DOI:10.32604/biocell.2022.017435
| BIOCELL DOI:10.32604/biocell.2022.017435 | |
| Article |
CDK5 inhibition promotes osteoblastic differentiation of MSCs and blocks the migration of osteosarcoma MG-63 cells
1College of Medicine, Southwest Jiaotong University, Chengdu, 610031, China
2Provincial Laboratory of Orthopaedic Engineering, Department of Bone and Joint Surgery, Affiliated Hospital of Southwest Medical University, Luzhou, 646000, China
*Address correspondence to: Ke Duan, keduan@swmu.edu.cn; Tailin Guo, tlguo@home.swjtu.edu.cn
Received: 10 May 2021; Accepted: 07 July 2021
Abstract: CDK5 belongs to the cyclin-dependent kinase family. CDK5 is multifunctional and plays an important role in neural differentiation. However, the role of CDK5 in osteoblastic differentiation remains unclear. The present study investigated functions and molecular mechanism of CDK5 in osteoblastic differentiation. It was found that, CDK5 inhibition promoted the expression of Runx2, ALP, OCN and OPN of MSCs and the mineralization of MC-3T3E1 cells and MSCs. CDK5 inhibition enhanced the development of F-actin, nuclear localization of β-catenin and YAP, as well as the expression of RMRP RNA. When F-actin was suppressed by Blebbistatin, the nuclear localization of YAP and β-catenin, and expression of RMRP RNA as well as Runx2 and ALP were decreased. These indicate Seliciclib promotes osteoblastic differentiation mainly by F-actin. Moreover, Seliciclib also suppressed the migration of MG-63, suggesting a potential application for Seliciclib in bone defect repair and inhibition of the migration of osteosarcoma cells after osteosarcoma surgical resection.
Keywords: F-actin; YAP; Beta-catenin; Nuclear localization; MSC differentiation
CDK5 is a member of the cyclin-dependent kinase (CDK) family and shares 60% sequence identity to CDK1. Unlike other CDKs, CDK5 is the only CDK that does not play a role in the cell cycle. It is expressed ubiquitously and mostly expressed in the brain (Tsai et al., 1993).
Importantly, CDK5 is an upstream regulator of F-actin dynamics, affecting it differently under different situations. P35/CDK5 kinase can phosphorylate the serine/threonine kinase p21 (Rac1)-activated kinase1 (Pak1) on site T212; this leads to its inactivation in neurons and differentiating epithelial cells (Rashid et al., 2001; Zhong et al., 2003). Pak1 is a positive regulator for RAC family small GTPase 1, a kinase able to promote the polymerization of actin. CDK5 can be a negative regulator of Ras homolog family member A (RhoA). CDK5 phosphorylates DLC1 (deleted in liver cancer 1) on several sites (e.g., S120) to suppress RhoA (Tripathi et al., 2014). DLC1 is a GTPase-activating protein (GAP) that can down-regulate the activation of RhoA (Kim et al., 2007). CDK5 upregulates actin polymerization by phosphorylating, and thus suppressing, non-receptor tyrosine kinase Src. Src activates the Rho GTPase activating protein p190 (p190 RhoGAP), a suppressor of RhoA (Tripathi and Zelenka, 2009). However, the F-actin can promote the osteoblastic differentiation of MSCs (Honjo et al., 2012). The effects of CDK5 on F-actin dynamics in mesenchymal stem cells (MSCs) are unclear.
CDK5 also binds to β-catenin to reduce N-cadherin- mediated and calcium-dependent cell–cell adhesion (Kwon et al., 2000). Moreover, CDK5/p35 complex binds to β-catenin, thereby blocking its function as a transcription factor (Li et al., 2010). While during osteoblastic differentiation of MSCs, Wnt signaling pathways play important roles. Beta-catenin has many functions important for osteoblastic differentiation. It is both a transcription factor and a membrane skeleton junction protein. It is involved in the mechanotransduction-mediated osteoblastic differentiation (Yan et al., 2016). What’s more, Yes- associated Protein (YAP) is a transcriptional factor regulated by the Hippo pathway. YAP can promote osteogenesis and suppress the adipogenic differentiation of MSCs by interacting with β-catenin and maintaining its nuclear localization (Pan et al., 2018). Importantly, the nuclear localization of YAP can be promoted by the well-formed F-actin (Kim et al., 2016).
Of many discovered functions of CDK5, the most well-known one is its role in the normal development of cerebral cortex via promoting neuronal migration (Gilmore et al., 1998). CDK5 regulates cell migration by phosphorylating the talinhead (TH) domain (Huang et al., 2009). Phosphorylation of the TH domain helps the assembly of adhesion complexes, maintaining a balance between the assembly and disassembly of these complexes as well as a balanced lamellipodia stabilization-destabilization. Balance of lamellipodia stabilization-destabilization is needed for cell migration. Furthermore, as a result of its roles in cell migration and cell-cell adhesion, CDK5 is involved in the metastasis of many cancers (Feldmann et al., 2008; Petrosiute et al., 2011; Strock et al., 2006). Osteosarcoma, occurring frequently in adolescents, is the most common and highly metastatic primary bone tumor (Kager et al., 2003; Meyers and Gorlick, 1997). Most cancer deaths are caused by metastases rather than by primary tumor growth. Finding drag to inhibit the metastasis of osteosarcoma is essential to treat osteosarcoma.
Considering the association of CDK5 with F-actin, β-catenin, YAP, and cell migration, we hypothesized that CDK5 may play important roles in F-actin dynamics, β-catenin and YAP nuclear localization of the osteoblastic differentiation of MSCs, and also the migration of osteosarcoma cells. Therefore, this study was conducted to understand roles of CDK5 in these processes.
MSCs were extracted from the bone marrow of Sprague-Dawley rat of 2 weeks old. MSCs were cultured (95% relative humidity, 37°C, 5% CO2) in a proliferation medium [α-MEM, 10% fetal bovine serum (FBS), 1% antibiotic/antimycotic (all Hyclone)] with a medium changed every 2 days.
MC-3T3 cells (ATCC, Virginia, US) were cultured in an osteogenic induction medium (1 × 10−7 mol/L dexamethasone, 1 × 10−2 mol/L β-glycerol phosphate disodium, 5 × 10−2 mm/L vitamin C) with a medium changed every 2 days. MG-63 cells (ATCC, Virginia, US) were cultured in a high-glucose medium [DMEM, 10% FBS, 1% antibiotic/antimycotic (all Hyclone)] with a medium changed every 2 days.
For RT-qPCR, MSCs were seeded at 5 × 104, 3 × 104, 2 × 104 and 2 × 104 cells/well in 12-well plate and incubated for 1, 3, 5, and 7 days. For Alizarin red staining, MC-3T3E1 cells and MSCs were seeded in a 24-well plate at 3 × 104 and 4 × 104 cells/well, respectively. MC-3T3E1 and MSCs were incubated for 14 and 21 days, respectively. For F-actin staining and immunofluorescent staining of YAP and β-catenin, MSCs were seeded in a 48-well plate at 3 × 103 cells/well and incubated for 5 days. For migration analysis, MG-63 cells were seeded in a 24-well transwell (8 μm bore diameter, Corning) at 2 × 104 cells/well and incubated for 2 days. For cell survival rate analysis, MSCs or MG-63 cells were seeded in a 48-well plate at the concentration 1 × 104 cells/well and measured on days 1, 3, 7, and 10, respectively. Unless otherwise specified, the concentration of Seliciclib (CDK5 inhibitor, Med Chem Express) treated was 9 μM.
The concentration of Blebbistatin (Ble, myosin II inhibitor, Med Chem Express) treated was 5 μM. For nuclear protein extraction, 5 × 105 MSCs were plate in 6-well plate and cultured for 5 days. For western blot, 2 × 105 MSCs were plate in 6-well plate and cultured for 3 days. For siRNA transfection, 2 × 104 cells/cm2 MSCs were plated.
The sense and anti-sense sequence of siRNA targeting CDK5 were GGAGAUCUGUCUACUCAAA and UUUGAGUAGACAGAUCUCC (Qin Ke, Beijing, China). Mix 10 μm siCDK5 and DMEM in a ratio of 1:50. LipoRNAiMAX (ThermoFisher) transfection reagent and DMEM were mixed in a ratio of 1:50. Mix the mixture of siCDK5 and LipoRNAiMAX in a 1:1 ratio and mix well. It was incubated at room temperature for 15 min, and then added into the non-antibiotic/antimycotic medium to make the final concentration of siCDK5 30 nM. The non-antibiotic/antimycotic medium was replaced 6 h after transfection. Culture for 2 days for further analysis.
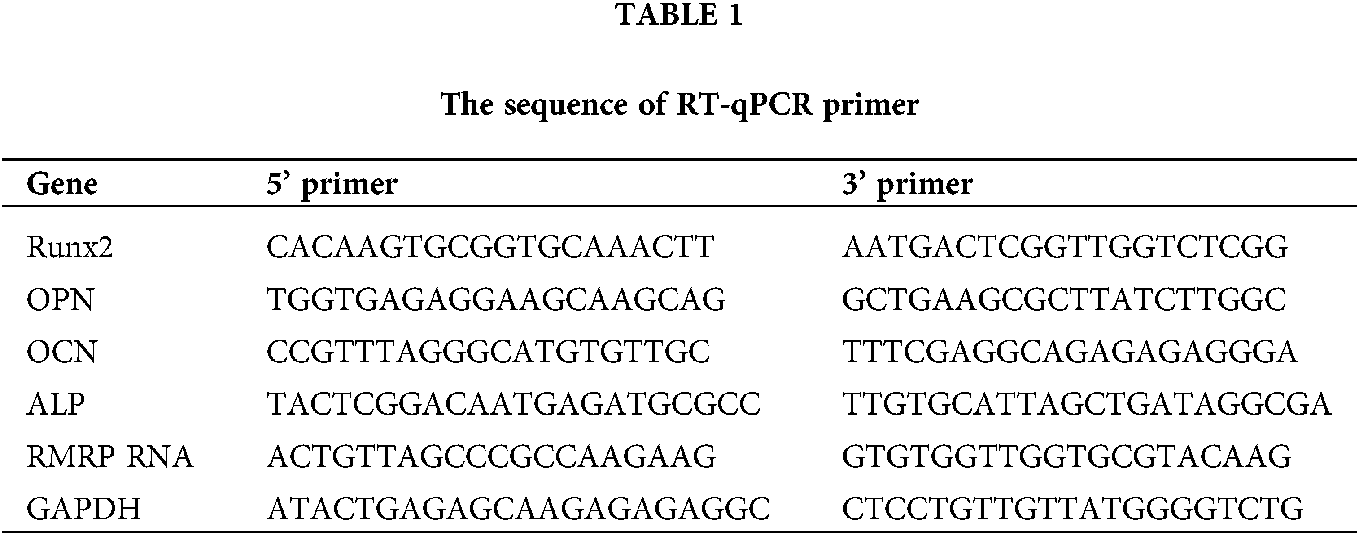
Cells were lysed with TRIzol (100 µL/well) (Invitrogen, Carlsbad, CA, USA) and transferred into Eppendorf tubes. After addition of 20 µL of chloroform, the tube was centrifuged (4°C, 1.2 × 104 g, 15 min). The supernatant was collected, mixed with 50 µL of 2-isopropanol, allowed to rest for 10 min, and centrifuged (4°C, 1.2 × 104 g, 10 min). The supernatant was discarded. Pellet was mixed with 100 µL of 75% ethanol, centrifuged (4°C, 7.5 × 103 g, 5 min). The pellet was dissolved in 20 µL of DNase/RNase-free water (Biosharp, Guangzhou, China). Then, the RNA was reverse transcribed into cDNA (Reverted First Strand cDNA Synthesis Kit, Bio-Rad, Hercules, CA, USA) and then amplified by real-time quantitative polymerase-chain reaction (RT-qPCR, SYBR Green PCR MasterMix Kit, Bio-Rad) using primers listed in Table 1. Relative gene expression was normalized to GAPDH expression. Throughout this study, each quantitative experiment included three parallel samples.
Immunofluorescent staining and F-actin staining
Cells were fixed for 3 h in PBS containing 5% glutaraldehyde, and then permeabilized for 5 min in PBS supplemented with 0.1% Triton X-100 (Bioforxx, German), and blocked for 40 min in PBS containing 5% bovine serum albumin (BSA). They were probed with YAP XR (R) Rabbit mAb (1:100; Cell Signaling Technology, Boston, USA) and Rb mAB to β-catenin (1:300; Abcam, Cambridge Science Park, UK) (4°C, overnight) and probed again with anti-rabbit IgG fAB2 Alexa Fluor (R) 647 Molecular Probes (1:500, Cell Signaling Technology; room temperature, 1 h), both in PBS supplemented with 1% BSA and 0.3% Triton X-100. Finally, nuclei were stained with DAPI (2 μg/mL, room temperature, 5 min; Sigma, St. Louis, Missouri, USA). Five cells were randomly selected from 3 independent experiments for calculating the percentage of nuclear localization using Image J (National Institutes of Health, New York, USA). And then calculate the relative nuclear localization using percentage of nuclear localization of experimental group relative to counterparts of that in control group.
Additionally, after permeation with Triton X-100, some samples were stained for F-actin by treatment with 20 μg/mL rhodamine-conjugated phalloidin (room temperature, 40 min; Sigma, St. Louis, Missouri, USA) and stained with DAPI. All samples were observed under a fluorescence microscope (Vert.A1, Car Zeiss AG, Jena, Germany) and a confocal microscope (*/A1R+, Nikon, Tokyo, Japan).
MC-3T3E1 cells or MSCs were fixed in 95% ethanol (20 min), incubated in 0.1% Alizarin red (Solarbio, Beijing, China) for 20 min, and then observed under a stereo microscope (SMZ7 45, Nikon, Tokyo, Japan).
Cell migration analysis and survival rate analysis
High-glucose DMEM (600 μL) supplemented with 10% FBS was added to the lower chamber of Transwell (8 μM bore diameter; Corning); 100 μL of serum-free DMEM containing 1 × 105 MG-63 cells were added to the upper chamber. Subsequently, 10 μL of 1% BSA was added to the upper chamber to maintain an equivalent osmotic pressure. After incubation for 48 h, the upper chamber was treated with 4% paraformaldehyde for 20 min, stained with 0.1% crystal violet solution for 20 min, and observed under a microscope (Vert.A1, Car Zeiss AG, Jena, Germany). Five fields of view were randomly captured for cell counting. MSCs and MG-63 cells survival rate analyses were carried out according to the manufacturer instruction of Cell Counting Kit-8 (CCK8, Dojindo, Kumamoto, Kyushu, Japan).
Western blot and nuclear protein extraction
Cells were lysed with RIPA buffer (Solarbio, Beijing, China). Lysed samples were separated by SDS-PAGE using an 10% bis-acrylamide gel and then transferred to PVDF membrane. Membranes were blocked with 1× TBST/5% non-fat dry milk for 40 min and incubated in 1× TBST/5% non-fat dry milk containing YAP XR (R) rabbit mAb (1:1000, 4°C), Rb mAB to β-catenin (1:5000, 4°C), Mo a beta-actin (1:5000; Bioss, Beijing, China), Mo a Lamin B (1:1000, Bioss), Runx2 (F12, 1:200, Santa Cruz, California, USA), ALP (B10, 1:200, Santa Cruz), OCN (D11, 1:200, Santa Cruz) and OPN (Akm2A1, 1:200, Santa Cruze) overnight. Nuclear protein extraction was carried out according to the manufacturer instruction of Nuclear Protein Extraction Kit (Solarbio, Beijing, China).
Characterization of MSCs were determined by flow cytometry with FITC-, APC- or PE-conjugated anti-CD45, CD29, CD44, CD90, CD11B and CD31 (All from Invitrogen, California, USA). MSCs were analyzed using an EPICS-ALTRA flow cytometry (Beckman Coulter, California, USA).
Data from three independent experiments were analyzed by independent t-test using GraphPad Prism 7 (GraphPad Prism 7, Graph Pad, San Diego, CA, USA). A P-value < 0.05 was considered statistically significant.
Effects of Seliciclib on osteoblastic differentiation and survival rate of MSCs
On days 1, 5, and 7, the expression of Runx2 was significantly higher in the Seliciclib-treated group than in the control group. On day 3, the difference between the two groups was negligible. The expression of ALP was significantly higher in the Seliciclib-treated group (vs. control) on days 1, 3 and 7. On day 5, the difference was minimal. The expression of OCN was significantly higher in the Seliciclib-treated group on days 1, 5, and 7, and was significantly lower in that group (both vs. control) on day 3. The expression of OPN was significantly higher in the Seliciclib-treated group than in the control on days 1 and 7, but it was significantly lower on days 3 and 5. Overall, the relative expression of four genes (i.e., Seliciclib-treated/control) generally increased between days 1–7 (Fig. 1A). WB showed the same tendence that expression of Runx2, ALP, OCN, and OPN in the Seliciclib-treated group were increased to 1.4, 1.78, 1.51, and 1.3 times, respectively (Fig. 1D). Mineralization is an indicator of osteoblastic differentiation. Alizarin red staining found that, after cultured in the osteogenic induction medium for 14 days, the Seliciclib-treated and siCDK5 group stained more intensely than did the control group. Both MC-3T3E1 and MSCs showed the same tendence (Fig. 1B). The survival rates of MSCs were 98.04%, 98.55%, 98.22%, 98.74% on days 1, 3, 7, and 10, respectively when 3 μM Seliciclib were used. When it came to 6 and 9 μM Seliciclib, the survival rate increased with the time extension. The survival rates of 6 μM Seliciclib-treated group were 91.32%, 94%, 94.18%, and 97.60% on days 1, 3, 7, and 10, respectively. The counterparts of 9 μM Seliciclib-treated group were 80.01%, 82.00%, 91.29% and 95.93% (Fig. 1C). The survival rates all were over 90% on day 7 and all were over 95.00% on day 10 with 3, 6, 9 μM Seliciclib treated, suggesting a low toxicity of Seliciclib to MSCs in the concentration range of 3 to 9 μM Seliciclib.
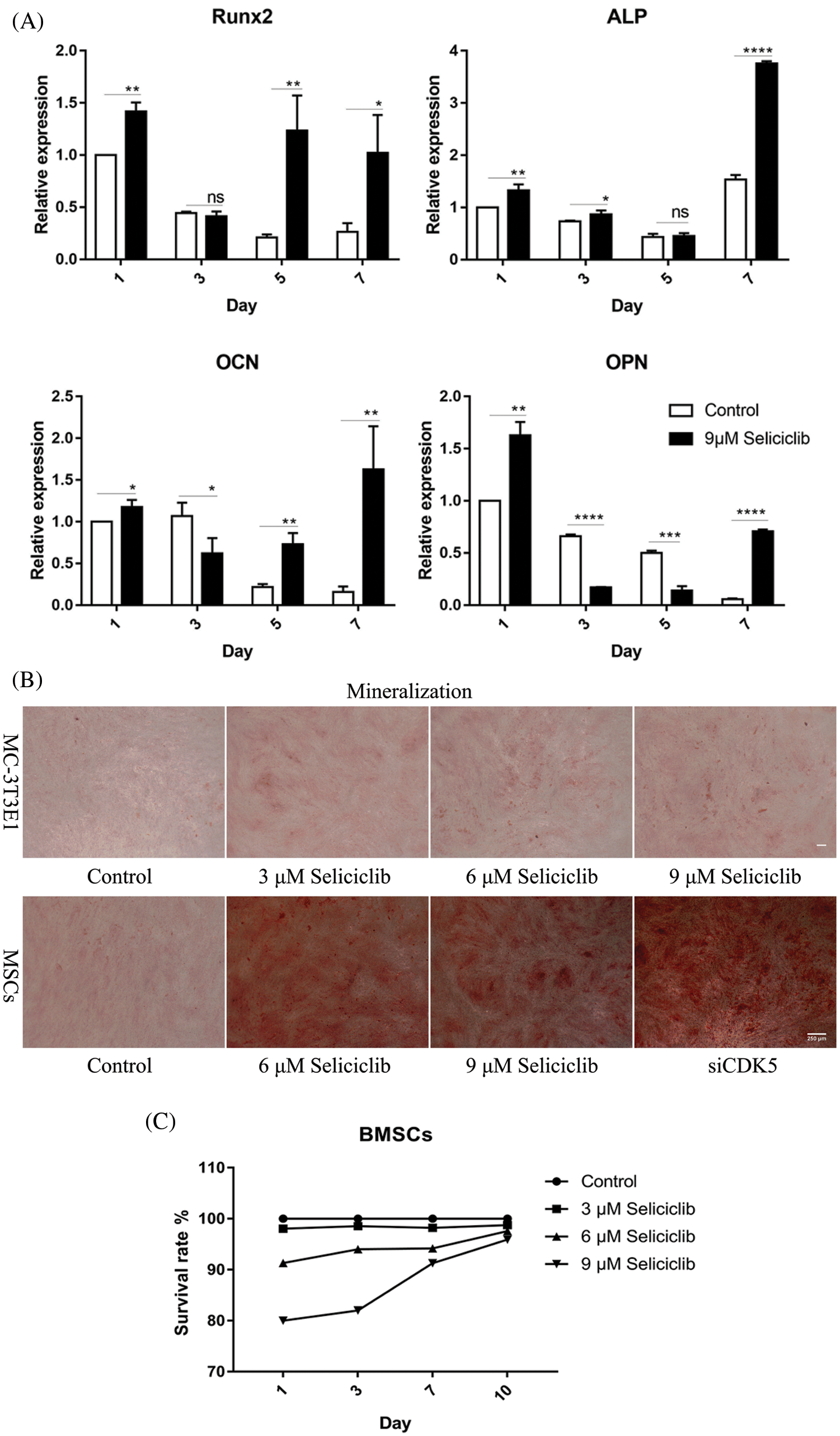
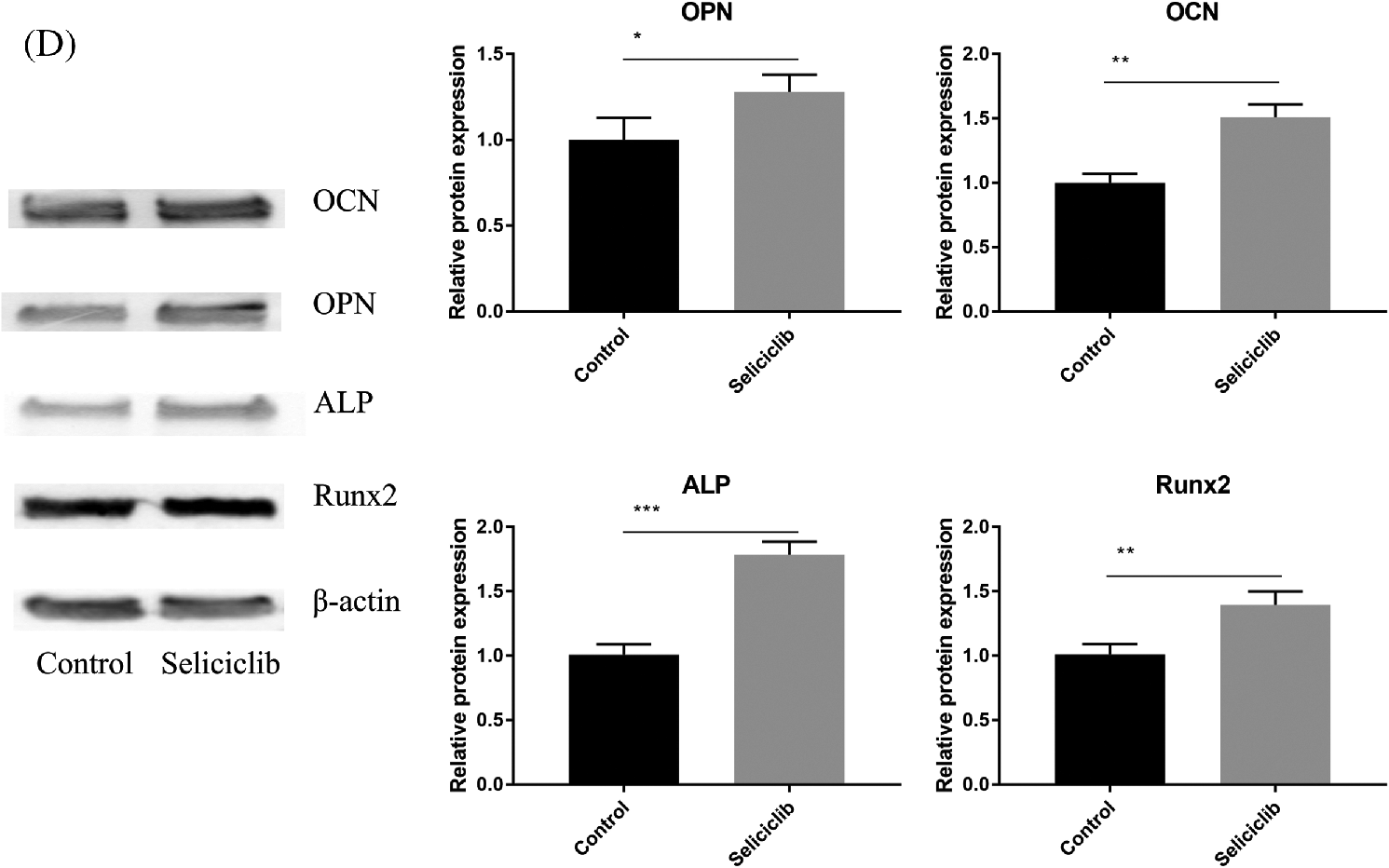
Figure 1: The expression of 4 osteoblastic differentiation marker genes of MSCs of rats, mineralization of MC-3T3E1 and survival rate of MSCs after Seliciclib treated. (A) The expression of Runx2, ALP, OCN and OPN (left to right) between Seliciclib (9 μM) treated group (black) and control (white) from day 1 to 7. Fold changes are relative to control of day 1. (B) Seliciclib and siCDK5 promote the mineralization of MC-3T3E1 and MSCs at different concentrations of 3, 6 and 9 μM. Scale bar: 200 μm. (C) The survival rate of MSCs after 3, 6, and 9 μM Seliciclib treated. (D) WB shows the expression of Runx2, ALP, OCN, and OPN after Seliciclib treated. *P < 0.05.
Effects of CDK5 inhibition on F-actin formation, nuclear localization of YAP and β-catenin, and RMRP RNA expression
Fluorescence microscopy revealed that, compared with the control group, the F-actin fibers formed in the Seliciclib-treated and siCDK5 group appeared longer and more well-formed (Fig. 2A). Moreover, F-actin fibers in the Seliciclib-treated group exhibited clearer, whereas F-actin fibers were vague in the control group. Confocal microscopy showed that, in the Seliciclib-treated group, β-catenin was dominantly distributed in the nucleus (Fig. 2B). In comparison, in the control group, it appeared to be homogeneously distributed in the cell. Same tendencies were observed for YAP in the Seliciclib-treated and control groups (Fig. 2C). Five randomly selected cells from three independent experiments were used to calculate the relative nuclear localization. Bar graphs showed that, compared with the control, nuclear localization of β-catenin and YAP in the Seliciclib-treated group increased about 1.9 and 2.3 times (Fig. 2E). WB showed that nuclear localization of YAP and β-catenin of Seliciclib-treated groups increased to 1.55 and 1.60 times (vs. control) respectively. While cytoplasm localization of YAP and β-catenin decreased to 0.97 and 1.9 times (vs. control), respectively (Fig. 2G). RT-qPCR found that, compared with the control, the expression of RMRP RNA in the Seliciclib-treated group was significantly lower on day 1 but was significantly higher on days 3, 5, and 7 (Fig. 2D). Furthermore, the intragroup difference in the expression of RMRP RNA increased between days 1–7. The siCDK5 decreased the expression of CDK5 to 0.41 times compared to the control group (Fig. 2F).
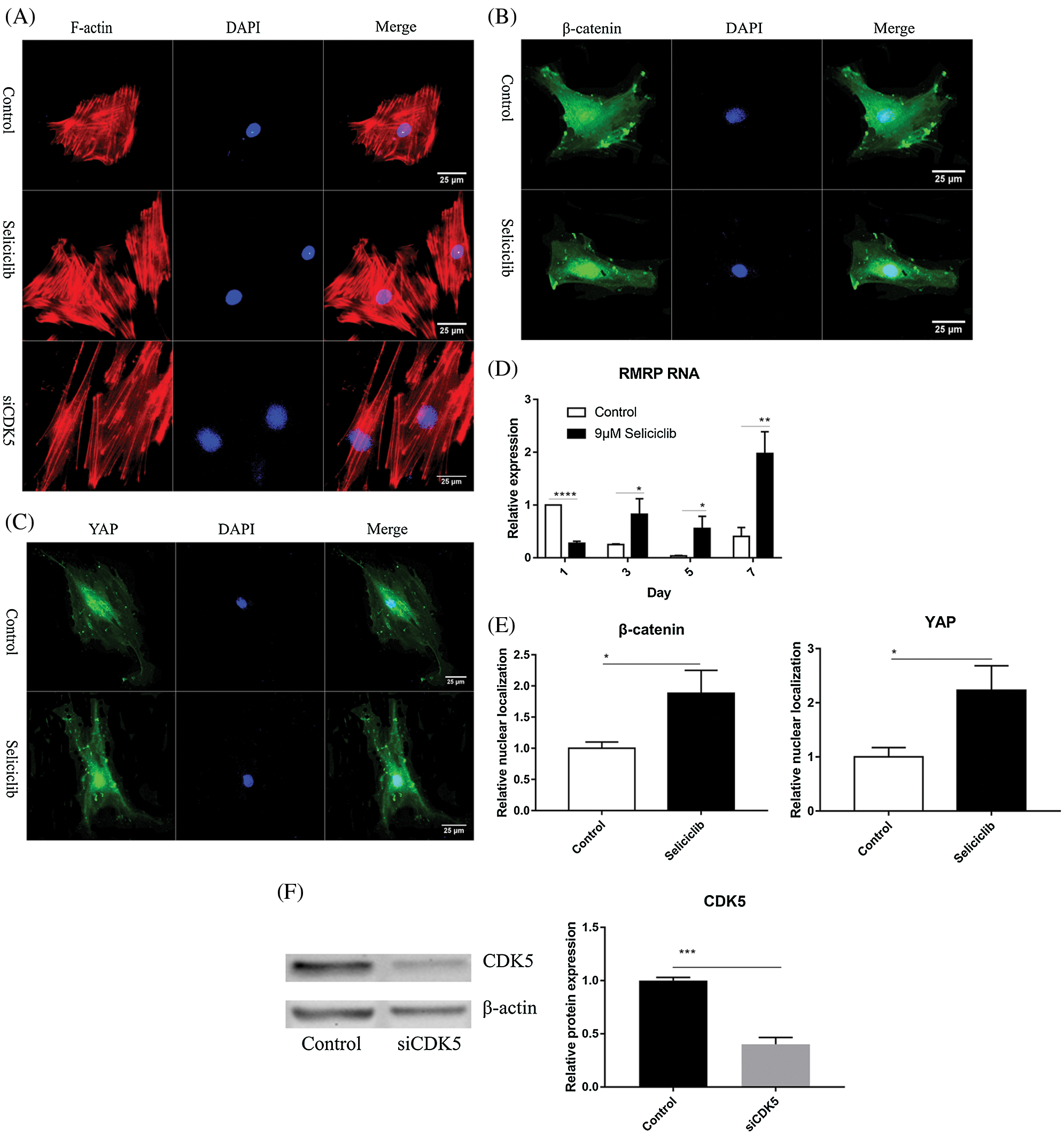
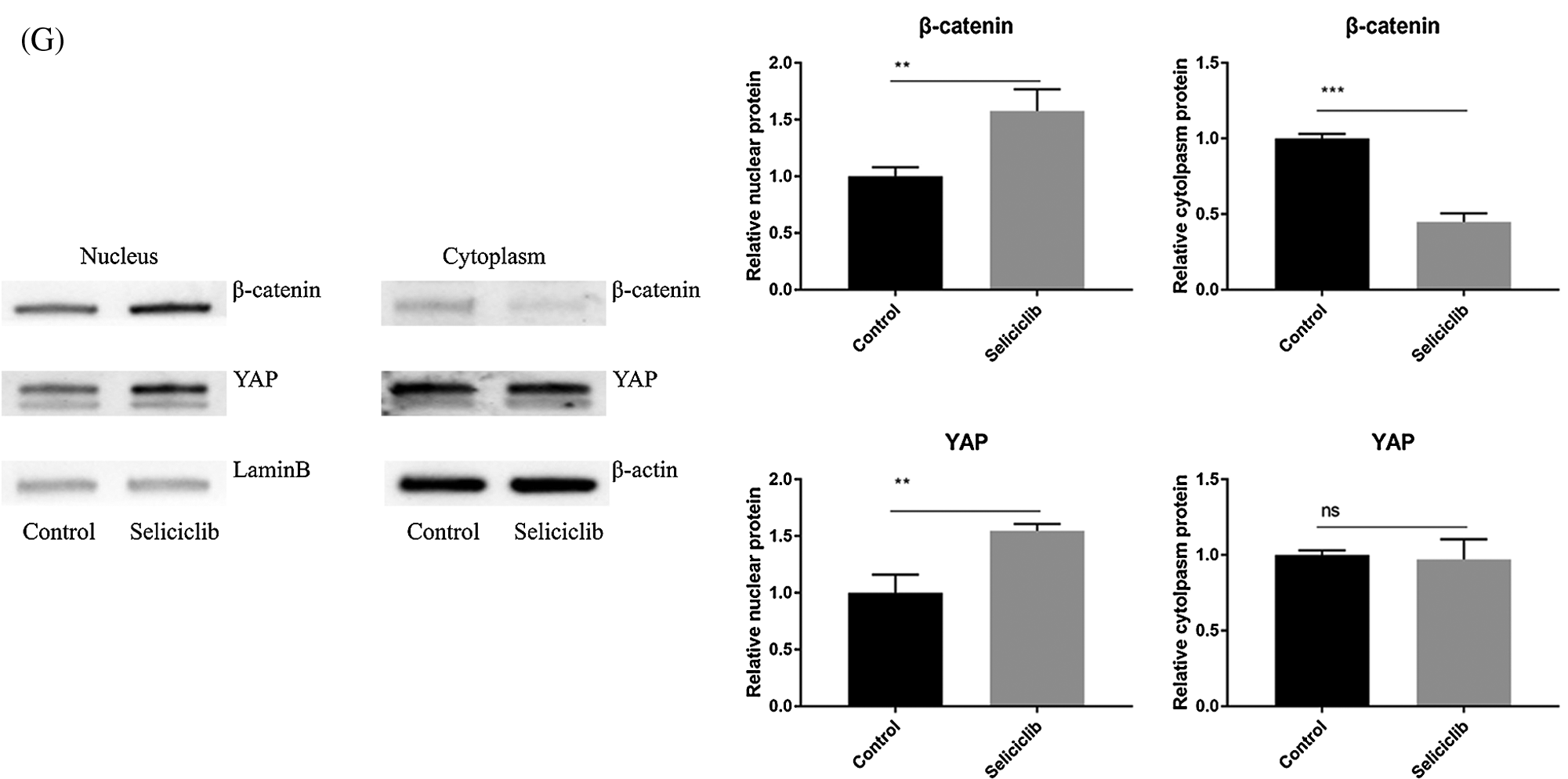
Figure 2: The F-actin development, YAP and β-catenin nuclear localization and the expression of RMRP RNA after Seliciclib-treated. (A) The F-actin staining shows the development of Seliciclib (9 μM)–treated (middle), siCDK5-treated group (below) and control group (above). (B and C) Immunofluorescent staining of β-catenin and YAP after Seliciclib (9 μM) treated (below) and control group (above). (D) RMRP RNA expression of Seliciclib-treated group (black) and control (white) from day 1 to day 7. Fold changes are relative to control of day 1. (E) Relative nuclear localization of YAP and β-catenin between Seliciclib-treated and control group. (F) WB shows the siCDK5 Silencing efficiency. (G) WB shows nuclear and cytoplasm localization of YAP and β-catenin. *p < 0.05.
Effects of F-actin inhibition on nuclear localization of YAP and β-catenin and osteoblastic differentiation
In the control group (treated with Seliciclib alone), many well-formed F-actin were observed (Fig. 3A). In comparison, in the group simultaneously treated with Blebbistatin and Seliciclib, actin was distributed throughout the cell with the formation of a small number of fibers. Additionally, in the control group, YAP and β-catenin exhibited evident nuclear localization (Figs. 3B and 3C), whereas such trends were not found in the simultaneously treated group. Five randomly selected cells were used to calculate the relative nuclear localization. The bar graph showed that, compared with Seliciclib-treated group, nuclear localization of β-catenin in Seliciclib+Ble-treated group and Ble-treated group decreased to about 0.6 and 0.7 times, respectively (Fig. 3D). Counterparts of YAP in Seliciclib+Ble-treated and Ble-treated groups decreased to about 0.5 and 0.48 times, respectively (Fig. 3D). WB showed that, compared to Seliciclib-treated group, nuclear localization of β-catenin in Seliciclib+Ble-treated and Ble-treated groups decreased to 0.54 and 0.56 times, respectively (Fig. 3F). Counterparts of YAP in Seliciclib+Ble-treated and Ble-treated groups decreased to 0.9 and 0.834 times, respctively (Fig. 3F). Cytoplasm localization of YAP in Seliciclib+Ble-treated and Ble-treated groups did not change significantly (vs. control). Counterparts of β-catenin in Seliciclib+Ble-treated and Ble-treated groups increased to 1.66 and 1.9, respectively (Fig. 3F). Interestingly, MSCs in the control group appeared spindle-like, whereas those in the simultaneously treated and only Ble-treated group were branched, resembling neurons. The expression levels of RMRP RNA, Runx2, and ALP in the simultaneously treated group were significantly lower than those in the control group (Fig. 3E), indicating that the down- regulation of CDK5 promoted RMRP RNA expression and osteoblastic differentiation at least via facilitating the development of F-actin.
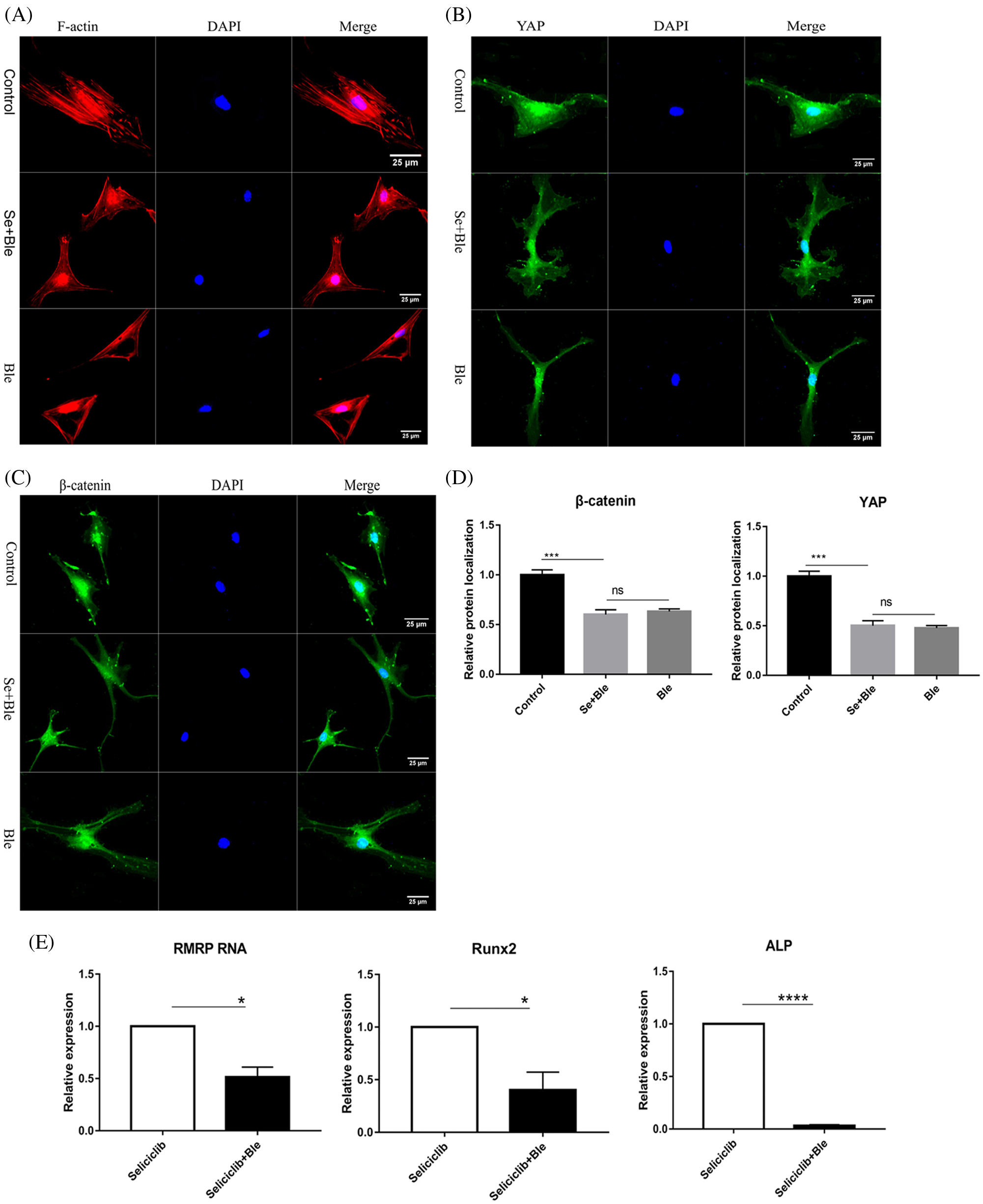
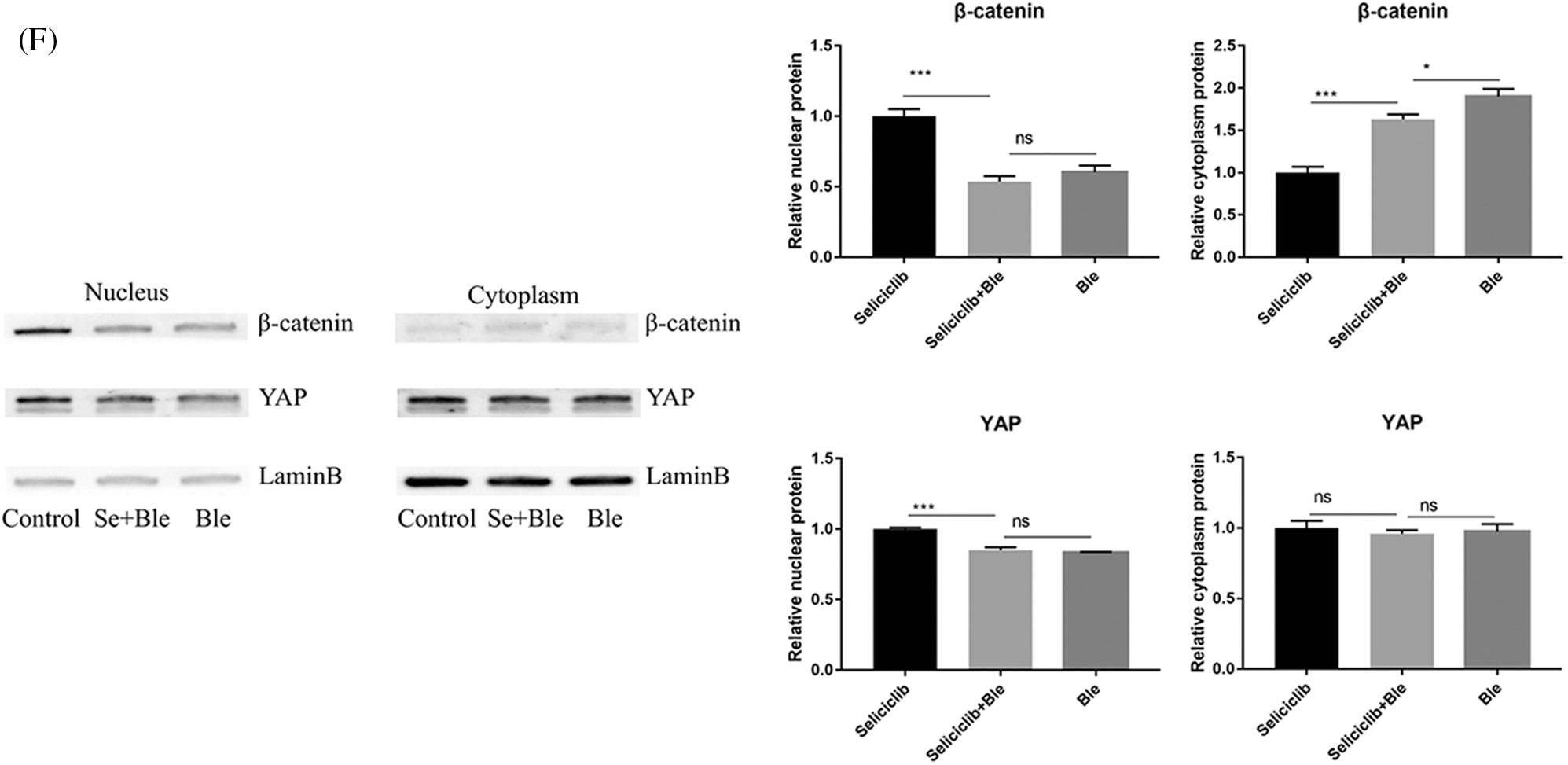
Figure 3: YAP and β-catenin nuclear localization and expression of RMRP RNA, Runx2 and ALP after F-actin impressed. (A) F-actin development of Seliciclib (9 μM)-treated group (above) Seliciclib (9 μM)/Ble (5 μM)-treated group (middle) and Ble(5 μM)-treated group. (B and C) Immunofluorescent staining of YAP and β-catenin shows their nuclear localization between Seliciclib (9 μM)-treated group (above) and Seliciclib (9 μM)/Ble (5 μM)-treated group (blow). (D) Relative nuclear localization of YAP and β-catenin of Seliciclib-treated, Seliciclib+Ble treated group and Ble-treated group. (E) Expression of RMRP RNA, Runx2 and ALP of Seliciclib (9 μM)-treated group (white) and Seliciclib (9 μM)/Ble (5 μM)-treated group (black). Fold changes are relative to Seliciclib-treated group. (F) WB shows nuclear and cytoplasm localization of YAP and β-catenin of Seliciclib-treated group, Seliciclib+Ble treated group and Ble-treated group. *p < 0.05.
Effect of Seliciclib on migration of MG-63 cells and spreading of MSCs
Crystal violet staining showed that, Seliciclib treatment reduced the migration of MG-63 cells in a concentration-dependent manner (Fig. 4A). Analysis of 5 randomly selected fields of view found that, compared with the control, the number of migratory cells decreased 25%, 45%, and 55% at 3, 6, and 9 µM, respectively (Fig. 4B). Survival rates of MG-63 of 3 µM Seliciclib-treated group were 97.95%, 98.69%, 99.07% and 98.86% on days 1, 4, 7, and 10, respectively. Counterparts of MG-63 of 6 µM Seliciclib-treated group were 93.74%, 95.87%, 98.21% and 98.43% on days 1, 4, 7, and 10, respectively. When it came to 9 µM Seliciclib, survival rates were 85.78%, 92.57%, 96.70% and 97.15% on days 1, 4, 7, and 10 (Fig. 4C). The difference between any two groups was statistically significant. F-actin and DAPI staining showed that cell spreading compared with the control group, in the Seliciclib-treated group was less and the cell shape became small and spindle-like (Fig. 4D). Cell nuclei, compared with the control group, also showed the same change with smaller and spindle-like shape in the Seliciclib-treated group.
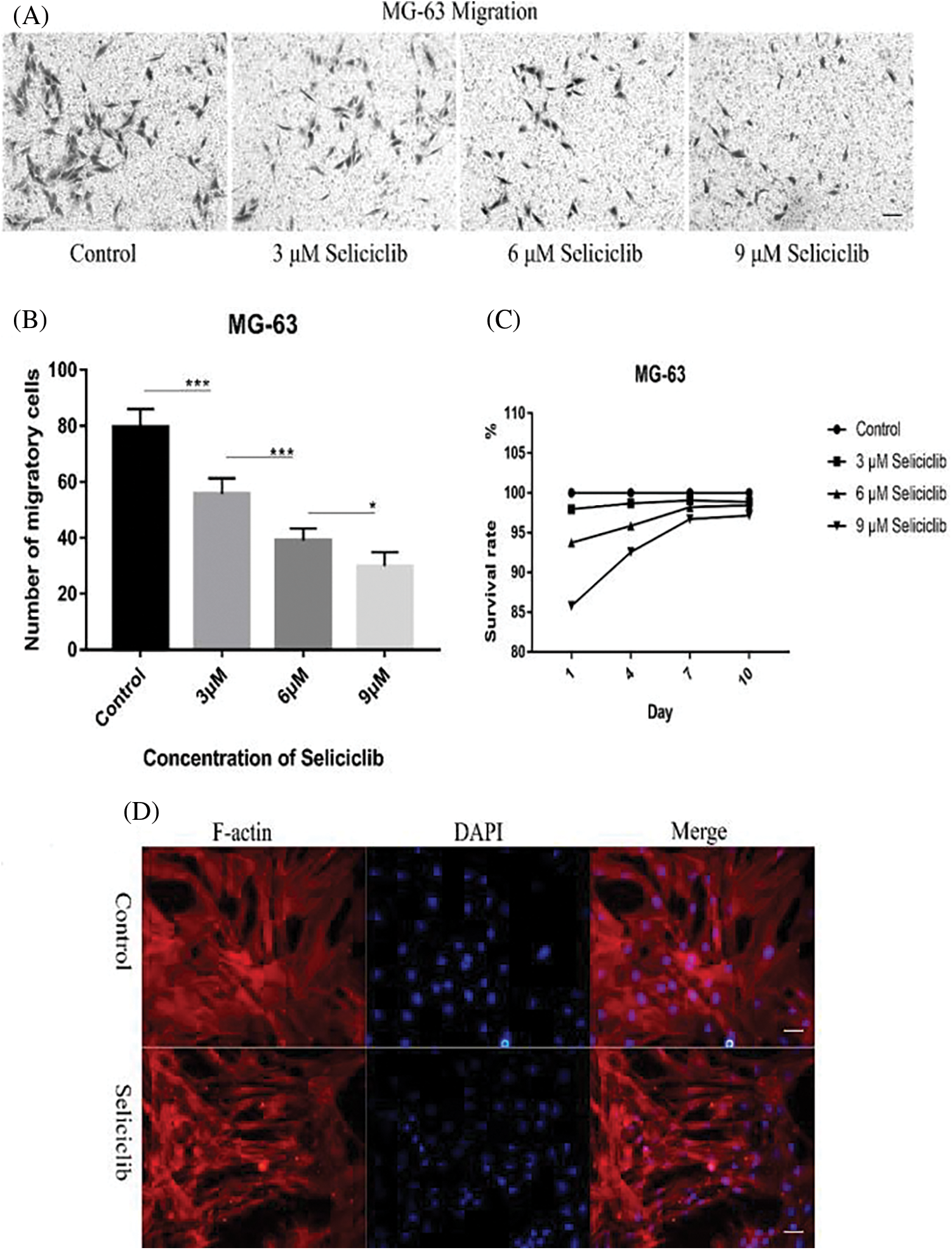
Figure 4: Migration of MG-63 cells and cell spreading of MSCs after Seliciclib treated. (A) Crystal violet staining shows the migration of MG-63 of Seliciclib (3 µM, 6 µM and 9 µM)-treated group (three on the right) and control (left). Scal bar 100 µm. (B) Bar graph shows average migratory cells from Five randomly selected areas of different concentrations of Seliciclib (0, 3 µM, 6 µM and 9 µM from left to right). Date represents mean ± SEM. (C) Survival rates of MG-63 after 3, 6 and 9 µM Seliciclib treated. (D) F-actin staining shows MSCs spreading between control and Seliciclib treated group. Scal bar 200 µm. *p < 0.05.
Here we identify CDK5 as a negative regulator in the osteoblastic differentiation. And CDK5 inhibition promotes the osteoblastic differentiation by increasing the polymerization of F-actin which then promotes the nuclear localization of YAP and β-catenin, as well as the expression of RMRP RNA, Runx2 and ALP. CDK5 has been proved to directly inhibit β-catenin by combining with β-catenin. Here we further proved that CDK5 negatively regulates nuclear localization of β-catenin through F-actin dynamics. Moreover, we found CDK5 inhibition suppressed the migration of MG-63.
Inhibition of CDK5 may enhance F-actin development via two pathways. First, inhibited CDK5 reduces the phosphorylation of Pak1 on T212, resulting in Rac1 activation (Rashid et al., 2001; Zhong et al., 2003). Additionally, CDK5 inhibition may reduce the phosphorylation of DLC1 on S120, S205, S422, and S509 sites, leading to its inactivation (Tripathi et al., 2014). Because DLC1 is a GAP that can down-regulate the activation of RhoA (Wong et al., 2005; Wong et al., 2008), CDK5 inhibition activates RhoA. Combined with myosin II, F-actin forms stress fibers. In the early stage of cell adhesion, activated Rac1 promotes the formation of the leading edge through generating a flat lamella. Subsequently, RhoA is activated to facilitate the development (i.e., formation-thickening) of stress fibers and generation of stress via RhoA-ROCK-RLCs signaling (Lee et al., 2013; Woods et al., 2005). Generation of stress leads to cell spreading and shape change. Depolymerization of F-actin can cause the incomplete of stress fiber. This will lead to a cell spreading.
In the present study, CDK5 inhibition created thicker and longer F-actin (compared with control) (Fig. 1A). Moreover, the cell spreading was less, and cell shape became small and spindle-like in the Seliciclib-treated group (with 86 cells covering less area than the control group with 58 cells) (Fig. 4C), also suggesting a tendence of aggregation of MSCs in Seliciclib-treated group. And MSCs nucleus and MG-63 showed the same change (Fig. 4A). When the F-actin almost totally became actin, MSCs spreading increased and became branched and similar to neurons (Fig. 3C). These suggest that, in the Seliciclib-treated group, F-actin may be formed primarily via the second pathway.
RMRP RNA, a long non-coding RNA, is a component of small nucleolar ribonucleoprotein (snoRNP) particle named RNase MRP. Mutations of RMRP RNA at different sites are sources of skeletal dysplasias such as cartilage-hair hypoplasia (Thiel and Rauch, 2011; Young and Bannasch, 2008). Reduced RMRP RNA can downregulate Runx2 and ALP, and impair mineralization of bone-related cells, indicating RMRP RNA to be a positive regulator of osteoblastic differentiation (Steinbusch et al., 2017). However, molecular mechanisms underlying how RMRP RNA promotes osteoblastic differentiation are unknown. Beta-catenin and YAP promote the expression of RMRP RNA, both separately and synergistically (Jinjoo and Sunjoo, 2015). CDK5/p35 complex can combine with β-catenin and then reduce N-cadherin-mediated and calcium-dependent cell-cell adhesion (Kwon et al., 2000). So CDK5 inhibition may also blocks the migration of MG-63 by releasing β-catenin and then increases the cell-cell adhesion. Additionally, CDK5/p35 complex combines with, and thus inactivates, β-catenin. In the current study, CDK5 inhibition by Seliciclib treatment increased nuclear localization of YAP and β-catenin and up-regulated RMRP RNA expression (vs. control). Additionally, when CDK5 and F-actin were both inhibited, the nuclear localization of β-catenin was reduced (Figs. 3B and 3E), indicating that β-catenin nuclear localization is F-actin dependent. YAP plays an important role in maintaining the nuclear localization of β-catenin during osteoblastic differentiation (Pan et al., 2018). Thus, after CDK5 inhibition, β-catenin is released. Meanwhile, nuclear localization of β-catenin might be enhanced by the increased nuclear localization of YAP. And these indicate CDK5 is an upstream and negative regulator of RMRP RNA (graphical abstract).
After surgical resection of osteosarcoma, osteosarcoma cells are prone to recurrence and metastasis, and bone repair is also required. CDK5 inhibitor Seliciclib can promote osteoblastic differentiation. The inhibition of MG-63 cell migration elevated when Seliciclib concentration was increased from 3 to 9 μM. This means that Seliciclib has a significant future in the treatment of osteosarcoma after surgical resection. The amount of Seliciclib used to kill cancer in clinical trials is far greater than the amount used to promote osteoblastic differentiation, and the target of Seliciclib used to kill cancer is mainly CDK1/2 or ERK1/2 (Iurisci et al., 2006). The IC50 of Seliciclib for CDK5/P35, CDC2/cyclinB, CDK2/cyclinA, ERK1 and ERk2 in vitro is 0.16, 0.65, 0.7, 34, and 14 μM, respectively (Iurisci et al., 2006). CDC2/cyclinB and CDK2/cyclinA are not involved in the F-actin dynamics. And the 3, 6, and 9 μM of Seliciclib used in promoting osteoblastic differentiation showed a low toxicity (Fig. 1C). Flow cytometry analysis showed that positive rates of CD90, CD29 and CD44 of MSCs were 98.8%, 98.5%, and 84.6%, respectively. Positive rates of CD45, CD11B and CD31 of MSCs were 0.11%, 1.49%, and 0.66%, respectively (Suppl. Fig. S1).
Although these results reveal that, the downregulation of CDK5 promotes the osteoblastic differentiation of MSCs partially by enhancing the development of F-actin, the mechanisms responsible for CDK5 down-regulation are unknown and needed to be identified. CDK5, RhoA, YAP, β-catenin and RMRP RNA play important roles in the cancer cell migration, invasion, proliferation and morphological change of tumor cells (Feldmann et al., 2008; Morin, 1999; Wang et al., 2019; Do et al., 2019; Wong et al., 2008). Results of those study suggest a link between CDK5, RhoA, YAP, β-catenin, and RMRP RNA and new insights for understanding the molecular mechanisms of some carcinogenesis.
Author Contribution: The authors confirm contribution to the paper as follows: study conception and design: Hong Fu; data collection: Hong Fu, Haoyu Zhao; analysis and interpretation of results: Hong Fu, Yali Yang, Siyu Wang; draft manuscript preparation: Hong Fu, Ke Duan; project supervision: Tailin Guo. All authors reviewed the results and approved the final version of the manuscript.
Ethics Approval: Scientific Research Ethics Committee of General Hospital of Western Theater Command has approved and consented the rat we used to extract MSCs on March 13, 2019.
Availability of Data and Materials: All data generated or analyzed during this study are included in this published article (and its supplementary information files).
Funding Statement: This work was supported by the National Natural Science Foundation of China under Grant (32071343); Fundamental Research Funds for the Central Universities under Grant (2682020ZT80); Sichuan Science and Technology Program under Grant (21YYJC3323).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Do KK, Eun KK, Da-Woon J, Jin K (2019). Cytoskeletal alteration modulates cancer cell invasion through rhoa-yap signaling in stromal fibroblasts. PLos One 14: e0214553. [Google Scholar]
Feldmann G, Mishra A, Strock C, Ball D, Maitra A, Nelkin B (2008). CDK5 inhibition diminishes pancreatic cancer tumorigenesis and metastasis. Pancreas 37: 469–470. [Google Scholar]
Gilmore EC, Ohshima T, Goffinet AM, Kulkarni AB, Herrup K (1998). Cyclin-dependent kinase 5-deficient mice demonstrate novel developmental arrest in cerebral cortex. Journal of Neuroscience 18: 6370–6377. [Google Scholar]
Honjo T, Kubota S, Kamioka H, Sugawara Y, Ishihara Y et al. (2012). Promotion of Ccn2 expression and osteoblastic differentiation by actin polymerization, which is induced by laminar fluid flow stress. Journal of Cell Communication and Signaling 6: 225–232. [Google Scholar]
Huang C, Rajfur Z, Yousefi N, Chen Z, Jacobson K, Ginsberg MH (2009). Talin phosphorylation by Cdk5 regulates Smurf1-mediated talin head ubiquitylation and cell migration. Nature Cell Biology 11: 624–630. [Google Scholar]
Iurisci I, Filipski E, Reinhardt J, Bach S, Gianella-Borradori A et al. (2006). Improved tumor control through circadian clock induction by Seliciclib, a cyclin-dependent kinase inhibitor. Cancer Research 66: 10720–10728. [Google Scholar]
Jinjoo P, Sunjoo J (2015). Wnt activated β-catenin and YAP proteins enhance the expression of non-coding RNA component of RNase MRP in colon cancer cells. OncoTargets and Therapy 6: 34658–34668. [Google Scholar]
Kager L, Zoubek A, PöTschger U, Kastner U, Flege S et al. (2003). Primary metastatic osteosarcoma: Presentation and outcome of patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. Journal of Clinical Oncology 21: 2011–2018. [Google Scholar]
Kim MH, Kim J, Hong H, Lee SH, Lee JK et al. (2016). Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP/TAZ activation. EMBO Journal 35: 462–478. [Google Scholar]
Kim TY, Lee JW, Kim HP, Jong HS, Kim TY et al. (2007). DLC-1, a GTPase-activating protein for Rho, is associated with cell proliferation, morphology, and migration in human hepatocellular carcinoma. Biochemical and Biophysical Research Communications 355: 72–77. [Google Scholar]
Kwon YT, Gupta A, Zhou Y, Nikolic M, Tsai LH (2000). Regulation of N-cadherin-mediated adhesion by the p35-Cdk5 kinase. Current Biology 10: 363–372. [Google Scholar]
Lee JS, Kim YH, Lee JH (2013). Involvement of RhoA/ROCK signaling for alteration of stress fiber via lymphotoxin β receptor stimulation in fibroblastic reticular cell isolated from lymph node. Animal Cells and Systems 17: 421–428. [Google Scholar]
Li Q, Liu X, Zhang M, Ye G, Qiao Q et al. (2010). Characterization of a novel human CDK5 splicing variant that inhibits Wnt/β-catenin signaling. Molecular Biology Reports 37: 2415–2421. [Google Scholar]
Meyers PA, Gorlick R (1997). Osteosarcoma. Pediatric Clinics of North America 44: 973–989. [Google Scholar]
Morin PJ (1999). β-catenin signaling and cancer. Bioessays 21: 1021–1030. [Google Scholar]
Pan JX, Xiong L, Zhao K, Zeng P, Wang B et al. (2018). YAP promotes osteogenesis and suppresses adipogenic differentiation by regulating β-catenin signaling. Bone Research 6: 18. [Google Scholar]
Petrosiute A, Huang A, Nthale J, Myers J, Davis K (2011). Modulation of CDK5 affects local invasion and metastasis of medulloblastoma. Pediatric Blood & Cancer 56: 961 (Poster 309). [Google Scholar]
Rashid T, Banerjee M, Nikolic M (2001). Phosphorylation of Pak1 by the p35/Cdk5 kinase affects neuronal morphology. Journal of Biological Chemistry 276: 49043–49052. [Google Scholar]
Steinbusch MM, Caron MM, Surtel DA, Friedrich F, Lausch E et al. (2017). Expression of RMRP RNA is regulated in chondrocyte hypertrophy and determines chondrogenic differentiation. Scientific Reports 7: 6440. [Google Scholar]
Strock CJ, Park JI, Nakakura EK, Bova GS, Isaacs JT et al. (2006). Cyclin-dependent kinase 5 activity controls cell motility and metastatic potential of prostate cancer cells. Cancer Research 66: 7509–7515. [Google Scholar]
Thiel CT, Rauch A (2011). The molecular basis of the cartilage-hair hypoplasia-anauxetic dysplasia spectrum. Best Practice & Research Clinical Endocrinology & Metabolism 25: 131–142. [Google Scholar]
Tripathi BK, Qian X, Mertins P, Wang D, Papageorge AG et al. (2014). CDK5 is a major regulator of the tumor suppressor DLC1. Journal of Cell Biology 207: 627–642. [Google Scholar]
Tripathi BK, Zelenka PS (2009). Cdk5-dependent regulation of Rho activity, cytoskeletal contraction, and epithelial cell migration via suppression of Src and p190RhoGAP. Molecular and Cellular Biology 29: 6488–6499. [Google Scholar]
Tsai LH, Takahashi T, Caviness VS, Harlow E (1993). Activity and expression pattern of cyclin-dependent kinase 5 in the embryonic mouse nervous system. Development 119: 1029–1040. [Google Scholar]
Wang J, Xiao T, Zhao M (2019). MicroRNA-675 directly targets MAPK1 to suppress the oncogenicity of papillary thyroid cancer and is sponged by long non-coding RNA RMRP. OncoTargets and Therapy 12: 7307. [Google Scholar]
Wong CC, Wong CM, Ko FC, Chan LK, Ching YP, Yam JW, Ng IO (2008). Deleted in liver cancer 1 (DLC1) negatively regulates Rho/ROCK/MLC pathway in hepatocellular carcinoma. PLoS One 3: e2779. [Google Scholar]
Wong CM, Yam JWP, Ching YP, Yau TO, Leung THY et al. (2005). Rho GTPase-activating protein deleted in liver cancer suppresses cell proliferation and invasion in hepatocellular carcinoma. Cancer Research 65: 8861–8868. [Google Scholar]
Woods A, Wang G, Beier F (2005). RhoA/ROCK signaling regulates Sox9 expression and actin organization during chondrogenesis. Journal of Biological Chemistry 280: 11626–11634. [Google Scholar]
Yan Y, Sun H, Gong Y, Yan Z, Zhang X et al. (2016). Mechanical strain promotes osteoblastic differentiation through integrin-β1-mediated β-catenin signaling. International Journal of Molecular Medicine 38: 594–600. [Google Scholar]
Young AE, Bannasch DL (2008). SNPs in the promoter regions of the canine RMRP and SHOX genes are not associated with canine chondrodysplasia. Animal Biotechnology 19: 1–5. [Google Scholar]
Zhong JL, Banerjee MD, Nikolic M (2003). Pak1 and its T212 phosphorylated form accumulate in neurones and epithelial cells of the developing rodent. Developmental Dynamics: An Official Publication of the American Association of Anatomists 228: 121–127. [Google Scholar]
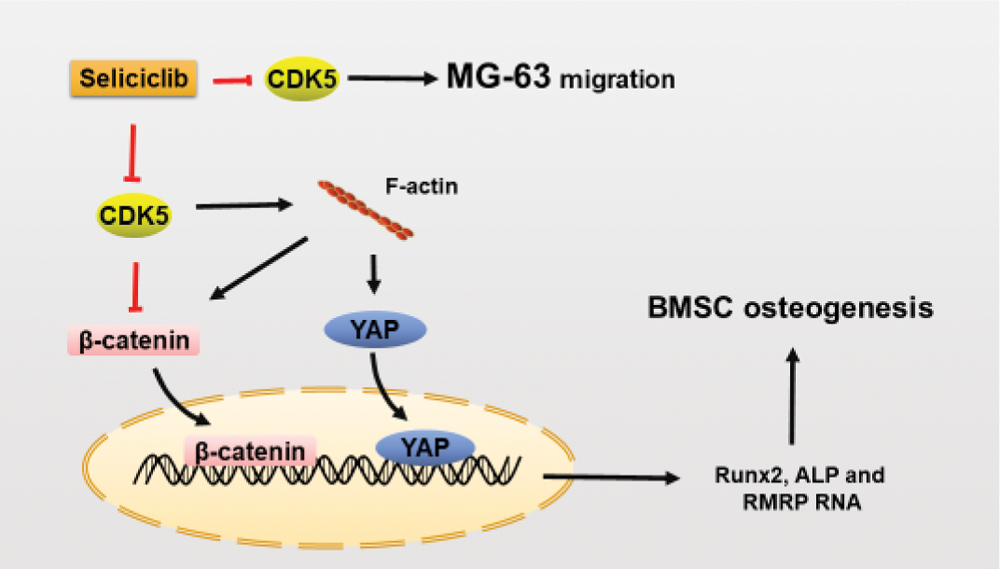
GRAPHICAL ABSTRACT: CDK5 inhibition promotes osteoblastic differentiation and blocks the migration of MG-63.
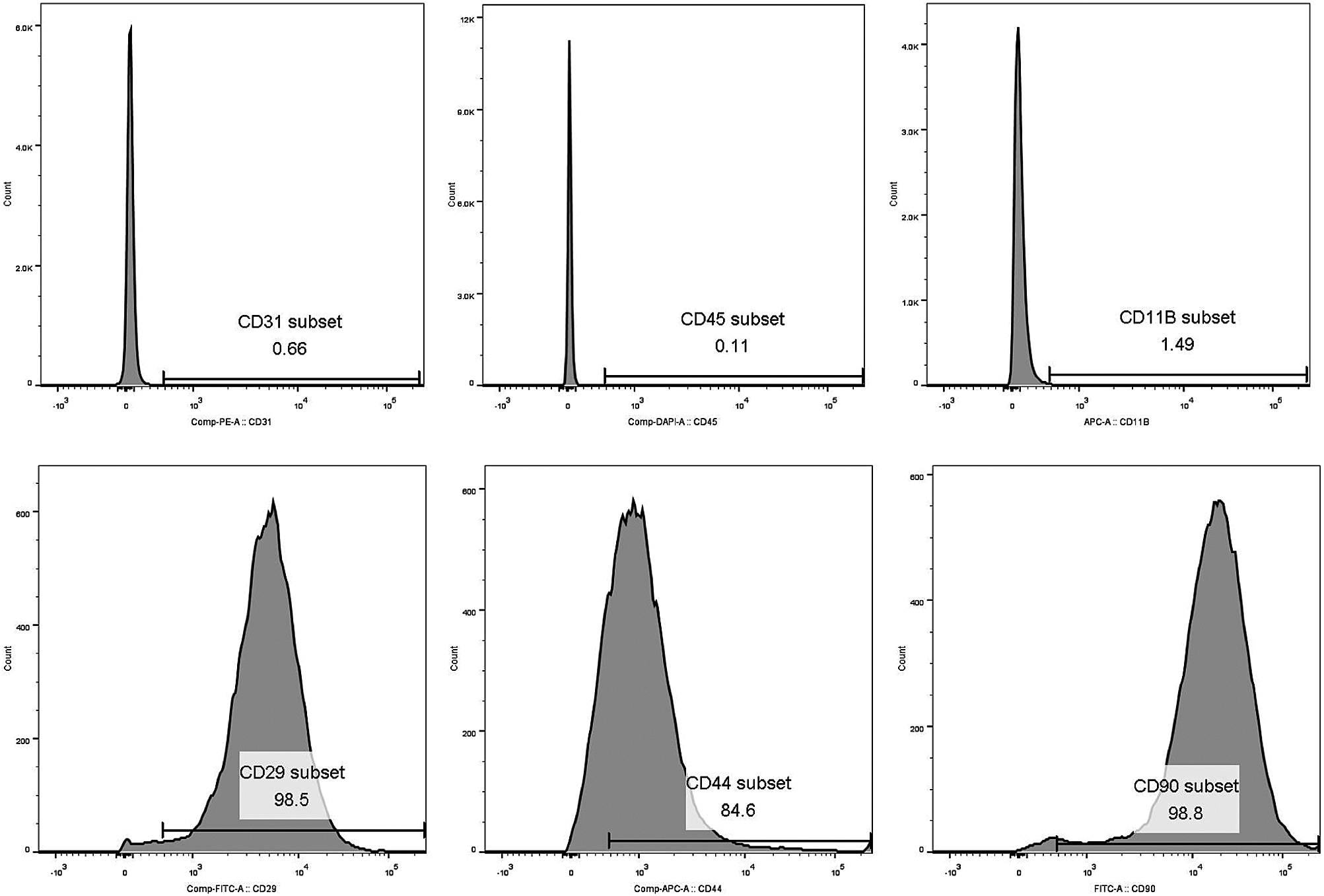
SUPPLEMENTARY FIGURE S1: BMSCs characteration. CD31, CD45 and CD11B characteration of MSCs showed negative results. CD90, CD29 and CD44 characteration of MSCs showed positive results.
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |