DOI:10.32604/biocell.2021.012901
| BIOCELL DOI:10.32604/biocell.2021.012901 | |
| Article |
Ang-(1-7) exerts anti-inflammatory and antioxidant activities on high glucose-induced injury by prohibiting NF-κB-IL-1β and activating HO-1 pathways in HUVECs
1Department of Cardiovascular Medicine, Dongguan Songshan Lake Center Hospital, Dongguan Cardiovascular Institute, Dongguan Shilong People’s Hospital Affiliated to Southern Medical University, Dongguan, 523326, China
2Department of Ultrasound, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou, China
3Department of Cardiology, Huangpu Division of the First Affiliated Hospital, Sun Yat-sen University, Guangzhou, 510700, China
4Department of Oncology, Dongguan Third People’s Hospital, Dongguan, 523326, China
5Second Ward of Internal Medicine, Dongguan Eighth People’s Hospital, Dongguan Children’s Hospital, Dongguan, 523003, China
*Address correspondence to: Jun Lan, sam19680415@126.com; Jingfu Chen, chenjf-sums@hotmail.com
#These authors contributed equally to this work
Received: 17 July 2020; Accepted: 18 December 2020
Abstract: Previous reports have suggested that Ang-(1-7) may have a protective effect in endothelial cells against high glucose (HG)-induced cell injury thanks to a modulatory mechanism in the NF-κB signaling pathway. In this study, we have examined whether NF-κB-IL-1β and Heme oxygenase-1 (HO-1) pathways contribute to the protection of Ang-(1-7) against hyperglycemia-induced inflammation and oxidative stress in human umbilical vein endothelial cells (HUVECs). Our results indicate that time-varying exposures of HUVECs, from 1 h to 24 h, to high glucose concentrations result in an increased expression of phosphorylated (p)-p65 and HO-1 in a time-dependent manner. As an inhibitor of NF-κB, pyrrolidinedithiocarbamic acid (PDTC) suppressed IL-1β production induced by HG. Of note, HUVECs previously treated with Ang-(1-7) (2 μM) for 30 min before being exposed to HG concentrations significantly ameliorated the HG-increased in p-p65 and IL-1β expression; whereas obviously up-regulated the level of HO-1, along with inhibition of oxidative stress, inflammation, and the HG-induced cytotoxicity. Importantly, when HUVECs were previously treated either with PDTC or IL-1Ra for 30 min before being exposed to HG, it significantly prevented damages caused by high glucose concentrations mentioned above, while the treatment of HO-1 inhibitor Sn-protoporphyrin (SnPP) before exposure to both HG and Ang-(1-7) significantly blocked the protective effect exerted by Ang-(1-7) on endothelial cells against injuries induced by HG mentioned above. To conclude, the data of this study showed that activation and inhibition of the NF-κB-IL-1β pathway and HO-1 pathway may constitute an important defense mechanism against endothelial cell damage caused by HG concentrations. We additionally gave new evidence showing that exogenous Ang-(1-7) exerts a protective effect on HUVECs against the HG-induced cell injury via the inhibition and the activation of the NF-κB-IL-1β pathway and the HO-1 pathway, respectively.
Keywords: Angiotensin-(1-7); High glucose; Human umbilical vein endothelial cells; NF-κB; IL-1β; HO-1
| Abbreviations | |
| ACE2: | Angiotensin-converting enzyme 2 |
| Ang-(1-7): | Angiotensin-(1-7) |
| Ang-II: | Angiotensin-II |
| ANOVA: | Analysis of variance |
| CCK-8: | Cell counter kit-8 |
| COX-2: | Cyclooxygenase-2 |
| CV: | Coefficients of variation |
| DCFH-DA: | 2’,7’-dichlorodihydrofluorescein diacetate |
| DM: | Diabetes mellitus |
| ECL: | Enhanced chemiluminescence |
| ELISA: | Enzyme-linked immunosorbent assay |
| FBS: | Fetal bovine serum |
| HO-1: | Heme oxygenase-1 |
| iNOS: | Inducible nitric oxide synthase |
| MAN: | Mannitol |
| MFI: | Mean fluorescence intensity |
| OD: | Optical density |
| PBS: | Phosphate-buffered saline |
| PDTC: | Pyrrolidinedithiocarbamic acid |
| PVDF: | Polyvinylidene difluoride |
| ROS: | Reactive oxygen species |
| SDS-PAGE: | Sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| SOD: | Superoxide dismutase |
| STZ: | Streptozotocin |
| TBS: | Tris-buffered saline |
| VCAM-1: | Vascular cell adhesion molecule 1 |
The predominance of diabetes mellitus (DM) is constantly incrementing, and the number of new cases is predicted to approximate 591.9 million in 2035 around the world (Guariguata et al., 2014; Shaw et al., 2010). DM almost damages every organ of the human body. It is important to stress that cardiovascular complications represent the most common cause of death in diabetic patients. Consequently, diabetes-induced vascular endothelium injury is a central player in the pathogenesis of medical complications of diabetes mellitus, with vascular endothelial cells playing a key role in preserving endothelial dysfunction (Capellini et al., 2010; Taguchi et al., 2014). Several studies have indicated that DM causes multiple pathological alterations in vascular endothelial cells; damages include inflammation, oxidative stress, and augmented apoptosis (Bhatt et al., 2013; Zhang et al., 2016b; Zhao et al., 2016a). However, the molecular mechanism of endothelial cell injury induced by hyperglycemia in DM is still unclear.
The peptide hormone Ang-(1-7) is recognized as a new-found member family of the hormonal cascade renin-angiotensin system (RAS), and its related biological active effects have challenged the public’s knowledge of the traditional RAS. The membrane-bound aminopeptidase, angiotensin-converting enzyme 2 (ACE-2), cleaves angiotensin II (ANG II) to produce bioactive heptapeptides Ang-(1-7). Previous research has revealed that Ang-(1–7) is an anti-mediator of Ang-II, which exerts protection of organs and cells by anti-inflammatory, antioxidation, anti-apoptosis effects (Lin et al., 2016; Meneses et al., 2015; Zhang et al., 2016a). Ang-(1–7) seems, by all accounts, to have a crucial role in protecting against cardiovascular diseases (Chang et al., 2016; Hisatake et al., 2017; Patel et al., 2016; Zhang et al., 2015a). Therefore, the public pays more and more attention to the protection provided by Ang-(1-7) against cardiovascular complications resulting from hyperglycemia. The above finding is supported by the following: first, previous research has indicated that the level of Ang-(1-7) is low in plasma of diabetic-induced cardiac dysfunction patients (Hao et al., 2013). Second, ACE2 expression levels are diminished in diabetic patients (Reich et al., 2008). Third, increased expression of ACE2 contributes to the improvement of blood glucose control and the alleviation in streptozotocin (STZ)-induced diabetic mice exhibiting glomerular lesions (Liu et al., 2011). Fourth, Ang-(1-7) partly controls blood sugar levels by regulating insulin production, insulin resistance, and pancreatic β-cell survival (He et al., 2015a). Fifth, Ang-(1-7) can exert protection against diabetes-related cardiovascular events (Benter et al., 2007; Yousif et al., 2012). Furthermore, a recent study has indicated that upregulation of Ang-(1-7)-mediated signaling pathways confers protection of endothelial function via suppression of oxidative stress in diabetic mice (Zhang et al., 2015b). These studies indicate that Ang-(1-7) is strongly related to endothelial dysfunction caused by hyperglycemia.
Nonetheless, the potential contribution of endogenous Ang (1-7) in preserving the functionality of endothelial cells in DM patients is still poorly understood. According to a previous study, Ang-(1-7) inhibits the activation of NF-κB in HUVECs (Liang et al., 2015) and IL-1β in vascular diseases (Jin et al., 2012). As a stress-responsive protein, HO-1 is induced by oxidative stress and has cytoprotective and anti-inflammatory functions in cardiovascular disease (Chen et al., 2018; Peiro et al., 2017). The HO-1 pathway is also associated with Ang II-induced production of reactive oxygen species (ROS) in human late endothelial progenitor cells during angiogenesis (Mai et al., 2014). Furthermore, IL-1β, NF-κB, and HO-1 are linked to hyperglycemia- or diabetes-induced endothelial dysfunction in vivo and in vitro (Guan et al., 2014; He et al., 2015b; Mylroie et al., 2015; Wang et al., 2015; Wenzel et al., 2015; Zhu et al., 2015). However, the mechanism behind how NF-κB, IL-1β, and HO-1 regulate high glucose-induced endothelial cell functions is not completely clear and still needs to be further explored. In this study, we hypothesized that inhibition of both NF-κB and IL-1β and upregulation of HO-1 protect HUVECs against hyperglycemia-elicited oxidative stress and endothelial inflammation.
Mannitol, Angiotensin-(1-7), PDTC, IL-1Ra (inhibitor of IL-1β receptor), superoxide dismutase (SOD) assay kit, glutathione assay kit, and 2’-7’dichlorodihydrofluorescein diacetate (DCFH-DA) were obtained from Sigma-Aldrich (St Louis, MO, USA). SnPP was bought from Calbiochem (La Jolla, CA, USA). The cell counting kit-8 (CCK-8) was obtained from Dojindo Lab (Kumamoto, Japan). Fetal bovine serum (FBS) and DMEM medium were purchased from Gibco BRL (Grand Island, NY, USA). Phosphorylated (p)- p65 antibody, total (t)-p65 antibody, Heme oxygenase-1 (HO-1) antibody, inducible nitric oxide synthase (iNOS) antibody, Bcl-2 antibody, Bax antibody, cleaved caspase-3 antibody, cyclooxygenase-2 (COX-2) antibody, vascular cell adhesion molecule 1 (VCAM-1, also known as CD54) antibody and intercellular cell adhesion molecule-1 (ICAM-1) antibody were purchased from Cell Signaling Technology (Boston, MA, USA). Enzyme-linked immunosorbent assay (ELISA) kits used for the identification of IL-1β and TNF-α were acquired from Wuhan Boster Biological Technology, Ltd. (Wuhan, China). BCA protein assay kit and HRP-conjugated secondary antibody were bought from KangChen Biotech, Inc. (Shanghai, China). Enhanced chemiluminescent (ECL) solution was obtained from KeyGen Biotech (Nanjing, China).
HUVECs were provided by the Experimental Animal Center of Sun Yat-sen University (Guangzhou, China). HUVECs were grown in DMEM medium containing 10% FBS and incubated in a 37°C atmosphere with 5% CO2 and 95% relative humidity.
To create high glucose conditions, HUVECs were cultured using DMEM medium containing normal glucose concentration (5.5 mM) for 12 h and then cultured in medium with high glucose concentration (40 mM) for 24 h as previously described (Chen et al., 2018). Cells in control groups were cultured in a medium with 5.5 mM glucose for 24 h. To assess whether exogenous Ang-(1-7) confers a protective effect against damages induced by elevated glucose levels, HUVECs were exposed to 40 mM glucose (high glucose, HG) in the absence or presence of Ang-(1-7) for 24 h. To explore whether the NF-κB-IL-1β or HO-1 pathway contributed to the protective effects of Ang-(1-7), HUVECs were cultured in HG, 2 μM Ang-(1-7), 40 μM SnPP, 20 μM SnPP, 100 μM PDTC, or 20 ng/mL IL-1Ra.
Mechanical HUVECs harvesting was performed using a cell scraper, and then cells were lysed with cell lysis buffer at 4°C for 30 min after the indicated treatments. Nuclear and cytoplasmic proteins were separated by using NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, Cleveland, OH, USA). Protein quantitation was performed using the BCA protein assay kit. Loading buffer was added into the cytosolic extracts to increase the density of the sample and boiled for 5 min. Protein separation was performed by loading 30 mg protein from each sample in a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto polyvinylidene difluoride (PVDF) membranes. Once the transfer was finished, the PVDF membranes (Millipore, Burlington, MA, USA) were blocked, using fresh TBS-T (0.1% Tween20 in Tris-buffered saline) blocking buffer containing non-fat dry milk (5%) at room temperature (RT) for 60 min. After the blocking step, primary antibody solutions were prepared by diluting them to the recommended concentrations in newly prepared TBS-T containing 3% free-fat milk as follows: p-p65 (1:1000 dilution) antibody, t-p65 (1:1000 dilution) antibody, HO-1 (1:1000 dilution) antibody, Bax (1:1000 dilution) antibody, Bcl-2 (1:1000 dilution) antibody, cleaved caspase-3 (1:1000 dilution) antibody, iNOS (1:1000 dilution) antibody, COX-2 (1:1000 dilution) antibody, VCAM-1 (1:1000 dilution) antibody and ICAM-1 (1:1000 dilution) antibody, GAPDH (1:1000 dilution) antibody and Histone H3 (1:1000 dilution) antibody. The membranes were incubated with their primary antibody solution overnight at 4°C with gentle agitation. PVDF membranes were washed with TBS-T for 15 min to remove any unbound primary antibody. Incubation with the diluted horseradish peroxidase (HRP)-conjugated secondary antibody (Kangchen Biotech, Shanghai, China) at a concentration of 1:3000 in TBS-T containing 3% fat-free milk was performed at RT for 90 min. Subsequently, the PVDF membranes were rinsed thrice for 5 min each with TBS-T. The detection of proteins was performed using the ECL detection system. Finally, the intensity of bands was analyzed by Image J 1.47i software for protein quantification. The experiment was repeated thrice.
CCK-8 assay was performed to measure cell viability. Approximately 5 × 103 HUVECs were seeded into 96-well plates. 100 μL of CCK8 solution purchased from Dojindo Molecular Technologies (Rockville, MD, USA) were added to each well of the plate before being incubated for 24, 48, 72, and 96 h, respectively. The optical density (OD) at 450 nm was measured utilizing a microplate reader (Molecular Devices, Sunnyvale, CA, USA). The percentage of the viable cells was calculated according to the following formula: (OD treatment group/OD control group) × 100%. The cell viability assay was repeated five times. The intra- and inter-assay coefficients of variation (CVs) were 3.4% and 4.2%, respectively.
Measurement of intracellular ROS production
Intracellular ROS production was assessed via the oxidation of cell-permeable DCFH-DA to the highly fluorescent DCF. HUVECs were grown in glass slides with DMEM medium. The slides were washed two times with phosphate-buffered saline (PBS) prior to incubation in a serum-free medium containing 10 µmol/L of DCFH-DA solution at 37°C for 30 min. Next, cells were rinsed five times with PBS before measuring DCF fluorescence over the entire field of view using a fluorescence microscope connecting to the BX50-FLA imaging system (Olympus, Tokyo, Japan). Mean fluorescence intensity (MFI) was determined from 5 random fields using Image J 1.47i software (NIH Image, Bethesda, MD, USA). The results obtained served as an index of intracellular ROS levels. The experiment was repeated five times. The intra- and inter-assay CVs were 2.9% and 3.6%, respectively.
Superoxide dismutase (SOD) activity assay
The qualitative determination of SOD enzymatic activity was tested by the SOD assay kit. HUVECs were rinsed with PBS before being lysed with ice-cold lysis buffer [0.1 M Tris/HCl (pH 7.4)] with 5 mmol/L β-mercaptoethanol, 0.5% Triton and 0.1 mg/mL phenylmethylsulfonyl fluoride. Subsequently, lysate clearance was performed by centrifugation (14,000 × g, 5 min), and the supernatant was collected. Superoxide dismutase activity was assessed using the ‘SOD Assay Kit’ in accordance with the manufacturer’s instructions (Sigma, USA). This was followed by reading the absorbance values (optical density (OD) at 450 nm in a microplate reader. The experiment was repeated thrice. The intra- and inter-assay CVs were 1.2% and 2.4%, respectively.
ELISA detection of IL-1β and TNF-α in culture supernatants
An ELISA test was performed to measure the IL-1β and TNF-α levels from culture media of HUVECs cultured in 96-well plates. The assay was conducted following the manufacturer’s instructions. The experiments were carried out five times. The intra- and inter-assay CVs were 2.6% and 3.3%, respectively.
Determination of intracellular glutathione (GSH)
1 × 106 HUVECs were plated in a 10 cm-dish (Corning Inc., Corning, NY, USA) and cultured 24 h prior to treatments. Then cells were first collected and deproteinized using 5% 5-sulfosalicylic acid solution. Assessment of intracellular glutathione levels was done using the Glutathione Assay Kit from Sigma-Aldrich in accordance with the manufacturer’s protocol. The experiment was repeated thrice. The intra- and inter-assay CVs were 1.5% and 3.1%, respectively.
All data from the present study are shown as the mean ± standard error of the mean (SEM). SPSS 13.0 software (SPSS, Chicago, IL, USA) was utilized to determine any difference statistically significant among the groups by conducting a one-way analysis of variance (ANOVA) followed by LSD post hoc comparison test. If a P-value was less than 0.05, it was regarded as statistically significant.
High glucose activates NF-κB pathway and increases NF-κB transcription factor translocation into the nucleus in HUVECs
As can be seen in Figs. 1a and 1b, when HUVECs were exposed to high glucose concentrations (40 mM) for different times (3, 6, 12, and 24 h), they exhibited a significant increase of NF-κB p65 subunit phosphorylation levels while the expression level of total (t) p65 NF-κB remained unaltered. Meantime, p65 phosphorylation did not vary with time in low glucose conditions (5.5 mM) (Suppl. Figs. S1a and S1b). Additionally, our data revealed decreased expression levels of cytoplasmic (t) p65 (Figs. 1c and 1d). Conversely, an increase of nuclear (t) p65 levels was observed (Figs. 1e and 1f). Besides, 40 mM mannitol used for the osmotic control of high glucose did not affect the expression and translocation of NF-κB in HUVECs (Figs. 1a–1f). These results indicate that HG promotes NF-κB translocation into the nucleus and, therefore, activation of the NF-κB pathway in HUVECs.
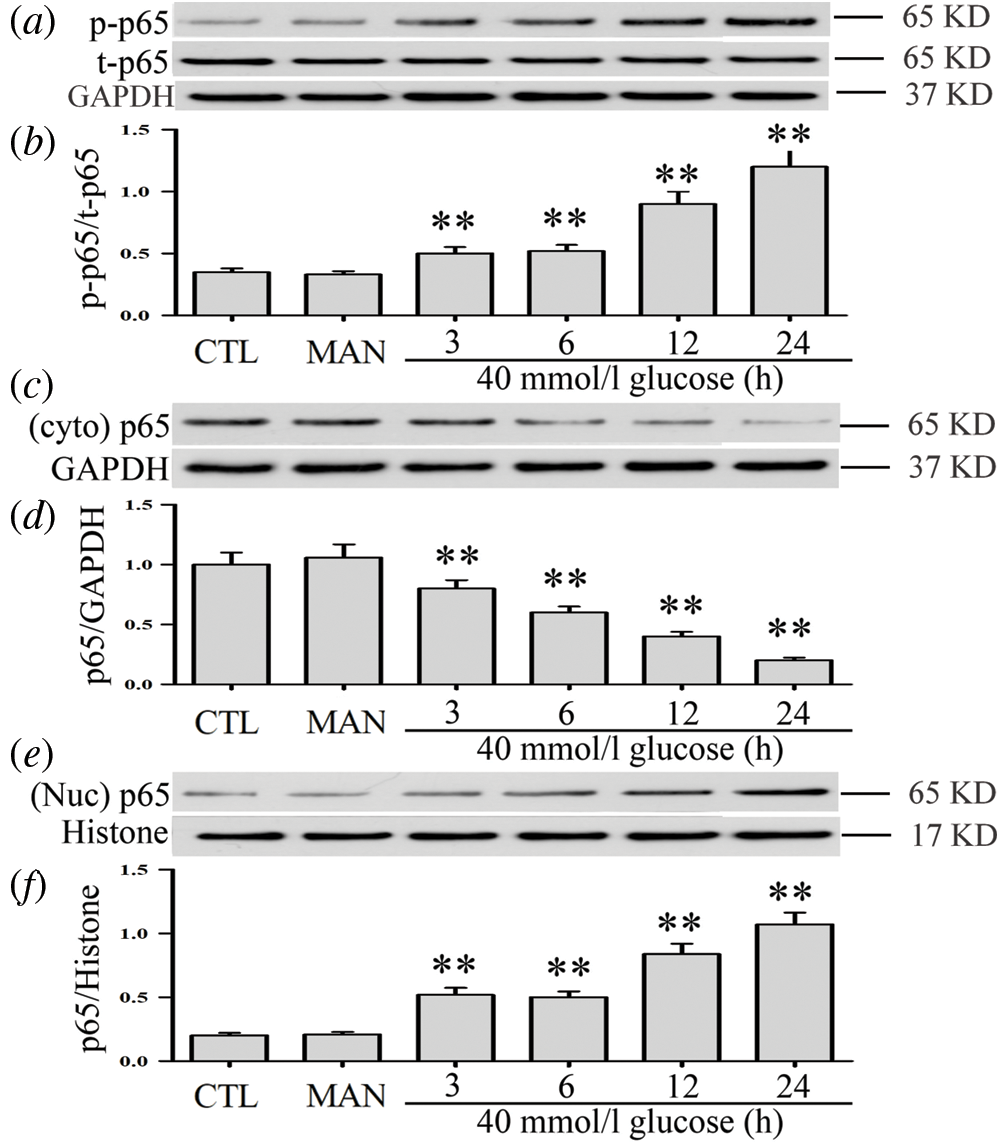
Figure 1: High glucose activates the NF-κB pathway and increases NF-κB transcription factor translocation into the nucleus in HUVECs.
HUVECs were grown in a medium containing high glucose levels (40 mM) for a given period of time (3, 6, 12 and 24 h) in order to evaluate the effects of HG on NF-ΚB p65 phosphorylation. Cytoplasm and nuclear extracts were extracted. (a, c and f) Expression levels of p-p65 and t-p65 were analyzed by Western blot. (b, d and f) Densitometric analysis of western blots for p65 expression levels was performed with ImageJ 1.47i software. Results are the mean ± SEM (N = 3). **P < 0.01 vs. control group. CTL, control; MAN, mannitol; Cyto, cytoplasm; Nuc, nuclear; p, phosphorylated; t, total.
Ang-(1-7) inhibits HG-induced NF-κB signaling activation and translocation of NF-κB into the nucleus in HUVECs
When HUVECs were previously treated with 2 μM Ang-(1-7) for 30 min before high-glucose exposure (40 mM), HG-increased p-p65 levels were significantly inhibited (Figs. 2a and 2b). Moreover, Ang-(1-7) treatment led to an increase in the cytoplasmic (t) p65 levels (Figs. 2c and 2d), whereas it decreased the nuclear (t)p65 levels in HUVECs (Figs. 2e and 2f). The above data suggest that Ang-(1-7) suppresses the activation of the NF-κB pathway and nuclear translocation of NF-κB transcription factor induced under hyperglycemia conditions in HUVECs.
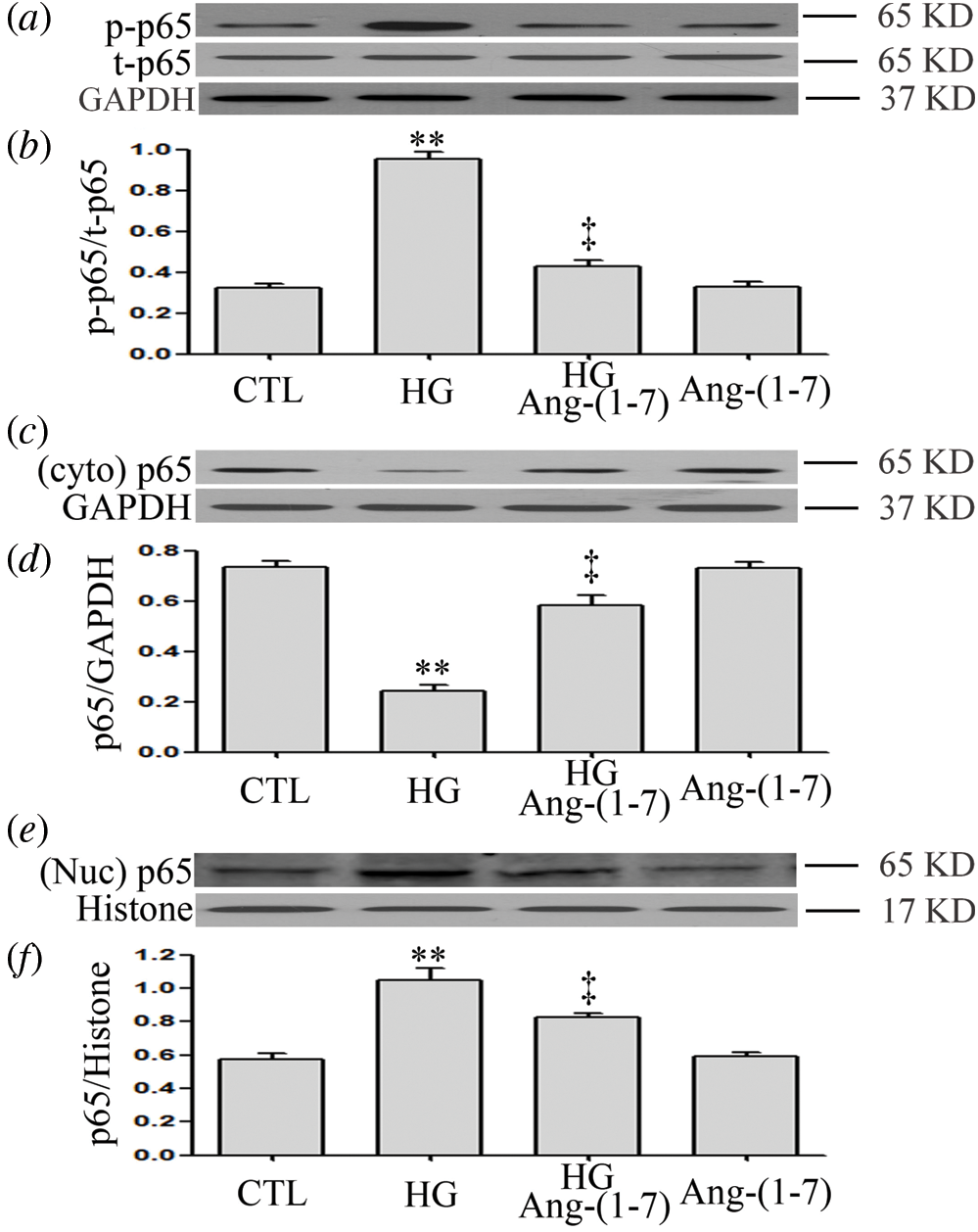
Figure 2: Ang-(1-7) inhibits HG-induced NF-κB signaling activation and translocation of NF-κB into the nucleus in HUVECs.
HUVECs were exposed to high glucose concentrations (40 mM) for 24 h treated either with 2 μM Ang-(1-7) or not for 30 min prior to exposure to HG. Cytoplasmic and nuclear extracts were obtained. (a, c and e) Endogenous expression of NF-κB p65 was assessed by Western blot. (b, d and f) Quantitative analysis of Western blot data in a, c and e was performed by densitometric analysis using ImageJ 1.47 I software. Results are the mean ± SE (N = 3). **P < 0.01 vs. control group; ‡P < 0.01 vs. HG-treated group. Ang-(1-7), Angiotensin-(1-7); CTL, control; HG, high glucose; t, total; Nuc, nuclear; Cyto, cytoplasm; p, phosphorylated.
Ang-(1-7) enhances HG-increased HO-1 levels in HUVECs
As can be seen from Figs. 3a and 3b, when HUVECs were exposed to 40 mM glucose for different intervals of time (3, 6, 12, and 24 h), the expression level of HO-1 was markedly up-regulated. In addition, a 30-min pretreatment with 2 μM Ang-(1-7) before exposure to 40 mM glucose markedly further enhanced HO-1 expression in HUVECs (Figs. 3c and 3d).
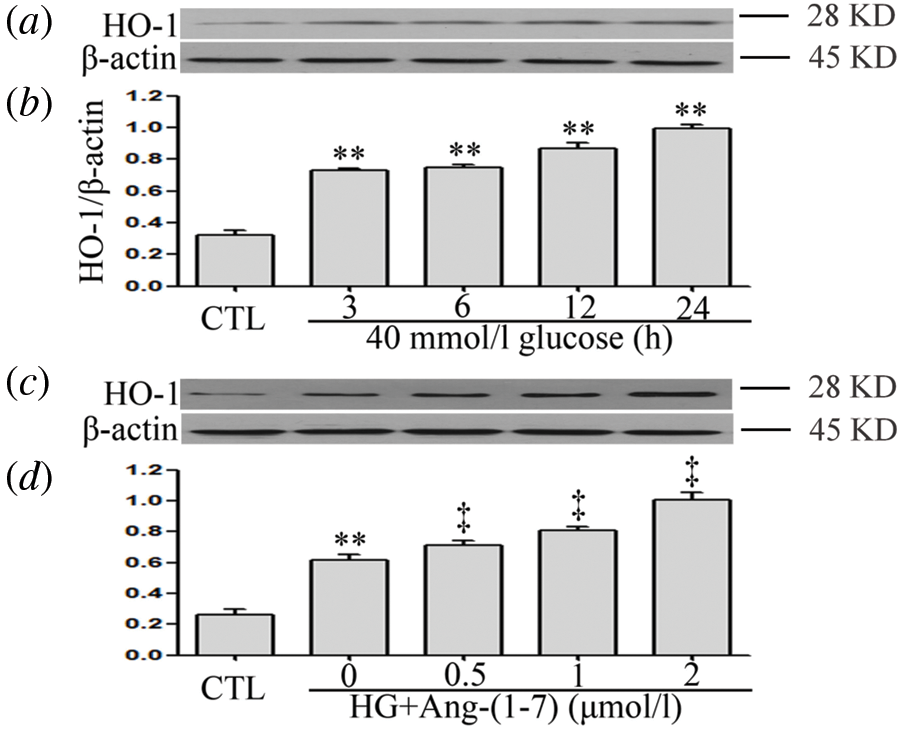
Figure 3: Ang-(1-7) enhances HG-increased HO-1 levels in HUVECs. (a and b) HUVECs were cultured in a medium containing high glucose concentration (40 mM) for specified times (3, 6, 12 and 24 h, respectively). (c and d) HUVECs were cultured in high glucose levels (40 mM) for 24 h with or without being previously treated with increasing concentrations of Ang-(1-7) corresponding to 0.5, 1, and 2 μM, respectively, for 30 min. (a–d) HO-1 expression levels were measured by Western blot assay. Data are the mean ± SEM (N = 3). **P < 0.01 vs. control group; ‡P < 0.01 vs. HG-treated group. CTL, control; HG, high glucose; Ang-(1-7), Angiotensin-(1-7); HO-1, heme oxygenase-1.
Ang-(1-7) and PDTC suppress hyperglycemia-induced cytotoxicity in HUVECs
As seen in Fig. 4a, HG significantly decreased cell viability, whereas 40 mM mannitol had no effect on HUVEC viability (Suppl. Fig. S2). In addition, HG markedly increased levels of apoptotic proteins (Bax and cleaved caspase-3) and decreased Bcl-2 expression to induce cell apoptosis (Figs. 4b and 4c). By contrast, different concentrations (0.5, 1, and 2 μM) of Ang-(1-7) treatments of HUVECs exposure to HG obviously mitigated the HG-induced cytotoxicity index. Our previous research has indicated that 100 μM PDTC (inhibits NF-κB signaling) suppresses high glucose-induced injuries in cultured cardiomyocytes. Similarly, 100 μM PDTC treatment of HUVECs cultured with HG notably blocked HG-induced cytotoxicity index (Figs. 4d–4f).
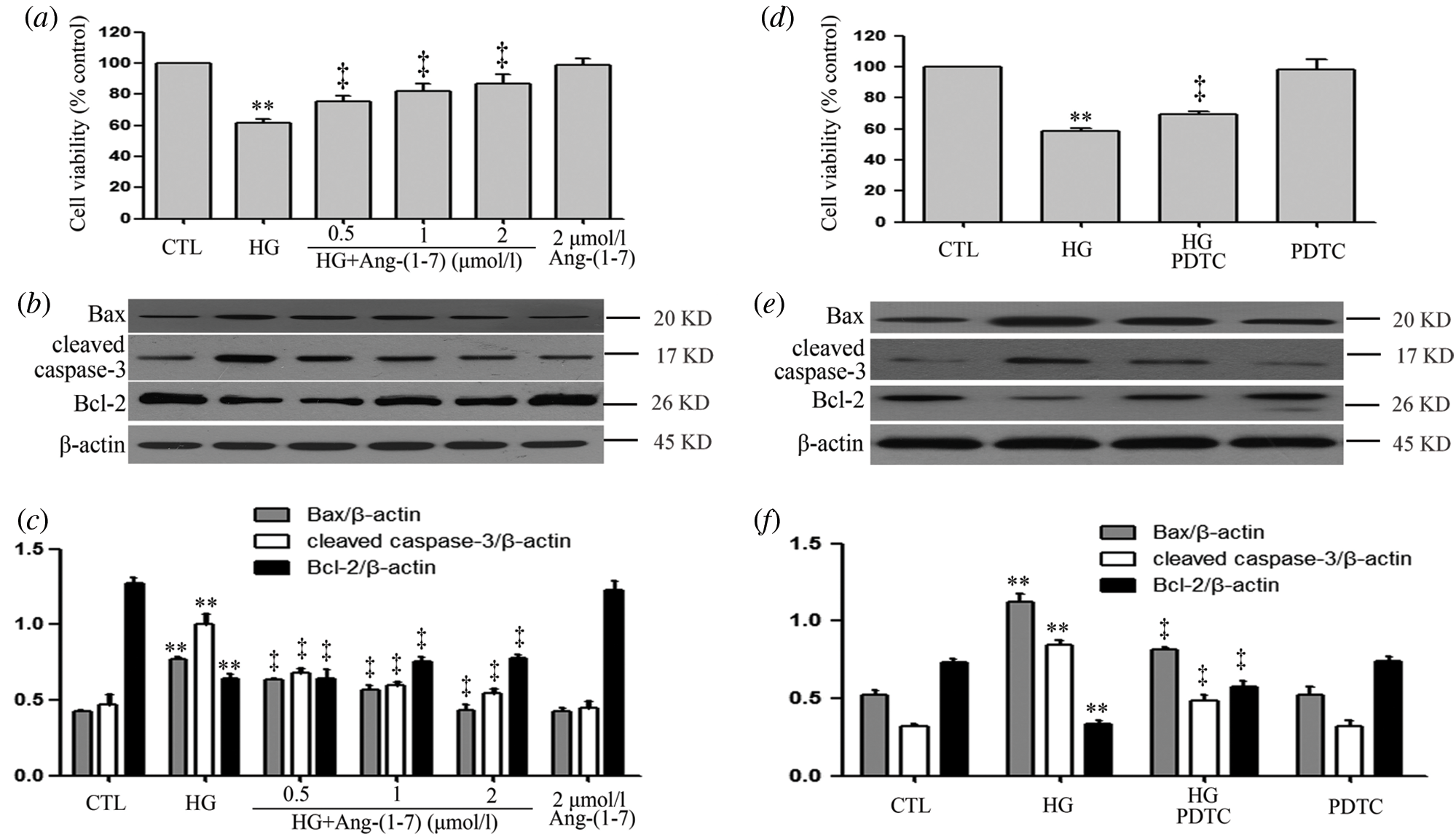
Figure 4: Ang-(1-7) and PDTC suppress hyperglycemia-induced cytotoxicity in HUVECs.
(a, b and c) HUVECs were grown in high glucose concentrations (40 mM) for 24 h with or without being previously treated with increasing concentrations of Ang-(1-7) corresponding to 0.5, 1, and 2 μM, respectively, for 30 min. (d, e, and f) HUVECs were grown in high glucose concentrations (40 mM) for 24 h with or without being previously treated 100 μM PDTC for 30 min. (a and d) Cell viability was evaluated using the CCK-8. (b and e) Expression levels of pro-apoptotic proteins: Bax, cleaved caspase-3 and Bcl-2, evaluated by Western blot analysis. (c and f) Densitometric analysis of Western blots from b and e. Data are the mean ± SEM (N = 3). **P < 0.01 vs. control group; ‡P < 0.01 vs. HG-treated group. CTL, control; HG, high glucose; Ang-(1-7), Angiotensin-(1-7); PDTC, pyrrolidine dithiocarbamate.
SnPP promotes HG-induced cytotoxicity and partly blocks the cardioprotective effects of Ang-(1-7) in HUVECs
Because Ang-(1-7) enhances HG-increased HO-1 levels, we further identified the role of HO-1 in a parallel treatment with Ang-(1-7). Under high glucose culture conditions and different concentrations (10, 20, and 40 μM) of SnPP (an inhibitor of HO-1 pathway), there was enhanced HG-decreased cell viability (Fig. 5a). Our preceding data showed that culturing HUVECs with high glucose media containing 2 μM Ang-(1-7) reduced HG-decreased cell viability as well as HG-induced cell apoptosis (Figs. 5b–5d). However, if HUVEC cultures are treated with high glucose, Ang-(1-7) and SnPP, it partly blocks the above effects of Ang-(1-7) (Figs. 5b–5d). These results suggest that HO-1 is involved in cardioprotection of Ang-(1-7) for HUVECs cultured with HG.
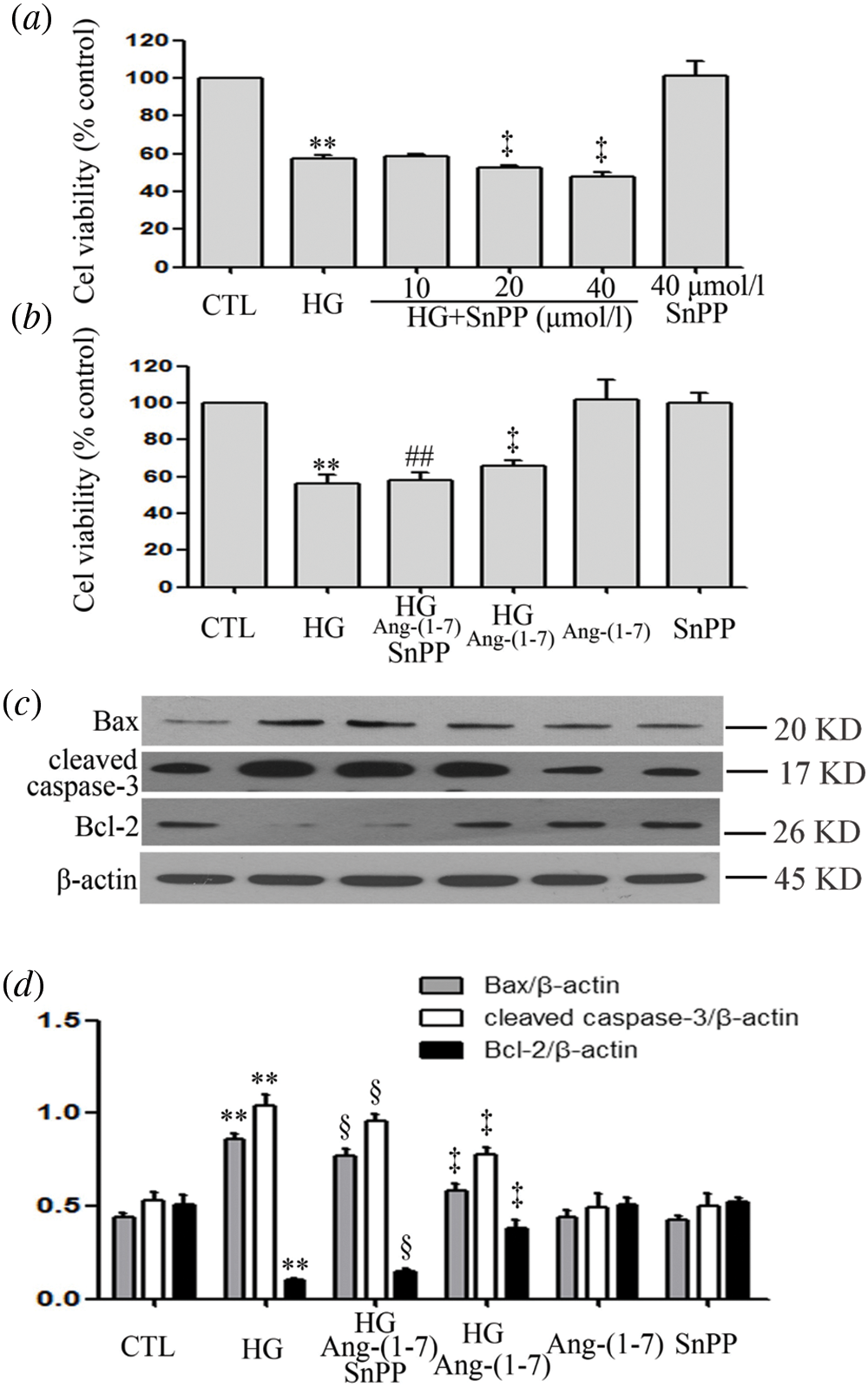
Figure 5: SnPP promotes HG-induced cytotoxicity and partly blocks the cardioprotective effects of Ang-(1-7) in HUVECs.
(a) HUVECs were grown in high glucose concentrations (40 mM) for 24 h with or without being previously treated with increasing concentrations of SnPP (10, 20 and 40 μM, respectively) for 30 min. (b, c, and d) HUVECs were cultured in high glucose concentrations (40 mM) for 24 h with or without being previously treated with the stated concentrations of Ang-(1-7) or SnPP, 2 µM or 40 μM, respectively, for 30 min. (a and b) Cell viability was evaluated using the CCK-8. (c) Expression levels of pro-apoptotic proteins: Bax, cleaved caspase-3 and Bcl-2, evaluated by Western blot analysis. (d) Densitometric analysis of Western blot from c. Results are the mean ± SEM (N = 3). **P < 0.01 vs. control group; ‡P < 0.01 vs. the HG treated group; §P < 0.01 vs. the HG + Ang-(1-7) treated group. CTL, control; HG, high glucose; Ang-(1-7), Angiotensin-(1-7).
Ang-(1-7) and PDTC counteract HG-induced oxidative stress in HUVECs
Results revealed that HUVECs grown under HG culture conditions significantly increased in oxidative stress elicited by an enhanced ROS production and decrease in the activation of SOD and GSH (Figs. 6a–6f). However, HG-induced oxidative stress was alleviated by Ang-(1-7) or PDTC (Figs. 6a–6f). Furthermore, treatment of HUVECs alone with Ang-(1-7) or PDTC did not alter oxidative stress (Figs. 6a–6f).
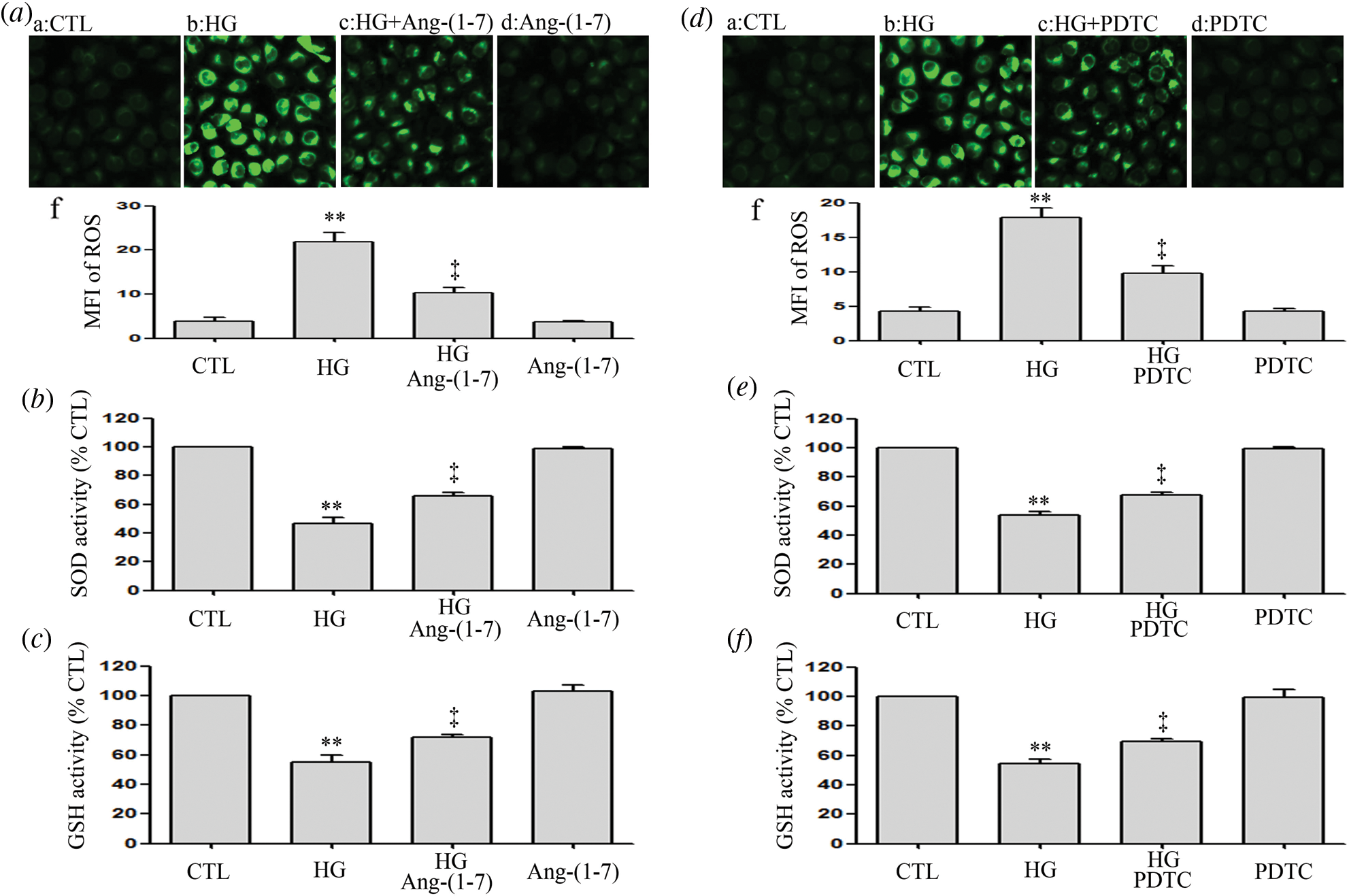
Figure 6: Ang-(1-7) and PDTC counteract HG-induced oxidative stress in HUVECs.
(a, b and c) HUVECs were cultured in high glucose concentrations (40 mM) for 24 h pre-treated either with 2 μM Ang-(1-7) or not for 30 min. (d, e and f) HUVECs were exposed to high glucose concentrations (40 mM) for 24 h with or without being previously treated with PDTC (100 μM) for 30 min. (a and d) Levels of intracellular ROS were measured by DCFH-DA staining and followed by photofluorography. (a–f and d–f) Mean fluorescence intensity (MFI) of DCFH-DA calculated by Image J 1.47i software for quantitative analysis. Results are the mean ± SEM (N = 3). **P < 0.01 vs. control group; ‡P < 0.01 vs. the HG-treated group. CTL, control; HG, high glucose; Ang-(1-7), Angiotensin-(1-7); PDTC, pyrrolidine dithiocarbamate.
SnPP enhances HG-induced oxidative stress and partly blocks the anti-oxidation effect of Ang-(1-7) in HUVECs
Conversely to Ang-(1-7) or PDTC treatment, the treatment of HUVECs with HG and 40 μmol/L SnPP further promoted HG-induced oxidative stress (Figs. 7a–7c). Our preceding data showed that culturing HUVECs with high glucose media containing 2 μmol/L Ang-(1-7) eased HG-induced oxidative stress, which seems to indicate that Ang-(1-7) exerts an antioxidant effect on HUVECs (Figs. 7a–7c). However, this effect was, in part, blocked by SnPP (Figs. 7a–7c).
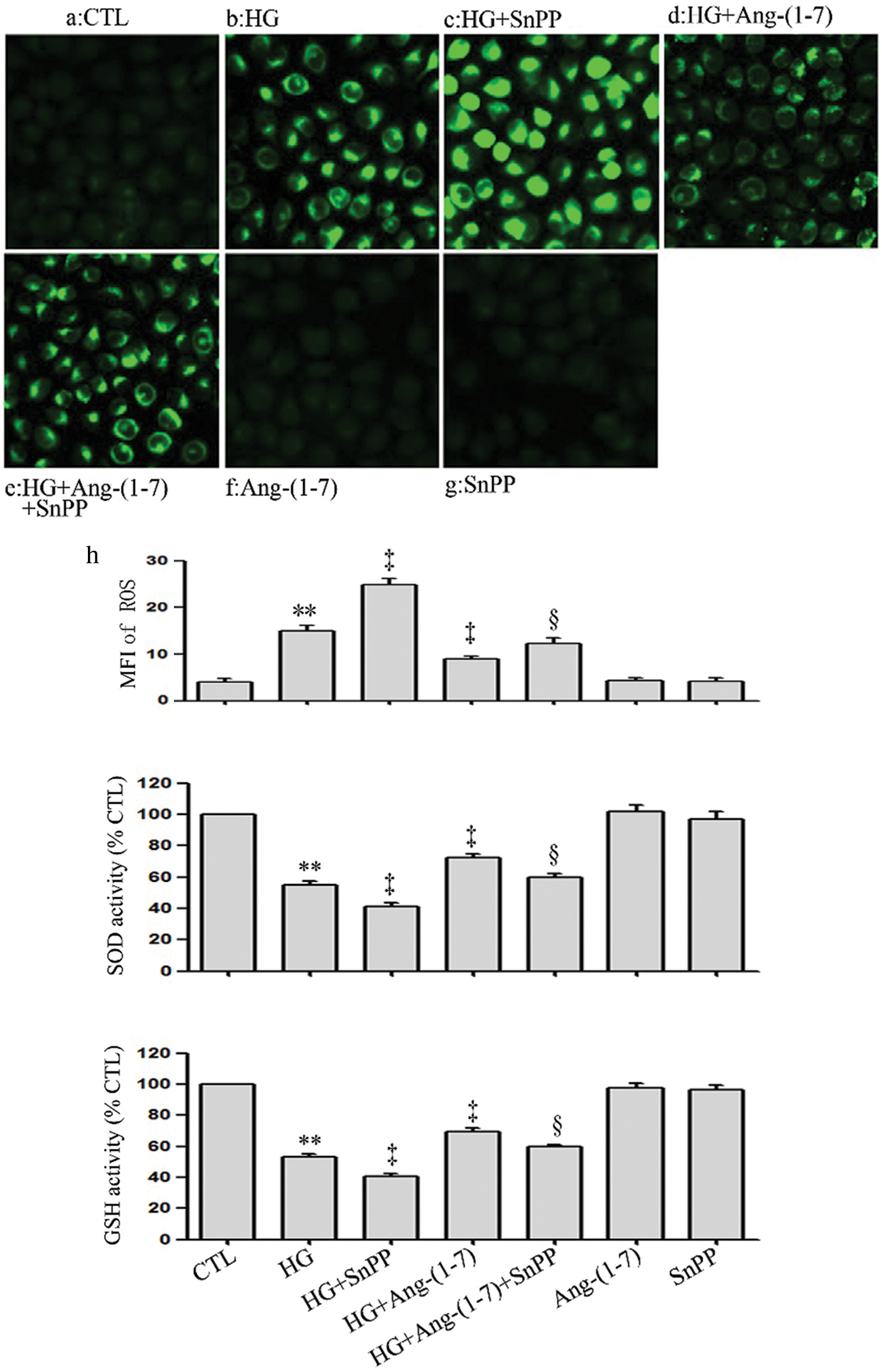
Figure 7: SnPP enhances HG-induced oxidative stress and partly blocks the anti-oxidation effect of Ang-(1-7) in HUVECs. HUVECs were exposed to high glucose concentrations (40 mM) for 24 h treated either with 2 µM Ang-(1-7) or 40 μM SnPP for 30 min, respectively. (a) Levels of intracellular ROS were assessed by DCFH-DA staining followed by photofluorography. (a–h) Mean fluorescence intensity (MFI) of DCFH-DA calculated by Image J 1.47i software. (b and c) SOD enzymatic activity and GSH conversion were assessed by ELISA. Results are the mean ± SEM (N = 3). **P < 0.01 vs. control group; ‡P < 0.01 vs. the HG-treated group. §P < 0.01 vs. the HG + Ang-(1-7) treated group. CTL, control; HG, high glucose; Ang-(1-7), Angiotensin-(1-7); SnPP, Sn-protoporphyrin IX.
Ang-(1-7) and PDTC suppress HG-induced inflammation, along with expression levels of VCAM-1 and ICAM-1 in HUVECs
As can be seen from Fig. 8, HG dramatically upregulated pro-inflammatory cytokines (IL-1β and TNF-α), enzymes (iNOS and COX-2), VCAM-1 and ICAM-1 (Figs. 8a–8f). On the contrary, the above index was inhibited by Ang-(1-7) or PDTC (Figs. 8a–8f). Moreover, Ang-(1-7) or PDTC did not alter the above profile in HUVECs cultured in normal conditions (Figs. 8a–8f).
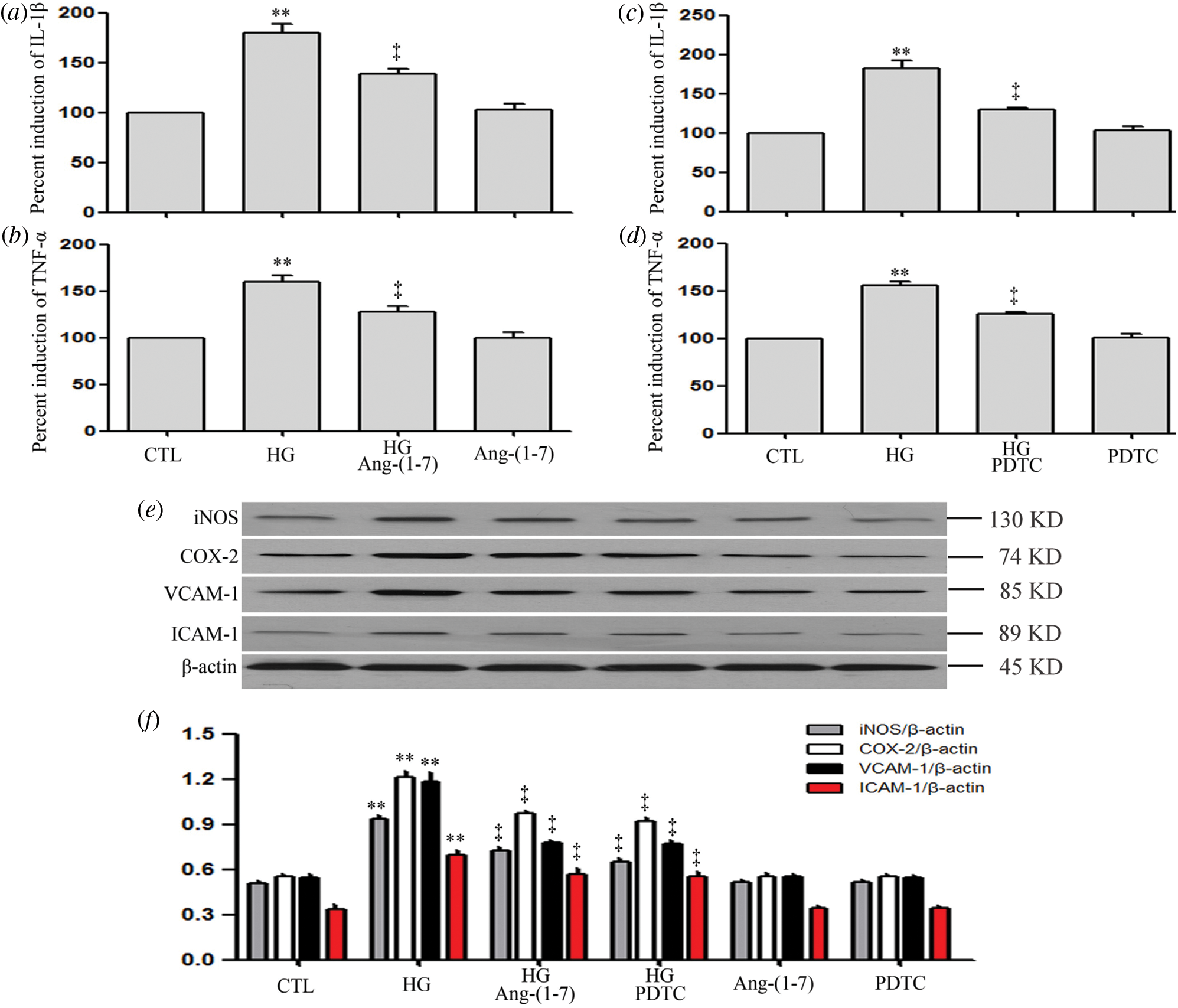
Figure 8: Ang-(1-7) and PDTC suppress HG-induced inflammation, along with expression levels of VCAM-1 and ICAM-1 in HUVECs.
(a and b) HUVECs were grown in high glucose concentrations (40 mM) for 24 h and then treated either with 2 µM Ang-(1-7) or not for 30 min. (c and d) HUVECs were exposed to high glucose concentrations (40 mM) for 24 h and then treated either with 100 μM PDTC or not for 30 min. (e and f) HUVECs were exposed to high glucose concentrations (40 mM) for 24 h with or without being treated with 2 μM Ang-(1-7) or 100 μM PDTC for 30 min. Levels of IL-1β (a and c) and TNF-α (b and d) in cell supernatants were assessed by enzyme-linked immunosorbent assay (ELISA). (e) Expression levels of iNOS, COX-2, VCAM-1, and ICAM-1, measured by Western blot assay. (f) Densitometric analysis from the Western blot of e. Results are the mean ± SEM (N = 3). **P < 0.01 vs. control group; ‡P < 0.01 vs. the HG-treated group. CTL, control; HG, high glucose; Ang-(1-7), Angiotensin-(1-7); PDTC, pyrrolidine dithiocarbamate.
SnPP increases HG-induced inflammation and partly blocks the anti-inflammation of Ang-(1-7) in HUVECs
Contrarily to Ang-(1-7) or PDTC treatment, treatment of HUVECs with HG plus SnPP ulteriorly upregulated pro-inflammatory cytokines expression (IL-1β and TNF-α), enzymes (iNOS and COX-2), VCAM-1 and ICAM-1 (Figs. 9a–9d). However, no increase was observed in levels of VCAM-1, ICAM-1, or pro-inflammatory cytokines and enzymes when HUVECs were cultured in normal glucose medium containing SnPP (Figs. 9a–9d).
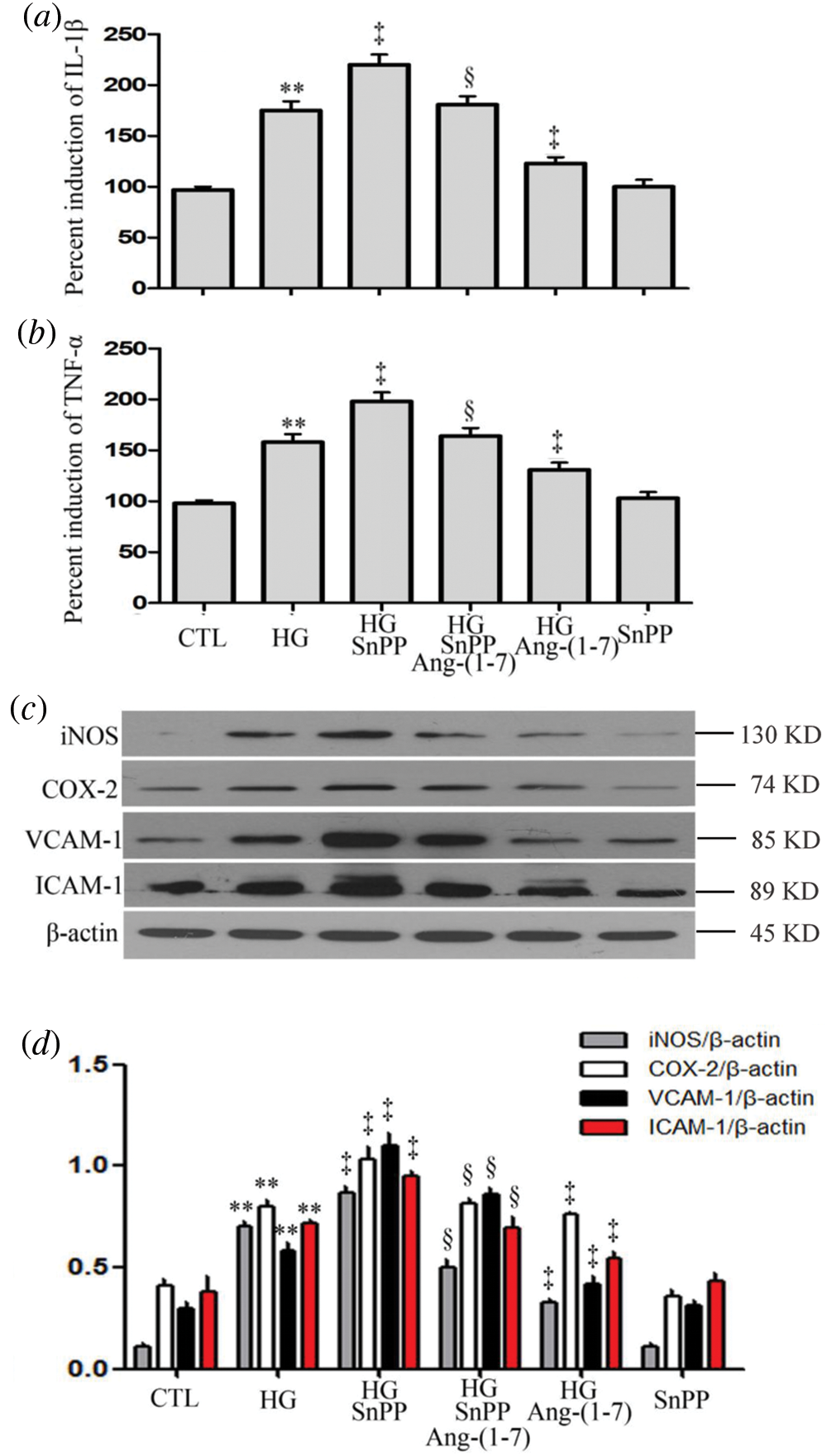
Figure 9: SnPP increases HG-induced inflammation and partly blocks the anti-inflammation effect of Ang-(1-7) in HUVECs.
(a–d) HUVECs were cultured in high glucose concentrations (40 mM) for 24 h with or without being treated with 2 µM Ang-(1-7) or 40 μM SnPP for 30 min, respectively. Levels of IL-1β (a) and TNF-α (b) in cell supernatants were assessed by enzyme-linked immunosorbent assay (ELISA). (c) Expression levels of iNOS, COX-2, VCAM-1, and ICAM-1 were measured by Western blot assay. (d) Densitometric analysis from the Western blot of c. Results are the mean ± SEM (N = 3). **P < 0.01 vs. the control group; ‡P < 0.01 vs. the HG-treated group. §P < 0.01 vs. the HG + Ang-(1-7) treated group. CTL, control; HG, high glucose; Ang-(1-7), Angiotensin-(1-7); SnPP, Sn-protoporphyrin IX.
IL-1Ra inhibits HG-induced oxidative stress and cytotoxicity in HUVECs
The above data showed that hyperglycemia triggered cytotoxicity and overproduction of ROS in HUVECs. However, the oxidative stress and cytotoxicity were attenuated by IL-1Ra (an inhibitor of IL-1β receptor) (Figs. 10a and 10b). These data suggest that HG may induce oxidative stress and cytotoxicity by regulating the inflammation process in HUVECs.
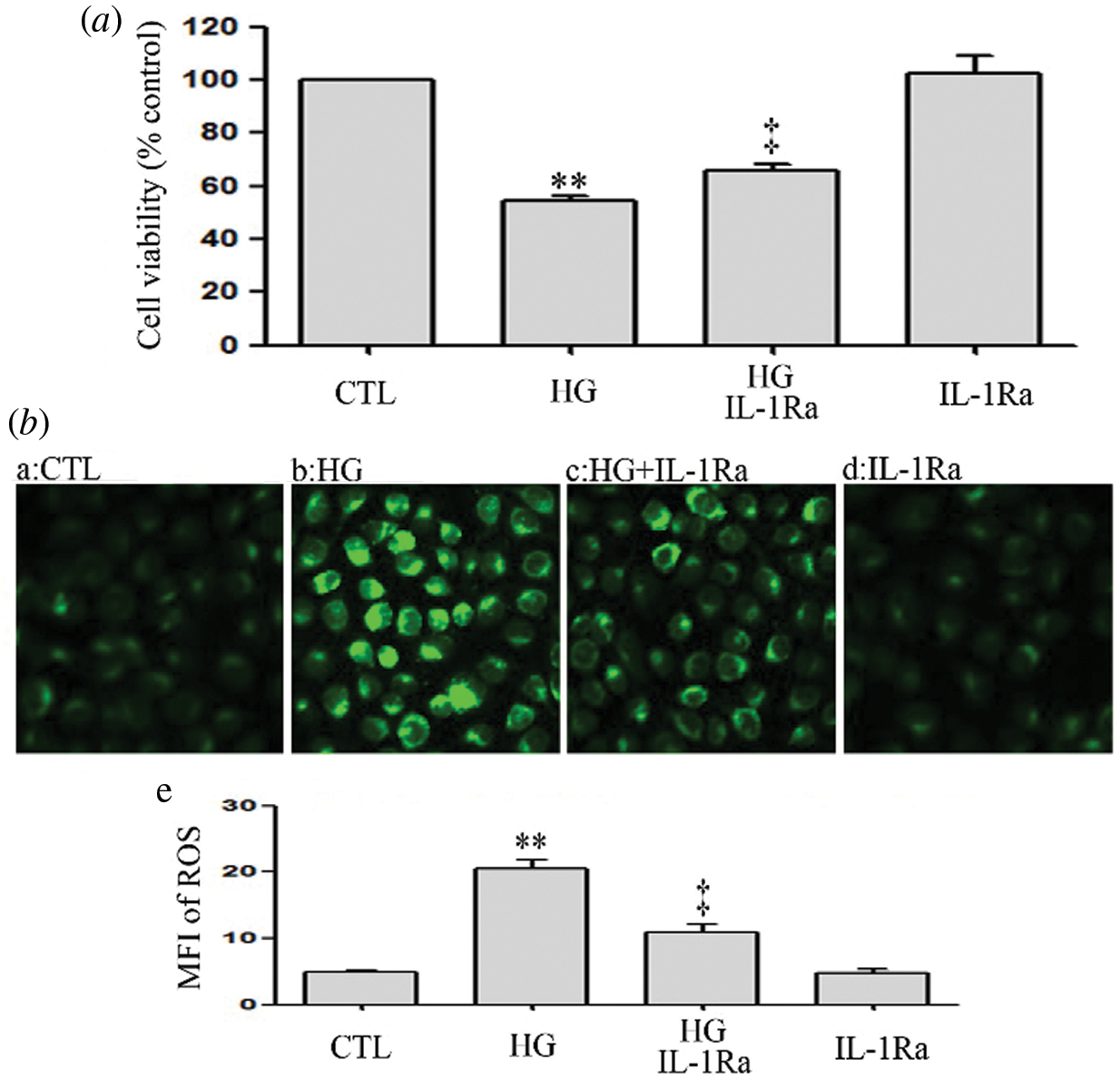
Figure 10: IL-1Ra inhibits HG-induced oxidative stress and cytotoxicity in HUVECs.
(a and b) HUVECs were cultured in high glucose concentrations (40 mM) for 24 h and then treated either with 20 ng/mL IL-1Ra or not for 30 min. (a) Cell viability was assessed using the CCK-8. (b) Levels of intracellular ROS were evaluated by DCFH-DA staining and followed by photofluorography. (b-e) Mean fluorescence intensity (MFI) of DCFH-DA calculated by Image J 1.47i software. Results are the mean ± SEM (N = 3). CTL, control; HG, high glucose; IL-1Ra, Interleukin-1 receptor antagonist.
This study revealed several novel findings related to the possible mechanisms by which high glucose levels trigger an increase of ROS production and inflammation and the protective role that Ang-(1-7) exerts against endothelial dysfunction caused by hyperglycemia in HUVECs. First, in HUVECs, the NF-κB-IL-1β signaling pathway was activated in response to the oxidative stress and inflammation caused by high glucose levels. Second, there was an up-regulation of the HO-1 signaling pathway to exert a protective effect against the oxidative stress and inflammation caused by high glucose concentrations. Third, Ang-(1-7) seemed to have a protective effect on HG-induced oxidative stress and inflammation. Last, Ang-(1-7) exerted its protective effects through the activation and inhibition of the HO-1 pathway and NF-κB-IL-1, respectively.
Our results suggest that exposure of HUVECs to HG increases oxidative stress and inflammation, which were consistent with previous researches (Guan et al., 2014; He et al., 2015b; Wang et al., 2015; Zhang et al., 2015b; Zhu et al., 2015). These data suggest that the injury made by HG-induced ROS production and inflammation was strongly associated with endothelial dysfunction. However, the underlying mechanism is still undefined.
NF-κB is an initiation factor of oxidative stress and inflammation in HG-induced HUVECs. Generally, NF-κB is located at the cytoplasm and activated by the stimulus. Under stimulation, NF-κB is activated from its stable complex and transfers to the nucleus from the cytoplasm. This activated process allows NF-κB (p65) to bind to the promoter of target genes. In the present study, our data found that p-p65 NF-κB was up-regulated and activated to be translocated into the nucleus. These results indicate that the NF-κB signaling pathway is activated under high glucose concentrations in HUVECs. Our data was supported by other studies (Williams et al., 2012; You et al., 2016). Early research has found that the NF-κB pathway serves as a pivotal mediator in hyperglycemia-induced endothelial cell oxidative stress (Chu et al., 2017) and inflammation (Fratantonio et al., 2017). Our findings suggested that inhibition of the NF-κB pathway in HUVECs, when cultured in the presence of PDTC and HG, distinctly suppressed HG-elicited apoptosis, oxidative stress, and inflammation. These findings suggest that the activation of the NFκB pathway is implicated in HG-induced dysfunction in HUVECs. Some previous researches have shown that the NF-κB-IL-1β pathway mediates inflammation (Li et al., 2015a; Li et al., 2014; Li et al., 2015b; Liu et al., 2014). However, whether the NF-κB-IL-1β pathway mediates HG-induced dysfunction in HUVECs is unknown. We conducted further investigations to validate this hypothesis and found that: (1) PDTC significantly inhibited HG-induced IL-1β; (2) PDTC suppressed HG-induced cell cytotoxicity, oxidative stress, and inflammation; and (3) IL-1Ra mitigated HG-decreased cell viability and HG-increased ROS. These results demonstrated that IL-1β acts as a downstream molecule in the NF-κB pathway in HUVECs under hyperglycemia conditions and the NF-κB-IL-1β pathway orchestrates the HG-elicited endothelial dysfunction in HUVECs.
However, some research has suggested that hyperglycemic conditions alone are not able to induce NF-κB activation in vascular cells. For example, Lafuente et al. (2008) suggested that high glucose enhances a pre-existing inflammatory response rather than the direct activator of NF-κB and subsequent inflammation in diabetes (Lafuente et al., 2008). Moreover, Peiro et al. (2017) found that excess glucose uptake occurred following activation with the NF-κB-IL-1β pathway in human vascular cells (Peiro et al., 2017). Thus, high glucose may not a direct cause of vascular endothelial injury but only enhance this injury through NF-κB-mediated inflammation.
Evidence has determined the endothelial protection of HO-1. Hypoxia can boost HO-1 expression in endothelial cells (Lin et al., 2017), which efficiently ameliorates various stimulus-induced endothelial dysfunction (He et al., 2015b; Lai et al., 2015; Lin et al., 2017; Zhao et al., 2016b; Zhong and Tang, 2016). More recently, studies suggest that HO-1 exerts protective effects in the hyperglycemia-treated endothelial cells (He et al., 2015b; Zhong and Tang, 2016). However, the potential relationship between the HG-induced endothelial dysfunction and HO-1 was still not completely clear. Upregulation of HO-1 provides protection against HG-induced oxidative stress in endothelial cells (He et al., 2015b). Consistent with the early findings (Tian et al., 2014; Zhong and Tang, 2016), our results demonstrated that HG time-dependently enhanced HO-1 expression level in HUVECs. To further investigate the role of HO-1 in hyperglycemia-elicited injury, HUVECs were cultured in HG medium added with SnPP. Our results showed that when HUVECs are treated with SnPP in high glucose levels, cytotoxicity, oxidative stress, and inflammation are significantly exacerbated. These results suggested that HG-induced HUVECs injuries are associated with the HO-1 pathway.
Another novel finding in this study revealed the existence of a relationship between the inhibition of the NF-κB-IL-1β signaling pathway, the activation of the HO-1 pathway, and the protective effects of exogenous Ang-(1-7) against endothelial oxidative stress and inflammation caused by high glucose. As a newly discovered RAS family member and anti-mediator of Ang-II, Ang-(1-7) executes anti-inflammatory, anti-oxidation, anti-apoptotic effects (Lin et al., 2016; Meneses et al., 2015; Zhang et al., 2016a). In the past few years, more attention was given to the protective effects of Ang-(1-7) on DM-related cardiovascular damages. As reported, Ang-(1-7) exerts a protective effect on DM-related cardiovascular events (Benter et al., 2007; Yousif et al., 2012). Additionally, a recent study has indicated that the Ang-(1-7)-mediated signaling pathway has a protective role in the endothelial function via suppressing oxidative stress in diabetic mice (Zhang et al., 2015b). As a matter of fact, Ang-(1-7) is strongly associated with hyperglycemia-induced endothelial dysfunction. Nonetheless, the potential role of endogenous Ang (1-7) in maintaining endothelial function in diabetic patients is still poorly understood. It has been communicated that Ang-(1-7) can impede the activation of NF-κB in HUVECs (Liang et al., 2015) and IL-1β in vascular diseases (Jin et al., 2012). HO-1 pathway has also been described for taking part in Ang II-derived ROS during angiogenesis in human late endothelial progenitor cells (Zhu et al., 2015), and this research showed the involvement of NF-κB-IL-1β and HO-1 pathways in the oxidative stress and inflammation induced by hyperglycemia in HUVECs, which impelled us to further examine the functions of these two pathways in the endothelial protective effect that exogenous Ang-(1-7) exerts in the context of oxidative stress and inflammation produced by high glucose. In agreement with earlier research (Benter et al., 2007; Yousif et al., 2012; Zhang et al., 2015b), our results showed that exogenous Ang-(1-7) confers protection against the HG-induced oxidative stress and inflammation, leading to increased cell viability, SOD and GSH activity, and decreased cell apoptosis, ROS generation, and inflammation. Importantly, exogenous Ang-(1-7) markedly blocked HG-induced p-p65, IL-1β, and HO-1. In order to explore the function of both NF-κB-IL-1β and HO-1 pathways in the cardioprotection effect of exogenous Ang-(1-7) against the HG-induced oxidative stress and inflammation, HUVECs were treated with either an inhibitor of NF-κB (PDTC) or an antagonist of IL-1β receptor (IL-1Ra) or an inhibitor of HO-1 (SnPP) before being exposed to Ang-(1-7) and HG, respectively. The findings of this study revealed that SnPP markedly reversed the endothelial protective effect of exogenous Ang-(1-7) against the oxidative stress and inflammation elicited by hyperglycemia. Ang-(1-7) impeded the activation of the NF-κB-IL-1β signaling pathway, and both PDTC and IL-1Ra significantly suppressed HG-induced oxidative stress and inflammation. These findings suggest that inhibiting NF-κB-IL-1β and activating the HO-1 pathway play important roles in the endothelial protective effects of exogenous Ang-(1-7) against the HG-induced oxidative stress and inflammation.
In summary, the present study gives the first evidence that NF-κB-IL-1β and HO-1 pathways contribute to the protective effects of exogenous Ang-(1-7) in endothelial cells against hyperglycemia-elicited injuries, including cytotoxicity, oxidative stress, and inflammation. Further study on animal models will probably help to understand better these two signaling pathways and their recently found roles in the pathophysiology of diabetes-associated cardiovascular complications.
Availability of Data and Materials: The data of this study are available from the corresponding author upon reasonable request.
Author Contribution: The authors confirm contribution to the paper as follows: study conception and design: FC, YQD, JL, JFC; data collection: FC, YQD, WZ, YLZ, JL; analysis and interpretation of results: FC, YQD, WZ, YLZ, JL, SHL, CT, GHL; draft manuscript preparation: FC, YQD, JL, JFC. All authors reviewed the results and approved the final version of the manuscript.
Funding Statament: This study was supported by the Technology Planning Project of Huangpu District (201544-01), Medical Scientific Research Foundation of Guangdong Province (A2015287), Science and Technology Planning Project of Guangdong Province (2017ZC0474), Natural Science Foundation of Guangdong Province (2015A030313690) and General Project of Dongguan City (Nos. 201950715024922 and 2018507150241344).
Conflicts of Interest: The authors declare no conflicts of interest.
Benter IF, Yousif MH, Cojocel C, Al-Maghrebi M, Diz DI (2007). Angiotensin-(1-7) prevents diabetes-induced cardiovascular dysfunction. American Journal of Physiology-Heart and Circulatory Physiology 292: H666–H672. DOI 10.1152/ajpheart.00372.2006. [Google Scholar] [CrossRef]
Bhatt MP, Lim YC, Hwang J, Na S, Kim YM, Ha KS (2012). C-peptide prevents hyperglycemia-induced endothelial apoptosis through inhibition of reactive oxygen species-mediated transglutaminase 2 activation. Diabetes 62: 243–253. DOI 10.2337/db12-0293. [Google Scholar] [CrossRef]
Capellini VK, Celotto AC, Baldo CF, Olivon VC, Viaro F et al. (2010). Diabetes and vascular disease: Basic concepts of nitric oxide physiology, endothelial dysfunction, oxidative stress and therapeutic possibilities. Current Vascular Pharmacology 8: 526–544. DOI 10.2174/157016110791330834. [Google Scholar] [CrossRef]
Chang RL, Lin JW, Kuo WW, Hsieh DJ, Yeh YL et al. (2016). Angiotensin-(1-7) attenuated long-term hypoxia-stimulated cardiomyocyte apoptosis by inhibiting HIF-1alpha nuclear translocation via Mas receptor regulation. Growth Factors 34: 11–18. DOI 10.3109/08977194.2016.1155150. [Google Scholar] [CrossRef]
Chen J, Zhang W, Xu Q, Zhang J, Chen W et al. (2018). Ang-(1-7) protects HUVECs from high glucose-induced injury and inflammation via inhibition of the JAK2/STAT3 pathway. International Journal of Molecular Medicine 41: 2865–2878. [Google Scholar]
Chu P, Han G, Ahsan A, Sun Z, Liu S et al. (2017). Phosphocreatine protects endothelial cells from Methylglyoxal induced oxidative stress and apoptosis via the regulation of PI3K/Akt/eNOS and NF-κB pathway. Vascular Pharmacology 91: 26–35. DOI 10.1016/j.vph.2016.08.012. [Google Scholar] [CrossRef]
Fratantonio D, Speciale A, Canali R, Natarelli L, Ferrari D et al. (2017). Low nanomolar caffeic acid attenuates high glucose-induced endothelial dysfunction in primary human umbilical-vein endothelial cells by affecting NF-κB and Nrf2 pathways. BioFactors 43: 54–62. DOI 10.1002/biof.1312. [Google Scholar] [CrossRef]
Guan G, Han H, Yang Y, Jin Y, Wang X, Liu X (2014). Neferine prevented hyperglycemia-induced endothelial cell apoptosis through suppressing ROS/Akt/NF-κB signal. Endocrine 47: 764–771. DOI 10.1007/s12020-014-0186-1. [Google Scholar] [CrossRef]
Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE (2014). Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Research and Clinical Practice 103: 137–149. DOI 10.1016/j.diabres.2013.11.002. [Google Scholar] [CrossRef]
Hao PP, Chen YG, Liu YP, Zhang MX, Yang JM et al. (2013). Association of plasma angiotensin-(1-7) level and left ventricular function in patients with type 2 diabetes mellitus. PLoS One 8: e62788. DOI 10.1371/journal.pone.0062788. [Google Scholar] [CrossRef]
He J, Yang Z, Yang H, Wang L, Wu H et al. (2015a). Regulation of insulin sensitivity, insulin production, and pancreatic beta cell survival by angiotensin-(1-7) in a rat model of streptozotocin-induced diabetes mellitus. Peptides 64: 49–54. DOI 10.1016/j.peptides.2014.12.012. [Google Scholar] [CrossRef]
He M, Nitti M, Piras S, Furfaro AL, Traverso N et al. (2015b). Heme oxygenase-1-derived bilirubin protects endothelial cells against high glucose-induced damage. Free Radical Biology and Medicine 89: 91–98. DOI 10.1016/j.freeradbiomed.2015.07.151. [Google Scholar] [CrossRef]
Hisatake S, Kiuchi S, Kabuki T, Oka T, Dobashi S, Ikeda T (2017). Serum angiotensin-converting enzyme 2 concentration and angiotensin-(1-7) concentration in patients with acute heart failure patients requiring emergency hospitalization. Heart and Vessels 32: 303–308. DOI 10.1007/s00380-016-0877-z. [Google Scholar] [CrossRef]
Jin HY, Song B, Oudit GY, Davidge ST, Yu HM et al. (2012). ACE2 deficiency enhances angiotensin II-mediated aortic profilin-1 expression, inflammation and peroxynitrite production. PLoS One 7: e38502. DOI 10.1371/journal.pone.0038502. [Google Scholar] [CrossRef]
Lafuente N, Matesanz N, Azcutia V, Romacho T, Nevado J et al. (2008). The deleterious effect of high concentrations of D-glucose requires pro-inflammatory preconditioning. Journal of Hypertension 26: 478–485. DOI 10.1097/HJH.0b013e3282f331fb. [Google Scholar] [CrossRef]
Lai TH, Shieh JM, Tsou CJ, Wu WB (2015). Gold nanoparticles induce heme oxygenase-1 expression through Nrf2 activation and Bach1 export in human vascular endothelial cells. International Journal of Nanomedicine 10: 5925–5939. [Google Scholar]
Li XQ, Lv HW, Wang ZL, Tan WF, Fang B, Ma H (2015a). MiR-27a ameliorates inflammatory damage to the blood-spinal cord barrier after spinal cord ischemia: Reperfusion injury in rats by downregulating TICAM-2 of the TLR4 signaling pathway. Journal of Neuroinflammation 12: 25. DOI 10.1186/s12974-015-0246-3. [Google Scholar] [CrossRef]
Li XQ, Wang J, Fang B, Tan WF, Ma H (2014). Intrathecal antagonism of microglial TLR4 reduces inflammatory damage to blood-spinal cord barrier following ischemia/reperfusion injury in rats. Molecular Brain 7: 28. DOI 10.1186/1756-6606-7-28. [Google Scholar] [CrossRef]
Li YH, Yu JZ, Xin YL, Feng L, Chai Z et al. (2015b). Protective effect of a novel Rho kinase inhibitor WAR-5 in experimental autoimmune encephalomyelitis by modulating inflammatory response and neurotrophic factors. Experimental and Molecular Pathology 99: 220–228. DOI 10.1016/j.yexmp.2015.06.016. [Google Scholar] [CrossRef]
Liang B, Wang X, Zhang N, Yang H, Bai R et al. (2015). Angiotensin-(1-7) attenuates angiotensin II-Induced ICAM-1, VCAM-1, and MCP-1 expression via the MAS receptor through suppression of P38 and NF-κB pathways in HUVECs. Cellular Physiology and Biochemistry 35: 2472–2482. DOI 10.1159/000374047. [Google Scholar] [CrossRef]
Lin HC, Su SL, Lu CY, Lin AH, Lin WC et al. (2017). Andrographolide inhibits hypoxia-induced HIF-1alpha-driven endothelin 1 secretion by activating Nrf2/HO-1 and promoting the expression of prolyl hydroxylases 2/3 in human endothelial cells. Environmental Toxicology 32: 918–930. DOI 10.1002/tox.22293. [Google Scholar] [CrossRef]
Lin L, Liu X, Xu J, Weng L, Ren J et al. (2016). Mas receptor mediates cardioprotection of angiotensin-(1-7) against Angiotensin II-induced cardiomyocyte autophagy and cardiac remodelling through inhibition of oxidative stress. Journal of Cellular and Molecular Medicine 20: 48–57. DOI 10.1111/jcmm.12687. [Google Scholar] [CrossRef]
Liu CX, Hu Q, Wang Y, Zhang W, Ma ZY et al. (2011). Angiotensin-converting enzyme (ACE) 2 overexpression ameliorates glomerular injury in a rat model of diabetic nephropathy: A comparison with ACE inhibition. Molecular Medicine 17: 59–69. DOI 10.2119/molmed.2010.00111. [Google Scholar] [CrossRef]
Liu YK, Huang LJ, Zhao S, Lin W, He JB et al. (2014). Effect of Xuebijing injection on TLR4--NF-κB--IL-1beta pathway of myocardial hypoxia/reoxygenation in rats. Zhongguo Ying Yong Sheng Li Xue Za Zhi 30: 55–59. [Google Scholar]
Mai J, Qiu Q, Lin YQ, Luo NS, Zhang HF et al. (2014). Angiotensin II-derived reactive oxygen species promote angiogenesis in human late endothelial progenitor cells through heme oxygenase-1 via ERK1/2 and AKT/PI3K pathways. Inflammation 37: 858–870. DOI 10.1007/s10753-013-9806-9. [Google Scholar] [CrossRef]
Meneses C, Morales MG, Abrigo J, Simon F, Brandan E, Cabello-Verrugio C (2015). The angiotensin-(1-7)/Mas axis reduces myonuclear apoptosis during recovery from angiotensin II-induced skeletal muscle atrophy in mice. Pflügers Archiv-European Journal of Physiology 467: 1975–1984. DOI 10.1007/s00424-014-1617-9. [Google Scholar] [CrossRef]
Mylroie H, Dumont O, Bauer A, Thornton CC, Mackey J et al. (2015). PKCepsilon-CREB-Nrf2 signaling induces HO-1 in the vascular endothelium and enhances resistance to inflammation and apoptosis. Cardiovasc Research 106: 509–519. DOI 10.1093/cvr/cvv131. [Google Scholar] [CrossRef]
Patel VB, Zhong JC, Grant MB, Oudit GY (2016). Role of the ACE2/Angiotensin 1-7 axis of the renin-angiotensin system in heart failure. Circulation Research 118: 1313–1326. DOI 10.1161/CIRCRESAHA.116.307708. [Google Scholar] [CrossRef]
Peiro C, Romacho T, Azcutia V, Villalobos L, Fernandez E et al. (2017). Erratum to: Inflammation, glucose, and vascular cell damage: The role of the pentose phosphate pathway. Cardiovascular Diabetology 16: 82. DOI 10.1186/s12933-017-0502-1. [Google Scholar] [CrossRef]
Reich HN, Oudit GY, Penninger JM, Scholey JW, Herzenberg AM (2008). Decreased glomerular and tubular expression of ACE2 in patients with type 2 diabetes and kidney disease. Kidney International 74: 1610–1616. DOI 10.1038/ki.2008.497. [Google Scholar] [CrossRef]
Shaw JE, Sicree RA, Zimmet PZ (2010). Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Research and Clinical Practice 87: 4–14. DOI 10.1016/j.diabres.2009.10.007. [Google Scholar] [CrossRef]
Taguchi K, Sakata K, Ohashi W, Imaizumi T, Imura J, Hattori Y (2014). Tonic inhibition by G protein-coupled receptor kinase 2 of Akt/endothelial nitric-oxide synthase signaling in human vascular endothelial cells under conditions of hyperglycemia with high insulin levels. Journal of Pharmacology and Experimental Therapeutics 349: 199–208. DOI 10.1124/jpet.113.211854. [Google Scholar] [CrossRef]
Tian S, Ge X, Wu K, Yang H, Liu Y (2014). Ramipril protects the endothelium from high glucose-induced dysfunction through CaMKKbeta/AMPK and heme oxygenase-1 activation. Journal of Pharmacology and Experimental Therapeutics 350: 5–13. DOI 10.1124/jpet.114.212928. [Google Scholar] [CrossRef]
Wang D, Wang Q, Yan G, Qiao Y, Sun L et al. (2015). High glucose and interleukin 1beta-induced apoptosis in human umbilical vein endothelial cells involves in down-regulation of monocarboxylate transporter 4. Biochemical and Biophysical Research Communications 466: 607–614. DOI 10.1016/j.bbrc.2015.09.016. [Google Scholar] [CrossRef]
Wenzel P, Rossmann H, Muller C, Kossmann S, Oelze M et al. (2015). Heme oxygenase-1 suppresses a pro-inflammatory phenotype in monocytes and determines endothelial function and arterial hypertension in mice and humans. European Heart Journal 36: 3437–3446. DOI 10.1093/eurheartj/ehv544. [Google Scholar] [CrossRef]
Williams CR, Lu X, Sutliff RL, Hart CM (2012). Rosiglitazone attenuates NF-κB-mediated Nox4 upregulation in hyperglycemia-activated endothelial cells. American Journal of Physiology-Cell Physiology 303: C213–C223. DOI 10.1152/ajpcell.00227.2011. [Google Scholar] [CrossRef]
You Q, Wu Z, Wu B, Liu C, Huang R et al. (2016). Naringin protects cardiomyocytes against hyperglycemia-induced injuries in vitro and in vivo. Journal of Endocrinology 230: 197–214. DOI 10.1530/JOE-16-0004. [Google Scholar] [CrossRef]
Yousif MH, Dhaunsi GS, Makki BM, Qabazard BA, Akhtar S, Benter IF (2012). Characterization of Angiotensin-(1-7) effects on the cardiovascular system in an experimental model of type-1 diabetes. Pharmacological Research 66: 269–275. DOI 10.1016/j.phrs.2012.05.001. [Google Scholar] [CrossRef]
Zhang F, Liu J, Li SF, Song JX, Ren JY, Chen H (2015a). Angiotensin-(1-7New perspectives in atherosclerosis treatment. Journal of Geriatric Cardiology 12: 676–682. [Google Scholar]
Zhang F, Ren X, Zhao M, Zhou B, Han Y (2016a). Angiotensin-(1-7) abrogates angiotensin II-induced proliferation, migration and inflammation in VSMCs through inactivation of ROS-mediated PI3K/Akt and MAPK/ERK signaling pathways. Scientific Reports 6: 72. DOI 10.1038/srep34621. [Google Scholar] [CrossRef]
Zhang Y, Liu J, Luo JY, Tian XY, Cheang WS et al. (2015b). Upregulation of Angiotensin (1-7)-mediated signaling preserves endothelial function through reducing oxidative stress in diabetes. Antioxidants & Redox Signaling 23: 880–892. DOI 10.1089/ars.2014.6070. [Google Scholar] [CrossRef]
Zhang Y, Liu T, Chen Y, Dong Z, Zhang J et al. (2016b). CD226 reduces endothelial cell glucose uptake under hyperglycemic conditions with inflammation in type 2 diabetes mellitus. Oncotarget 7: 12010–12023. DOI 10.18632/oncotarget.7505. [Google Scholar] [CrossRef]
Zhao H, Ma T, Fan B, Yang L, Han C et al. (2016a). Protective effect of trans-delta-viniferin against high glucose-induced oxidative stress in human umbilical vein endothelial cells through the SIRT1 pathway. Free Radical Research 50: 68–83. DOI 10.3109/10715762.2015.1108412. [Google Scholar] [CrossRef]
Zhao R, Feng J, He G (2016b). Hypoxia increases Nrf2-induced HO-1 expression via the PI3K/Akt pathway. Frontiers in Bioscience (Landmark Edition) 21: 385–396. DOI 10.2741/4395. [Google Scholar] [CrossRef]
Zhong ZY, Tang Y (2016). Upregulation of periostin prevents high glucose-induced mitochondrial apoptosis in human umbilical vein endothelial cells via activation of Nrf2/HO-1 signaling. Cellular Physiology and Biochemistry 39: 71–80. DOI 10.1159/000445606. [Google Scholar] [CrossRef]
Zhu D, Wang H, Zhang J, Zhang X, Xin C et al. (2015). Irisin improves endothelial function in type 2 diabetes through reducing oxidative/nitrative stresses. Journal of Molecular and Cellular Cardiology 87: 138–147. DOI 10.1016/j.yjmcc.2015.07.015. [Google Scholar] [CrossRef]
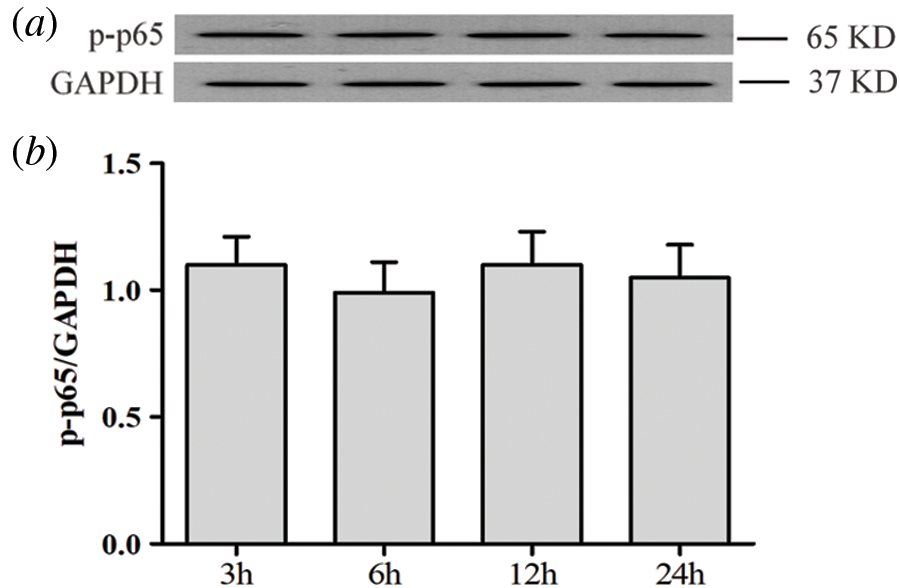
SUPPLEMENTARY FIGURE S1: Activation of the NF-κB pathway is not affected by low glucose in HUVECs.
HUVECs were grown in a medium containing low glucose (5.5 mM) for a given period of time (3, 6, 12 and 24 h) in order to evaluate the effects of low glucose on NF-ΚB p65 phosphorylation. Expression levels of p-p65 were analyzed by Western blot. (a) Densitometric analysis of western blots for p65 expression levels was performed with ImageJ 1.47i software (b). Results are the mean ± SEM (N = 3).
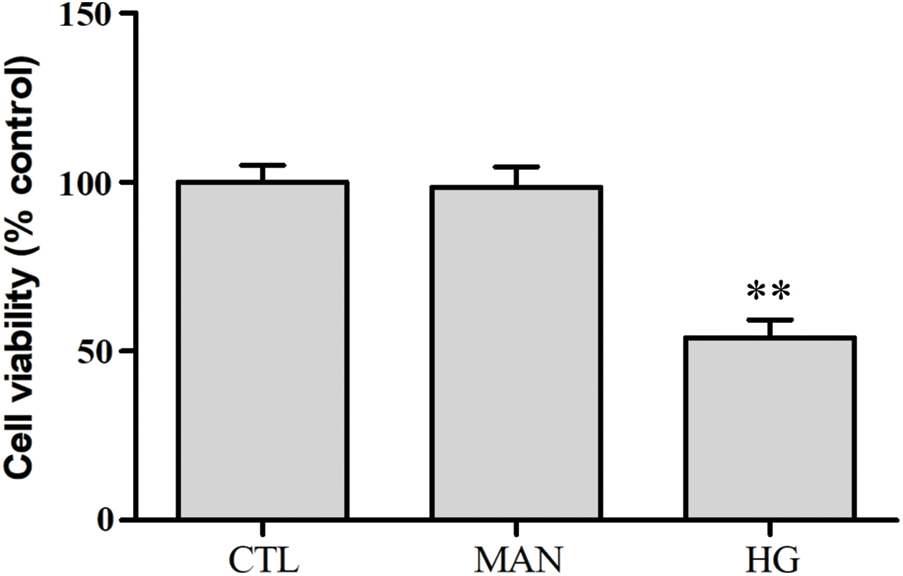
SUPPLEMENTARY FIGURE S2: Mannitol has no effect on cytotoxicity in HUVECs.
HUVECs were grown in high glucose concentrations (40 mM) and mannitol (40 mM) for 24 h. Cell viability was evaluated using the CCK-8. Data are the mean ± SEM (N = 3). **P < 0.01 vs. control group. CTL, control; HG, high glucose; MAN, mannitol.
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |