DOI:10.32604/biocell.2022.016607
| BIOCELL DOI:10.32604/biocell.2022.016607 | |
| Article |
Human β-defensin 2 enhances IL-1β production and pyroptosis through P2X7-mediated NLRP3 expression in macrophages
1Department of Periodontology, Guanghua School and Hospital of Stomatology, Sun Yat-sen University, Guangzhou, China
2Guangdong Provincial Key Laboratory of Stomatology, Guanghua School and Hospital of Stomatology, Sun Yat-sen University, Guangzhou, China
3Department of Oral Implantology, Delun Dental, Guangzhou, China
*Address correspondence to: Chuanjiang Zhao, zhaochj@mail.sysu.edu.cn
Received: 31 March 2021; Accepted: 02 May 2021
Abstract: Periodontal disease is the leading cause of tooth loss, which is also a high-risk factor for other diseases including oral cancer and cardiovascular disease. Periodontitis is one of the most common type of periodontal diseases. Interleukin-1β (IL-1β) plays a key role in the pathogenesis of periodontitis. However, the mechanism how IL-1β is produced during periodontitis is still unclear. In the present study, we found that human β-defensin 2 (hBD2) enhances IL-1β production through an LPS-primed human acute monocytic leukemia (THP-1) macrophage model. Inhibition of P2X purinoceptor 7 (P2X7) reduced hBD2-enhanced IL-1β production. Incubation of LPS-primed THP-1 macrophages with potassium chloride also suppressed hBD2-enhanced IL-1β production. Silence of inflammasome adaptor Nod-like receptor family pyrin domain containing 3 (NLRP3) led to reduced hBD2-enhanced IL-1β production. Likewise, inhibition of caspase-1 also resulted in the decrease of IL-1β. Moreover, an ethidium bromide uptake test indicated that hBD2-activated caspase-1 mediated pyroptotic pore formation. Subsequent lactate dehydrogenase detection and flow cytometric analysis indicated that hBD2 also induced pyroptosis. In brief, these findings illustrated not only the mechanism of hBD2 in enhancing the inflammatory response, but also provided novel therapeutic targets for periodontitis.
Keywords: Periodontitis; Human β-defensin 2; IL-1β; Signal transduction; Pyroptosis
Periodontitis, affects the gingiva and the supporting connective tissue and alveolar bone, is among the most common human disorders (Williams, 1990). Periodontitis develops with accumulation of dental plaque, disturbances in the oral microbiota, immune response of the host, tissue destruction and ultimate tooth loss (Michaud et al., 2017). Systemic diseases, environmental and psychological factors are considered as modulators for periodontitis (de Morais et al., 2018). Moreover, periodontitis is a high risk factor for other diseases including oral cancer, cardiovascular disease, diabetes mellitus and rheumatoid arthritis (Beck et al., 1996; de Molon et al., 2019; Geismar et al., 2006; Lalla and Papapanou, 2011; Michaud et al., 2017).
Macrophages are amongst the first immune cells respond to bacteria and their products in periodontitis through potently activating caspase-1, producing large amounts of proinflammatory cytokines including interleukin (IL)-1β and IL-18, releasing lactate dehydrogenase (LDH), and initiating the pyroptotic cell death (Fleetwood et al., 2017). IL-1β is significantly increased in gingival biopsy samples and gingival crevicular fluid (GCF) in patients with periodontitis (Howells, 1995; Offenbacher et al., 2018). Many studies have revealed that IL-1β plays a key role in periodontal disease through upregulating adhesion molecules on leucocytes, stimulating the production of chemokines and inducing expression of other inflammatory mediators (e.g., matrix metalloproteinases, MMPs) for subsequent inflammatory responses (Graves, 2008; Preshaw and Taylor, 2011). In addition, IL-1β also induces bone resorption and connective tissue degradation (Birkedal-Hansen, 1993). Essentially, periodontitis is caused by the dysregulated immune response mediated by proinflammatory cytokines (e.g., IL-1β, IL-6 and tumor necrosis factor-α) in periodontal tissue (Preshaw and Taylor, 2011).
It is well known that two signals are required for IL-1β production. First, exogenous stimuli (e.g., lipopolysaccharide, LPS) induce transcription of pro-IL-1β through the Toll-like receptor (TLR). Second, extracellular stimuli including ATP initiate the assembly of an inflammasome via NOD-like receptors (NLRs, e.g., NLRP3 and AIM2) to activate IL-1β by caspase-1 (Latz, 2010). During this process, ATP-activated purinergic receptor P2X7 is essential for inducing assembly of NLRP3 inflammasome, activation of caspase-1 and mature IL-1β production (Kayagaki et al., 2011; Mariathasan et al., 2006). However, the mechanism how IL-1β is produced in periodontitis is still unclear. Human β-defensin (hBD) expressed by human normal gingival epithelial cells plays a vital role in the innate immune response and protects human from infection (Liu et al., 2014; Yang et al., 1999), which also has antimicrobial effect in gingiva of periodontitis patients (Wang et al., 2015). Our previous work in patients with periodontitis has indicated that the anti-microbial ability of hBD2 is stronger than hBD1 and hBD3 (Wang et al., 2015). Other studies further demonstrated that hBD2 modulates human immunity through inducing monocytes including macrophage productions of chemokines and proinflammatory cytokines including IL-1β (Chaly et al., 2000; Rohrl et al., 2010a). To date, the role of hBD2 on IL-1β production in periodontitis is not known.
In addition, IL-1β could promote caspase-1/Gasdermin D (GSDMD)-induced pyroptosis, which is critical for controlling infection (Bergsbaken et al., 2009; Miao et al., 2010; Wang et al., 2017b). Pyroptosis in macrophage during innate immune response might be a regulatory mechanism to protect human from infection in periodontitis and subsequently maintain the normal number of immune cells to avoid excessive inflammatory responses (Fleetwood et al., 2017). GSDMD, a substrate of both caspase-1 and caspase-11/4/5, leads to non-selected membrane pore formation which can be detected by EtBr or 7-AAD staining (Fink and Cookson, 2006; Zhang et al., 2018). However, whether hBD2 could induce pyroptosis of macrophage through IL-1β is unclear.
Thus, the primary aim of this study is to investigate whether hBD2 enhances IL-1β production and pyroptosis in periodontitis using LPS-primed THP-1 macrophage model.
Synthetic hBD-2 (ab243124) was purchased from Abcam (Cambridge, MA, USA); ethidium bromide (EtBr) (#E8751), potassium chloride (KCl) (#P5405), apyrase (#A6410), KN-62 (#SLBR3900V), oxidized ATP (oATP) (#061M1721V), LPS from Escherichia coli serotype O1101:B4 (#L4391) and phorbol 12-myristate 13-acetate (PMA) (#061M1721V) were obtained from Sigma-Aldrich (St. Louis, MO, USA); ATP (#10127531001) was from Roche Applied Science (Indianapolis, IN, USA); YVAD-cmk (#400012) was purchased from Merck Millipore (Darmstadt, Germany); SYTO42 (#S7563) was obtained from Life Technologies (Grand Island, NY, USA); NLRP3 (#ab91413), caspase-1 (#ab238972), nuclear factor kappa-B (NF-κB) (#ab32360), GSDMD (#ab215203)) and GAPDH (#ab8245) antibodies were from Abcam; NLRP3 small interfering RNA (siRNA) and scrambled siRNA were purchased from Sangon Biotech (Shanghai, China); LDH-Cytotoxicity Colorimetric Assay kit (#K311) was obtained from Biovsion (Milpitas, CA, USA); Annexin V (#A13201) was from Thermo Fisher Scientific (Rockford, IL, USA).
THP-1 cells were maintained in RPMI1640 medium (# 61870044) (Thermo Fisher Scientific) containing with 10% FBS (#10099) (Thermo Fisher Scientific), 100 U/mL penicillin and 100 mg/mL streptomycin (#15070063) (Thermo Fisher Scientific) at 37°C. NLRP3 siRNA was transfected into THP-1 cells to silence NLRP3 expression using Lipofectamine 2000 (#11668019) (Thermo Fisher Scientific) according to the manufacturer’s instruction.
To perform experiments, THP-1 cells were plated in 24-well plates and subsequently differentiated for 12 h cultured by RPMI1640 medium supplemented with 100 nM PMA and 10% FBS (Suppl. Fig. S1). Next, medium was replaced with RPMI1640 medium without FBS. After 48 h, THP-1 cells were treated with or without 100 ng/mL LPS in RPMI1640 medium without FBS for 3 h. Then THP-1 cells were washed twice using fresh RPMI1640 medium followed by culture with 5 mM ATP for 1 h or 0.02, 0.2, 2, 5, 10, 20, or 40 μg/mL hBD2 for 8 h (Fig. 1A). For different subsequent experiments, THP-1 cells were incubated with 130 mM KCl, 20 U/mL Apyrase, 20 mM KN-62, 900 mM oATP or 100 mM YVAD-cmk (Merck Millipore, Germany) in RPMI1640 medium for 30 min before the treatment of ATP or hBD2. After this, the cell-free supernatants and THP-1 cells were collected for further analysis.
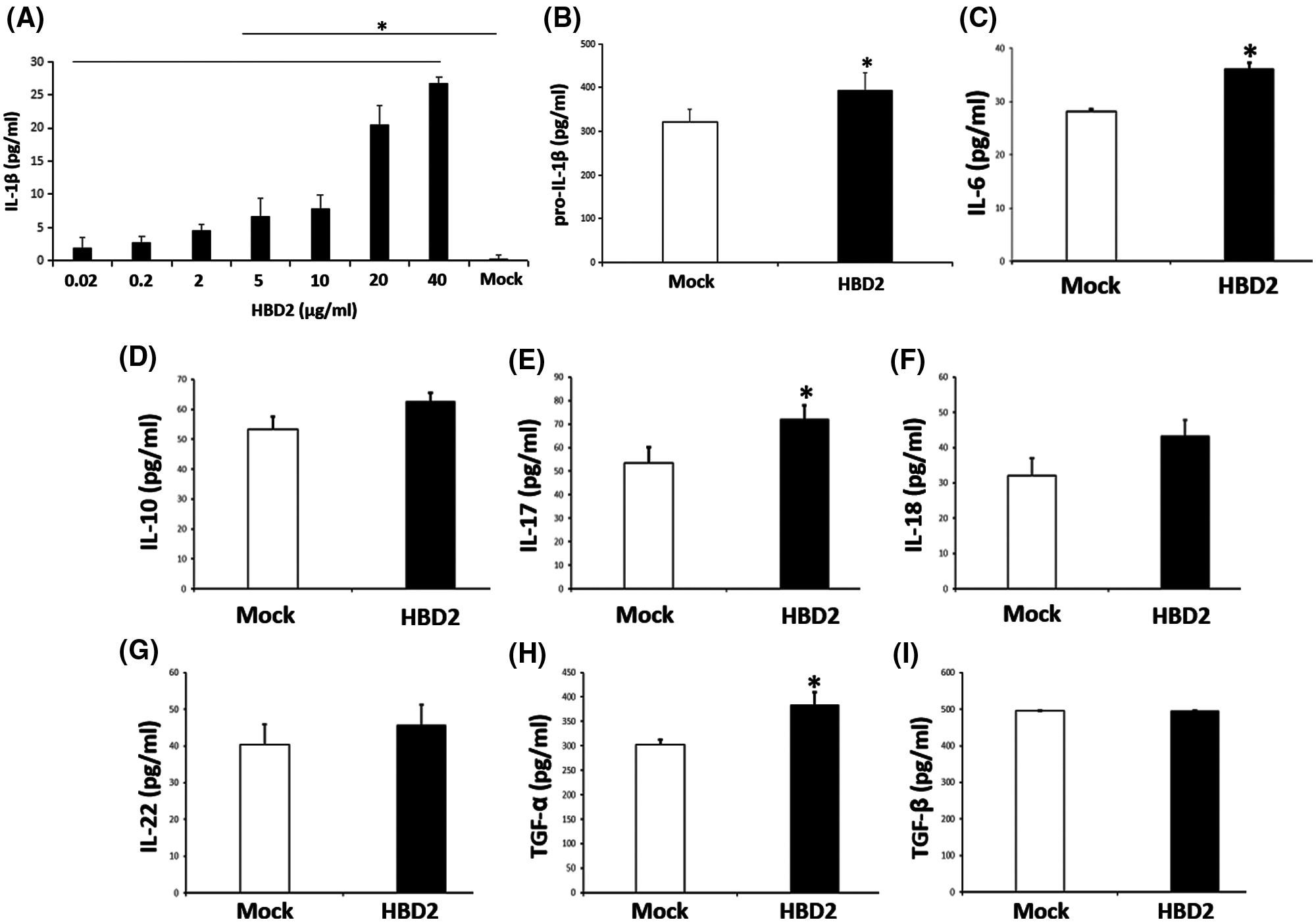
Figure 1: hBD2 treatment of LPS-primed THP-1 cells increases IL-1β production.
(A) The concentration of released IL-1β in the cell medium measured by ELISA (N = 3 per group). (B) The cellular level of pro-IL-1β measured by ELISA (N = 3 per group). (C-I) The concentration of released IL-6, IL-10, IL-17, IL-18, IL-22, TGF-α, and TGF-β in the cell medium measured by ELISA (N = 9 per group). Statistical analysis is based on a one-way ANOVA with Tukey’s multiple-comparison test. *P < 0.05.
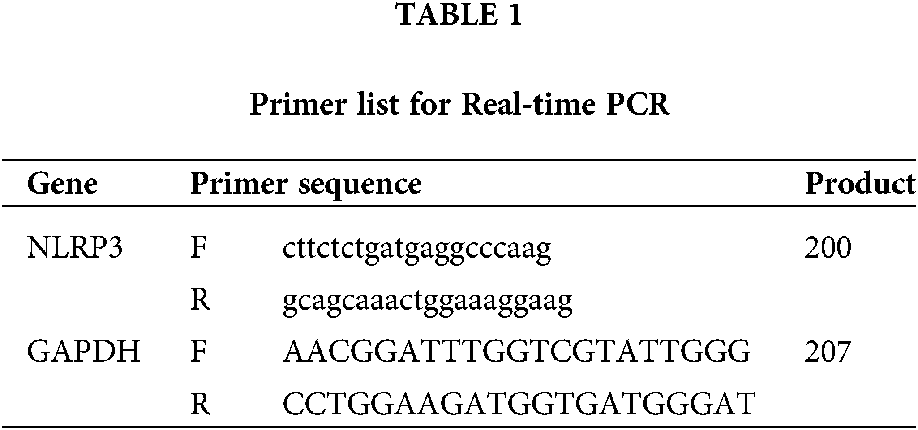
After the transfection of NLRP3 siRNA, real-time PCR was performed to verify the efficiency of NLRP3 knock down in THP-1 cell. Total mRNA was isolated from THP-1 cells using the Trizol reagent (#15596018) (Life Technologies) and then reversed transcribed by the QuantiTect Reverse Transcription Kit (#205313) (Qiagen, Shanghai, China). Real-Time PCR was performed by the StepOnePlus system (Applied Biosystem) using Thermo Fisher Scientific Maxima SYBR Green/ROX qPCR Master Mix assay (2X) (#K0221). Primer sequences were showed in Table 1.
The released IL-1β, IL-6, IL-10, IL-17, IL-18, IL-22, transforming growth factor-β (TGF-β), TGF-α, and pro-IL-1β were measured by ELISA kits obtained from R&D Systems (Minneapolis, MN, USA) according to the manufacturer’s instruction. Of these, released IL-1β in the cell-free supernatants was detected by Human IL-1 beta/IL-1F2 Quantikine ELISA Kit (#DLB50) while pro-IL-1β in cells was measured using Human Pro-IL-1 beta/IL-1F2 Quantikine ELISA Kit (#DLBP00).
To detect cellular level of target proteins, protein extracted from THP-1 cells were detected by Western Blot as previously described (Pan et al., 2018). Briefly, equal amounts of protein (30 μg) from each group were loaded and separated by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently transferred onto Millipore Immobilon-P membranes (Shanghai, China). Subsequently, the membranes were blocked with 5% nonfat milk for 2 h at room temperature (RT) and then incubated with the primary antibodies in 1% nonfat milk overnight at 4°C. Following primary antibody incubation, the membranes were washed 4 times by Tris-buffered saline contained 0.1% Tween20 (TBST) and subsequently exposed to secondary antibody (1:10,000, Aksomics, Shanghai, China). The signals of targeted proteins were detected by the SuperSignal West Femto Maximum Sensitivity Substrate kit obtained from Thermo Scientific (Rockford, USA). Primary antibodies used in the present study were listed as follow: caspase-1-p10 (1:500), NLRP3 (1:500), NF-κB (1:500), GSDMD (1:400) and GAPDH (1:3,000). All primary antibodies were purchased from Abcam (Boston, USA).
Pore formation was determined by EtBr stain permeability as described previously (Fink and Cookson, 2006). Briefly, THP-1 cells were plated on coverslips in the well of 24-well plate. Subsequently, cells were differentiated by PMA and stimulated using LPS as described above. After discarding the medium, coverslips were covered by 15 mL PBS supplemented with 10 mM SYTO 42 (blue) and 25 mg/mL EtBr (red) for 5 min in dark following the treatment with various compounds (5 mM ATP, 20 μg/mL hBD2, 100 mM YVAD-cmk). Images were acquired for further analysis. Pore formation was investigated by the percentage of EtBr positive-stained cells.
THP-1 cells plated in 24-well plates were differentiated by PMA and stimulated using LPS as described above. After the treatment with various compounds (5 mM ATP, 20 μg/mL hBD2 (according to optimization of concentration in Fig. 1A), or 100 mM YVAD-cmk), the culture medium was collected for the measurement of released LDH according to the manufacturer’s instruction.
Flow cytometric analysis for programmed cell death
THP-1 cells plated in 24-well plates were differentiated by PMA and stimulated using LPS as described above. After the treatment with various compounds (5 mM ATP, 20 μg/mL hBD2, 100 mM YVAD-cmk), cells were collected and washed twice with incubation buffer (10 mmol/L HEPES/NaOH, pH 7.4, 140 mmol/L NaCl, 5 mmol/L CaCl2). Subsequently, cells were resuspended and stained with 1.5 μg/mL Annexin V and moderate propidium iodide (PI) (Thermo Scientific) in 100 μL PBS at room temperature (RT) for 15 min in the dark. After washing twice by PBS, cells were resuspended into 400 μL incubation buffer and analyzed by flow cytometry.
All experiments in this study were repeated three times and average values of three experiments were presented as the mean ± SD calculated by STDEV formula in Excel. The significance of all data was estimated by a Tukey’s multiple-comparison test in the ANOVA analysis using the SigmaStat 3.5 software. Importantly, statistical significance was accepted when P < 0.05.
hBD2 treatment enhances IL-1β production in LPS-primed THP-1 macrophages
To identify whether hBD2 enhances IL-1β production in periodontitis, the LPS-primed THP-1 macrophage model was used. Results showed that hBD2 promoted mature IL-1β secretion from LPS-primed THP-1 macrophages in a dose-dependent way (Fig. 1A). The concentration of 20 μg/mL hBD2 was applied in this study. Meanwhile, hBD2 treatment also increased cellular level of pro-IL-1β (Fig. 1B). These data suggested that hBD2 enhanced IL-1β production in LPS-primed THP-1 macrophages. To further reveal the role of hBD2 in periodontitis, other released cytokines related to periodontitis were detected. Besides IL-1β, hBD2 enhanced the secretion of IL-6, IL-17, TGF-α but not IL-10, IL-18, IL-22 and TGF-β (Figs. 1C–1I), suggesting that hBD2 might contribute to periodontitis through modulating these cytokines.
hBD2 treatment promotes IL-1β production through P2X7 in LPS-primed THP-1 macrophages
Previous studies have indicated the effect of ATP on the secretion of IL-1β is mediated by P2X7, which can be inhibited by KN-62 and oATP (Elssner et al., 2004; Ferrari et al., 2006). Results indicated that inhibition of P2X7 by oATP and KN-62 suppressed IL-1β production in LPS-primed THP-1 macrophages treated with ATP (Fig. 2A) or hBD2 (Fig. 2B). Above data suggested that hBD2 treatment of LPS-primed THP-1 macrophages leading to increased IL-1β production was mediated by P2X7.
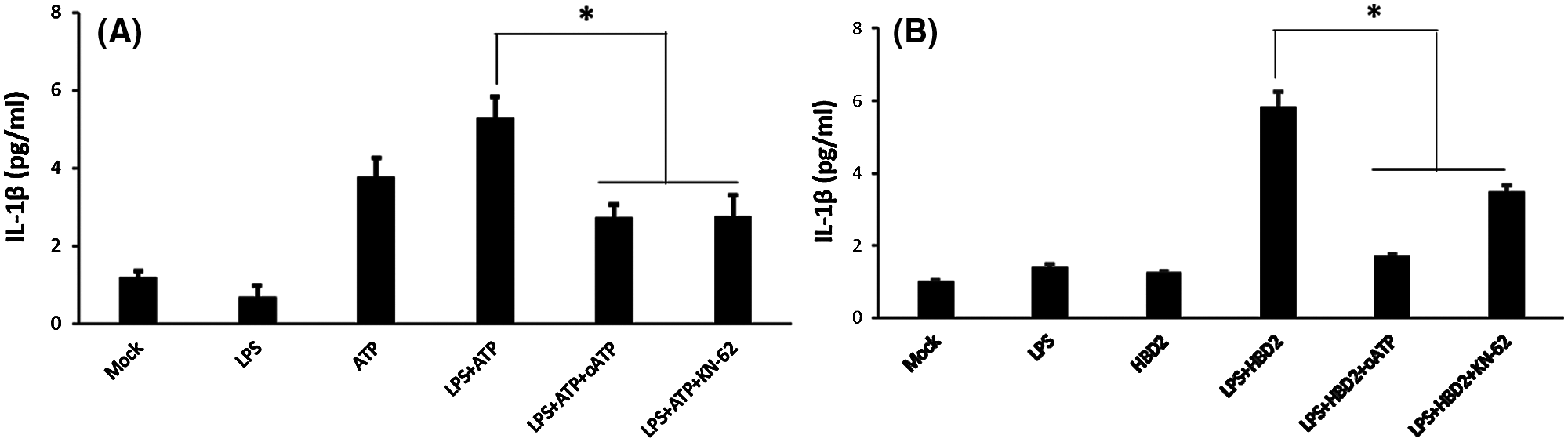
Figure 2: hBD2 treatment of LPS-primed THP-1 cells increases IL-1β production via P2X7.
(A and B) The concentration of released IL-1β in the cell medium measured by ELISA (N = 3 per group). Statistical analysis is based on a one-way ANOVA with Tukey’s multiple-comparison test. *P < 0.05.
K+ efflux is necessary for hBD2-enhanced IL-1β production in LPS-primed THP-1 macrophages
A previous study has indicated that defensins promote ATP efflux for subsequent immune response (Vylkova et al., 2007). To address whether hBD2 enhances IL-1β production through promoting ATP efflux in LPS-primed THP-1 macrophages, the autocrine ATP inhibitor apyrase was used. It was indicated that apyrase did not influence the effect of hBD2 on IL-1β production (Fig. 3).
In addition, cytosolic K+ efflux induced by ATP-activated P2X7 is required for inflammasome assembly and following caspase-1 activation and IL-1β mature processing (Kahlenberg and Dubyak, 2004). To identify whether K+ efflux contributes to IL-1β production after hBD2 treatment, LPS-primed THP-1 macrophages were cultured with RPMI1640 medium supplemented with 130 mM KCl. Results showed that hBD2-enhanced IL-1β production was abrogated by KCl, while prevention of cytosolic K+ efflux also block ATP-induced IL-1β production (Fig. 3). All these results suggested that ATP-independent K+ efflux was required for hBD2-enhanced IL-1β production in LPS-primed THP-1 macrophages.
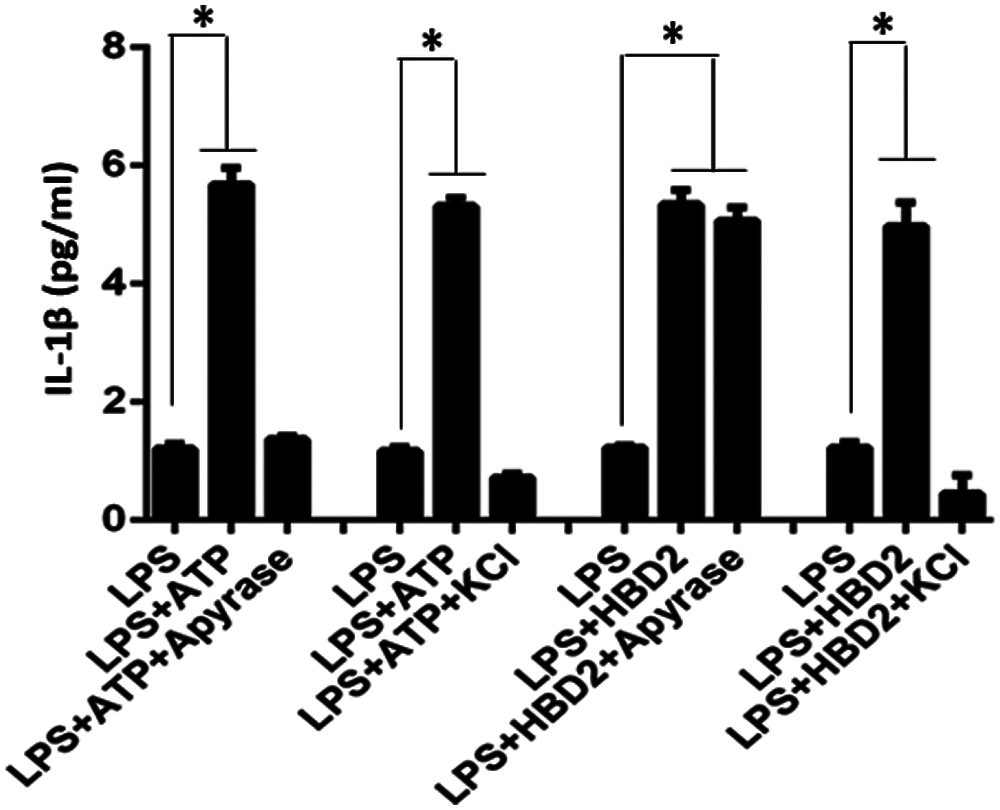
Figure 3: K+ efflux is required for IL-1β production in hBD2-treated LPS-primed THP-1 cells. The concentration of released IL-1β in the cell medium measured by ELISA (N = 3 per group.). Statistical analysis is based on a one-way ANOVA. *P < 0.05.
hBD2 treatment enhances IL-1β production through NLRP3 in LPS-primed THP-1 macrophages
NLRP3 expression is necessary for the production and release of IL-1β (Latz, 2010). hBD2 treatment in LPS-primed THP-1 macrophages significantly increased protein levels of NF-κB which is a vital transcript for NLRP3 expression (Broz and Dixit, 2016), NLRP3 and activated caspase-1 (caspase-1 p10) (Figs. 4A and 4B). To further identify the role of NLRP3 in hBD2-induced IL-1β production, siRNA was used to knockdown NLRP3 (Fig. 4C). Results indicated that NLRP3 knockdown reduced activation of caspase-1 and led to decrease of IL-1β from LPS-primed THP-1 macrophages treated with hBD2 (Figs. 4D and 4F). Above data together suggested that hBD2 enhanced IL-1β production through NLRP3 inflammasome in LPS-primed THP-1 macrophages.

Figure 4: NLRP3 is necessary for IL-1β production in hBD2-treated LPS-primed THP-1 cells.
(A) Protein abundance of activated caspase-1 (caspase-1 p10), NF-κB and NLRP3 in THP-1 cells treated with or without LPS (N = 3 per group). (B) In the bar graphs, levels of caspase-1 p10, NF-κB and NLRP3 were assessed and normalized by GAPDH level (N = 3 per group). (C) Relative expression level of NLRP3 in hBD2-treated LPS-primed THP-1 cells transfected with NLRP3 siRNAs (N = 3 per group). (D) The concentration of released IL-1β in the cell medium from in hBD2-treated LPS-primed THP-1 cells (N = 3 per group). (E) Protein abundance of caspase-1 in hBD2-treated LPS-primed THP-1 cells transfected with or without NLRP3 siRNA. (F) In the bar graphs, levels of caspase-1 were assessed and normalized by GAPDH level (N = 3 per group). Statistical analysis is based on a one-way ANOVA with Tukey’s multiple-comparison test. *P < 0.05.
hBD2 treatment enhances IL-1β production through caspase-1 activation in LPS-primed THP-1 macrophages
Activation of caspase-1 regulated by numerous factors mentioned above is a critical step for IL-1β mature and release (Schroder and Tschopp, 2010). As observed in LPS-primed THP-1 macrophages, hBD2 treatment increased the level of activated caspase-1 (Figs. 4A and 4B). To further address the effect of hBD2 on caspase-1 activation, YVAD-cmk (Merck Millipore) (caspase-1 inhibitor) was used. Results showed that inhibition of caspase-1 by YVAD-cmk (Merck Millipore) dramatically suppressed IL-1β production in LPS-primed THP-1 macrophages treated with ATP (positive control) or hBD2 (Figs. 5A and 5B). Above data suggested that caspase-1 activation is critical for hBD2-enhanced IL-1β production in LPS-primed THP-1 macrophages.
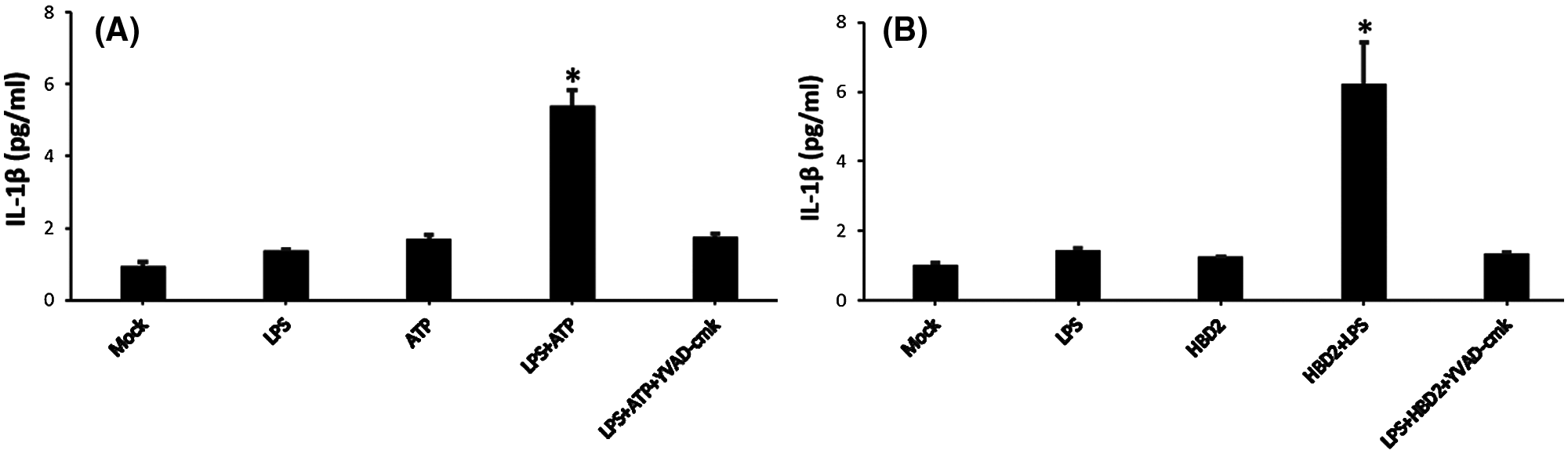
Figure 5: hBD2 treatment of LPS-primed THP-1 cells increases IL-1β production through caspase-1 activation.
The concentration of released IL-1β in the cell medium measured by ELISA (N = 3 per group). Statistical analysis is based on a one-way ANOVA with Tukey’s multiple-comparison test. *P < 0.05.
Activated caspase-1 involves in pore formation in LPS-primed THP-1 macrophages treated with hBD2
Previous study has revealed that activated caspase-1 triggers membrane pore formation and subsequent pyroptosis in LPS-primed macrophages (Bergsbaken et al., 2009). Moreover, pyroptotic pores allow the passage of small molecules including EtBr, but not the large molecules such as EthD2 (Fink and Cookson, 2006; Zhang et al., 2018). Results showed that hBD2 treatment significantly enhanced EtBr uptake in LPS-primed THP-1 macrophages (Fig. 6A). However, inhibition of caspase-1 prevented the increase of EtBr enhanced by hBD2 (Fig. 6A). These results were consistent with those in ATP treatment of LPS-primed THP-1 macrophages (Fig. 6B). Thus, above results suggested that activated caspase-1 was involved in pyroptotic pore formation in LPS-primed THP-1 macrophages treated with hBD2.

Figure 6: Activated caspase-1 is essential for pore formation in hBD2-treated LPS-primed THP-1 cells.
Pore formation was detected by the percentage of the cells stained with EtBr. Blue: DAPI; Red: EtBr. (A) Representative images and quantitative statistics of EtBr staining cells. THP-1 cells were treated by 100 ng/mL LPS, 5 mM ATP, 100 mM YVAD-cmk, respectively. (B) Representative images of EtBr staining 0.20 μg/mL hBD2, 5 mM ATP, 100 mM YVAD-cmk, respectively. Scale bar: 50 μm. At least three images were analyzed per group. *P < 0.05.
hBD2 treatment induces pyroptosis in LPS-primed THP-1 macrophages
To further investigate whether hBD2 induced pyroptosis in LPS-primed THP-1 macrophages, LDH release assay and flow cytometric analysis were performed. In LPS-primed THP-1 macrophages, hBD2 treatment significantly increased the level of cleaved GSDMD which is required for pyroptosis (Wang et al., 2017b) (Figs. 7A and 7B). However, suppression of caspase-1 prevented the increase of cleaved GSDMD induced by hBD2 (Figs. 7A and 7B). In addition, hBD2 treatment dramatically elevated the released LDH (a marker for pyroptosis) in LPS-primed THP-1 macrophages, whereas inhibition of caspase-1 alleviated the effect of hBD2 (Fig. 7C). More importantly, hBD2 induced programmed cell death of LPS-primed THP-1 macrophages while caspase1 inactivation reduced cell death triggered by hBD2 (Figs. 7D and 7I). These results suggested that hBD2 treatment induced pyroptosis through activating caspase 1 in LPS-primed THP-1 macrophages.

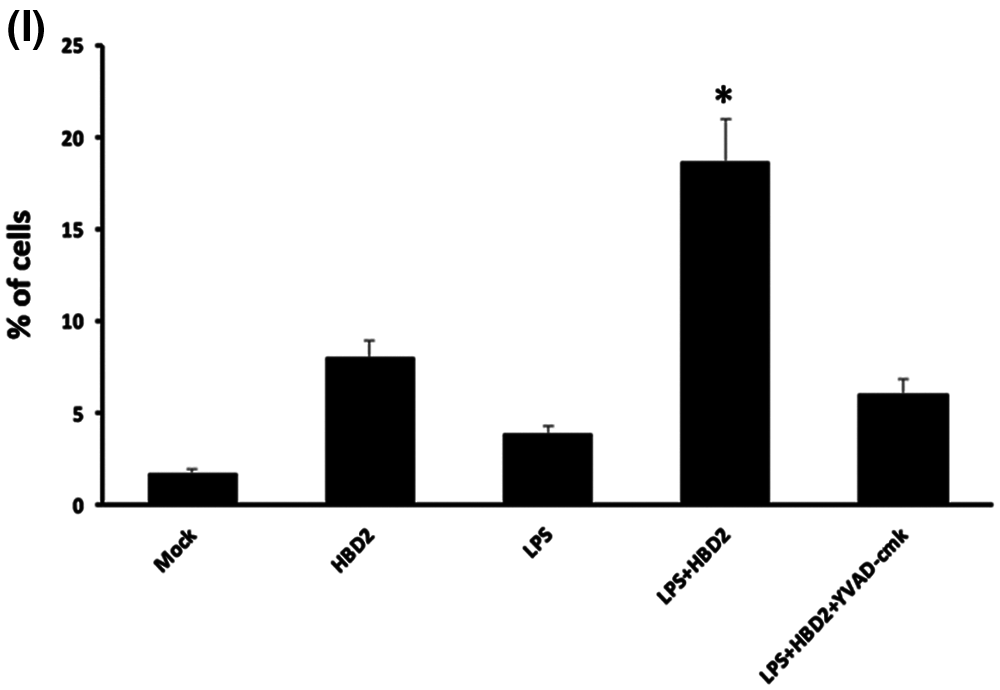
Figure 7: hBD2 treatment triggers pyroptosis in LPS-primed THP-1 macrophages.
(A) Protein abundance of cleaved GSDMD in THP-1 cells treated with LPS, HBD2 or YVAD-cmk. (B) In the bar graphs, cleaved GSDMD level was assessed and normalized by GAPDH level (N = 3 per group). (C) Level of released LDH in THP-1 cells treated with LPS, HBD2 or YVAD-cmk (N = 3 per group). (D–H) Representative of flow cytometric analysis of programmed cell death in THP-1 cells treated with LPS, HBD2 or YVAD-cmk. (I) The bar graphs indicated numbers of death cells in different groups (N = 3 per group.). Statistical analysis is based on a one-way ANOVA with Tukey’s multiple-comparison test. *P < 0.05.
Periodontal disease is the leading cause of tooth loss (Michaud et al., 2017), and the high risk factor for other diseases including oral cancer and cardiovascular disease coronary heart disease (Beck et al., 1996; Geismar et al., 2006; Michaud et al., 2017). IL-1β plays a key role in the pathogenesis of periodontal disease (Graves, 2008; Preshaw and Taylor, 2011). However, the mechanism how IL-1β is produced during periodontal disease is still unclear. Using LPS-primed THP-1 macrophages model, we found that hBD2 induced NLRP3 expression and caspase-1 activation through P2X7 while enhancing cytosolic K+ efflux to promote IL-1β production and pyroptosis.
Alarmins including defensins can attract and activate macrophages to promote innate and adaptive immune responses (Oppenheim and Yang, 2005; Yang et al., 2009). To date, growing evidence have revealed that defensins exert influences on immune responses by numerous immune-related receptors. It has been demonstrated that Gαi protein-coupled receptors contained CC chemokine receptor (CCR) 2 and CCR6 mediate the chemotactic activity of defensins (Rohrl et al., 2010a; Rohrl et al., 2010b). Moreover, human β-defensin 3 (hBD3) activated dendritic cells and primary epithelial cells to mediate the production of cytokines through TLR1/TLR2 receptors (Biragyn et al., 2002; Inthasin et al., 2018), while murine β-defensin 2 can activate dendritic cells via the TLR4 receptor (Biragyn et al., 2002). Here, we found that hBD2 can activate LPS-primed THP-1 macrophages to enhance the production of IL-1β by P2X7 receptor. A recent study also indicated that hBD2 and hBD3 enhance expression of pro-inflammatory cytokines in normal THP-1 macrophages in a P2X7 dependent manner (Wanke et al., 2016). As the member of α-defensin, human neutrophil peptide (HNP)-1 induces IL-8 production in lung and intestinal epithelial cells via the purinergic receptor P2Y6 (Ibusuki et al., 2015; Khine et al., 2006). These findings together suggested that alarmin or defensins may bind to different receptors in different cells to regulate pro-inflammatory cytokine production and activate the immune system.
Cytosolic K+ efflux induced by ATP-activated P2X7 is required for NLRP3 expression and subsequent caspase-1 activation and IL-1β mature processing (Kahlenberg and Dubyak, 2004). It was demonstrated that hBD2 may regulate K+ efflux through P2X7. However, hBD2 may directly regulate cytosolic K+ efflux. Recent study has revealed that voltage-gated K+ (Kv) channel S1-S2 linker is a novel receptor site for hBD2, which anchors in the channel S1–S2 linker whereas modulates channel activation by electrostatic repulsion via an adjacent S4 helix (Feng et al., 2015). Furthermore, hBD2 can regulate K+ channel activation through binding with channel extracellular pore region (Yang et al., 2015). More importantly, it was revealed that hBD2-enhanced IL-1ß production did not require ATP release while a previous study has indicated that hBD2 enhances pro-inflammatory cytokine expression through ATP-release in a P2X7R dependent manner (Wanke et al., 2016). Thus, hBD2 may directly regulate cytosolic K+ efflux to induce IL-1β production in a P2X7-independent manner.
It was showed that hBD2 could induce the protein level of caspase 1, NLRP3, NF-kB and pro-IL-1β, suggesting that hBD2 may have the effect on protein synthesis of inflammatory cytokines. However, no study has indicated this effect of hBD2. As a non-coding RNA, miRNAs regulate protein expression of target genes at post-transcriptional level (Krol et al., 2010; Trionfini and Benigni, 2017). Limited studies have showed that defensin may be associated with microRNA regulation in the immune defense. All these studies together suggested that hBD2 may regulate protein level of inflammatory cytokines through miRNA (Duskova et al., 2013; Liao et al., 2017).
Activation of caspase-1 plays multifunctional roles. Besides mediating IL-1β production, activated caspase-1 is also crucial for pyroptosis (Bergsbaken et al., 2009). In fact, data concerning the relationship between human defensins and cell death are limited. A recent study has revealed that hBD3 induces apoptosis in airway smooth muscle cells (Wang et al., 2017a). To date, no report mentions the role of human defensins in pyroptosis. It was revealed that hBD2 treatment induced caspase-1-mediated pyroptotic pore formation in LPS-primed macrophages. It has been reported that P2X7-K+ efflux-caspase-1 signaling pathway mediates pyroptotic pore formation with HNP-1 in LPS-primed macrophages (Chen et al., 2014). Other studies further indicated that P2X7 induces pore formation in IL-1β production independent manner (Bergsbaken et al., 2011; Le Feuvre et al., 2002). Therefore, hBD2 may induce pyroptosis through an IL-1β- independent manner. We thought that hBD2 may trigger innate immune response to protect human from infection in periodontitis and subsequently maintain the normal number of immune cells to avoid excessive inflammatory responses through pyroptosis.
Several issues needed to be focused on in the future study. First, it remains to be determined whether hBD2 associated with P2X7 to mediate IL-1β production in hBD2 treatment of LPS-primed THP-1 macrophages, though a previous study has demonstrated that alarmins bind to P2X7 (Chen et al., 2014). Second, the mechanism how hBD2 regulates cytosolic K+ efflux and the pathway involved in hBD2-induced pyroptosis still needed to be identified. Third, the gingival hBD2 level and IL-1β level should be detected simultaneously for analyzing the relationship between hBD2 and IL-1β in periodontitis patients and the periodontitis model in mice. Finally, the role of hBD2 and IL-1β in periodontitis and pyroptosis should be further identified by the experimental mice model of periodontitis using LPS injections into the gingival tissue to mimic periodontal disease.
In summary, this study demonstrated that hBD2 induced NLRP3 expression to promote IL-1β production and pyroptosis through ATP-activated P2X7 in LPS-primed THP-1 macrophages. The present study illustrated the mechanism by which hBD2 promotes the inflammatory response and following pyroptosis and provided novel therapeutic targets for the treatment of periodontal disease.
Availability of Data and Materials: The data of the present study are available from the corresponding author upon reasonable request.
Authors’ Contribution: The authors confirm contribution to the paper as follows: study conception and design: PPW, CJZ; data collection: PPW, GL, LG; analysis and interpretation of results: PPW, GL, LG, CJZ; draft manuscript preparation: PPW, CJZ. All authors reviewed the results and approved the final version of the manuscript.
Funding Statement: This work was supported by National Natural Science Foundation of China (NSFC) (81500871) and Natural Science Foundation of Guangdong Province (2016A030310214).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Beck J, Garcia R, Heiss G, Vokonas PS, Offenbacher S (1996). Periodontal disease and cardiovascular disease. Journal of Periodontology 67: 1123–1137. DOI 10.1902/jop.1996.67.10s.1123. [Google Scholar] [CrossRef]
Bergsbaken T, Fink SL, Cookson BT (2009). Pyroptosis: Host cell death and inflammation. Nature Reviews Microbiology 7: 99–109. DOI 10.1038/nrmicro2070. [Google Scholar] [CrossRef]
Bergsbaken T, Fink SL, Den Hartigh AB, Loomis WP, Cookson BT (2011). Coordinated host responses during pyroptosis: Caspase-1-dependent lysosome exocytosis and inflammatory cytokine maturation. Journal of Immunology 187: 2748–2754. DOI 10.4049/jimmunol.1100477. [Google Scholar] [CrossRef]
Biragyn A, Ruffini PA, Leifer CA, Klyushnenkova E, Shakhov A et al. (2002). Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science 298: 1025–1029. DOI 10.1126/science.1075565. [Google Scholar] [CrossRef]
Birkedal-Hansen H (1993). Role of matrix metalloproteinases in human periodontal diseases. Journal of Periodontology 64: 474–484. [Google Scholar]
Broz P, Dixit VM (2016). Inflammasomes: Mechanism of assembly, regulation and signalling. Nature Reviews Immunology 16: 407–420. DOI 10.1038/nri.2016.58. [Google Scholar] [CrossRef]
Chaly YV, Paleolog EM, Kolesnikova TS, Tikhonov II, Petratchenko EV (2000). Neutrophil alpha-defensin human neutrophil peptide modulates cytokine production in human monocytes and adhesion molecule expression in endothelial cells. European Cytokine Network 11: 257–266. [Google Scholar]
Chen Q, Jin Y, Zhang K, Li H, Chen W et al. (2014). Alarmin HNP-1 promotes pyroptosis and IL-1beta release through different roles of NLRP3 inflammasome via P2X7 in LPS-primed macrophages. Innate Immunity 20: 290–300. DOI 10.1177/1753425913490575. [Google Scholar] [CrossRef]
de Molon RS, Rossa C, Thurlings RM, Cirelli JA, Koenders MI (2019). Linkage of periodontitis and rheumatoid arthritis: Current evidence and potential biological interactions. International Journal of Molecular Sciences 20: 4541. DOI 10.3390/ijms20184541. [Google Scholar] [CrossRef]
de Morais EF, Pinheiro JC, Leite RB, Santos PPA, Barboza CAG, Freitas RA (2018). Matrix metalloproteinase-8 levels in periodontal disease patients: A systematic review. Journal of Periodontal Research 53: 156–163. DOI 10.1111/jre.12495. [Google Scholar] [CrossRef]
Duskova K, Nagilla P, Le HS, Iyer P, Thalamuthu A, Martinson J, Bar-Joseph Z, Buchanan W, Rinaldo C, Ayyavoo V (2013). MicroRNA regulation and its effects on cellular transcriptome in Human Immunodeficiency Virus-1 (HIV-1) infected individuals with distinct viral load and CD4 cell counts. BMC Infectious Diseases 13: 250. DOI 10.1186/1471-2334-13-250. [Google Scholar] [CrossRef]
Elssner A, Duncan M, Gavrilin M, Wewers MD (2004). A novel P2X7 receptor activator, the human cathelicidin-derived peptide LL37, induces IL-1β processing and release. Journal of Immunology 172: 4987–4994. DOI 10.4049/jimmunol.172.8.4987. [Google Scholar] [CrossRef]
Feng J, Yang W, Xie Z, Xiang F, Cao Z et al. (2015). Kv channel S1-S2 linker working as a binding site of human β-defensin 2 for channel activation modulation. Journal of Biological Chemistry 290: 15487–15495. DOI 10.1074/jbc.M115.639500. [Google Scholar] [CrossRef]
Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A et al. (2006). The P2X7 receptor: A key player in IL-1 processing and release. Journal of Immunology 176: 3877–3883. DOI 10.4049/jimmunol.176.7.3877. [Google Scholar] [CrossRef]
Fink SL, Cookson BT (2006). Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cellular Microbiology 8: 1812–1825. DOI 10.1111/j.1462-5822.2006.00751.x. [Google Scholar] [CrossRef]
Fleetwood AJ, Lee MKS, Singleton W, Achuthan A, Lee MC et al. (2017). Metabolic remodeling, inflammasome activation, and pyroptosis in macrophages stimulated by Porphyromonas gingivalis and its outer membrane vesicles. Frontiers in Cellular and Infection Microbiology 7: 351. [Google Scholar]
Geismar K, Stoltze K, Sigurd B, Gyntelberg F, Holmstrup P (2006). Periodontal disease and coronary heart disease. Journal of Periodontology 77: 1547–1554. [Google Scholar]
Graves D (2008). Cytokines that promote periodontal tissue destruction. Journal of Periodontology 79: 1585–1591. [Google Scholar]
Howells GL (1995). Cytokine networks in destructive periodontal disease. Oral Diseases 1: 266–270. [Google Scholar]
Ibusuki K, Sakiyama T, Kanmura S, Maeda T, Iwashita Y et al. (2015). Human neutrophil peptides induce interleukin-8 in intestinal epithelial cells through the P2 receptor and ERK1/2 signaling pathways. International Journal of Molecular Medicine 35: 1603–1609. [Google Scholar]
Inthasin N, Wongprompitak P, Boonwong C, Ekpo P (2018). Role of Toll-like receptor 2 in mediating the production of cytokines and human beta-defensins in oral mucosal epithelial cell response to leptospiral infection. Asian Pacific Journal of Allergy and Immunology 37: 198–204. DOI 10.12932/AP-100518-100308. [Google Scholar]
Kahlenberg JM, Dubyak GR (2004). Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. American Journal of Physiology Cell Physiology 286: C1100–C1108. [Google Scholar]
Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479: 117–121. [Google Scholar]
Khine AA, Del Sorbo L, Vaschetto R, Voglis S, Tullis E et al. (2006). Human neutrophil peptides induce interleukin-8 production through the P2Y6 signaling pathway. Blood 107: 2936–2942. [Google Scholar]
Krol J, Loedige I, Filipowicz W (2010). The widespread regulation of microRNA biogenesis, function and decay. Nature Reviews Genetics 11: 597–610. [Google Scholar]
Lalla E, Papapanou PN (2011). Diabetes mellitus and periodontitis: A tale of two common interrelated diseases. Nature Reviews Endocrinology 7: 738–748. [Google Scholar]
Latz E (2010). The inflammasomes: Mechanisms of activation and function. Current Opinion in Immunology 22: 28–33. [Google Scholar]
Le Feuvre RA, Brough D, Iwakura Y, Takeda K, Rothwell NJ (2002). Priming of macrophages with lipopolysaccharide potentiates P2X7-mediated cell death via a caspase-1-dependent mechanism, independently of cytokine production. Journal of Biological Chemistry 277: 3210–3218. [Google Scholar]
Liao X, Yang L, Zhang Q (2017). microRNA expression changes after lipopolysaccharide treatment in gills of amphioxus Branchiostoma belcheri. Developmental & Comparative Immunology 70: 39–44. [Google Scholar]
Liu J, Chen J, Du X, Hu L, Chen L (2014). The expression of hBDs in the gingival tissue and keratinocytes from healthy subjects and periodontitis patients. Archives of Oral Biology 59: 193–198. [Google Scholar]
Mariathasan S, Weiss DS, Newton K, Mcbride J, O’rourke K et al. (2006). Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440: 228–232. DOI 10.1038/nature04515. [Google Scholar] [CrossRef]
Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M et al. (2010). Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nature Immunology 11: 1136–1142. DOI 10.1038/ni.1960. [Google Scholar] [CrossRef]
Michaud DS, Fu Z, Shi J, Chung M (2017). Periodontal disease, tooth loss, and cancer risk. Epidemiologic Reviews 39: 49–58. DOI 10.1093/epirev/mxx006. [Google Scholar] [CrossRef]
Offenbacher S, Jiao Y, Kim SJ, Marchesan J, Moss KL et al. (2018). GWAS for interleukin-1β levels in gingival crevicular fluid identifies IL37 variants in periodontal inflammation. Nature Communications 9: 3686. DOI 10.1038/s41467-018-05940-9. [Google Scholar] [CrossRef]
Oppenheim JJ, Yang D (2005). Alarmins: Chemotactic activators of immune responses. Current Opinion in Immunology 17: 359–365. DOI 10.1016/j.coi.2005.06.002. [Google Scholar] [CrossRef]
Pan H, Ding Y, Yan N, Nie Y, Li M, Tong L (2018). Trehalose prevents sciatic nerve damage to and apoptosis of Schwann cells of streptozotocin-induced diabetic C57BL/6J mice. Biomedcine & Pharmacotherapy 105: 907–914. DOI 10.1016/j.biopha.2018.06.069. [Google Scholar] [CrossRef]
Preshaw PM, Taylor JJ (2011). How has research into cytokine interactions and their role in driving immune responses impacted our understanding of periodontitis? Journal of Clinical Periodontology 38: 60–84. DOI 10.1111/j.1600-051X.2010.01671.x. [Google Scholar] [CrossRef]
Rohrl J, Yang D, Oppenheim JJ, Hehlgans T (2010a). Human β-defensin 2 and 3 and their mouse orthologs induce chemotaxis through interaction with CCR2. Journal of Immunology 184: 6688–6694. DOI 10.4049/jimmunol.0903984. [Google Scholar] [CrossRef]
Rohrl J, Yang D, Oppenheim JJ, Hehlgans T (2010b). Specific binding and chemotactic activity of mBD4 and its functional orthologue hBD2 to CCR6-expressing cells. Journal of Biological Chemistry 285: 7028–7034. DOI 10.1074/jbc.M109.091090. [Google Scholar] [CrossRef]
Schroder K, Tschopp J (2010). The inflammasomes. Cell 140: 821–832. DOI 10.1016/j.cell.2010.01.040. [Google Scholar] [CrossRef]
Trionfini P, Benigni A (2017). MicroRNAs as master regulators of glomerular function in health and disease. Journal of the American Society of Nephrology 28: 1686–1696. [Google Scholar]
Vylkova S, Sun JN, Edgerton M (2007). The role of released ATP in killing Candida albicans and other extracellular microbial pathogens by cationic peptides. Purinergic Signalling 3: 91–97. [Google Scholar]
Wang P, Duan D, Zhou X, Li X, Yang J et al. (2015). Relationship between expression of human gingival beta-defensins and levels of periodontopathogens in subgingival plaque. Journal of Periodontal Research 50: 113–122. [Google Scholar]
Wang W, Qu X, Dang X, Shang D, Yang L et al. (2017a). Human β-defensin-3 induces IL-8 release and apoptosis in airway smooth muscle cells. Clinical & Experimental Allergy 47: 1138–1149. [Google Scholar]
Wang Y, Gao W, Shi X, Ding J, Liu W et al. (2017b). Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547: 99–103. [Google Scholar]
Wanke D, Mauch-Mucke K, Holler E, Hehlgans T (2016). Human beta-defensin-2 and -3 enhance pro-inflammatory cytokine expression induced by TLR ligands via ATP-release in a P2X7R dependent manner. Immunobiology 221: 1259–1265. [Google Scholar]
Williams RC (1990). Periodontal disease. The New England Journal of Medicine 322: 373–382. [Google Scholar]
Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ et al. (1999). Beta-defensins: Linking innate and adaptive immunity through dendritic and T cell CCR6. Science 286: 525–528. [Google Scholar]
Yang D, De La Rosa G, Tewary P, Oppenheim JJ (2009). Alarmins link neutrophils and dendritic cells. Trends in Immunology 30: 531–537. [Google Scholar]
Yang W, Feng J, Xiang F, Xie Z, Zhang G et al. (2015). Endogenous animal toxin-like human β-defensin 2 inhibits own K+ channels through interaction with channel extracellular pore region. Cellular and Molecular Life Sciences 72: 845–853. [Google Scholar]
Zhang Y, Chen X, Gueydan C, Han J (2018). Plasma membrane changes during programmed cell deaths. Cell Research 28: 9–21. DOI 10.1038/cr.2017.133. [Google Scholar] [CrossRef]

SUPPLEMENTARY FIGURE S1: THP-1 cells differentiate into macrophage by PMA treatment.
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |