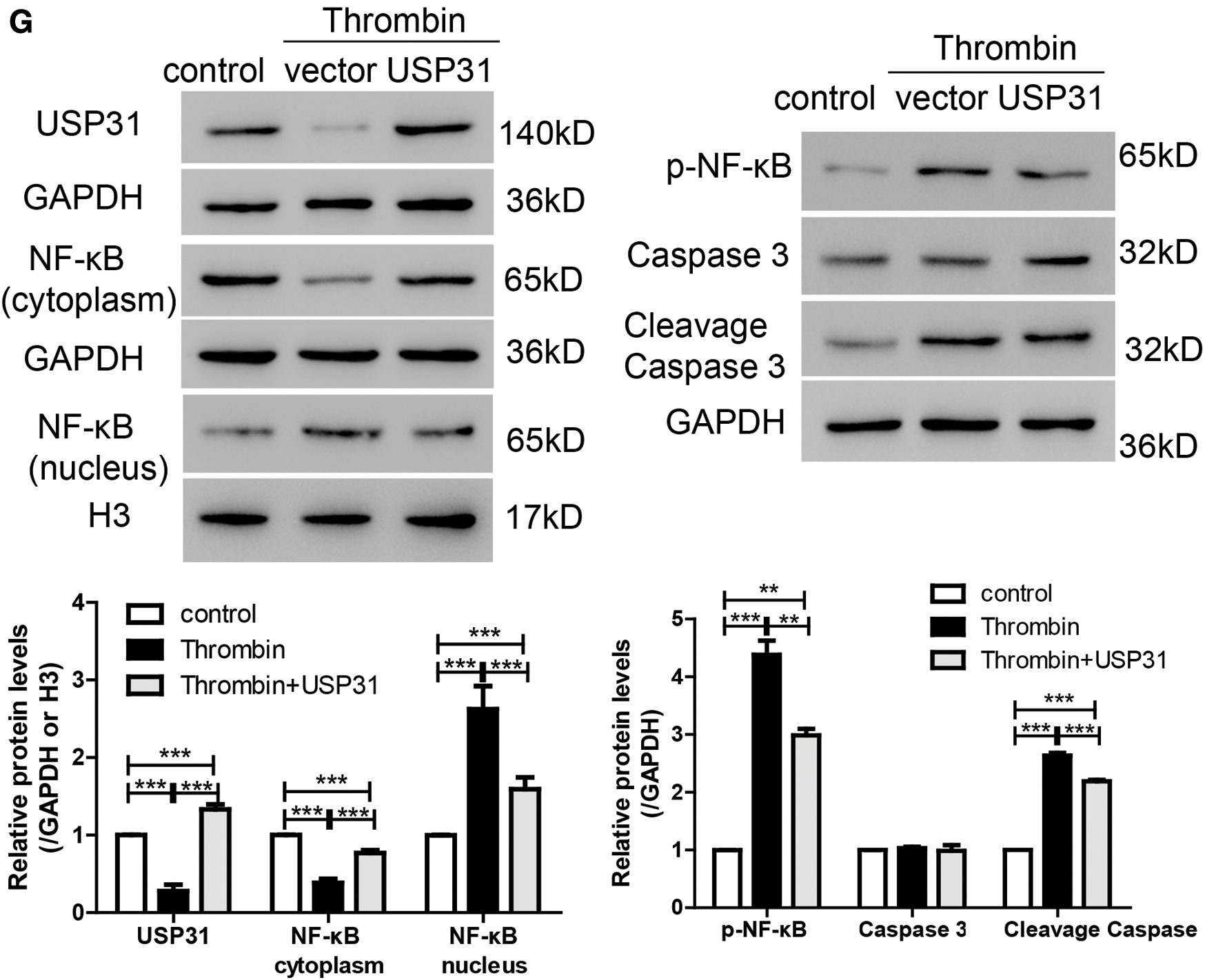
Figure 3: UPS31 overexpression alleviates the effects induced by thrombin. (A) BV-2 cells were transfected with USP31 overexpression plasmid and then analyzed the transfection efficiency. ***P < 0.001 vs. control. (B–F) BV-2 cells were treated with thrombin and/or transfected with USP31 overexpression plasmid. Cell apoptosis (B), ROS(C) and production of inflammatory cytokines including IL-1β (D), TNF-α (E), and MCP-1 (F) were analyzed by flow cytometry, DCFH-DA and ELISA, respectively. (G) Western blot was used to detect the protein levels of USP31, NF-κB, p- NF-κB, caspase 3, and cleavage caspase 3. ***P < 0.001.<
Silencing of USP31 induces apoptosis, ROS, and inflammatory cytokines via NF-κB pathway
To further understand the underlying mechanisms of USP31 in ICH, siRNAs against USP31 were transfected into BV-2 cells. The USP31 level was significantly downregulated by siRNAs (Fig. 4A). As shown in Figs. 4B–4G, USP31 knockdown increased apoptosis, ROS level and the release of IL-1β, TNF-α, and MCP-1. Based on the results shown in Fig. 3G, USP31 could alleviate the increase of nuclear NF-κB, we hypothesized that NF-κB might be involved in this process. To this purpose, PDTC, an inhibitor of NF-κB signaling pathway, was used to treat BV-2 cells. As expected, PDTC blocked the increase of apoptosis, ROS level and the release of IL-1β, TNF-α, and MCP-1 induced by USP31 knockdown (Figs. 4A–4F). Furthermore, the increase of nuclear NF-κB was due to the USP31 silencing (Fig. 4H). To further validate the results, BV-2 cells were then treated with SN50, a NF-κB competitor. As indicated, the effects induced by USP31 knockdown was partially rescued by SN50 treatment, which in turn increased the apoptosis, the ROS level, and the release of IL-1β, TNF-α, and MCP-1 (Fig. 5). Taking together, silencing of USP31 induces apoptosis, ROS, and inflammatory cytokines via NF-κB pathway.
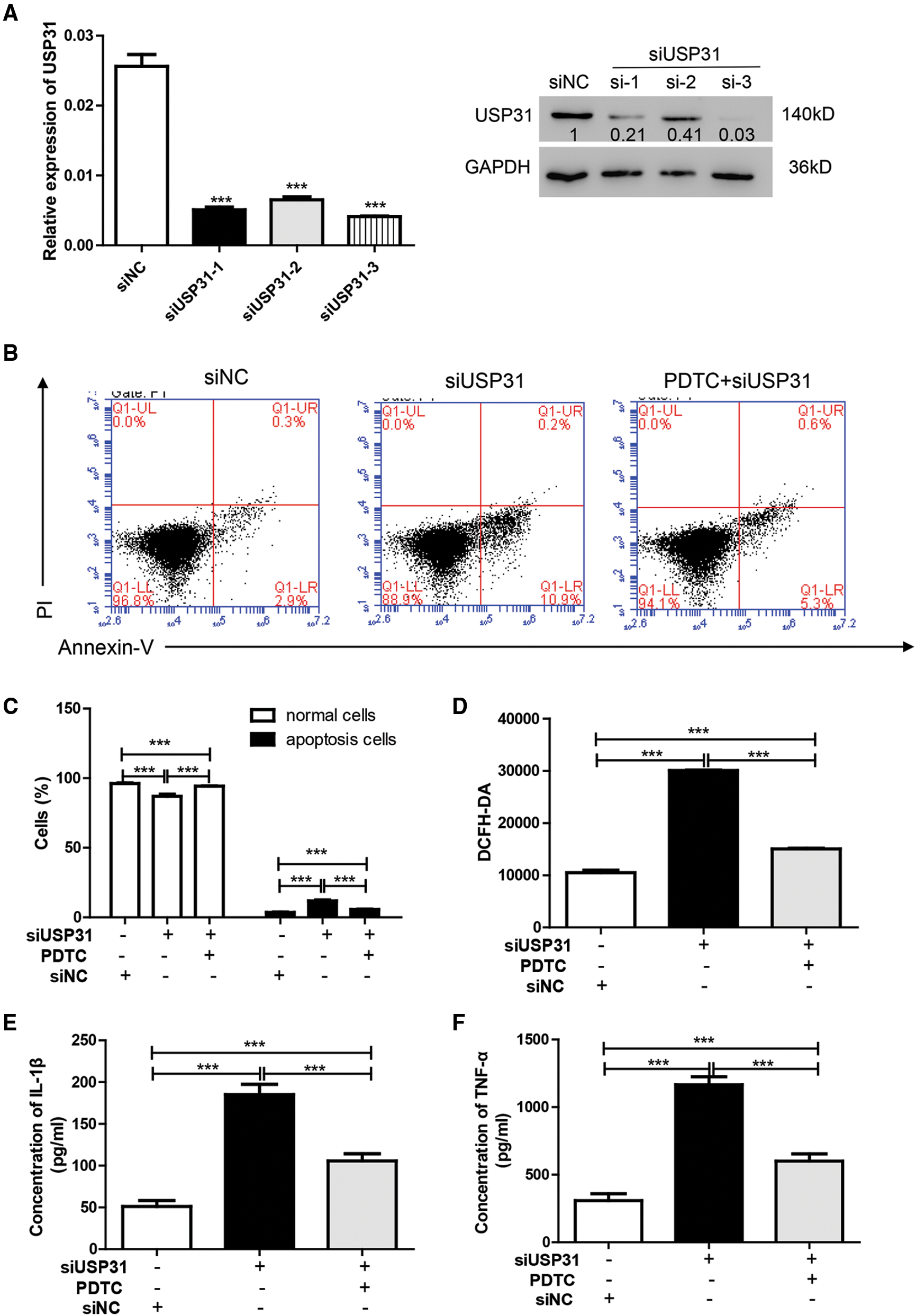
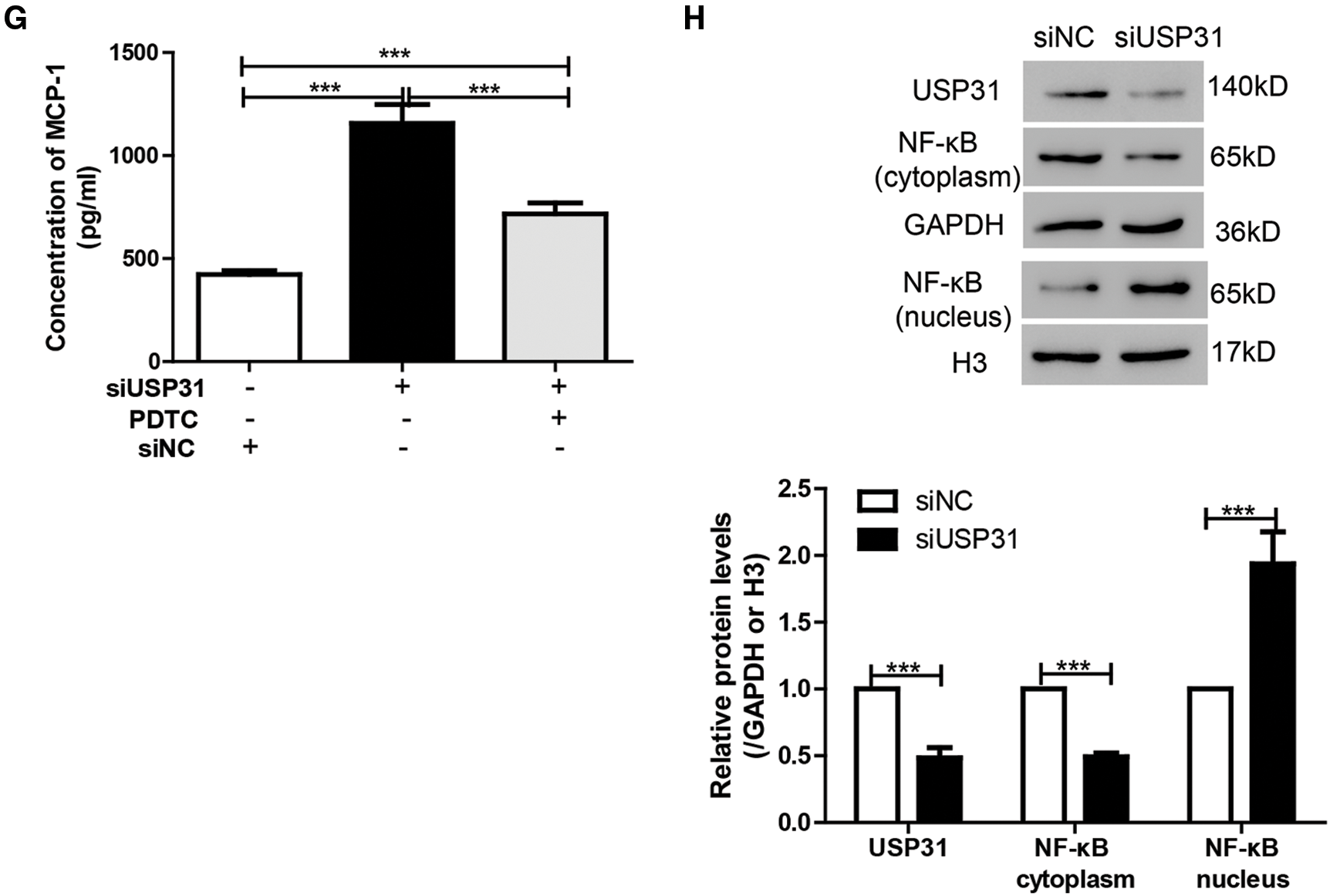
Figure 4: PDTC blocks USP31 knockdown induced apoptosis, ROS, and the productions of inflammatory cytokines. (A) BV-2 cells were transfected with siRNA targeting USP31 and then analyzed the transfection efficiency. ***P < 0.001 vs. siNC. (B–G) BV-2 cells were treated with PDTC and/or transfected with siUSP31. Cell apoptosis (B), ROS(D) and release of inflammatory cytokines including IL-1β (E), TNF-α (F) and MCP-1 (G) were analyzed by flow cytometry, DCFH-DA and ELISA, respectively. (C) Apoptosis rates of different groups. (H) Western blot was used to detect the protein level of NF-κB. ***P < 0.001.
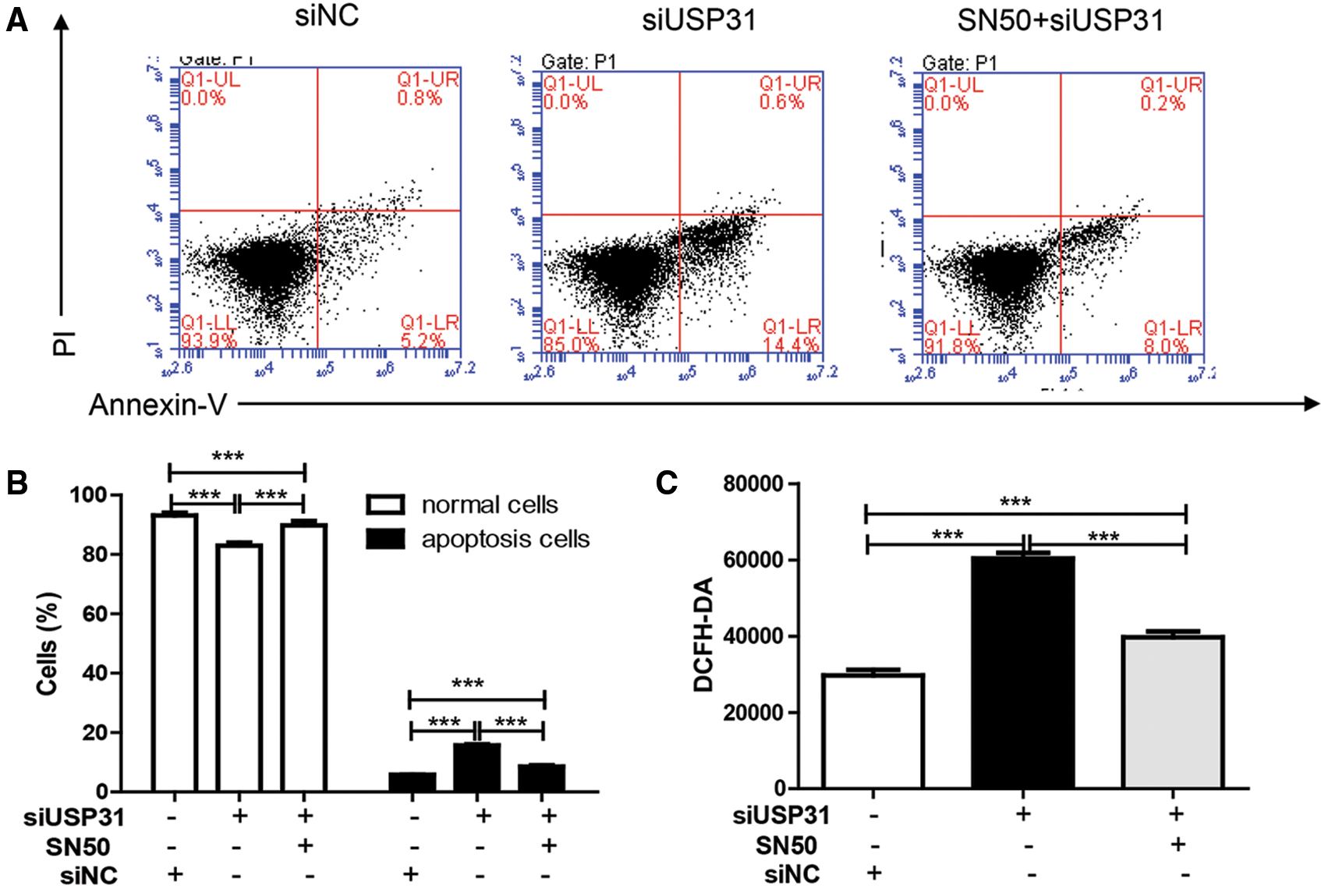
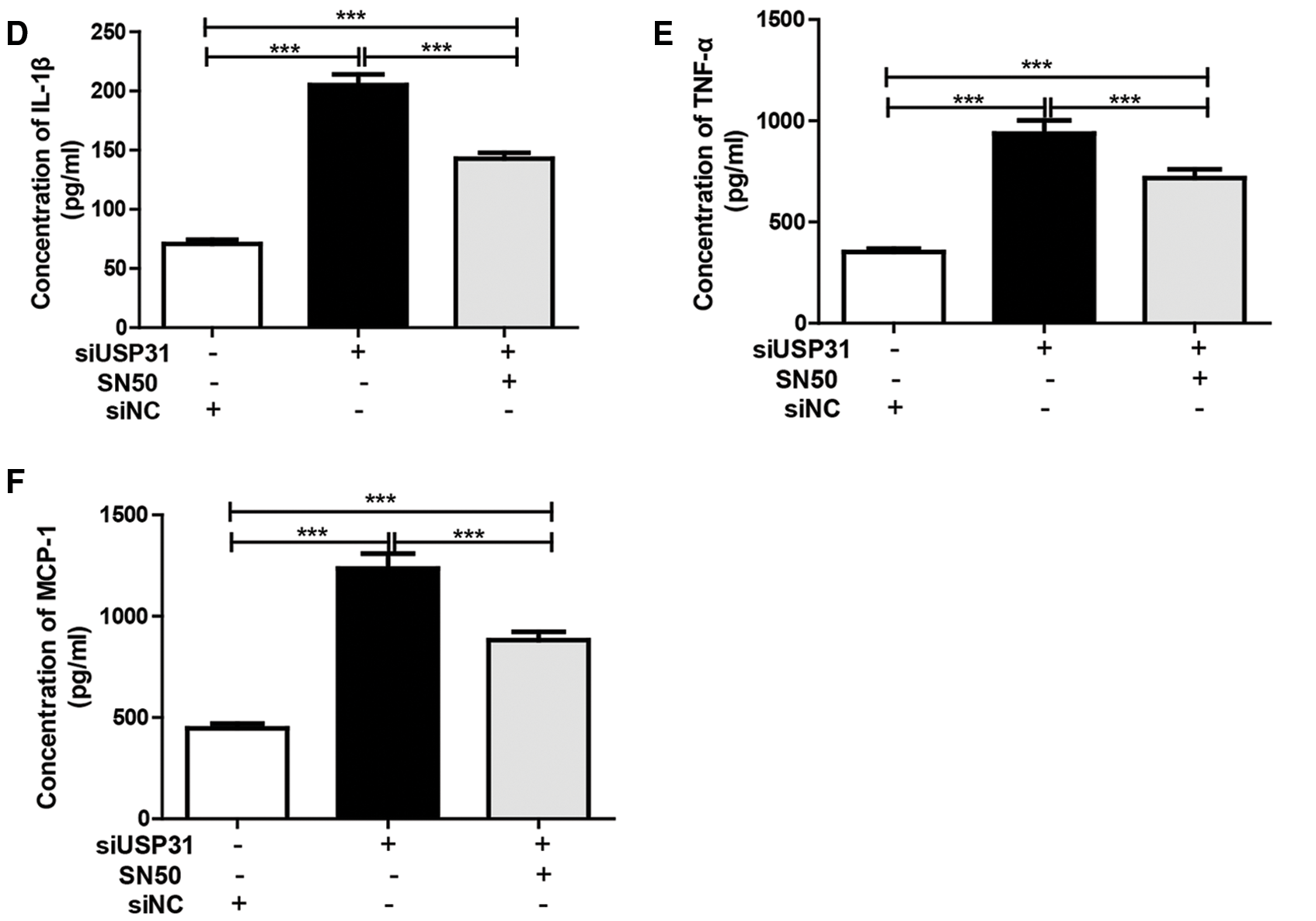
Figure 5: SN50 blocks USP31 knockdown induced apoptosis, ROS, and inflammatory cytokines. BV-2 cells were treated with SN50 and/or transfected with siUSP31. Cell apoptosis (A), ROS (C) and release of inflammatory cytokines including IL-1β (D), TNF-α (E), and MCP-1 (F) were analyzed by flow cytometry, DCFH-DA and ELISA, respectively. (B) Apoptosis rates of different groups. ***P < 0.001 vs. siNC.
USP31 inhibits the activity of NF-κB pathway by decreasing the ubiquitination of IκBα
To explore the association between USP31 and IκBα, BV-2 cells were infected with lentivirus depleting USP31. The results showed that depletion of USP31 depressed the level of IκBα, which can be rescued by MG132, a proteasome inhibitor, treatment (Fig. 6A). To study the association of USP31 and IκBα, immunoprecipitation (IP) was performed, and we found that there is an interaction between USP31 and IκBα (Fig. 6B). Because USP31 belongs to deubiquitinating enzymes family, we then sought to detect the ubiquitination of IκBα. As shown in Fig. 6C, USP31 overexpression decreased the ubiquitination level of IκBα. In agreement, a higher level of the ubiquitination of IκBα in response to the thrombin treatment was observed compared with that of the control group (Fig. 6D). Taking together, these results suggest that USP31 inhibits the activity of NF-κB pathway by decreasing the ubiquitination level of IκBα.
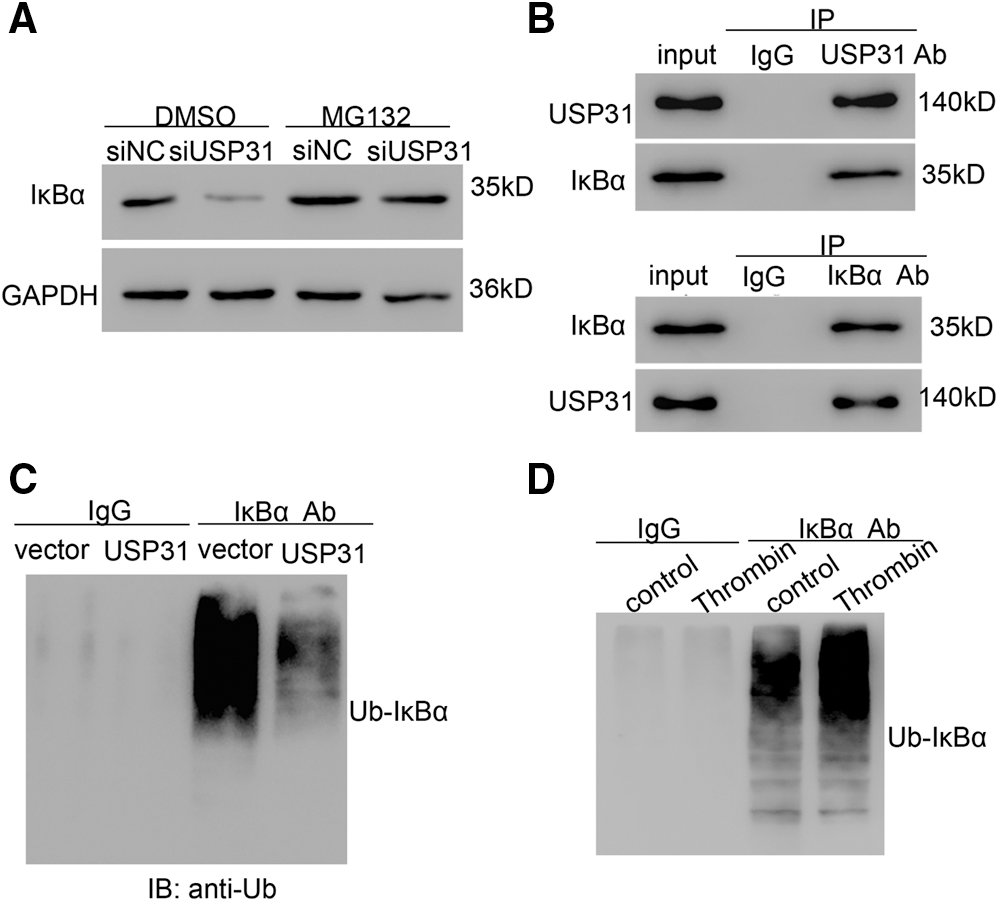
Figure 6: USP31 decreasing the ubiquitination level of IκBα. (A) BV-2 cells were transfected with siUSP31 lentivirus for 24 h and then treated with DMSO or 10 μM of MG132 for 24 h. The protein level of IκBα was measured. (B) IP was performed to measure the association between USP31 and IκBα. (C) BV-2 cells transfecting vector or USP31 were IP with IκBα or IgG, and IB with a-Ubiquitin (Ub). (D) Thrombin treated BV-2 cells were IP with κBα or IgG, and IB with a-Ubiquitin (Ub).
To our knowledge, this study is for the first time to explore the role of USP31 in ICH. USP31 is a novel member of USP family which participate in many biological processes by controlling protein ubiquitination cycle (Finley, 2009). Previous studies demonstrated that USP31 negatively regulates NF-κB pathway (Tzimas et al., 2006) and inhibits sarcomagenesis (Ye et al., 2018). In this study, lower level of USP31 in ICH patients indicates that it might play a protective role in ICH, which is a common but serious brain disease in older people. When ICH happens, the production of thrombin immediately increases to a high level, resulting in early brain injury (Ohnishi et al., 2007). Thrombin has been found to induce microglia via secreting proinflammatory cytokines, and finally contribute to brain injury (Moller et al., 2000). In the current study, we demonstrated that treatment with thrombin stimulated the productions of IL-1β, TNF-α and MCP-1 (Fig. 1). Meanwhile, thrombin inhibited the expression of USP31 in BV-2 cells (Fig. 1). Overexpression of USP31 could alleviate thrombin-induced cell apoptosis, ROS, and release of IL-1β, TNF-α, and MCP-1, indicating that USP31 might participate in thrombin-stimulated microglial activation.
The activity of NF-κB signaling pathway affects many cellular progresses. In this study, we found that thrombin increased the activity of NF-κB which can be alleviated by USP31 overexpression (Fig. 3G). NF-κB inhibitor reversed the apoptosis, ROS and the productions of inflammatory cytokines induced by silence of USP31 (Fig. 4). These results suggested that USP31 was involved in regulating NF-κB activity. Due to USP31 is a deubiquitinating enzyme and ubiquitination is a critical physiological process, we then detected USP31 mediated ubiquitination and found USP31 decreased the ubiquitination level of IκBα (Fig. 6). Usually, NF-κB is inactivated by binding with IκBα. Thus, NF-κB can be activated by the degradation of IκBα. As reported, TNFAIP3 knockout stimulates the activation of NF-κB by enhancing IκBα degration (Meng et al., 2017). Taken together, USP31 inhibited the activity of NF-κB signaling pathway by decreasing the ubiquitination level of IκBα in BV-2 cells. However, USP31 is found to reverse the ubiquitination level of TRAF2 and/or TRAF6, resulting in the dysregulation of NF-κB activation in 293T cells (Tzimas et al., 2006). Further studies are need to comfirm the mechnism of USP31 in other cell types.
Availability of Data and Materials: The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Authors’ Contributions: The authors confirm contribution to the paper as follows: study conception and design: S-JL; data collection: S-YP, HZ, YT, JS, and M-NY; analysis and interpretation of results: S-YP, HZ, YT, JS, and M-NY; draft manuscript preparation: S-YP. All authors reviewed the results and approved the final version of the manuscript.
Ethics Approval: This study was approved by Shanghai Xuhui Dahua Hospital Ethics Committee (LLW-FO-401, August 28th, 2019) and written informed consent were signed by all participants.
Funding Statement: The authors received no specific funding for this study.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Aronowski J, Zhao X (2011). Molecular pathophysiology of cerebral hemorrhage: Secondary brain injury. Stroke 42: 1781–1786. DOI 10.1161/STROKEAHA.110.596718. [Google Scholar] [CrossRef]
Burguillos MA, Deierborg T, Kavanagh E, Persson A, Hajji N et al. (2011). Caspase signalling controls microglia activation and neurotoxicity. Nature 472: 319–324. DOI 10.1038/nature09788. [Google Scholar] [CrossRef]
Ehlers MD (2003). Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nature Neuroscience 6: 231–242. DOI 10.1038/nn1013. [Google Scholar] [CrossRef]
Finley D (2009). Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annual Review of Biochemistry 78: 477–513. DOI 10.1146/annurev.biochem.78.081507.101607. [Google Scholar] [CrossRef]
Gilgun-Sherki Y, Melamed E, Offen D (2001). Oxidative stress induced-neurodegenerative diseases: The need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 40: 959–975. DOI 10.1016/S0028-3908(01)00019-3. [Google Scholar] [CrossRef]
Hayden MS, Ghosh S (2004). Signaling to NF-κB. Genes & Development 18: 2195–2224. DOI 10.1101/gad.1228704. [Google Scholar] [CrossRef]
Henn A, Lund S, Hedtjarn M, Schrattenholz A, Porzgen P, Leist M (2009). The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX: Alternatives to Animal Experimentation 26: 83–94. DOI 10.14573/altex.2009.2.83. [Google Scholar] [CrossRef]
Hua Y, Wu J, Keep RF, Nakamura T, Hoff JT, Xi G (2006). Tumor necrosis factor-α increases in the brain after intracerebral hemorrhage and thrombin stimulation. Neurosurgery 58: 542–550. DOI 10.1227/01.NEU.0000197333.55473.AD. [Google Scholar] [CrossRef]
Keep RF, Hua Y, Xi G (2012). Intracerebral haemorrhage: Mechanisms of injury and therapeutic targets. Lancet Neurology 11: 720–731. DOI 10.1016/S1474-4422(12)70104-7. [Google Scholar] [CrossRef]
Liu C, Liu C, Liu H, Gong L, Tao T et al. (2017). Increased expression of ubiquitin-specific protease 4 participates in neuronal apoptosis after intracerebral hemorrhage in adult rats. Cellular and Molecular Neurobiology 37: 427–435. DOI 10.1007/s10571-016-0375-y. [Google Scholar] [CrossRef]
Lockhart PJ, Hulihan M, Lincoln S, Hussey J, Skipper L et al. (2004). Identification of the human ubiquitin specific protease 31 (USP31) gene: Structure, sequence and expression analysis. DNA Sequence 15: 9–14. DOI 10.1080/10855660310001638197. [Google Scholar] [CrossRef]
Meng Z, Zhao T, Zhou K, Zhong Q, Wang Y et al. (2017). A20 ameliorates intracerebral hemorrhage-induced inflammatory injury by regulating TRAF6 polyubiquitination. Journal of Immunology 198: 820–831. DOI 10.4049/jimmunol.1600334. [Google Scholar] [CrossRef]
Moller T, Hanisch UK, Ransom BR (2000). Thrombin-induced activation of cultured rodent microglia. Journal of Neurochemistry 75: 1539–1547. DOI 10.1046/j.1471-4159.2000.0751539.x. [Google Scholar] [CrossRef]
Moore DF, Li H, Jeffries N, Wright V, Cooper RAJr. et al. (2005). Using peripheral blood mononuclear cells to determine a gene expression profile of acute ischemic stroke: A pilot investigation. Circulation 111: 212–221. DOI 10.1161/01.CIR.0000152105.79665.C6. [Google Scholar] [CrossRef]
Morotti A, Goldstein JN (2016). Diagnosis and management of acute intracerebral hemorrhage. Emergency Medicine Clinics of North America 34: 883–899. DOI 10.1016/j.emc.2016.06.010. [Google Scholar] [CrossRef]
Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM et al. (2005). A genomic and functional inventory of deubiquitinating enzymes. Cell 123: 773–786. DOI 10.1016/j.cell.2005.11.007. [Google Scholar] [CrossRef]
O’donnell MJ, Xavier D, Liu L, Zhang H, Chin SL et al. (2010). Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE studyA case-control study. Lancet 376: 112–123. DOI 10.1016/S0140-6736(10)60834-3. [Google Scholar] [CrossRef]
Ohnishi M, Katsuki H, Fujimoto S, Takagi M, Kume T, Akaike A (2007). Involvement of thrombin and mitogen-activated protein kinase pathways in hemorrhagic brain injury. Experimental Neurology 206: 43–52. DOI 10.1016/j.expneurol.2007.03.030. [Google Scholar] [CrossRef]
Saccani S, Marazzi I, Beg AA, Natoli G (2004). Degradation of promoter-bound p65/RelA is essential for the prompt termination of the nuclear factor κB response. Journal of Experimental Medicine 200: 107–113. DOI 10.1084/jem.20040196. [Google Scholar] [CrossRef]
Sang M, Wang X, Zhang H, Sun X, Ding X et al. (2017). Gene expression profile of peripheral blood mononuclear cells in response to intracerebral hemorrhage. DNA and Cell Biology 36: 647–654. DOI 10.1089/dna.2017.3650. [Google Scholar] [CrossRef]
Shapira S, Harb OS, Caamano J, Hunter CA (2004). The NF-κB signaling pathway: Immune evasion and immunoregulation during toxoplasmosis. International Journal for Parasitology 34: 393–400. DOI 10.1016/j.ijpara.2003.12.005. [Google Scholar] [CrossRef]
Tzimas C, Michailidou G, Arsenakis M, Kieff E, Mosialos G et al. (2006). Human ubiquitin specific protease 31 is a deubiquitinating enzyme implicated in activation of nuclear factor-κB. Cellular Signalling 18: 83–92. DOI 10.1016/j.cellsig.2005.03.017. [Google Scholar] [CrossRef]
van Asch CJ, Luitse MJ, Rinkel GJ, van der Tweel I, Algra A et al. (2010). Incidence, case fatality, and functional outcome of intracerebral haemorrhage over time, according to age, sex, and ethnic origin: A systematic review and meta-analysis. Lancet Neurology 9: 167–176. DOI 10.1016/S1474-4422(09)70340-0. [Google Scholar] [CrossRef]
Wang J, Dore S (2007). Inflammation after intracerebral hemorrhage. Journal of Cerebral Blood Flow & Metabolism 27: 894–908. DOI 10.1038/sj.jcbfm.9600403. [Google Scholar] [CrossRef]
Xi G, Reiser G, Keep RF (2003). The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: Deleterious or protective? Journal of Neurochemistry 84: 3–9. DOI 10.1046/j.1471-4159.2003.01268.x. [Google Scholar] [CrossRef]
Xu Z, Li X, Chen J, Zhao J, Wang J et al. (2016). USP11, deubiquitinating enzyme, associated with neuronal apoptosis following intracerebral hemorrhage. Journal of Molecular Neuroscience 58: 16–27. DOI 10.1007/s12031-015-0644-0. [Google Scholar] [CrossRef]
Ye S, Lawlor MA, Rivera-Reyes A, Egolf S, Chor S et al. (2018). YAP1-mediated suppression of USP31 enhances NFκB activity to promote sarcomagenesis. Cancer Research 78: 2705–2720. DOI 10.1158/0008-5472.CAN-17-4052. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |