DOI:10.32604/biocell.2022.017538
| BIOCELL DOI:10.32604/biocell.2022.017538 | |
| Article |
INTS-MFS: A novel method to predict microRNA-disease associations by integrating network topology similarity and microRNA function similarity
1College of Information and Electronic Engineering, Hunan City University, Yiyang, 413000, China
2College of Computer Science and Electronic Engineering, Hunan University, Changsha, 410082, China
*Address correspondence to: Buwen Cao, cbwhj@126.com; Jiawei Luo, luojiawei@hnu.edu.cn
Received: 19 May 2021; Accepted: 24 June 2021
Abstract: Identifying associations between microRNAs (miRNAs) and diseases is very important to understand the occurrence and development of human diseases. However, these existing methods suffer from the following limitation: first, some disease-related miRNAs are obtained from the miRNA functional similarity networks consisting of heterogeneous data sources, i.e., disease similarity, protein interaction network, gene expression. Second, little approaches infer disease-related miRNAs depending on the network topological features without the functional similarity of miRNAs. In this paper, we develop a novel model of Integrating Network Topology Similarity and MicroRNA Function Similarity (INTS-MFS). The integrated miRNA similarities are calculated based on miRNA functional similarity and network topological characteristics. INTS-MFS obtained AUC of 0.872 based on five-fold cross-validation and was applied to three common human diseases in case studies. As a results, 30 out of top 30 predicted Prostatic Neoplasm-related miRNAs were included in the two databases of dbDEMC and PhenomiR2.0. 29 out of top 30 predicted Lung Neoplasm-related miRNAs and Breast Neoplasm-related miRNAs were included in dbDEMC, PhenomiR2.0 and experimental reports. Moreover, INTS-MFS found unknown association with hsa-mir-371a in breast cancer and lung cancer, which have not been reported. It provides biologists new clues for diagnosing breast and lung cancer.
Keywords: Disease-related miRNA; MiRNA-disease association; Functional similarity; Network topological similarity
MicroRNAs (miRNAs) are approximately 22-nucleotide noncoding RNAs, act as an important regulator involved in posttranscriptional regulation of gene expression (Bartel, 2004). Recently, increasing evidence has showed that the development and progression of various complex human diseases result from the mutation and functional disorders (Alvarez-Garcia and Miska, 2005; Lynam-Lennon et al., 2009; Wu et al., 2021). Furthermore, more and more studies have indicated that miRNAs could influence multiple stages of the biological processes (Chen et al., 2018a; Lee et al., 1993), such as differentiation (Karp and Ambros, 2005), cell development (Miska, 2005), and viral infection (Miska, 2005). Therefore, it is obvious that miRNAs have critical impact on the human diseases. However, disease-associated miRNA identifying methods based on biological experiments is costly and time-consuming. It is necessary to reveal novel types of disease-related miRNAs with computational methods.
In recent years, many computational methods have been developed for miRNA-disease association prediction (Xuan et al., 2015; You et al., 2017; Zeng et al., 2016; Zou et al., 2016). Jiang et al. (2010) proposed a novel computational method to predict latent miRNA-disease associations by integrating miRNA functional similarity data, phenotype similarity data, and experimentally validated disease-miRNA association to evaluate the probability that a miRNA may be included in a specific disease. However, the accuracy of this method was serious restricted by predicted miRNA-target interactions only with the information of miRNA neighbors. Shi et al. (2013) developed a random walk analysis method to rank miRNA-disease pairs by searching for functional associations between miRNAs targets and diseases genes in protein-protein interaction network. Chen et al. (2017) proposed a model named RKNNMDA (Ranking-based K-Nearest Neighbors for MiRNA–Disease Association prediction) to search the k-nearest neighbors of miRNAs and diseases. Zeng et al. (2018) developed a structural perturbation method on the bilayer network of the miRNA-disease to predict latent miRNA-disease associations. Liu et al. (2021) proposed a collaborative filtering method based on neural network to identify the miRNA-disease association. Experimental results showed that the proposed method could effectively prioritize the miRNA associated with disease and obtain the AUC value of 0.921. Zhang et al. (2019) introduced a meta-pathway method to select miRNAs for candidate diseases. The miRNA functional similarity network was reconstructed by family information, miRNA cluster information, experimental verified miRNA target association, and the information between disease and miRNA. The comprehensive data network and reasonable factors ensure the high performance of the method. Dong et al. (2019) proposed a method to predict potential miRNA-disease associations based on edge perturbation, a feature vector is designed to describe the structural Hamiltonian information of each edge of the graph, and the extracted features were used to train a multi-layer perception model to predict candidate disease-miRNA associations. The leave one cross validation and case analysis illustrated the effectiveness of the proposed method. Wang et al. (2019) developed an integrated framework for the identification of potential miRNA-disease based on a new negative sample extraction strategy. Qin et al. (2015) proposed a novel miRNA-disease association recognition method based on domains, the functional and structural blocks of proteins. Experimental results on real datasets demonstrated the high performance of the proposed method. Ding et al. (2018) predicted disease-related miRNAs in a multi-layer heterogeneous network by combining miRNA target gene information and heterogeneous flow. Li et al. (2018) proposed a new computational model to identify potential miRNA-disease association, the model uses Kronecker product or Kronecker and a larger miRNA-disease space by combining miRNA space and disease space. Li et al. (2017) proposed a similarity-based miRNA-disease prediction method that calculates similarities within a miRNA-disease association network. Experimental results and case studies validated the effectiveness of the proposed method. Chen et al. (2018b) proposed a global similarity method based on a two-tier random walk and designed a Laplacian score of graphs to calculate the global similarity of networks, which revealed the correlation between miRNAs and diseases. Experimental results reveal that this method is better than existing approaches in terms of overall prediction accuracy, a case study further showed this method is feasible. Chen et al. (2012) developed a Random Walk with Restart for MiRNA-Disease Association (RWRMDA) to predict latent miRNA-disease interactions by implanting random walk on the miRNA-miRNA functional similarity network. Differing from classical local network similarity measures, the global network similarity measures is introduced in this study. Chen et al. (2016), a novel computational model of WBSMDA is presented to discover disease-miRNA associations by taking advantage of within and between scores of each candidate disease-miRNA pair, which effectively integrated Gaussian interaction profile kernel similarity. The corresponding experiment demonstrated the effectiveness of this method.
However, majority of existing methods suffer from the following limitations. On the one hand, some disease-related miRNAs are obtained from the miRNA functional similarity networks consisting of heterogeneous data sources, i.e., disease similarity, protein interaction network, gene expression. On the other hand, little approaches infer disease-related miRNAs depending on the network topological features without the functional similarity of miRNAs. Notably, when the functional similarity network of miRNAs is constructed, it also has the characteristics of complex network (Cao et al., 2021; Luo and Xiao, 2017).
To tackle the above problems, inspired by cwMINE (Cao et al., 2016a), we proposed a novel computational method of Integrating Network Topology Similarity and MicroRNA Function Similarity for miRNA-disease association prediction (INTS-MFS). This model exploited not only the known miRNA functional similarity but also the network topological similarity of the miRNA functional similarity network. To evaluate the effectiveness of INTS-MFS, five-fold cross-validation was carried out the known miRNA-disease association data downloaded from HMDD V2.0 (Li et al., 2014). Furthermore, three diseases (Breast Neoplasm, Lung Neoplasm, Prostatic Neoplasm) of case studies were used to evaluate the prediction ability of implementing INTS-MFS on the data collected from HMDD V2.0. All the candidate miRNAs of these three diseases were ranked according to their prediction score, respectively. Then the top 30 predicted miRNAs of these three diseases were examined in dbDEMC (https://www.picb.ac.cn/dbDEMC/) (Yang et al., 2010), PhenomiR2.0 (Ruepp et al., 2010), and published literature. As a result, 30 out of top 30 predicted Prostatic Neoplasm-related miRNAs were included in the two databases of dbDEMC and PhenomiR2.0. 29 out of top 30 predicted Lung Neoplasm-related miRNAs were included in dbDEMC, PhenomiR2.0 and experimental reports. For Breast Neoplasm, 29 of top 30 predicted miRNAs were included in dbDEMC, PhenomiR2.0 and published literatures. Experimental results and case studies demonstrated the model of INTS-MFS with a reliable performance could be help for miRNA–disease association prediction. Fig. 1 shows the overall workflow of INTS-MFS method.
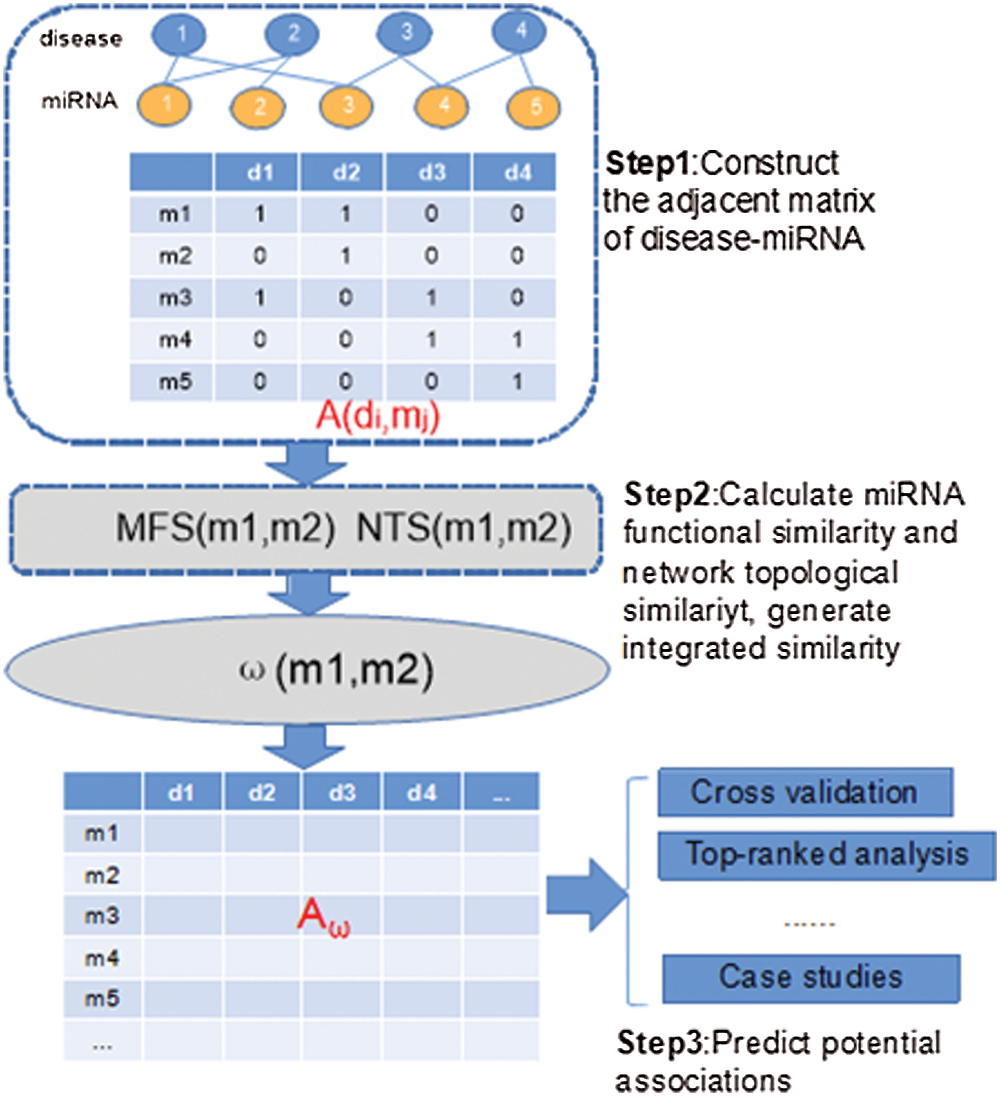
Figure 1: Overall flowchart of INTS-MFS for identifying latent miRNA–disease associations.
The model of INTS-MFS is based on the combination of functional similarity and network topological characteristics of miRNA functional similarity network. The miRNA-disease association identification method mainly consists of three steps: (1) data collection; (2) similarity calculation and (3) association identification. To validate the candidate miRNA associations, the two public databases, i.e., dbDEMC and PhenomiR2.0, are employed to evaluate the candidate predictions with case studies.
Human miRNA–disease associations
The disease-miRNA association dataset was downloaded from the HMDD v2.0 database, which includes 5424 distinct experimentally confirmed associations between 378 diseases and 495 miRNAs. It constructs the adjacency matrix A,
1) MiRNA functional similarity
The miRNA functional similarity was computed based on the following assumption (Cheng et al., 2012; Zhou et al., 2011): if a disease is associated with a miRNA, the other diseases similar to that disease will also possibly be related to the miRNA, and vice versa. In this study, The miRNA functional similarity data is downloaded from http://www.cuilab.cn/files/images/cuilab/misim.zip (Wang et al., 2010). According to Wang et al. (2010), the matrix MFS is constructed to represent the miRNA functional similarity. The element
where
2) Network topological similarity
Considering that the miRNA functional similarity network has the characteristics of complex network, in this study, we compute miRNA functional similarity by integrating miRNA functional similarity and network topological similarity, which is described as follows (Cao et al., 2016b):
where NTS (m1, m2) denotes the network topological similarity of miRNA pair (m1, m2). Nm1 denotes the neighbors of miRNA m1.
3) Association identification
To make full use of functional similarity and network topology similarity of miRNA similarity network, we construct the following formula:
where
The algorithm of INTS-MFS mainly consists of three steps. In the first step, the adjacent matrix of disease-miRNA is constructed through the disease-miRNA associations (Lines 1–8). In the second step, INTS-MFS calculates function similarity for each miRNA pair, which provides the basis for the calculation of network topological similarity (Lines 9–18). Finally, when the combined similarity is computed, INTS-MFS generates the miRNA-disease association matrix and predicts the potential miRNA-disease associations (Lines 19–26), these results will be evaluated with the five-fold cross-validation. To identify the latent association between miRNAs and diseases, the top-ranked results analysis is executed, the case studies of diseases are further demonstrated.

To systematically evaluate the prediction accuracy of our method, five-fold cross-validation was implemented on the basis of disease-miRNA associations downloaded from the HMDD database. For five-fold cross-validation, we randomly partition all known associations of each disease into five disjointed subsections, four of which are used as testing samples and the remaining one is used as a training sample through multiple iterations. To avoid data bias caused by random selection, five-fold cross-validation was repeated 5 times.
For those disease related to only a few miRNAs, it is insufficient to evaluate the performance of the identification method, 22 human diseases, which are associated with at least 60 miRNAs, are employed to test the capacity of the prediction approaches. Since the integrated miRNA similarity for diseases and miRNAs are calculated on the basis of known disease-miRNA associations, the integrated similarity should be recomputed for each iteration of the cross-validation experiments when the known associations change. The area under the ROC (Receiver Operating Characteristics, ROC) curve (AUC) was used to assess the quality of the predicted associations.
Comparison with different similarities
To compare the performance of the proposed method, we first investigated the effect of different similarity strategies imposed on INTS-MFS. In this paper, we compared two types of similarities, i.e., functional similarity, network topological similarity, which is widely applied in the literature. Table 1 shows the comparison results using INTS-MFS with different similarities.
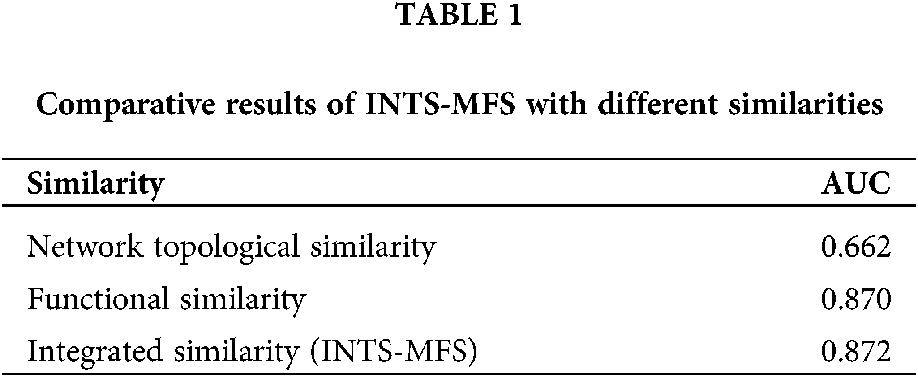
From Table 1, we can clearly see that the integrated similarity outperforms the other two types of similarities. It demonstrates that our method can effectively improve the performances of the identification of associations between miRNAs and diseases. It further shows the integrated similarity can compensate for the shortcomings of the network topological similarity or the functional similarity.
Prediction performance evaluation
To show the effectiveness of the INTS-MFS algorithm, we compared our method with five state-of-the-art algorithms, namely, DeepWalk (Li et al., 2017), RWRMDA (Chen et al., 2012), MIDP (Xuan et al., 2015b), WBSMDA (Chen et al., 2016), KMDR-KS (Li et al., 2018). Notably, RWRMDA and MIDP were applied to the old version of HMDD, we recalculated the similarity of diseases or miRNAs pair with the newest version of HMDD.
Table 2 describes the prediction results for INTS-MFS and the other five methods by the five-fold cross-validation. For AUC, the value of 1 represented a perfect prediction, while the value of 0.5 indicated a purely random performance. As shown in Table 2, the average AUC value of INTS-MFS, DeepWalk, RWRMDA, MIDP, WBSMDA, and KMDR-KS for all 22 diseases are 87.2%, 80.1%, 83.3%, 82.9%, 83.2%, respectively. INTS-MFS achieves the best performance for all the 22 diseases except squamous cell carcinoma and urinary bladder neoplasms and the AUC is 7.3% 8.2%, 4.4%, 4.5%, 4.7% higher than the other five approaches, respectively. Moreover, INTS-MFS outperformed the other five methods with the correctly identified disease–miRNA associations (Fig. 2). These prediction results demonstrate that our method obtains effective prediction performance, it results from the integrated miRNA similarity including miRNA function similarity and network topology similarity. Fig. 3 presents the ROC curves of each method using five-fold cross-validation. The X-axis of the ROC graph is TPR (the true positive rate) while the Y-axis is FPR (the false positive rate). It further demonstrates the effectiveness of our proposed method.
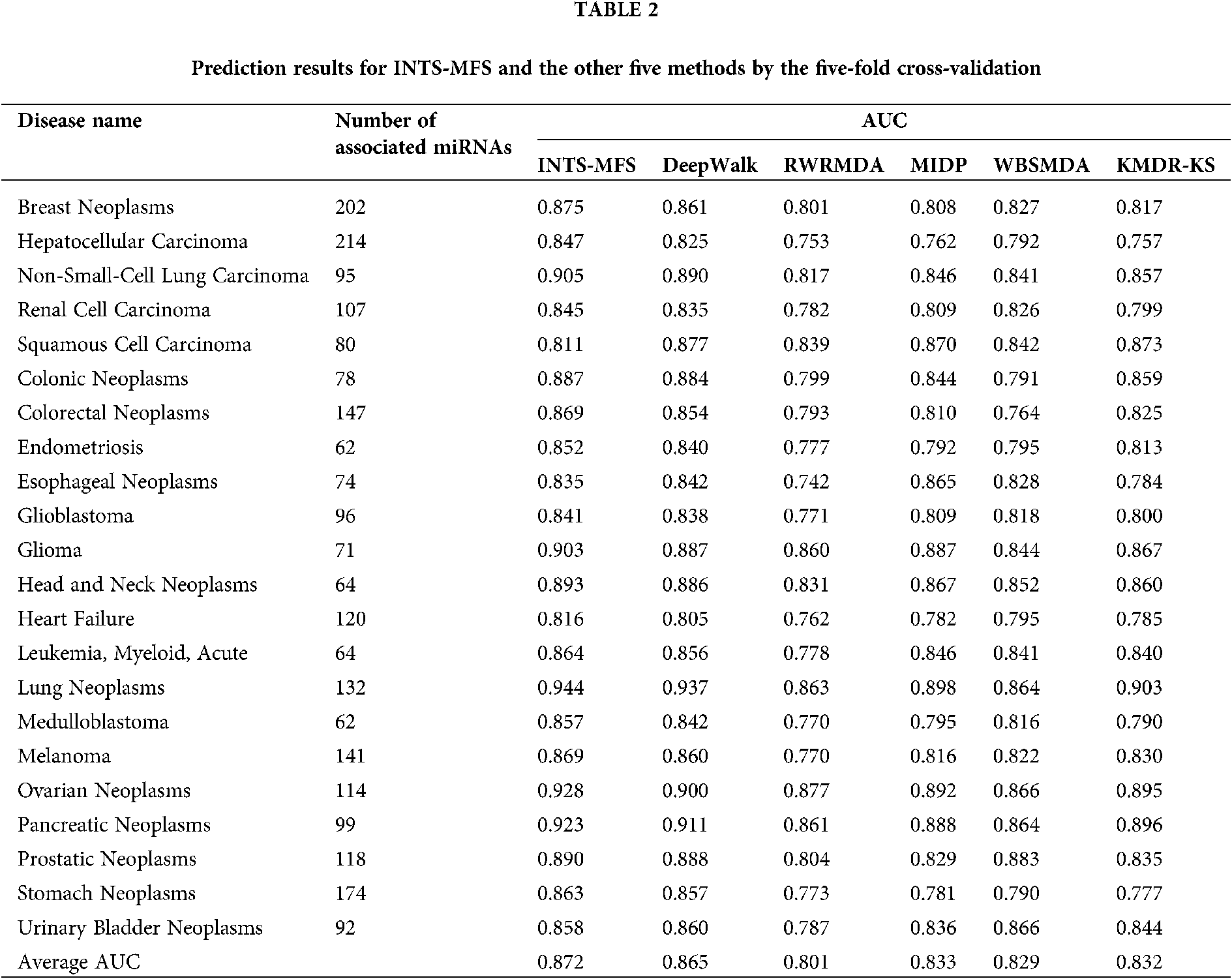
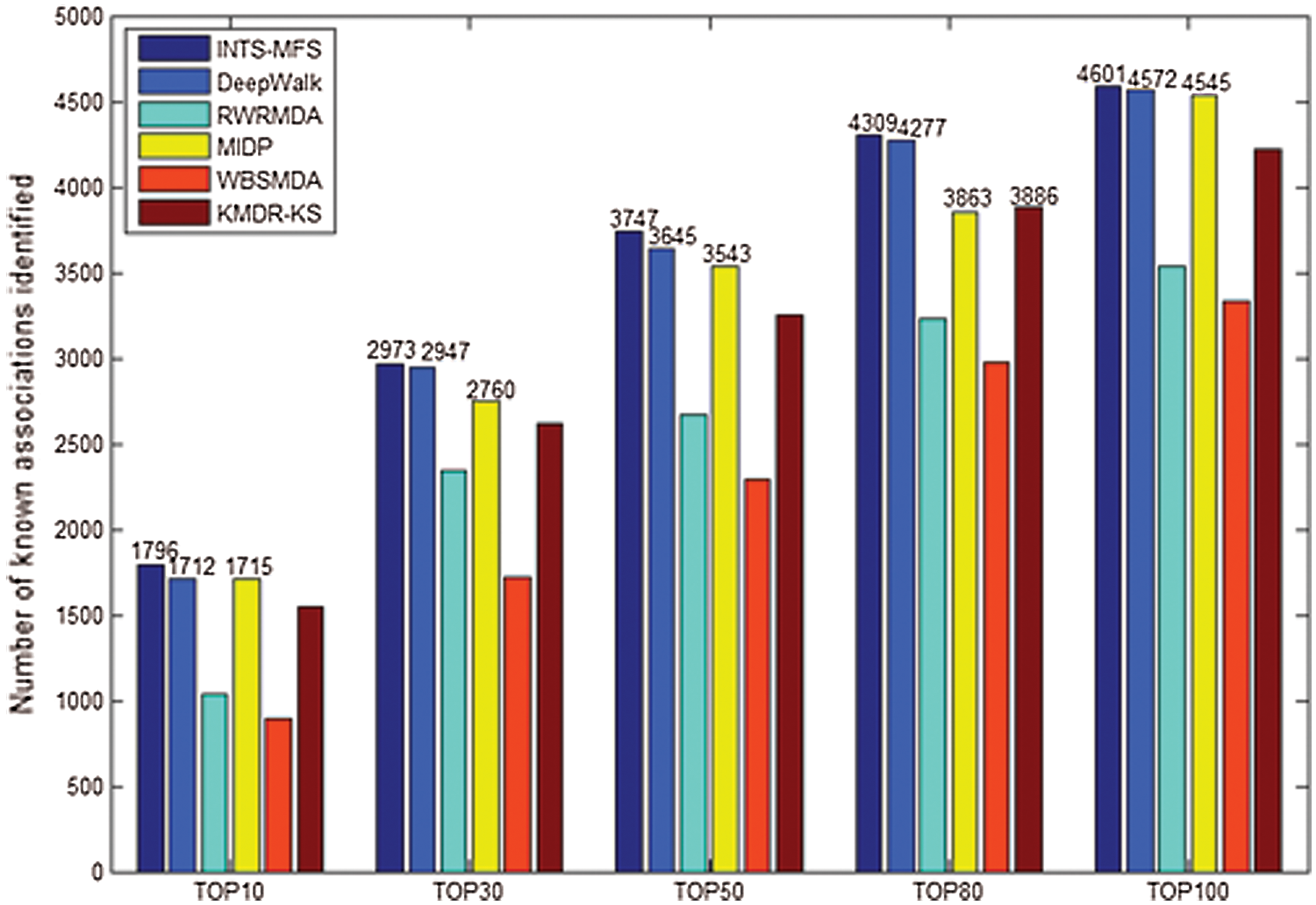
Figure 2: Number of correctly identified disease–miRNA associations by different methods.
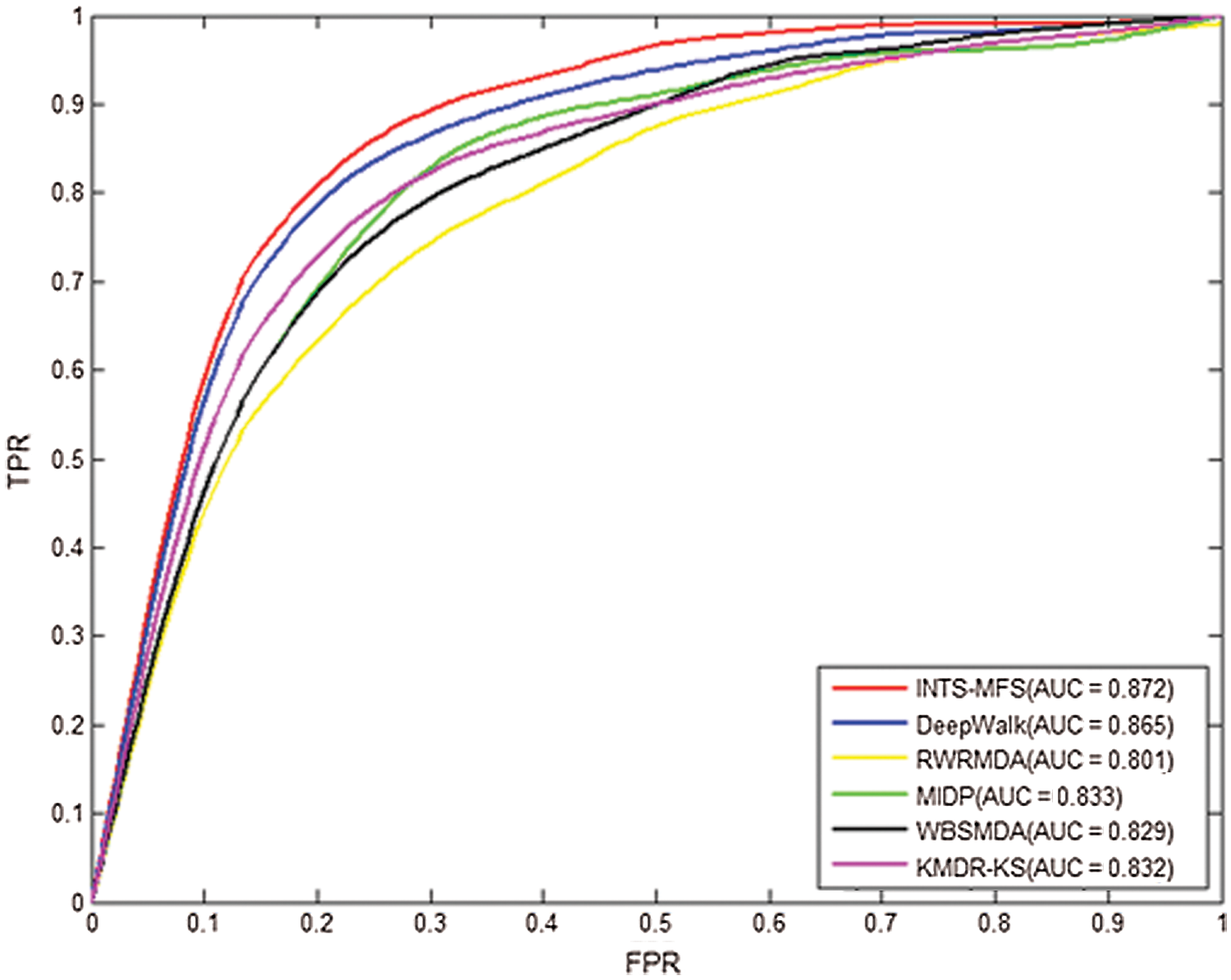
Figure 3: The ROC curves and average AUCs of INTS-MFS and other five methods for 22 diseases.
Generally, the top-ranked associations are more important for some disease. The number of correctly identified known disease-miRNA interactions under different top selections is shown in Fig. 2. In our study, among the 5425 known disease-miRNA associations, INTS-MFS correctly identified 3747 (69.08%) known associations in the top 50 predictions. The result shows the effectiveness of INTS-MFS in identifying confirmed disease-miRNA interactions.
To further validate the ability of INTS-MFS to mine new miRNA-disease associations, the case studies of several important diseases (Breast Neoplasm, Lung Neoplasm, Prostatic Neoplasm) are presented. All known associations included in the HMDD database are taken as the training set, and the miRNA non-related to each disease are ranked according to the similarity score of each miRNA. The corresponding results were validated based on two independent databases, namely, dbDEMC and PhenomiR2.0, and research literature.
The first case study is breast cancer, which causes women’s cancer deaths, especially in developed countries. Recently, more and more evidence has shown that many miRNAs are related to the cancers comprised of breast neoplasms. The American Cancer Society had presented that there were about 2.1 million newly diagnosed female breast cancer cases, which accounts for almost 1 in 4 cancer cases among women in 2018 (Bray et al., 2018). Identifying more miRNAs associated with breast cancer will help to assess clinical results accurately. As a result, our method directly showed 15 out of the top 15 (100%) associations predicted by our proposed method were supported by dbDEMC and PhenomiR2.0 database, 28 out of the top 30 potentially related miRNAs to be linked with beast neoplasms through dbDEMC (as shown in Table 3). And, some predicted miRNAs were verified by previously published literature, i.e., hsa-miR-378a. Notably, hsa-miR-371a (22nd in the prediction list) is not confirmed with all evidence proposed in this study. However, no association between hsa-miR-371a and breast cancer has been reported.
Lung cancer (lung neoplasms) has the poorest prognosis among cancers and is the largest threat to people’s health and life, it accounts for 18.4% of the total cancer deaths (Bray et al., 2018). As shown in Table 4, 15 out of the top 15 (100%) and 28 out of the top 30 latently associated miRNAs were validated by the two aforementioned databases. In addition, a candidate of hsa-mir-378a (24th in the prediction list) is supported by previously published literature. Especially, the association between hsa-miR-371a (22nd in the prediction list) and lung cancer is not validated.
Hsa-miR-371a is an important miRNA that forms a cluster with hsa-miR-371b, hsa-miR-372, hsa-miR-373 within 1.1 kb on chromosome 19 (http://www.mirbase.org/), the sequence shown that represents the most commonly cloned form from large-scale cloning study (Landgraf et al., 2007). Although hsa-miR-371a has been associated with 8, 12 diseases recorded in HMDD and HMDD V3.2, respectively, no association with breast cancer and lung cancer has been recorded so far. The association, between hsa-miR-371a and breast cancer and lung cancer, needs to be further explored by biologists.
The final case study in this study is prostatic cancer, which is the second major cause of male cancer to deaths in developed countries. We implemented our method to prioritize latent prostatic neoplasm associated miRNAs, the results show that 15 and 30 out of the top 15 and top 30 identified miRNAs were validated in dbDEMC and PhenomiR2.0, which is shown in Table 5.
In conclusion, experimental results of cross validation and case studies full illustrate that our proposed method achieves satisfaction prediction performance. Interestingly, we identified the miRNA of hsa-miR-371a, which is ranked top 22 and 20 in the breast and lung neoplasm, respectively, the association with breast and lung cancer is unconfirmed in existing research. According to dbDEMC, hsa-mir-371a is associated with other diseases, such as Azoospermia, Glioma, Lupus Nephritis, and so on. Therefore, we explored the role of hsa-miR-371a in breast cancer and lung cancer from the diagnosis of known diseases, in the hope that the associations between hsa-miR-371a and more diseases will be validated by future biological experiments.
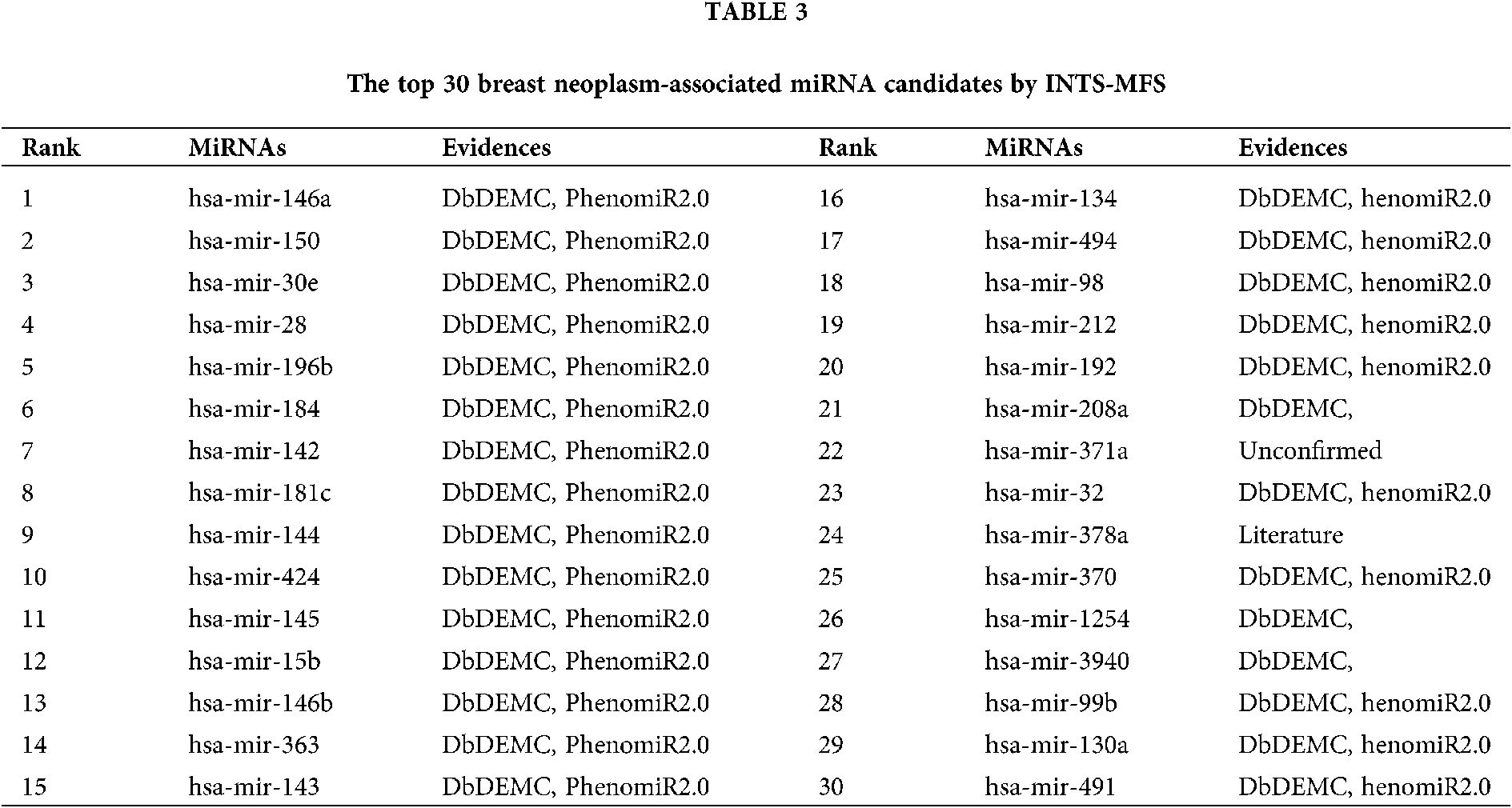
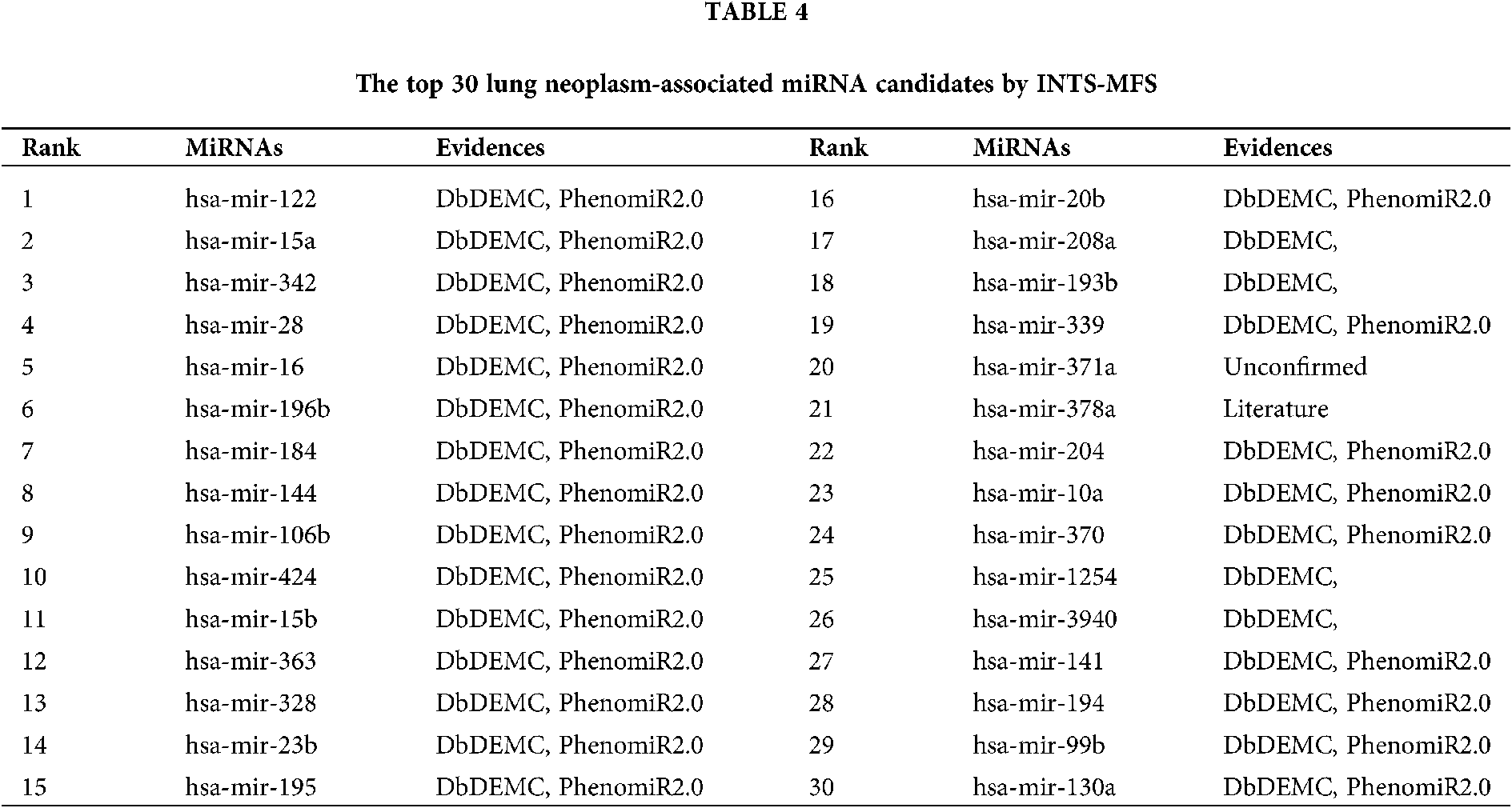
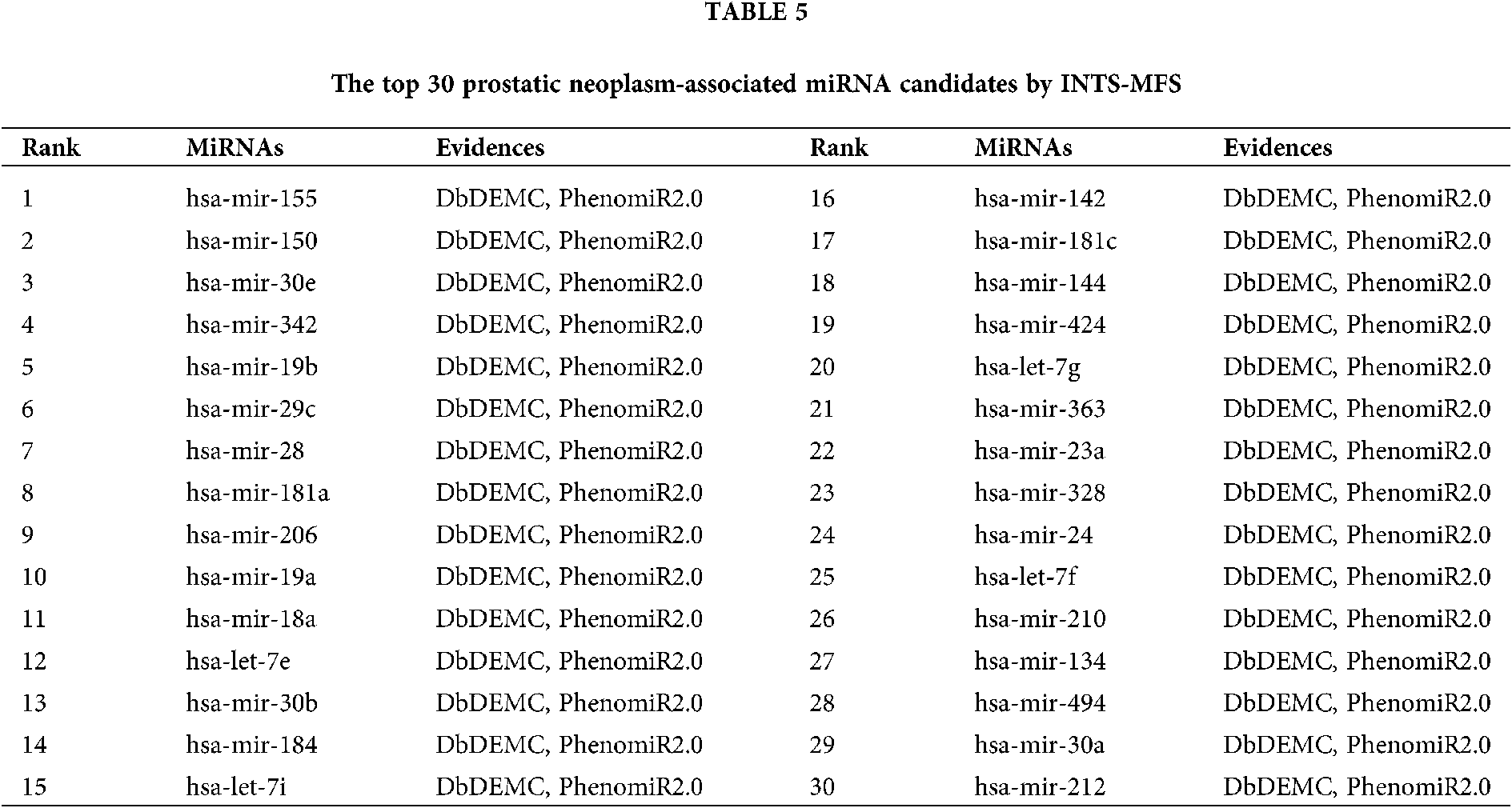
The identification of potential miRNA-disease associations would help us understand the pathogenesis of disease and promote the treatment of diseases. In this paper, we developed a model of Integrating Network Topology Similarity and MicroRNA Function Similarity (INTS-MFS) for identifying miRNA-disease association. In model of INTS-MFS, the integrated miRNA function similarity and network topology similarity were combined to calculate the prediction score of each miRNA–disease pair. The AUC of INTS-MFS is 0.872 based on five-fold cross-validation, which showed a better performance than previous methods. Furthermore, the predicted disease-related miRNAs of three major human diseases: breast neoplasm, lung neoplasm and prostatic neoplasm were respectively confirmed by the human disease databases and experimental reports.
Despite the successful exploitation of integrated similarity through application of INTS-MFS for miRNA-disease association prediction, there are also inevitable limitations that affect the performance of INTS-MFS, and in the hope that these shortcomings will to be improved in future research. First, the proposed method fails to predict associations between new diseases and miRNAs that do not exist within the similarity network, this is because our method is only executed by known miRNA-disease associations. Second, the material including miRNA functional similarity possibly contains noise and outlier, the prediction accuracy of associations is affected to some extent. Finally, the existing known miRNA-disease associations are insufficient. Therefore, a heterogeneous network integrating disease-gene, miRNA-gene associations and protein-protein interaction network can be used for the prediction of miRNA-disease association. It will further potentially improve prediction results.
Availability of Data and Materials: The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Author Contribution: Study conception and design: BC, JL. Data collection and process: BC, SX. Analysis and interpretation of results: KZ, SY. Draft manuscript preparation: BC, SX. All authors reviewed the results and approved the final version of the manuscript.
Funding Statement: This work was supported in part by the National Natural Science Foundation of China under Grants 61873089, 62032007 and the Key Project of the Education Department of Hunan Province under Grant 20A087, the Innovation Platform Open Fund Project of Hunan Provincial Education Department under Grant 20K025.
Conflicts of Interest: The authors declare that there is no conflict of interest regarding the publication of the paper.
Alvarez-Garcia I, Miska EA (2005). MicroRNA functions in animal development and human disease. Development 132: 4653–4662. [Google Scholar]
Bartel DP (2004). MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116: 281–297. [Google Scholar]
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians 68: 394–424. [Google Scholar]
Cao B, Deng S, Qin H, Luo J, Li G, Liang C (2021). Inferring microRNA-disease associations based on the identification of a functional module. Journal of Computational Biology 28: 33–42. [Google Scholar]
Cao B, Luo J, Liang C, Wang S (2016a). Identifying protein complexes by combining network topology and biological characteristics. Journal of Computational and Theoretical Nanoscience 13: 7666–7675. [Google Scholar]
Cao B, Luo J, Liang C, Wang S, Ding P (2016b). PCE-FR: A novel method for identifying overlapping protein complexes in weighted protein-protein interaction networks using pseudo-clique extension based on fuzzy relation. IEEE Transactions on Nanobioscience 15: 728–738. [Google Scholar]
Chen M, Liao B, Li Z (2018a). Global similarity method based on a two-tier random walk for the prediction of microRNA-disease association. Scientific Reports 8: 6481. [Google Scholar]
Chen X, Liu MX, Yan GY (2012). RWRMDA: Predicting novel human microRNA-disease associations. Molecular BioSystems 8: 2792–2798. [Google Scholar]
Chen X, Wang L, Qu J, Guan NN, Li JQ (2018b). Predicting miRNA-disease association based on inductive matrix completion. Bioinformatics 34: 4256–4265. [Google Scholar]
Chen X, Wu QF, Yan GY (2017). RKNNMDA: Ranking-based KNN for MiRNA-disease association prediction. RNA Biology 14: 952–962. [Google Scholar]
Chen X, Yan CC, Zhang X, You ZH, Deng L et al. (2016). WBSMDA: Within and between score for MiRNA-disease association prediction. Scientific Reports 6: 21106. [Google Scholar]
Cheng F, Liu C, Jiang J, Lu W, Li W et al. (2012). Prediction of drug-target interactions and drug repositioning via network-based inference. PLoS Computational Biology 8: e1002503. [Google Scholar]
Ding P, Luo J, Liang C, Xiao Q, Cao B (2018). Human disease MiRNA inference by combining target information based on heterogeneous manifolds. Journal of Biomedical Informatics 80: 26–36. [Google Scholar]
Dong Y, Sun Y, Qin C, Zhu W (2019). EPMDA: Edge perturbation based method for miRNA-disease association prediction. IEEE/ACM Transactions on Computational Biology and Bioinformatics 17: 2170–2175. [Google Scholar]
Jiang Q, Hao Y, Wang G, Juan L, Zhang T et al. (2010). Prioritization of disease microRNAs through a human phenome-microRNAome network. BMC Systems Biology 4: S2. [Google Scholar]
Karp X, Ambros V (2005). Encountering microRNAs in cell fate signaling. Science 310: 1288–1289. [Google Scholar]
Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N et al. (2007). A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129: 1401–1414. [Google Scholar]
Lee RC, Feinbaum RL, Ambros V (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75: 843–854. [Google Scholar]
Li GH, Luo JW, Xiao Q Liang C, Ding P, Cao B (2017). Predicting microRNA-disease associations using network topological similarity based on deepwalk. IEEE Access 5: 24032–24039. [Google Scholar]
Li G, Luo J, Xiao Q, Liang C, Ding P (2018). Prediction of microRNA-disease associations with a Kronecker kernel matrix dimension reduction model. RSC Advances 8: 4377–4385. [Google Scholar]
Li Y, Qiu C, Tu J, Geng B, Yang J et al. (2014). HMDD v2. 0: A database for experimentally supported human microRNA and disease associations. Nucleic Acids Research 42: D1070–D1074. [Google Scholar]
Liu Y, Wang SL, Zhang JF, Zhang W, Li W (2021). A neural collaborative filtering method for identifying miRNA-disease associations. Neurocomputing 422: 176–185. [Google Scholar]
Luo J, Xiao Q (2017). A novel approach for predicting microRNA-disease associations by unbalanced bi-random walk on heterogeneous network. Journal of Biomedical Informatics 66: 194–203. [Google Scholar]
Lynam-Lennon N, Maher SG, Reynolds JV (2009). The roles of microRNA in cancer and apoptosis. Biological Reviews 84: 55–71. [Google Scholar]
Miska EA (2005). How microRNAs control cell division, differentiation and death. Current Opinion in Genetics & Development 15: 563–568. [Google Scholar]
Qin GM, Li RY, Zhao XM (2015). Inferring the miRNA-disease associations based on domain-disease associations. IFAC-PapersOnLine 48: 7–11. [Google Scholar]
Ruepp A, Kowarsch A, Schmidl D, Buggenthin F, Brauner B et al. (2010). PhenomiR: A knowledgebase for microRNA expression in diseases and biological processes. Genome Biology 11: R6. [Google Scholar]
Shi H, Xu J, Zhang G, Xu L, Li C et al. (2013). Walking the interactome to identify human miRNA-disease associations through the functional link between miRNA targets and disease genes. BMC Systems Biology 7: 101. [Google Scholar]
Wang CC, Chen X, Yin J, Qu J (2019). An integrated framework for the identification of potential miRNA-disease association based on novel negative samples extraction strategy. RNA Biology 16: 257–269. [Google Scholar]
Wang D, Wang J, Lu M, Song F, Cui Q (2010). Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 26: 1644–1650. [Google Scholar]
Wu Q, Zhao J, Zheng Y, Xie X, He Q et al. (2021). Associations between common genetic variants in microRNAs and Hirschsprung disease susceptibility in Southern Chinese children. Journal of Gene Medicine 23: e3301. [Google Scholar]
Xuan P, Han K, Guo Y, Li J, Li X et al. (2015). Prediction of potential disease-associated microRNAs based on random walk. Bioinformatics 31: 1805–1815. [Google Scholar]
Yang Z, Ren F, Liu C, He S, Sun G et al. (2010). dbDEMC: A database of differentially expressed miRNAs in human cancers. BMC Genomics 11: S5. [Google Scholar]
You ZH, Huang ZA, Zhu Z, Yan GY, Li ZW et al. (2017). PBMDA: A novel and effective path-based computational model for miRNA-disease association prediction. PLoS Computational Biology 13: e1005455. [Google Scholar]
Zeng X, Liu L, Lü L, Zou Q (2018). Prediction of potential disease-associated microRNAs using structural perturbation method. Bioinformatics 34: 2425–2432. [Google Scholar]
Zeng XX, Zhang X, Zou Q (2016). Integrative approaches for predicting microRNA function and prioritizing disease-related microRNA using biological interaction networks. Briefings in Bioinformatics 17: 193–203. [Google Scholar]
Zhang X, Zou Q, Rodriguez-Paton A, Zeng X (2019). Meta-path methods for prioritizing candidate disease miRNAs. IEEE/ACM Transactions on Computational Biology and Bioinformatics 16: 283–291. [Google Scholar]
Zhou T, Ren J, Medo M, Zhang YC (2011). Bipartite network projection and personal recommendation. 2011 International Conference on Applied Social Science, vol. III. [Google Scholar]
Zou Q, Li J, Song L, Zeng X, Wang G (2016). Similarity computation strategies in the microRNA-disease network: A survey. Briefings in Functional Genomics 15: 55–64. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |