DOI:10.32604/biocell.2022.018144
| BIOCELL DOI:10.32604/biocell.2022.018144 | |
| Viewpoint |
AAV-based gene therapy approaches for genetic forms of tauopathies and related neurogenetic disorders
1Faculty of Medicine and University Hospital Cologne, Institute of Human Genetics, University of Cologne, Cologne, 50931, Germany
2Faculty of Medicine and University Hospital Cologne, Center for Molecular Medicine Cologne (CMMC), University of Cologne, Cologne, 50931, Germany
3Faculty of Medicine and University Hospital Cologne, Department of Neurology, University of Cologne, Cologne, 50937, Germany
4Faculty of Medicine and University Hospital Cologne, Center for Rare Diseases, University of Cologne, Cologne, 50937, Germany
5Faculty of Medicine and University Hospital Cologne, Center Anatomy, Department of Molecular and Translational Neuroscience, University of Cologne, Cologne, 50931, Germany
*Address correspondence to: Hans Zempel, hans.zempel@uk-koeln.de; hzempel@uni-koeln.de
Received: 02 July 2021; Accepted: 03 September 2021
Abstract: Tauopathies comprise a spectrum of genetic and sporadic neurodegenerative diseases mainly characterized by the presence of hyperphosphorylated TAU protein aggregations in neurons or glia. Gene therapy, in particular adeno-associated virus (AAV)-based, is an effective medical approach for difficult-to-treat genetic diseases for which there are no convincing traditional therapies, such as tauopathies. Employing AAV-based gene therapy to treat, in particular, genetic tauopathies has many potential therapeutic benefits, but also drawbacks which need to be addressed in order to successfully and efficiently adapt this still unconventional therapy for the various types of tauopathies. In this Viewpoint, we briefly introduce some potentially treatable tauopathies, classify them according to their etiology, and discuss the potential advantages and possible problems of AAV-based gene therapy. Finally, we outline a future vision for the application of this promising therapeutic approach for genetic and sporadic tauopathies.
Keywords: Tauopathies; Neurogenetic diseases; AAV-based gene therapy; Neurodegeneration; Alzheimer disease; TAU
| Abbreviations | |
| AAV: | Adeno-associated virus |
| AD: | Alzheimer’s Disease |
| AGD: | Argyrophilic Grain Disease |
| ASO: | Antisense oligonucleotide |
| CBD: | Corticobasal Degeneration |
| CDK5: | Cyclin-dependent kinase 5 |
| CNS: | Central nervous system |
| DM: | Myotonic Dystrophy |
| ELISA: | Enzyme-linked Immunosorbent Assay |
| FDA: | US Food and Drug Administration |
| FTLD-TAU: | Frontotemporal Lobar Degeneration with Tauopathy |
| GoF: | Gain-of-function |
| iPSCs: | Induced Pluripotent Stem Cells |
| ITR: | Inverted terminal repeats |
| LoF: | Loss-of-function |
| MAP: | Microtubule-associated proteins |
| PiD: | Pick’s Disease |
| PSP: | Progressive Supranuclear Palsy |
| rAAV: | Recombinant adeno-associated virus |
| RNAi: | RNA interference |
| RPE65: | Retinal-pigment epithelium-specific-65-kDa-protein |
| scAAV: | Self-complementary AAV |
| shRNA: | Short hairpin RNA |
| siRNA: | Small interfering RNA |
| VUS: | Variant of unknown significance |
TAU is a microtubule binding protein encoded in humans by the gene MAPT, which is alternatively spliced to produce eight isoforms, six of which are expressed in the human central nervous system (CNS). Under normal conditions, TAU is sorted into the axons likely due to several sorting mechanisms (Zempel and Mandelkow, 2019), where it promotes microtubule assembly and stability. However, in disease conditions (e.g., Alzheimer’s Disease (AD), pathological TAU mutations, etc.) these sorting mechanisms malfunction, which leads to mislocalization of TAU into the soma and dendrites. Under pathological conditions, TAU missorting is associated with TAU hyperphosphorylation and subsequent dissociation from microtubules. Hyperphosphorylated TAU can form insoluble aggregates called neurofibrillary tangles, the hallmark of several neurodegenerative diseases known collectively as tauopathies, the most frequent of which is AD (Zempel and Mandelkow, 2014).
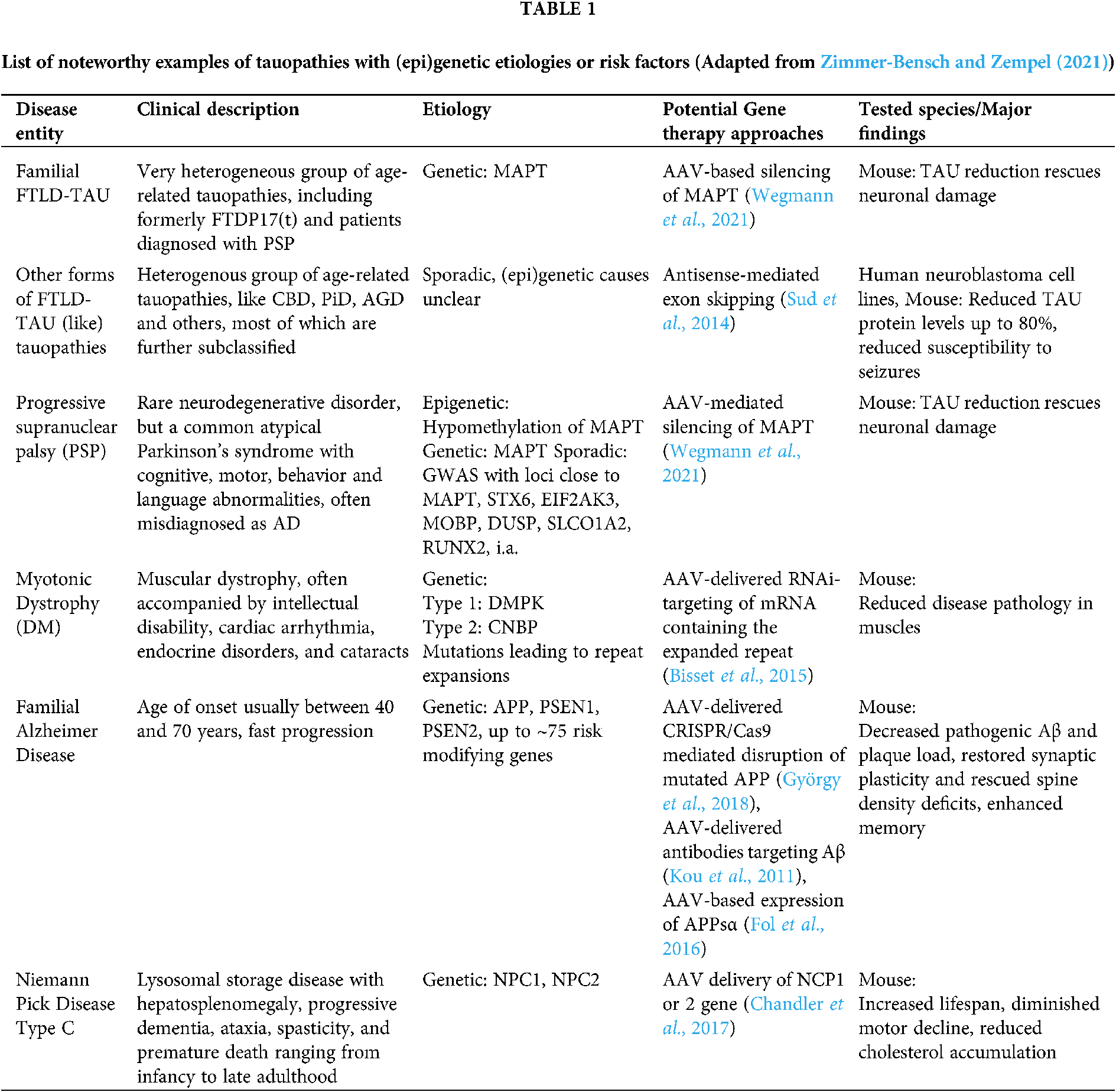
Tauopathies encompass a spectrum of neurodegenerative diseases whose main feature is the presence of aggregated deposits of TAU protein in the form of neurofibrillary or gliofibrillary tangles (Goedert and Spillantini, 2017). Most tauopathies present clinically as syndromes of cognitive deterioration or movement disorders, or both (Murley et al., 2020). Several systems to classify tauopathies have been proposed, but the diversity of their etiologies, pathomechanisms and phenotypes leads to overlapping classifications. The distinction between primary and secondary tauopathies is often blurry, as in the case of AD, which is traditionally classified as a secondary tauopathy, but mounting evidence suggests a central role of TAU pathology in driving the pathomechanisms of the disease. Only 1–2% of AD cases are familial, presenting a clear genetic inheritance, and while the causes of the rest of AD cases are poorly understood, the disease starts almost universally with the accumulation of Aβ plaques and TAU tangles in the brain, leading to neurodegeneration and loss of cognitive function (Long and Holtzman, 2019). Moreover, imbalanced or altered isoform expression alone of TAU can be causative for an AD-like form of Frontotemporal Dementia (FTD), Frontotemporal Lobar Degeneration with tauopathy (FTLD-TAU), and is observed in several FTD-associated tauopathies, i.a. Progressive Supranuclear Palsy (PSP), Corticobasal Degeneration (CBD), Pick’s Disease (PiD), Argyrophilic Grain Disease (AGD) (Park et al., 2016). Here, we subdivide major tauopathies into either genetic diseases, in which proven inherited genetic mutations are the cause of the disease, or sporadic or idiopathic diseases, in which clear genetic causes are absent (Table 1) (For a more extensive list see Zimmer-Bensch and Zempel, 2021), and outline potential AAV-based gene therapy approaches.
Gene therapy aims to correct a genetic problem at its roots, and focuses on gene modification to treat genetic diseases by repairing or suppressing defective genes or reintroducing functional ones (Kaji and Leiden, 2001). The delivery of the therapeutic genetic material is usually achieved via vectors, the majority of which are of viral origin, although other non-viral methods do exist (e.g., naked DNA, electroporation, lipoplexes, etc.), albeit with reduced levels of transfection and therapeutic efficiency (Ramamoorth and Narvekar, 2015).
As of 2021, over 3,180 gene therapy clinical trials were conducted, with more than half of them in phase I. In 263 of these trials (approximately 8.3% of the total number of gene therapy clinical trials), AAV has been used as the vector of choice for gene transfer, with 24 trials relevant to neurogenetic diseases (Gene Therapy Clinical Trials Worldwide Database. The Journal of Gene Medicine. Wiley 2021). Following the approval of Spark Therapeutics’ Luxturna (for the treatment of Retinal-pigment epithelium-specific-65-kDa-protein(RPE65)-mutation-induced blindness/retinitis pigmentosa) by the U.S. Food and Drug Administration (FDA) in 2017 as the first AAV vector-based gene therapy, several gene therapies have also received FDA approval, with Novartis’ Zolgensma (to treat spinal muscular atrophy) being the second FDA-approved AAV-based gene therapy.
The recombinant adeno-associated virus (rAAV) is the standard vehicle of choice when it comes to AAV-based gene therapy, renowned for its safety and efficacy. It is a 4.8 kb single-stranded DNA virus that comprises two inverted terminal repeats (ITR) framing the expression cassette, which contains either a constitutive or a tissue-specific promoter that drives the transgene expression, and a polyA sequence (Le Bec and Douar, 2006).
The current line of treatment for tauopathies is generally supportive, aiming at symptom alleviation. A variety of efforts have been made to develop drugs that manipulate TAU post translational modifications or aggregation, or target TAU immunologically via antibodies, but most of these trials have shown varying, and sometimes disappointing, levels of success (Coughlin and Irwin, 2017). The promise of gene therapy is to cure the disease, improve symptoms, and stop disease progression. Several studies have demonstrated that TAU knockout mice have no obvious phenotype, with Microtubule-associated proteins (MAP)/microtubule functions being preserved probably via compensation by upregulation of other MAPs (van Hummel et al., 2016). Therapeutically, reducing TAU levels or its toxic gain-of-function can be achieved by inhibiting TAU translation or even by inducing alternative splicing in favor of one isoform or the other, via the use of small interfering RNA fragments (siRNA) or antisense oligonucleotides (ASOs) (DeVos et al., 2017; Sud et al., 2014; Xu et al., 2014). While potentially promising, delivery of siRNA and ASOs remains challenging, and effects are limited to a few weeks or months, requiring several administrations per year. Viral vectors, such as AAV, can present an optimal medium to deliver not only RNAi (RNA interference), but also serve as a vector for gene replacement therapy with long lasting expression.
With approximately 150 clinical trials completed (~50% with met clinical safety and endpoints), more than 3000 treated patients, only 9 serious adverse events and no related deaths (Kuzmin et al., 2021), AAVs are the best choice for difficult-to-treat neurological disorders, like genetic forms of tauopathy. AAVs are not pathogenic, and some of their serotypes have a natural tropism for the CNS (Serotypes 4, 5, 8, 9). Also, AAV expression can persist for decades in neurons and other long lasting cells like cardiomyocytes (which is relevant for tauopathies that also affect the heart, e.g., Myotonic Dystrophy (DM) type 1 and 2), unlike mitotically active cells in which AAV expression is lost overtime (Sun and Roy, 2021). Tauopathies with clear genetic causes would be prime targets for AAV-based gene therapy; similar approaches have been tested in animal models of other neurodegenerative diseases like Huntington’s disease (Franich et al., 2008), and AAV2/8 have already been used to deliver anti-TAU antibodies into the brain of P301S-tg-mice, a model of frontotemporal dementia (Ising et al., 2017). These AAV-mediated gene transfer methods can be employed to deliver shRNAs (short hairpin RNA) based on siRNAs that suppressed the expression of P301S-mutated human TAU in mouse primary neurons, leading to amelioration of behavioural deficits in this mouse model of tauopathies (Xu et al., 2014).
The potential therapeutic benefits of AAV-based therapy are not exclusive to genetic tauopathies, but may be extended to sporadic forms of those diseases, if pathomechanistic workup reveals clear targets. AAV-delivered RNAi interference (RNAi), e.g., targeting of Cyclin-dependent kinase 5 (CDK5), a major TAU kinase that contributes to pathological TAU hyperphosphorylation, decreased the numbers of neurofibrillary tangles in the brains of AD mice (Piedrahita et al., 2010).
Naturally, there are limitations (For notable advantages and limitations of AAV-based gene therpay, see Box 1): Although AAVs are considered non pathogenic, activation of the host immune response can occur. Neutralizing antibodies or other forms of immunity against certain serotypes (AAV1, AAV2) are present in up to 70% of the population (Mingozzi and High, 2013). Although very young children are naive to AAV exposure, maternal antibodies may restrict the use of peripherally delivered AAVs to the age of approximately 7–11 months (Calcedo et al., 2011). However, hardly any severe adverse effects have been noticed in AAV-based gene therapy clinical trials, with transient, and usually asymptomatic, hepatitis being the most severe side effect (Kuzmin et al., 2021; Büning and Schmidt, 2015). Another issue is diseases that require a high proportion of transduced cells in the body, and for which much higher virus doses are needed to achieve beneficial results. Such high doses can be toxic and lead to liver failure and shock (Hinderer et al., 2018).
The diagnosis of pediatric forms of tauopathies and neurogenetic diseases is usually based on unclear genetic evidence, which makes pinpointing a specific target for gene therapy an exhausting task. On the other hand, in age-associated tauopathies, brain damage that has already happened at the disease onset is unlikely to be reversed with AAV-based gene therapy. Yet, given the probable ability of atrophic neurons to regenerate their normal function (Huang et al., 2014), the timing of initiation of the treatment would be a crucial factor in its success, with patients with known familial tauopathies treated in the presymptomatic phase having the highest chance of benefiting from treatment (Martier and Konstantinova, 2020). This complicates clinical studies due to necessary long-term follow up.
Several issues must be addressed for AAV-based gene therapy to become useful for genetic forms of tauopathy and related disorders.
Host immunity: One way to overcome host immunity when using AAVs is to focus on recombinant viruses derived from AAV-serotypes that i) are not serotype 1/2, and ii) already have a natural tropism for the CNS (i.e., serotypes 4,5,8,9, and for certain neuromuscular diseases with muscle involvement also skeletal/cardiac muscle, i.e., serotypes 6,7,8,9), and unconventional/novel AAVs already in clinical use/trials (e.g., AAVrh10, AAVrh74, LK200, AAVHSC15, SPK100, AAVhu37). To further reduce the danger of neutralizing antibodies/immunity, in case of pre-existing immunity against certain serotypes, standard Enzyme-linked Immunosorbent Assay (ELISA)-based serotesting or antibody titration of patients for existing antibodies against the specified AAV-serotypes could be used to identify the therapeutic serotype window on a patient-by-patient basis, where adequate.

Toxicity and tropism: To further reduce the (already low) risk of peripheral immune response or other possible peripheral side effects (such as hepatic toxicity), and to reduce the necessary amount of virus (also reducing production cost/time), intrathecal delivery should be the preferred route of administration for CNS specific disease. Other points to consider in order to avoid administering very high doses of AAV can include designing new promotors and capsids that enhance respectively the transgene expression and target tissue specificity, dose adjustment based on patient-specific factors and genetic predispositions, and the calculated administration of immunosuppressive agents to eliminate neutralizing antibodies (He et al., 2021). Further, modifying cap-proteins could constitute an approach to set up a library of viruses redundantly targeting CNS cells: Cap gene of AAV encodes three capsid proteins, which interact to form the capsid. These capsid proteins contain 12 hypervariable regions, and their serotype determines the tropism of the AAV virus (Gao et al., 2003). Consequently, hybrid AAV particles, in which the capsid is provided by one strain and the genome by another or the capsid itself is the result of hybridized capsids from different strains, can achieve more controlled and higher specific tissue targeting (Burger et al., 2004). This will provide a redundant battery of viruses from all specified serotypes with different (CNS)-tropisms, with up to a 1000-fold higher delivery capacity for specific cells compared to native AAVs (Ravindra Kumar et al., 2020), dramatically reducing titer necessity, potential toxicity and production cost/time.
Delivery: Interestingly, the tropism of different AAV serotypes is not solely controlled by the capsid proteins, but can also be influenced by the conditions of administration. Different serotypes tend to show different tropism depending on the route of delivery. For example, AAV9 showed higher transduction efficiency of cardiomyocytes when injected via the mouse tail vein, but this efficiency was reduced in comparison to AAV6 when both were injected into the left ventricle of the heart (Zincarelli et al., 2010). Moreover, age of the host at the time of AAV administration can affect the biodistribution of AAV particles; changing the time of injection from the day of birth to later stages of development shifted the tropism of several AAV serotypes from neuronal to non-neuronal, respectively (Chakrabarty et al., 2013). These factors should be considered carefully when designing clinical trials for AAV-based gene therapy.
Human specific disease-relevant neuronal assay systems: In case of necessity for a patient-specific genetic intervention (e.g., diseases with gain-of-function (GoF) mutations where silencing of the single nucleotide-mutated allele is necessary), the use of induced Pluripotent Stem Cells (iPSC)-derived CNS-cells expressing the mutated transgene could help to validate patient-tailored knockdown efficiency and functional consequences. Functional tests in iPSC-derived CNS-cells (e.g., allele specific knockdown) could also resolve candidate genetic alterations in case of a clear disease entity but unclear genetic pathogenic cause, and also serve as the functional readout for the genetic intervention (for scheme see Fig. 1). The arguments raised above, the current methodologies that allow testing AAVs on human cells of specific lineages, and the potentially patient specific shRNA design or gene replacement strategies all speak against the notion of routinely using non-human primates to test the safety of the developed viruses. With cell-type specific tropism of engineered AAVs, and human specific RNAi/gene expression paradigms, experiments in primates appear to us unnecessary and unhelpful due to unpredictable side/off-target effects simply due to interspecies differences. We strongly discourage the routine testing of all AAV-based gene therapy approaches in primates.

Figure 1: Scheme for envisioned gene therapy approach for tauopathies and neurogenetic diseases (Abbreviations: LoF, loss of function; GoF, gain of function; VUS, variant of unknown significance; iPSC, induced pluripotent stem cells; AAV, adeno-associated virus).
In conclusion, AAV-based gene therapy is a potentially powerful tool to cure hereditary diseases. Genetic tauopathies and related neurogenetic diseases are prime targets for this kind of therapy, especially since there is no traditional therapy in sight, and promising data have been obtained from a number of clinical trials for other neurogenetic and neurodegenerative diseases. Nonetheless, strategies to adapt current AAV-based gene therapy approaches to target the heterogeneous group of genetic or sporadic, pediatric or age-related tauopathies need to be developed and implemented in order to establish a safe, effective and personalized AAV-based gene therapy for specific tauopathies.
Authors’ Contribution: The authors confirm contribution to the paper as follows: Concept and initial drafting of the manuscript: MAAK, HZ; Refinement of conceptionalization, proofreading, scientific and clinical context: MAAK, CK, GW and HZ. All authors reviewed the results and approved the final version of the manuscript.
Funding Statement: This work was supported by funding from Else-Kröner-Fresenius-Stiftung and by the Köln Fortune Program/ Faculty of Medicine, University of Cologne.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present viewpoint.
Bisset DR, Stepniak-Konieczna EA, Zavaljevski M, Wei J, Carter GT, Weiss MD, Chamberlain JR (2015). Therapeutic impact of systemic AAV-mediated RNA interference in a mouse model of myotonic dystrophy. Human Molecular Genetics 24: 4971–4983. DOI 10.1093/hmg/ddv219. [Google Scholar] [CrossRef]
Büning H, Schmidt M (2015). Adeno-associated vector toxicity—To be or not to be? Molecular Therapy 23: 1673–1675. DOI 10.1038/mt.2015.182. [Google Scholar] [CrossRef]
Burger C, Gorbatyuk OS, Velardo MJ, Peden CS, Williams P, Zolotuchin S, Reier PJ, Mandel RJ, Muzyczka N (2004). Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Molecular Therapy 10: 302–317. DOI 10.1016/j.ymthe.2004.05.024. [Google Scholar] [CrossRef]
Calcedo R, Morizono H, Wang L, McCarter R, He J, Jones D, Batshaw ML, Wilson JM (2011). Adeno-associated virus antibody profiles in newborns, children, and adolescents. Clinical and Vaccine Immunology 18: 1586–1588. DOI 10.1128/CVI.05107-11. [Google Scholar] [CrossRef]
Chakrabarty P, Rosario A, Cruz P, Siemienski Z, Ceballos-Diaz C et al. (2013). Capsid serotype and timing of injection determines AAV transduction in the neonatal mice brain. PLOS ONE 8: e67680. DOI 10.1371/journal.pone.0067680. [Google Scholar] [CrossRef]
Chandler RJ, Williams IM, Gibson AL, Davidson CD, Incao AA, Hubbard BT, Porter FD, Pavan WJ, Venditti CP (2017). Systemic AAV9 gene therapy improves the lifespan of mice with Niemann-Pick disease, type C1. Human Molecular Genetics 26: 52–64. DOI 10.1093/hmg/ddw367. [Google Scholar] [CrossRef]
Coughlin D, Irwin DJ (2017). Emerging diagnostic and therapeutic strategies for tauopathies. Current Neurology and Neuroscience Reports 17: 72. DOI 10.1007/s11910-017-0779-1. [Google Scholar] [CrossRef]
DeVos SL, Miller RL, Schoch KM, Holmes BB, Kebodeaux S et al. (2017). Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Science Translational Medicine 9: 1–30. DOI 10.1126/scitranslmed.aag0481.Tau. [Google Scholar] [CrossRef]
Fol R, Braudeau J, Ludewig S, Abel T, Weyer SW et al. (2016). Viral gene transfer of APPsα rescues synaptic failure in an Alzheimer’s disease mouse model. Acta Neuropathologica 131: 247–266. DOI 10.1007/s00401-015-1498-9. [Google Scholar] [CrossRef]
Franich NR, Fitzsimons HL, Fong DM, Klugmann M, During MJ, Young D (2008). AAV vector-mediated RNAi of mutant Huntingtin expression is neuroprotective in a novel genetic rat model of Huntington’s disease. Molecular Therapy 16: 947–956. DOI 10.1038/mt.2008.50. [Google Scholar] [CrossRef]
Gao G, Alvira MR, Somanathan S, Lu Y, Vandenberghe LH, Rux JJ, Calcedo R, Sanmiguel J, Abbas Z, Wilson JM (2003). Adeno-associated viruses undergo substantial evolution in primates during natural infections. Proceedings of the National Academy of Sciences of the United States of America 100: 6081–6086. DOI 10.1073/pnas.0937739100. [Google Scholar] [CrossRef]
Goedert M, Spillantini MG (2017). Propagation of Tau aggregates. Molecular Brain 10: 1–9. DOI 10.1186/s13041-017-0298-7. [Google Scholar] [CrossRef]
György B, Lööv C, Zaborowski MP, Takeda S, Kleinstiver BP et al. (2018). CRISPR/Cas9 mediated disruption of the Swedish APP allele as a therapeutic approach for early-onset Alzheimer’s Disease. Molecular Therapy—Nucleic Acids 11: 429–440. DOI 10.1016/j.omtn.2018.03.007. [Google Scholar] [CrossRef]
He X, Urip BA, Zhang Z, Ngan CC, Feng B (2021). Evolving AAV-delivered therapeutics towards ultimate cures. Journal of Molecular Medicine 99: 593–617. DOI 10.1007/s00109-020-02034-2. [Google Scholar] [CrossRef]
Hinderer C, Katz N, Buza EL, Dyer C, Goode T, Bell P, Richman LK, Wilson JM (2018). Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Human Gene Therapy 29: 285–298. DOI 10.1089/hum.2018.015. [Google Scholar] [CrossRef]
Huang Z, Ha GK, Petitto JM, Petitto J (2014). Reversal of neuronal atrophy: Role of cellular immunity in neuroplasticity and aging the neuroprotective effects of cellular immunity. Journal of Neurological Disorders 2: 1000170. DOI 10.4172/2329-6895.1000170. [Google Scholar] [CrossRef]
Ising C, Gallardo G, Leyns CEG, Wong CH, Jiang H et al. (2017). Correction: AAV-mediated expression of anti-tau scFvs decreases tau accumulation in a mouse model of tauopathy. Journal of Experimental Medicine 214: 2163–2163. DOI 10.1084/jem.2016212505192017c. [Google Scholar] [CrossRef]
Kaji EH, Leiden JM (2001). Gene and stem cell therapies. Journal of the American Medical Association 285: 545–550. DOI 10.1001/jama.285.5.545. [Google Scholar] [CrossRef]
Kou J, Kim H, Pattanayak A, Song M, Lim JE, Taguchi H, Paul S, Cirrito JR, Ponnazhagan S, Fukuchi KI (2011). Anti-amyloid-β single-chain antibody brain delivery via AAV reduces amyloid load but may increase cerebral hemorrhages in an Alzheimer’s disease mouse model. Journal of Alzheimer’s Disease 27: 23–28. DOI 10.3233/JAD-2011-110230. [Google Scholar] [CrossRef]
Kuzmin DA, Shutova MV, Johnston NR, Smith OP, Fedorin VV et al. (2021). The clinical landscape for AAV gene therapies. Nature Reviews. Drug Discovery 20: 173–174. DOI 10.1038/d41573-021-00017-7. [Google Scholar] [CrossRef]
Le Bec C, Douar AM (2006). Gene therapy progress and prospects—Vectorology: Design and production of expression cassettes in AAV vectors. Gene Therapy 13: 805–813. DOI 10.1038/sj.gt.3302724. [Google Scholar] [CrossRef]
Long JM, Holtzman DM (2019). Alzheimer disease: An update on pathobiology and treatment strategies. Cell 179: 312–339. DOI 10.1016/j.cell.2019.09.001. [Google Scholar] [CrossRef]
Martier R, Konstantinova P (2020). Gene therapy for neurodegenerative diseases: Slowing down the ticking clock. Frontiers in Neuroscience 14: 580179. DOI 10.3389/fnins.2020.580179. [Google Scholar] [CrossRef]
McCarty DM, Monahan PE, Samulski RJ (2001). Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Therapy 8: 1248–1254. DOI 10.1038/sj.gt.3301514. [Google Scholar] [CrossRef]
Mingozzi F, High KA (2013). Immune responses to AAV vectors: Overcoming barriers to successful gene therapy. Blood 122: 23–36. DOI 10.1182/blood-2013-01-306647. [Google Scholar] [CrossRef]
Murley AG, Coyle-Gilchrist I, Rouse MA, Simon Jones P, Li W et al. (2020). Redefining the multidimensional clinical phenotypes of frontotemporal lobar degeneration syndromes. Brain 143: 1555–1571. DOI 10.1093/brain/awaa097. [Google Scholar] [CrossRef]
Park SA, Il Ahn S, Gallo JM (2016). Tau mis-splicing in the pathogenesis of neurodegenerative disorders. BMB Reports 49: 405–413. DOI 10.5483/BMBRep.2016.49.8.084. [Google Scholar] [CrossRef]
Piedrahita D, Hernández I, López-Tobón A, Fedorov D, Obara B et al. (2010). Silencing of CDK5 reduces neurofibrillary tangles in transgenic Alzheimer’s mice. Journal of Neuroscience 30: 13966–13976. DOI 10.1523/JNEUROSCI.3637-10.2010. [Google Scholar] [CrossRef]
Ramamoorth M, Narvekar A (2015). Non viral vectors in gene therapy—An overview. Journal of Clinical and Diagnostic Research 9: GE01–GE06. DOI 10.7860/JCDR/2015/10443.5394. [Google Scholar] [CrossRef]
Ravindra Kumar S, Miles TF, Chen X, Brown D, Dobreva T et al. (2020). Multiplexed Cre-dependent selection yields systemic AAVs for targeting distinct brain cell types. Nature Methods 17: 541–550. DOI 10.1038/s41592-020-0799-7. [Google Scholar] [CrossRef]
Sud R, Geller ET, Schellenberg GD (2014). Antisense-mediated exon skipping decreases Tau protein expression: A potential therapy for tauopathies. Molecular Therapy—Nucleic Acids 3: 1–11. DOI 10.1038/mtna.2014.30. [Google Scholar] [CrossRef]
Sun J, Roy S (2021). Gene-based therapies for neurodegenerative diseases. Nature Neuroscience 24: 297–311. DOI 10.1038/s41593-020-00778-1. [Google Scholar] [CrossRef]
van Hummel A, Bi M, Ippati S, Van Der Hoven J, Volkerling A et al. (2016). No overt deficits in aged tau-deficient C57Bl/6.Mapttm1(EGFP)kit GFP knockin mice. PLoS One 11: 1–14. DOI 10.1371/journal.pone.0163236. [Google Scholar] [CrossRef]
Wegmann S, DeVos SL, Zeitler B, Marlen K, Bennett RE et al. (2021). Persistent repression of tau in the brain using engineered zinc finger protein transcription factors. Science Advances 7: 1–20. DOI 10.1126/sciadv.abe1611. [Google Scholar] [CrossRef]
Xu H, Rösler TW, Carlsson T, de Andrade A, Fiala O, Hollerhage M, Oertel WH, Goedert M, Aigner A, Höglinger GU (2014). Tau silencing by siRNA in the P301S mouse model of tauopathy. Current Gene Therapy 14: 343–351. DOI 10.2174/156652321405140926160602. [Google Scholar] [CrossRef]
Yan Z, Zhang Y, Duan D, Engelhardt JF (2000). Trans-splicing vectors expand the utility of adeno-associated virus for gene therapy. Proceedings of the National Academy of Sciences of the United States of America 97: 6716–6721. DOI 10.1073/pnas.97.12.6716. [Google Scholar] [CrossRef]
Zempel H, Mandelkow E (2014). Lost after translation: Missorting of Tau protein and consequences for Alzheimer disease. Trends in Neurosciences 37: 721–732. DOI 10.1016/j.tins.2014.08.004. [Google Scholar] [CrossRef]
Zempel H, Mandelkow E (2019). Mechanisms of axonal sorting of tau and influence of the axon initial segment on tau cell polarity. Advances in Experimental Medicine and Biology 1184: 69–77. DOI 10.1007/978-981-32-9358-8_6. [Google Scholar] [CrossRef]
Zimmer-Bensch G, Zempel H (2021). DNA methylation in genetic and sporadic forms of neurodegeneration: Lessons from Alzheimer’s, related tauopathies and genetic tauopathies.Preprints: 2021050717. DOI 10.20944/preprints202105.0717.v1. [Google Scholar] [CrossRef]
Zincarelli C, Soltys S, Rengo G, Koch WJ, Rabinowitz JE (2010). Comparative cardiac gene delivery of adeno-associated virus serotypes 1–9 reveals that AAV6 mediates the most efficient transduction in mouse heart. Clinical and Translational Science 3: 81–89. DOI 10.1111/j.1752-8062.2010.00190.x. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |